

Abstract
Fat-specific protein 27 gene (FSP27), isolated by screening for genes specifically expressed in fully differentiated mouse adipocytes, belongs to the cell death-inducing DNA fragmentation factor, alpha subunit-like effector family. FSP27 is induced in not only adipose tissue but also the liver of ob/ob mice, and it promotes the development of fatty liver. The FSP27 gene is expressed in a fatty liver-specific manner and is not detected in the normal mouse liver. FSP27 expression is directly regulated by the induction of the hepatic peroxisome proliferator-activated receptor γ (PPARγ) in ob/ob fatty liver. In the present study, expression of hepatic FSP27 mRNA was determined in non-genetic fatty liver models. The FSP27 gene was markedly induced in the high-fat- or methionine- and choline-deficient (MCD) diet-induced fatty liver, but it was not elevated in alcohol-induced fatty liver. Interestingly, the induction of FSP27 mRNA due to the MCD diet was independent of PPARγ levels and completely absent in the liver from PPARγ-null mice. These results suggest that FSP27 mRNA expression in the liver depends on the etiology of fatty liver.
Keywords: fat-specific protein 27, peroxisome proliferator-activated receptor, fatty liver
Fat-specific protein 27 (FSP27) was originally isolated from fully differentiated mouse adipocytes by screening for specifically expressed genes.1,2) The human homolog of FSP27, i.e., cell death-inducing DNA fragmentation factor, alpha subunit-like effector (CIDE) C, was also isolated from a human liver cDNA library.3) Identification of protein sequence and domain structure classified FSP27 as a member of the CIDE protein family, which also includes CIDEA, CIDEB, and CIDEC/FSP27. Adipocyte FSP27 was recently reported to be a lipid droplet (LD)-associated protein that promotes the formation of unilocular LDs.4) Furthermore, FSP27 knockout mice demonstrated a lean phenotype with atrophic adipose tissue due to high-energy expenditure; this mouse line was also resistant to diet-induced obesity and insulin.5,6)
In a previous study, we demonstrated that ob/ob mice that are deficient in the nuclear receptor peroxisome proliferator-activated receptor γ (PPARγ), specifically in the liver (ob/ob-PPARγKO), showed dramatic improvement in severe fatty liver disease, suggesting that hepatic PPARγ promotes the generation of fatty liver.7) To understand the mechanism of PPARγ-dependent fatty liver formation, subtractive cDNA cloning was used to compare downstream target genes of hepatic PPARγ in ob/ob-PPARγKO and ob/ob-PPARγ wild-type mice (ob/ob-PPARγWT); FSP27 cDNA was isolated, and the FSP27 gene was found to be regulated by hepatic PPARγ. FSP27 expression markedly decreased in ob/ob-PPARγKO liver. Furthermore, forced expression of FSP27 by adenovirus in hepatocytes in vivo and in vitro resulted in an increase of LDs through elevated triglyceride (TG) levels, whereas knock-down of FSP27 in ob/ob mice resulted in the loss of hepatic TG.8) These results strongly suggest that FSP27 is directly associated with hepatic TG accumulation.9)
The ob/ob mouse is a well-known genetically leptin-deficient mouse and a typical model for type II diabetes, obesity, and fatty liver. Recently, FSP27 expression was also observed in fatty liver of a new insulin resistant model mouse line, ddY-H.10) Although FSP27 is highly expressed in ob/ob and other mouse models of fatty liver, it remains largely unknown whether other etiologies are involved in the expression of FSP27 in fatty livers. To address this question, the expression of hepatic FSP27 was examined in several fatty liver models, including db/db mice, as well as high-fat (HF)-, alcohol (AL)-, and methionine- and choline-deficient (MCD) diet-fed mice. The FSP27 gene was markedly induced in db/db, and in HF-and MCD-induced fatty liver, but not in AL-induced fatty liver. However, hepatic PPARγ, a master regulator of FSP27, was induced in db/db, and in HF and AL fatty livers, but not in MCD fatty liver. Interestingly, the induction of FSP27 in MCD fatty liver was not observed in liver-specific PPARγKO mouse liver. Thus, our studies reveal new findings on the regulation of FSP27 in fatty liver and suggest that FSP27 expression in fatty liver is influenced by the etiology of this condition.
MATERIALS AND METHODS
Animals
PPARγ liver-specific knockout mice on an ob/ob or normal genetic background were generated using a floxed PPARγ allele and Cre recombinase under the control of the albumin promoter, as previously described.7) Female diabetic db/db mice (C57BL/KsJ-leprdb) and female C57BL/6J wild-type mice were obtained at 8 weeks of age from Nippon CREA (Tokyo, Japan). All animal protocols and studies were performed according to guidelines from the Center for Experimental Animals at Fukuoka University.
