

Abstract
Background
Disorders of sex development (DSD) can result from congenital defect in sex determining pathway. Mitogen‐activated protein kinase kinase kinase 1 (MAP3K1) is one of the commonest genes that has been identified to cause 46, XY DSD. It can present as complete or partial gonadal dysgenesis even within the same kindred. Few mutations in this gene have previously been identified in a high proportion of individuals with 46, XY gonadal dysgenesis.
Methods and Results
We report three siblings with same novel variant in MAP3K1 gene presenting with variable degrees of partial gonadal dysgenesis. Clinical and genetic assessments were performed for the three siblings, while endocrine evaluation was done for two of them. The identified mutation (p.Thr657Arg) was previously classified as a pathogenic variant, although apparently there are no reported humans with this mutation.
Conclusion
This report adds to the genotype‐phenotype correlation, highlighting the clinical importance of considering MAP3K1 gene defects as part of the differential diagnosis for complete or partial gonadal dysgenesis especially with multiple affected family members.
Short abstract
We describe in detail the clinical phenotypes of three affected siblings, with same novel variant in MAP3K1 gene presenting with variable degrees of partial gonadal dysgenesis. In this report, we declare the identification of a new disease causing missense variant in MAP3K1 gene which was not apparently described in humans before. Our report adds to the genotype‐phenotype correlation, highlighting the clinical importance of considering MAP3K1 gene defects as part of the differential diagnosis for complete or partial gonadal dysgenesis especially with multiple affected family members.
1. INTRODUCTION
Disorders of sex development (DSD) are congenital conditions in which development of chromosomal, gonadal, or anatomic sex is atypical (Granados et al., 2017). There are no clear estimates of the incidence rate of subjects presenting with ambiguous genitalia at birth, however, it has been estimated to be approximately 1 in 4500–5500 birth. The incidence rate among subjects with 46, XY to have a DSD has been estimated to be 1 in 20,000 births (Lee et al., 2016). The process of sex development in human embryo composed of three stages. The indifferent stage, from fertilization to 6 weeks gestation, is where both XY and XX embryos are morphologically indistinguishable. This is followed by sex determination stage where the bipotential gonads form testes or ovaries. This stage is controlled by several determining genes. These genes comprise two signaling pathways, SRY (NM_003140.2) and its downstream target SOX9 (NM_000346.3) in the testis determination and WNT4‐CTNNB1 (β‐catenin, NM_001098209.1) signaling pathway in the ovary determination (León et al., 2019; Xue et al., 2019). The expression of the SRY gene on the Y chromosome causes the undifferentiated gonad to develop into a testis. Whereas in the absence of a Y chromosome and expression of SRY, the undifferentiated gonad develops as an ovary and the Wolffian ducts regress (Ono & Harley, 2013). The third stage is sex differentiation where the gonads produce hormones that differentiate the internal and external genitalia (León et al., 2019). Sertoli cells in the developing testis produce Anti‐Müllerian Hormone (AMH), leading to regression of the Müllerian ducts, while production of testosterone by the Leydig cells promotes differentiation of Wolffian duct structures into male internal genitalia.
DSDs are classified into three subclasses: sex chromosome DSDs, 46, XY DSDs and 46, XX DSDs. The 46, XY DSDs comprise disorders of gonadal development, disorders of androgen synthesis and action, persistent Müllerian duct syndrome, and other unclassified disorders (León et al., 2019). Most individuals with DSD lack a known specific genetic aetiology; however, exome sequencing has been used to identify a likely genetic aetiology in up to 43% of patients with 46, XY DSD (Baxter et al., 2015; Eggers et al., 2016). Here, we report three siblings with a heterozygous missense MAP3K1 variant causing variable degrees of 46, XY partial gonadal dysgenesis.
2. PATIENTS AND METHODS
2.1. Ethical compliance
This study was approved by our regional research ethical committee. Ref. No.: AA/AJ/733.
2.2. Patients
Three siblings with ambiguous genitalia, were evaluated by the Genetic and Endocrine teams at Tawam Hospital, Al Ain, United Arab Emirates. Molecular characterization of the aetiology was made possible after one of the siblings referred for genetic evaluation before proceeding with surgical correction. Those siblings were born to a non‐consanguineous couple (Figure 1). There was no history of miscarriages, neonatal deaths, infertility, developmental delay, or endocrine or sex reversal disorders. A maternal first cousin was reported to have had hypospadias, but no other information was available about him.
