

Abstract
Voltage-gated sodium (NaV) channels are pore-forming transmembrane proteins that play essential roles in excitable cells, and they are key targets for antiepileptic, antiarrhythmic, and analgesic drugs. We implemented a heterobivalent design strategy to modulate the potency, selectivity, and binding kinetics of NaV channel ligands. We conjugated μ-conotoxin KIIIA, which occludes the pore of the NaV channels, to an analogue of huwentoxin-IV, a spider-venom peptide that allosterically modulates channel gating. Bioorthogonal hydrazide and copper-assisted azide–alkyne cycloaddition conjugation chemistries were employed to generate heterobivalent ligands using polyethylene glycol linkers spanning 40–120 Å. The ligand with an 80 Å linker had the most pronounced bivalent effects, with a significantly slower dissociation rate and 4–24-fold higher potency compared to those of the monovalent peptides for the human NaV1.4 channel. This study highlights the power of heterobivalent ligand design and expands the repertoire of pharmacological probes for exploring the function of NaV channels.
Introduction
Voltage-gated sodium (NaV) channels are fundamental for the generation and propagation of action potentials in excitable cells, and they are important therapeutic targets for antiepileptic, antiarrhythmic, and analgesic drugs.1−3 Humans have nine NaV channel subtypes denoted NaV1.1–NaV1.9. NaV1.1–NaV1.3 and NaV1.6 are expressed in both the central nervous system (CNS) and the peripheral nervous system (PNS), while NaV1.7–NaV1.9 are found primarily in peripheral sensory neurons.2 NaV1.4 and NaV1.5 are predominantly located in skeletal and cardiac muscles, respectively, where they play critical roles in muscle contraction.2
NaV channels are large transmembrane proteins composed of a pore-forming α-subunit in complex with one or two auxiliary β-subunits that modulate their expression, localization, gating, kinetics, and pharmacology (Figure 1).3,4 The α-subunit (∼260 kDa) folds into four homologous but non-identical domains (denoted DI–DIV) joined by intracellular linkers, with each domain containing six transmembrane segments (S1–S6). The S1–S4 segments within each domain form a voltage-sensing domain (VSD), while the S5 and S6 segments from each domain come together in a circular fashion to form the central pore of the channel (Figure 1A).1,2,4 The VSDs allow the channel to respond to changes in the membrane electrical potential, causing it to cycle (or “gate”) among three distinct states: a closed/resting state in which the channel can be activated by membrane depolarization, an open ion-conducting state, and a nonconducting inactivated state.1,2,4
Figure 1.

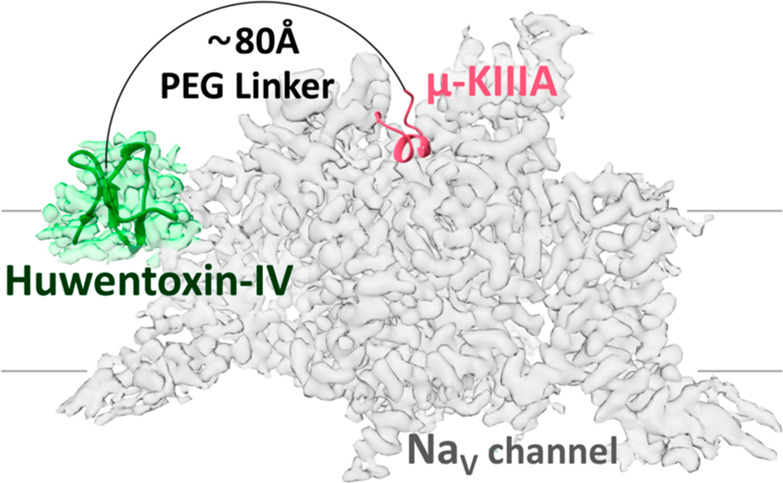
NaV channel architecture and overview of the bivalent inhibitor strategy. (A) Topology of NaV channel α- and β-subunits. The α-subunit comprises four domains (denoted I–IV), with each domain containing six transmembrane segments (S1–S6). Segments S1–S4 in each domain form a voltage-sensing domain (VSD, gray), while S5, S6, and the membrane-penetrant pore loops (P-loops) form the pore domain (white).1,4 (B) Schematic of the bivalent ligand strategy. Initial binding of either a gating modifier peptide (green) or a pore-blocking peptide (magenta) should bring the other peptide close to the channel, thereby enhancing binding kinetics (red arrows) and potency compared to those of monovalent ligands. The dotted line illustrates the spatial limit of the local concentration effect of the conjugated gating modifier when the pore blocker is bound. (C) Cryo-electron microscopy structure of hNaV1.7-β1 in the presence of HwTx-IV and μ-KIIIA. The hNaV1.7-β1 structure was used to determine the distance between the two peptides as this channel is our target of interest and because this structure contains HwTx-IV. A triple-mutant variant of HwTx-IV (E1G, E4G, Y33W; m3-HwTx-IV) was placed in the HwTx-IV density in a random orientation due to the unknown interaction sites with the channel. The distance between the center of the m3-HwTx-IV density and the N-terminus of μ-KIIIA is ∼50 Å in a direct line (dotted line) (i.e., if steric overlap is ignored) and ∼80 Å considering the length of a half-circle (solid line) that comfortably avoids steric overlap with the channel (PDB entries 5T3M, 6J8E, and 6J8G and EMD entry 9781).18,27 Figures were generated using UCSF Chimera, version 1.13.1.
Although NaV channels are important drug targets, their therapeutic potential is far from fulfilled. Many venom peptides from arachnids,4,5 cone snails,6−8 sea anemones,9 and other venomous animals target NaV channels with high potency and selectivity, and consequently have attracted interest both as pharmacological tools and as lead compounds for new analgesic, antiepileptic, and antiarrhythmic drugs.10−14 These peptides can be divided into two broad classes based on their mechanism of action: (i) pore blockers that bind to the outer vestibule of the channel, thereby sterically preventing the entry of Na+ into the channel pore, and (ii) allosteric modulators known as “gating modifiers” that interact with one or more of the VSDs and alter the gating and kinetics of the channel.15
A new era in NaV channel research began with the determination of the first three-dimensional structures of vertebrate NaV channels, namely, NaV1.4 from both electric eel16 and humans,17 and human NaV1.7.18 The muscle-specific NaV1.4 channel has been the subject of extensive functional and mechanistic studies, and mutations in this channel have been linked with muscle channelopathies such as paramyotonia congenita and hyperkalemic periodic paralysis.19,20 NaV1.7 is of particular interest as a potential analgesic target because of its strong genetic association with pain. Loss-of-function mutations in the gene encoding NaV1.7 lead to a congenital insensitivity to pain, whereas gain-of-function mutations underlie disorders such as erythromelalgia and paroxysmal extreme pain disorder that are characterized by severe episodic pain.21,22
As part of our ongoing attempts to develop new pharmacological probes and therapeutic leads for human NaV channels,10,23−26 we devised and tested in this study a bivalent linker design with a focus on NaV1.4 and NaV1.7 due to their (patho)physiological relevance and experimentally determined structures. Our strategy was to covalently link a pore blocker toxin with a gating modifier toxin using variable-length polyethylene glycol (PEG) linkers to simultaneously target two binding sites of the channel, thereby potentially enhancing binding kinetics, potency, and subtype selectivity (Figure 1B,C). We show that joining monovalent ligands with an optimal-length PEG linker leads to a bivalent ligand with significantly enhanced potency at NaV1.4 due to a greatly reduced rate of dissociation from the channel.
Results
Bivalent Ligand Design
For the pore blocker, we chose μ-conotoxin KIIIA (hereafter μ-KIIIA), a peptide isolated from venom of the marine cone snail Conus kinoshitai, with well-established pharmacology at NaV1.4 and NaV1.7, and extensive structure–activity relationship (SAR) information.28 μ-KIIIA is a 16-residue peptide with an α-helical core stabilized by three disulfide bonds with CysI–CysV, CysII–CysIV, and CysIII–CysVI connectivity (Figure 2).29 It preferentially blocks rat (r) NaV1.2 (IC50 = 5 nM) and rNaV1.4 (IC50 = 48 nM) over rNaV1.7 (IC50 = 147 nM).30,31 Cryo-electron microscopy (cryo-EM) studies of μ-KIIIA bound to hNaV1.227 in combination with SAR studies8,28 revealed that residues K7, W8, R10, D11, and R14 are functionally important for the pore blocking of NaV channels, while the N-terminus can be modified without abrogating binding. Thus, we added an ε-azido-l-lysine to the N-terminus of μ-KIIIA (AzK-KIIIA) to make it suitable for bioorthogonal copper-catalyzed azide–alkyne cycloaddition (CuAAC) chemistry,32 while retaining the free N-terminal α-amino group as it might affect the peptide’s binding kinetics.31
Figure 2.

3D structures and sequences of KIIIA, HwTx-IV, and analogues. (A) NMR-derived structures of μ-KIIIA (PDB entry 2LXG),29 HwTx-IV (PDB entry 1MB6),33 and the triple mutant m3-HwTx-IV (PDB entry 5T3M)36 are displayed in cartoon style with the disulfide bonds colored yellow. Images were generated using PyMol version 2.3.2 (http://pymol.org). (B) Amino acid sequence and disulfide connectivity of μ-KIIIA, HwTx-IV, and their analogues. Peptides used for constructing bivalent ligands are shown in bold. The three amino acid mutations in m3-HwTx-IV are colored red, and N-terminal modifications are bolded. Asterisks indicate C-terminal amidation.
