

Abstract
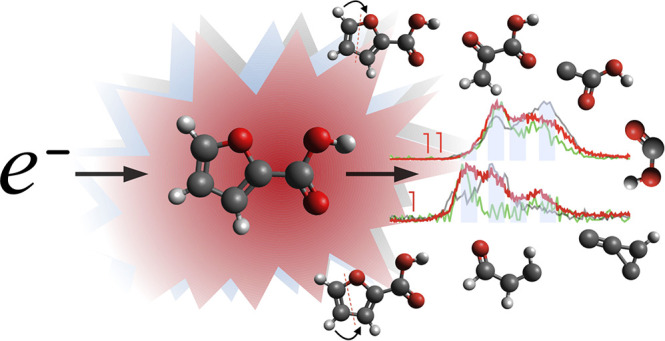
We report a dissociative electron attachment study to 2-furoic acid (C5H4O3) isolated in a gas phase, which is a model molecule consisting of a carboxylic group and a furan ring. Dissociation of furan by low energy electrons is accessible only via electronic excited Feshbach resonances at energies of incident electrons above 5 eV. On the other hand, carboxylic acids are well-known to dissociate via attachment of electrons at subexcitation energies. Here we elucidate how the electron and proton transfer reactions induced by carboxylation influence stability of the furan ring. Overlap of the furan and carboxyl π orbitals results in transformation of the nondissociative π2 resonance of the furan ring to a dissociative resonance. The interpretation of hydrogen transfer reactions is supported by experimental studies of 3-methyl-2-furoic and 5-methyl-2-furoic acids (C6H6O3) and density functional theory (DFT) calculations.
1. Introduction
Electron scattering by furan-containing molecules has attracted significant attention in recent years (e.g., refs (1−9)). This is caused by a more general interest in electron interactions with biomolecular systems due to their involvement in the radiation chemistry of living tissues.10−13 The molecular structure of furan can be considered as a building block of biomolecules, particularly sugar and more complex structures such as nucleic acids. Carboxylic acids are studied as a model components of complex biomolecules as well,14−16 particularly as important prototypical systems for hydrogen bonds.17 It has been proposed theoretically18,19 and demonstrated experimentally for the formic acid,20 oxalic acid,21 or pyruvic acid22 dimers that the formation of a double hydrogen bond via carboxylic groups can induce ultrafast proton transfer between two bonded monomers. The double hydrogen bond structure of dimer has also been observed for 2-furoic acid23−25 (2FA)—a molecule studied here, which possesses both a heterocyclic ring and a carboxylic group.
Substitution on a furan ring can significantly affect its ability to attach low energy electrons as well as its susceptibility to fragment. Already the first studies of the electron attachment to furan, tetrahydrofuran, and fructose1 demonstrated that fructose is a much better electron scavenger than bare or hydrogenated furan. A large (>10 × 10–20 m2) cross section for electron attachment to deoxyribose was then reported by Ptasińska et al.26 Another example is the severe damage caused to the furan ring after electron transfer from cytidine in a complex nucleoside, deoxycytidine monophosphate.27 Substitution of furan hydrogens by electronegative fluorine atoms not only enhances its ability to attach low energy electrons at subexcitation energies,28 but also enables breakage of the ring by these species.29 Moreover, several examples of chemical intramolecular mechanisms of hydrogen migration in the charged state of the furan and its substituents were observed.30,31 Finally, substitution can affect the chemical and biochemical interaction of the ring structures.32,33 It is therefore interesting to study how substituion of the ring influences the interaction with low energy electrons, particularly for 2FA with several important applications.
2FA, C5H4O3, is also known as pyromucic acid or α-furancarboxylic acid or furan-2-carboxylic acid. 2FA can be synthesized by the oxidation of either furfuryl alcohol or furfural.34 An industrial way of achieving 2FA involves the Cannizzaro disproportionation reaction of furfural in an aqueous sodium hydroxide solution.35,36 The main industrial applications of 2FA include pharmaceuticals, agrochemicals, fragrances, and flavors.37−39 Due to its highly nonlinear optical properties, 2FA finds its application in optic technologies.40,41 Of particular importance for the present study is a proposed antitumor activity of 2FA.42 Electron affinic antitumor drugs exhibit strong synergistic effects with radiation, which have been subscribed to the interaction with secondary low energy electrons.43
Presently, in the literature, there is a paucity of experimental electron scattering studies with 2FA, likely because of its low volatility and sensitivity to air, complicating its handling and sublimation for gas phase studies. The use of a direct insertion probe technique for sample sublimation in the present experiment significantly improves the handling of 2FA and may be advantageous also for other studies of low volatility samples.
In addition to 2FA measurements we investigated 3-methyl-2-furoic acid (3M-2FA) and 5-methyl-2-furoic acid (5M-2FA) molecules. These measurements helped us to better understand the mechanism of 2FA dissociation upon electron attachment.
2. Experimental Section
The electron attachment experiments were performed using the TEM–QMS (trochoidal electron monochromator–quadrupole mass spectrometer) setup originally constructed by the Michael Allan group at the University of Freiburg44 and used for studies of isolated molecules. After its transport to the J. Heyrovský Institute of Physical Chemistry (Czech Republic), it has been modified for studies with low volatility compounds.45 Here, we briefly describe the apparatus and focus more on the procedure used to prepare an isolated gas phase target of 2FA.
The TEM–QMS used in the present study was placed in a vacuum chamber with a background pressure of 10–7 mTorr, and it was able to measure the energy dependent and mass-selected yields of stable anions forming after electron attachment. The TEM–QMS employs two main parts: (i) a trochoidal electron monochromator (TEM) that delivers a monochromatic electron beam (20–40 nA) and (ii) a detection system based on a quadrupole mass spectrometer (QMS) capable of separating formed negative ions according to their m/z. The electron beam was steered by means of electrostatic potentials and an axial collimating magnetic field into the collision chamber filled with the gas sample. Anions were drawn from the collision region, at right angle to the electron beam, into the detector by an extraction field of electrostatic lens system, which directed them into a quadrupole mass filter. The resolution of the quadrupole was ∼100 M/ΔM. Considering that H– is an important fragmentation channel of carboxylic acids,46,47 it is important to say that the combination of ion optics and quadrupole mass filter in the present experiment does not allow us to detect this fragmentation channel. The collision chamber vapor pressures during the course of the experiment were in the range 1 × 10–4–5 × 10–4 mTorr as monitored by a Penning gauge. The electron energy calibration was made by measuring the 4.4 eV band of O– production from CO2.48 The presence of low energy electrons was verified by measuring the parent anion signal from SF6, which has an extremely strong signal in this range and offers a reliable energy reference.49,50
The sample of 2FA, a beige crystalline powder, was obtained from Sigma-Aldrich and had a stated purity of 98%. The methyl-substituted 3-methyl-2-furoic acid and 5-methyl-2-furoic acid were also purchased from Sigma-Aldrich with stated purity of 97%. Loading the powder sample into the probe for measurements was done in the AtmosBag glovebag filled with Ar (Messer, purity 99.998%), which ensured an isolated and controlled environment during the filling in.