FIGURE 1.
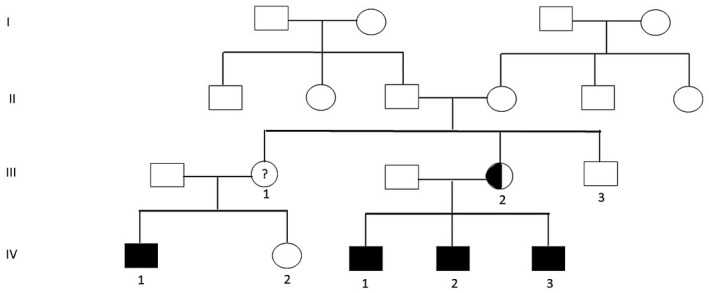
Family pedigree. Circles and squares are females and males, respectively; filled symbols are affected members; half‐filled symbols are carrier members; Roman numbers indicate the generations; and Arabic numbers indicate offspring
2.3. Endocrine evaluation
All hormonal investigations done using chemoluminescence immunoassay tested on Roche 8000i. Normal reference range of the hormonal levels in this paper was as reported for age by our laboratory.
2.4. Whole exome sequencing
Blood samples were collected from the patients and the parents after written informed consent was obtained. DNA was extracted from peripheral white blood cells using Flexigene DNA extraction kit (Qiagen Gmbh, Germany) according to the manufacturer's protocol. Whole‐exome sequencing was carried out by CENTOGENE AG laboratory in Rostock, Germany (www.centogene.com). The sample was processed on the Ion Proton Platform (Life Technologies, Renfrew, United Kingdom). Approximately 36.5 Mb of coding exons were converted as described by Consensus Coding Sequences. An in‐house bioinformatics pipeline, including read alignment to GRCh37/hg19 genome assembly, variant calling (single nucleotide and small deletion/insertion variants), annotation and comprehensive variant filtering is applied. All variants with minor allele frequency (MAF) of less than 1% in gnomAD database, and disease‐causing variants reported in HGMD®, in ClinVar or in CentoMD® are considered. The investigation for relevant variants is focused on coding exons and flanking ±20 intronic nucleotides of genes with a clear genotype phenotype correlation (based on OMIM® information).
3. RESULTS
3.1. Clinical description
Patient 1 is a 2‐year‐old and 6‐month‐old male, born at term by a Caesarean section due to previous Caesarean section, with a birth weight of 3160 g (28th%ile), a length of 51 cm (40th%ile) and a head circumference of 36 cm (45th%ile). His antenatal history was unremarkable. At birth he was noted to have perineal hypospadias with penile length of 3 cm with severe chordee and bifid scrotum with both testes palpable in the scrotum (Figure 2). His scrotum ultrasound visualized testicles within the scrotal sac and described as more of retractile testicles. His pelvic and abdominal ultrasound were reported as normal with no abnormal internal structures. His initial electrolytes were normal with acceptable baseline gonadotropin and androgen levels for his age (4 days old): Follicle‐stimulating hormone (FSH) 7.3 mIU/ml (normal for age ≤3.3), Luteinizing hormone (LH) 6.0 mIU/ml (normal for age ≤0.4), testosterone (T) 2.78 nmol/L (normal for age 13.6–26.0), dihydrotestosterone (DHT) 126 pg/ml (normal for age ≤1200), dehydroepiandrosterone sulfate (DHEAS) 9.7 µmol/L (normal for age 2.93–16.5), and androstenedione 11 nmol/L (normal for age 1–11.5). Adrenocorticotropic hormone (ACTH) 19 pmol/L (normal range 0–10.1), and 17 hydroxyprogesterone 15.4 pmol/L (normal range 1–10) which were mildly high and normalized by 1 month of age while Anti‐Müllerian Hormone (AMH) was 116.1 pmol/L (reference range 5.5–103). As T level was not matching with higher level of LH and FSH, Beta human chorionic gonadotropin (HCG) stimulation test was done at 3 weeks of age using 500 IU daily for 3 days and it showed a significant increase in both T level and DHT level up to 13.08 nmol/L and 1140 pg/ml, respectively, with T/DHT ratio of <12. Chromosomal karyotype confirmed 46, XY and chromosomal microarray was normal. Whole exome sequencing showed a variant NM_005921.2 (MAP3K1):c.1970C>G (p.Thr657Arg) in exon 11 of MAP3K1 gene causing an amino acid change from Threonine to Arginine at position 657. This variant was classified as a variant of uncertain significance according to ACMG guidelines, which was reclassified recently as pathogenic variant by Chamberlin et al. (2019). He underwent surgical correction for his ambiguous genitalia as two stages repair at 14 months and 20 months of age.