For the gating modifier, we chose an optimized analogue of μ-theraphotoxin-Hs2a [HwTx-IV; optimized analogue m3-HwTx-IV (Figure 2)] originally identified in the venom of the tarantula Cyriopagopus schmidti (formerly Haplopelma schmidti).33 m3-HwTx-IV has three mutations relative to the native toxin (E1G, E4G, Y33W), which makes it an exceptionally potent inhibitor of human (h) NaV1.7 (IC50 = 0.4 nM).34 m3-HwTx-IV is a 35-residue peptide containing an inhibitor cystine knot (ICK) motif35 in which a double-stranded antiparallel β-sheet is stabilized by three disulfide bonds with CysI–CysIV, CysII–CysV, and CysIII–CysVI connectivity, with a three-dimensional (3D) structure highly similar to that of native HwTx-IV (Figure 2).33,36 m3-HwTx-IV also inhibits NaV1.1–NaV1.3 and NaV1.6 with low nanomolar potency and is a moderately potent inhibitor of hNaV1.4 (IC50 = 370 nM).36 Mutational and cryo-EM structural studies show that HwTx-IV binds to the DII VSD domain of NaV1.7.18 Residues W30 and K32 are critical for its activity, and while the N-terminus can be extended with polar or nonpolar residues without a loss of potency, the C-terminal amide is essential for potent inhibition of NaV1.7.34 On the basis of this information, we introduced an N-terminal serine residue (S-m3-HwTx-IV) that can be selectively converted into an aldehyde, thereby making it suitable for bioorthogonal hydrazone ligation.37
We selected linker lengths for toxin conjugation that would allow simultaneous binding of the two peptides to their respective NaV channel binding sites based on the cryo-EM structures of hNaV1.7 in complex with HwTx-IV18,38 and hNaV1.2 bound to μ-KIIIA27 (Figure S1). Ideally, binding of either peptide to its binding site should bring the second peptide into the proximity of its binding site, resulting in enhanced potency and altered binding kinetics and subtype selectivity. The distance between the two binding sites was estimated to be ∼50 Å in a direct line and ∼80 Å considering a half-circle (Figure 1C). We thus decided on a systematic series of PEG linkers ranging in length from 40 to 120 Å. We included a shorter 40 Å linker, two linkers spanning the distance predicted from the cryo-EM structures (60 and 80 Å), and a longer linker of 120 Å. The linker lengths of 40–120 Å are approximate values determined using Avogadro software.39 We hypothesized that the shorter 40 Å linker would not provide any bivalent effects as it does not span the two toxin binding sites, that the longer 120 Å linker might provide less optimal bivalent binding effects due to being too dynamic, and that the 60 and 80 Å linkers should yield pronounced and observable bivalent binding effects because they are within the optimal length to span the two toxin binding sites.40
Heterobifunctionalized PEG linkers were designed to tether the two toxins together using a hydrazide function compatible with bioorthogonal hydrazone ligation and an alkyne function compatible with bioorthogonal CuAAC chemistry.32,37,41 PEG is a nontoxic amphiphilic polymer that is monodisperse at the lengths employed here. PEG has good aqueous solubility and has been successfully used as a linker in many applications.42−45
Synthesis, Folding, and Bioactivity of Unconjugated Pore Blocker and Gating Modifier Peptides
μ-KIIIA and AzK-KIIIA were assembled using manual 9-fluorenylmethyloxycarbonyl (Fmoc) solid-phase peptide synthesis (SPPS),46 followed by oxidative folding. This yielded two distinct isomers with identical masses for both μ-KIIIA (observed monoisotopic mass, 1882.62 Da; calculated, 1882.64 Da), as reported previously,29 and AzK-KIIIA (observed monoisotopic mass, 2036.70 Da; calculated, 2036.71 Da) (Figure S2). The ability of each analogue to inhibit hNaV1.7 was determined by whole-cell patch-clamp electrophysiology; the major isomer from oxidative folding of AzK-KIIIA potently inhibited the channel (IC50 = 96 nM), whereas the minor isomer did not (IC50 = 934 nM). We therefore selected the major isomer for bioorthogonal conjugation (Table S1). S-m3-HwTx-IV was assembled using automated microwave-assisted Fmoc-SPPS, after which oxidative folding yielded a single isomer (observed monoisotopic mass, 4070.27 Da; calculated, 4069.91 Da) (Figure S3).
We compared the inhibitory potency of AzK-KIIIA and S-m3-HwTx-IV on both hNaV1.4 and hNaV1.7 to assess the impact of the modifications (Table 1). μ-KIIIA inhibited hNaV1.7 with a potency [IC50 = 132 ± 37 nM (Table 1)] similar to what was previously reported for inhibition of rNav1.7 (IC50 = 147 nM).31 Addition of the AzK residue to μ-KIIIA slightly enhanced the potency against both channels (from 132 ± 37 to 96 ± 41 nM on hNaV1.7 and from 48 ± 6 to 32 ± 10 nM on hNaV1.4), confirming that bioactivity was retained upon N-terminal modification. Addition of the N-terminal serine residue to m3-HwTx-IV was also well tolerated; this change improved potency at hNaV1.4 (IC50 decreased from 369 ± 196 to 212 ± 20 nM) but reduced potency at hNaV1.7, although it still exhibited excellent potency on this subtype (IC50 increased from 0.4 ± 0.1 to 4 ± 0.3 nM).
Table 1. Inhibitory Potencies of the Parent, Precursors, and Bivalent Ligands at hNaV1.4 and hNaV1.7a.
| Ligand | IC50 (nM ± SEM) | IC50 relative to 1 | n | |
|---|---|---|---|---|
| hNaV1.4 | ||||
| Bivalent ligands | ||||
| 1 | [m3-HwTx-IV]-[PEG80]-[K-KIIIA] | 9 ± 1 | 1 | 3 |
| 2 | [m3-HwTx-IV]-[PEG60]-[K-KIIIA] | 14 ± 1 | 1.6× ↑ | 3 |
| 3 | [m3-HwTx-IV]-[PEG120]-[K-KIIIA] | 13 ± 3 | 1.4× ↑ | 3 |
| Controls | ||||
| 4 | [m3-HwTx-IV]-[PEG40]-[K-KIIIA] | 29 ± 5 | 3.2× ↑ | 4 |
| 5 | AzK-KIIIA/S-m3-HwTx-IV | 33 ± 8 | 3.7× ↑ | 3 |
| 6 | AzK-KIIIA | 32 ± 10 | 3.6× ↑ | 3 |
| 7 | μ-KIIIA30 | 48 ± 6b | 5.3× ↑ | 3 |
| 8 | [K-KIIIA]-[PEG80] | 144 ± 46 | 16.0× ↑ | 4 |
| 9 | S-m3-HwTx-IV | 212 ± 20 | 23.6× ↑ | 4 |
| 10 | m3-HwTx-IV36 | 369 ± 196c | 41.0× ↑ | 6 |
| 11 | HwTx-IV49 | 400 ± nab | 44.4× ↑ | 3 |
| 12 | [m3-HwTx-IV]-[PEG80] | 409 ± 64 | 45.4× ↑ | 4 |
| hNaV1.7 | ||||
| Bivalent ligand | ||||
| 1 | [m3-HwTx-IV]-[PEG80]-[K-KIIIA] | 6 ± 0.1 | 1 | 3 |
| Controls | ||||
| 4 | [m3-HwTx-IV]-[PEG40]-[K-KIIIA] | 6 ± 2 | no change | 4 |
| 5 | AzK-KIIIA/S-m3-HwTx-IV | 6 ± 1 | no change | 3 |
| 6 | AzK-KIIIA | 96 ± 41 | 16.0× ↑ | 3 |
| 7 | μ-KIIIA | 132 ± 37 | 22.0× ↑ | 4 |
| 9 | S-m3-HwTx-IV | 4 ± 0.3 | 1.5x ↓ | 4 |
| 10 | m3-HwTx-IV34 | 0.4 ± 0.1 | 15.0× ↓ | 6 |
| 11 | HwTx-IV34 | 17 ± 2 | 2.8× ↑ | 10 |
| 12 | [m3-HwTx-IV]-[PEG80] | 8 ± 1 | 1.3× ↑ | 4 |
n is the number of cells, with each cell considered an independent experiment.
IC50 values were determined on rat NaV channel.
The IC50 value was determined for the peptide with no C-terminal amidation. Legend: n.a., not available; ↑, fold increase in IC50 relative to ligand 1; ↓, fold decrease in IC50 relative to ligand 1.
Linker Synthesis and Heterobivalent Ligand Assembly
Four PEG linkers ranging in length from 40 to 120 Å (PEG40/60/80/120) with N-terminal alkyne and C-terminal hydrazide functionalities were synthesized manually on solid support (Scheme 1 and Figures S4 and S5).
Scheme 1. Synthetic Strategy for the Production of Heterobifunctionalized (Alkyne and Hydrazide) PEG Linkers.

Reaction conditions: (i) 5% (v/v) hydrazine hydrate in DMF, 25 °C, 45 min; (ii) 10% (v/v) methanol in DMF, 25 °C, 10 min; (iii) HCTU, DIPEA, Fmoc-NH-PEG4-CH2CH2COOH (1.2 equiv), 25 °C, 2 h; (iv) 30% (v/v) piperidine in DMF, 25 °C, 2 min; (v) HCTU, DIPEA, Fmoc-l-propargylglycine (4 equiv), 25 °C, 2 h; (vi) 90% (v/v) TFA/H2O, 25 °C, 30 min.
2-Chlorotrityl chloride (2-CTC) resin was converted to 2-chlorotrityl hydrazine resin.47 Unreacted sites were capped with methanol. Repeated couplings of Fmoc-protected PEG4 (2× PEG4 for PEG40, 3× PEG4 for PEG60, 4× PEG4 for PEG80, and 6× PEG4 for PEG120) were carried out using standard Fmoc-SPPS protocols.46 Fmoc-l-propargylglycine was used as the final amino acid to incorporate the alkyne moiety. The PEG linkers were cleaved with TFA and purified using RP-HPLC. The linker is illustrated as PEG[Å], where Å indicates the linker length estimated using Avogadro software.39
To conjugate these linkers to the peptides, the N-terminal Ser in m3-HwTx IV was first oxidized with sodium periodate (1.5 equiv) in sodium phosphate buffer (10 mM, pH 7.0) for 2 min at 25 °C. The individual PEG linkers were then ligated to the N-terminal aldehyde of m3-HwTx-IV in sodium citrate buffer (100 mM, pH 4.5) for 24 h at −20 °C.37 Under these low-temperature conditions, slow-growing ice crystals produce locally high concentrations of reactants, which favors hydrazone bond formation.48 AzK-KIIIA was then conjugated to the alkyne moiety of the linker via CuAAC chemistry32 with a 70/30 (v/v) H2O/tBuOH mixture, copper sulfate (1.4 equiv), and ascorbic acid (5 equiv) for 1 h at 25 °C, yielding a triazole linkage (Scheme 2 and Figure S6).