Many low volatility compounds require thermal treatment (degassing) prior to their sublimation for gas phase studies. Construction of the direct insertion probe (see Figure 1) allows both degassing and sublimation of the sample. In the probe, a solid state sample is placed in a glass reservoir inside a copper sublimation oven, which is separated from the collision region only by an ∼1 cm capillary, which significantly reduces the travel path of the compound and offers a direct sample temperature control by means of the resistively heated oven. The sublimation of the sample for gas phase studies in the present experiment was achieved by heating the oven to 80 °C. After we place the sample inside the chamber, we wait until the base pressure, at 10–7 mbar, is stable. Then, we slowly heat the sample, at the same time monitoring the increasing pressure by a residual gas analyzer (electron impact ionization MS) to identify the composition of the vapor. We have not observed any sample impurities with the studied molecules. An additional inlet line from outside the vacuum can be used for gases and volatile samples, connected to the reaction chamber through the same capillary. This inlet is advantageous for calibration of the instrument. The insertion probe is equipped with an auxiliary valve (see detail in Figure 1) with the connection to the fore-line pump that enables filling up the sample without a need to vent the vacuum chamber to air.
Figure 1.
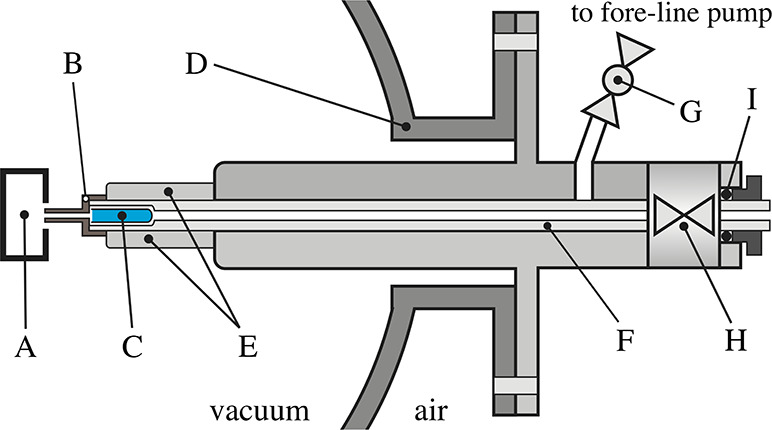
Schematic diagram of the direct insertion probe: A, collision chamber; B, thermocouple; C, glass reservoir with sample; D, vacuum chamber; E, sublimation oven; F, movable insertion probe; G, fore-line pump ball vacuum valve; H, vacuum valve; I, rubber sealing O-ring.
3. Theoretical Methods
The present experimental work is supported by density functional theory (DFT) quantum chemical calculations performed with the Gaussian 09 software.51 First, 2FA geometries were optimized using the hybrid B3LYP method52,53 and standard cc-PVTZ or aug-cc-PVTZ basis set. Further, threshold energies were estimated for the dissociative electron attachment (DEA) reactions at the same level of theory. The reliability of the approach is well-known from previous studies (e.g., refs (22) and (54)). Individual threshold energy calculations were based on the formula
| 1 |
where EMa– and EMb are the energies
of the breakup products in the DEA process,
for a newly formed stable negative ion and its neutral part, respectively. EM is the energy of the parent molecule. In the
reactions involving more than two neutral fragments (n > 1), the neutral part Mb can be replaced by a sum 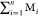 . As
will be discussed in more detail in section 4.3, this method
can provide information on resonance energies and enable one to trace
possible reaction pathways. For several fragments, surprisingly complex
rearrangement reactions are required so that the calculated threshold
correctly agrees with the onset of the experimental signal. Individual
threshold energies are then obtained from the sums of electronic and
zero-point energies.
. As
will be discussed in more detail in section 4.3, this method
can provide information on resonance energies and enable one to trace
possible reaction pathways. For several fragments, surprisingly complex
rearrangement reactions are required so that the calculated threshold
correctly agrees with the onset of the experimental signal. Individual
threshold energies are then obtained from the sums of electronic and
zero-point energies.
4. Results and Discussion
Theoretically, we explored four minimum energy conformers for 2FA. First, the carboxylic moiety can be oriented in two ways with respect to the furan ring, and second, the hydrogen atom in the carboxylic acid can be oriented either toward or away from the ring. Graphical representations of these conformers and calculated relative energy levels are shown in Table 1. Choosing the conformer does not affect much the position of the theoretical threshold energy. For the purpose of our analysis it is not critical to choose which energy conformer we use in the calculations. The calculated energy thresholds between two, most energetically separated conformers introduce the difference only by ∼230–280 meV, depending on the basis set. For DEA threshold calculations, we use the lowest energy conformer of 2FA.
Table 1. Relative Electronic Energies (in kJ mol–1) of 2FA Acid Conformers Compared with Available Theoretical Results from Halasa et al.55.

The DEA fragmentation processes to 2FA lead to a large number of anionic products. Generally, DEA is a two-step process that can be described by the following equation:
| 2 |
where we can distinguish creation of a transient parent anion M#– of a target molecule M and then fragmentation into a detected negative fragment ion Ma– and an undetected neutral fragment Mb. In the following sections, we will describe the mechanisms of partial dissociation channels of the 2FA after electron attachment.
4.1. Hydrogen Loss Channel
The dominant fragmentation channel results in the formation of the stable, closed shell anion, (M–H)−, by the loss of a neutral hydrogen atom:
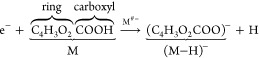 |
3 |
The DEA reaction resulting in hydrogen loss from a molecule itself56,57 or from the hydroxyl moiety of a carboxylic group is a well-known process.3,22,58 In the present case, it occurs primarily at an electron energy of ∼1.2 eV as demonstrated in the energy dependent ion yields in Figure 2. In 2FA, there are four hydrogen atoms which may be cleaved from the parent ion. The threshold energy calculations in Table 3 confirm that only the hydrogen in the carboxylic acid group fulfills the energy onset of the low energy resonance.
Figure 2.
