

Abstract
Background
Superficial epidermolytic ichthyosis (SEI), known as ichthyosis bullosa of Siemens (IBS; OMIM No. 146800) before, is a type of keratinopathic ichthyosis due to the KRT2 mutations (NM_000423.3; OMIM No. 600194). Here, we report the first case of SEI caused by a KRT2 mosaic mutation.
Methods
We presented the clinical data of a 5‐year‐old Chinese boy who suffered from SEI. The histopathological examination and immunofluorescence were performed to rule out immunobullous skin diseases and diseases with subepidermal blisters. Genomic DNA samples were extracted from the lesion tissue and next‐generation sequencing was performed. We also confirmed the variant allele frequency (VAF) in different tissues by an Ultra‐Deep Sequencing technology.
Results
The patient presented with blisters on the lower extremities and linear, superficially hyperkeratotic lesions. Immunofluorescence of IgG, IgA, IgM, C3, C4, and C1q were negative, and the histopathological results showed intraepidermal blisters containing lymphocytes and eosinophils. A heterozygous missense mutation, c.G1459A (p. Glu487Lys), in exon 7 of the KRT2 gene was detected at a 31.17% allele frequency. The same mutation p. Glu487Lys has been described several times in the literature.
Conclusion
Thus, in our patient, the mosaic mutation explains the blaschkoid ichthyosiform phenotype. To our knowledge, this is the first case of SEI with a KRT2 mosaic mutation.
Keywords: KRT2, somatic mosaicism, superficial epidermolytic ichthyosis, Ultra‐Deep Sequence
We reported the first case of a mosaic SEI caused by a somatic p.E487 K mutation of KRT2 with a severe phenotype. We also confirmed the variant allele frequency (VAF) in different tissues by an Ultra‐Deep Sequencing technology.
1. INTRODUCTION
Superficial epidermolytic ichthyosis (SEI), known as ichthyosis bullosa of Siemens (IBS; OMIM No. 146800) before, is a type of keratinopathic ichthyosis due to the KRT2 mutations (NM_000423.3). KRT2 encodes keratin 2, which constitutes the cytoskeleton and plays an important role in normal tissue structure and function (Hotz et al., 2016). Distinct pairs of type I and type II keratins heterodimerize to form keratin intermediate filaments that increase the mechanical resilience of epithelial cells. Keratins are formed in a strict differentiation stage‐dependent manner by keratinocytes. Expression of keratin 2 is restricted to the uppermost suprabasal layers of the epidermis where it colocalizes with keratin 10 (Fischer et al., 2014). The phenotype is often mild because the loss of function of keratin 2 can be partially compensated for by the up‐regulation of keratin 1 expression.
Clinical features of SEI include mild epidermal hyperkeratosis over the flexural areas, blisters at birth, and the development of superficial peeling of hyperkeratotic skin. Generally, lesions such as erythroderma regresses almost completely with age. Keratotic lichenification and focal areas of denudation may concentrate on the joint areas of arms and legs, showing the typical Mauserung phenomenon (Takeichi & Akiyama, 2016). Herein, we report a case of a mosaic SEI caused by a somatic p.E487 K mutation of KRT2 with a severe phenotype.
2. MATERIALS AND METHODS
2.1. Ethical compliance
The patient's parents all signed informed consent before the study. This study was approved by the ethics committees of Shanghai Jiaotong University School of Medicine and conducted in accordance with the principles of the Declaration of Helsinki.
2.2. atients
This study concerned the clinical and molecular study of an SEI patient presenting with blisters on the lower extremities and linear, superficially hyperkeratotic lesions (Figure 1).
FIGURE 1.