MCD and HF Diet Study
Female C57BL/6J mice were randomly divided into 2 groups at 8 weeks of age. One group (n=4) of mice was fed a MCD (Oriental Yeast, Japan) or HF (HFD32; CLEA, Japan) diet ad libitum, while the control group was fed normal chow. Mice were fed with MCD or HF diet for 1 or 2 months, respectively.11,12) Plasma levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were measured using commercial transaminase CII-test Wako kit (Wako Pure Chemical Industries, Ltd.).
Alcohol Diet Study
AL diet was administered according to previous studies.13) Female C57BL/6J mice were randomly divided into 2 groups at 8 weeks of age and fed a 4% ethanol-containing liquid diet (F2LEW; Oriental Yeast, Japan). For controls, mice were fed the same volume of a control liquid diet (F2LCW; Oriental Yeast), prepared by replacing ethanol with isocaloric sucrose. The mice were fed the AL diet for 2 months.
RNA Extraction and Quantitative Real-Time Polymerase Chain Reaction (PCR)
Mouse RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA), and quantitative polymerase chain reaction (QPCR) was performed using complementary DNA (cDNA) generated from 1 µg of total RNA with an AffinityScript QPCR cDNA Synthesis kit (Agilent Technologies). Primer sequences used are listed as follows: FSP27: forward, 5′-ATG AAG TCT CTC AGC CTC CTG-3′ and reverse, 5′-AAG CTG TGA GCC ATG ATG C-3′; PPARγ: forward, 5′-CAT GGC CAT TGA GTG CCG AGT-3′ and reverse, 5′-ACA TCC CCA CAG CAA GGC AC-3′; adipocyte fatty acid-binding protein (aP2): forward, 5′-GAT GCC TTT GTG GGA ACC TG-3′ and reverse, 5′-GAA TTC CAC GCC CAG TTT GA-3′; phosphoenolpyruvate carboxykinase (PEPCK): forward, 5′-CAT ATG CTG ATC CTG GGC ATA AC-3′ and reverse, 5′-CAA ACT TCA TCC AGG CAA TGT C-3′; fatty acid synthase (FAS): forward, 5′-GGA GGT GGT GAT AGC CGG TAT-3′ and reverse, 5′-TGG GTA ATC CAT AGA GCC CAG-3′; sterol regulatory element-binding transcription factor1c (SREBP1c): forward, 5′-GGA GCC ATG GAT TGC ACA TT-3′ and reverse, 5′-AGG AAG GCT TCC AGA GAG GA-3′; cluster of differentiation 36 (CD36): forward, 5′-TGG CCT TAC TTG GGA TTG G-3′ and reverse, 5′-CCA GTG TAT ATG TAG GCT CAT CCA-3′; and acidic ribosomal phosphoprotein P0 (36B4): forward, 5′-AAA CTG CTG CCT CAC ATC CG-3′ and reverse, 5′-TGG TGC CTC TGG AGA TTT TCG-3′ QPCR reactions were carried out using Brilliant III Ultra-Fast SYBR Green QPCR Master Mix (Agilent Technologies) in an Mx3005P Real-Time PCR System (Agilent Technologies). Values of sample mRNAs were normalized to 36B4 mRNA.
Hepatic TG Measurement
To measure liver TG concentrations, total lipids were extracted using the following protocol. Hepatic TG was extracted from 0.1 g of liver and homogenized in 1 mL of 0.1 m KCl. In a new tube, 50 µL of homogenates was diluted by adding 50 µL of 0.1 m KCl, followed by 0.375 mL of chloroform–methanol (1 : 2). Following vortexing, the mixture was kept at room temperature for 5 min, after which 0.125 mL of chloroform was added to the tube and vortexed. Next, 0.125 mL of water was added followed by vortexing again. After centrifugation at 16000×g for 15 min, the organic layer was transferred to another tube. For complete extraction of liver lipid, an additional extraction was repeated using 100 µL of chloroform. Lipids dissolved in organic solvents were dried down and re-suspended in the enzymatic kit buffers prior to TG assays performed using commercial kits (Sekisui Medical).
Statistical Analysis
Experimental values are expressed as mean±standard error of the mean (SEM). Statistical analysis was performed by Student’s t-test for unpaired data, with p<0.05 considered statistically significant.
