

Abstract
Introduction
Statins are known to lower CRP, and this reduction has been suggested to contribute to the established efficacy of these drugs in reducing cardiovascular events and outcomes. However, the exact mechanism underlying the CRP-lowering effect of statins remains elusive.
Methods
In order to test the possibility of direct interaction, we performed an in silico study by testing the orientation of the respective ligands (statins) and phosphorylcholine (the standard ligand of CRP) in the CRP active site using Molecular Operating Environment (MOE) software.
Results
Docking experiments showed that all statins could directly interact with CRP. Among statins, rosuvastatin had the strongest interaction with CRP (pKi = 16.14), followed by fluvastatin (pKi = 15.58), pitavastatin (pKi = 15.26), atorvastatin (pKi = 14.68), pravastatin (pKi = 13.95), simvastatin (pKi = 7.98) and lovastatin (pKi = 7.10). According to the above-mentioned results, rosuvastatin, fluvastatin, pitavastatin and atorvastatin were found to have stronger binding to CRP compared with the standard ligand phosphocholine (pKi = 14.55).
Conclusions
This finding suggests a new mechanism of interaction between statins and CRP that could be independent of the putative cholesterol-lowering activity of statins.
Keywords: C-reactive protein, statins, inflammation, docking, interaction
C-reactive protein (CRP) is a liver protein associated with inflammation. Being readily assayable in the circulation it has an established clinical role in the evaluation of the inflammatory status in generalized inflammatory diseases. Among these, foremost is rheumatoid arthritis, followed by chronic diseases such as spondylitis, lupus and others [1, 2]. CRP levels rise after any microbial caused inflammation, from tonsils to subcutaneous tissues, as well as after traumas [3].
The major novelty in the field of inflammatory disorders has been the recognition that generalized changes associated with CRP levels 40–200 mg/l are just one face of the pathophysiological role of CRP [4]. Indeed, detection of CRP rises only measurable by high-sensitivity testing (hs-CRP) provided unexpected information on the role of hs-CRP as a marker of diseases not at the classical inflammation sites, but rather at the cardiovascular level [5].
Hs-CRP > 2 mg/l is associated with major cardiovascular risk markers, such as elevated lipids, in particular low-density lipoprotein (LDL)-cholesterol, as well as in general with the progression of cardiovascular lesions [6]. In this particular case, hs-CRP > 2 mg/l becomes a major marker for early detection of arterial lesions and, more importantly, for the monitoring of agents reducing cardiovascular risk, in particular lipid-lowering medications. Clinical studies on the major HMG-CoA reductase inhibitors, i.e. statins, have shown that patients with coronary disease not associated with marked hypercholesterolemia benefit from the reduction of this inflammatory marker induced by statins [7].
Recently questions have been raised on the actual biomarker role of CRP after a study on genetic loci associated with hs-CRP levels failed to detect a clear association between these and the occurrence of CV events, in contrast to neighboring loci such as IL-6R or the APOCI-CII cluster [8]. A later Mendelian randomization study [9] from a coronary heart disease genetic collaboration, investigating individuals from 47 epidemiological studies in 15 countries, detected four genes tagging single nucleotide polymorphisms in the CRP gene. Variants of these were associated with an up to 30% per allele difference in hs-CRP concentrations but were unrelated to other coronary risk factors. None of these alleles was additive to the classical risk scores for coronary heart disease. In spite of the altogether not informative genetic characterization, it is unquestionable that the reduction of hs-CRP is associated with a definite CV benefit. Indeed, by using canakinumab, a selective antagonist of interleukin-1b in the CANTOS Study on post-myocardial infarction patients, a clear benefit in cardiovascular risk was associated with a highly significant (-35–40%) reduction of hs-CRP [10].
In the still ongoing debate on the clinical significance of CRP reduction induced by lipid-lowering or other treatments, the newly developed PCSK9 antagonists offer a good case in point. The major end point trials do not indicate anti-inflammatory activity of either bococizumab [11] or evolocumab [12], which may be responsible for the observed coronary prevention. The impressive LDL-cholesterol lowering induced by these agents was not associated with changes in hs-CRP levels [13]. These findings confirm the results of a careful meta-analysis from many trials with PCSK9i, showing a lack of effect on CRP levels [14]. An effect of these drugs on tissue cholesterol reduction, leading to a lack of inflammasome activation consequent to cholesterol crystal deposition [15], would thus not be supported. This major difference between statins, leading to reduced tissue cholesterol and lower inflammasome activation [16], and PCSK9 antagonists, has been underlined in recent reports attempting to explain the mortality difference in statin vs. PCSK9 trials [17] and the clearly reduced mortality in the CANTOS trial [8].
Since at present only indirect antagonist action of statins on CRP production has been hypothesized [18], i.e. consequent to inhibited protein geranylgeranyltransferase [19], a mechanistically more effective antagonism of the CRP activity as exerted by statins should be evaluated. This will be of major importance for the definition of an active site of CRP binding, allowing one to hypothesize the molecular structure of agents potentially able to inhibit the activity of CRP. In order to achieve this goal, active ligands, i.e. statins, were studied and compared to phosphorylcholine, the standard ligand of CRP [20]. Appropriate docking experiments were designed showing that all statins and phosphorylcholine could directly interact with CRP in the active site, providing reliable information on the most appropriate design of drugs potentially antagonizing CRP. In addition, the present study could potentially clarify whether the antagonist action on the CRP docking site may go in parallel with the well-assessed dose-related plasma LDL-C reduction or may be independent activity.