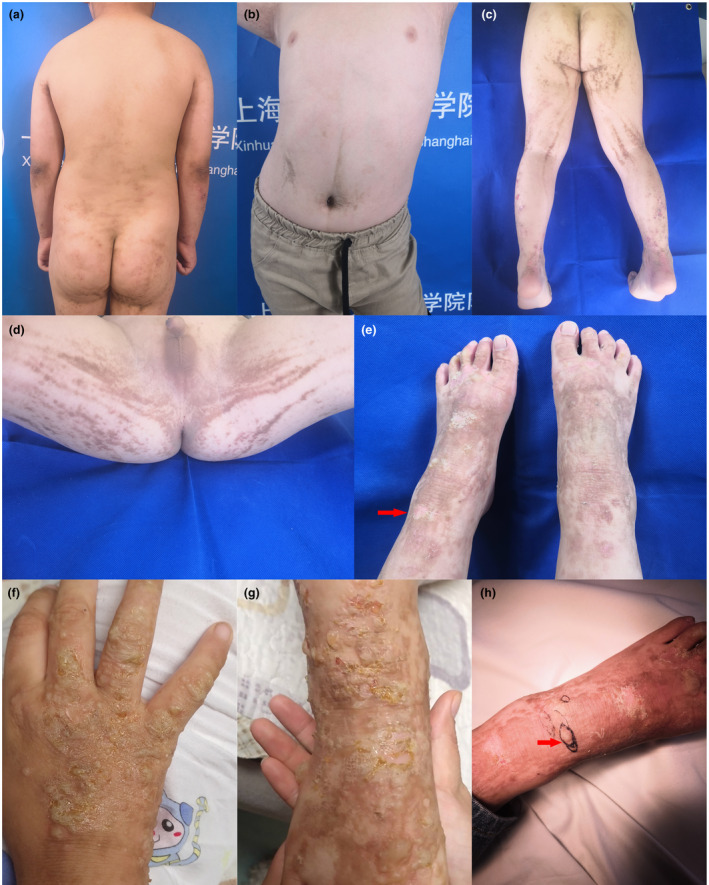
(a,b) A small number of linear hyperkeratotic lesions on the trunk. (c,d) Greyish‐brown linear hyperkeratotic lesions on the buttocks. (e) Hyperkeratotic plaques and focal areas of peeling (Mauserung phenomenon) on the left foot. (f,g) Pustules and blisters on the upper and lower extremities. (h) Lesion tissue from which DNA was extracted.
2.3. Next‐generation sequencing
To determine the lesion's genetic cause, genomic DNA samples were extracted from the lesion tissue after informed consent. We performed a next‐generation sequencing‐based multigene panel including 569 genetic loci of genodermatoses. Genomic DNA samples were extracted from the lesion tissue with standard methods (Qiagen Blood DNA Kit, Germany) and sent to Anbailong Biotech (China) for sequencing. The targeted exon was enriched and then sequenced on an Illumina NovaSeq platform (Illumina, USA) according to the manufacturer's instructions. We removed reads with mapping quality below 20 and potential duplicates. An average of 1 GB of mappable sequences was obtained with a mean coverage of 250×; 99% of the targeted bases were covered sufficiently for variant calling (>10×).
2.4. Ultra‐Deep sequencing
To confirm the variant allele frequency (VAF) in different tissues, an Ultra‐Deep Sequencing was performed (NovaSeq, Illumina, USA). DNA was isolated from the patient's blood, oral mucosa, hair follicles, normal skin, and his parents’ blood using standard methods. Amplicon covering the disease‐causing mutation c.G1459A/p.Glu487Lys (NM_000423.3) in the KRT2 gene were designed using Ion AmpliSeq Designer: forward PCR primer, AACAAGTTGAATGACCTGGA; reverse PCR primer, TGTCA GTTCTCAGTYGTCAG; primer binding sequences for the second round of PCR were added to 5'‐end: forward PCR primer, ACACGACGCTCTTCCGATCTAACAA GTTGAATGACCTGGA; reverse PCR primer, TTCCTTGGCACCCGAGAATTCCA TGTCAGTTCTCAGTYG TCAG.The library was loaded onto the Illumina NovaSeq for initiation of cluster generation and a pair‐end 150 cycle sequencing protocol. The sequencing depth varied from 4108 to 182334 reads.
2.5. Sanger sequencing
Sanger sequencing was carried out for the tissue sample in order to see whether the variant was detectable by this method (ABI PRISM®3730). By comparing with the hg38 human genome reference, repeated sequences and identified genetic variants were excluded.
