

Abstract
Introduction:
Phase I and II trials provide the initial human safety and tolerability data for new drugs. However, the methods for presenting toxicity data are not standardized. Clinicians often first encounter these data at professional conferences. We sought to characterize how the burden of adverse events (AE) is reported at the largest professional conference in clinical oncology.
Methods:
We collected toxicity data from all lung cancer-associated phase I and II trial presentations and posters at the American Society for Clinical Oncology annual meetings 2017-2019. We captured AE features including the minimum incidence utilized for reporting; whether AEs shown were treatment-emergent or treatment-related, grouped by organ system or separated by individual descriptors; whether combined or separated across dose levels when a dose escalation component was included; and whether dose-limiting toxicities, serious AE, dose reduction rules and denominators for laboratory tests were described.
Results:
209 trials were analyzed. There was wide variability in toxicity reporting practices. Six different thresholds for reporting AE of any grade were used. Treatment-related AEs were reported twice as frequently as treatment-emergent AEs. Toxicities were as likely to be reported across dose level as by dose level. Terms such as dose-limiting toxicity and serious AE were rarely defined. Dose reduction rules and denominators for laboratory tests were never defined.
Conclusion:
Standardization of methods for reporting toxicities could improve the quality and ease of comparability of data on adverse effects in early phase therapeutic trials. A minimal AE data disclosure template is proposed.
Keywords: lung cancer, toxicity, adverse effects, conference
Introduction
The clinical influence of early (phase I-II) trial data in oncology has dramatically increased with the advent of accelerated drug approval and break-through drug classification within the Food and Drug Administration (FDA) Safety and Innovation Act (SIA) in 2012, which allowed drug approval based upon the results of compelling, if limited, initial clinical data. 1 National guideline organizations, such as the National Comprehensive Cancer Network (NCCN), have also produced recommendations on the basis of early phase trial data. 2,3 Professional conferences are a major avenue for dissemination of early phase (phase I-II) trial data, as well as a key source of continuing medical education for oncologists who increasingly rely on early phase trial data to understand not just the efficacy but the safety and tolerability (‘toxicity’) of novel therapies. Recently there has been a call to improve communication of toxicities for novel therapies, particularly in oncology. 4
Despite this, there is no systematic description of current practices for reporting toxicities at conferences and there may be large variation in how safety and tolerability are presented in early phase trials. Listings of treatment-emergent adverse events (AEs), which do not include a filter of causality introduced by the investigators, would be expected to be associated with higher rates of AE reporting. In contrast, listings of treatment-related AEs do include such a filter and would be expected to be associated with lower rates of AE reporting from the same trial. In order to facilitate practical displays of AEs, frequency filters may also be used, such that only AEs occurring in a certain number or percentage of patients may be shown. The use or lack of use of combined AE terms (for example combining impairment in memory and concentration as “cognitive effects”) in the presence of frequency filters could cause toxicities to fall above or below the presentation threshold and facilitate over or under-representation of toxicities, respectively. Finally, when multiple different doses of an agent or agents are being explored in the same trial, showing aggregated toxicity across all dose levels, rather than only at the subsequently designated ‘recommended phase II dose and schedule’ (RP2D), could be considered appropriate as a greater total number of patients will have contributed to the dataset. Alternatively, such an approach may be criticized because it could over-estimate toxicity through the contributions from cohorts dosed above the RP2D or under-estimate it from the dilutional impact of data from cohorts dosed below the RP2D.
The American Society for Clinical Oncology (ASCO) annual meeting is the largest conference in oncology, with approximately 5,500 US-based oncologists attending the 2019 ASCO annual meeting, which is equivalent to 40% of the 13,216 medical hematologists/oncologists registered with Medicare, together with large numbers of international oncologists. 5,6 We characterized parameters used to display toxicity in lung cancer-associated trials at the ASCO annual meetings 2017-2019.
Materials and methods
We reviewed all abstracts on phase I and II medical trials for the years 2017-2019 available at the ASCO virtual meeting website under the sections “Lung cancer” and “Developmental therapeutics.” Abstracts must have had a poster or slides available in order to assess detailed toxicity information. We downloaded all materials for presentations on phase I-II trials that presented toxicity data and included at least one patient with lung cancer. We excluded presentations that did not provide toxicity data or investigated radiation alone or in combination due to the different toxicity profile of radiation compared to systemic agents (see Figure 1).
