

Abstract

Anthracycline anticancer drugs doxorubicin and aclarubicin have been used in the clinic for several decades to treat various cancers. Although closely related structures, their molecular mode of action diverges, which is reflected in their biological activity profile. For a better understanding of the structure–function relationship of these drugs, we synthesized ten doxorubicin/aclarubicin hybrids varying in three distinct features: aglycon, glycan, and amine substitution pattern. We continued to evaluate their capacity to induce DNA breaks, histone eviction, and relocated topoisomerase IIα in living cells. Furthermore, we assessed their cytotoxicity in various human tumor cell lines. Our findings underscore that histone eviction alone, rather than DNA breaks, contributes strongly to the overall cytotoxicity of anthracyclines, and structures containing N,N-dimethylamine at the reducing sugar prove that are more cytotoxic than their nonmethylated counterparts. This structural information will support further development of novel anthracycline variants with improved anticancer activity.
Introduction
Anthracyclines comprise one of the most successful classes of natural product chemotherapeutic agents. Two archetypal anthracyclines are doxorubicin (1) and aclarubicin (12, Figure 1), both effective anticancer agents isolated from nature.1,2 Doxorubicin has been in use in the clinic for more than five decades and is prescribed worldwide to about a million patients annually for the treatment of a variety of cancers.3−5 Aclarubicin in contrast is prescribed exclusively in Japan and China, mainly for the treatment of acute myeloid leukemia (AML). Although doxorubicin is very effective, its use coincides with cardiotoxicity, formation of secondary tumors, and infertility.6−9 Therefore, clinical use with doxorubicin is generally limited to a cumulative dose of 450–550 mg/m2.7,10,11 The formation of reactive oxygen species (ROS) by these drugs has been considered as a major mechanism mediating anthracycline-induced cardiotoxicity.12,13 However, aclarubicin, which has a higher redox potential than doxorubicin,14 displays fewer cardiotoxic side effects, and recent findings in our labs suggested that this difference in cardiotoxicity relates to significant differences in the mode of action of these two compounds.15 Doxorubicin causes chromatin damage by inducing histone eviction, as well as the formation of DNA double-strand breaks by poisoning topoisomerase IIα (TopoIIα).16,17 Aclarubicin is capable of evicting histones as well, but targets TopoIIα without inducing DNA double-strand breaks.17−19 In addition, it has been shown that aclarubicin affects cell viability by reducing the mitochondrial respiratory activity.20 Histone eviction induced by anthracycline drugs results in epigenetic and transcriptional changes, which are thought to then induce apoptosis.17 We recently showed that anthracyclines that induce both DNA double-strand break formation and histone eviction are cardiotoxic. Aclarubicin and N,N-dimethyldoxorubicin (3) both lack DNA damage activity but are able to induce histone eviction, and can thus be used as effective anticancer drugs without cardiotoxicity.15 The structural basis causing this difference in biological activities, however, is still lacking. Therefore, better insight into the structure–function relationship of these molecules is needed.
Figure 1.
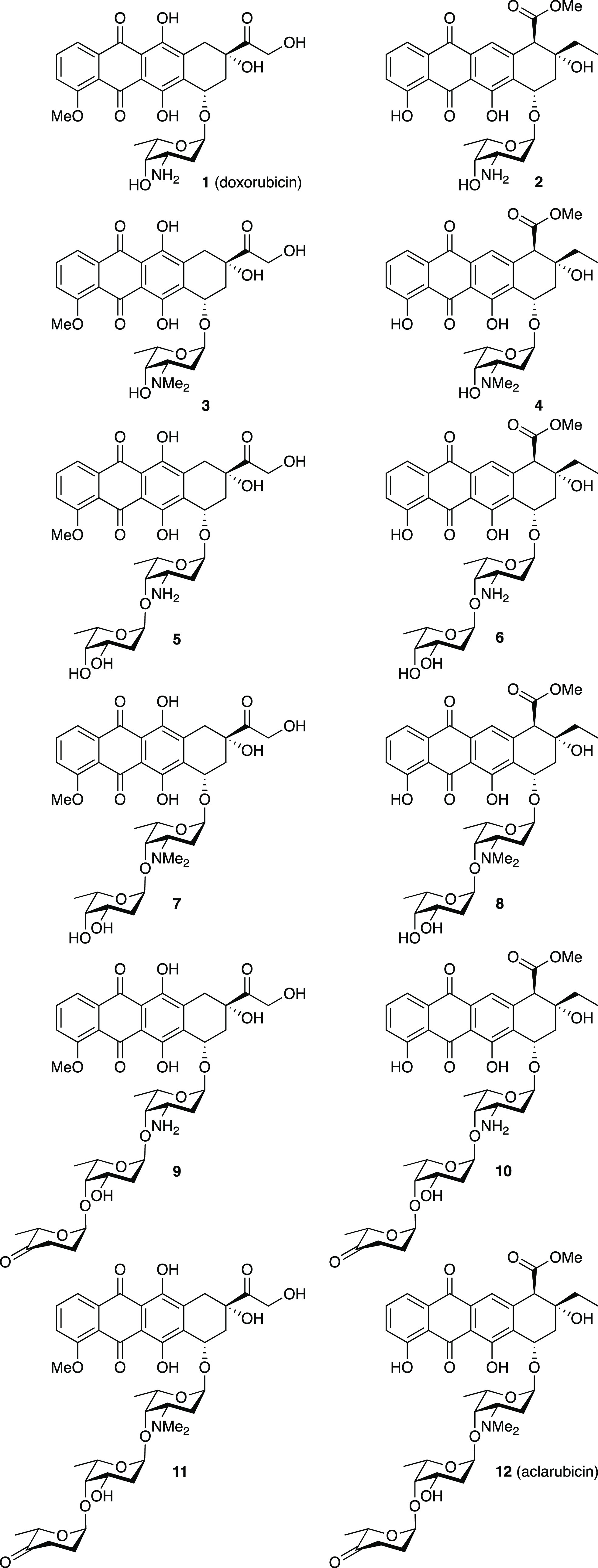
Chemical structures of doxorubicin (1), aclarubicin (12), and hybrid structures (2–11), subject of the here-presented studies.
In addition to the treatment-limiting side effects, development of resistance constitutes to be a frequent clinical limitation for the treatment of patients with anthracycline drugs.21,22 Common mechanisms of resistance toward anthracycline drugs are reduced expression or activity of TopoIIα and overexpression of membrane transporters such as P-glycoprotein (P-gp) and multidrug resistance-associated protein (MRP), both of which decrease the cellular accumulation of the drugs via increased drug export.23−25
Although the structures of doxorubicin (1) and aclarubicin (12) are quite similar (they both contain an anthraquinone and a sugar containing a basic amine), three differences can be identified: (i) variation in the substitution and oxidation pattern of the anthraquinone aglycon, (ii) variation in the size of the carbohydrate part, and (iii) the methylation pattern of the amine of the first sugar attached to the anthraquinone. Doxorubicin features an α-l-daunosamine as the single monosaccharidic carbohydrate appendage, while aclarubicin features an α-l-rhodosamine (N,N-dimethyldaunosamine) that is further glycosylated at the 4-hydroxyl with a disaccharide composed of α-l-oliose and α-l-cinerulose A. Thousands of analogues of doxorubicin and aclarubicin have been isolated from bacterial sources or prepared through organic synthesis.26 In spite of this, the chemical space between doxorubicin and aclarubicin has not been fully explored. Although some doxorubicin/aclarubicin hybrids have been prepared (including compounds 2,273,15,284,298,3010,31 and 11(32)), the reported methods of synthesis are fragmented and the complete set, as shown in Figure 1, has not been evaluated in the context of the different modes of action described above. We therefore set out to generate a comprehensive set of doxorubicin/aclarubicin hybrid structures, systematically varying the structural elements in which the two anthracyclines differ. Based on these structural differences between doxorubicin and aclarubicin, we envisaged the set of doxorubicin/aclarubicin hybrids 2–11 (Figure 1) that comprises anthracyclines composed of either of the two aglycons, additionally featuring either a monosaccharide, a disaccharide, or a trisaccharide glycan composed of the sugar configurations also found in the parent structures, and bearing either no or two N-methyl substituents. Altogether, they fill the chemical space between doxorubicin (1) and aclarubicin (12). Furthermore, we probed this coherent set of anthracycline hybrid structures for their DNA damaging, TopoIIα relocalization, histone evicting, and cytotoxic activities to get a better understanding of the structural basis underlying the observed difference for the anticancer activity of these compounds. These new insights could ultimately lead to the development of new anthracycline variants with improved anticancer activity.
Results
Synthesis of Doxorubicin/Aclarubicin Hybrid Monosaccharides 2 and 4
For the assembly of the set of anthracyclines, we used Biao Yu’s gold(I)-mediated condensation33 of the glycans and aglycons, as these mild glycosylation conditions are compatible with the lability and reactivity of the deoxy sugars that are to be appended to the anthraquinones. The anthraquinone aglycons were readily obtained by acidic hydrolysis of the drugs doxorubicin (1) and aclarubicin (12). This yielded aklavinone (14)34 and, following protection of the primary alcohol in doxorubicinone as the tert-butyldimethylsilyl (TBS) ether, 14-O-TBS-doxorubicinone 16(35) (Scheme 1). Condensation of daunosaminyl cyclopropylethynylbenzoate (ABz) 13 (see Schemes S1 and S2 (Supporting Information) for a complete description of the syntheses of the building blocks) and aklavinone (14) under Yu’s conditions provided anthracycline 15 in a stereoselective manner (Scheme 1). The stereoselectivity of this glycosylation can be accounted for by long-range participation36,37 of the allyl carbamate, as well as the conformation of the intermediate oxocarbenium ion that can be substituted in a stereoselective manner on the α-face.38 The yield of this glycosylation reaction (73%) compares favorably to the yields (50–60%) reported by Pearlman et al., who used glycal donors in combination with Brønsted acid catalysis.39 The N-Alloc group in 15 was then removed using a catalytic amount of Pd(PPh3)4 and N,N-dimethylbarbituric acid (NDMBA) as the allyl scavenger.40 This was followed by desilylation using an HF·pyr complex to give the first hybrid structure 2.41 The corresponding dimethylamine 4 could be prepared by performing reductive alkylation with formaldehyde and NaBH(OAc)3 after the removal of the Alloc functionality, and finally a desilylation. The third monosaccharide anthracycline 3 was obtained as we previously described.15
Scheme 1. Synthesis of Hybrid Monosaccharide Anthracyclines 2–4.