RESULTS
FSP27 Expression in the ob/ob Mouse Is Induced by PPARγ and Depends on Fatty Liver Formation
To evaluate and quantify the levels of FSP27 gene expression in ob/ob-PPARγKO and ob/ob-PPARγWT livers, QPCR was carried out on cDNAs from each genotype. Expression of FSP27 mRNA observed in ob/ob-PPARγWT liver was 5-fold higher than normal mouse PPARγWT or KO livers. Expression of FSP27 mRNA in ob/ob-PPARγKO liver was only 25% of levels in ob/ob-PPARγWT liver (Fig. 1A). While hepatic PPARγ mRNA in ob/ob-PPARγWT liver was approximately 3.5-fold higher than normal mouse PPARγWT liver (Fig. 1B). FSP27 expression was markedly lower in ob/ob-PPARγKO liver relative to normal mouse PPARγWT liver. To evaluate the potency of PPARγ as an FSP27 inducer, PPARγ was overexpressed in liver-derived AML-12 cells by using a PPARγ-adenoviral vector; AML-12 cells do not constitutively express PPARγ or FSP27. Expression of FSP27 mRNA in AML-12 cells was induced approximately 7-fold and 200-fold without and with the PPARγ-specific ligand, rosiglitazone, respectively, compared with control adenoviral vector (data not shown). These results suggest that FSP27 is a fatty liver-specific gene and that PPARγ expression in the liver is a critical trigger for the ectopic induction of FSP27 mRNA.
Fig. 1. FSP27 Gene Expression Depends on Hepatic PPARγ Expression.

QPCR analyses of FSP27 (A) and PPARγ (B) mRNAs were performed in each genotyped mouse liver. Expression was normalized to 36B4 mRNA, and each bar represents the average±S.E.M. of three separate experiments. Normal, normal genetic background mice; ob/ob, leptin-deficient mice; WT, PPARγ wild-type mice liver; KO, PPARγKO mice liver. Note: ob/ob-PPARγWT mice have fatty liver, whereas ob/ob-PPARγKO mice are normal or have much less fat.7) Significant differences from PPARγWT liver: * p<0.01, ** p<0.001.
FSP27 mRNA Is Induced in db/db, HF, and MCD Fatty Livers, but Not in AL Fatty Liver
The expression of FSP27 mRNA was low in normal liver, but high in ob/ob fatty liver, raising the question of whether FSP27 is also expressed in the fatty livers as a result of different etiologies. To address this question, we used different fatty liver mouse models, namely, HF-, AL-, and MCD diet-fed and leptin receptor-mutated db/db mice. Biochemical parameters for each fatty liver model are summarized in Table 1. TG levels in all livers examined were >2-fold higher than that in the control groups.
Table 1.
Biochemical Parameters of Each Fatty Liver Model Mouse
| Parameters | db/m | db/db | Cont (HF) | HF | Cont (AL) | AL | Cont (MCD) | MCD |
|---|---|---|---|---|---|---|---|---|
| Body weight (g) | 23±0.1 | 55±0.8*** | 22±0.3 | 38±3.3* | 30±0.3 | 24±1.5* | 21±0.7 | 13±0.0*** |
| Liver weight (g) | 11±0.0 | 2.7±0.1** | 0.98±0.0 | 1.4±0.2 | 1.1±0.0 | 1.2±0.1 | 0.87±0.0 | 0.38±0.0*** |
| Liver TG (mg/g liver) | 2.4±0.1 | 6.0±0.9* | 6.6±0.7 | 14±2.0* | 11±1.0 | 22±5.1* | 4.0±0.2 | 11±2.1* |
| AST (IU/L) | 114±8.0 | 146±25 | N.D. | N.D. | N.D. | N.D. | 25±0.59 | 43±5.5* |
| ALT (IU/L) | 30±8.7 | 49±2.7 | N.D. | N.D. | N.D. | N.D. | 2.9±0.34 | 7.4±4.3** |
Each group contains 3–4 mice. Cont, untreated control mice; TG, triglyceride; AST, aspartate aminotransferase activity; ALT, alanine aminotransferase activity. N.D., not determined. Values are mean±S.E.M. Significant differences from db/m mice or control diet:
p<0.05
p<0.01
p<0.001.
The db/db, HF, and MCD fatty livers showed a 12-, 5-, and fold increase, respectively, in FSP27 mRNA compared to control groups (Figs. 2A, B, D). However, no significant differences in FSP27 mRNA were observed between control and AL groups (Fig. 2C). To elucidate the induction mechanism of FSP27 in the fatty livers, PPARγ expression was also evaluated in these samples. The db/db and HF fatty livers displayed approximately a 1.6- and 1.8-fold increase in PPARγ mRNA, which positively correlated with FSP27 mRNA expression (Figs. 2A, B). AL fatty liver showed approximately a 2.8-fold increase in PPARγ mRNA, whereas FSP27 mRNA remained unchanged (Fig. 2C). Conversely, no significant differences in PPARγ mRNA were observed between the control and MCD groups (Fig. 2D); nevertheless, FSP27 mRNA was markedly induced by this diet. Furthermore, aP2 and CD36 mRNAs, which are known as typical targets of PPARγ significantly increased in the db/db, HF, and AL fatty livers. Interestingly, the expression of these genes in MCD fatty liver was unchanged. These findings results suggest that FSP27 gene expression is not always dependent on increases in PPARγ expression, and potentially represent a distinct etiology of fatty liver.