FIGURE 2.
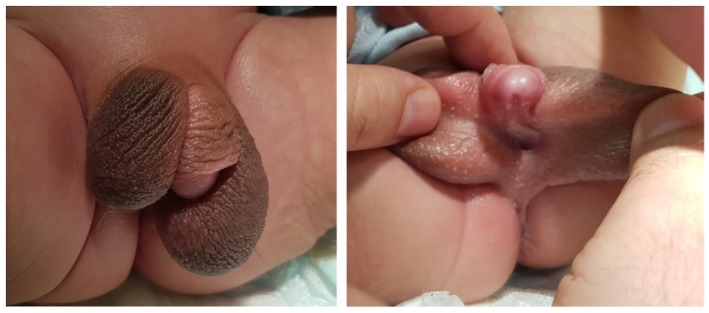
Patient's 1 genitalia, consisting of perineal hypospadias, severe chordee and bifid scrotum with both testes palpable in the scrotum
Patient 2 is a 4‐year‐old male, born at term by Caesarean section due to failed induction of labor, with a birth weight of 3270 g (20th%ile), a birth length of 53 cm (79.9th%ile) and a head circumference of 36 cm (45th%ile). The pregnancy history was remarkable for controlled gestational diabetes on diet and an antenatal ultrasound showed mild to moderate polyhydramnios. After delivery, he was found to have coronal hypospadias (with normal penial length; 3.5 cm) with severe chordee and both testes palpable within normally developed scrotum. He was nondysmorphic. The patient was not assessed at that time by either one of our teams; Endocrine or Genetics, that's why no hormonal studies and anatomical imaging of the internal anatomy were done that time. He was assessed by Pediatric Surgery and undergone hypospadias and chordee repair surgery at age of 16 months. At 2 years of age, he had another surgery for incomplete circumcision and retention penile cyst. He was evaluated in the Genetics clinic after his younger sibling (patient 1) evaluation and he was found to have the same variant in the MAP3K1 gene.
Patient 3, the youngest sibling in this family, is 7 months old. He was born at term by an emergency Caesarian section due to fetal distress with a birth weight of 2800 g (8.38th%ile), a birth length of 46.5 cm (5th%ile) and a head circumference of 35 cm (26.4th%ile). After birth, he was found to have micropenis (length of 1.5 cm), perineal urethra, no vaginal opening with bifid empty scrotum, and no palpable gonads (Figure 3). A pelvic ultrasound showed a uterus/cervix like structure seen posterior to the urinary bladder measuring grossly about 2.4 × 1.4 × 1 cm with no ovaries visualized. The external genitalia were scanned showing soft tissue echogenicity structures surrounded by minimal fluid, most likely represent bilateral inguinal hernias more on the right side, likely containing bowel. Midline soft tissue structure, most likely represent enlarged clitoris. No definite penile or testicular structures could be seen by the ultrasound. His initial electrolytes were normal. At 1 week of age, baseline AMH was 93.3 pmol/L, LH was 0.8 mIU/ml and FSH was 3.6 mIU/ml. In link with that, estradiol was 47 pmol/L (normal range 41.4–159), random testosterone 1.99 nmol/L (normal range 13.6–26). Adrenal workup revealed no abnormalities that could highlight a steroid synthesis defect: 17‐hydroxyprogesterone 35.4 nmol/L, ACTH 13.4 pmol/L, DHEA 3.18 µmol/L, and androstenedione 9.13 nmol/L. At 2 weeks of age, beta HCG stimulation test with 500 units daily for 3 days showed suboptimal response with T level change from baseline of 1.04 nmol/L to post‐stimulation level of 2.5 nmol/L, while the DHT was undetectable <50 pg/ml at baseline and increased post‐stimulation to 149 pg/ml. Diagnostic laparoscopy and bilateral inguinal hernia repair showed that the right gonad was inside the hernial sac and the left gonad was close to the internal ring. The patient has what looks like normal uterus with two fallopian tubes which were close to the gonads (Figure 4). The two gonads were mobilized inside the abdomen by pulling the gonads outside the hernial sac, adhesions were released and hernia was repaired. No biopsy was taken from the gonads.