Reagents and conditions: (a) 0.2 M aqueous (aq) HCl, 90 °C, quant.; (b) PPh3AuNTf2 (10 mol %), dichloromethane (DCM), −20 °C, 73% (>20:1 α/β); (c) (i) Pd(PPh3)4, NDMBA, DCM, (ii) HF·pyridine, pyr., 40% over two steps; (d) (i) Pd(PPh3)4, NDMBA, DCM, (ii) aq CH2O, NaBH(OAc)3, EtOH, (iii) HF·pyridine, pyr., 43% over three steps; (e) (i) aq HCl, 90 °C, (ii) TBS-Cl, imidazole, dimethylformamide (DMF), 97% over two steps.
Synthesis of Hybrid Disaccharides 5–8
We then turned our attention to the four disaccharidic antracyclines 5–8. This required the synthesis of disaccharide donor 21, which is depicted in Scheme 2A. Compound 21 was constructed through an iodonium di-collidinium perchlorate (IDCP)42-mediated glycosylation of l-olioside thioglycoside donor 18, protected as the tetraisopropyldisiloxane ether, which effectively shields the β-face to facilitate the stereoselective introduction of the desired α-linkage. The reaction between donor 18 and acceptor 17 delivered the desired disaccharide 19 in excellent yield and stereoselectivity. Triphenylphosphine was added to the reaction mixture to reduce the in situ formed sulfenamide that was formed from the Alloc carbamate and the generated phenylsulfenyl iodide.43,44 The chemoselective removal of the anomeric p-methoxyphenolate (PMP) protective group in 19 was achieved using silver(II) hydrogen dipicolinate (Ag(DPAH)2),45,46 and the anomeric alcohol thus liberated was then condensed with carboxylic acid 20 under Steglich conditions,47 to deliver the disaccharide alkynylbenzoate donor 21. The coupling to the two aglycone acceptors 14 and 16 is outlined in Scheme 2B. Treatment of a mixture of donor 21 and doxorubicinone acceptor 16 with PPh3AuNTf2 proceeded stereoselectively to give 22 in 64% yield. Ensuing Alloc removal proceeded quantitatively to give 23, after which HF·pyridine-mediated desilylation yielded the first disaccharide anthracycline 5. To introduce the dimethylamino functionality, amine 23 was treated with formaldehyde and a substoichiometric amount of NaBH(OAc)3 to prevent reduction of the hydroxyketone function on the aglycone.28 A final desilylation resulted in dimethylated 7. Subjecting donor 21 and aklavinone 14 to gold(I)-mediated glycosylation also proceeded stereoselectively to give the protected disaccharide anthracycline, of which the Alloc group was removed to give 24 in 87% yield over the two steps. Removal of the disiloxane moiety with HF·pyridine then gave disaccharide anthracycline 6. A double-reductive N-methylation was performed on fully deprotected 6 to give 8.
Scheme 2. (A) Synthesis of Disaccharide Alkynylbenzoate Donor 21; (B) Synthesis of Hybrid Disaccharide Anthracyclines 5–8.

Reagents and conditions: (a) IDCP, Et2O, 1,2-dichloroethane (DCE) (4:1 v/v), then PPh3, 89%; (b) (i) Ag(II)(hydrogen dipicolinate)2, NaOAc, MeCN, H2O, 0 °C, (ii) 20, EDCI·HCl, N,N-diisopropylethylamine (DIPEA), 4-dimethylaminopyridine (DMAP), DCM, 84% over two steps (1:8 α/β).
Reagents and conditions: (c) 16, PPh3AuNTf2 (10 mol %), DCM, 64% (>20:1 α/β); (d) Pd(PPh3)4, NDMBA, DCM, quant.; (e) HF·pyridine, pyr., 76% for 5, 81% for 7; (f) aq CH2O, NaBH(OAc)3, EtOH, 71%; (g) (i) 14, PPh3AuNTf2 (10 mol %), −20 °C, DCM, (ii) Pd(PPh3)4, NDMBA, DCM, 87% over two steps (>20:1 α/β); (h) HF·pyridine, pyr., 41%; (i) aq CH2O, NaBH(OAc)3, EtOH, 34%.
Synthesis of Hybrid Trisaccharides 9–11
To complete the set of target compounds, trisaccharide anthracyclines 9–11 were prepared. These required trisaccharide alkynylbenzoate donor 30, the synthesis of which is shown in Scheme 3A. First, protected daunosaminyl acceptor 17 and oliosyl donor 25 were condensed using the conditions described for the synthesis of disaccharide 18 to provide disaccharide 26. This glycosylation proceeded with excellent stereoselectivity, which can be attributed to the structure of the intermediate oxocarbenium ion.38 Removal of the benzoyl protective group in 26 gave acceptor 27.
Scheme 3. (A) Synthesis of Trisaccharide Alkynylbenzoate Donor 30; (B) Synthesis of Hybrid Trisaccharide Anthracyclines 9–11.

Reagents and conditions: (a) IDCP, Et2O/DCE (4:1 v/v), then PPh3; (b) NaOMe, MeOH, 78% over two steps (>20:1 α/β); (c) IDCP, Et2O/DCE (4:1 v/v), then PPh3, 100% (>20:1 α/β); (d) (i) NaOMe, MeOH, 85%, (ii) Dess–Martin periodinane, NaHCO3, CH2Cl2, 97%; (e) (i) Ag(II)(hydrogen dipicolinate)2, NaOAc, MeCN/H2O (1:1, v/v), 0 °C, (ii) 20, EDCI·HCl, DIPEA, DMAP, CH2Cl2, 75% over the two steps (1:7 α/β).
Reagents and conditions: (f) (i) 16, PPh3AuNTf2 (10 mol %), DCM, (ii) 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ), DCM, pH 7 phosphate buffer (18:1, v/v), 57% over two steps (>20:1 α/β); (g) Pd(PPh3)4, NDMBA, DCM, 81% from 31, 61% for 10; (h) HF·pyridine, pyr., 73% for 9, 73% for 11; (i) aq CH2O, NaBH(OAc)3, EtOH, 52%; (j) 14, PPh3AuNTf2 (10 mol %), DCM, −20 °C, 71% (>20:1 α/β); (k) DDQ, DCM/pH 7 phosphate buffer (18:1, v/v), 90%.
Elongation of this disaccharide was achieved using an IDCP-mediated glycosylation using l-rhodinoside donor 28 to stereoselectively provide the protected trisaccharide. Removal of the benzoyl ester gave the alcohol, which was oxidized using a Dess–Martin oxidation to install the required ketone functionality in 29. The trisaccharide was converted to the corresponding Yu donor with the oxidation–Steglich esterification sequence, as described earlier, to give 30. Of note, the silver(II) reagent used to remove the anomeric para-methoxyphenol moiety left the para-methoxybenzyl-protecting group unscathed. Treatment of aglycon 16 and donor 30 with PPh3AuNTf2 led to the stereoselective formation of the first protected trisaccharide anthracycline, of which the para-methylbenzyl (PMB) group was removed to give partially protected anthracycline 31 in 57% yield, over two steps (Scheme 3B). This represents a significant improvement over a previous synthesis, reported by Tanaka et al.,32 who combined a trisaccharide bromide and the aglycone acceptor in a TBABr/collidine-mediated glycosylation to give the trisaccharide anthracycline in 22% yield. Removal of the Alloc group and desilylation of 31 then afforded 9. A double-reductive amination on 31 followed by desilylation provided hybrid anthracycline 11. For the synthesis of 10, a mixture of 30 and 14 was treated with PPh3AuNTf2 at −20 °C to afford 32 as a single diastereoisomer in 71% yield. Removal of the Alloc and PMB groups finally gave 10. The analytical data for the compounds described previously in the literature (2,273,284,298,3010,311132) were in good agreement with the reported data.
DNA Double-Strand Breakage and Histone Eviction
Since the main difference in biological activity between doxorubicin and aclarubicin is their capacity to induce DNA double-strand breaks, we tested the ability of hybrid structures 2–11 in comparison to their parental drugs 1 and 12 to induce DNA damage. Anthracyclines are often used in the treatment of acute myeloid leukemia; therefore, human chronic myelogenous leukemia cells (K562 cells) were incubated for 2 h with 10 μM 1–12, and etoposide as a positive control for DNA double-strand break formation.48,49 These concentrations are corresponding to physiological serum peak levels of cancer patients at standard treatment.17,50 DNA break formation was analyzed by measuring phosphorylation of H2AX (γH2AX), a well-known marker for DNA double-strand breaks, by Western blot (Figure 2A,B) as well as by constant-field gel electroporation (Figure 2C).51 Only doxorubicin (1) and hybrid structure 9 induced DNA double-strand breaks, as is evident from both assays (Figure S1A–C, Supporting Information). None of the other compounds induced phosphorylated H2AX and thus resemble the activity of aclarubicin (12). Subsequently, compounds 1–12 were tested for their ability to induce histone eviction. To visualize histone eviction, the release of photoactivated green fluorescent protein-labeled histone H2A (PAGFP-H2A) was followed in the adherent human melanoma MelJuSo cell line using time-lapse confocal microscopy, as previously described.15,17 Compounds 3, 8, and 11 are equally potent at evicting histones to their parent structures doxorubicin (1) and aclarubicin (12). Compounds 4, 6, and 7 are able to evict histones, but do so less efficiently than 1 and 12, while compounds 2, 5, 9, and 10 fail to evict histones (Figures 2D and S2).
Figure 2.

Evaluation of DNA break capacity and histone evicting activity of hybrid structures 2–11 and parent compounds doxorubicin (1) and aclarubicin (12). (A) K562 cells were treated for 2 h with 10 μM of the indicated drugs, etoposide was used as a positive control for DNA double-strand breaks. γH2AX levels were examined by Western blot. Actin was used as a loading control, and molecular weight markers are as indicated. (B) Quantification of the γH2AX signal normalized to actin. Results are presented as mean ± standard deviation (SD) of three independent experiments. Ordinary one-way analysis of variance (ANOVA) with Dunnett’s multiple comparison test; ns, not significant; ****P < 0.0001. (C) Quantification of broken DNA relative to intact DNA as analyzed by constant-field gel electrophoresis (CFGE). Etoposide was used as a positive control for DNA double-strand breaks. Results are presented as mean ± SD of three independent experiments. Ordinary one-way ANOVA with Dunnett’s multiple comparison test; *P < 0.05, ****P < 0.0001 is indicated, all others are not significant. (D) Quantification of the release of fluorescent PAGFP-H2A from the photoactivated nuclear regions after administration of 10 μM of the indicated drugs. Results are shown as mean ± SD of 10–20 cells from at least three independent experiments. Ordinary two-way ANOVA with Dunnett’s multiple comparison test; ns, not significant; ****P < 0.0001. See also Figures S1 and S2.