Scheme 2. Bivalent Ligand Assembly Strategy for [m3-HwTx-IV]-[PEG[Å]]-[K-KIIIA] Constructs.

Selective oxidation of the N-terminal serine of S-m3-HwTx-IV to an aldehyde via sodium periodate treatment, followed by hydrazone ligation (colored blue). Final conjugation of AzK-KIIIA to the alkyne moiety of the linkers via copper-catalyzed azide–alkyne cycloaddition to form a triazole-linked conjugate (colored red). KIIIA is colored magenta, and m3-HwTx-IV is colored green. Disulfide bonds are shown as yellow sticks.
Structural Ligand Integrity of Bivalent Constructs and Their Precursors
One-dimensional (1D) 1H nuclear magnetic resonance (NMR) spectra were recorded to examine the structural integrity of AzK-KIIIA, S-m3-HwTx-IV, and the PEG-linked conjugates. Secondary Hα chemical shifts of AzK-KIIIA aligned well with published values for μ-KIIIA,29 except near the N-terminus where the AzK residue was added (Figure 3A). The negative secondary Hα shifts for residues 8–13 of AzK-KIIIA confirmed the presence of an α-helix in this region that is part of the toxin pharmacophore.27,28 The fingerprint regions of the 1D 1H NMR spectra of the PEG conjugate [m3-HwTx-IV]-[PEG80]-[K-KIIIA] overlapped well with the corresponding spectra of the S-m3-HwTx-IV and AzK-KIIIA precursors, indicating that the individual toxins retained their disulfide-stabilized 3D structures after PEG ligation (Figure 3B).
Figure 3.

NMR analysis of AzK-KIIIA, S-m3-HwTx-IV, and [m3-HwTx-IV]-[PEG80]-[K-KIIIA]. (A) Secondary Hα chemical shifts of μ-KIIIA29 and AzK-KIIIA obtained from sequence-specific resonance assignments using two-dimensional TOCSY and NOESY spectra. The secondary Hα shifts aligned throughout Asn3–Cys17. The N-terminal modification with AzK had chemical shift differences of 0.37 ppm for Cys1 Hα and 0.12 ppm for Cys2 Hα. The x-axis shows the sequence of AzK-KIIIA. The hash (#) indicates AzK, and the asterisk (*) indicates C-terminal amidation. (B) 1H NMR spectra [600 MHz, 25 °C, 90/10% (v/v) H2O/D2O] of AzK-KIIIA, S-m3-HwTx-IV, and [m3-HwTx-IV]-[PEG80]-[K-KIIIA]. The spectrum of [m3-HwTx-IV]-[PEG80]-[K-KIIIA] was the sum of the spectra of its individual peptide components as is exemplified for the tryptophan ε-NH region of AzK-KIIIA (magenta dots) and S-m3-HwTx-IV (green dots). Additional signals in the fingerprint region of the conjugate correspond to five secondary amide protons present in the synthetic PEG80 linker, and the C5 proton in the 1,2,3-triazole occurring in the bivalent compound.
Inhibition of hNaV1.4 and hNaV1.7 by Bivalent Ligands
We compared the inhibitory potency of the bivalent ligands and the monovalent precursors (individually and as equimolar mix) at hNaV1.4 and hNaV1.7 using patch-clamp electrophysiology to reveal any observable bivalent effects in terms of potency and selectivity (Figure 4 and Table 1).
Figure 4.

Inhibitory activity of monovalent and bivalent ligands at hNaV1.4 and hNaV1.7. (A) Concentration–response curves for bivalent PEG40, PEG60, PEG80, and PEG120 constructs, and an equimolar mixture of AzK-KIIIA and S-m3-HwTx-IV. (B) Concentration–response curves for bivalent PEG80 and AzK-KIIIA or S-m3-HwTx-IV conjugated to PEG80. (C) Concentration–response curves for bivalent PEG80 and monovalent peptides AzK-KIIIA and S-m3-HwTx-IV. For these experiments, we used HEK293 cells that stably express either NaV1.7 or NaV1.4 along with the β1 auxiliary subunit. Data are from experiments in which five to seven increasing concentrations of ligand were added to cells. Data points are means ± SEM of three or four independent experiments. (Note that for some concentrations the error bar is smaller than the symbol.) Fitting of the normalized concentration–response data yielded the IC50 values listed in Table 1.
At hNaV1.4, [m3-HwTx-IV]-[PEG80]-[K-KIIIA] was the most potent inhibitor (IC50 = 9 ± 1 nM), with 3.6-fold higher potency than AzK-KIIIA and the equimolar (1/1) AzK-KIIIA/S-m3-HwTx-IV mixture (Figure 4A). The bivalent ligands [m3-HwTx-IV]-[PEG60]-[K-KIIIA] and [m3-HwTx-IV]-[PEG120]-[K-KIIIA] were 2.4-fold more potent than AzK-KIIIA and AzK-KIIIA/S-m3-HwTx-IV, but the improvement in potency was not as pronounced as for [m3-HwTx-IV]-[PEG80]-[K-KIIIA] (3.6-fold more potent), suggesting that 80 Å is closer to the optimal linker length. The equimolar AzK-KIIIA/S-m3-HwTx-IV mixture yielded no improvement in potency compared to that of AzK-KIIIA; this was not surprising, considering that in an equimolar mixture the more potent ligand drives inhibition at lower concentrations and without a covalent linker bringing the other ligand into the proximity of its binding site no additive effects should be observed. [m3-HwTx-IV]-[PEG40]-[K-KIIIA] had a potency similar to those of AzK-KIIIA and AzK-KIIIA/S-m3-HwTx-IV, indicating an absence of bivalent effects presumably because this linker is too short to span the two toxin binding sites.
We also studied the potency impact of the PEG80 linker when attached to AzK-KIIIA or S-m3-HwTx-IV to exclude the possibility of the linker being responsible for the observed effects. Linker attachment caused a 4.5-fold decrease in inhibitory potency on NaV1.4 for [K-KIIIA]-[PEG80] compared to AzK-KIIIA, and a 1.9-fold decrease in potency for [m3-HwTx-IV]-[PEG80] compared to S-m3-HwTx-IV (Figure 4B and Table 1). This further confirmed that there is a significant bivalent effect on potency, because [m3-HwTx-IV]-[PEG80]-[K-KIIIA] is 16-fold more potent than [K-KIIIA]-[PEG80] and 45-fold more potent than [m3-HwTx-IV]-[PEG80].
At hNaV1.7, we did not observe any bivalent potency effects for [m3-HwTx-IV]-[PEG80]-[K-KIIIA], with the IC50 value (6 ± 0.1 nM; n = 3) being similar to that of [m3-HwTx-IV]-[PEG40]-[K-KIIIA], the AzK-KIIIA/S-m3-HwTx-IV mixture, and [m3-HwTx-IV]-[PEG80] (Figure 4A and Table 1). This observation prompted us to examine the binding kinetics of the monovalent and bivalent ligands at hNaV1.4 and hNaV1.7.
The bivalent ligands had reduced subtype selectivity, because ligand binding was primarily determined by the most potent ligand for each NaV subtype (μ-KIIIA for hNaV1.4 and m3-HwTx-IV for hNaV1.7). While m3-HwTx-IV had an ∼1000-fold preference for hNaV1.7 over hNaV1.4, and μ-KIIIA a 3-fold preference for hNaV1.4 over hNaV1.7, [m3-HwTx-IV]-[PEG80]-[K-KIIIA] was nearly equipotent at both channels with only 2-fold selectivity for hNaV1.7 over hNaV1.4 (Figure 4A,C and Table 1).
Ligand Binding Kinetics at hNaV1.4 and hNaV1.7
Ligand binding affinity is characterized by the equilibrium dissociation constant (Kd) and is determined from the ratio of kinetic rate constants that reflect formation of the ligand–receptor complex (association rate constant, kon) and its dissociation (dissociation rate constant, koff), with the equation Kd = koff/kon. Experimentally, we determined kon and koff using ligand wash-in and washout periods, described by the time constant τ, using the formulas kon = (1/τon – koff)/[ligand] and koff = 1/τoff.23,50koff could not be determined accurately for some ligands due to the poor reversibility of binding, and kon was calculated as the observed kon (kon*), described by the equation kon* = 1/τon. Kinetic data for precursors and bivalent ligands were determined at concentrations 10-fold higher than their respective IC50 values using patch-clamp electrophysiology to identify potential bivalent effects (Figure 5 and Table 2).
Figure 5.

Kinetics of NaV inhibition by monovalent and bivalent ligands at hNaV1.4 and hNaV1.7. (A) On-rate measurements for S-m3-HwTx-IV and AzK-KIIIA alone or conjugated to the PEG80 linker, and bivalent ligands [m3-HwTx-IV]-[PEG60]-[K-KIIIA] and [m3-HwTx-IV]-[PEG80]-[K-KIIIA]. On-rates were calculated from the fitted exponentials (dashed lines). (B) Off-rate measurements of precursor peptides S-m3-HwTx-IV and AzK-KIIIA and bivalent ligands [m3-HwTx-IV]-[PEG60]-[K-KIIIA] and [m3-HwTx-IV]-[PEG80]-[K-KIIIA] over hNaV1.4 and S-m3-HwTx-IV, AzK-KIIIA, and bivalent ligand [m3-HwTx-IV]-[PEG80]-[K-KIIIA] over hNaV1.7. Off-rates were calculated from the fitted exponentials (dashed lines). (C) Channel currents measured at the end of a 25 min ligand washout period. For these experiments, we used HEK293 cells that stably express either NaV1.7 or NaV1.4 along with the β1 auxiliary subunit. Data points are the mean ± SEM of three to five independent experiments. Statistical significance was determined by one-way analysis of variance followed by Tukey’s multiple-comparison test. ****P < 0.0001; *P = 0.019.