Methods
The orientation of the respective ligands and phosphorylcholine in the CRP active site was examined by a Molecular Operating Environment (MOE, Chemical Computing Group Inc. Montreal, http://www.chemcomp/com) docking experiment (Table I). Phosphorylcholine bound to CRP crystal structure was downloaded from the RCSB Protein Data Bank (PDB entry: 1B09). All the computational procedures were carried out with MOE. The molecular structures of the fluvastatin, pravastatin, pitavastatin, rosuvastatin, atorvastatin, simvastatin and lovastatin and phosphorylcholine (as the standard ligand) were prepared by MOE Builder and minimized energy was calculated using Hamiltonian-Force Field-MMFF94x by MOE. The docking procedure was performed with the default settings of the MOE-DOCK. The final docking scores were evaluated using the Generalized-Born Volume Integral/Weighted Surface area (GBVI/WSA) dG scoring function with the GBVI [21]. The GBVI/WSA dG is a forcefield-based scoring function, which estimates the free energy of binding of the ligand from a given pose. The dissociation constant (Ki) was computed through the binding free energy estimated with the GBVI/WSA dG scoring function according to the following equation: ΔG = RTln(Ki), where R represents the gas constant and T the temperature in kelvin. The Ki was computed from the binding free energy values at a fixed temperature (300 K).
Table I.
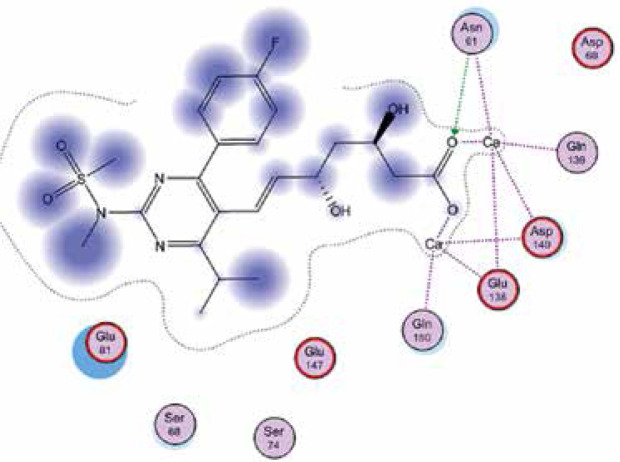