3. RESULTS
3.1. Clinical data
A 5‐year‐old Chinese boy was referred to our outpatient department with blisters and linear, superficially hyperkeratotic lesions following the lines of Blaschko in the lower extremities. The lesions were first noticed 40 days after birth. At the onset, lesions presented as symmetric, erythematous, hyperkeratotic plaques following the lines of Blaschko, and were situated mainly over the lower extremities and buttocks. A few lesions were distributed sporadically on the trunk. The lesions later changed into greyish‐brown, linear, hyperkeratotic plaques, and focal areas of peeling (Mauserung phenomenon) could be observed. No treatment had been provided. There was no definite history of blistering until 4 months prior. At that time, pea‐sized pustules and blisters appeared continuously on the upper and lower extremities, with intense pruritus (Figure 1). Topical glucocorticoids were ineffective, while oral glucocorticoids had an immediate effect. However, the lesions would relapse after withdrawal of the treatment. Blisters could not be observed after mechanical trauma on normal skin. Neither of his parents had a similar phenotype. The patient has no siblings, and his family history is unremarkable. A biopsy was performed to rule out other skin diseases. The histopathological results showed hyperkeratosis, papillomatous epidermal hyperplasia, intra‐ and intercellular edema in the upper epidermis, and intraepidermal blisters containing lymphocytes and eosinophils, thus ruling out diseases with subepidermal blisters, such as bullous pemphigoid and linear IgA bullous dermatosis (Figure 2). Immunofluorescence of IgG, IgA, IgM, C3, C4, and C1q were negative, ruling out immunobullous skin diseases such as pemphigus.
FIGURE 2.
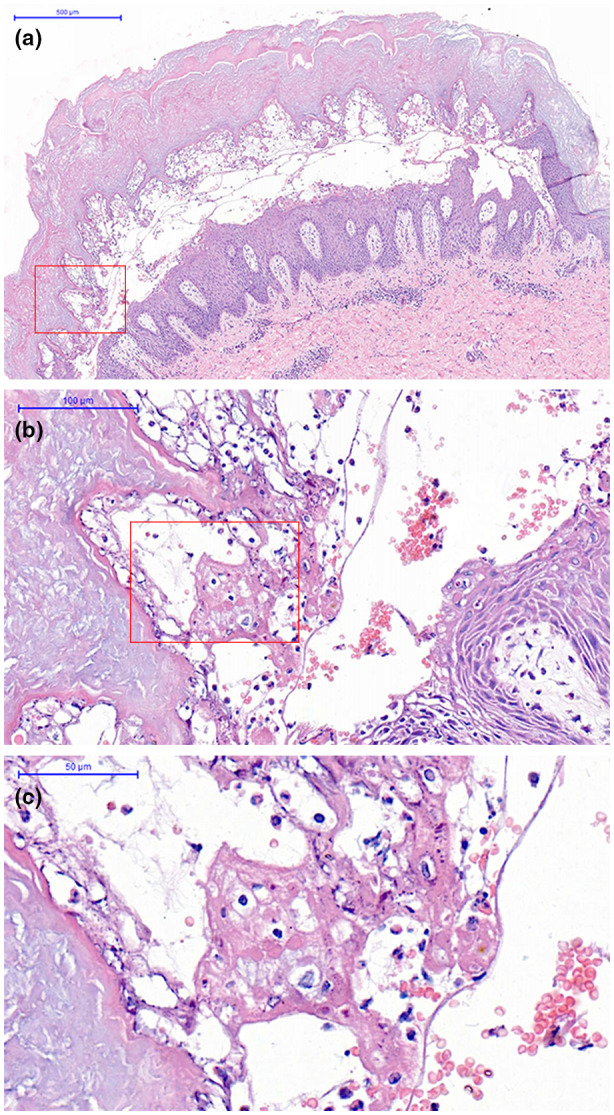
(a) Intraepidermal blisters containing lymphocytes and eosinophils (original magnification, ×50). (b,c) the intercellular separation (original magnification, ×250, ×500).