FIGURE 3.
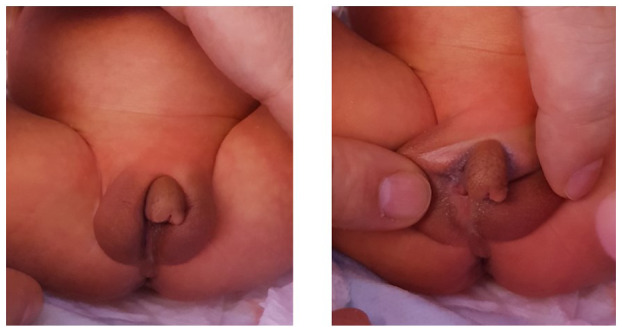
Patient's 3 external genitalia, consisting of micropenis, perineal urethra, no vaginal opening with bifid empty scrotum, and no palpable gonads
FIGURE 4.
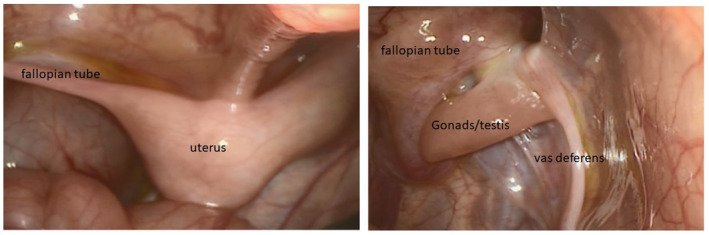
Intra‐operation findings of patient 3
Chromosomal karyotype confirmed 46, XY. Fluorescence in Situ Hybridization (FISH) was applied using DXZ1 and DYZ3, DNA probes, specific for chromosomes X and Y, respectively; it showed one signal was obtained from each of chromosomes X and Y specific DNA probes, indicating a male sex compliment. Molecular testing revealed that he also has the same variant, c.1970C>G p.(Thr657Arg) in MAP3K1 gene.
Parental testing confirmed that the mother has the c.1970C>G p.(Thr657Arg) in MAP3K1 gene variant while the father is negative. The aunt who has a child with reported hypospadias has not yet been tested, neither has her son.
4. DISCUSSION
Gonadal dysgenesis is one of 46, XY DSD causes. It is characterized by incomplete or defective formation of the gonads (ovary or testis) due to either structural or numerical anomalies of the sex chromosomes or mutations in the genes involved in the development of the gonads (McCann‐Crosby et al., 2014). According to the gonadal morphology, it can be classified as either complete gonadal dysgenesis (CGD) or partial gonadal dysgenesis (PGD). Although 45,X/46, XY is the most common karyotype seen in PGD, 46, XY and other form of mosaicism involving the Y chromosomes can also be seen (McCann‐Crosby et al., 2014).
Along with the SRY gene, several genes and signaling pathways involved in sex determination associated with a wide phenotypic spectrum of DSD have been identified. Mitogen‐activated protein kinase kinase kinase 1 (MAP3K1; NM_005921.2) and MAPK‐signaling pathway is one of the genetic pathways that control normal development of the testes. Recent studies suggest that MAP3K1 is one of the more commonly mutated genes in 46, XY DSD, with mutations reported in 13%–18% of patients (Ostrer, 2014). The gene consists of 20 exons in length and the reported mutations occurred at well‐conserved sites in exons 2, 3, 13, and 14 and have the characteristic of being in‐frame alterations, either non‐conservative single‐nucleotide variants or familial splice acceptor site variants that resulted in in‐frame insertions or deletions. (See Table 1). The net result of these mutations is gain‐of‐function effect, causing increased phosphorylation of the downstream targets, p38 and ERK1/2, and increased binding of cofactors RHOA (NM_001313941.1), MAP3K4 (NM_001291958.1), FRAT1 (NM_005479.3), and AXIN1. These downstream effects tilt the balance of gene expression in the testis‐determining pathway causing decreased expression of SOX9 and its downstream targets, FGF9 (NM_002010.2) and FGFR2 (NM_022976.1), and increased expression of B‐catenin and its downstream target, FOXL2 (NM_023067.3) (Ostrer, 2014). Adding new insights into MAPK related DSD pathogenesis, it has been reported that effects on protein binding depend on the domain in which the mutations occur (Chamberlin et al., 2019).