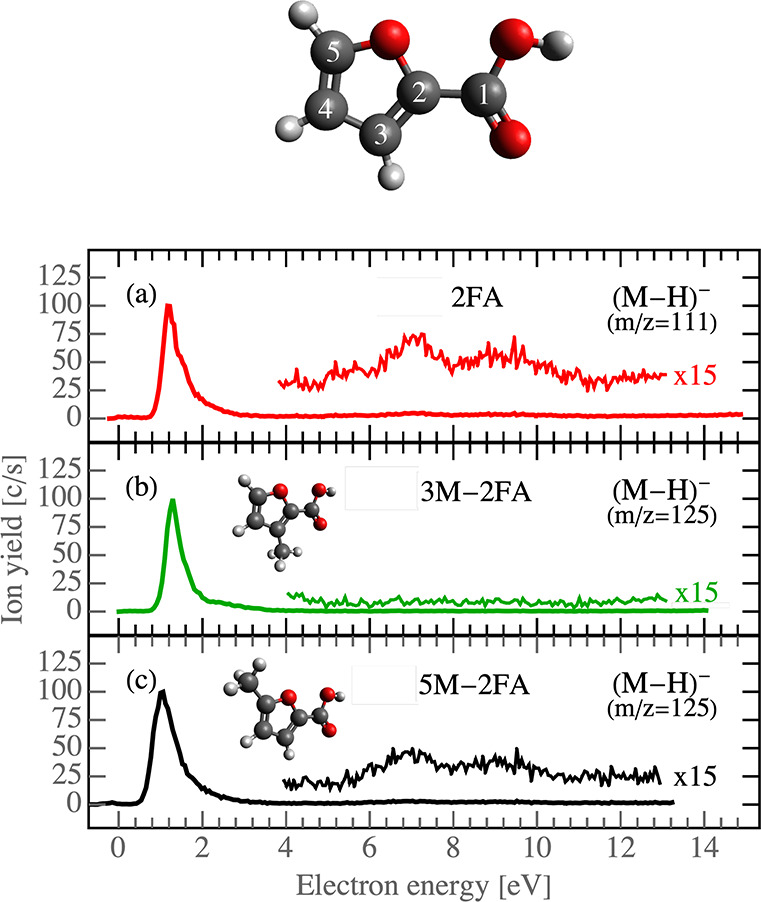
2FA molecule model with depicted carbon numbers (top); ion yields for the most prominent DEA fragments of 2FA, 3M-2FA, and 5M-2FA (bottom).
Table 3. B3LYP/aug-cc-pVTZ Threshold Energies for Individual DEA Fragmentation Channels.

Cleavage of hydrogen atoms from the furan ring can be achieved only by significant energy input of above 2.5 eV. Such fragmentation can occur via core excited Feshbach resonances observed as a broader and overlapping structures in the (M–H)− anion yield from 5 to 11 eV (Figure 2). Based on the calculations, cleavage of any of the hydrogen atoms of the furan ring is possible at these energies. However, our supporting experiments with methyl-substituted compounds indicate that only the hydrogen from position 3 is cleaved. We can see that Feshbach resonances are well visible in the spectra of 2FA and 5M-2FA (Figure 2a,c), but they disappear in the spectrum of 3M-2FA (Figure 2b), where the hydrogen in position 3 is replaced with a methyl group.
The differences in computed energies for 2FA indicate that hydrogen loss from position 3 may be favored by ≈0.5 eV in comparison to the other ones. While the energy may explain the preference for the channel in the case of ergodic dissociation of the transient anion, it does not explain why other channels are closed. An alternative explanation may be that the (M–H)− anion of 3M-2FA, at these higher energies, is metastable and undergoes dissociation to smaller anionic fragments as will be discussed in sections 4.2 and 4.3.
4.2. Electronically Excited Feshbach Resonances
In Figure 3, we present the ion yield curves for DEA channels of the three targets together, in decreasing intensity order. In Figure 3, the values of the calculated threshold energies for the individual dissociation channels are included to enable comparisons with the theoretical evaluations of the dissociation thresholds. Table 3 presents the threshold energies calculated for several possible reaction pathways for each detected DEA channel. The uncertainty in the identification of the reaction pathways is caused by the fact that the energy threshold depends on the neutral fragmentation products. Hence, finding an agreement with the experimental signal onset requires calculation of several reaction pathways and often an inclusion of complex rearrangement reactions.
Figure 3.
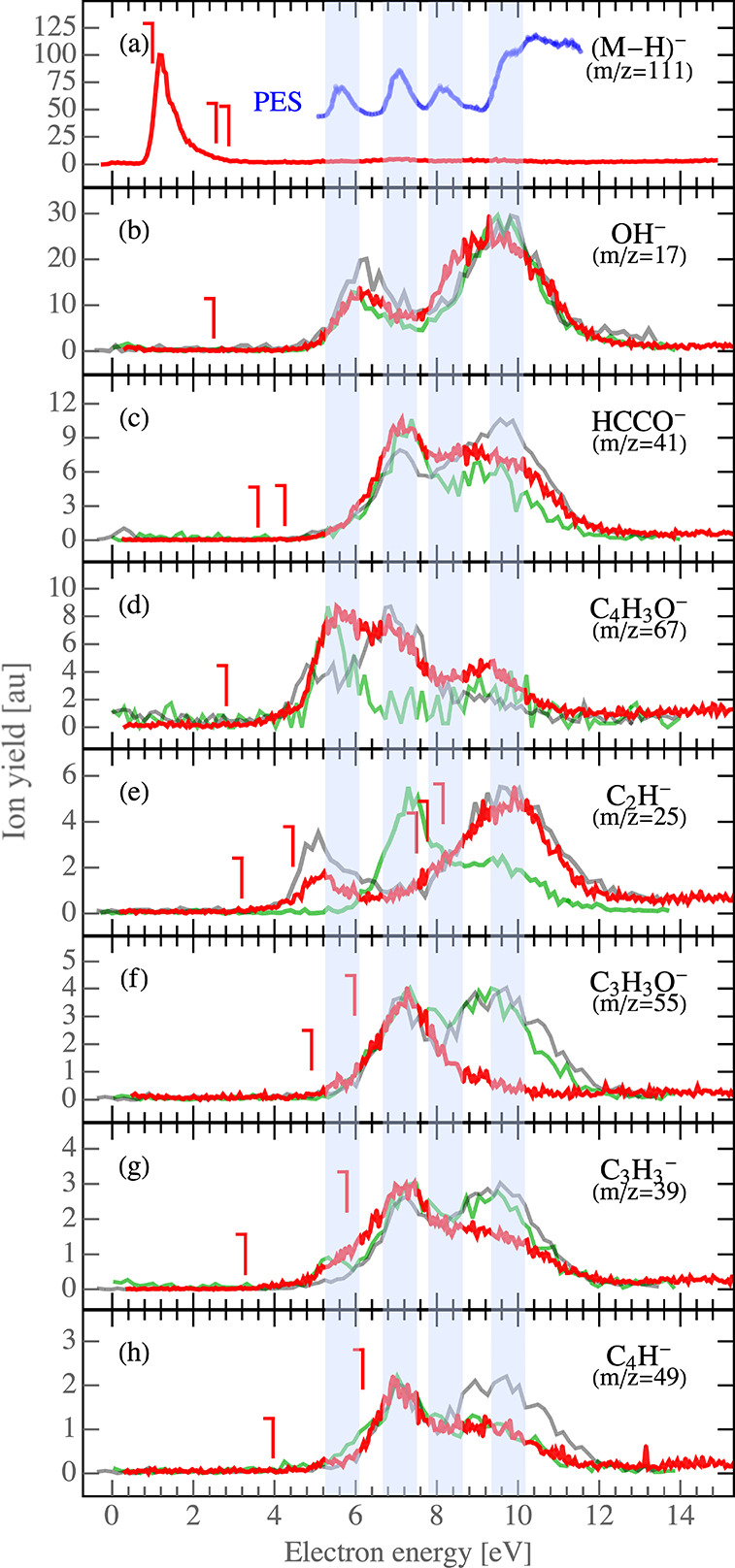
Ion yields for individual DEA fragments of 2FA (red), 3M-2FA (semitransparent green), and 5M-2FA (semitransparent black). The calculated threshold energies (from Tables 3 and 4) for individual dissociation channels refer to 2FA. The blue curve in panel a is a photoelectron spectrum from ref (59) shifted by 4 eV. The blue transparent vertical stripes are to enhance the relation between the PES and the DEA spectra.