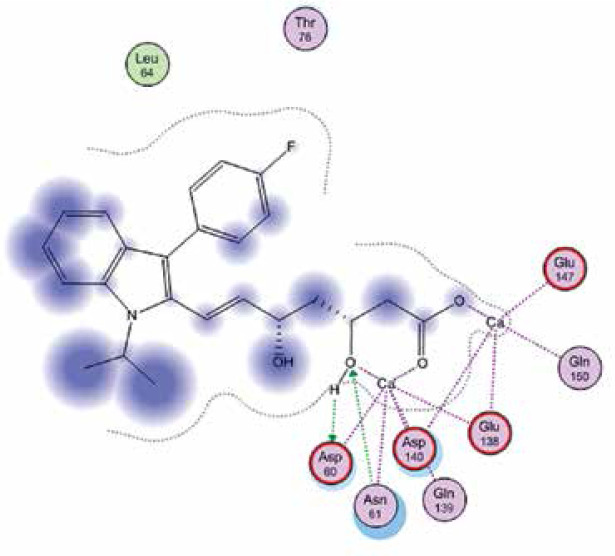
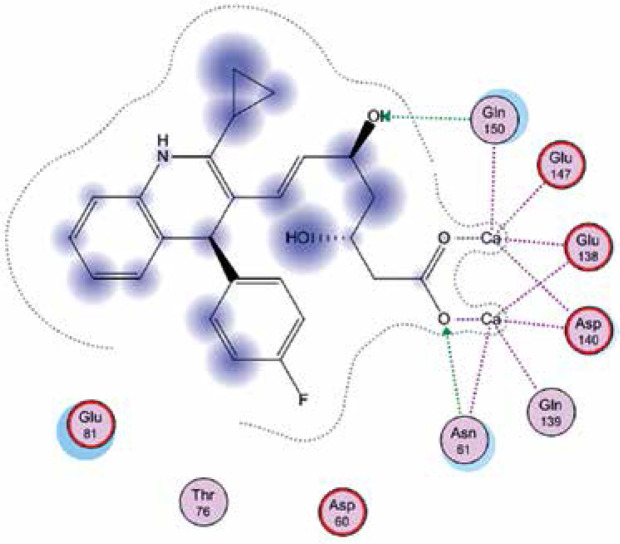
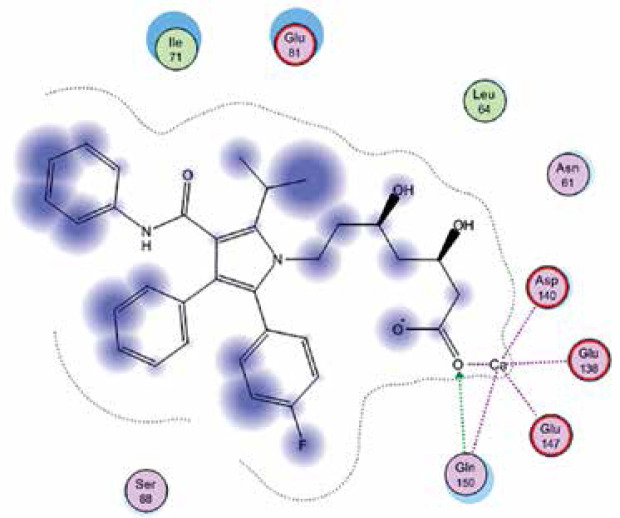
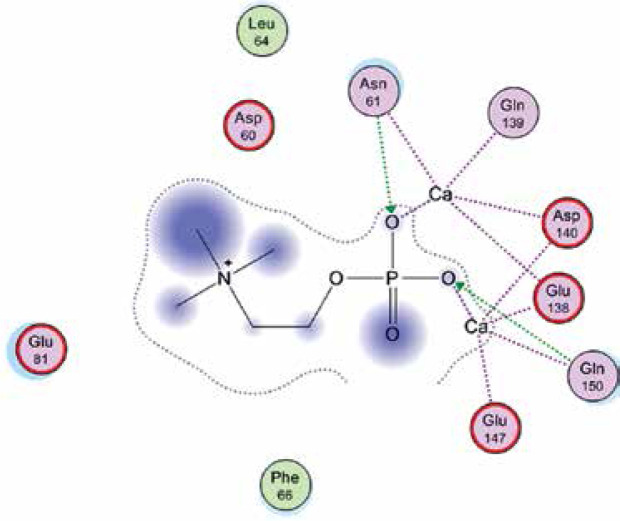
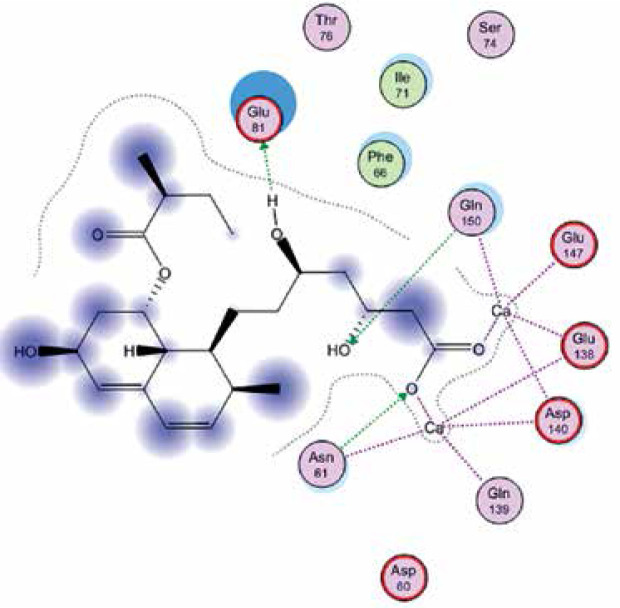
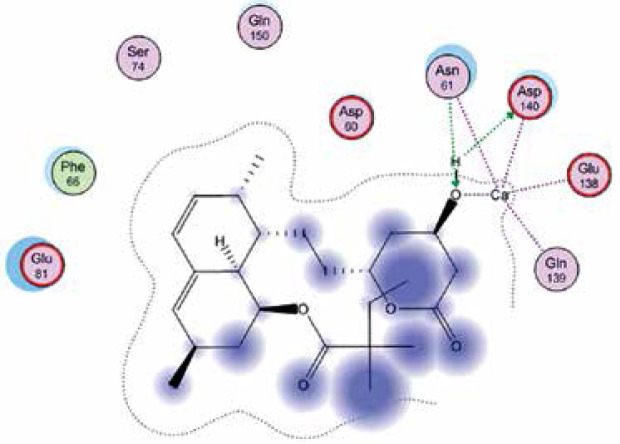
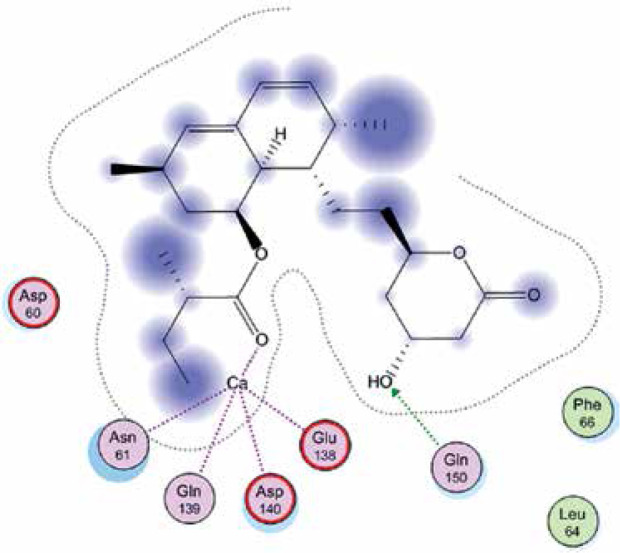
Right column – 3D-docking of phosphorylcholine and respective ligands with the CRP molecular target, Ca2+ (brown) is seen at the center. Left column – Docking of CRP with a-h compounds in 2D diagram. A – rosuvastatin, B – fluvastatin, C – pitavastatin, D – atorvastatin, E – phosphorylcholine, F – pravastatin, G – simvastatin, H – lovastatin
| Ligands | Docking 3D representation | Docking 2D representation |
|---|---|---|
| (A) | 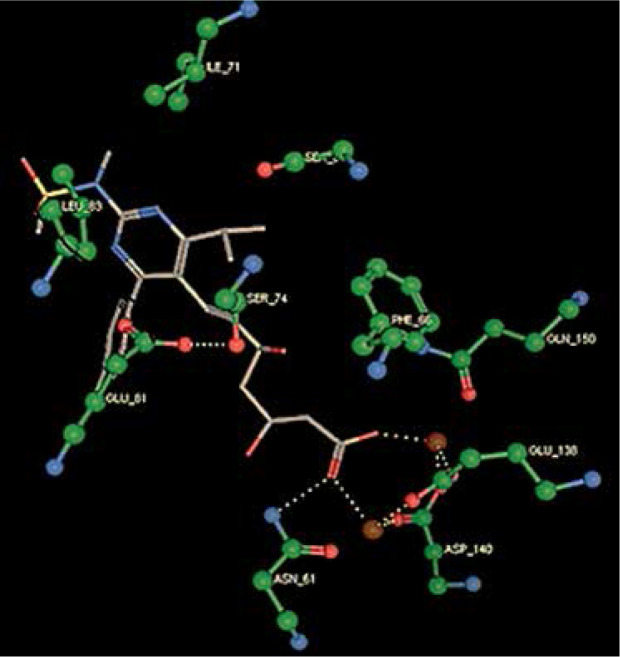 |
 |
| (B) | 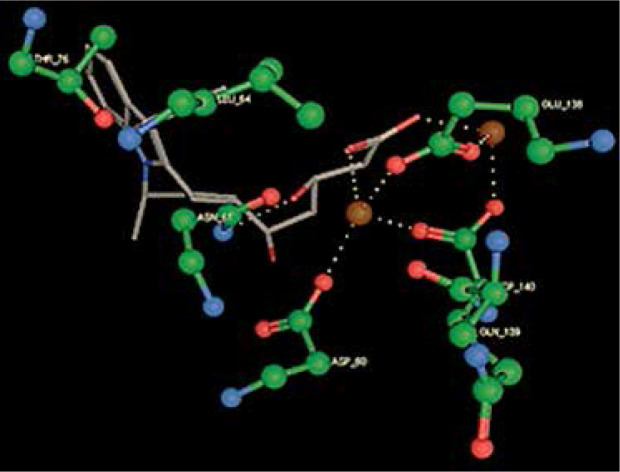 |
 |
| (C) | 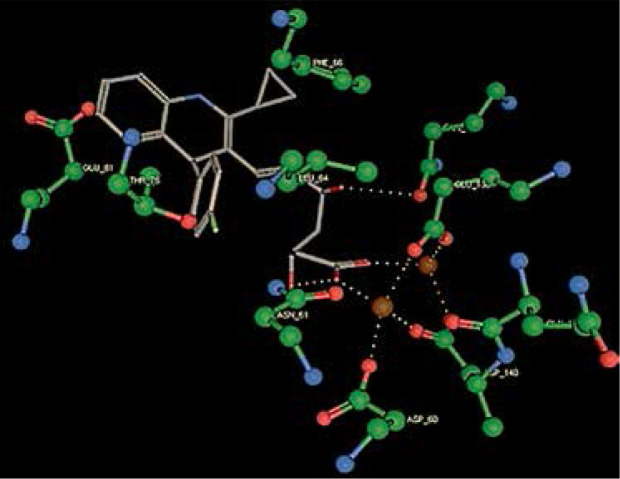 |
 |
| (D) | 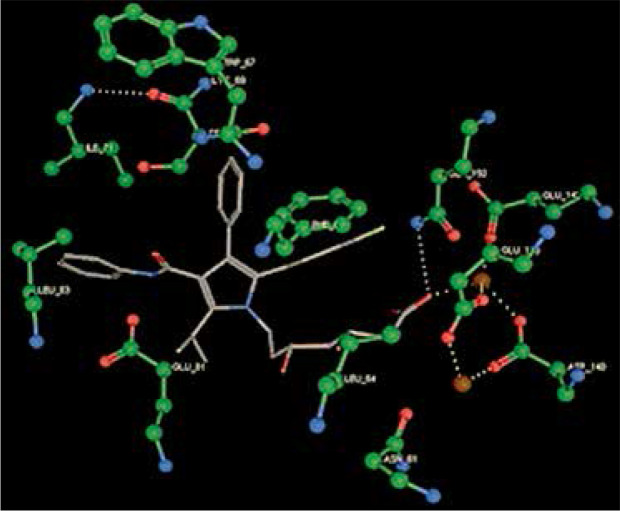 |
 |
| (E) | 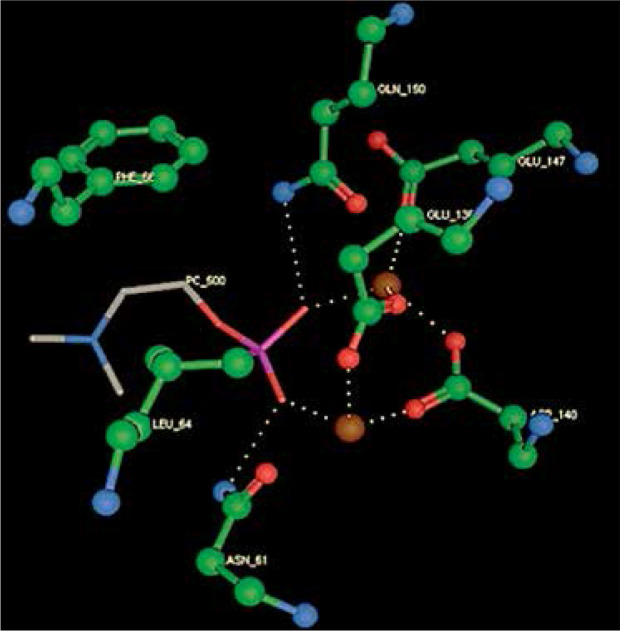 |
 |
| (F) | 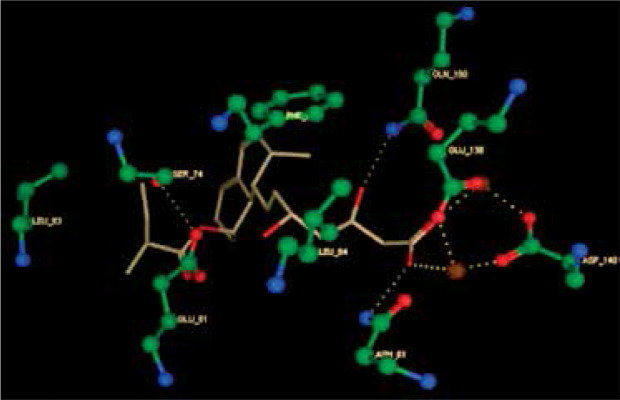 |
 |
| (G) | 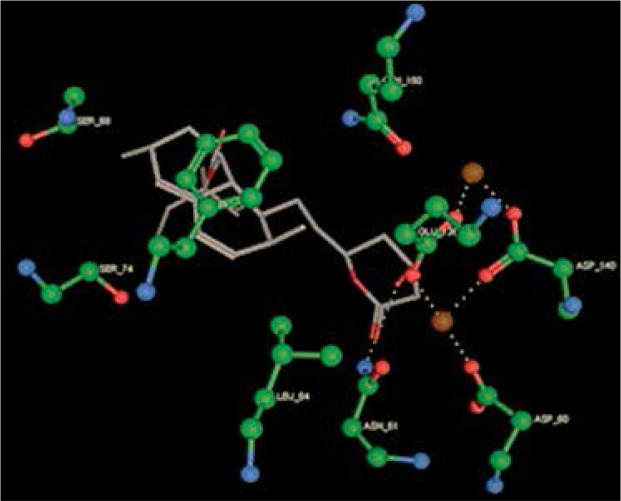 |
 |
| (H) | 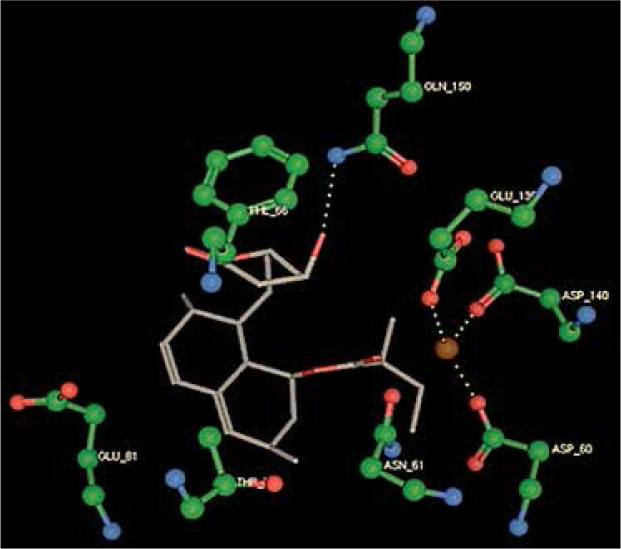 |
 |
 | ||
Results
We studied the affinity of seven statins (rosuvastatin, fluvastatin, pitavastatin, atorvastatin, pravastatin, simvastatin and lovastatin) as well as the standard ligand phosphorylcholine to CRP. Docking experiments showed that all statins could directly interact with CRP (Table I). Among statins, rosuvastatin had the strongest