Cytotoxicity and Cellular Uptake
To test the cell cytotoxicity of the panel of hybrid anthracyclines, K562 cells were treated for 2 h with compounds 1–12 at physiological relevant concentrations, and cell survival was measured 72 h post-treatment using a CellTiter-Blue assay (Figure 3A,B).17,50 Compounds 3, 8, and 11 were effectively killing K562 cells. While compounds 3 and 8 showed cytotoxicity in the same range as their parental drugs doxorubicin (1) and aclarubicin (12), respectively, compound 11 was ∼13 times more cytotoxic than doxorubicin and 2.5 times more than aclarubicin. Compounds 4, 7, 9, and 10 were only effective at higher concentrations, while compounds 2, 5, and 6 did not show any cytotoxicity (Figures 3A,B and S3A). The observed cytotoxicity is not specific for this acute myeloid leukemia cell line (K562) because similar toxicity profiles were observed for these compounds when tested in the melanoma cell line MelJuSo, the colorectal carcinoma cell line HCT116, the two prostate cancer cell lines PC3 and DU145, and the glioblastoma cell line U87 (Figure 3C–G). To validate that the differences in DNA damage, chromatin damage induction, and effective cytotoxicity are not caused by differences in cellular uptake of the different hybrid structures, we performed drug uptake experiments for compounds 1–12 utilizing the inherent fluorescent property of the anthraquinone moieties found in the anthracycline drugs.52 K562 and MelJuSo cells were treated with 1 μM of the indicated compounds for 2 h, and fluorescence was then measured by flow cytometry (Figure S3B–E, Supporting Information). The fractional increase/decrease in fluorescence was compared to the parental drugs with that of the corresponding anthraquinone aglycon—the fluorophore within the anthracyclines. Significant differences in uptake of the different hybrid structures were observed. Compounds 3 and 11 are taken up ∼6 and 4 times more efficiently than doxorubicin (1), respectively, while compounds 5, 7, and 9 were more poorly taken up by K562 cells compared to doxorubicin (1). A similar observation is made for compounds 4, 6, 8, and 10, which were taken up more efficiently than aclarubicin (12), whereas uptake of compound 2 is significantly less compared to aclarubicin (12). Nevertheless, when drug uptake is plotted against the IC50 in K562 cells or drug uptake in MelJuSo cells against histone eviction speed, no correlation between uptake of the hybrid structures with cytotoxicity or histone eviction was observed (Figure S3F,G, Supporting Information). Of note here is that, while the uptake of compound 5 is similar to that of doxorubicin (1), this compound is not able to induce DNA double-strand breaks or evict histones. Consequently, this compound is one of the least cytotoxic hybrids from this set of compounds (Figure 3H). As anthracycline drugs target TopoII, we decided to validate if the lack of cytotoxicity of compound 5 can be caused by the loss of ability to interfere with the catalytic cycle of TopoII. Therefore, we transiently overexpressed GFP-tagged TopoIIα in MelJuSo cells and followed the protein localization over time upon treatment with 10 μM of the different doxorubicin/aclarubicin hybrid compounds. At steady state, TopoIIα is localized in the nucleus where it accumulates in nucleoli, but upon treatment with the hybrid anthracyclines, the protein rapidly relocalizes (Figure S4A,B). While most of the hybrid compounds are able to relocate TopoIIα, compound 5 does not. Furthermore, relocalization of TopoIIα by compounds 2, 6, and 10 was less efficient than by the other compounds, which might explain why these four are the least cytotoxic hybrid variants from this set of compounds.
Figure 3.

Cytotoxicity of compounds 1–12. (A, B) K562 cells were treated for 2 h at the indicated doses (higher doses in (A), lower doses in (B)) of the various hybrid compounds followed by drug removal. Cell survival in MelJuSo (C), human colorectal carcinoma cell line HCT116 (D), human prostate tumor cell line PC3 (E) and DU145 (F), and human glioblastoma cell line U87 (G). Cells were treated for 2 h at indicated dose followed by drug removal. Cell viability was measured by a Cell-Titerblue assay 72 h post-treatment. Data are shown as mean ± SD from three different experiments. (H) Table showing the IC50 values for the different doxorubicin/aclarubicin hybrid compounds for the indicated cell lines. See also Figure S3A, Supporting Information.
Correlation between N,N-Dimethylation and Cytotoxicity
Although no clear correlation is observed between the structural features of the compounds and their IC50 values (Figure S5A–C, Supporting Information), there is a strong relationship between the rate of histone eviction and cell toxicity (Figure 4A,B). In general, N,N-dimethylation of the sugar attached to the anthraquinone strongly improves histone eviction and enhances cytotoxicity of these compounds (Figure 4C). This observation could be very useful in the development of more effective anthracycline drugs, since (with the exception of aclarubicin) all anthracycline drugs currently used in the clinic (doxorubicin, daunorubicin, epirubicin, and idarubicin) contain a primary amine on their sugar moiety.
Figure 4.

Cytotoxicity correlates with N,N-dimethylation and efficiency of histone eviction. (A) Histone eviction speed (time at which 25% of the initial signal is reduced) versus IC50 of the various hybrid compounds is plotted. Two-tailed Spearman r correlation, *P < 0.05. (B) Zoom-in of data plotted in (A). (C) N,N-Dimethylation of the first sugar over no methylation gives improved IC50 in K562 cells (1 versus 3/2 versus 4/5 versus 7/6 versus 8/9 versus 11/10 versus 12). IC50 is plotted for the corresponding hybrid structures without (no; N) and with (yes; Y) N,N-dimethylation. The fold change of IC50 improvement as a result of N,N-dimethylation is indicated above the bars. IC50 could not be determined for compounds 2, 5, and 6 (gray bars) and was therefore depicted as the highest concentration tested (10 μM).
Discussion and Conclusions
Although anthracycline anticancer drugs are known to induce severe side effects, these effective chemotherapeutic drugs have been one of the cornerstones in oncology for over five decades. Following the discovery of doxorubicin, many anthracycline variants have been isolated, prepared, and evaluated with the aim of reducing their toxicity, but this has not led to any effective and less cardiotoxic variants to enter clinical practice other than aclarubicin (12). Remarkably, this drug is only used in Japan and China.3 It has long been thought that the cytotoxic activity of anthracyclines was due to their DNA double-strand breaking capacity;53 however, we have previously shown that histone eviction activity is likely the main mechanism of cytotoxicity.15,17−19 Here, we have developed synthetic chemistry to assemble a complete set of doxorubicin/aclarubicin hybrid structures varying at the anthraquinone aglycon, the nature of the carbohydrate portion, and the alkylation pattern of the amine on the first sugar moiety. The set of doxorubicin/aclarubicin hybrids was assembled using Yu’s gold-catalyzed glycosylation of the anthracycline aglycons, which in all cases proceeded with excellent stereoselectivity. The required di- and trisaccharides were generated using fully stereoselective IDCP-mediated glycosylations. Overall, the developed synthetic strategy proved to be broadly applicable and delivered the set of anthracyclines in a highly efficient manner. Furthermore, we have subjected these hybrid structures to detailed biological evaluation, including cellular uptake, TopoIIα relocalization capacity, DNA damage, and histone eviction assays. Although no clear correlation was found between the anthraquinone aglycon moiety and the number of carbohydrate fragments with the observed cytotoxicity of the compounds, a clear relationship between histone eviction efficiency and cytotoxicity was revealed. The coherent set of hybrid structures yielded three compounds that were more cytotoxic than doxorubicin (3, 8, and 11). Across the board, N,N-dimethylation of the carbohydrate appended to the anthraquinone aglycon considerably improved cytotoxicity (3 and 4 outperform 1 and 2; 7 and 8 outperform 5 and 6, and 11 and 12 outperform 9 and 10). How exactly N,N-dimethylation of the amino sugar improves cytotoxicity is not yet fully understood, but the addition of the methyl groups makes those compounds slightly more hydrophobic, which might influence their uptake. Furthermore, it has been shown that N-methylation of anthracyclines modulates their transport by the membrane transporter P-glycoprotein (P-gp).54 It has been suggested that the steric hindrance created by the methyl groups can impair the interaction between the positively charged amino group with the active site of the P-gp exporter, which leads to better intracellular drug accumulation. This would also indicate that the various N,N-dimethylated hybrid variants might be effective drugs for the treatment of multidrug-resistant tumors, in which elevated expression of the P-gp exporter is often observed.23,55 A third option for the enhanced effectivity of the N,N-dimethylation amino sugar variants might be a change in the interaction dynamics of the anthracycline drugs with the DNA. It is known that doxorubicin–DNA aminal adducts can form between the 3′-NH2 of the doxorubicin sugar, the N2 of the guanine base, and formaldehyde.56−59 The addition of two methyl groups to the critical amino sugar might convert these drugs from a covalent DNA intercalator into a reversible DNA intercalator, affecting the dynamics by which these drugs perturb the DNA–histone organization.
In addition to N,N-dimethylation of the sugar moiety, the doxorubicin anthraquinone aglycon appears to be slightly better than the aclarubicin anthraquinone aglycon and the aclarubicin trisaccharide improves cytotoxicity over the doxorubicin monosaccharide. A combination of these structural features is found in compound 11, the most cytotoxic compound in the focused library, being 13 times more cytotoxic than doxorubicin and 2.5 times more than aclarubicin in K562 cells. Histone eviction by compound 11 is approximately three times as fast as doxorubicin and twice as fast as for aclarubicin. The subsequent difference in cytotoxicity between compound 11 and doxorubicin or aclarubicin can therefore only partially be explained by the enhanced histone eviction efficacy. However, besides the difference in histone eviction efficacy, it has been shown that various anthracycline drug can have selectivity for distinct (epi-)genomic regions (and can therefore be considered different drugs because of different genomic targets).18 The different targeted (epi-)genomic regions by these drugs can subsequently have divergent downstream effects, which may explain the improved cytotoxicity for compound 11 over doxorubicin (1) and aclarubicin (12).