Table 2. Kinetic Data for Monovalent and Bivalent Ligands at hNaV1.4 and hNaV1.7 Determined via Patch-Clamp Electrophysiology.
| Ligand | kon*a (s–1) | kon (nM–1 s–1) | koff (s–1) | Kd (nM) | |
|---|---|---|---|---|---|
| hNaV1.4 | |||||
| 1 | [m3-HwTx-IV]-[PEG80]-[K-KIIIA] | (1.48 ± 0.04) × 10–2 | nd | irreversibleb | nd |
| 2 | [m3-HwTx-IV]-[PEG60]-[K-KIIIA] | (1.52 ± 0.12) × 10–2 | nd | irreversibleb | nd |
| 6 | AzK-KIIIA | (1.39 ± 0.15) × 10–2 | 3.97 × 10–5 | (1.18 ± 0.18) × 10–3 | 2.97 × 10 |
| 9 | S-m3-HwTx-IV | (6.25 ± 1.02) × 10–3 | 1.02 × 10–6 | (4.11 ± 0.36) × 10–3 | 4.03 × 103 |
| 8 | [K-KIIIA]-[PEG80] | (9.75 ± 0.89) × 10–3 | nd | nd | nd |
| 12 | [m3-HwTx-IV]-[PEG80] | (9.19 ± 0.79) × 10–3 | nd | nd | nd |
| hNaV1.7 | |||||
| 1 | [m3-HwTx-IV]-[PEG80]-[K-KIIIA] | (4.28 ± 0.48) × 10–3 | nd | irreversibleb | nd |
| 6 | AzK-KIIIA | (1.60 ± 0.03) × 10–2 | 1.62 × 10–5 | (3.92 ± 2.54) × 10–4 | 2.42 × 10 |
| 9 | S-m3-HwTx-IV | (1.43 ± 0.06) × 10–2 | 3.57 × 10–4 | (8.12 ± 0.04) × 10–7 | 2.28 × 10–3 |
| 12 | [m3-HwTx-IV]-[PEG80] | (1.32 ± 0.11) × 10–2 | nd | nd | nd |
τon is the time constant wash-in. kon* = 1/τon. kon = (1/τon – koff)/[ligand]. koff = 1/τoff, determined within an experimental washout time of 25 min. Kd = koff/kon. Kinetic data were determined using peptide concentrations equivalent to 10 times their IC50 values, and kon* and koff are given as the mean ± SEM of three to five independent experiments.
koff is less than the lowest valid measurement under the chosen experimental conditions.
At hNaV1.4, we observed a strong bivalent effect for the dissociation rate of [m3-HwTx-IV]-[PEG80]-[K-KIIIA] compared to monovalent AzK-KIIIA and S-m3-HwTx-IV, but no bivalent effect in terms of association rate (Figure 5A,B and Table 2). [m3-HwTx-IV]-[PEG80]-[K-KIIIA] and [m3-HwTx-IV]-[PEG60]-[K-KIIIA] had association rates similar to that of AzK-KIIIA but faster than that of S-m3-HwTx-IV. The PEG80 linker, when attached to the individual ligands, had little impact on the wash-in kinetics of [K-KIIIA]-[PEG80] compared to that of AzK-KIIIA, and for [m3-HwTx-IV]-[PEG80] compared to S-m3-HwTx-IV (Figure 5 and Table 2).
By contrast, the dissociation rates of the bivalent ligands [m3-HwTx-IV]-[PEG80]-[K-KIIIA] and [m3-HwTx-IV]-[PEG60]-[K-KIIIA] were substantially slower than those of the monovalent ligands (Figure 5B,C). Inhibition of hNaV1.4 by the monovalent ligands AzK-KIIIA and S-m3-HwTx-IV was not completely reversible, with sodium currents restored to ∼40% and ∼55%, respectively, of the maximum peak current (Imax) within the washout period of 25 min. Inhibition by [m3-HwTx-IV]-[PEG80]-[K-KIIIA] and [m3-HwTx-IV]-[PEG60]-[K-KIIIA] was nearly irreversible with recoveries of only 2.6 ± 0.2% and 9.5 ± 0.1%, respectively, of currents at the end of the washout period (Figure 5C and Table 2).
The comparison of the remaining hNaV1.4 currents at the end of the washout period revealed that [m3-HwTx-IV]-[PEG80]-[K-KIIIA] and [m3-HwTx-IV]-[PEG60]-[K-KIIIA] allowed significantly (P < 0.0001) slower recovery of inward currents compared to the monovalent ligands. Of the two bivalent ligands, [m3-HwTx-IV]-[PEG80]-[K-KIIIA] exerted a more pronounced bivalent effect and allowed significantly (P = 0.019) slower recovery compared to [m3-HwTx-IV]-[PEG60]-[K-KIIIA]. Although the koff values could not be determined accurately due to the nearly irreversible nature of both bivalent ligands [R2 < 0.66 (data not shown)] under the chosen experimental conditions, the washout of [m3-HwTx-IV]-[PEG80]-[K-KIIIA] was significantly (P < 0.0001) slower compared to that with the monovalent ligands (Figure 5B,C), consistent with the enhanced potency of the bivalent ligand.
At hNaV1.7, we did not observe any bivalent effects of [m3-HwTx-IV]-[PEG80]-[K-KIIIA] (Figure 5 and Table 2). This can be explained by the washout results revealing that the monovalent ligand S-m3-HwTx-IV is already a nearly irreversible binder at this channel [koff = (8.12 ± 0.04) × 10–7 s–1], leaving little room for improvement in terms of the dissociation rate for the bivalent ligand (Figure 5B,C). Although the koff and Kd values for this bivalent ligand remain to be calculated for an accurate comparison to the monovalent ligands, the comparison of the remaining hNaV1.7 currents at the end of the washout period revealed that S-m3-HwTx-IV and [m3-HwTx-IV]-[PEG80]-[K-KIIIA] had significantly (P < 0.0001) slower recovery of inward currents compared to that of the monovalent ligand AzK-KIIIA (Figure 5C). Inhibition by [m3-HwTx-IV]-[PEG80]-[K-KIIIA], S-m3-HwTx-IV, and AzK-KIIIA had recoveries of 2.2 ± 0.8%, 3.6 ± 0.3%, and 12.6 ± 1.2%, respectively, normalized to Imax at the end of the washout period (Figure 5C and Table 2).
Discussion
Conjugation of ligands that target the same ion channel via distinct modulatory mechanisms and binding sites is an innovative strategy for expanding the pharmacological toolbox available to study these channels. Bivalent or multivalent ligands often increase the effective concentration in the vicinity of the target, which can translate into various observable multivalent effects, including enhanced potency and binding kinetics.51,52 For example, an engineered homobivalent protein kinase inhibitor had 100-fold higher potency for a particular subgroup of kinases,53 and a homobivalent agonist targeting oxytocin receptor homodimers displayed potency that was ∼1000-fold greater than that of its monovalent counterpart.54 Heterobivalent and multivalent ligands with improved potency have also been developed against the 5-HT3 receptor55 and the nicotinic acetylcholine receptor,56 respectively.
Here, we conjugated the pore-blocking conotoxin μ-KIIIA to the optimized gating modifier spider toxin m3-HwTx-IV via bioorthogonal ligation with different length PEG linkers (40–120 Å) and characterized the inhibitory potency, subtype selectivity, and binding kinetics of the bivalent and monovalent ligands at hNaV1.4 and hNaV1.7. Both venom peptides in the bivalent ligand [m3-HwTx-IV]-[PEG80]-[K-KIIIA] retained their overall 3D structure (Figure 3B), which was reflected in their bioactivity (Table 1). The dependence of bivalent effects on linker length was consistent with the structural model used to design the bivalent ligands (Figure 1C). The bivalent ligand with the PEG80 linker produced the most pronounced bivalent effects, reflecting the measured half-circle length of 80 Å, an important finding that informs the appropriate linker lengths for future design strategies. The bivalent ligands with shorter (60 Å) and longer (120 Å) linkers displayed bivalent effects that were less pronounced than those of the 80 Å linker, and as predicted, the bivalent ligand with a 40 Å linker did not display any bivalent effects as it should not be able to span the two targeted binding sites.
The strongest bivalent effect was observed with [m3-HwTx-IV]-[PEG80]-[K-KIIIA] at hNaV1.4, which had 3.6- and 16-fold enhanced potency compared to those of AzK-KIIIA and [K-KIIIA]-[PEG80] and 24- and 45-fold enhanced potency compared to those of S-m3-HwTx-IV and [m3-HwTx-IV]-[PEG80], respectively. This improvement in potency seems to be driven by a greatly reduced dissociation rate of the bivalent ligand (<5% current recovered after a 25 min washout period) when compared to those of the monovalent constituents (40–55% recovered), while having similar on-rates (kon*) despite the larger size of the bivalent ligand (Table 2). Binding of the bivalent ligand at hNaV1.4 is driven by the more potent KIIIA moiety; however, the presence of HwTx-IV is crucial as it acts as a tether, converting what would be full dissociation events for KIIIA into rapid rebinding events, resulting in a nearly irreversible (within the washout period) bivalent inhibitor with measurably higher potency (Figures 4 and 5).
We did not observe any bivalent effects in terms of potency or binding kinetics for [m3-HwTx-IV]-[PEG80]-[K-KIIIA] at hNaV1.7, which can be explained by the binding kinetics of the bivalent and monovalent ligands. At hNaV1.4, bivalency enhanced potency by slowing dissociation. At hNaV1.7, this is not possible, because monovalent S-m3-HwTx-IV is already a nearly irreversible binder (koff of 8.12 × 10–7 s–1 compared to a value of >10–3 s–1 at hNaV1.4). This hypothesis is supported by a recent study that investigated a similar heterobivalent ligand design comprising μ-KIIIA enzymatically ligated via a different linker to spider-venom peptide PaurTx3 (also known as β-TRTX-Ps1a).57 PaurTx3 is a reversible binder at hNaV1.7 (in contrast to S-m3-HwTx-IV), and therefore, in this case, the heterobivalent ligand yielded improved potency along with slower dissociation compared to those for the monovalent ligands. It is important to note that [m3-HwTx-IV]-[PEG80]-[K-KIIIA] might still have therapeutically beneficial bivalent effects at hNaV1.7, which could not be observed with the washout period that we used but could become apparent in vivo, for example, through longer analgesic effects due to slower koff rates compared to that of m3-HwTx-IV.