interaction with CRP (pKi = 16.14) through one H bonding interaction of its carbonyl group to ASN_61 residues of CRP, followed by fluvastatin (pKi = 15.58; two H bonding interactions of its hydroxyl group to ASP_60 and ASN_61 residues), pitavastatin (pKi = 15.26; two H bonding interactions of its hydroxyl group to GLN_150 and ASN_61 residues), atorvastatin (pKi = 14.68; one H bonding interaction of its carbonyl group to GLN_150 residue), pravastatin (pKi = 13.95; three H bonding interactions of its hydroxyl group to GLN_150, ASN_61 and GLU_81 residues), simvastatin (pKi = 7.98; two H bonding interactions of its hydroxyl group to ASN_61 and ASP_140 residues), and lovastatin (pKi = 7.10; one H bonding interaction of its hydroxyl group to GLN_150 residue) (Table II). The standard ligand phosphorylcholine had interaction with CRP with a pKi value of 14.55 through two H bonding interactions of its hydroxyl group to GLN_150 and ASN_61 residues (Table I). Docking results indicated that each of the ligands [A-H] had an H-bonding interaction between the oxygen atom and ASN_61 residue except atorvastatin and lovastatin. Likewise, all ligands [A-H] had an H-bonding interaction between the oxygen atom and GLN-150 residue except simvastatin, fluvastatin and rosuvastatin (Table I). According to the above-mentioned results, rosuvastatin, fluvastatin, pitavastatin and atorvastatin with pki values of 16.14, 15.58, 15.26 and 14.68 were found to have stronger binding to CRP compared with the standard ligand phosphocholine (pKi = 14.55).