In summary, in this study, we have developed highly effective and broadly applicable synthetic chemistry, which was used to prepare a set of ten doxorubicin/aclarubicin hybrid structures and studied their specific biological activities in cells. This has given us better insights into the structure–activity relationship for this extensively used group of chemotherapeutics, which can help to direct the development of new effective anticancer drugs. Interestingly, the most potent compounds identified from the systematic library of compounds (3, 8, and 11) do not exert their activity through the induction of DNA double-strand break formation following inhibition of TopoIIα, but rather through the induction of histone eviction, indicating that histone eviction by anthracyclines could be the dominant factor for the cytotoxicity of this class of anticancer drugs.
Experimental Section
Chemistry
Doxorubicin was obtained from Accord Healthcare Limited, U.K., aclarubicin from Santa Cruz Biotech, and etoposide from Pharmachemie, Haarlem, The Netherlands. For the synthesis of the doxorubicin/aclarubicin hybrid compounds, all reagents were of commercial grade and used as received. Traces of water from reagents were removed by coevaporation with toluene in reactions that required anhydrous conditions. All moisture/oxygen-sensitive reactions were performed under an argon atmosphere. DCM used in the glycosylation reactions was dried with flamed 4 Å molecular sieves before being used. Reactions were monitored by thin-layer chromatography (TLC) analysis with detection by UV (254 nm) and, where applicable, by spraying with 20% sulfuric acid in EtOH or with a solution of (NH4)6Mo7O24·4H2O (25 g/L) and (NH4)4Ce(SO4)4·2H2O (10 g/L) in 10% sulfuric acid (aq) followed by charring at ∼150 °C. Flash column chromatography was performed on silica gel (40–63 μm). 1H and 13C spectra were recorded on Bruker AV 400 and Bruker AV 500 spectrometers in CDCl3, CD3OD, pyridine-d5, or D2O. Chemical shifts (δ) are given in parts per million (ppm) relative to tetramethylsilane (TMS) as internal standard (1H NMR in CDCl3) or the residual signal of the deuterated solvent. Coupling constants (J) are given in hertz. All 13C spectra are proton-decoupled. Column chromatography was carried out using silica gel (0.040–0.063 mm). Size-exclusion chromatography was carried out using a Sephadex LH-20, using DCM/MeOH (1:1, v/v) as the eluent. Neutral silica was prepared by stirring regular silica gel in aqueous ammonia, followed by filtration, washing with water, and heating at 150 °C overnight. High-resolution mass spectrometry (HRMS) analysis was performed with an LTQ Orbitrap mass spectrometer (Thermo Finnigan), equipped with an electrospray ion source in positive mode (source voltage, 3.5 kV; sheath gas flow, 10 mL/min; capillary temperature, 250 °C) with resolution R = 60 000 at m/z 400 (mass range m/z = 150–2000) and dioctyl phthalate (m/z = 391.28428) as a “lock mass”, or with a Synapt G2-Si (Waters), equipped with an electrospray ion source in positive mode (electrospray ionization time-of-flight (ESI-TOF)), injection via NanoEquity system (Waters), with LeuEnk (m/z = 556.2771) as “lock mass”. Eluents used: MeCN/H2O (1:1 v/v) supplemented with 0.1% formic acid. The high-resolution mass spectrometers were calibrated prior to measurements with a calibration mixture (Thermo Finnigan). Purity of all compounds is >95%, as determined by 1H NMR.
Syntheses of the monosaccharide donors/acceptors are described in the Supporting Information.
General Procedure A: p-Methoxyphenolate Oxidative Deprotection
To a solution of p-methoxyphenyl glycoside in 1:1 MeCN/H2O (0.02 M, v/v) were added NaOAc (10 equiv) and then Ag(DPAH)2·H2O60 (2.1 equiv for trisaccharides, 4 equiv for monosaccharides) portionwise over 30 min at 0 °C. The mixture was stirred until disappearance of the starting material, after which it was poured into sat. aq NaHCO3. This was then extracted with DCM thrice, dried over MgSO4, and concentrated in vacuo to give the crude lactols.
General Procedure B: Alkynylbenzoate Esterification
A solution of ortho-cyclopropylethynylbenzoic acid methyl ester47 in tetrahydrofuran (THF) (5 mL/mmol) and 1 M NaOH (5 mL/mmol) was stirred at 50 °C for at least 5 h. It was then poured into 1 M HCl (6 mL/mmol) and extracted with DCM thrice. The combined organic layers were then dried over MgSO4 and concentrated in vacuo. The resultant acid was then used without further purification.
To a solution of the above crude lactol in DCM (0.1 M) were added DIPEA (9 equiv), DMAP (1 equiv), EDCI·HCl (3.2 equiv), and the above carboxylic acid (3 equiv). After stirring overnight, the mixture was diluted with DCM and washed with sat. aq NaHCO3 and brine. Drying over MgSO4, concentration in vacuo, and column chromatography of the residue (EtOAc/pentane) gave the alkynylbenzoates.
General Procedure C: Au(I)-Catalyzed Glycosylation
To a solution of the glycosyl donor and the required anthracycline acceptor (1–2 equiv) in DCM (0.05 M), activated molecular sieves (4 Å) were added. The mixture was stirred for 30 min. Subsequently, a freshly prepared 0.1 M DCM solution of PPh3AuNTf2 (prepared by stirring 1:1 PPh3AuCl and AgNTf2 in DCM for 30 min) (0.1 equiv) in DCM was added dropwise at the designated temperature. After stirring for 30 min (for room temperature (RT)) or overnight (−20 °C or lower), the mixture was filtered and concentrated in vacuo. Column chromatography (EtOAc/pentane or Et2O/pentane and then acetone/toluene) followed by (if required) size-exclusion chromatography (Sephadex LH-20, 1:1 DCM/MeOH v/v) gave the glycosides.
Synthesis of Anthracycline Monosaccharides 2–4
The synthesis of 3 is described in ref (15).
7-[3-N-Allyloxycarbonyl-2,3-dideoxy-α-l-fucopyranoside]-aklavinone (15)
Prepared according to General Procedure C from donor 13 and aklavinone 14 (2 equiv) at RT to give after column chromatography (4:96 Et2O/pentane and then 1.5:98.5 acetone/toluene) the title compound as a yellow solid (149 mg, 0.201 mmol, 73%). 1H NMR (400 MHz, chloroform-d) δ 12.66 (s, 1H), 12.04 (s, 1H), 7.83 (dd, J = 7.5, 1.2 Hz, 1H), 7.77–7.64 (m, 2H), 7.31 (dd, J = 8.4, 1.2 Hz, 1H), 5.86 (ddt, J = 16.3, 10.8, 5.6 Hz, 1H), 5.46 (d, J = 3.8 Hz, 1H), 5.28–5.12 (m, 3H), 4.63 (d, J = 8.8 Hz, 1H), 4.58–4.41 (m, 2H), 4.21 (s, 1H), 4.15–4.01 (m, 2H), 3.86 (dq, J = 8.7, 4.1 Hz, 1H), 3.78 (s, 1H), 3.69 (s, 3H), 2.50 (dd, J = 15.0, 4.4 Hz, 1H), 2.34 (d, J = 15.0 Hz, 1H), 1.92 (td, J = 12.8, 4.1 Hz, 1H), 1.81–1.68 (m, 2H), 1.49 (dq, J = 14.3, 7.3 Hz, 1H), 1.36–1.18 (m, 3H), 1.08 (t, J = 7.3 Hz, 3H), 0.99 (t, J = 7.9 Hz, 9H), 0.66 (qd, J = 7.9, 2.1 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 192.9, 181.5, 171.6, 162.7, 162.3, 155.2, 142.9, 137.5, 133.7, 133.0, 132.9, 131.3, 124.9, 121.1, 120.3, 117.8, 115.9, 114.8, 101.6, 71.5, 71.4, 71.1, 67.6, 65.6, 57.2, 52.6, 47.4, 34.0, 32.2, 30.4, 17.6, 7.2, 6.8, 5.4. HRMS: [M + Na]+ calcd for C38H49NO12SiNa 774.2533; found 774.2525.
7-[α-l-Rhodosamino]-aklavinone (4)
To a solution of 15 (23.7 mg, 0.032 mmol) in DCM (3.2 mL) were added N,N-dimethylbarbituric acid (15 mg, 0.096 mmol, 3 equiv) and tetrakis(triphenylphosphine)palladium(0) (1.8 mg, 1.6 μmol, 0.05 equiv). After stirring for 2.5 h, the mixture was concentrated in vacuo. Column chromatography (DCM; 2:98 MeOH/DCM) gave the crude amine. This was then redissolved in EtOH (7.7 mL), and 37% aq CH2O (79 μL, 30 equiv) was added NaBH(OAc)3 (67 mg, 0.32 mmol, 10 equiv). The mixture was stirred for 2.5 h before being quenched by addition of sat. aq NaHCO3. It was then poured into H2O and extracted with DCM, dried over Na2SO4, and concentrated in vacuo to give the crude dimethylated amine. This was then redissolved in pyridine (3.2 mL) in a poly(tetrafluoroethylene) (PTFE) tube, after which HF·pyr complex (70 wt % HF, 125 μL) was added at 0 °C. Over the course of 4 h, additional HF·pyr complex (70 wt % HF, 125 μL each time) was added five times. Solid NaHCO3 was added to quench, and the mixture was stirred until cessation of effervescence. It was then filtered off, and the filtrate was partitioned between DCM and H2O. The organic layer was dried over Na2SO4 and concentrated in vacuo. Column chromatography on neutral silica (DCM; 20:80 MeOH/DCM) gave the title compound as a yellow solid (7.9 mg, 13.9 μmol, 43% over three steps). 1H NMR (500 MHz, chloroform-d) δ 12.70 (s, 1H), 12.01 (s, 1H), 7.83 (dd, J = 7.5, 1.1 Hz, 1H), 7.77–7.66 (m, 2H), 7.31 (dd, J = 8.4, 1.2 Hz, 1H), 5.55 (d, J = 3.9 Hz, 1H), 5.29–5.20 (m, 1H), 4.27 (s, 1H), 4.16–4.03 (m, 2H), 3.87 (s, 1H), 3.70 (s, 3H), 2.54 (dd, J = 15.2, 4.5 Hz, 1H), 2.45 (s, 6H), 2.33 (d, J = 15.2 Hz, 1H), 2.05 (td, J = 13.1, 12.6, 4.2 Hz, 1H), 1.89 (dd, J = 12.9, 4.6 Hz, 1H), 1.76 (dq, J = 14.6, 7.3 Hz, 1H), 1.52 (dq, J = 14.5, 7.3 Hz, 1H), 1.38 (dd, J = 6.5, 2.1 Hz, 3H), 1.09 (t, J = 7.3 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 192.9, 181.4, 171.3, 162.8, 162.3, 142.8, 137.6, 133.6, 133.1, 131.2, 125.0, 121.1, 120.4, 115.9, 114.9, 101.1, 71.9, 71.4, 67.0, 65.8, 61.1, 57.2, 52.7, 42.0, 34.0, 32.2, 27.8, 17.0, 6.8. HRMS: [M + H]+ calcd for C30H36NO10 570.2339; found 570.2921.