Figure 1:
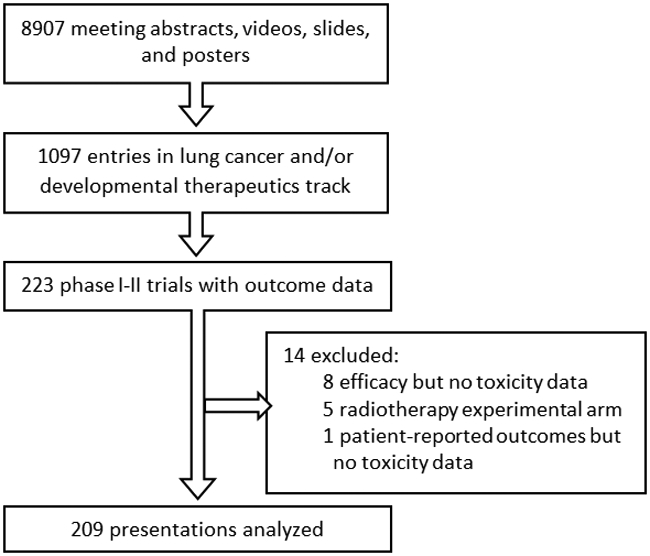
Consort diagram
We developed a list of parameters that could potentially affect interpretation of toxicity results based on experience with implementing early phase clinical trials. This list was iteratively revised and expanded after exploratory analysis of one year of ASCO presentations, resulting in the reporting elements listed in Table 1. We categorized trials labeled as phase I/II as phase I if the trial included a dose escalation component and as phase II if the trial did not include dose escalation. Due to the increased sensitivity of a small absolute number of patients compared to an incidence threshold for reporting toxicities and the qualitative difference in number of toxicities reported by trials that used a threshold of 1-2 patients compared to an incidence, we categorized reporting thresholds as 1-2 patients and then remainder of thresholds for reporting as 5-10%, 15-20% and ≥25%. We considered a term to represent consolidated conditions if an organ system term (e.g. gastrointestinal disorders) was used, if the author specifically annotated that a term combined different AEs, or if the author referenced use of standard terminology (see below) but reported more generalized terms (e.g. elevated liver function in place of elevated AST or ALT.) We considered terminology standardized if terms were consistent with Common Terminology Criteria for Adverse Events (CTCAE) or the Medical Dictionary for Regulatory Activities (MedDRA) preferred terms.
Table 1:
Elements of reporting toxicities and outcome assignments
| Reporting element | Outcome assignments |
|---|---|
| What minimum incidence was utilized for reporting toxicities of any grade? | 1 patient, 2-3 patients, 5% incidence, 10% incidence, 15-20% incidence, ≥25% incidence, not reported |
| What minimum incidence was utilized for reporting toxicities grade ≥ 3? | 1 patient, 2-3 patients, 5% incidence, 10% incidence, 15-20% incidence, ≥25% incidence, not reported |
| Were treatment-related toxicities reported? | Y/N/Not reported |
| Were treatment-emergent toxicities reported? | Y/N/Not reported |
| Was a dose-escalation component included? | Y/N |
| Were adverse events combined across dose levels if dose-escalation was included? | Y/N |
| Were dose-limiting toxicities reported if the trial included a dose-escalation component? | Y/N |
| Were key dose-limiting toxicity criteria described? | Y/N |
| Were serious adverse events reported? | Y/N |
| Were key serious adverse event criteria described? | Y/N |
| Were adverse events leading to treatment change reported? | Y/N |
| Were standardized terminology (CTCAE or MedDRA) utilized? | Y/N |
| Were grouped adverse event terms utilized? | Y/N |
| Was overall incidence of adverse events at organ system level reported? | Y/N |
| Was the total number of patients with any adverse event any grade reported? | Y/N |
| Was the total number of patients with any adverse event grade ≥ 3 reported? | Y/N |
We utilized unordered multinomial logistic regressions to evaluate if the minimum incidence for reporting toxicities varied by phase (dichotomized as phase I and phase II) or trial size (dichotomized as <40 patients and ≥40 patients), using 1-2 patients as the referent group. Phase and trial size were evaluated separately. For all other elements listed in Table 1, we utilized logistic regression to evaluate if these characteristics varied by phase (using phase I as referent group) or trial size (using 1-2 patients as referent group) and evaluated the magnitude of effect. The p-values reported are not corrected for multiple hypothesis testing. The Bonferroni p-value corrected for multiple hypothesis testing of the 16 elements in Table 1 is 0.003, thus only regression results with p<0.003 would have met rigorous standards for detecting differences between groups, which was not considered practical given the dataset available for analysis.