Table II.
Data for the docking interactions of phosphorylcholine and statin molecules at the active sites of C-reactive protein (CRP) as the molecular target
| No. of target protein (PDB-ID) | Ligand | Docking score | pKi | H–bonding interactions with amino acid residues |
|---|---|---|---|---|
| 1B09 | Rosuvastatin | –22.0777 | 16.14 | ASN_61 |
| Fluvastatin | –21.3116 | 15.58 | ASP_60 ASN_61 |
|
| Pitavastatin | –20.8739 | 15.26 | ASN_61 GLN_150 |
|
| Atorvastatin | –20.0896 | 14.68 | GLN_150 | |
| XRay Phosphorylcholine | –19.9102 | 14.55 | ASN_61 GLN_150 |
|
| Pravastatin | –19.0900 | 13.95 | ASN_61 GLN_150 GLU_81 |
|
| Simvastatin | –10.9197 | 7.98 | ASN_61 ASP_140 |
|
| Lovastatin | –9.7223 | 7.10 | GLN_150 |
Discussion
The effect of lipid-lowering drugs on inflammation is receiving growing interest, particularly after the observation that PCSK9 antagonists, apparently devoid of significant antiinflammatory activity, at least as witnessed by a lack of reduction of hs-CRP [14], also affect to a lesser extent fatal CV events vs. statins [13, 17]. The present study, based on an in silico approach by molecular docking, on the interaction between CRP and different statins on the putative binding site of CRP is thus of potential high clinical interest and may lead to the development of newer agents, statins or others, with more powerful activity in binding inhibition.