7-[α-l-Daunosamino]-aklavinone (2)
To a solution of 15 (60 mg, 0.081 mmol) in DCM (8.1 mL) were added N,N-dimethylbarbituric acid (38 mg, 0.24 mmol, 3 equiv) and tetrakis(triphenylphosphine)palladium(0) (4.6 mg, 4.1 μmol, 0.05 equiv). After stirring for 2.5 h, the mixture was concentrated in vacuo. Column chromatography (DCM; 2:98 MeOH/DCM) gave the crude amine. This was then redissolved in pyridine (6 mL) in a PTFE tube, after which HF·pyr complex (70 wt % HF, 710 μL) was added at 0 °C. After 3.5 and 5.5 h, additional HF·pyr complex (70 wt % HF, 355 μL each time) was added. After stirring for a total of 6.5 h, solid NaHCO3 was added to quench, and the mixture was stirred until cessation of effervescence. It was then filtered off, and the filter cake was rinsed thoroughly with MeOH/DCM (9:1 v/v). The combined filtrates were then concentrated in vacuo. Column chromatography (DCM; 20:80 MeOH/DCM) gave the title compound as a yellow solid (18 mg, 33 μmol, 41% over two steps). 1H NMR (500 MHz, methanol-d4) δ 7.77–7.61 (m, 2H), 7.53 (s, 1H), 7.31–7.20 (m, 1H), 5.49 (s, 1H), 5.14 (d, J = 4.7 Hz, 1H), 4.27 (q, J = 6.5 Hz, 1H), 4.08 (s, 1H), 3.73 (s, 2H), 3.67 (d, J = 2.8 Hz, 1H), 3.57–3.47 (m, 1H), 2.52 (dd, J = 15.0, 5.2 Hz, 1H), 2.32 (d, J = 15.0 Hz, 1H), 2.03 (td, J = 12.9, 4.0 Hz, 1H), 1.99–1.90 (m, 1H), 1.76 (dq, J = 14.7, 7.4 Hz, 1H), 1.56 (dq, J = 13.9, 7.1 Hz, 1H), 1.31 (d, J = 6.6 Hz, 3H), 1.11 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, MeOD) δ 193.6, 182.3, 172.6, 163.7, 143.8, 138.5, 134.7, 134.0, 125.8, 121.2, 120.8, 117.0, 115.8, 101.7, 72.5, 72.1, 68.4, 68.1, 58.2, 53.0, 49.8, 48.4, 35.8, 33.3, 30.1, 17.0, 7.1. HRMS: [M + H]+ calcd for C28H32NO10 542.2026; found 542.2031.
Synthesis of Anthracycline Disaccharides 5–8
p-Methoxyphenyl-2-deoxy-3,4-tetraisopropyldisiloxyl-α-l-fucopyranosyl-(1 → 4)-3-N-allyloxycarbonyl-2,3-dideoxy-α-l-fucopyranoside (19)
To a solution of the glycosyl acceptor 17 (901 mg, 2.67 mmol, 1 equiv) and the glycosyl donor 18 (1.80 g, 3.73 mmol, 1.3 equiv) in Et2O/DCE (70 mL, 4:1 v/v), activated molecular sieves (4 Å) were added. The mixture was stirred for 30 min, and then, at 10 °C, iodonium dicollidine perchlorate (5.00 g, 10.7 mmol, 4 equiv) was added. After 30 min, triphenylphosphine (1.40 g, 5.34 mmol, 2 equiv) was added, and the mixture was stirred for an additional hour. It was then diluted with EtOAc and filtered; washed with 10% aq Na2S2O3, 1 M CuSO4 solution twice, and H2O; and then dried over MgSO4. Concentration in vacuo and column chromatography (5:95–10:90 EtOAc/pentane) of the residue gave the title compound as a white foam (1.69 g, 2.38 mmol, 89%). 1H NMR (500 MHz, chloroform-d) δ 7.05–6.93 (m, 2H), 6.93–6.70 (m, 2H), 6.16 (d, J = 7.9 Hz, 1H), 5.92 (ddt, J = 16.1, 10.9, 5.6 Hz, 1H), 5.52 (d, J = 3.2 Hz, 1H), 5.30 (dq, J = 17.2, 1.6 Hz, 1H), 5.20 (dq, J = 10.5, 1.4 Hz, 1H), 4.93 (d, J = 3.7 Hz, 1H), 4.58 (qdt, J = 13.3, 5.6, 1.4 Hz, 2H), 4.41 (ddd, J = 12.2, 4.6, 2.5 Hz, 1H), 4.37–4.25 (m, 1H), 4.14–4.04 (m, 2H), 4.01 (s, 1H), 3.77 (s, 3H), 3.54 (s, 1H), 2.19–2.05 (m, 2H), 1.99 (dd, J = 12.6, 4.6 Hz, 1H), 1.89 (td, J = 12.7, 3.5 Hz, 1H), 1.34 (d, J = 6.4 Hz, 3H), 1.18 (d, J = 6.5 Hz, 3H), 1.14–0.83 (m, 28H). 13C NMR (126 MHz, CDCl3) δ 155.6, 154.4, 150.9, 132.8, 117.4, 117.2, 114.4, 101.8, 96.2, 81.2, 73.0, 69.8, 68.0, 67.4, 65.4, 55.5, 46.4, 33.1, 31.5, 17.6, 17.5, 17.4, 17.3, 17.3, 17.2, 17.2, 17.2, 17.1, 17.1, 14.1, 13.9, 13.0, 12.4. HRMS: [M + Na]+ calcd for C35H59NO10Si2Na 732.35752; found 732.3587.
o-Cyclopropylethynylbenzoyl-2-deoxy-3,4-tetraisopropyldisiloxane-α-l-fucopyranosyl-(1 → 4)-3-N-allyloxycarbonyl-2,3-dideoxy-l-fucopyranoside (21)
Prepared according to General Procedure A and B from 19 (1.69 g, 2.38 mmol) to give after column chromatography (10:90–20:80 EtOAc/pentane) the title compound as a white foam (1.54 g, 1.99 mmol, 84% over two steps, α/β 1:8). 1H NMR (500 MHz, chloroform-d) δ 8.00–7.85 (m, 1H), 7.47 (dd, J = 7.8, 1.4 Hz, 1H), 7.41 (ddd, J = 9.1, 6.0, 1.4 Hz, 1H), 7.35–7.24 (m, 1H), 6.35 (d, J = 7.6 Hz, 1H), 5.99 (dd, J = 10.0, 2.3 Hz, 1H), 5.96–5.84 (m, 1H), 5.36–5.15 (m, 2H), 4.93 (d, J = 3.9 Hz, 1H), 4.56 (qdt, J = 13.3, 5.6, 1.5 Hz, 2H), 4.45 (ddd, J = 12.1, 4.5, 2.5 Hz, 1H), 4.11–4.06 (m, 1H), 4.01 (d, J = 2.5 Hz, 1H), 3.87 (dddd, J = 12.1, 7.1, 4.1, 2.6 Hz, 1H), 3.85–3.79 (m, 1H), 3.48–3.44 (m, 1H), 2.22 (ddd, J = 11.9, 4.1, 2.2 Hz, 1H), 2.14 (td, J = 12.4, 4.0 Hz, 1H), 1.99 (dd, J = 12.4, 4.6 Hz, 1H), 1.85 (td, J = 12.3, 10.0 Hz, 1H), 1.51 (tt, J = 7.2, 5.7 Hz, 1H), 1.36–1.30 (m, 6H), 1.13–0.81 (m, 28H). 13C NMR (126 MHz, CDCl3) δ 164.3, 155.8, 134.2, 133.0, 132.0, 131.1, 130.8, 127.0, 125.1, 117.7, 102.3, 99.8, 93.2, 80.6, 74.5, 73.3, 73.0, 69.9, 68.4, 65.7, 50.1, 33.3, 32.2, 17.8, 17.8, 17.6, 17.5, 17.5, 17.5, 17.4, 17.3, 14.3, 14.2, 13.2, 12.7, 9.0, 8.9, 0.8. HRMS: [M + Na]+ calcd for C40H61NO10Si2Na 794.37317; found 794.3749.
7-[2-Deoxy-3,4-tetraisopropyldisiloxyl-α-l-fucopyranosyl-(1 → 4)-3-N-allyloxycarbonyl-2,3-dideoxy-α-l-fucopyranoside]-14-O-tert-butyldimethylsilyl-doxorubicinone (22)
Prepared according to General Procedure C from donor 21 (722 mg, 1.00 mmol) and 14-O-tert-butyldimethylsilyl-doxorubicinone 16 (793 mg, 1.50 mmol, 1.5 equiv) to give after column chromatography (5:95–20:80 EtOAc/pentane–4:96 acetone/toluene) the title compound as a red solid (714 mg, 0.640 mmol, 64%). 1H NMR (500 MHz, chloroform-d) δ 13.83 (s, 1H), 13.09 (s, 1H), 7.93 (dd, J = 7.7, 1.0 Hz, 1H), 7.72 (t, J = 8.1 Hz, 1H), 7.43–7.32 (m, 1H), 6.07 (d, J = 7.8 Hz, 1H), 5.91–5.78 (m, 1H), 5.50 (d, J = 3.8 Hz, 1H), 5.27–5.18 (m, 2H), 5.13 (dq, J = 10.5, 1.4 Hz, 1H), 4.98–4.86 (m, 3H), 4.61–4.37 (m, 4H), 4.13 (q, J = 6.5 Hz, 1H), 4.05 (d, J = 24.2 Hz, 6H), 3.90–3.77 (m, 1H), 3.55 (s, 1H), 3.09 (dd, J = 18.8, 2.0 Hz, 1H), 2.81 (d, J = 18.7 Hz, 1H), 2.29 (d, J = 14.6 Hz, 1H), 2.22–2.05 (m, 2H), 2.05–1.95 (m, 1H), 1.92 (dd, J = 13.1, 4.5 Hz, 1H), 1.78 (td, J = 12.9, 4.0 Hz, 1H), 1.30 (dd, J = 16.4, 6.4 Hz, 6H), 1.16–0.82 (m, 37H), 0.15 (d, J = 2.7 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 211.4, 186.8, 186.4, 161.0, 156.3, 155.7, 135.7, 135.3, 134.0, 133.9, 132.9, 120.7, 119.8, 118.5, 117.5, 111.3, 111.2, 101.9, 101.0, 81.0, 73.2, 69.9, 69.7, 68.2, 68.0, 66.7, 65.5, 56.7, 46.6, 35.7, 34.0, 33.3, 31.3, 26.0, 18.7, 17.8, 17.7, 17.6, 17.5, 17.5, 17.5, 17.4, 17.3, 17.2, 14.3, 14.1, 13.1, 12.6, −5.2, −5.3. HRMS: [M + Na]+ calcd for C55H83NO17Si3Na 1136.48665; found 1136.4866.