Fig. 2. FSP27 Gene Expression in Fatty Livers Developed by Different Methods.
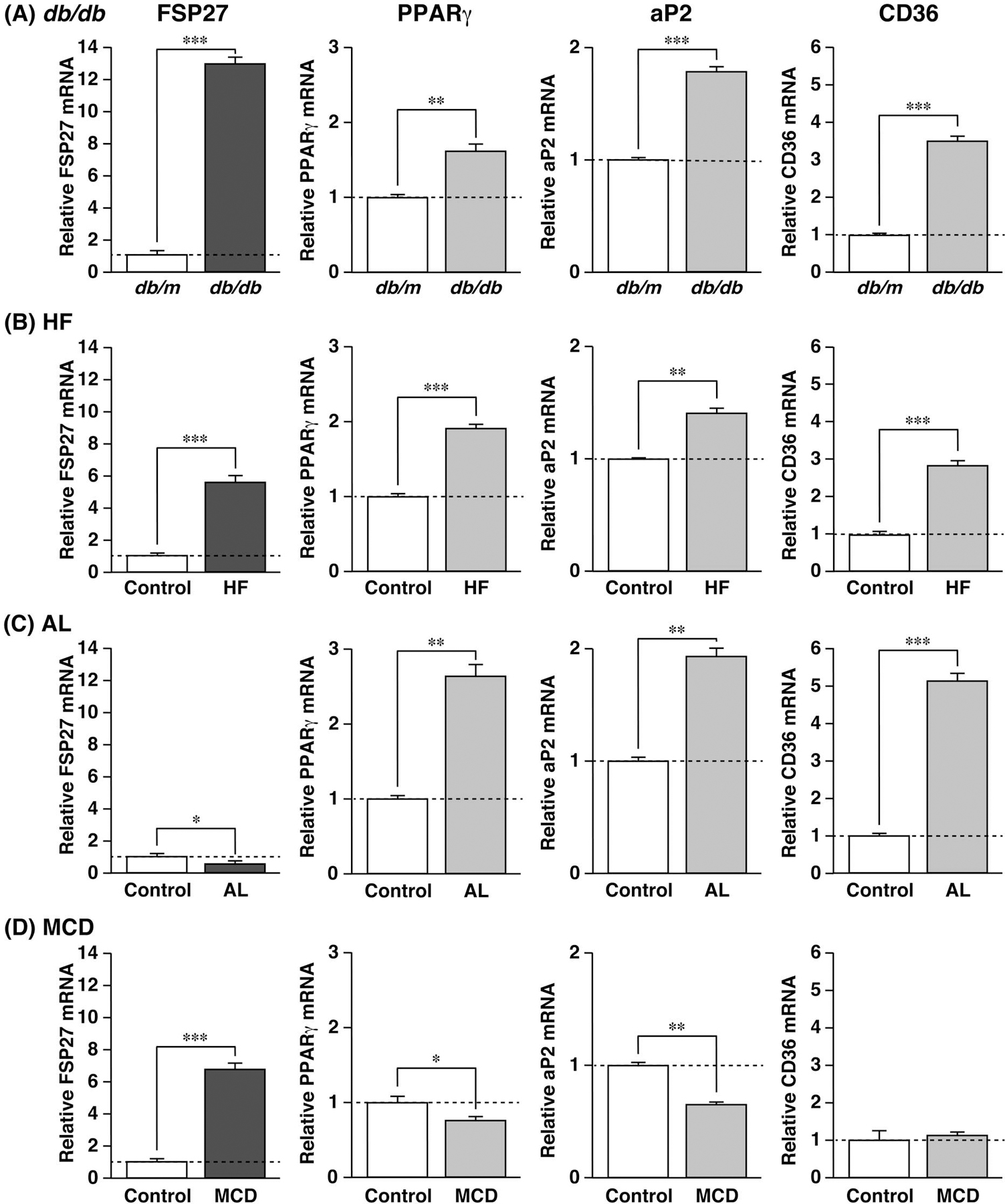
QPCR analyses of FSP27, PPARγ, aP2, and CD36 mRNAs were performed using liver samples from each group. Expression of each mRNA was examined in genetically modified (A) leptin receptor-mutated mice (db/db), as well as in mice fed diets comprising either (B) high fat (HF), (C) alcohol (AL), or (D) lacking methionine and choline (MCD). Expression was normalized to 36B4 mRNA, and each bar represents the average±S.E.M. of 3 individual experiments. Significant differences from db/m mice or control diet: * p<0.05, ** p<0.01, *** p<0.001.