Results
The ASCO 2017-2019 Annual Meetings provided 8907 meeting slides and posters. Of these, 1370 presentations were on lung cancer or developmental therapeutics. Phase I-II trials with outcome data comprised 223 entries, of which 209 disclosed toxicity data and were included in our analysis (see Figure 1). Of these 209 presentations, 120 (57%) were phase I and 89 (43%) were phase II trials (see Table 2). Trials that broadly enrolled patients on the basis of having a solid tumor, that included at least one patient with lung cancer, were the subject of 68/209 (33%) presentations while the remaining 141/209 (67%) presentations were more specific in including patients with lung cancer. Median trial size was 38 patients; phase I trials enrolled a median of 34 patients while phase II trials enrolled a median of 49 patients (mean number of patients was 53 and 86 patients, respectively). While trial phase and trial size were associated, they were not colinear and occasionally had unique associations with other trial characteristics as evaluated below.
Table 2:
Trial characteristics (N=209)
| Characteristic | Number of trials (%) | Median number of patients (mean) |
|---|---|---|
| All trials | 209 (100%) | 38 (range 5-550); mean 67 |
| <40 patients | 109 (52%) | 24 (mean 25) |
| ≥ 40 patients | 100 (48%) | 80 (mean 114) |
| Cancer type | ||
| Lung | 141 (67%) | 39 (mean 74) |
| Multiple cancer types | 68 (33%) | 37 (mean 53) |
| Phase | ||
| I | 120 (57%) | 34 (mean 53) |
| II | 89 (43%) | 49 (mean 86) |
Frequency of AEs
There was wide variability in thresholds used for reporting AEs, with 12 different thresholds used to report AEs of any grade that ranged from 1 patient to ≥40% incidence. These were simplified to the five categories in Table 3. Utilizing unordered multinomial logistic regression and the five categories of reporting thresholds in Table 3, larger trial size (≥40 patients) was associated with a 1.6 to 2 fold increase in the odds of using a reporting threshold of 5-10% or 15-20% incidence compared to 1-2 patients for AE any grade (OR 1.6 and 2.0 respectively, overall p<0.001). Phase II trials did not tend to use higher incidence (i.e. less sensitive) thresholds for reporting compared to phase I trials (OR −0.3 and 0.6 respectively for odds of using a reporting threshold of 5-10% or 15-20% incidence compared to 1-2 patients, overall p=0.03 using unordered multinomial regression).
Table 3:
Threshold for reporting AE any grade (n=209)
| Number of patients with AE |
Number of trials with AE any grade by threshold |
Association with trial size and phase (OR, 95% CI)* |
|
|---|---|---|---|
| Phase II vs I | Trial size ≥40 vs <40 |
||
| 1-2 patients | 46 (22%) | - | - |
| 5-10% incidence | 66 (32%) | −.3 (−1.0-0.5) | 1.6 (0.7-2.4) |
| 15-20% incidence | 30 (14%) | 0.6 (−0.3-1.6) | 2.0 (0.9-3.0) |
| >=25% incidence | 5 (2%) | −15 (−1713-1684) | 0.9 (−1.0-2.8) |
| Not reported | 62 (30%)* | 0.3 (−0.5-1.0) | 1.2 (0.4-2.1) |
| Total | 209 | ||
Bold indiates p<0.05
Among the 141/209 trials that provided the thresholds for reporting both AEs of any grade and grade ≥3, two different methods were used for displaying toxicity counts of different grades: a) 100/141 trials reported grade ≥3 AE incidence only where the overall incidence across grades reached a given threshold (see column 2 of Table 4) and b) 41/141 trials used a lower incidence threshold for reporting grade ≥3 AEs compared to AEs of any grade (see columns 3 and 4 of Table 4). As an example of the former method, a trial reported AEs of any grade that reached an incidence of 10% and reported how many of those events were grade ≥3. Consequently, this excluded AEs grade ≥3 that did not meet a 10% incidence overall. In the latter method, a trial reported AEs of any grade that reached 10% incidence but also reported all AEs grade 3 or higher (i.e. a threshold of 1 patient).