7-[2-Deoxy-3,4-tetraisopropyldisiloxyl-α-l-fucopyranosyl-(1 → 4)-3-amino-2,3-dideoxy-α-l-fucopyranoside]-14-O-tert-butyldimethylsilyl-doxorubicinone (23)
A solution of 22 (704 mg, 0.631 mmol) and N,N-dimethylbarbituric acid (440 mg, 2.84 mmol, 4.5 equiv) in DCM (63 mL) was degassed for 5 min. Then, Pd(PPh3)4 (36.5 mg, 0.032 mmol, 0.05 equiv) was added and the mixture was allowed to stir for 20 min. It was then directly subjected to column chromatography (pentane, then 0:100–50:50 acetone/toluene) to give the title compound as a red solid (650 mg, 0.631 mmol, 100%). 1H NMR (500 MHz, chloroform-d) δ 7.93 (dd, J = 7.8, 1.0 Hz, 1H), 7.73 (t, J = 8.1 Hz, 1H), 7.42–7.33 (m, 1H), 5.53–5.41 (m, 1H), 5.21 (dd, J = 4.1, 2.2 Hz, 1H), 4.98 (d, J = 3.7 Hz, 1H), 4.96–4.81 (m, 2H), 4.65 (s, 1H), 4.42 (ddd, J = 12.1, 4.6, 2.5 Hz, 1H), 4.15 (q, J = 6.5 Hz, 1H), 4.10–3.93 (m, 5H), 3.53 (s, 1H), 3.40–3.20 (m, 3H), 3.18–3.00 (m, 2H), 2.82 (d, J = 18.7 Hz, 1H), 2.29 (dt, J = 14.8, 2.2 Hz, 1H), 2.21–2.09 (m, 2H), 2.05–1.93 (m, 1H), 1.76 (ddd, J = 27.6, 14.0, 4.2 Hz, 1H), 1.29 (d, J = 6.5 Hz, 3H), 1.23 (d, J = 6.5 Hz, 3H), 1.13–0.75 (m, 36H), 0.15 (d, J = 1.4 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 211.2, 186.7, 186.4, 161.0, 156.3, 155.6, 135.7, 135.3, 134.0, 132.1, 132.1, 128.6, 120.7, 119.7, 118.5, 111.3, 101.3, 101.1, 81.5, 73.3, 70.1, 69.6, 68.3, 67.8, 66.6, 56.7, 46.8, 35.6, 33.8, 33.4, 25.9, 18.7, 17.7, 17.7, 17.6, 17.6, 17.5, 17.5, 17.4, 17.3, 17.2, 14.2, 14.1, 13.1, 12.6. HRMS: [M + H]+ calcd for C51H80NO15Si3 1030.48358; found 1030.4855.
7-[2-Deoxy-α-l-fucopyranosyl-(1 → 4)-3-amino-2,3-dideoxy-α-l-fucopyranoside]-doxorubicinone (5)
To a solution of 23 (30.5 mg, 29.6 μmol) in pyridine (3.0 mL) in a PTFE tube was added HF·pyr complex (70 wt % HF, 232 μL) at 0 °C. Over the course of 4 h, two additional such portions of HF·pyr complex were added. Then, solid NaHCO3 was added to quench, and the mixture was stirred until cessation of effervescence. It was then filtered off and concentrated in vacuo. Column chromatography on neutral silica (0:100–20:80 MeOH/DCM) gave the title compound as a red solid (15.1 mg, 22.4 μmol, 76%). 1H NMR (500 MHz, pyridine-d5) δ 7.78 (d, J = 7.7 Hz, 1H), 7.46 (t, J = 8.1 Hz, 1H), 7.14 (d, J = 8.4 Hz, 1H), 5.52 (d, J = 3.0 Hz, 1H), 5.17 (d, J = 3.9 Hz, 1H), 5.12 (d, J = 2.3 Hz, 2H), 5.06 (d, J = 3.8 Hz, 1H), 4.36 (dt, J = 12.1, 3.9 Hz, 1H), 4.33–4.19 (m, 2H), 3.80 (d, J = 2.9 Hz, 1H), 3.68 (s, 3H), 3.54 (s, 1H), 3.41 (t, J = 8.7 Hz, 1H), 3.34–3.12 (m, 2H), 2.51 (d, J = 14.4 Hz, 1H), 2.30 (td, J = 12.2, 3.9 Hz, 1H), 2.22 (dd, J = 14.3, 5.1 Hz, 1H), 2.08 (dd, J = 12.3, 4.9 Hz, 1H), 1.97 (dd, J = 9.2, 2.8 Hz, 2H), 1.27 (d, J = 6.4 Hz, 3H), 1.06 (d, J = 6.4 Hz, 3H). 13C NMR (126 MHz, Pyr) δ 215.4, 187.5, 161.9, 157.5, 156.2, 135.2, 121.6, 120.1, 119.9, 112.3, 112.0, 101.9, 101.9, 81.6, 77.1, 72.4, 70.9, 69.0, 68.8, 66.7, 66.2, 57.1, 48.0, 37.9, 34.6, 34.4, 34.2, 18.1. HRMS: [M + H]+ calcd for C33H40NO14 674.24488; found 674.2456.
7-[2-Deoxy-α-l-fucopyranosyl-(1 → 4)-3-dimethylamino-2,3-dideoxy-α-l-fucopyranoside]-doxorubicinone (7)
To a solution of 23 (102 mg, 99 μmol) in EtOH (20 mL) and 37% aq CH2O (245 μL, 30 equiv) was added NaBH(OAc)3 (40 mg, 0.193 mmol, 1.95 equiv). The mixture was stirred for 1.5 h before being poured into sat. aq NaHCO3. This was extracted with DCM, dried over Na2SO4, and concentrated in vacuo. Column chromatography chromatography (3:97 acetone/toluene) gave the dimethylated amine as a red solid (75 mg, 70.9 μmol, 71%). 1H NMR (500 MHz, chloroform-d) δ 13.92 (s, 1H), 13.24 (s, 1H), 8.01 (dd, J = 7.7, 1.0 Hz, 1H), 7.77 (t, J = 8.1 Hz, 1H), 7.43–7.37 (m, 1H), 5.54 (d, J = 3.8 Hz, 1H), 5.25 (dd, J = 4.1, 2.1 Hz, 1H), 5.01 (d, J = 3.4 Hz, 1H), 4.98–4.84 (m, 2H), 4.79 (s, 1H), 4.49–4.34 (m, 2H), 4.09 (s, 3H), 3.95 (t, J = 1.8 Hz, 1H), 3.91 (q, J = 6.5 Hz, 1H), 3.75 (s, 1H), 3.38–3.35 (m, 1H), 3.18 (dd, J = 18.9, 1.9 Hz, 1H), 2.98 (d, J = 18.8 Hz, 1H), 2.32 (dt, J = 14.7, 2.3 Hz, 1H), 2.19 (s, 6H), 2.17–2.06 (m, 3H), 2.06–1.96 (m, 2H), 1.89 (td, J = 12.8, 4.0 Hz, 1H), 1.80 (dd, J = 13.0, 4.1 Hz, 1H), 1.26 (d, J = 6.6 Hz, 3H), 1.19 (d, J = 6.4 Hz, 3H), 1.07 (ddt, J = 9.4, 7.4, 4.6 Hz, 24H), 0.96 (s, 9H), 0.14 (d, J = 2.9 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 211.4, 187.2, 186.8, 161.1, 156.6, 156.0, 135.8, 135.6, 134.3, 134.2, 121.0, 119.9, 118.5, 111.5, 111.4, 101.5, 99.9, 74.1, 73.8, 70.6, 69.6, 68.8, 67.3, 66.7, 61.8, 56.8, 43.5, 35.7, 34.1, 33.4, 26.0, 18.1, 17.8, 17.8, 17.7, 17.6, 17.6, 17.5, 17.5, 17.4, 17.4, 14.4, 14.3, 13.2, 12.7. HRMS: [M + H]+ calcd for C53H84NO15Si3 1058.51488; found 1058.51488. To a solution of the above compound (38 mg, 35.9 μmol) in pyridine (3.6 mL) in a PTFE tube was added HF·pyr complex (70 wt % HF, 282 μL) at 0 °C. Over the course of 4.5 h, three additional such portions of HF·pyr complex were added. Then, solid NaHCO3 was added to quench, and the mixture was stirred until cessation of effervescence. It was then filtered off and concentrated in vacuo. Column chromatography on neutral silica (DCM; 10:90 MeOH/DCM) gave the title compound as a red solid (20.3 mg, 28.9 μmol, 81%). 1H NMR (500 MHz, chloroform-d + MeOD) δ 8.02 (d, J = 7.6 Hz, 1H), 7.81 (t, J = 8.0 Hz, 1H), 7.42 (t, J = 7.3 Hz, 1H), 5.55 (d, J = 4.0 Hz, 1H), 5.28 (s, 1H), 5.05 (d, J = 3.9 Hz, 1H), 4.76 (d, J = 5.6 Hz, 2H), 4.41 (q, J = 6.6 Hz, 1H), 4.14–4.03 (m, 4H), 3.97 (q, J = 6.6 Hz, 1H), 3.83 (d, J = 6.5 Hz, 1H), 3.24 (dd, J = 18.9, 5.9 Hz, 1H), 3.02 (dd, J = 19.2, 6.3 Hz, 1H), 2.39–2.08 (m, 8H), 2.07–1.80 (m, 4H), 1.29 (d, J = 6.7 Hz, 3H), 1.21 (d, J = 6.6 Hz, 3H). 13C NMR (126 MHz, CDCl3 + MeOD) δ 213.6, 187.2, 186.8, 161.1, 155.9, 155.3, 135.9, 135.4, 133.8, 133.5, 120.8, 119.8, 118.6, 111.6, 111.4, 100.9, 99.2, 73.6, 71.0, 69.2, 68.6, 66.6, 65.4, 65.2, 61.7, 56.6, 43.0, 35.5, 33.8, 32.3, 28.7, 17.9, 16.6. HRMS: [M + H]+ calcd for C35H44NO14 702.27619; found 702.2769.