3.2. Gene sequencing
NGS reveals a heterozygous missense mutation, c.G1459A, in exon 7 of the KRT2 gene. This G to A transition resulted in the change in glutamate to lysine at amino acid position 487 (p. Glu487Lys). The same mutation p. Glu487Lys has been described several times in the literature. The transition appears to be a hot point mutation of KRT2, which is likely due to the deamination of 5‐methyl cytosine to thymine at a CpGdi nucleotide in exon 7 (Whittock, Ashton, Griffiths, Eady, & McGrath, 2001). This missense mutation was confirmed by Sanger sequencing (Figure 3b). The resulting VAF values are shown in Figure 3. The proband displayed a VAF of 12.94% to 31.17%, with the highest VAF found in lesion tissues and the lowest in the patient's blood. The VAF of his parents’ blood was approximately that of the healthy controls (Figure 3a). The differences in mutation frequencies suggest that the KRT2 mutation is somatic, leading to postzygotic mosaicism in the affected skin, which explains the blaschkoid ichthyosiform erythrodermic phenotype. About 16% variant allele was detected in the normal skin. There is a possibility that the proportion of mutant cells in normal skin does not reach a pathogenic threshold, so the typical SEI phenotype did not appear.
FIGURE 3.
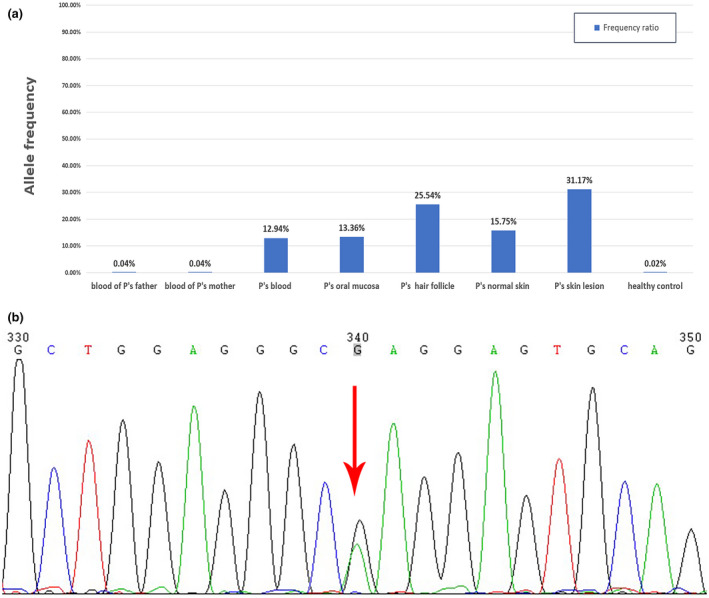
(a) The variant allele frequencies in different samples. (b) Direct sequencing of the probands showing a heterozygous mutation of c.G1459A in exon 7 of the KRT2 gene.
4. DISCUSSION
To our knowledge, the mosaic SEI caused by a somatic mutation has not before been reported. The term mosaic is used in medicine to define organisms composed of at least two different genetic populations originating from a homogeneous zygote (Prieto‐Barrios et al., 2018). Mosaicism manifests in cutaneous disease via two primary mechanisms. Type 1 segmental mosaicism results from a dominant, heterozygous, early postzygotic mutation in an otherwise healthy embryo, and manifests as a stripe or patch of affected skin. Type 2 segmental mosaicism originates in a heterozygous embryo and reflects the early postzygotic loss of the corresponding wild‐type allele, generating areas of more severely affected skin (Happle., 2016). In our case, the patient belongs to type 1 segmental mosaicism.
The mosaic phenotype would depend on several factors, the type of mutation, the moment during embryogenesis at which the mutation occurs, the differentiation potential of the mutated progenitor cell, and the distribution of the mutated cells (Prieto‐Barrios et al., 2018). Generally, the phenotype will not be inherited unless germ cells are affected. Therefore, the DNA sequence of sperm should be determined for the patient before he marries, avoiding germline transmission in his offspring. Genetic counseling and prenatal diagnosis are necessary.