Hepatic PPARγ in MCD Fatty Liver Regulates FSP27 Expression
The MCD diet is frequently used for the generation of fatty liver and nonalcoholic steatohepatitis.14) Unlike the ob/ob, db/db, and HF fatty liver mouse models, mice fed a MCD diet showed a tendency toward low hepatic PPARγ and high FSP27 mRNA levels relative to control mice (Fig. 2D). Thus, to evaluate the potential of hepatic PPARγ as an inducer of FSP27 mRNA in MCD fatty liver, PPARγKO mice from a normal genetic background were administered a MCD diet. This treatment markedly induced the expression of FSP27 mRNA in PPARγWT mice, but not in PPARγKO mice, which displayed expression levels similar to those in mice fed the control diet (Figs. 3A, B). These results suggest that PPARγ in MCD fatty liver is functional and regulates FSP27 mRNA expression.
Fig. 3. Induction of FSP27 mRNA in MCD Fatty Liver Is Potentially Regulated by Hepatic PPARγ.

QPCR analysis of FSP27 (A) and PPARγ (B) mRNAs were performed using liver samples from each genotyped mouse. Expression was normalized to 36B4 mRNA, and each bar represents the average±S.E.M. of 3 individual experiments. Significant differences from PPARγWT liver: * p<0.001.
FSP27 Induced in MCD Fatty Liver Is Not Associated with Hepatic TG Levels
Previous reports demonstrated that FSP27 induced in ob/ob fatty liver promotes the development of fatty liver by stimulating the accumulation of hepatic TG.8) To examine the association between FSP27 and hepatic TG content, TG levels were measured in the livers of PPARγKO mice that had been fed a MCD diet. No significant differences in hepatic TG content were observed between PPARγWT and KO mice that were fed a MCD diet (Fig. 4A).
Fig. 4. Hepatic TG Content in MCD Fatty Liver Is Independent of FSP27 Levels.

(A) Hepatic TG content in PPARγWT and PPARγKO mice fed a MCD diet. Each bar represents the average±S.E.M. of 3 individual experiments. (B) QPCR analysis to assess the effects of PPARγ deficiency on hepatic gene expression. QPCR analyses of FAS, SREBP1c, aP2, and PEPCK mRNAs were performed using liver samples for each genotyped mouse. Expression was normalized to 36B4 mRNA, and each bar represents the average±S.E.M. of 3 individual experiments. Note: no significant differences on all data were observed between PPARγWT and PPARγKO mice fed a MCD diet.
The expression of genes involved in de novo lipogenesis, such as FAS and SREBP1c, or those involved glucose metabolism, such as PEPCK, remained largely unchanged in PPARγWT and KO mice fed a MCD diet (Fig. 4B). Interestingly, mRNA of the aP2 gene also remained unchanged in these mice (Fig. 4B). The MCD diet promotes steatohepatitis and fibrosis in mice; hence, mRNAs levels of the fibrosis marker genes alpha-smooth-muscle actin, tissue inhibitor of metalloproteinase 1, and collagen type I were measured. However, these mRNAs remained unchanged in PPARγWT and PPARγKO mice that had been fed the MCD diet (data not shown).
DISCUSSION
PPARγ expression was the highest in adipose tissue,15,16) and was present at measurable levels in the colon epithelium17,18) and macrophages.19) PPARγ is induced in ob/ob mouse liver, and it is critical for the development of fatty liver.7) Elevated PPARγ levels in ob/ob fatty liver induces FSP27 expression; the induced FSP27 then coordinates with factors in the lipogenic pathway to elevate hepatic triglyceride levels.8) From these earlier studies, we concluded that activation of the PPARγ-FSP27 signal that leads to TG accumulation in the liver is triggered by elevated PPARγ expression in the liver, although the molecular mechanism responsible for elevated hepatic PPARγ expression levels remains unclear. In the present study, we demonstrated that the expression of hepatic FSP27 depends on the etiology of fatty liver. Indeed, FSP27 expression remained unchanged in AL fatty liver, despite elevated PPARγ mRNA. Contrary to the observations in AL fatty liver, it is noteworthy that FSP27 was highly expressed in MCD fatty liver without the elevation of PPARγ mRNA. In addition, the results from liver-specific PPARγ-null mice revealed that FSP27 induced in MCD fatty liver is regulated by constitutively expressed hepatic PPARγ.20)
Because all commercial FSP27 antibodies obtained from the 4 companies were not available for the Western blot analysis, a customized FSP27 antibody was newly prepared from the synthesized FSP27 peptides. Unfortunately, we could not estimate the native FSP27 protein level in MCD fatty liver because the antibody was not specific. However, we believe that the MCD diet induces not only FSP27 mRNA level but also FSP27 protein level as compared with the control diet.