TABLE 1.
Reported mutations in MAP3K1 gene
| DNA change | Protein change | Number of patients | Reference |
|---|---|---|---|
| c.634‐8T>A | — | 10 | Pearlman et al. (2010) |
| c.1846G>A | p.Gly616Arg | 5 | |
| c.566T>G | p.Leu189Pro | 2 | |
| c.2416G>A | p.D806N | 4 | Das et al. (2013) |
| c.3084A>G | p.Q1028Q | 1 | |
| c. 1284G>A | p.T428T | 1 | |
| c. 2822_2824insCAA | p. 942insT | 2 | |
| c.458C>T | p.Pro153Leu | 1 | Loke et al. (2014) |
| c.2180‐2A>G | p.Gly727_Ile761del | 1 | |
| c.1846G>A | p.(Gly616Arg) | 2 | Baxter et al. (2015) |
| c.1016G>A | p.(Arg339Gln) | 1 | |
| c.770C>T | p.(Pro257Leu) | 1 | |
| c.566T>G | p.(Leu189Arg) | 2 | Eggers et al. (2016) |
| c.2071T>C | p.(Cys691Arg) | 1 | |
| c.4328C>T | p.(Ala1443Val) | 2 | |
| c.934A>T | p.(Met312Leu) | 4 | |
| c.710A>G | p.(Gln237Arg) | 1 | |
| c.394G>C | p.(Asp132His) | 1 | |
| c.14_16insCGG | p.(Ala5dup) | 2 | Granados et al. (2017) |
| c.1760T>A | p.(Leu587His) | 2 | |
| c.566T>A | p.(Leu189Gln) | 1 | |
| c.2291T>G | p.(Leu764Arg) | 1 | |
| c.2072G>A | p.(Cys691Tyr) | 2 | Kopylova IV et al. (2018) |
| c.2858_2872del CAACAACAACAACAA | p.944_948del | 1 | |
| c.2117T>G | p.(L706R) | 1 | Xue et al. (2019) |
| c.1970C>G | p.(Thr657Arg) | 3 |
Current report |
Our mutation is tested to be pathogenic as per this study.
MAP3K1 mutations were first described in 2010 in two large families with 46, XY DSD in an autosomal dominant, sex‐limited pattern of transmission, as well as in 2 of 11 sporadic cases of 46, XY gonadal dysgenesis. In the large families that were reported, there were variable phenotypic presentations ranging from complete gonadal dysgenesis, sometimes with gonadoblastoma, to partial gonadal dysgenesis like hypospadias and micropenis with cryptorchidism (Pearlman et al., 2010). 46, XX individuals carrying pathogenic variants in MAP3K1 gene are completely asymptomatic. Therefore, it was recommended by Granados and his colleagues that testing for MAP3K1 variants should be considered in patients with 46, XY complete or partial gonadal dysgenesis, particularly in families with multiple affected individuals with 46, XY DSD (Granados et al., 2017).
Table 1 summarizes all reported mutations of MAP3K1 gene and the number of cases with each one. The variant (c.634‐8T>A) is so far the most common variant in MAP3K1 associated DSD. We are reporting a heterozygous missense MAP3K1 variants in three siblings causing partial gonadal dysgenesis with variable severities. To the best of our knowledge, this variant is reported for the first time in human and both family segregation analysis and endocrine hormonal functional studies are strongly suggestive that this variant is pathogenic. Recently, this variant was reported to be pathogenic by Chamberlin et al. (2019) who used actual patient samples, structural modeling, and functional data to understand alterations of the MAP3K1 protein and the effects on protein folding, binding and downstream target phosphorylation.