As mentioned above, SEI is characterized by mild clinical manifestations. Symptoms usually improve with age and the phenotype should be milder as a mosaic SEI. However, this patient's symptoms are severe. The continuous pustules and blisters will not subside unless oral glucocorticoids are applied. We reviewed the literature to determine the correlation between genotype and phenotype.
As with all intermediate filament proteins, keratins were composed of four α‐helical regions (1A, 1B, 2A and 2B), each possessing a repeating heptad amino acid motif (a–b–c–d–e–f–g)n (Whittock et al., 2001). The p. Glu487Lys is located at the end of the 2B domain of keratin 2, which represents the highly conserved helix termination peptide (Rothnagel et al., 1994). This mutation was located in position d as well. Therefore, the nonconservative glutamic acid‐to‐lysine substitution may lead to a severe alteration in forming the heterodimer in the region of the tail domain of the rod (Suga et al., 2000). In addition, the substitution changes the charge of the side chain and presumably affects the function of the 2B domain, resulting in the instability of filament. Early studies of epidermolytic ichthyosis cases have suggested that substitutions in the H1 domain of keratin 1 bring about a less‐severe clinical presentation than those located at the beginning of the 1A or at the end of the 2B rod domains (Nishizawa et al., 2007). Mutations in the KRT1 gene affecting positions a and d were associated with a severe phenotype (Akasaka et al., 2015). Nevertheless, there are no previous reports of SEI that demonstrate variety in clinical phenotype associated with the heptad amino acid position. A severe phenotype caused by a germline mutation at this position has been reported (Shimada et al., 2018). The phenotype and histology of a patient's biopsy in Shimada et al. show a lot of similarities compared to the patient presented in this manuscript. But our patient's lesions are distributed along the Blaschko lines while theirs did not show. A postzygotic mutation may happen at the early time during embryogenesis if they were mosaicism.
We hypothesize that mutations affecting position d are associated with a severe phenotype. Nonetheless, phenotypic heterogeneity has been reported in patients with the same p. Glu487Lys mutation (formerly p. Glu493Lys; Whittock et al., 2001). Further functional studies on the p. Glu487Lys mutant may be necessary to understand the correlation between genotype and phenotype in SEI patients.
CONFLICTS OF INTEREST
All authors state that no financial and personal relationships with other people or organizations that could inappropriately influence (bias) their work.
AUTHORS CONTRIBUTION
Li Ming and Yao Zhirong participated in the overall design and revising the manuscript. Li Yue and Cheng Ruhong investigated the family history and collected the clinical data of the patient. Cheng Ruhong participated in the experimental data analysis. Li Yue and Liang Jianying participated in the experimental operation and drafting the manuscript. Li Ming and Yao Zhirong are corresponding authors of this manuscript. All authors read and approved the final manuscript.
ADDITIONAL CONTRIBUTIONS
We thank the patient's parents for granting permission to publish this information.
ACKNOWLEDGEMENTS
We thank all subjects for their ongoing participation in this study. This work was supported by a grant from the Biomedical Engineering Cross Research Foundation of Shanghai Jiaotong University (YG2017MS73), a grant from the Shanghai Pujiang Talent Program (18PJ1407300) and a grant from Shanghai Health System Excellent Academic Leader Training Project (2018BR22).
Li Y, Cheng R, Liang J, Yao Z, Li M. The first case of a mosaic superficial epidermolytic ichthyosis diagnosed by Ultra‐Deep Sequence. Mol Genet Genomic Med. 2020;8:e1457 10.1002/mgg3.1457
Yue Li and Ruhong Cheng contributed equally to this paper.
Contributor Information
Zhirong Yao, Email: zryaosmu@sohu.com.