PPARγ and other PPARγ-targets, aP2 and CD36 mRNAs, showed an increase in AL fatty liver, but FSP27 mRNA was unchanged. While, no significant differences in PPARγ, aP2, and CD36 mRNAs were observed between the control and MCD groups, FSP27 mRNA was markedly induced by this diet. The reason for the discrepancy between AL and MCD fatty liver expression of FSP27 are not currently understood. However, our results suggest that the transcriptional regulation of FSP27 by PPARγ differs from that of other PPARγ targets such as aP2 and CD36. We assume that the regulation of hepatic FSP27 requires PPARγ as well as an unknown factor other than PPARγ, and this unknown factor may be repressed in AL fatty liver. It is possible, therefore, that FSP27 mRNA in AL fatty liver remains unchanged even when aP2 and CD36 mRNA levels are elevated. Additionally, this factor is likely to potentially accelerate the function of hepatic PPARγ because FSP27 mRNA levels in MCD fatty liver increased without any increase in PPARγ mRNA levels. Recently, it has been reported that hepatic FSP27 is directly regulated by cAMP response element-binding protein (CREB), independent of regulation by PPARγ.21) It remains unclear whether CREB associates with FSP27 expression in AL fatty liver. We propose that the regulation of FSP27 may be more complicated with the participation of multiple factors.
Recently, a novel regulatory mechanism of hepatic FSP27 through mitogen-activated protein kinase (MAPK) phosphatase-1 (MKP-1) was reported. The study showed that FSP27 mRNA was repressed in fatty livers of both db/db and HF-fed mice lacking MKP-1.22) The function of PPARγ is controlled by multiple factors, including ligand binding and phosphorylation by the MAPKs. Specifically, extracellular signal-regulated kinase (ERK) 1/2 and c-Jun N-terminal kinase (JNK) phosphorylate PPARγ1 and PPARγ2 on Ser84 and Ser112, respectively, resulting in decreased PPARγ ligand binding and transcriptional activity.23,24) Thus, dephosphorylation by MKP-1 restores PPARγ function by decreasing MAPK-dependent phosphorylation of PPARγ, leading to increased FSP27 mRNA levels. However, the mechanism for MKP-1-mediated upregulation of FSP27 is not likely to be involved in the increase in FSP27 mRNA levels in MCD fatty liver observed in this study, because the MCD diet generally induces an increase in the levels of MAPK activity that promotes phosphorylation by JNK and ERK.25,26)
The physiological function of FSP27 in MCD fatty liver remains unclear. We demonstrated that FSP27 expression in the ob/ob fatty liver leads to increased in vitro or in vivo triglyceride levels and that FSP27 is a direct mediator of PPARγ-dependent fatty liver generation.8) However, the present study shows that hepatic TG levels in PPARγWT mice relative to the PPARγKO mice, both of which received the MCD diet, remained unchanged, whereas FSP27 in PPARγKO mouse liver dramatically decreased compared to that in PPARγWT mouse liver. Unlike the situation in ob/ob fatty liver, this discrepancy may be due to a large influence of unknown TG accumulation pathways, with the exception of the PPARγ-FSP27 signaling pathway activity in MCD fatty liver. For example, it is generally known that methionine and choline deficiencies result in the accumulation of TG because of the decrease of very low-density lipoprotein (VLDL) synthesis.14) This mechanism greatly contributes toward TG accumulation, and the decrease of FSP27 in MCD fatty liver may therefore be a negligible influence. While a function of FSP27 other than TG accumulation induced in MCD fatty liver cannot be ruled out, it has been reported that FSP27 induces apoptosis via caspase-3, caspase-7, and caspase-9, and triggers the release of cytochrome c from the mitochondria, which implies that the mitochondrial pathway is involved in FSP27-induced apoptosis.27–29) Indeed, the consumption of a MCD diet induced caspase-3 activity and apoptosis in liver.30) More studies are needed to elucidate the function of FSP27 in MCD fatty liver.
Recent studies have demonstrated CIDEC (human homolog of mouse FSP27) gene expression in the livers of obese human subjects before and after undergoing gastric bypass surgery.31) This procedure reduced obesity through loss of body weight. One year after surgery, the hepatic steatosis grade of subjects significantly decreased relative to pre-surgery conditions, and CIDEC expression in the liver showed a decline of >60%. Further, PPARγ expression was significantly decreased in the post-surgery liver. These data suggest a positive correlation between human CIDEC expression and hepatic steatosis grade. Elucidation of the physiological function of the CIDEC/FSP27 pathway potentially may lead to new therapeutic opportunities for inhibiting TG accumulation in the liver.
Acknowledgments
This work was supported by a Grant from the Mochida Memorial Foundation for Medical and from KAKENHI (22590253).
Footnotes
The authors declare no conflict of interest.