The observed anionic fragments, with the exception of (M–H)− (1.2 eV resonance) and C2H– (discussion in section 4.3), are formed above 5 eV. In Figure 3, we can distinguish three dominant energy bands, centered around 5.3, 6.7, and 9.4 eV, which correspond to core-excited resonances. These can fall into two main classes: the Feshbach resonances with a cationic core and two electrons in the s2 Rydberg orbital or core-excited shape resonances (possibly supported by a dipole moment of the parent excited state). The first class has been shown to be operative in a broad range of molecules;60−62 the second class has been shown to be operative in selected organic molecules.63 In the case of simple amides, the question of which mechanism is operative sparked a recent discussion.64−66 A useful tool to identify the contribution of the Feshbach resonances is to compare the DEA spectra with an available photoelectron spectrum (PES) and semiempirical molecular orbital (MO) calculations for 2FA. The relationships between energies available from photoelectron and electron attachment spectroscopies is well-known. The spectra are typically shifted by a constant energy of a few electronvolts.2,22,67−70 The PES data reveals three distinct bands in the region of 9–13 eV. We use the same MO notation as Klapstein et al.59 to denote these bands. The first band in the PES, located at 9.32 eV, is related to ionization from the ring π3 orbital. The second, composed of two overlapping bands, the ring π2 orbital and the nO′ band of the carbonyl group, appears at 10.74 eV. The third band, due to out-of-plane lone-pair orbital on the hydroxyl oxygen, nO, is located at 11.90 eV. The next higher, unassigned, band is visible in the experimental photoelectron spectrum at 13.5 eV. These bands were predicted by MO calculation using the modified neglect of diatomic overlap (MNDO) method, and by the HAM/3 method.59 Here we focus only on comparisons to the experimental PES. When we collate the DEA bands above 5 eV with the PES, we can assign bands as in Table 2.
Table 2. Assignment of the Bands from the PES Spectrum to DEA2FA; DEAFN Band Locations from Furana.
| DEA2FA → PES | (assignment) | DEAFN |
|---|---|---|
| 5.3 → 9.32 | (π3) | 6 |
| 6.7 → 10.74 | (nO′, π2) | |
| 9.4 → 13.50 | 10.5 |
All data in electronvolts.
The energy differences between corresponding DEA and PES bands can be estimated to ∼4 eV. Typically, previous reports found the energy shift in the range 3.3–4.5 eV depending on the molecular target.2,22,60 The PES band assigned to the nO″ band is not visible in the DEA spectrum; however, it may contribute to the adjacent ion yield signals, making them broader. Using the same energy shift, as for other bands, we can estimate its position to be at ∼7.9 eV in the DEA spectrum. The correlation with the photoelectron spectrum indeed suggests that the observed DEA bands are mediated by the formation of Feshbach resonances with two electrons in the same Rydberg orbitals. It should be noted that for a molecule of this size with a complicated excited structure other core-excited processes can be operative at overlapping energies.
We can compare the present results with those of furan (FN)1 to gain more insight. FN fragments into three pronounced DEA channels of C2HO– (m/z = 41), (FN–H)− (m/z = 67), and C3H3– (m/z = 39) fragments, in decreasing intensity order. The present work allows us to observe the quantitative agreement in theses anionic fragments between FN and 2FA; the same ion yield intensity order can be observed in Figure 3, panels c, d, and g, respectively. Concerning the energetics, the FN ion yields band peaks at 6 and 10.5 eV. The first band is sharp and intense, and the latter is less intense and broader. Such bands are observed also in the spectrum of 2FA, slightly shifted toward lower energies and observed as 5.3 and 9.4 eV bands, respectively (see Table 2). However, we can see in Figure 3 that, except for the m/z = 67 fragment corresponding to the loss of carboxyl group from 2FA, the intensity of the first 5.3 eV band is negligible. For all ions, a new and intense band appears in the spectrum of 2FA (in Figure 3) at an energy of 6.7 eV, which corresponds to the overlapping π2 orbital of the ring and carbonyl oxygen as discussed above. We can see that initially bound excited state corresponding to electron removal from the π2 orbital in FN is transformed to dissociative state after carboxylation in 2FA.
In addition to the fragments, which were previously observed for furan, we also observe new fragments in 2FA. The most intense is OH– (m/z 17) resulting from dissociation of the carboxylic group. Another fragment that can be directly assigned to the carboxylic group is COOH–. However, this fragment was observed with low intensity at the detection limit of our setup, so the spectrum is not shown here. Even though one can expect formation of these ions at energies typical for DEA to formic acid,46 this is not the case. The main Feshbach resonance in formic acid peaks at 7.6 eV71−73 corresponding to the electron removal from the nO″ orbital of the hydroxyl oxygen. The OH– and COOH– fragments in the present case, however, peak at 6 and 9 eV, where bare HCOOH dissociates with only low yields.73 Either such electronic states localized on HCOOH group become dissociative in 2FA or the entrance states are that of the furan ring. Correct assignment of these bands will require high level ab initio calculations.
Substitution of the carboxylic group then also opens several new fragmentation channels of the furan ring. The most intense of these channels results in the formation of C2H– (m/z 25) followed by C3H3O– (m/z 55) and C4H– (m/z 49). Most of these fragmentation channels can be described by a simple reaction mechanism as shown in Table 3.
DEA to 3M-2FA and 5M-2FA results in similar anionic products. When comparing the yields for 3- and 5-methylated furoic acid with that of 2FA, we can see similar resonance trends, both quantitatively and qualitatively. Increase of the signal intensity in the case of m/z 39 may be assigned to competition of this fragmentation channel with that of m/z 25, and a similar increase in the case of m/z 55 may be assigned to competition with the channel resulting in the formation of m/z 41 anion. The COOH loss, m/z 67 for 2FA, corresponds to m/z 81 anions observed for methylated molecules. However, substitutions cause significant changes in the signal of m/z 67 and 25 anions, which will be discussed in section 4.3.
4.3. Hydrogen Migration
The anionic channel leading to the formation of C2H– from 2FA shows two distinctive resonance bands in the ion yield spectrum peaking at ∼5 eV and around 9 eV. The higher energy band was previously assigned as a Feshbach resonance. Fragmentation resulting in a stable anion and two neutral fragments predict energy thresholds of 8.15, 7.77, and 7.49 eV, for reactions 1, 2, and 3 in Table 4. These are good estimates for this band. However, the experimental onset for the lower band appears at ≈4 eV.
Table 4. Possible Dissociation Channels for C2H– DEA Channel of the 2FA Moleculea.

Dissociation schemes 1, 2, and 3 depict elementary fragmentation of the molecule, whereas more complex reaction channels where hydrogen migrates from/to carboxyl group are presented in reactions 2a, 3a, and 3b or within the ring, reactions 4 and 5. The threshold energies are calculated using B3LYP/aug-cc-pVTZ.
First, what is the nature of this band? The band seems to be slightly shifted with respect to the main 5.3 eV band contribution observed in the spectrum of other anions. This apparent shift may be caused by the overlap with higher energy bands for all the anions except C2H–, which is well separated. However, such overlap does not explain the low energy onset of the resonance.