7-[2-Deoxy-3,4-tetraisopropyldisiloxyl-α-l-fucopyranosyl-(1 → 4)-3-amino-2,3-dideoxy-α-l-fucopyranoside]-aklavinone (24)
Prepared according to General Procedure C from donor 21 (623 mg, 0.806 mmol) and aklavinone 14 (665 mg, 1.61 mmol, 2.00 equiv) at −20 °C to give after column chromatography (10:90 EtOAc/pentane and then 2:98–10:90 acetone/toluene) of the residue an inseparable mixture of the disaccharide anthracycline and acceptor, which was continued to the next step. A solution of the above mixture and N,N-dimethylbarbituric acid (562 mg, 3.60 mmol, 2.2 equiv) in DCM (81 mL) was degassed for 5 min. Then, Pd(PPh3)4 (23 mg, 0.040 mmol, 0.025 equiv) was added and the mixture was allowed to stir for 30 min. It was then directly subjected to column chromatography (pentane, then 0:100–25:75 acetone/toluene) to give the title compound as a yellow solid (636 mg, 0.700 mmol, 86% over two steps). 1H NMR (400 MHz, chloroform-d) δ 7.76 (d, J = 7.5 Hz, 1H), 7.70–7.58 (m, 2H), 7.25 (d, J = 8.4 Hz, 1H), 5.47 (d, J = 2.8 Hz, 1H), 5.25 (dd, J = 4.1, 1.8 Hz, 1H), 4.97 (d, J = 3.6 Hz, 1H), 4.42 (ddd, J = 12.0, 4.7, 2.5 Hz, 1H), 4.19–4.05 (m, 3H), 4.00 (d, J = 2.6 Hz, 1H), 3.70 (s, 3H), 3.51 (d, J = 2.5 Hz, 1H), 3.24 (qt, J = 9.3, 6.6, 5.6 Hz, 1H), 2.52 (dd, J = 15.0, 4.3 Hz, 1H), 2.36–2.28 (m, 1H), 2.17–2.08 (m, 1H), 2.01 (dd, J = 12.3, 4.6 Hz, 1H), 1.86–1.68 (m, 3H), 1.49 (dd, J = 14.1, 7.0 Hz, 1H), 1.30 (d, J = 6.4 Hz, 3H), 1.23 (d, J = 6.5 Hz, 3H), 1.17–0.85 (m, 31H).13C NMR (101 MHz, CDCl3) δ 192.6, 181.2, 171.4, 162.5, 162.1, 142.7, 137.4, 133.4, 132.9, 131.2, 124.8, 120.9, 120.2, 115.7, 114.6, 101.7, 101.1, 81.7, 73.3, 71.6, 70.9, 70.2, 68.1, 67.8, 57.1, 52.6, 46.8, 33.9, 33.4, 32.2, 17.7, 17.7, 17.6, 17.5, 17.5, 17.4, 17.3, 17.3, 14.3, 14.1, 13.1, 12.6, 6.8. HRMS: [M + H]+ calcd for C46H68NO14Si2 914.4178; found 914.4173.
7-[2-Deoxy-α-l-fucopyranosyl-(1 → 4)-3-amino-2,3-dideoxy-α-l-fucopyranoside]-aklavinone (6)
To a solution of 24 (91 mg, 0.10 mmol) in pyridine (10 mL) in a PTFE tube was added HF·pyr complex (70 wt % HF, 393 μL) at 0 °C. Over the course of 4.5 h, three additional such portions of HF·pyr complex were added. Then, solid NaHCO3 was added to quench, and the mixture was stirred until cessation of effervescence. It was then filtered off and partitioned between DCM and H2O. The organic layer was washed with brine, dried over Na2SO4, and concentrated in vacuo. Column chromatography on neutral silica (DCM; 20:80 MeOH/DCM) followed by size-exclusion chromatography (Sephadex LH-20; eluent, DCM/MeOH, 1:1) gave the title compound as a yellow solid (27.5 mg, 40.9 μmol, 41%). 1H NMR (400 MHz, chloroform-d + MeOD) δ 7.79 (dd, J = 7.5, 1.3 Hz, 1H), 7.74–7.57 (m, 2H), 7.32–7.23 (m, 1H), 5.47 (t, J = 2.5 Hz, 1H), 5.27–5.20 (m, 1H), 4.97 (d, J = 3.5 Hz, 1H), 4.20–4.01 (m, 4H), 3.70 (s, 3H), 3.64 (d, J = 3.0 Hz, 2H), 3.61–3.52 (m, 2H), 3.11 (dd, J = 10.6, 6.7 Hz, 1H), 2.53 (dd, J = 15.0, 4.4 Hz, 1H), 2.27 (d, J = 15.0 Hz, 1H), 1.97 (ddd, J = 22.5, 12.3, 4.2 Hz, 2H), 1.86–1.64 (m, 3H), 1.50 (dt, J = 14.6, 7.4 Hz, 1H), 1.28 (d, J = 6.4 Hz, 3H), 1.23 (d, J = 6.5 Hz, 3H), 1.07 (q, J = 7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 192.6, 181.4, 171.4, 162.5, 162.0, 142.6, 137.5, 133.5, 132.9, 131.1, 124.9, 121.0, 120.3, 115.8, 114.7, 101.3, 100.8, 81.1, 71.6, 70.9, 70.8, 68.0, 67.4, 65.4, 57.0, 52.6, 46.7, 34.1, 33.2, 32.7, 32.2, 17.3, 16.9, 6.7. HRMS: [M + H]+ calcd for C34H42NO13 672.2656; found 672.2645.
7-[2-Deoxy-α-l-fucopyranosyl-(1 → 4)-3-dimethylamino-2,3-dideoxy-α-l-fucopyranoside]-aklavinone (8)
To a solution of 6 (26.2 mg, 37.4 μmol) in EtOH (3.7 mL) and 37% aq CH2O (200 μL, 60 equiv) was added NaBH(OAc)3 (85 mg, 0.374 mmol, 10 equiv). The mixture was stirred for 2.5 h before being poured into sat. aq NaHCO3. This was extracted with DCM, dried over Na2SO4, and concentrated in vacuo. Column chromatography on neutral silica (3:97–10:90 MeOH/DCM) gave the title compound as a yellow solid (8.8 mg, 12.6 μmol, 34%). 1H NMR (500 MHz, chloroform-d) δ 12.69 (s, 1H), 12.04 (s, 1H), 7.83 (dd, J = 7.5, 1.2 Hz, 1H), 7.78–7.60 (m, 2H), 7.31 (dd, J = 8.4, 1.2 Hz, 1H), 5.51 (d, J = 3.7 Hz, 1H), 5.27 (dd, J = 4.3, 1.9 Hz, 1H), 5.01 (s, 1H), 4.53 (dd, J = 14.2, 7.7 Hz, 1H), 4.17–4.05 (m, 2H), 4.00 (q, J = 6.6 Hz, 1H), 3.74 (d, J = 8.5 Hz, 1H), 3.63 (d, J = 3.1 Hz, 1H), 2.52 (dd, J = 15.0, 4.3 Hz, 1H), 2.29 (dd, J = 16.9, 9.2 Hz, 1H), 2.25–2.11 (m, 6H), 2.07 (dt, J = 10.9, 5.4 Hz, 1H), 1.87–1.79 (m, 1H), 1.75 (dq, J = 14.6, 7.7, 7.3 Hz, 1H), 1.51 (dq, J = 14.3, 7.2 Hz, 1H), 1.28 (d, J = 6.5 Hz, 3H), 1.20 (d, J = 6.5 Hz, 3H), 1.09 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 192.9, 181.5, 162.7, 162.3, 142.8, 137.5, 133.6, 133.1, 124.9, 121.1, 120.3, 116.0, 114.8, 101.7, 99.2, 71.8, 71.7, 70.8, 68.5, 66.3, 66.0, 61.7, 57.3, 52.7, 43.4, 33.9, 33.2, 32.3, 18.0, 16.8, 6.8. HRMS: [M + H]+ calcd for C36H46NO13 700.2969; found 700.2966.