REFERENCES
- 1).Danesch U, Hoeck W, Ringold GM. Cloning and transcriptional regulation of a novel adipocyte-specific gene, FSP27. CAAT-enhancer-binding protein (C/EBP) and C/EBP-like proteins interact with sequences required for differentiation-dependent expression. J. Biol. Chem, 267, 7185–7193 (1992). [PubMed] [Google Scholar]
- 2).Williams PM, Chang DJ, Danesch U, Ringold GM, Heller RA. CCAAT/enhancer binding protein expression is rapidly extinguished in TA1 adipocyte cells treated with tumor necrosis factor. Mol. Endocrinol, 6, 1135–1141 (1992). [DOI] [PubMed] [Google Scholar]
- 3).Liang L, Zhao M, Xu Z, Yokoyama KK, Li T. Molecular cloning and characterization of CIDE-3, a novel member of the cell-death-inducing DNA-fragmentation-factor (DFF45)-like effector family. Biochem. J, 370, 195–203 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4).Puri V, Konda S, Ranjit S, Aouadi M, Chawla A, Chouinard M, Chakladar A, Czech MP. Fat-specific protein 27, a novel lipid droplet protein that enhances triglyceride storage. J. Biol. Chem, 282, 34213–34218 (2007). [DOI] [PubMed] [Google Scholar]
- 5).Nishino N, Tamori Y, Tateya S, Kawaguchi T, Shibakusa T, Mizunoya W, Inoue K, Kitazawa R, Kitazawa S, Matsuki Y, Hiramatsu R, Masubuchi S, Omachi A, Kimura K, Saito M, Amo T, Ohta S, Yamaguchi T, Osumi T, Cheng J, Fujimoto T, Nakao H, Nakao K, Aiba A, Okamura H, Fushiki T, Kasuga M. FSP27 contributes to efficient energy storage in murine white adipocytes by promoting the formation of unilocular lipid droplets. J. Clin. Invest, 118, 2808–2821 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6).Toh SY, Gong J, Du G, Li JZ, Yang S, Ye J, Yao H, Zhang Y, Xue B, Li Q, Yang H, Wen Z, Li P. Up-regulation of mitochondrial activity and acquirement of brown adipose tissue-like property in the white adipose tissue of fsp27 deficient mice. PLoS ONE, 3, e2890 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7).Matsusue K, Haluzik M, Lambert G, Yim SH, Gavrilova O, Ward JM, Brewer B Jr, Reitman ML, Gonzalez FJ. Liver-specific disruption of PPARgamma in leptin-deficient mice improves fatty liver but aggravates diabetic phenotypes. J. Clin. Invest, 111, 737–747 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8).Matsusue K, Kusakabe T, Noguchi T, Takiguchi S, Suzuki T, Yamano S, Gonzalez FJ. Hepatic steatosis in leptin-deficient mice is promoted by the PPARgamma target gene Fsp27. Cell Metab, 7, 302–311 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9).Okumura T Role of lipid droplet proteins in liver steatosis. J. Physiol. Biochem, 67, 629–636 (2011). [DOI] [PubMed] [Google Scholar]
- 10).Satoh H, Ide N, Kagawa Y, Maeda T. Hepatic steatosis with relation to increased expression of peroxisome proliferator-activated receptor-γ in insulin resistant mice. Biol. Pharm. Bull, 36, 616–623 (2013). [DOI] [PubMed] [Google Scholar]
- 11).Sugimoto H, Okada K, Shoda J, Warabi E, Ishige K, Ueda T, Taguchi K, Yanagawa T, Nakahara A, Hyodo I, Ishii T, Yamamoto M. Deletion of nuclear factor-E2-related factor-2 leads to rapid onset and progression of nutritional steatohepatitis in mice. Am. J. Physiol. Gastrointest. Liver Physiol, 298, G283–G294 (2010). [DOI] [PubMed] [Google Scholar]
- 12).Yokota T, Kinugawa S, Hirabayashi K, Matsushima S, Inoue N, Ohta Y, Hamaguchi S, Sobirin MA, Ono T, Suga T, Kuroda S, Tanaka S, Terasaki F, Okita K, Tsutsui H. Oxidative stress in skeletal muscle impairs mitochondrial respiration and limits exercise capacity in type 2 diabetic mice. Am. J. Physiol. Heart Circ. Physiol, 297, H1069–H1077 (2009). [DOI] [PubMed] [Google Scholar]
- 13).Nakajima T, Kamijo Y, Tanaka N, Sugiyama E, Tanaka E, Kiyosawa K, Fukushima Y, Peters JM, Gonzalez FJ, Aoyama T. Peroxisome proliferator-activated receptor alpha protects against alcohol-induced liver damage. Hepatology, 40, 972–980 (2004). [DOI] [PubMed] [Google Scholar]
- 14).Hebbard L, George J. Animal models of nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol, 8, 35–44 (2011). [DOI] [PubMed] [Google Scholar]