A major observation stemming from the present study is that the inhibitory action of major statins on the CRP binding site appears to be to a large extent related to the lipid-lowering power of these molecules. As reported in a very recent review on the correlation between cholesterol lowering and inflammatory changes [22], CRP reductions occurring after statin treatment in trials of different length ranged between –9.8 and 89-3%. Interestingly, the largest reductions occurred in a study with either atorvastatin or pravastatin being given to individuals with extreme CRP elevations [23]. It should be noted that in this last quoted study published in 2005, CRP levels were apparently not evaluated by a high sensitivity assay; CRP reductions were, respectively –89.3 for atorvastatin and –82.4 for pravastatin. It is important to note that when evaluating studies on high-intensity statins, these gave the highest CRP reductions: in a direct comparative study on an IVUS evaluation of coronary plaque progression, atorvastatin treatment reduced CRP by 35.6% vs. 5.2% for pravastatin (p < 0.01) [24]. In the 2-year JUPITER study on rosuvastatin, the statin with the highest power, CRP, evaluated by the hs technique, was lowered by 57.1% [25]. Finally, a lower percent reduction of CRP (–27%) was found in the very large Heart Protection Study investigating the preventive activity of simvastatin [26]. In this study, differently from others, the reduction in vascular death appeared to be independent of baseline CRP levels. Another study evaluating effects of atorvastatin vs. simvastatin on atherosclerosis progression showed that hs-CRP after 1 year of treatment was decreased by 44.9% in the atorvastatin 80 mg group vs. 14.0% in the simvastatin 40 mg group. The same trend was maintained until the end of the study: –40.1% in the atorvastatin 80 mg group vs. 19.7% in the simvastatin 40 mg group [27].
Docking studies may be thus of crucial value since up to now no real correlation studies have monitored hs-CRP reduction vis-à-vis the chemical structure and inhibition of docking activity of statins for the CRP putative binding site. As shown by Kinlay [28], in a meta-analysis including 23 studies with statins, 89–98% of the CRP reduction (mean –28%) was related to the degree of LDL-C lowering; however, the event reduction was more significantly related to the reduction of LDL-C.
Finally, while there is no doubt that statins can improve systemic and vascular inflammation, also based on genome-wide association studies, controversial findings indicate that CRP may or may not play a significant role in the pathogenesis of atherosclerosis and particularly of acute CAD manifestations. The genome-wide association study [8] showed a lack of concordance between the effects on coronary risk of CRP genotypes and CRP levels, arguing against a causal association of CRP with coronary disease. Very recently Diederichsen et al. [29] in a vascular prediction study in 1,179 middle-aged subjects, concluded that, when adjusted for traditional risk factors and coronary calcium score, hs-CRP did not appear to add significantly to risk prediction.
In addition to CRP reduction or inhibited CRP binding, other anti-atherogenic mechanisms of statins should not be forgotten. Statins possess numerous pleiotropic actions [30–42] and the relative role of each one of these in coronary prevention is difficult to establish. Hs-CRP, because of the relative straightforward determination and clear association with CAD risk, is a well-established CV risk marker, and inhibitory mechanisms may certainly prove beneficial.
The present study is limited by its in silico nature which necessitates further supportive evidence from experimental studies. In addition, future studies are warranted to test whether any direct interaction between statins and CRP structures could be considered at the protein binding sites other than the phosphorylcholine binding site.
In conclusion, the present study evaluated, by a docking technology, the inhibition of the binding of CRP to its putative active side by different statins. This led to a classification of statins by power of inhibitory activity, apparently in the same range as the CRP reduction associated with the lowering of CAD events. A limitation of the present study is obviously that the inhibitory action of statins on CRP binding to its site is linked to the amount of statins reaching this site in vivo, information not available by means of the present technology and, in addition, to the individual metabolism of statins allowing them to reach this site. Despite this limitation the study has the innovative potential of providing a tool for the study of different molecules, assessing their inhibitory effect on CRP binding, and thus of clear value in cardiovascular event reduction.
Acknowledgments
Neda Shakour and Massimiliano Ruscica contributed equally to this study.