Therefore it is possible that the band results from a shape resonance. We explored the Rydberg states of the system using the semiempirical formula of Gallup;74 however, we did not find any high lying shape resonances (LUMO → 0.07 eV, LUMO + 1 → 2.24 eV, LUMO + 2 → 3.29 eV). Still, the dipole supported inner shell resonances may be possible as described by Khvostenko et al.63 In furan the resonance is positioned at 5.55 eV,63 which is ∼0.5 eV below the corresponding S1 state. A similar shift was proposed for amides in the recent study of Li et al.,75 and the same shift due to a dipole interaction may also well describe the low lying C2H– resonance in the present case. Irrespective of the nature of this resonance, we can see from Table 4 that it cannot be explained by any direct dissociation channel, so a more complex reaction must be involved.
Several complex dissociation channels resulting in formation of C2H– anions are listed in Table 4. These include reaction channels where hydrogen migrates from/to carboxyl group, reactions 2a, 3a, and 3b, or within the ring, reactions 4 and 5. The lowest threshold of 3.19 eV was estimated for reaction 5 with only one neutral cofragment. Any of these reactions can explain the lowest energy band in the C2H– anion signal, but identifying the most likely will require further experiments or an estimation of the different reaction barriers.
The second interesting observation is the formation of m/z 67 anion from the methyl-substituted species. The anion formed by simple cleavage of the carboxyl group corresponds to m/z 66. Formation of m/z 67 anion therefore requires a more complex fragmentation pathway, with hydrogen migration from the carboxyl group to the furan ring.
It is now interesting to explore how methyl substitution at different positions in the molecule influences hydrogen migration. The ion yield for C4H3O– (m/z 67) in Figure 3d significantly changes with methyl substitutions. The three main bands positioned at 5.3, 6.7, and 9.4 eV are visible in 2FA. For 3M-2FA the band located around 6.7 eV is significantly suppressed, whereas for 5M-2FA a decrease in ion yield is observable to the band around 5.3 eV, leaving the middle band (6.7 eV) the strongest. Methylation also results in a slight decrease of the intensity of the 9.4 eV band.
When exploring the yields of C2H– (m/z 25) in Figure 3e, we can see only two resonances in the case of 2FA around 5 and 9 eV, which do not change upon substitution at the 5 position of the ring. However, substitution at the 3 position of the ring results in complete disappearance of the low 5.3 eV resonance and in the formation of a new peak in the anion yield around 6.7 eV.
The present experimental results do not allow us to unambiguously identify what causes these strong changes in the hydrogen migration after methyl substitution at the 3 position of the ring. We can only speculate about the mechanisms involved.
The disappearance of the 5.3 eV resonance in the yield of the m/z 25 anion after 3-methylation may be partially described by the new signal at this energy in the yield of m/z 39 anion, corresponding to the C3H3– anion due to the attached methyl group. This gives us only two options for how the C2H– can be extracted from the furan ring at low energies. The carbons of the resulting C2H– anion are, in the first option, the 2 and 3 carbons of the furan ring and, in the second option, the 3 and 4 carbons of the ring. Returning to Table 4, only reaction mechanisms 2a, 3a, and 3b are consistent with this explanation. The appearance of a new resonance in the yield of m/z 25 anion at 6.7 eV after 3-methylation can be then linked to the disappearance of this resonance in the yield of m/z 67 anion. It seems that, after cleavage of the carboxylic group from 3M-2FA, the anion becomes highly unstable and dissociation via release of C2H– will be much easier than stabilization of the ring. As the mechanism requires hydrogen migration, the low stability of the 3M-2FA anion may be an explanation also for the disappearance of the signal of Feshbach resonances in the (M–H)− anion signal described in section 4.1.
As a further note, our experimental measurements are performed under conditions which are similar to our earlier work on pyruvic acid,22 where we observed the formation of dimer anions and consequently parent anions. In the present study, we did not observe the creation of stable molecular parent anions, nor dimers. This again indicates a strong interaction of the ring with the COOH group. Consequently, the ability of the carboxyl group to form intermolecular hydrogen bonds is reduced. The binding energy of the carboxyl group bonded dimer of 2FA is ∼0.26 eV as reported by Ghalla et al.,24 which is less than half of the energy for formic acid (∼0.62 eV76).
5. Conclusions
In the present work, we have measured dissociative electron attachment in 2-furoic acid, 3-methyl-2-furoic acid, and 5-methyl-2-furoic acid. A rich fragmentation pattern has been revealed, with the strongest dissociation channel for all three compounds coming from the cleavage of the hydroxyl bond and production of the (M–H)− anion at electron energies around 1.2 eV. At higher energies (M–H)− anions are formed via core excited Feshbach resonances that are not detected for 3M-2FA. The behavior can be explained by exclusive formation of the (M–H)− only from the 3 position of the molecule or low stability of the 3M-2FA dehydrogenated anion at these energies, which undergoes further fragmentation.
Assignment of individual core excited Feshbach resonances to individual Rydberg states of the molecule based on PES indicate that the nondissociative π–1s2 resonance of furan becomes dissociative in 2FA upon overlapping the orbital with that of the carbonyl group. This is important with respect to electron transfer reactions after irradiation of complex molecular systems.
DFT calculations of energetic thresholds for individual DEA reaction channels of 2FA are in good agreement with a direct bond cleavage mechanism for most of the observed fragments. However, complex rearrangement and fragmentation of the neutral cofragments may also occur during the DEA. Experiments with methyl-substituted molecules help us to better identify the most probable reaction pathways, including hydrogen migration.
Acknowledgments
The project was supported by Czech Science Foundation grant 19-01159S. M.Z. acknowledges the Academic Computer Centre in Gdańsk (TASK) for a computational grant and support from National Science Centre (Poland) research grant 2018/02/X/ST2/01946. We would like to thank Juraj Fedor for fruitful discussion of the manuscript.
The authors declare no competing financial interest.