In terms of selectivity, [m3-HwTx-IV]-[PEG80]-[K-KIIIA] was nearly equipotent at both channels (IC50 values of 9 nM for hNaV1.4 and 6 nM for hNaV1.7), because binding was driven by the most potent ligand subunit for each channel (KIIIA for hNaV1.4 and m3-HwTx-IV for hNaV1.7). This might be of interest for molecular probe development where such modulation of selectivity could be an advantage of devising new pharmacological tools to study the effects of multiple subtypes simultaneously. It also highlights that ligand selection is critical, particularly for heterobivalent drug development, because reduced selectivity can translate into undesirable off-target effects.
Our results highlight the importance of investigating potency at the level of kon and koff rates in the design and engineering of bivalent ligands. Ligand binding kinetics are particularly important for therapeutic development because they define the target interaction, length of effects, dosing, and therapeutic window.58,59 Ligands with slow dissociation rates, especially peptides with high selectivity, are often preferred drug leads because this translates into an increased target residence time, extended therapeutic effects, and improved patient compliance due to a lower frequency of drug administration.60−62 The design of such long-acting ligands, however, remains challenging, and targeting two binding sites on a single channel via bivalent ligand design, as demonstrated in this work, represents an elegant strategy for delivering such long-acting therapeutic leads.
Conclusion
In summary, we report the design, synthesis, and pharmacological characterization of a series of heterobivalent peptide ligands targeting hNaV1.4 and hNaV1.7. We developed a synthetic strategy that employed bioorthogonal ligation chemistry to conjugate a pore-blocking peptide to a gating modifier peptide using a panel of different length PEG linkers. We identified a heterobivalent ligand with improved potency, a switch from reversible to nearly irreversible binding, and new channel selectivity. This work highlights the power of heterobivalent ligand design to decrease the ligand–channel dissociation rate, which can translate into more potent and longer-lasting therapeutic effects. It furthermore provides important insights for future bivalent design strategies, including ligand- and linker-length selection. The strategy described here is expected to be broadly applicable to other ligands and ion channels, adding to the chemical repertoire of ion channel probes and drug leads.
Experimental Section
Synthesis of Peptides
KIIIA peptides were manually synthesized by Fmoc-SPPS on a 0.2 mmol scale on Rink Amide aminomethyl-polystyrene resin (0.69 mmol/g; Rapp Polymere GmbH, Tuebingen, Germany) using 4 equiv of Fmoc-l-protected amino acids (Iris Biotech GmbH, Marktredwitz, Germany). Terminal amino acid coupling for AzK-KIIIA was performed with 2 equiv of N-α-(Fmoc-ε-azido-l-lysine) (Iris Biotech). Amide couplings were carried out using O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU, 4 equiv) (Chem-Implex International Inc., Wood Dale, IL) in the presence of N,N-diisopropyl-ethylamine (DIPEA, 4 equiv) (Auspep Pty. Ltd., Melbourne, Australia) in N,N-dimethylformamide (DMF) (RCL Labscan, Bangkok, Thailand). Couplings were performed for 30 min. N-terminal Fmoc deprotection was performed with 30% (v/v) piperidine (Chem-Supply Pty. Ltd., Gillman, Australia) in DMF (3 × 4 mL) for 5 min. After each coupling, washing was carried out with 50% (v/v) dichloromethane (DCM) (Chem-Supply Pty. Ltd.) in DMF, followed by DMF (3 × 4 mL). S-m3-HwTx-IV was synthesized automatically by microwave-assisted Fmoc-SPPS on a CEM Liberty Prime synthesizer at a 0.1 mmol scale on Rink Amide ProTide resin (LL) (0.19 mmol/g; CEM Corp., Matthews, NC).
Cleavage and Purification of Reduced Peptides
KIIIA peptides were cleaved from the resin via a 2 h treatment with 90% trifluoroacetic acid (TFA) (Chem-Supply Pty. Ltd.), 5% H2O (Milli-Q, Millipore, Milford, MA), and 5% (v/v) triisopropylsilane (TIPS) (Alfa Aesar by Thermo Fischer Scientific, Kandel, Germany) at 25 °C with agitation. S-m3-HwTX-IV peptide was cleaved with 92.5% TFA, 2.5% TIPS, 2.5% H2O, and 2.5% (v/v) 2,2′-(ethylenedioxy)diethanethiol (DODT) (Sigma-Aldrich, St. Louis, MO) for 30 min at 38 °C using a CEM RAZOR cleavage system. Cleaved peptides were filtered, concentrated by evaporation under N2, precipitated, and washed with cold diethyl ether (Et2O, 3 × 10 mL) (RCL Labscan). Peptides were isolated by centrifugation at 4032g (5000 rpm) for 3 min at 4 °C, dissolved in 50% (v/v) acetonitrile (ACN) (Lichrosolv, Merck, Darmstadt, Germany) in H2O, and lyophilized.
The linear peptides were purified by preparative RP-HPLC using a Waters (Milford, MA) 600E HPLC system with a Zorbax Eclipse XDB-C18 column (PrepHT, 21.2 mm × 250 mm, 7 μm) (Agilent Technologies, Santa Clara, CA) and eluted with a linear gradient from 5% to 40% solvent B over 35 min, where solvent A was 0.05% TFA in H2O and solvent B was 0.043% TFA in 90/10% (v/v) ACN/H2O. The flow rate was 15 mL/min, and the ultraviolet (UV) absorbance was monitored at 214 nm.
Oxidative Folding of Peptides
Oxidative folding of KIIIA peptides was performed as described previously.29 Oxidative folding of S-m3-huwentoxin-IV was accomplished by glutathione-assisted folding at 25 °C overnight under the following conditions: 15 μM reduced peptide in 0.1 M Tris-HCl (pH 8.0, Amresco, Solon, OH), 10% (v/v) isopropanol (Chem-Supply Pty. Ltd.), and 5 mM reduced and 1 mM oxidized glutathione (Sigma-Aldrich). The reaction was quenched by decreasing the pH to 2 using an ACN/TFA/H2O mixture [1/1/1 (v/v/v)].
Synthesis of PEG Linkers
PEG linkers were synthesized by solid-phase synthesis on a 2-chlorotrityl chloride resin [2-Cl-(Trt)-Cl] (1.58 mmol/g; Iris Biotech GmbH) on a 0.55 mmol scale. First, hydrazination with hydrazine hydrate (Sigma-Aldrich) of 2-Cl-(Trt)-Cl resin was performed as described previously.45 The hydrazide resin [2-Cl-(Trt)-NHNH2] was directly used for the first coupling with Fmoc-NH-PEG4-CH2CH2COOH (Fmoc-PEG4, 1.2 equiv) (ChemPep Inc., Wellington, FL), O-(6-chlorobenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HCTU, 1.2 equiv; Chem-Impex), and DIPEA (1.2 equiv) in DMF. The coupling was performed overnight at 25 °C, followed by washing and Fmoc deprotection as described above. Further couplings were performed for 2 h at 25 °C. After a successful second coupling of Fmoc-PEG4, the resin was split and one-quarter of the resin was transferred into a new reaction vessel. The remaining 75% of the resin was again coupled with Fmoc-PEG4, and one-quarter of the resin was split and transferred again. This Fmoc-PEG4 coupling and transfer was continued until there was one-quarter of the resin left in the initial reaction vessel. This resulted in four reaction vessels: one each with 2× Fmoc-PEG4 couplings, 3× Fmoc-PEG4, 4× Fmoc-PEG4, and 6× Fmoc-PEG4 coupling. Each resin was coupled with 4 equiv of Fmoc-l-propargylglycine (AnaSpec Inc., Fremont, CA) for 30 min to incorporate the alkyne group.
Cleavage of PEG Linkers
All synthesized PEG linkers were cleaved with 90% (v/v) TFA in H2O for 1 h at 25 °C with agitation. The cleaved linkers were filtered, concentrated by evaporation, and precipitated with cold Et2O for 2 h at −80 °C. After 2 h, linkers were isolated by centrifugation at 4032g (5000 rpm) for 15 min at 4 °C, dissolved in 50% ACN (v/v) in H2O, and lyophilized.
Serine to Aldehyde Conversion of S-m3-HwTx-IV
The N-terminal serine of S-m3-HwTx IV was oxidized with sodium periodate to an N-terminal aldehyde moiety in 10 mM sodium phosphate (pH 7). The sodium periodate stock solution was freshly prepared at 100 mM in H2O. The reaction was performed with 0.5 mM peptide and a 1.5-fold molar excess of sodium periodate (0.75 mM). The periodate solution was incubated for 2 min at 25 °C in the dark. Oxidation was terminated by the addition of N-α-Fmoc-l-serine (Iris Biotech GmbH) to a final concentration of 5 mM.
Hydrazone Ligation
Ligation of the hydrazide-PEG linker with the aldehyde moiety of m3-HwTx-IV was performed with 100 mM sodium citrate (pH 4.5) using a peptide concentration of 1 mg/mL (130 μM) and a 2-fold molar excess of hydrazide-PEG linker (260 μM). The reaction was allowed to proceed at –20 °C in the dark for 24 h. The product was confirmed by matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS) on a TOF/TOF 5800 mass spectrometer (AB SCIEX, Framingham, MA) and analytical RP-HPLC. For MALDI-TOF MS, the peptide sample was diluted to approximately 1 pmol/μL in a MALDI solvent [0.05% TFA in 50% ACN (v/v) in H2O], mixed [1/1 (v/v)] with an α-cyano-4-hydroxycinnamic acid matrix [5 mg/mL in 50% ACN (v/v) in H2O], and spotted onto an Opti-TOF 384-well (123 mm × 81 mm) MALDI plate (AB Sciex). For RP-HPLC, an analytical Kromasil 100-3.5-C18 column (2.1 mm × 150 mm, 3.5 μm; Merck) with a flow rate of 0.2 mL/min was used with a linear gradient from 0% to 45% solvent B over 45 min, where solvent A was 0.05% TFA in H2O and solvent B was 0.043% TFA in 90/10% (v/v) ACN/H2O. The UV absorbance was monitored at 214 and 280 nm.