Managing these patients will require a multidisciplinary team approach to address the urgent medical, surgical, and psychological needs. Sex assignment is a critical management issue that needs addressing. Although this might be obvious in some patients, such as in Patient 2, it might be a more challenging decision as in patient 3 with female internal organs and poor Beta HCG stimulation test response which later on may necessitate revising the male sex rearing chosen by the family.
5. CONCLUSION
Our three reported siblings have a novel heterozygous missense variant in MAP3K1 gene expanding the spectrum of pathogenic mutations causing 46, XY gonadal dysgenesis. Furthermore, our report adds to the genotype‐phenotype correlation, highlighting the clinical importance of considering MAP3K1 gene defects as part of the differential diagnosis for complete or partial gonadal dysgenesis especially with multiple affected family members.
CONFLICT OF INTERESTS
The authors declare that they have no competing interests.
AUTHORS’ CONTRIBUTION
AA‐S, NA‐H, MH, RA, and AM contributed to conception and design. AA‐S drafted the manuscript. All authors contributed to acquisition, revised manuscript, and agreed to be accountable for all aspects of work ensuring integrity and accuracy. All authors read and approved the final manuscript.
CONSENT FOR PUBLICATION
Consent for publication was obtained from the parent of the reported patients.
ACKNOWLEDGEMENTS
The authors thank the patients and their parents for taking part in this study and for giving permission to share their data.
How to cite this article: Al Shamsi A, Al Hassani N, Hamchou M, Almazrouei R, Mhanni A. A novel missense heterozygous mutation in MAP3K1 gene causes 46, XY disorder of sex development: case report and literature review. Mol. Genet. Genomic Med.. 2020;8:e1514. 10.1002/mgg3.1514
References
- Baxter, R. M. , Arboleda, V. A. , Lee, H. , Barseghyan, H. , Adam, M. P. , Fechner, P. Y. , Bargman, R. , Keegan, C. , Travers, S. , Schelley, S. , Hudgins, L. , Mathew, R. P. , Stalker, H. J. , Zori, R. , Gordon, O. K. , Ramos‐Platt, L. , Pawlikowska‐Haddal, A. , Eskin, A. , Nelson, S. F. , & Vilain, E. (2020). Exome sequencing for the diagnosis of 46,XY disorders of sex development. The Journal of Clinical Endocrinology & Metabolism, 100(2), E333–E344. [PubMed: 25383892]. 10.1210/jc.2014-2605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlin, A. , Huether, R. , Machado, A. Z. , Groden, M. , Liu, H.‐M. , Upadhyay, K. , O, V. , Gomes, N. L. , Lerario, A. M. , Nishi, M. Y. , Costa, E. M. F. , Mendonca, B. , Domenice, S. , Velasco, J. , Loke, J. , & Ostrer, H. (2019). Mutations in MAP3K1 that cause 46, XY disorders of sex development disrupt distinct structural domains in the protein. Human Molecular Genetics, 28(10), 1620–1628. 10.1093/hmg/ddz002 [DOI] [PubMed] [Google Scholar]
- Das, D. K. , Rahate, S. G. , Mehta, B. P. , Gawde, H. M. , & Tamhankar, P. M. (2013). Mutation analysis of mitogen activated protein kinase 1 gene in Indian cases of 46,XY disorder of sex development. Indian Journal of Human Genetics, 19(4), 437–442. 10.4103/0971-6866.124372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggers, S. , Sadedin, S. , van den Bergen, J. A. , Robevska, G. , Ohnesorg, T. , Hewitt, J. , Lambeth, L. , Bouty, A. , Knarston, I. M. , Tan, T. Y. , Cameron, F. , Werther, G. , Hutson, J. , O’Connell, M. , Grover, S. R. , Heloury, Y. , Zacharin, M. , Bergman, P. , Kimber, C. , …, Sinclair, A. H. (2016). Disorders of sex development: insights from targeted gene sequencing of a large international patient cohort. Genome Biology, 17, 243 [PubMed: 27899157]. 