References
- Sulzer P.; Ptasińska S.; Zappa F.; Mielewska B.; Milosavljevic A. R.; Scheier P.; Märk T. D.; Bald I.; Gohlke S.; Huels M. A.; et al. Dissociative electron attachment to furan, tetrahydrofuran, and fructose. J. Chem. Phys. 2006, 125, 044304. 10.1063/1.2222370. [DOI] [PubMed] [Google Scholar]
- Janečková R.; May O.; Milosavljević A. R.; Fedor J. Partial cross sections for dissociative electron attachment to tetrahydrofuran reveal a dynamics-driven rich fragmentation pattern. Int. J. Mass Spectrom. 2014, 365, 163–168. 10.1016/j.ijms.2014.01.017. [DOI] [Google Scholar]
- Baccarelli I.; Bald I.; Gianturco F. A.; Illenberger E.; Kopyra J. Electron-induced damage of DNA and its components: Experiments and theoretical models. Phys. Rep. 2011, 508, 1–44. 10.1016/j.physrep.2011.06.004. [DOI] [Google Scholar]
- Milosavljević A. R.; Kočišek J.; Papp P.; Kubala D.; Marinković B. P.; Mach P.; Urban J.; Matejčík Š. Electron impact ionization of furanose alcohols. J. Chem. Phys. 2010, 132, 104308. 10.1063/1.3352422. [DOI] [PubMed] [Google Scholar]
- Maljković J. B.; Blanco F.; Čurík R.; García G.; Marinković B. P.; Milosavljević A. R. Absolute cross sections for electron scattering from furan. J. Chem. Phys. 2012, 137, 064312. 10.1063/1.4742759. [DOI] [PubMed] [Google Scholar]
- Khakoo M. A.; Muse J.; Ralphs K.; da Costa R. F.; Bettega M. H. F.; Lima M. A. P. Low-energy elastic electron scattering from furan. Phys. Rev. A: At., Mol., Opt. Phys. 2010, 81, 062716. 10.1103/PhysRevA.81.062716. [DOI] [Google Scholar]
- Dampc M.; Linert I.; Zubek M. Ionization and fragmentation of furan molecules by electron collisions. J. Phys. B: At., Mol. Opt. Phys. 2015, 48, 165202. 10.1088/0953-4075/48/16/165202. [DOI] [Google Scholar]
- Regeta K.; Allan M. Absolute cross sections for electronic excitation of furan by electron impact. Phys. Rev. A: At., Mol., Opt. Phys. 2015, 91, 012707. 10.1103/PhysRevA.91.012707. [DOI] [PubMed] [Google Scholar]
- Wolff W.; Rudek B.; da Silva L. A.; Hilgers G.; Montenegro E. C.; Homem M. G. P. Absolute ionization and dissociation cross sections of tetrahydrofuran: Fragmentation-ion production mechanisms. J. Chem. Phys. 2019, 151, 064304. 10.1063/1.5115403. [DOI] [Google Scholar]
- Boudaiffa B.; Cloutier P.; Hunting D.; Huels M. A.; Sanche L. Resonant Formation of DNA strand breaks by low-energy (3 to 20 eV) electrons. Science 2000, 287, 1658–1660. 10.1126/science.287.5458.1658. [DOI] [PubMed] [Google Scholar]
- Gorfinkiel J. D.; Ptasińska S. Electron scattering from molecules and molecular aggregates of biological relevance. J. Phys. B: At., Mol. Opt. Phys. 2017, 50, 182001. 10.1088/1361-6455/aa8572. [DOI] [Google Scholar]
- Alizadeh E.; Orlando T. M.; Sanche L. Biomolecular damage induced by ionizing radiation: the direct and indirect effects of low-energy electrons on DNA. Annu. Rev. Phys. Chem. 2015, 66, 379–398. 10.1146/annurev-physchem-040513-103605. [DOI] [PubMed] [Google Scholar]
- Low-Energy Electron Scattering from Molecules, Biomolecules and Surfaces; Čársky P., Čurik R., Eds.; CRC Press: Boca Raton, FL, 2012. [Google Scholar]
- Vasil’ev Y. V.; Figard B. J.; Voinov V. G.; Barofsky D. F.; Deinzer M. L. Resonant electron capture by some amino acids and their methyl esters. J. Am. Chem. Soc. 2006, 128, 5506–5515. 10.1021/ja058464q. [DOI] [PubMed] [Google Scholar]
- Bald I.; Langer J.; Tegeder P.; Ingólfsson O. From isolated molecules through clusters and condensates to the building blocks of life. Int. J. Mass Spectrom. 2008, 277, 4–25. 10.1016/j.ijms.2008.06.013. [DOI] [Google Scholar]
- Fabrikant I. I.; Eden S.; Mason N. J.; Fedor J. Recent progress in dissociative electron attachment: From diatomics to biomolecules. Adv. At., Mol., Opt. Phys. 2017, 66, 545. 10.1016/bs.aamop.2017.02.002. [DOI] [Google Scholar]
- Kambara O.; Tominaga K.; Nishizawa J.; Sasaki T.; Wang H.; Hayashi M. Mode assignment of vibrational bands of 2-furoic acid in the terahertz frequency region. Chem. Phys. Lett. 2010, 498, 86–89. 10.1016/j.cplett.2010.08.047. [DOI] [Google Scholar]
- Bachorz R. A.; Harańczyk M.; Dąbkowska I.; Rak J.; Gutowski M. Anion of the formic acid dimer as a model for intermolecular proton transfer induced by a π* excess electron. J. Chem. Phys. 2005, 122, 204304. 10.1063/1.1899144. [DOI] [PubMed] [Google Scholar]
- Keolopile Z. G.; Gutowski M.; Buonaugurio A.; Collins E.; Zhang X.; Erb J.; Lectka T.; Bowen K. H.; Allan M. Importance of Time Scale and Local Environment in Electron-Driven Proton Transfer. The Anion of Acetoacetic Acid. J. Am. Chem. Soc. 2015, 137, 14329–14340. 10.1021/jacs.5b08134. [DOI] [PubMed] [Google Scholar]
- Allan M. Electron Collisions with Formic Acid Monomer and Dimer. Phys. Rev. Lett. 2007, 98, 123201. 10.1103/PhysRevLett.98.123201. [DOI] [PubMed] [Google Scholar]
- Keolopile Z. G.; Ryder M. R.; Calzada B.; Gutowski M.; Buytendyk A. M.; Graham J. D.; Bowen K. H. Electrophilicity of oxalic acid monomer is enhanced in the dimer by intermolecular proton transfer. Phys. Chem. Chem. Phys. 2017, 19, 29760–29766. 10.1039/C7CP00474E. [DOI] [PubMed] [Google Scholar]
- Zawadzki M.; Ranković M.; Kočišek J.; Fedor J. Dissociative electron attachment and anion-induced dimerization in pyruvic acid. Phys. Chem. Chem. Phys. 2018, 20, 6838–6844. 10.1039/C7CP07472G. [DOI] [PubMed] [Google Scholar]
- Flakus H. T.; Jabłońska M.; Kusz J. An anomalous linear dichroic effect in the polarized IR spectra of 2-furancarboxylic acid crystals: Proton transfer induced by co-operative interactions involving hydrogen bonds. Vib. Spectrosc. 2009, 49, 174–182. 10.1016/j.vibspec.2008.07.001. [DOI] [Google Scholar]
- Ghalla H.; Issaoui N.; Oujia B. Theoretical study of the polarized infrared spectra of the hydrogen bond in 2-furoic acid crystal dimer. Int. J. Quantum Chem. 2012, 112, 1373–1383. 10.1002/qua.23117. [DOI] [Google Scholar]
- Ghalla H.; Issaoui N.; Castillo M. V.; Brandán S. A.; Flakus H. T. A complete assignment of the vibrational spectra of 2-furoic acid based on the structures of the more stable monomer and dimer. Spectrochim. Acta, Part A 2014, 121, 623–631. 10.1016/j.saa.2013.11.001. [DOI] [PubMed] [Google Scholar]