Table 4:
Concordance of reporting thresholds between AEs any grade and grade≥3 (n=141*)
| Number of patients with AE |
Same threshold AE any grade and grade ≥3 (N=100) |
Discordant thresholds between AE any grade and grade ≥3 (N=41)** |
|
|---|---|---|---|
| Any grade | Grade ≥3 | ||
| 1-2 patients | 39 (39%) | 5 (12%)† | 32 (78%) |
| 5-10% incidence | 44 (44%) | 19 (46%) | 9 (21%) |
| 15-20% incidence | 15 (15%) | 14 (34%) | - |
| >=25% incidence | 2 (2%) | 3 (7%) | - |
| Total | 100 | 41 | 41 |
141/209 (67%) displayed reporting thresholds for both AEs of any grade and grade ≥3. 68 trials did not disclose the threshold used for reporting either or both AEs of any grade and grade ≥3.
41 studies had different thresholds for AEs of any grade versus grade≥3 which are represented in both columns.
These 5 studies used a threshold of 2 patients to report AEs of any grade and 1 patient to report AEs grade ≥3
Attribution of AEs
182/209 (87%) of presentations indicated whether AEs were treatment-emergent or treatment-related (see Table 5). Treatment-related AEs were reported in 139/209 (67%) presentations. We found no evidence that the odds of reporting treatment-related AEs were affected by phase or trial size (p = 0.08 and 0.49, respectively). Overall treatment-emergent AEs were included in 70/209 (33%) presentations. Phase II trials were associated with a lower odds of reporting treatment-emergent AEs compared to phase I trials (OR 0.3, 95% CI 0.2-0.6, p<0.01.) 39/209 (19%) reported both treatment-emergent and treatment-related AEs; 27/209 (13%) did not specify if AE were treatment-related or emergent.
Table 5:
Reporting element results and association with trial phase and size
| Odds ratio (95% CI)* | |||
|---|---|---|---|
| Reporting element | Yes/Total trial number (%) |
Phase II vs I | Trial size ≥40 vs <40 |
| Were treatment-related toxicities reported?** | 139/209 (67%) | 0.5 (0.3-1.1); p=0.08 | 0.8 (0.4-1.6); p=0.49 |
| Were treatment-emergent toxicities reported?** | 70/209 (33%) | 0.3 (0.2-0.6); p<0.01 | 1.0 (0.5-1.8); p=0.94 |
| Was a dose-escalation component included? | 99/209 (47%) | N/A† | 0.4 (0.2-0.7); p<0.01 |
| Were adverse events combined across dose levels if dose-escalation was included? (n=99) | 46/99 (46%) | N/A† | 1.9 (0.8-4.5); p=0.12 |
| Were dose-limiting toxicities reported if the trial included a dose-escalation component? (n=99) | 67/99 (68%) | N/A† | 0.7 (0.3-1.7); p=0.45 |
| Were key dose-limiting toxicity criteria described? | 21/67 (31%) | N/A† | 1 (0.3-3.1); p=0.95 |
| Were serious adverse events reported? | 68/209 (33%) | 0.4 (0.2-0.7); p<0.01 | 0.9 (0.5-1.6); p=0.65 |
| Were key serious adverse event criteria described? | 0/68 (0%) | - | - |
| Were adverse events leading to treatment change reported? | 89/209 (43%) | 0.9 (0.5-1.5); p=0.59 | 2.5 (1.4-4.4); p=<0.01 |
| Were standardized terminology (CTCAE or MedDRA) utilized? | 180/209 (86%) | 1.1 (0.5-2.3); p=0.89 | 1 (0.4-2.1); p=0.96 |
| Were grouped adverse event terms utilized? | 48/209 (23%) | 1.6 (0.9-3.2); p=0.13 | 1.9 (1-3.7); p=0.05 |
| Was overall incidence of adverse events at organ system level reported? | 7/209 (3%) | 1.8 (0.4-8.4); p=0.43 | 2.8 (0.5-14.9); p=0.20 |
| Was the total number of patients with any adverse event any grade reported? | 92/209 (44%) | 0.8 (0.5-1.5); p=0.54 | 1.7 (1-3); p=0.05 |
| Was the total number of patients with any adverse event grade ≥3 reported? | 92/209 (44%) | 1 (0.6-1.7); p=0.96 | 2.6 (1.5-4.5); p<0.01 |
Using unadjusted logistic regression. Bold indicates p-values less than 0.05.