CuAAC Chemistry
Aqueous stock solutions of 20 mM AzK-KIIIA, 50 mM copper(II) sulfate (CuSO4), and 100 mM ascorbic acid were prepared. The CuAAC chemistry reaction was performed with 4.5 mM AzK-KIIIA, 10 mM alkyne-PEG[Å], CuSO4 (1.4 equiv), and ascorbic acid (5 equiv) in a H2O/tBuOH [70/30% (v/v)] solution. The mixture was stirred in a closed vessel for 2 h at 25 °C.
Peptide and PEG Linker Purification
A LC-20AT HPLC system (Shimadzu Corp., Tokyo, Japan) was used for all peptide purification. Oxidized peptides and bioorthogonal reaction products were purified via a semipreparative RP-HPLC system using a Zorbax 300 SB-C18 column (9.4 mm × 250 mm, 5 μm; Agilent Technologies) with a linear gradient from 10% to 40% solvent B over 30 min, where solvent A was 0.05% TFA in H2O and solvent B was 0.043% TFA in 90/10% (v/v) ACN/H2O. The flow rate was 4 mL/min, and the UV absorbance was monitored at 215 and 280 nm. Fractions corresponding to the peptide of interest were collected, pooled, and lyophilized.
Small-scale purifications were performed via analytical RP-HPLC using either a Zorbax 300 SB-C18 column (4.6 mm × 150 mm, 3.5 μm; Agilent Technologies) with a flow rate of 1 mL/min or an analytical Kromasil 100-3.5-C18 column (2.1 mm × 150 mm, 3.5 μm; Merck) with a flow rate of 0.2 mL/min. A linear gradient from 0% to 45% solvent B over 45 min was used, where solvent A was 0.05% TFA in H2O and solvent B was 0.043% TFA in 90/10% (v/v) ACN/H2O. The UV absorbance was monitored at 214 and 280 nm.
Mass Spectrometry
The mass and purity of peptides and bioorthogonal reaction products were determined using liquid chromatography-coupled MS (LC-MS) using a high-resolution API Qstar Pulsar mass spectrometer (PerkinElmer Sciex, Foster City, CA) or a high-resolution TripleTOF 5600 mass spectrometer system (AB Sciex). LC with the API Qstar MS system was performed with an Atlantis T3-C18 column (2.1 mm × 100 mm, 3 μm; Waters), and LC with the TripleTOF 5600 MS system was carried out with a Zorbax RRHD 300 SB-C18 column (2.1 mm × 100 mm, 1.8 μm; Agilent Technologies). Chromatographic separation was performed using a linear gradient from 5% to 40% solvent B, where solvent A was 0.1% formic acid (FA) (Sigma-Aldrich) in H2O and solvent B was 0.1% FA in 90/10% (v/v) ACN/H2O. The flow rate was 0.2 mL/min. Eluted compounds were detected by electrospray ionization in positive ion mode.
Analysis of Peptides, Linkers, and Bioorthogonal Reaction Products
The purity of the tested ligands was >95% as determined by analytical RP-HPLC on a LC-20AT chromatography system (Shimadzu Corp.). A Waters Atlantis T3 C18 column was used with a flow rate of 0.2 mL/min and a linear gradient from 0% to 45% solvent B over 45 min, where solvent A was 0.05% TFA in H2O and solvent B was 0.043% TFA in 90/10% (v/v) ACN/ H2O. The UV absorbance was monitored at 214 and 280 nm, with the 214 nm absorbance used to calculate purity. Data were recorded and processed with LabSolutions software (Shimadzu Corp.).
Structural Analysis by NMR
S-m3-HwTx-IV, AzK-KIIIA, and [m3-HwTx-IV]-[PEG80]-[K-KIIIA] were dissolved in 90/10% (v/v) H2O/D2O to a concentration of 1 mg/mL. 1D 1H, two-dimensional (2D) 1H–1H total correlated spectroscopy (TOCSY), and 2D 1H–1H nuclear Overhauser effect spectroscopy (NOESY) spectra were recorded on a Bruker Avance 600 MHz NMR spectrometer equipped with a cryogenically cooled probe (cryoprobe) at 25 °C. Spectra were processed using TopSpin (Bruker), and sequence-specific resonance assignments were made using CCPNMR Analysis 2.4.1.63
Cell Culture
Cell culture reagents were from Life Technologies Corp. unless otherwise stated. Human embryonic kidney (HEK) 293 cells co-expressing either hNaV1.4 or hNaV1.7 and the β1 auxiliary subunit (SB Drug Discovery, Glasgow, U.K.) were maintained at 37 °C in a humidified 5% CO2 incubator in Minimal Essential Medium (Sigma-Aldrich) supplemented with 10% FBS, 100 units/mL penicillin, 100 μg/mL streptomycin, 2 mM l-glutamine, and variable concentrations of blasticidin, geneticin, and zeocin according to the manufacturer’s protocols. Replicating cells were subcultured every 3–4 days in a 1/5 ratio using 0.25% trypsin/EDTA.
Patch-Clamp Electrophysiology
Sodium currents were recorded using an automated whole-cell patch-clamp system (QPatch 16X; Sophion Bioscience, Ballerup, Denmark) as described previously.23 The extracellular solution comprised 1 mM CaCl2, 1 mM MgCl2, 5 mM HEPES, 3 mM KCl, 140 mM NaCl, and 20 mM TEA-HCl at pH 7.3 (320 mOsm), and the intracellular solution comprised 140 mM CsF, 1 mM EGTA/5 mM CsOH, 10 mM HEPES, and 10 mM NaCl at pH 7.3 (320 mOsm). The elicited currents were sampled at 25 kHz and filtered at 4 kHz. Cells were maintained at a holding potential −80 mV, and Na+ currents elicited by 20 ms voltage steps to 0 mV from a −120 mV conditioning pulse applied for 200 ms. To obtain concentration–response curves, cells were incubated for 5 min with increasing concentrations of precursor peptides or bivalent ligands. This incubation period should be sufficient to obtain accurate IC50 values for even the most potent ligands described in this study, as it has been used previously to study exceptionally potent inhibitors of NaV1.7 (IC50 < 1 nM).23 However, we cannot exclude the possibility that the IC50 might be overestimated for the most potent ligands, but this would change none of our conclusions.
For on-rate experiments, Na+ currents were measured at 15 s intervals over 15 min immediately following addition of peptide at a concentration equivalent to 10 times its IC50 for the NaV subtype being analyzed. For koff measurements, cells were incubated with peptide for 10 min at a concentration equivalent to 10 times its IC50 for the NaV subtype being analyzed, and Na+ currents were assessed at 10 s intervals during 25 min saline washes. The kon, koff, and Kd values were calculated using the equation Kd = koff/kon (nM), where koff = 1/τoff (s–1) and kon = (1/τon – koff)/[ligand] (nM–1 s–1). Data were analyzed using Assay software (Sophion Biosciences), and Na+ currents (INa) plotted as I/Imax.
Data Analysis
For the in vitro electrophysiological recordings, curve fitting was performed using GraphPad Prism version 10 (GraphPad Software, San Diego, CA) using nonlinear regression with log inhibitor versus normalized response and variable Hill slope for dose–responses and IC50 determination, and exponential one-phase association and dissociation for on- and off-rate analysis, respectively. Data are means ± SEM.
Acknowledgments
The authors thank the members of the Muttenthaler group and Zoltan Dekan for their support, training, and help with some of the data acquisitions. The authors acknowledge funding support from the Australian National Health & Medical Research Council (Program Grant APP1072113 and Principal Research Fellowship APP1136889 to G.F.K.) and The University of Queensland (UQ Postdoctoral Fellowship to A.A.W.). M.M. was supported by the European Research Council under the European Union’s Horizon 2020 research and innovation program (714366) and by the Australian Research Council (DP190101667).
Glossary
Abbreviations Used
- ACN
acetonitrile
- tBuOH
tert-butanol
- 2-CTC
2-chlorotrityl chloride
- CuAAC
copper-catalyzed azide–alkyne cycloaddition
- CuSO4
copper(II) sulfate
- Et2O
diethyl ether
- DIPEA
N,N-diisopropyl-ethylamine
- DMF
N,N-dimethylformamide
- Imax
maximum peak current
- μ-KIIIA
μ-conotoxin KIIIA
- kon
association rate constant
- kon*
observed association rate constant
- koff
dissociation rate constant
- Kd
equilibrium dissociation constant
- m3-HwTx-IV
E1G, E4G, Y33W huwentoxin-IV
- MALDI-TOF
matrix-assisted laser desorption ionization time-of-flight
- MS
mass spectrometry
- NaV
voltage-gated sodium channel
- NMR
nuclear magnetic resonance
- PDB
Protein Data Bank
- PEG
polyethylene glycol
- RP-HPLC
reversed-phase high-performance liquid chromatography
- SEM
standard error of the mean
- SPPS
solid-phase peptide synthesis
- TFA
trifluoroacetic acid
- TIPS
triisopropylsilane
- VSD
voltage-sensing domain
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c01107.
Author Contributions
A.P. and F.C.C. contributed equally to this work. A.P. synthesized precursor peptides and PEG linkers, performed the bivalent ligand assembly, NMR analysis, MS, and HPLC analysis, and wrote the first draft of the manuscript. F.C.C. conducted and analyzed the electrophysiology. A.A.W., T.D., and P.E.D. planned the bivalent ligand design, and A.A.W. and T.D. provided critical reviews of the manuscript. M.R.L.S. assisted with the PEG and conjugation chemistry and peptide purification, aided data analysis, and edited the manuscript. N.B.E. contributed to the synthesis of the precursor peptides and helped with data analysis. M.M. and G.F.K. conceived, funded, and supervised the project and wrote the manuscript with A.P. and F.C.C. All authors read the manuscript and provided feedback.
The authors declare no competing financial interest.