- Ptasińska S.; Denifl S.; Scheier P.; Märk T. D. Inelastic electron interaction (attachment/ionization) with deoxyribose. J. Chem. Phys. 2004, 120, 8505. 10.1063/1.1690231. [DOI] [PubMed] [Google Scholar]
- Kopyra J. Low energy electron attachment to the nucleotide deoxycytidine monophosphate: direct evidence for the molecular mechanisms of electron-induced DNA strand breaks. Phys. Chem. Chem. Phys. 2012, 14, 8287–8289. 10.1039/c2cp40847c. [DOI] [PubMed] [Google Scholar]
- Kočišek J.; Janečková R.; Fedor J. Long-lived transient anion of c-C4F8O. J. Chem. Phys. 2018, 148, 074303. 10.1063/1.5017478. [DOI] [PubMed] [Google Scholar]
- Sommerfeld T.; Davis M. C. Ring-opening attachment as an explanation for the long lifetime of the octafluorooxolane anion. J. Chem. Phys. 2018, 149, 084305. 10.1063/1.5045088. [DOI] [PubMed] [Google Scholar]
- Wąsowicz T. J.; Łabuda M.; Pranszke B. Charge transfer, complexes formation and furan fragmentation induced by collisions with low-energy helium cations. Int. J. Mol. Sci. 2019, 20, 6022. 10.3390/ijms20236022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdmann E.; Łabuda M.; Aguirre N. F.; Díaz-Tendero S.; Alcamí M. Furan fragmentation in the gas phase: new Insights from statistical and molecular dynamics calculations. J. Phys. Chem. A 2018, 122, 4153. 10.1021/acs.jpca.8b00881. [DOI] [PubMed] [Google Scholar]
- Foroumadi A.; Soltani F.; Moallemzadeh-Haghighi H.; Shafiee A. Synthesis, in vitro- antimycobacterial activity and cytotoxicity of some alkyl α-(5-aryl-1, 3, 4-thiadiazole-2-ylthio)acetates. Arch. Pharm. 2005, 338, 112–116. 10.1002/ardp.200400926. [DOI] [PubMed] [Google Scholar]
- Romano E.; Ladetto M. F.; Brandán S. A. Structural and vibrational studies of the potential anticancer agent, 5-difluoromethyl-1, 3, 4-thiadiazole-2-amino by DFT calculations. Comput. Theor. Chem. 2013, 1011, 57–64. 10.1016/j.comptc.2013.01.016. [DOI] [Google Scholar]
- Hurd C. D.; Garrett J. W.; Osborne E. N. Furan reactions. IV. Furoic acid from furfural. J. Am. Chem. Soc. 1933, 55, 1082–1084. 10.1021/ja01330a032. [DOI] [Google Scholar]
- Mariscal R.; Maireles-Torres P.; Ojeda M.; Sádaba I.; López Granados M. Furfural: a renewable and versatile platform molecule for the synthesis of chemicals and fuels. Energy Environ. Sci. 2016, 9, 1144–1189. 10.1039/C5EE02666K. [DOI] [Google Scholar]
- Carraher C. E. Jr Synthesis of furfuryl alcohol and furoic acid. J. Chem. Educ. 1978, 55, 269–270. 10.1021/ed055p269. [DOI] [Google Scholar]
- Aaron C. S.; Harbach P. R.; Wiser S. K.; Grzegorczyk C. R.; Smith A. L. The in vitro unscheduled DNA synthesis (UDS) assay in rat primary hepatocytes: Evaluation of 2-furoic acid and 7 drug candidates. Mutat. Res., Genet. Toxicol. Test. 1989, 223, 163–169. 10.1016/0165-1218(89)90044-X. [DOI] [PubMed] [Google Scholar]
- Hucker B.; Varelis P. Thermal decarboxylation of 2-furoic acid and its implication for the formation of furan in foods. Food Chem. 2011, 126, 1512–1513. 10.1016/j.foodchem.2010.12.017. [DOI] [Google Scholar]
- Lopez G. M.; Martin A. D.. Furfural: An Entry Point of Lignocellulose in Biorefineries to Produce Renewable Chemicals, Polymers, and Biofuels; World Scientific Publishing Co. Pte. Ltd.: 2018; Vol. 2. [Google Scholar]
- Uma B.; Murugesan K. S.; Krishnan S.; Das S. J.; Boaz B. M. Optical and dielectric studies on organic nonlinear optical 2-furoic acid single crystals. Optik (Munich, Ger.) 2013, 124, 2754. 10.1016/j.ijleo.2012.08.075. [DOI] [Google Scholar]
- Uma B.; Das S. J.; Krishnan S.; Boaz B. M. Growth, optical and thermal studies on organic nonlinear optical crystal: 2-Furoic acid. Phys. B 2011, 406, 2834–2839. 10.1016/j.physb.2011.04.038. [DOI] [Google Scholar]
- Sajadi Z.; Abrishami M. M.; Paricher-Mohseni; Chapman J. M. Jr; Hall I. H. Synthesis and evaluation of the antitumor properties of esters of 2-furoic acid and 2-furylacrylic acid. J. Pharm. Sci. 1984, 73, 266–267. 10.1002/jps.2600730233. [DOI] [PubMed] [Google Scholar]
- Schürmann R.; Vogel S.; Ebel K.; Bald I. The physico-chemical basis of DNA radiosensitization: Implications for cancer radiation therapy. Chem. - Eur. J. 2018, 24, 10271–10279. 10.1002/chem.201800804. [DOI] [PubMed] [Google Scholar]
- Stepanović M.; Pariat Y.; Allan M. Dissociative electron attachment in cyclopentanone, γ-butyrolactone, ethylene carbonate, and ethylene carbonate-d4: Role of dipole-bound resonances. J. Chem. Phys. 1999, 110, 11376. 10.1063/1.479078. [DOI] [Google Scholar]
- Langer J.; Zawadzki M.; Fárník M.; Pinkas J.; Fedor J.; Kočišek J. Electron interactions with Bis(pentamethylcyclopentadienyl) titanium(IV) dichloride and difluoride. Eur. Phys. J. D 2018, 72, 112. 10.1140/epjd/e2018-80794-6. [DOI] [Google Scholar]
- Prabhudesai V. S.; Kelkar A. H.; Nandi D.; Krishnakumar E. Functional group dependent site specific fragmentation of molecules by low energy electrons. Phys. Rev. Lett. 2005, 95, 143202. 10.1103/PhysRevLett.95.143202. [DOI] [PubMed] [Google Scholar]
- Ptasińska S.; Bass A. D.; Sanche L. Low energy electron attachment to condensed formic acid. J. Phys. Conf. Ser. 2008, 115, 012018. 10.1088/1742-6596/115/1/012018. [DOI] [Google Scholar]
- Dressler R.; Allan M. Energy partitioning in the O–/CO2 dissociative attachment. Chem. Phys. 1985, 92, 449. 10.1016/0301-0104(85)85038-2. [DOI] [Google Scholar]
- Klar D.; Ruf M.-W.; Hotop H. Attachment of electrons to molecules at submillielectronvolt resolution. Chem. Phys. Lett. 1992, 189, 448. 10.1016/0009-2614(92)85230-8. [DOI] [Google Scholar]
- Matejčík Š.; Eichberger P.; Plunger B.; Kiendler A.; Stamatovic A.; Märk T. D. Dissociative electron attachment to SF6: production of SF5– at temperatures below 300 K. Int. J. Mass Spectrom. Ion Processes 1995, 144, L13–L17. 10.1016/0168-1176(95)04167-J. [DOI] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; et al. Gaussian 09, rev. D.01; Gaussian, Inc.: Wallingford, CT, 2009.