10.1186/s13059-016-1105-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granados, A. , Alaniz, V. I. , Mohnach, L. , Barseghyan, H. , Vilain, E. , Ostrer, H. , Quint, E. H. , Chen, M. , & Keegan, C. E. (2017). MAP3K1‐related gonadal dysgenesis: Six new cases and review of the literature. American Journal of Medical Genetics, Part C: Seminars in Medical Genetics, 175(2), 253–259. [PubMed: 28504475]. 10.1002/ajmg.c.31559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopylova, I. V. , Kuznetsova, E. S. , Chugunov, I. S. , Orlova, E. M. , Danilenko, O. S. , Brovin, D. N. , Kareva, M. A. , & Peterkova, V. A. (2018). Disorder of sex development 46, XY associated with mutations in the gene MAP3K1. The report of clinical cases. Problems of Endocrinology, 64(1), 45–49. 10.14341/probl8596 [DOI] [Google Scholar]
- Lee, P. A. , Nordenström, A. , Houk, C. P. , Ahmed, S. F. , Auchus, R. , Baratz, A. , Baratz Dalke, K. , Liao, L.‐M. , Lin‐Su, K. , Looijenga 3rd, L. H. J. , Mazur, T. , Meyer‐Bahlburg, H. F. L. , Mouriquand, P. , Quigley, C. A. , Sandberg, D. E. , Vilain, E. , & Witchel, S. ; the Global DSD Update Consortium . (2016). Global disorders of sex development update since 2006: Perceptions, approach and care. Hormone Research in Paediatrics, 85(3), 158–180. 10.1159/000442975 [DOI] [PubMed] [Google Scholar]
- León, N. Y. , Reyes, A. P. , & Harley, V. R. (2019). A clinical algorithm to diagnose differences of sex development. The Lancet Diabetes & Endocrinology, 7, 560–574. 10.1016/S2213-8587(18)30339-5 [DOI] [PubMed] [Google Scholar]
- Loke, J. , Pearlman, A. , Radi, O. , Zuffardi, O. , Giussani, U. , Pallotta, R. , Camerino, G. , & Ostrer, H. (2014). Mutations in MAP3K1 tilt the balance from SOX9/FGF9 to WNT/beta-catenin signaling. Human Molecular Genetics, 23, 1073–1083. [PubMed, 24135036]. 10.1093/hmg/ddt502 [DOI] [PubMed] [Google Scholar]
- McCann‐Crosby, B. , Mansouri, R. , Dietrich, J. E. , McCullough, L. B. , Sutton, V. R. , Austin, E. G. , Schlomer, B. , Roth, D. R. , Karaviti, L. , Gunn, S. , Hicks, M. J. , & Macias, C. G. (2014). State of the art review in gonadal dysgenesis: challenges in diagnosis and management. International Journal of Pediatric Endocrinology, 2014, 4 [PubMed, 24731683]. 10.1186/1687-9856-2014-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono, M. , & Harley, V. R. (2013). Disorders of sex development: new genes, new concepts. Nature Reviews Endocrinology, 9(2), 79–91. [PubMed, 23296159]. 10.1038/nrendo.2012.235 [DOI] [PubMed] [Google Scholar]
- Ostrer, H. (2014). Disorders of sex development (DSDs): An update. Journal of Clinical Endocrinology and Metabolism, 99(5), 1503–1509. [PubMed, 24758178]. 10.1210/jc.2013-3690 [DOI] [PubMed] [Google Scholar]
- Pearlman, A. , Loke, J. , Le Caignec, C. , White, S. , Chin, L. , Friedman, A. , Warr, N. , Willan, J. , Brauer, D. , Farmer, C. , Brooks, E. , Oddoux, C. , Riley, B. , Shajahan, S. , Camerino, G. , Homfray, T. , Crosby, A. H. , Couper, J. , David, A. , & Ostrer, H. (2010). Mutations in MAP3K1 cause 46, XY disorders of sex development and implicate a common signal transduction pathway in human testis determination. American Journal of Human Genetics, 87(6), 898–904. [PubMed, 21129722]. 10.1016/j.ajhg.2010.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue, M. , Wang, X. , Li, C. , Zhao, M. , He, F. , & Li, X. (2019). Novel pathogenic mutations in disorders of sex development associated genes cause 46,XY complete gonadal dysgenesis. Gene, 15(718), 144072 10.1016/j.gene.2019.144072 [DOI] [PubMed] [Google Scholar]