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Otterness IG. The value of C-reactive protein measurement in rheumatoid arthritis. Semin Arthritis Rheum. 1994;24:91–104. doi: 10.1016/s0049-0172(05)80003-4. [DOI] [PubMed] [Google Scholar]
- 2.Stumpf C, Sheriff A, Zimmermann S, et al. C-reactive protein levels predict systolic heart failure and outcome in patients with first ST-elevation myocardial infarction treated with coronary angioplasty. Arch Med Sci. 2017;13:1086–93. doi: 10.5114/aoms.2017.69327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ticinesi A, Lauretani F, Nouvenne A, et al. C-reactive protein (CRP) measurement in geriatric patients hospitalized for acute infection. Eur J Intern Med. 2017;37:7–12. doi: 10.1016/j.ejim.2016.08.026. [DOI] [PubMed] [Google Scholar]
- 4.Shadick NA, Cook NR, Karlson EW, et al. C-reactive protein in the prediction of rheumatoid arthritis in women. Arch Intern Med. 2006;166:2490–4. doi: 10.1001/archinte.166.22.2490. [DOI] [PubMed] [Google Scholar]
- 5.Tsimikas S, Willerson JT, Ridker PM. C-reactive protein and other emerging blood biomarkers to optimize risk stratification of vulnerable patients. J Am Coll Cardiol. 2006;47:C19–31. doi: 10.1016/j.jacc.2005.10.066. [DOI] [PubMed] [Google Scholar]
- 6.Koenig W. High-sensitivity C-reactive protein and atherosclerotic disease: from improved risk prediction to risk-guided therapy. Int J Cardiol. 2013;168:5126–34. doi: 10.1016/j.ijcard.2013.07.113. [DOI] [PubMed] [Google Scholar]
- 7.Everett BM, Glynn RJ, MacFadyen JG, Ridker PM. Rosuvastatin in the prevention of stroke among men and women with elevated levels of C-reactive proteIn: justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER) Circulation. 2010;121:143–50. doi: 10.1161/CIRCULATIONAHA.109.874834. [DOI] [PubMed] [Google Scholar]
- 8.Elliott P, Chambers JC, Zhang W, et al. Genetic loci associated with C-reactive protein levels and risk of coronary heart disease. JAMA. 2009;302:37–48. doi: 10.1001/jama.2009.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.C Reactive Protein Coronary Heart Disease Genetics Collaboration (CCGC) Wensley F, Gao P, Burgess S, et al. Association between C reactive protein and coronary heart disease: mendelian randomisation analysis based on individual participant data. BMJ. 2011;342:d548. doi: 10.1136/bmj.d548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119–31. doi: 10.1056/NEJMoa1707914. [DOI] [PubMed] [Google Scholar]
- 11.Ridker PM, Revkin J, Amarenco P, et al. Cardiovascular efficacy and safety of bococizumab in high-risk patients. N Engl J Med. 2017;376:1527–39. doi: 10.1056/NEJMoa1701488. [DOI] [PubMed] [Google Scholar]
- 12.Raal FJ, Stein EA, Dufour R, et al. PCSK9 inhibition with evolocumab (AMG 145) in heterozygous familial hypercholesterolaemia (RUTHERFORD-2): a randomised, double-blind, placebo-controlled trial. Lancet. 2015;385:331–40. doi: 10.1016/S0140-6736(14)61399-4. [DOI] [PubMed] [Google Scholar]
- 13.Ridker PM. Mortality differences associated with treatment responses in CANTOS and FOURIER: insights and Implications. Circulation. 2018;137:1763–6. doi: 10.1161/CIRCULATIONAHA.117.033254. [DOI] [PubMed] [Google Scholar]
- 14.Sahebkar A, di Giosia P, Stamerra A, et al. Effect of monoclonal antibodies to PCSK9 on high-sensitivity C-reactive protein levels: a meta-analysis of 16 randomized controlled treatment arms. Br J Clin Pharmacol. 2016;81:1175–90. doi: 10.1111/bcp.12905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duewell P, Kono H, Rayner KJ, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–61. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang S, Xie X, Lei T, et al. Statins attenuate activation of the NLRP3 inflammasome by oxidized-LDL or TNF-alpha in vascular endothelial cells through a PXR-dependent mechanism. Mol Pharmacol. 2017;92:256–64. doi: 10.1124/mol.116.108100. [DOI] [PubMed] [Google Scholar]
- 17.Ruscica M, Tokgozoglu L, Corsinoi A, Sirtori CR. PCSK9 antagonists and inflammation. Atherosclerosis. 2018;268:235–6. doi: 10.1016/j.atherosclerosis.2017.10.022. [DOI] [PubMed] [Google Scholar]
- 18.Arnaud C, Burger F, Steffens S, et al. Statins reduce interleukin-6-induced C-reactive protein in human hepatocytes: new evidence for direct antiinflammatory effects of statins. Arterioscler Thromb Vasc Biol. 2005;25:1231–6. doi: 10.1161/01.ATV.0000163840.63685.0c. [DOI] [PubMed] [Google Scholar]
- 19.Raiteri M, Arnaboldi L, McGeady P, et al. Pharmacological control of the mevalonate pathway: effect on arterial smooth muscle cell proliferation. J Pharmacol Exp Therap. 1997;281:1144–53. [PubMed] [Google Scholar]
- 20.Lee RT, Takagahara I, Lee YC. Mapping the binding areas of human C-reactive protein for phosphorylcholine and polycationic compounds relationship between the two types of binding sites. J Biol Chem. 2002;277:225–32. doi: 10.1074/jbc.M106039200. [DOI] [PubMed] [Google Scholar]
- 21.Wojciechowski M, Lesyng B. Generalized born model: analysis, refinement, and applications to proteins. J Phys Chem B. 2004;108:18368–76. [Google Scholar]
- 22.Catapano AL, Pirillo A, Norata GD. Vascular inflammation and low-density lipoproteins: is cholesterol the link? A lesson from the clinical trials. Br J Pharmacol. 2017;174:3973–85. doi: 10.1111/bph.13805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ridker PM, Morrow DA, Rose LM, et al. Relative efficacy of atorvastatin 80 mg and pravastatin 40 mg in achieving the dual goals of low-density lipoprotein cholesterol < 70 mg/dl and C-reactive protein < 2 mg/l: an analysis of the PROVE-IT TIMI-22 trial. J Am Coll Cardiol. 2005;45:1644–8. doi: 10.1016/j.jacc.2005.02.080. [DOI] [PubMed] [Google Scholar]