- Becke A. D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A: At., Mol., Opt. Phys. 1988, 38, 3098. 10.1103/PhysRevA.38.3098. [DOI] [PubMed] [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Janećková R.; May O.; Fedor J. Dissociative electron attachment to methylacetylene and dimethylacetylene: Symmetry versus proximity. Phys. Rev. A: At., Mol., Opt. Phys. 2012, 86, 052702. 10.1103/PhysRevA.86.052702. [DOI] [Google Scholar]
- Halasa A.; Lapinski L.; Reva I.; Rostkowska H.; Fausto R.; Nowak M. J. Three conformers of 2-Furoic acid: structure changes induced with near-IR laser light. J. Phys. Chem. A 2015, 119, 1037–1047. 10.1021/jp512302s. [DOI] [PubMed] [Google Scholar]
- Zawadzki M.; Čížek M.; Houfek K.; Čurík R.; Ferus M.; Civiš S.; Kočišek J.; Fedor J. Resonances and Dissociative Electron Attachment in HNCO. Phys. Rev. Lett. 2018, 121, 143402. 10.1103/PhysRevLett.121.143402. [DOI] [PubMed] [Google Scholar]
- Zawadzki M.; Wierzbicka P.; Kopyra J. Dissociative electron attachment to benzoic acid (C7H6O2). J. Chem. Phys. 2020, 152, 174304. 10.1063/1.5135383. [DOI] [PubMed] [Google Scholar]
- Kopyra J.; König-Lehmann C.; Illenberger E.; Warneke J.; Swiderek P. Low energy electron induced reactions in fluorinated acetamide - probing negative ions and neutral stable counterparts. Eur. Phys. J. D 2016, 70, 140. 10.1140/epjd/e2016-70143-4. [DOI] [Google Scholar]
- Klapstein D.; MacPherson C. D.; O’Brien R. T. The photoelectron spectra and electronic structure of 2-carbonyl furans. Can. J. Chem. 1990, 68, 747–754. 10.1139/v90-118. [DOI] [Google Scholar]
- Ibǎnescu B. C.; May O.; Monney A.; Allan M. Electron-induced chemistry of alcohols. Phys. Chem. Chem. Phys. 2007, 9, 3163–3173. 10.1039/B704656A. [DOI] [PubMed] [Google Scholar]
- Ibanescu B. C.; Allan M. A dramatic difference between the electron-driven dissociation of alcohols and ethers and its relation to Rydberg states. Phys. Chem. Chem. Phys. 2008, 10, 5232–5237. 10.1039/b806578k. [DOI] [PubMed] [Google Scholar]
- Ibanescu B. C.; Allan M. Selective cleavage of the C–O bonds in alcohols and asymmetric ethers by dissociative electron attachment. Phys. Chem. Chem. Phys. 2009, 11, 7640–7648. 10.1039/b904945b. [DOI] [PubMed] [Google Scholar]
- Khvostenko V. I.; Vorob’yov A. S.; Khvostenko O. G. Inter-shell resonances in the interactions of electrons and polyatomic molecules. J. Phys. B: At., Mol. Opt. Phys. 1990, 23, 1975. 10.1088/0953-4075/23/12/008. [DOI] [Google Scholar]
- Li Z.; Ryszka M.; Dawley M. M.; Carmichael I.; Bravaya K. B.; Ptasińska S. Dipole-supported electronic resonances mediate electron-induced amide bond cleavage. Phys. Rev. Lett. 2019, 122, 073002. 10.1103/PhysRevLett.122.073002. [DOI] [PubMed] [Google Scholar]
- Fedor J. Comment on “Dipole-supported electronic resonances mediate electron-induced amide bond cleavage”. Phys. Rev. Lett. 2020, 124, 199301. 10.1103/PhysRevLett.124.199301. [DOI] [PubMed] [Google Scholar]
- Panelli G.; Moradmand A.; Griffin B.; Swanson K.; Weber Th.; Rescigno T. N.; McCurdy C. W.; Slaughter D. S.; Williams J. B.. Investigating resonant low-energy electron attachment to formamide: dynamics of model peptide bond dissociation and other fragmentation channels. Phys. Rev. Res. 2020,Published in ArXiV, 2020.
- Sanche L.; Schulz G. Electron transmission spectroscopy: Rare gases. Phys. Rev. A: At., Mol., Opt. Phys. 1972, 5, 1672–1683. 10.1103/PhysRevA.5.1672. [DOI] [Google Scholar]
- Schulz G. J. Resonances in electron impact on diatomic molecules. Rev. Mod. Phys. 1973, 45, 423–486. 10.1103/RevModPhys.45.423. [DOI] [Google Scholar]
- Sanche L.; Schulz G. J. Electron transmission spectroscopy: Resonances in triatomic molecules and hydrocarbons. J. Chem. Phys. 1973, 58, 479–493. 10.1063/1.1679228. [DOI] [Google Scholar]
- Spence D. Prediction of low energy molecular Rydberg states from Feshbach resonance spectra. J. Chem. Phys. 1977, 66, 669–674. 10.1063/1.433941. [DOI] [Google Scholar]
- Prabhudesai V. S.; Nandi D.; Kelkar A. H.; Parajuli R.; Krishnakumar E. Dissociative electron attachment to formic acid. Chem. Phys. Lett. 2005, 405, 172–176. 10.1016/j.cplett.2005.01.128. [DOI] [Google Scholar]
- Pelc A.; Sailer W.; Scheier P.; Probst M.; Mason N. J.; Illenberger E.; Märk T. D. Dissociative electron attachment to formic acid (HCOOH). Chem. Phys. Lett. 2002, 361, 277–284. 10.1016/S0009-2614(02)00925-9. [DOI] [Google Scholar]
- Slaughter D. S.; Weber Th.; Belkacem A.; Trevisan C. S.; Lucchese R. R.; McCurdy C. W.; Rescigno T. N. Selective bond-breaking in formic acid by dissociative electron attachment. Phys. Chem. Chem. Phys. 2020, 22, 13893–13902. 10.1039/D0CP01522A. [DOI] [PubMed] [Google Scholar]
- Aflatooni K.; Gallup G. A.; Burrow P. D. Electron attachment energies of the DNA bases. J. Phys. Chem. A 1998, 102, 6205–6207. 10.1021/jp980865n. [DOI] [Google Scholar]
- Li Z.; Carmichael I.; Ptasińska S. Dissociative electron attachment induced ring opening in five-membered heterocyclic compounds. Phys. Chem. Chem. Phys. 2018, 20, 18271–18278. 10.1039/C8CP02718H. [DOI] [PubMed] [Google Scholar]
- Kollipost F.; Wugt Larsen R.; Domanskaya A. V.; Norenberg M.; Suhm M. A. Communication: The highest frequency hydrogen bond vibration and an experimental value for the dissociation energy of formic acid dimer. J. Chem. Phys. 2012, 136, 151101. 10.1063/1.4704827. [DOI] [PubMed] [Google Scholar]