- 15).Chawla A, Schwarz EJ, Dimaculangan DD, Lazar MA. Peroxisome proliferator-activated receptor (PPAR) gamma. adipose-predominant expression and induction early in adipocyte differentiation. Endocrinology, 135, 798–800 (1994). [DOI] [PubMed] [Google Scholar]
- 16).Tontonoz P, Hu E, Graves RA, Budavari AI, Spiegelman BM. mPPAR gamma 2: tissue-specific regulator of an adipocyte enhancer. Genes Dev, 8, 1224–1234 (1994). [DOI] [PubMed] [Google Scholar]
- 17).Lefebvre AM, Chen I, Desreumaux P, Najib J, Fruchart JC, Geboes K, Briggs M, Heyman R, Auwerx J. Activation of the peroxisome proliferator-activated receptor gamma promotes the development of colon tumors in C57BL/6J-APCMin/+ mice. Nat. Med, 4, 1053–1057 (1998). [DOI] [PubMed] [Google Scholar]
- 18).Saez E, Tontonoz P, Nelson MC, Alvarez JG, Ming UT, Baird SM, Thomazy VA, Evans RM. Activators of the nuclear receptor PPARgamma enhance colon polyp formation. Nat. Med, 4, 1058–1061 (1998). [DOI] [PubMed] [Google Scholar]
- 19).Moore KJ, Fitzgerald ML, Freeman MW. Peroxisome proliferator-activated receptors in macrophage biology: friend or foe? Curr. Opin. Lipidol, 12, 519–527 (2001). [DOI] [PubMed] [Google Scholar]
- 20).Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor gamma and promotes adipocyte differentiation. Cell, 83, 813–819 (1995). [DOI] [PubMed] [Google Scholar]
- 21).Vila-Brau A, De Sousa-Coelho AL, Goncalves JF, Haro D, Marrero PF. Fsp27/CIDEC is a CREB target gene induced during early fasting in liver and regulated by FA oxidation rate. J. Lipid Res, 54, 592–601 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22).Roth Flach RJ, Zhang L, Qin H, Bennett A. Loss of MAP kinase phosphatase-1 protects from hepatic steatosis by repression of CIDEC/fat-specific protein 27. J. Biol. Chem, 286, 22195–22202 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23).Burns KA, Vanden Heuvel JP. Modulation of PPAR activity via phosphorylation. Biochim. Biophys. Acta, 1771, 952–960 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24).Hu E, Kim JB, Sarraf P, Spiegelman BM. Inhibition of adipogenesis through MAP kinase-mediated phosphorylation of PPARgamma. Science, 274, 2100–2103 (1996). [DOI] [PubMed] [Google Scholar]
- 25).Schattenberg JM, Singh R, Wang Y, Lefkowitch JH, Rigoli RM, Scherer PE, Czaja MJ. JNK1 but not JNK2 promotes the development of steatohepatitis in mice. Hepatology, 43, 163–172 (2006). [DOI] [PubMed] [Google Scholar]
- 26).Min AK, Kim MK, Kim HS, Seo HY, Lee KU, Kim JG, Park KG, Lee IK. Alpha-lipoic acid attenuates methionine choline deficient diet-induced steatohepatitis in C57BL/6 mice. Life Sci, 90, 200–205 (2012). [DOI] [PubMed] [Google Scholar]
- 27).Yonezawa T, Kurata R, Kimura M, Inoko H. Which CIDE are you on? Apoptosis and energy metabolism. Mol. Biosyst, 7, 91–100 (2011). [DOI] [PubMed] [Google Scholar]
- 28).Liu K, Zhou S, Kim J-Y, Tillison K, Majors D, Rearick D, Lee JH, Fernandez-Boyanapalli RF, Barricklow K, Houston MS, Smas CM. Functional analysis of FSP27 protein regions for lipid droplet localization, caspase-dependent apoptosis, and dimerization with CIDEA. Am. J. Physiol. Endocrinol. Metab, 297, E1395–E1413 (2009). [DOI] [PubMed] [Google Scholar]
- 29).Kim J-Y, Liu K, Zhou S, Tillison K, Wu Y, Smas CM. Assessment of fat-specific protein 27 in the adipocyte lineage suggests a dual role for FSP27 in adipocyte metabolism and cell death. Am. J. Physiol. Endocrinol. Metab, 294, E654–E667 (2008). [DOI] [PubMed] [Google Scholar]
- 30).Dixon LJ, Berk M, Thapaliya S, Papouchado BG, Feldstein AE. Caspase-1-mediated regulation of fibrogenesis in diet-induced steatohepatitis. Lab. Invest, 92, 713–723 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31).Hall AM, Brunt EM, Klein S, Finck BN. Hepatic expression of cell death-inducing dffa-like effector C in obese subjects is reduced by marked weight loss. Obesity (Silver Spring), 18, 417–419 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]