- 24.Nissen SE, Tuzcu EM, Schoenhagen P, et al. Effect of intensive compared with moderate lipid-lowering therapy on progression of coronary atherosclerosis: a randomized controlled trial. JAMA. 2004;291:1071–80. doi: 10.1001/jama.291.9.1071. [DOI] [PubMed] [Google Scholar]
- 25.Ridker PM, Danielson E, Fonseca FAH, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359:2195–207. doi: 10.1056/NEJMoa0807646. [DOI] [PubMed] [Google Scholar]
- 26.Heart Protection Study Collaborative Group C-reactive protein concentration and the vascular benefits of statin therapy: an analysis of 20 536 patients in the Heart Protection Study. Lancet. 2011;377:469–76. doi: 10.1016/S0140-6736(10)62174-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Wissen S, trip MD, Smilde TJ, et al. Differential hs-CRP reduction in patients with familial hypercholesterolemia treated with aggressive or conventional statin therapy. Atherosclerosis. 2002;165:361–6. doi: 10.1016/s0021-9150(02)00280-0. [DOI] [PubMed] [Google Scholar]
- 28.Kinlay S. Low-density lipoprotein-dependent and-independent effects of cholesterol-lowering therapies on C-reactive proteIn: a meta-analysis. J Am Coll Cardiol. 2007;49:2003–9. doi: 10.1016/j.jacc.2007.01.083. [DOI] [PubMed] [Google Scholar]
- 29.Diederichsen MZ, Diederichsen SZ, Mickley H, et al. Prognostic value of suPAR and hs-CRP on cardiovascular disease. Atherosclerosis. 2018;271:245–51. doi: 10.1016/j.atherosclerosis.2018.01.029. [DOI] [PubMed] [Google Scholar]
- 30.Chruściel P, Sahebkar A, Rembek-Wieliczko M, et al. Impact of statin therapy on plasma adiponectin concentrations: a systematic review and meta-analysis of 43 randomized controlled trial arms. Atherosclerosis. 2016;253:194–208. doi: 10.1016/j.atherosclerosis.2016.07.897. [DOI] [PubMed] [Google Scholar]
- 31.Parizadeh SMR, Azarpazhooh MR, Moohebati M, et al. Simvastatin therapy reduces prooxidant-antioxidant balance: results of a placebo-controlled cross-over trial. Lipids. 2011;46:333–40. doi: 10.1007/s11745-010-3517-x. [DOI] [PubMed] [Google Scholar]
- 32.Sahebkar A, Kotani K, Serban C, et al. Statin therapy reduces plasma endothelin-1 concentrations: a meta-analysis of 15 randomized controlled trials. Atherosclerosis. 2015;241:433–42. doi: 10.1016/j.atherosclerosis.2015.05.022. [DOI] [PubMed] [Google Scholar]
- 33.Sahebkar A, Pecin I, Tedeschi-Reiner E, et al. Effects of statin therapy on augmentation index as a measure of arterial stiffness: a systematic review and meta-analysis. Int J Cardiol. 2016;212:160–8. doi: 10.1016/j.ijcard.2016.03.010. [DOI] [PubMed] [Google Scholar]
- 34.Sahebkar A, Ponziani MC, Goitre I, Bo S. Does statin therapy reduce plasma VEGF levels in humans? A systematic review and meta-analysis of randomized controlled trials. Metabolism Clin Exp. 2015;64:1466–76. doi: 10.1016/j.metabol.2015.08.002. [DOI] [PubMed] [Google Scholar]
- 35.Sahebkar A, Rathouska J, Derosa G, Maffuioli P, Nachtigal P. Statin impact on disease activity and C-reactive protein concentrations in systemic lupus erythematosus patients: a systematic review and meta-analysis of controlled trials. Autoimmun Rev. 2016;15:344–53. doi: 10.1016/j.autrev.2015.12.007. [DOI] [PubMed] [Google Scholar]
- 36.Sahebkar A, Rathouska J, Simental-Mendía LE, Nachtigal P. Statin therapy and plasma cortisol concentrations: a systematic review and meta-analysis of randomized placebo-controlled trials. Pharmacol Res. 2016;103:17–25. doi: 10.1016/j.phrs.2015.10.013. [DOI] [PubMed] [Google Scholar]
- 37.Sahebkar A, Serban C, Mikhailidis DP, et al. Association between statin use and plasma d-dimer levels: a systematic review and meta-analysis of randomised controlled trials. Thromb Haemost. 2015;114:546–57. doi: 10.1160/TH14-11-0937. [DOI] [PubMed] [Google Scholar]
- 38.Sahebkar A, Serban C, Ursoniu S, et al. The impact of statin therapy on plasma levels of von Willebrand factor antigen: systematic review and meta-analysis of randomised placebo-controlled trials. Thromb Haemost. 2016;115:520–32. doi: 10.1160/TH15-08-0620. [DOI] [PubMed] [Google Scholar]
- 39.Serban C, Sahebkar A, Ursoniu S, et al. A systematic review and meta-analysis of the effect of statins on plasma asymmetric. doi: 10.1038/srep09902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Katsiki N, Mikhailidis DP, Banach M. Effects of statin treatment on endothelial function, oxidative stress and inflammation in patients with arterial hypertension and normal cholesterol levels. J Hypertens. 2011;29:2493–4. doi: 10.1097/HJH.0b013e32834c7bc9. [DOI] [PubMed] [Google Scholar]
- 41.Banach M, Jankowski P, Jóźwiak J, et al. PoLA/CFPiP/PCS guidelines for the management of dyslipidaemias for family physicians 2016. Arch Med Sci. 2017;13:1–45. doi: 10.5114/aoms.2017.64712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bielecka-Dabrowa A, Mikhailidis DP, Rizzo M, et al. The influence of atorvastatin on parameters of inflammation left ventricular function, hospitalizations and mortality in patients with dilated cardiomyopathy: 5-year follow-up. Lipids Health Dis. 2013;12:47. doi: 10.1186/1476-511X-12-47. [DOI] [PMC free article] [PubMed] [Google Scholar]