Supplementary Material
References
- Catterall W. A. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron 2000, 26, 13–25. 10.1016/S0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- de Lera Ruiz M.; Kraus R. L. Voltage-gated sodium channels: structure, function, pharmacology, and clinical indications. J. Med. Chem. 2015, 58, 7093–7118. 10.1021/jm501981g. [DOI] [PubMed] [Google Scholar]
- Catterall W. A. Forty years of sodium channels: structure, function, pharmacology, and epilepsy. Neurochem. Res. 2017, 42, 2495–2504. 10.1007/s11064-017-2314-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klint J. K.; Senff S.; Rupasinghe D. B.; Er S. Y.; Herzig V.; Nicholson G. M.; King G. F. Spider-venom peptides that target voltage-gated sodium channels: pharmacological tools and potential therapeutic leads. Toxicon 2012, 60, 478–491. 10.1016/j.toxicon.2012.04.337. [DOI] [PubMed] [Google Scholar]
- Xu Y.; Sun J.; Liu H.; Sun J.; Yu Y.; Su Y.; Cui Y.; Zhao M.; Zhang J. Scorpion toxins targeting voltage-gated sodium channels associated with pain. Curr. Pharm. Biotechnol. 2018, 19, 848–855. 10.2174/1389201019666181105160744. [DOI] [PubMed] [Google Scholar]
- Akondi K. B.; Muttenthaler M.; Dutertre S.; Kaas Q.; Craik D. J.; Lewis R. J.; Alewood P. F. Discovery, synthesis, and structure-activity relationships of conotoxins. Chem. Rev. 2014, 114, 5815–5847. 10.1021/cr400401e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin A. H.; Muttenthaler M.; Dutertre S.; Himaya S. W. A.; Kaas Q.; Craik D. J.; Lewis R. J.; Alewood P. F. Conotoxins: chemistry and biology. Chem. Rev. 2019, 119, 11510–11549. 10.1021/acs.chemrev.9b00207. [DOI] [PubMed] [Google Scholar]
- Green B. R.; Olivera B. M. Venom peptides from cone snails: pharmacological probes for voltage-gated sodium channels. Curr. Top. Membr. 2016, 78, 65–86. 10.1016/bs.ctm.2016.07.001. [DOI] [PubMed] [Google Scholar]
- Moran Y.; Gordon D.; Gurevitz M. Sea anemone toxins affecting voltage-gated sodium channels—molecular and evolutionary features. Toxicon 2009, 54, 1089–1101. 10.1016/j.toxicon.2009.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow C.; Chin Y.; Walker A.; Guo S.; Blomster L.; Ward M.; Herzig V.; Rokyta D.; King G. Venom peptides with dual modulatory activity on the voltage-gated sodium channel NaV1.1 provide novel leads for development of antiepileptic drugs. ACS Pharmacol. Transl. Sci. 2020, 3, 119–134. 10.1021/acsptsci.9b00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israel M. R.; Tay B.; Deuis J. R.; Vetter I. Sodium channels and venom peptide pharmacology. Adv. Pharmacol. 2017, 79, 67–116. 10.1016/bs.apha.2017.01.004. [DOI] [PubMed] [Google Scholar]
- Kalia J.; Milescu M.; Salvatierra J.; Wagner J.; Klint J. K.; King G. F.; Olivera B. M.; Bosmans F. From foe to friend: using animal toxins to investigate ion channel function. J. Mol. Biol. 2015, 427, 158–175. 10.1016/j.jmb.2014.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens M.; Peigneur S.; Tytgat J. Neurotoxins and their binding areas on voltage-gated sodium channels. Front. Pharmacol. 2011, 2, 71. 10.3389/fphar.2011.00071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang A. H.; Sharma G.; Undheim E. A. B.; Jia X.; Mobli M. A complicated complex: ion channels, voltage sensing, cell membranes and peptide inhibitors. Neurosci. Lett. 2018, 679, 35–47. 10.1016/j.neulet.2018.04.030. [DOI] [PubMed] [Google Scholar]
- Shen H.; Li Z.; Jiang Y.; Pan X.; Wu J.; Cristofori-Armstrong B.; Smith J. J.; Chin Y. K. Y.; Lei J.; Zhou Q.; King G. F.; Yan N. Structural basis for the modulation of voltage-gated sodium channels by animal toxins. Science 2018, 362, eaau2596 10.1126/science.aau2596. [DOI] [PubMed] [Google Scholar]
- Yan Z.; Zhou Q.; Wang L.; Wu J.; Zhao Y.; Huang G.; Peng W.; Shen H.; Lei J.; Yan N. Structure of the NaV1.4-β1 complex from electric eel. Cell 2017, 170, 470–482. 10.1016/j.cell.2017.06.039. [DOI] [PubMed] [Google Scholar]
- Pan X.; Li Z.; Zhou Q.; Shen H.; Wu K.; Huang X.; Chen J.; Zhang J.; Zhu X.; Lei J.; Xiong W.; Gong H.; Xiao B.; Yan N. Structure of the human voltage-gated sodium channel NaV1.4 in complex with β1. Science 2018, 362, eaau2486 10.1126/science.aau2486. [DOI] [PubMed] [Google Scholar]
- Shen H.; Liu D.; Wu K.; Lei J.; Yan N. Structures of human NaV1.7 channel in complex with auxiliary subunits and animal toxins. Science 2019, 363, 1303–1308. 10.1126/science.aaw2493. [DOI] [PubMed] [Google Scholar]
- Jurkat-Rott K.; Holzherr B.; Fauler M.; Lehmann-Horn F. Sodium channelopathies of skeletal muscle result from gain or loss of function. Pfluegers Arch. 2010, 460, 239–248. 10.1007/s00424-010-0814-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loussouarn G.; Sternberg D.; Nicole S.; Marionneau C.; Le Bouffant F.; Toumaniantz G.; Barc J.; Malak O. A.; Fressart V.; Pereon Y.; Baro I.; Charpentier F. Physiological and pathophysiological insights of Nav1.4 and Nav1.5 comparison. Front. Pharmacol. 2016, 6, 314. 10.3389/fphar.2015.00314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King G. F.; Vetter I. No gain, no pain: NaV1.7 as an analgesic target. ACS Chem. Neurosci. 2014, 5, 749–751. 10.1021/cn500171p. [DOI] [PubMed] [Google Scholar]
- Bennett D. L.; Clark A. J.; Huang J.; Waxman S. G.; Dib-Hajj S. D. The role of voltage-gated sodium channels in pain signaling. Physiol. Rev. 2019, 99, 1079–1151. 10.1152/physrev.00052.2017. [DOI] [PubMed] [Google Scholar]
- Cardoso F. C.; Dekan Z.; Rosengren K. J.; Erickson A.; Vetter I.; Deuis J. R.; Herzig V.; Alewood P. F.; King G. F.; Lewis R. J. Identification and characterization of ProTx-III [μ-TRTX-Tp1a], a new voltage-gated sodium channel inhibitor from venom of the tarantula thrixopelma pruriens. Mol. Pharmacol. 2015, 88, 291–303. 10.1124/mol.115.098178. [DOI] [PubMed] [Google Scholar]
- Deuis J. R.; Dekan Z.; Wingerd J. S.; Smith J. J.; Munasinghe N. R.; Bhola R. F.; Imlach W. L.; Herzig V.; Armstrong D. A.; Rosengren K. J.; Bosmans F.; Waxman S. G.; Dib-Hajj S. D.; Escoubas P.; Minett M. S.; Christie M. J.; King G. F.; Alewood P. F.; Lewis R. J.; Wood J. N.; Vetter I. Pharmacological characterisation of the highly Na(V)1.7 selective spider venom peptide Pn3a. Sci. Rep. 2017, 7, 40883. 10.1038/srep40883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow C. Y.; Chin Y. K. Y.; Ma L.; Undheim E. A. B.; Herzig V.; King G. F. A selective NaV1.1 activator with potential for treatment of Dravet syndrome epilepsy. Biochem. Pharmacol. 2020, 113991. 10.1016/j.bcp.2020.113991. [DOI] [PubMed] [Google Scholar]
- Rupasinghe D. B.; Herzig V.; Vetter I.; Dekan Z.; Gilchrist J.; Bosmans F.; Alewood P. F.; Lewis R. J.; King G. F. Mutational analysis of ProTx-I and the novel venom peptide Pe1b provide insight into residues responsible for selective inhibition of the analgesic drug target NaV1.7. Biochem. Pharmacol. 2020, 114080. 10.1016/j.bcp.2020.114080. [DOI] [PubMed] [Google Scholar]
- Pan X.; Li Z.; Huang X.; Huang G.; Gao S.; Shen H.; Liu L.; Lei J.; Yan N. Molecular basis for pore blockade of human Na+ channel NaV1.2 by the μ-conotoxin KIIIA. Science 2019, 363, 1309–1313. 10.1126/science.aaw2999. [DOI] [PubMed] [Google Scholar]
- Zhang M. M.; Green B. R.; Catlin P.; Fiedler B.; Azam L.; Chadwick A.; Terlau H.; McArthur J. R.; French R. J.; Gulyas J.; Rivier J. E.; Smith B. J.; Norton R. S.; Olivera B. M.; Yoshikami D.; Bulaj G. Structure/function characterization of μ-conotoxin KIIIA, an analgesic, nearly irreversible blocker of mammalian neuronal sodium channels. J. Biol. Chem. 2007, 282, 30699–30706. 10.1074/jbc.M704616200. [DOI] [PubMed] [Google Scholar]
- Khoo K. K.; Gupta K.; Green B. R.; Zhang M. M.; Watkins M.; Olivera B. M.; Balaram P.; Yoshikami D.; Bulaj G.; Norton R. S. Distinct disulfide isomers of μ-conotoxins KIIIA and KIIIB block voltage-gated sodium channels. Biochemistry 2012, 51, 9826–9835. 10.1021/bi301256s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Haegen A.; Peigneur S.; Tytgat J. Importance of position 8 in μ-conotoxin KIIIA for voltage-gated sodium channel selectivity. FEBS J. 2011, 278, 3408–3418. 10.1111/j.1742-4658.2011.08264.x. [DOI] [PubMed] [Google Scholar]
- McArthur J. R.; Singh G.; McMaster D.; Winkfein R.; Tieleman D. P.; French R. J. Interactions of key charged residues contributing to selective block of neuronal sodium channels by μ-conotoxin KIIIA. Mol. Pharmacol. 2011, 80, 573–584. 10.1124/mol.111.073460. [DOI] [PubMed] [Google Scholar]
- Kolb H. C.; Finn M. G.; Sharpless K. B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. . [DOI] [PubMed] [Google Scholar]