Ming Li, Email: liming01@xinhuamed.com.cn.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
REFERENCES
- Akasaka, E. , Minakawa, S. , Rokunohe, D. , Toyomaki, Y. , Matsuzaki, Y. , Sawamura, D. , & Nakano, H. (2015). Superficial epidermolytic ichthyosis caused by a novel KRT2 mutation. Journal of Dermatological Science, 79(1), 86–88. 10.1016/j.jdermsci.2015.04.006 [DOI] [PubMed] [Google Scholar]
- Fischer, H. , Langbein, L. , Reichelt, J. , Praetzel‐Wunder, S. , Buchberger, M. , Ghannadan, M. , … Eckhart, L. (2014). Loss of keratin K2 expression causes aberrant aggregation of K10, hyperkeratosis, and inflammation. The Journal of Investigative Dermatology, 134(10), 2579–2588. 10.1038/jid.2014.197 [DOI] [PubMed] [Google Scholar]
- Happle, R. (2016). The categories of cutaneous mosaicism: A proposed classification. American Journal of Medical Genetics. Part A, 170A(2), 452–459. 10.1002/ajmg.a.37439 [DOI] [PubMed] [Google Scholar]
- Hotz, A. , Oji, V. , Bourrat, E. , Jonca, N. , Mazereeuw‐Hautier, J. , Betz, R. C. , … Fischer, J. (2016). Expanding the Clinical and Genetic Spectrum of KRT1, KRT2 and KRT10 Mutations in Keratinopathic Ichthyosis. Acta dermato‐venereologica, 96(4), 473–478. 10.2340/00015555-2299 [DOI] [PubMed] [Google Scholar]
- Nishizawa, A. , Toyomaki, Y. , Nakano, A. , Takeuchi, S. , Matsuzaki, Y. , Takeda, H. , … Nakano, H. (2007). A novel H1 domain mutation in the keratin 2 gene in a Japanese family with ichthyosis bullosa of Siemens. The British Journal of Dermatology, 156(5), 1042–1044. 10.1111/j.1365-2133.2007.07832.x [DOI] [PubMed] [Google Scholar]
- Prieto‐Barrios, M. , Llamas‐Martin, R. , Velasco‐Tamariz, V. , Calleja‐Algarra, A. , Ruano, Y. , Ortiz‐Romero, P. L. , & Rodriguez‐Peralto, J. L. (2018). Phacomatosis pigmentokeratotica: a case of HRAS mosaicism causing rhabdomyosarcoma. The British Journal of Dermatology, 179(5), 1163–1167. 10.1111/bjd.16435 [DOI] [PubMed] [Google Scholar]
- Rothnagel, J. A. , Traupe, H. , Wojcik, S. , Huber, M. , Hohl, D. , Pittelkow, M. R. , … Roop, D. R. (1994). Mutations in the rod domain of keratin 2e in patients with ichthyosis bullosa of Siemens. Nature Genetics, 7(4), 485–490. 10.1038/ng0894-485 [DOI] [PubMed] [Google Scholar]
- Shimada, H. , Takeo, N. , Saito‐Shono, T. , Ishikawa, K. , Sakai, T. , Goto, M. , … Kitajima, Y. (2018). Superficial epidermolytic ichthyosis concomitant with atopic dermatitis. European Journal of Dermatology : EJD, 28(1), 94–96. 10.1684/ejd.2017.3158 [DOI] [PubMed] [Google Scholar]
- Suga, Y. , Arin, M. J. , Scott, G. , Goldsmith, L. A. , Magro, C. M. , Baden, L. A. , … Roop, D. R. (2000). Hot spot mutations in keratin 2e suggest a correlation between genotype and phenotype in patients with ichthyosis bullosa of Siemens. Experimental Dermatology, 9(1), 11–15. 10.1034/j.1600-0625.2000.009001011.x [DOI] [PubMed] [Google Scholar]
- Takeichi, T. , & Akiyama, M. (2016). Inherited ichthyosis: Non‐syndromic forms. The Journal of Dermatology, 43(3), 242–251. 10.1111/1346-8138.13243 [DOI] [PubMed] [Google Scholar]
- Whittock, N. V. , Ashton, G. H. , Griffiths, W. A. , Eady, R. A. , & McGrath, J. A. (2001). New mutations in keratin 1 that cause bullous congenital ichthyosiform erythroderma and keratin 2e that cause ichthyosis bullosa of Siemens. The British Journal of Dermatology, 145(2), 330–335. 10.1046/j.1365-2133.2001.04327.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding authors upon reasonable request.