27 studies did not specify if AE were treatment-related or treatment-emergent; these 27 studies were excluded from logistic regression.
Dose-escalation by definition includes only phase I trials.
Grouping of AEs
The overall incidence of any AE of any grade was reported in 92/209 (44%) studies and overall incidence of AEs grade 3 or higher in 92/209 (44%) studies (see Table 5). 48/209 (23%) included a term that represented a consolidated group of disease entities, such as infection instead of component terms of urinary tract infection, lung infection, etc. Of these 48 studies, 7 presented AEs only by organ system (e.g. endocrine, skin, gastrointestinal, etc) without identifying components (see Table 5).
Dose level grouping, definitions of actionable AEs and relevant AE denominators
Standardized terminology, consistent with CTCAE or MedDRA preferred terms, was used in 180/209 (86%) presentations (Table 5). All but 4 studies reported toxicity grade. Of 99 trials that included a dose escalation component, 46 presented AEs only as a combined dataset across all dose levels, rather than separated out by individual dose levels. Dose-limiting toxicities (DLTs) were reported in 67/99 (68%) of trials with a dose-escalation component but only 21 of these 67 defined the criteria for DLT. Serious AEs were reported in 68/209 (33%) trials, none of which defined the trial-specific criteria for serious AE. AEs that resulted in dose decrease, delay or discontinuation were reported in 89/209 (43%) trials and were 2.5 times more likely to be reported in trials with ≥40 patients (95% CI 1.4-4.4, p<0.01). While the individual toxicities leading to decisions on dose interruptions were often noted, the trial-specific criteria for such decisions (e.g. grades ≥3, at 1 or greater than one occurrence in a patient, etc) were never provided in the available presentations. Although not part of our initial data element capture (Table 1), we noted that the denominator was not included for laboratory abnormalities and we could not distinguish whether all patients were tested for these laboratory parameters or only a subset based on other criteria, such as clinical suspicion.
Discussion
Early phase trial data at clinical conferences may represent both the first time that the efficacy and toxicity of a new drug or drug combination are shown to medical providers and the same dataset may well be the dominant one available for education when highly active drugs are rapidly licensed. Beginning with CONSORT recommendations in 1996, consistent efficacy reporting standards to permit cross-trial and cross-drug comparisons have been developed and periodically refined, such as the Response Evaluation Criteria in Solid Tumors (RECIST) guidelines and Response Assessment in Neuro-Oncology (RANO) for CNS efficacy endpoints.7-9 However, standardization of toxicity reporting has received less attention. Guidelines for reporting harms were published eight years after the first CONSORT recommendations, but these have been poorly adhered to in oncology trial publications. 10,11 No guidelines exist for presentation of toxicity data at conferences. ASCO provides recommendations for abstract submission, but of 26 recommendations for reporting data from phase I-III trials, the only recommendation related to safety and tolerability is that phase I trial abstracts should report dose-limiting toxicities, a criterion which was not met in nearly a third of ASCO presentations in our dataset.12 Variation in toxicity reporting practices could dramatically alter the initial perceptions of the safety and tolerability of new drugs and drug combinations.
Overall, we found highly variable practices for reporting toxicities in this analysis of presentations at the largest annual conference in clinical oncology. Trials reported treatment-related AEs twice as frequently as treatment-emergent AEs (67% vs 33%, respectively). Conceivably, utilization of treatment-related approaches might be a sensible way of improving the signal to noise ratio in early phase trials, particularly when an experimental approach is being given as part of a combination with a standard therapy with known associated toxicities of its own. Alternatively, novel interactions with the standard therapy which increase the standard therapy’s toxicity, or AEs that were not expected to be related to a novel agent by its known mechanism of action could be under-reported as a result of this approach. Arguably, treatment-emergent AEs are less subject to bias, whereas treatment-related AEs, may have particular value in differentiating the effects of the novel agent over and above those associated with other agents with known toxicities in a combination. However, in most cases, we were unable to determine exactly how ‘treatment-related AEs’ were defined within the trials of combination therapies studied among the ASCO presentations.