- Peng K.; Shu Q.; Liu Z.; Liang S. Function and solution structure of huwentoxin-IV, a potent neuronal tetrodotoxin (TTX)-sensitive sodium channel antagonist from Chinese bird spider Selenocosmia huwena. J. Biol. Chem. 2002, 277, 47564–47571. 10.1074/jbc.M204063200. [DOI] [PubMed] [Google Scholar]
- Revell J. D.; Lund P. E.; Linley J. E.; Metcalfe J.; Burmeister N.; Sridharan S.; Jones C.; Jermutus L.; Bednarek M. A. Potency optimization of huwentoxin-IV on hNaV1.7: a neurotoxin TTX-S sodium-channel antagonist from the venom of the Chinese bird-eating spider Selenocosmia huwena. Peptides 2013, 44, 40–46. 10.1016/j.peptides.2013.03.011. [DOI] [PubMed] [Google Scholar]
- Pallaghy P. K.; Nielsen K. J.; Craik D. J.; Norton R. S. A common structural motif incorporating a cystine knot and a triple-stranded β-sheet in toxic and inhibitory polypeptides. Protein Sci. 1994, 3, 1833–1839. 10.1002/pro.5560031022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahnama S.; Deuis J. R.; Cardoso F. C.; Ramanujam V.; Lewis R. J.; Rash L. D.; King G. F.; Vetter I.; Mobli M. The structure, dynamics and selectivity profile of a NaV1.7 potency-optimized huwentoxin-IV variant. PLoS One 2017, 12, e0173551 10.1371/journal.pone.0173551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoghegan K. F.; Stroh J. G. Site-directed conjugation of nonpeptide groups to peptides and proteins via periodate oxidation of a 2-amino alcohol. Application to modification at N-terminal serine. Bioconjugate Chem. 1992, 3, 138–146. 10.1021/bc00014a008. [DOI] [PubMed] [Google Scholar]
- Xu H.; Li T.; Rohou A.; Arthur C. P.; Tzakoniati F.; Wong E.; Estevez A.; Kugel C.; Franke Y.; Chen J.; Ciferri C.; Hackos D. H.; Koth C. M.; Payandeh J. Structural basis of NaV1.7 inhibition by a gating-modifier spider toxin. Cell 2019, 176, 702–715. 10.1016/j.cell.2018.12.018. [DOI] [PubMed] [Google Scholar]
- Hanwell M. D.; Curtis D. E.; Lonie D. C.; Vandermeersch T.; Zurek E.; Hutchison G. R. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J. Cheminf. 2012, 4, 17. 10.1186/1758-2946-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack E. T.; Snyder P. W.; Perez-Castillejos R.; Bilgicer B.; Moustakas D. T.; Butte M. J.; Whitesides G. M. Dependence of avidity on linker length for a bivalent ligand-bivalent receptor model system. J. Am. Chem. Soc. 2012, 134, 333–345. 10.1021/ja2073033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer-Durand N.; Salmain M.; Vessières A.; Jaouen G. A new bioorthogonal cross-linker with alkyne and hydrazide end groups for chemoselective ligation. Application to antibody labelling. Tetrahedron 2012, 68, 9638–9644. 10.1016/j.tet.2012.09.062. [DOI] [Google Scholar]
- Zalipsky S. Functionalized poly(ethylene glycol) for preparation of biologically relevant conjugates. Bioconjugate Chem. 1995, 6, 150–165. 10.1021/bc00032a002. [DOI] [PubMed] [Google Scholar]
- Goswami L. N.; Houston Z. H.; Sarma S. J.; Jalisatgi S. S.; Hawthorne M. F. Efficient synthesis of diverse heterobifunctionalized clickable oligo(ethylene glycol) linkers: potential applications in bioconjugation and targeted drug delivery. Org. Biomol. Chem. 2013, 11, 1116–1126. 10.1039/c2ob26968f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekladious I.; Colson Y. L.; Grinstaff M. W. Polymer-drug conjugate therapeutics: advances, insights and prospects. Nat. Rev. Drug Discovery 2019, 18, 273–294. 10.1038/s41573-018-0005-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alconcel S. N. S.; Baas A. S.; Maynard H. D. FDA-approved poly(ethylene glycol)–protein conjugate drugs. Polym. Chem. 2011, 2, 1442–1448. 10.1039/c1py00034a. [DOI] [Google Scholar]
- Behrendt R.; White P.; Offer J. Advances in Fmoc solid-phase peptide synthesis. J. Pept. Sci. 2016, 22, 4–27. 10.1002/psc.2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J. S.; Tang S.; Qi Y. K.; Wang Z. P.; Liu L. Chemical synthesis of proteins using peptide hydrazides as thioester surrogates. Nat. Protoc. 2013, 8, 2483–2495. 10.1038/nprot.2013.152. [DOI] [PubMed] [Google Scholar]
- Agten S. M.; Suylen D. P. L.; Hackeng T. M. Oxime catalysis by freezing. Bioconjugate Chem. 2016, 27, 42–46. 10.1021/acs.bioconjchem.5b00611. [DOI] [PubMed] [Google Scholar]
- Xiao Y.; Bingham J. P.; Zhu W.; Moczydlowski E.; Liang S.; Cummins T. R. Tarantula huwentoxin-IV inhibits neuronal sodium channels by binding to receptor site 4 and trapping the domain ii voltage sensor in the closed configuration. J. Biol. Chem. 2008, 283, 27300–27313. 10.1074/jbc.M708447200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mould J.; Yasuda T.; Schroeder C. I.; Beedle A. M.; Doering C. J.; Zamponi G. W.; Adams D. J.; Lewis R. J. The α2δ auxiliary subunit reduces affinity of ω-conotoxins for recombinant N-type (CaV2.2) calcium channels. J. Biol. Chem. 2004, 279, 34705–34714. 10.1074/jbc.M310848200. [DOI] [PubMed] [Google Scholar]
- Vauquelin G.; Charlton S. J. Exploring avidity: understanding the potential gains in functional affinity and target residence time of bivalent and heterobivalent ligands. Br. J. Pharmacol. 2013, 168, 1771–1785. 10.1111/bph.12106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valant C.; Robert Lane J.; Sexton P. M.; Christopoulos A. The best of both worlds? Bitopic orthosteric/allosteric ligands of g protein-coupled receptors. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 153–178. 10.1146/annurev-pharmtox-010611-134514. [DOI] [PubMed] [Google Scholar]
- Hah J. M.; Sharma V.; Li H.; Lawrence D. S. Acquisition of a “Group A“-selective Src kinase inhibitor via a global targeting strategy. J. Am. Chem. Soc. 2006, 128, 5996–5997. 10.1021/ja060136i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busnelli M.; Kleinau G.; Muttenthaler M.; Stoev S.; Manning M.; Bibic L.; Howell L. A.; McCormick P. J.; Di Lascio S.; Braida D.; Sala M.; Rovati G. E.; Bellini T.; Chini B. Design and characterization of superpotent bivalent ligands targeting oxytocin receptor dimers via a channel-like structure. J. Med. Chem. 2016, 59, 7152–7166. 10.1021/acs.jmedchem.6b00564. [DOI] [PubMed] [Google Scholar]
- Cappelli A.; Manini M.; Paolino M.; Gallelli A.; Anzini M.; Mennuni L.; Del Cadia M.; De Rienzo F.; Menziani M. C.; Vomero S. Bivalent ligands for the serotonin 5-HT3 receptor. ACS Med. Chem. Lett. 2011, 2, 571–576. 10.1021/ml2000388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan J.; Huang J. X.; Vetter I.; Mobli M.; Lawson J.; Tae H. S.; Abraham N.; Paul B.; Cooper M. A.; Adams D. J.; Lewis R. J.; Alewood P. F. α-Conotoxin dendrimers have enhanced potency and selectivity for homomeric nicotinic acetylcholine receptors. J. Am. Chem. Soc. 2015, 137, 3209–3212. 10.1021/jacs.5b00244. [DOI] [PubMed] [Google Scholar]
- Tran H. N. T.; Tran P.; Deuis J. R.; Agwa A. J.; Zhang A. H.; Vetter I.; Schroeder C. I. Enzymatic ligation of a pore blocker toxin and a gating modifier toxin: creating double-knotted peptides with improved sodium channel NaV1.7 inhibition. Bioconjugate Chem. 2020, 31, 64–73. 10.1021/acs.bioconjchem.9b00744. [DOI] [PubMed] [Google Scholar]
- Zeberg H.; Nilsson J.; Århem P. The importance of the dissociation rate in ion channel blocking. Front. Cell. Neurosci. 2018, 12, 33. 10.3389/fncel.2018.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonge P. J. Drug-target kinetics in drug discovery. ACS Chem. Neurosci. 2018, 9, 29–39. 10.1021/acschemneuro.7b00185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo D.; Heitman L. H.; IJzerman A. P. The added value of assessing ligand-receptor binding kinetics in drug discovery. ACS Med. Chem. Lett. 2016, 7, 819–821. 10.1021/acsmedchemlett.6b00273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuetz D. A.; de Witte W. E. A.; Wong Y. C.; Knasmueller B.; Richter L.; Kokh D. B.; Sadiq S. K.; Bosma R.; Nederpelt I.; Heitman L. H.; Segala E.; Amaral M.; Guo D.; Andres D.; Georgi V.; Stoddart L. A.; Hill S.; Cooke R. M.; De Graaf C.; Leurs R.; Frech M.; Wade R. C.; de Lange E. C. M.; IJzerman A. P.; Müller-Fahrnow A.; Ecker G. F. Kinetics for drug discovery: an industry-driven effort to target drug residence time. Drug Discovery Today 2017, 22, 896–911. 10.1016/j.drudis.2017.02.002. [DOI] [PubMed] [Google Scholar]
- Vauquelin G. Effects of target binding kinetics on in vivo drug efficacy: koff, kon and rebinding. Br. J. Pharmacol. 2016, 173, 2319–2334. 10.1111/bph.13504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vranken W. F.; Boucher W.; Stevens T. J.; Fogh R. H.; Pajon A.; Llinas M.; Ulrich E. L.; Markley J. L.; Ionides J.; Laue E. D. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins: Struct., Funct., Genet. 2005, 59, 687–696. 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.