The reporting of toxicity combined across dose levels, rather than broken down by dose level, occurred in nearly half of trials with a dose escalation component (46%). While in theory this could either over-estimate or under-estimate toxicities at the recommended dose because of contamination by patients treated above or below the recommended dose, in general it is probably more likely to under-represent AEs, as more patients are likely to have been treated at dose levels below the non-tolerated dose than at the non-tolerated dose.
The utilization of frequency filters to limit the presentation of toxicity data may be a rational way of presenting only the most relevant data but was highly variable ranging from 1 patient to ≥ 40% of patients for all grade toxicities and from 1 patient to ≥10% of patients for grade 3 or greater toxicities (Table 3). Excluding 62 trials that did not specify what frequency filter was used, the majority of trials (112/147, 76%) used a frequency filter of 1 patient to 10% of patients for AE any grade.
Data presented at professional conferences are not intended to supply the compendium of information required to fully inform treatment decisions. The material provided by posters and slides are incomplete snapshots of trial data in which authors may not have the space to present the amount of data needed to fully represent drug toxicity. These materials are also accompanied by oral presentations in which the authors may anticipate supplying the additional context needed to interpret their slides and posters. Nonetheless, conferences represent the first exposure to novel agents for many providers and researchers and have the potential to influence therapeutic and research choices. Furthermore, oral only disclosures of information are unlikely to be as widely promulgated as the ‘hard’ data accessed from the poster or slide presentation itself. We suspect that the content of early phase clinical presentations at the ASCO annual meetings in relation to lung cancer is likely to reflect information provided across multiple tumor types at many, if not most, other general oncology conferences such as the annual meetings of the European Society of Medical Oncology (ESMO), American Association of Cancer Research (AACR), and disease specific conferences such as the World Conference on Lung Cancer. Consequently, we believe our findings will be applicable to all communication of clinical trial research.
In general, a lack of standardization in toxicity data presentation leaves the field open to intentional or unintentional abuse. If any grade, or even high grade, toxicity events are not shown because they are variably defined as below the limit required for presentation, the opportunity to show key clinical data worthy of communication to future prescribers, decision-makers or other clinical influencers may be missed. Relevant toxicity may be pushed below such presentation thresholds simply through the utilization of treatment-related versus treatment-emergent attribution filters; by combining data across multiple dose levels potentially ‘diluting’ the appearance of the toxicity signal of the recommended dose level; or by not utilizing appropriate grouped terminologies such that highly-related toxic effects are fractionated such that each then appears below a pre-defined presentation threshold. As a recent example of the latter phenomenon, the ASCO 2018 phase II presentation of the toxicities associated with lorlatinib, a third generation ALK/ROS1 tyrosine kinase inhibitor, only presented treatment-related AEs occurring in ≥10% of patients. Lorlatinib is now known to have adverse impacts on numerous higher CNS functions, including memory, speech patterns, sleep patterns, seizures, mood, visual and auditory perceptions and cognition. In the phase I and II ASCO presentations on lorlatinib in 2017 and 2018, only some grouped terms were used, including cognitive effects (23.3% in 2018) and mood effects (16% in 2018).13 The final prescribing information states that overall CNS effects, including mood, cognition, speech, vision and sleep-related AEs occurred in 54% of patients receiving lorlatinib.14 Vision disorders, sleep effects and speech effects were not included in the initial ASCO presentation possibly because these fell below the 10% frequency threshold for the ASCO dataset or may not have been considered treatment-related at the time.
Data on dose delays, dose modifications and dose discontinuations are potentially highly useful as they could normalize the differences between reporting toxicities into pragmatic, clinically impactful rates of toxicity and were reported in 43% of trials. However, without knowing the specific dose modification rules within a given trial and the events that lead to these dose reductions, even these data can be misleading. In our analysis, the trial-specific criteria for dose modifications were never provided in the available presentations. The same challenges in toxicity presentation also extend to later phase trials. For example, the dose reduction rate for alectinib in the first-line ALEX trial versus crizotinib was 16% but was 29% for brigatinib in the comparable ALTA-1L trial.15,16 Pragmatically, this suggests that alectinib is better tolerated than brigatinib. However, 63% of the dose reductions for brigatinib in ALTA-1L were only for laboratory abnormalities defined as actionable within the protocol by specific blood levels rather than by symptoms.16 Without specification of whether, for example, a laboratory abnormality was symptomatic, whether all or just a subset of patients were tested for a given laboratory abnormality, and what the similarities or differences in protocol-mandated requirements for dose reduction are between studies, concluding one agent is more “tolerable” than another based on trial dose reduction rates, in fact, may or may not be valid. Dose reduction rates in the real world, post-licensing, may offer additional insights into tolerability if dose decision making in this setting were freer than the kind of constraints used in a trial’s protocol. Alternatively, other measures of ‘overall tolerability’ may be important such as health-related quality of life (HRQOL) scores. Intriguingly, in this regard, despite brigatinib’s higher dose reduction rate in ALTA-1L than that of alectinib in the ALEX trial, only brigatinib and not alectinib was able to show significantly superior benefit over crizotinib in terms of HRQOL.17,18
Importantly, during our analyses we noted that when laboratory abnormalities were presented, whether all patients were tested for these laboratory parameters or only a subset based on other criteria, such as clinical suspicion, was not described. To illustrate the potential need to define a relevant denominator (of all patients or only those tested) when quoting the percentage of patients with a laboratory abnormality, in the reports of reduced testosterone seen among patients treated with crizotinib, non-industry studies in both single and multi-center settings reported a drug-associated drop in 100% and 84%, respectively, of men in whom these levels were checked.19,20 In contrast, in the industry-provided medical information available from Prizer, the manufacturer of crizotinib, this rate is reported as occurring in <1% of patients (across both sexes combined) in both the PROFILE 1014 and 1007 studies.21 Beyond the obvious issue of including both sexes in the denominator, neither of these industry trial sources included routine assessments of testosterone in enrolled patients, suggesting the denominator used would be more informative if modified from all patients treated with the drug to only those assessed for the laboratory value, and, in this example, also separated out by the patient’s sex.22,23
We propose the introduction of minimum elements for adequate reporting of toxicities for phase I and II clinical trials, listed in Table 6. Such an approach could also be extended and optimized across later phase trial datasets and in any approved drugs prescribing information. By requiring authors to standardize or, at the very least, mandatorily disclose their AE reporting methodology, the quality of trial data shared at conferences and the resulting dissemination of relevant, comparable data could be significantly improved.
Table 6:
Suggested minimum disclosure elements for adverse event reporting in oncology clinical trials
|
Acknowledgements
Funding
The work of DRC was partially supported by the University of Colorado Lung Cancer SPORE (P50CA058187).
The work of DG and DES was partially supported by the University of Colorado Cancer Center Support Grant (P30CA046934).
Footnotes
Conflicts of interest
Dr. Simons has nothing to disclose.
Mr. Smith has nothing to disclose.
Dr. Gao has nothing to disclose.
Dr. Camidge reports Honoraria from Roche/Genentech and Takeda Clinical Trials (institutional) sponsored by Pfizer, Roche/Genentech and Takeda.
References
- 1.Sherman RE, Li J, Shapley S, Robb M, Woodcock J. Expediting drug development--the FDA's new “breakthrough therapy” designation. N Engl J Med. 2013;369(20):1877–1880. [DOI] [PubMed] [Google Scholar]
- 2.FDA approves third oncology drug that targets a key genetic driver of cancer, rather than a specific type of tumor [press release]. Silver Spring, MD: Food and Drug Administration, August 15, 2019. 2019. [Google Scholar]
- 3.Ettinger DS WD, et al. NCCN Guidelines Version 7.2019 Non-Small Cell Lung Cancer. Plymouth Meeting, PA: August 30, 2019. 2019. [Google Scholar]
- 4.Sacks CA, Miller PW, Longo DL. Talking about Toxicity - “What We've Got Here Is a Failure to Communicate”. N Engl J Med. 2019;381(15):1406–1408. [DOI] [PubMed] [Google Scholar]
- 5.ASCO. Previous Meeting Demographics. American Society of Clinical Oncology; https://meetings.asco.org/am/previous-meeting-demographics. Published 2019. Accessed October 17, 2019. [Google Scholar]
- 6.Physician Compare. Data.Medicare.Gov; 2019. https://data.medicare.gov/data/physician-compare. Accessed October 17, 2019.
- 7.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45(2):228–247. [DOI] [PubMed] [Google Scholar]
- 8.Wen PY, Chang SM, Van den Bent MJ, Vogelbaum MA, Macdonald DR, Lee EQ. Response Assessment in Neuro-Oncology Clinical Trials. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2017;35(21):2439–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Begg C, Cho M, Eastwood S, et al. Improving the quality of reporting of randomized controlled trials. The CONSORT statement. Jama. 1996;276(8):637–639. [DOI] [PubMed] [Google Scholar]
- 10.Sivendran S, Latif A, McBride RB, et al. Adverse event reporting in cancer clinical trial publications. J Clin Oncol. 2014;32(2):83–89. [DOI] [PubMed] [Google Scholar]
- 11.Ioannidis JPA, Evans SJW, Gøtzsche PC, et al. Better Reporting of Harms in Randomized Trials: An Extension of the CONSORT Statement. Annals of Internal Medicine. 2004;141(10):781–788. [DOI] [PubMed] [Google Scholar]
- 12.Abstract biostastical guidelines. American Society for Clinical Oncology; https://meetings/asco.org/am/abstract-biostatistical-guidelines. Accessed 11/11/2019. [Google Scholar]
- 13.Besse B SB, Felip E et al. 2. Lorlatinib in patients with previously treated ALK+ advanced non-small cell lung cancer (NSCLC): updated efficacy and safety. Paper presented at: ASCO 2018 Annual Meeting; June, 2018, 2018; Chicago, IL. [Google Scholar]
- 14.Inc P. Full prescribing information [Lorlatinib]. The Food and Drug Administration; https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/210868s000lbl.pdf. Published 2018. Accessed 11/13/2019. [Google Scholar]
- 15.Huber RM KD, Ahn MJ et al. Brigatinib in crizotinib-refractory ALK+ non-small cell lung cancer: efficacy updates in the exploratory analysis of CNS ORR and overall ORR by baseline brain lesion status. Paper presented at: ASCO 2018 Annual Meeting; June 2018, 2018; Chicago IL. [Google Scholar]
- 16.Camidge DR, Kim HR, Ahn MJ, et al. Brigatinib versus Crizotinib in ALK-Positive Non-Small-Cell Lung Cancer. N Engl J Med. 2018;379(21):2027–2039. [DOI] [PubMed] [Google Scholar]
- 17.Perol M, Pavlakis N, Levchenko E, et al. Patient-reported outcomes from the randomized phase III ALEX study of alectinib versus crizotinib in patients with ALK-positive non-small-cell lung cancer. Lung Cancer. 2019;138:79–87. [DOI] [PubMed] [Google Scholar]
- 18.Campelo RC LH, Perol M et al. Health-related quality of life (HRQoL) results from ALTA-1L: Phase 3 study of brigatinib vs crizotinib as first-line (1L) ALK therapy in advanced ALK+ non-small cell lung cancer (NSCLC). Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2019;37(15):1.30422740 [Google Scholar]
- 19.Weickhardt AJ, Rothman MS, Salian-Mehta S, et al. Rapid-onset hypogonadism secondary to crizotinib use in men with metastatic nonsmall cell lung cancer. Cancer. 2012;118(21):5302–5309. [DOI] [PubMed] [Google Scholar]
- 20.Weickhardt AJ, Doebele RC, Purcell WT, et al. Symptomatic reduction in free testosterone levels secondary to crizotinib use in male cancer patients. Cancer. 2013;119(13):2383–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pfizer. Product monograph: Xalkori crizotinib capsules. Pfizer; https://www.pfizer.ca/sites/default/files/201904/XALKORI_PM_215123_27March2019_E.pdf. Published 2019. Updated 27 March 2019 Accessed 1/13/2020, 2020. [Google Scholar]
- 22.Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368(25):2385–2394. [DOI] [PubMed] [Google Scholar]
- 23.Solomon BJ, Mok T, Kim DW, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. 2014;371(23):2167–2177. [DOI] [PubMed] [Google Scholar]