Synthesis of Trisaccharides 9–11
p-Methoxyphenyl-2-deoxy-3-O-p-methoxybenzyl-α-l-fucopyranosyl-(1 → 4)-3-N-allyloxycarbonyl-2,3-dideoxy-α-l-fucopyranoside (27)
To a solution of the glycosyl acceptor 17 (169 mg g, 0.5 mmol, 1 equiv) and the glycosyl donor 25 (325 mg, 0.7 mmol, 1.4 equiv) in 4:1 Et2O/DCE (15 mL, v/v), activated molecular sieves (4 Å) were added. The mixture was stirred for 30 min and then, at 10 °C, iodonium dicollidine perchlorate (937 mg, 2.00 mmol, 4 equiv) was added. After 30 min, triphenylphosphine (262 mg, 1.00 mmol, 2 equiv) was added, and the mixture was stirred for an additional hour. It was then diluted with EtOAc and filtered; washed with 10% aq Na2S2O3, 1 M CuSO4 solution twice, and H2O; and then dried over MgSO4. Concentration in vacuo and column chromatography (15:85–20:80 EtOAc/pentane) of the residue gave the disaccharide. This was then dissolved in MeOH (8.8 mL) and DCM (8.8 mL), after which NaOMe was added to pH 10. After stirring for a week, it was neutralized by addition of dry ice and concentrated in vacuo. Column chromatography (20:80–50:50 EtOAc/pentane) gave the title compound as a clear oil (232 mg, 0.39 mmol, 78% over two steps). 1H NMR (400 MHz, chloroform-d) δ 7.28 (d, J = 6.7 Hz, 2H), 7.05–6.96 (m, 2H), 6.96–6.87 (m, 2H), 6.87–6.77 (m, 2H), 6.21 (d, J = 8.2 Hz, 1H), 5.92 (ddt, J = 16.4, 10.9, 5.5 Hz, 1H), 5.51 (d, J = 3.1 Hz, 1H), 5.37–5.25 (m, 1H), 5.20 (dt, J = 10.4, 1.4 Hz, 1H), 5.00–4.92 (m, 1H), 4.62–4.52 (m, 4H), 4.39–4.25 (m, 1H), 4.11 (q, J = 7.8, 7.1 Hz, 1H), 4.08–4.01 (m, 1H), 3.97 (td, J = 8.4, 3.1 Hz, 1H), 3.86 (s, 1H), 3.81 (s, 3H), 3.77 (s, 3H), 3.56 (s, 1H), 2.21 (s, 1H), 2.13 (dd, J = 12.6, 4.5 Hz, 1H), 2.08–2.00 (m, 2H), 1.86 (td, J = 12.7, 3.5 Hz, 1H), 1.38 (d, J = 6.6 Hz, 3H), 1.17 (d, J = 6.5 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 159.6, 155.9, 154.7, 151.1, 133.0, 130.0, 129.5, 117.6, 117.5, 114.6, 114.1, 101.4, 96.4, 81.5, 72.7, 70.2, 68.2, 67.5, 67.2, 65.7, 55.8, 55.4, 46.6, 31.8, 30.3, 17.4, 16.8. HRMS: [M + Na]+ calcd for C31H41NO10Na 610.2628; found 610.2632.
p-Methoxyphenyl-2,3-dideoxy-4-ulo-α-l-fucopyranosyl-(1 → 4)-2-deoxy-3-O-p-methoxybenzyl-α-l-fucopyranosyl-(1 → 4)-3-azido-2,3-dideoxy-α-l-fucopyranoside (29)
To a solution of the glycosyl acceptor 27 (120 g, 2.04 mmol) and the glycosyl donor 28 (1.01 g, 2.86 mmol, 1.4 equiv) in 4:1 Et2O/DCE (62.5 mL, v/v), activated molecular sieves (4 Å) were added. The mixture was stirred for 30 min and then, at 10 °C, iodonium dicollidine perchlorate (3.82 g, 8.16 mmol, 4 equiv) was added. After 35 min, triphenylphosphine (1.07 g, 4.08 mmol, 2.00 equiv) was added, and the mixture was stirred for an additional hour. It was then diluted with EtOAc and filtered; washed with 10% aq Na2S2O3, 1 M CuSO4 solution twice, and H2O; and then dried over MgSO4. Concentration in vacuo and column chromatography (10:90–30:70 EtOAc/pentane) of the residue gave the trisaccharide benzoate as a thick clear oil (1.59 g, 1.97 mmol, 97%). 1H NMR (400 MHz, chloroform-d) δ 8.12–8.05 (m, 2H), 7.61–7.54 (m, 1H), 7.51–7.37 (m, 2H), 7.28 (d, J = 2.2 Hz, 2H), 7.04–6.94 (m, 2H), 6.92–6.85 (m, 2H), 6.85–6.76 (m, 2H), 6.16 (d, J = 8.3 Hz, 1H), 5.92 (ddt, J = 16.3, 10.8, 5.6 Hz, 1H), 5.49 (d, J = 2.7 Hz, 1H), 5.34–5.16 (m, 2H), 5.04 (s, 1H), 5.03–4.94 (m, 2H), 4.72–4.50 (m, 5H), 4.40–4.25 (m, 1H), 4.17–4.01 (m, 2H), 3.99–3.88 (m, 2H), 3.79 (s, 3H), 3.77 (s, 3H), 3.56 (s, 1H), 2.29–2.15 (m, 2H), 2.14–1.98 (m, 3H), 1.94 (d, J = 14.0 Hz, 1H), 1.88–1.76 (m, 2H), 1.31 (d, J = 6.5 Hz, 3H), 1.16 (d, J = 6.5 Hz, 3H), 0.89 (d, J = 6.5 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 166.3, 159.2, 155.9, 154.7, 151.1, 133.1, 130.6, 130.5, 129.8, 129.0, 128.5, 117.7, 117.6, 114.6, 113.9, 101.5, 98.7, 96.4, 81.1, 77.5, 77.4, 77.2, 76.8, 74.9, 72.7, 70.6, 70.3, 70.3, 68.8, 67.5, 65.7, 65.7, 65.7, 55.8, 55.4, 46.6, 31.8, 31.3, 24.5, 23.1, 17.5, 17.2. HRMS: [M + Na]+ calcd for C44H55NO13Na 828.3571; found 828.3586.
The above benzoate (1.20 g, 2.04 mmol) was dissolved in MeOH (40 mL) and DCM (40 mL), after which NaOMe was added to pH 10. After stirring for a week, it was neutralized by addition of dry ice and concentrated in vacuo. Column chromatography (50:50–75:25 EtOAc/pentane) gave the alcohol as a white foam (1.21 g, 1.72 mmol, 85%). 1H NMR (400 MHz, chloroform-d) δ 7.32–7.19 (m, 2H), 7.05–6.95 (m, 2H), 6.93–6.85 (m, 2H), 6.85–6.75 (m, 2H), 6.15 (d, J = 8.3 Hz, 1H), 5.97–5.86 (m, 1H), 5.49 (d, J = 3.1 Hz, 1H), 5.30 (dq, J = 17.2, 1.6 Hz, 1H), 5.20 (dq, J = 10.6, 1.5 Hz, 1H), 4.99 (q, J = 1.5 Hz, 1H), 4.92 (d, J = 3.2 Hz, 1H), 4.70–4.46 (m, 4H), 4.43–4.34 (m, 1H), 4.31 (dt, J = 7.8, 4.3 Hz, 1H), 4.09 (q, J = 6.3 Hz, 1H), 4.01 (q, J = 6.6 Hz, 1H), 3.96–3.86 (m, 2H), 3.80 (s, 3H), 3.77 (s, 3H), 3.54 (s, 1H), 3.52 (s, 1H), 2.17 (td, J = 12.1, 3.7 Hz, 1H), 2.12–1.90 (m, 4H), 1.82 (td, J = 12.6, 3.5 Hz, 1H), 1.78–1.66 (m, 3H), 1.29 (d, J = 6.6 Hz, 3H), 1.14 (d, J = 6.5 Hz, 3H), 0.91 (d, J = 6.5 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 159.2, 155.9, 154.6, 151.1, 133.0, 130.6, 129.0, 117.7, 117.5, 114.6, 113.8, 101.4, 98.7, 96.4, 81.0, 74.9, 72.7, 68.9, 67.6, 67.5, 66.6, 55.8, 55.4, 46.6, 31.8, 31.3, 25.8, 23.6, 17.5, 17.1. HRMS: [M + Na]+ calcd for C37H51NO12Na 724.3309; found 724.3322.
To a solution of the above alcohol (351 mg, 0.500 mmol) in DCM (20 mL) were added NaHCO3 (840 mg, 5.00 mmol, 10 equiv) and Dess–Martin periodinane (530 mg, 1.25 mmol, 2.5 equiv). After stirring for 1.5 h, 10% aq Na2S2O3 (20 mL) was added, and the mixture was stirred for a further 30 min. Then, it was washed with sat. aq NaHCO3, dried over MgSO4, and concentrated in vacuo. Size-exclusion chromatography (Sephadex LH-20; eluent, 1:1 DCM/MeOH) gave the title compound as a white solid (341 mg, 0.487 mmol, 97%). 1H NMR (400 MHz, chloroform-d) δ 7.32–7.20 (m, 2H), 7.06–6.99 (m, 2H), 6.92–6.85 (m, 2H), 6.85–6.76 (m, 2H), 6.16 (d, J = 8.2 Hz, 1H), 5.92 (ddd, J = 17.3, 10.6, 5.4 Hz, 1H), 5.50 (d, J = 3.1 Hz, 1H), 5.36–5.15 (m, 2H), 5.10 (t, J = 4.3 Hz, 1H), 5.00 (dd, J = 3.7, 1.7 Hz, 1H), 4.72–4.45 (m, 5H), 4.38–4.25 (m, 1H), 4.08 (dq, J = 13.3, 6.4 Hz, 2H), 4.03–3.88 (m, 2H), 3.79 (s, 3H), 3.76 (s, 3H), 3.56 (s, 1H), 2.60 (ddd, J = 15.0, 8.9, 5.7 Hz, 1H), 2.41 (ddd, J = 15.6, 7.6, 5.5 Hz, 1H), 2.30 (ddt, J = 14.1, 8.9, 5.2 Hz, 1H), 2.25–1.99 (m, 4H), 1.84 (td, J = 12.7, 3.5 Hz, 1H), 1.33 (d, J = 6.5 Hz, 3H), 1.15 (d, J = 6.4 Hz, 3H), 0.97 (d, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 210.7, 158.9, 155.4, 154.3, 150.7, 132.7, 130.0, 128.7, 117.3, 117.2, 114.2, 113.5, 101.1, 97.6, 96.0, 80.7, 74.7, 72.1, 71.5, 69.9, 68.2, 67.1, 65.3, 55.4, 55.0, 46.2, 33.6, 31.4, 30.7, 29.1, 17.1, 17.0, 14.5. HRMS: [M + Na]+ calcd for C37H49NO12Na 722.3153; found 722.3165.
o-Cyclopropylethynylbenzoyl-2,3-dideoxy-4-ulo-α-l-fucopyranosyl-(1 → 4)-2-deoxy-3-O-p-methoxybenzyl-α-l-fucopyranosyl-(1 → 4)-3-azido-2,3-dideoxy-l-fucopyranoside (30)
Prepared according to General Procedures A and B from 29 (1.06 g, 1.51 mmol) to give the title compound as a white foam (872 mg, 1.14 mmol, 75% over two steps, α/β 1:7). Spectral data for the β-anomer: 1H NMR (400 MHz, chloroform-d) δ 7.94 (dd, J = 7.9, 1.4 Hz, 1H), 7.48 (dd, J = 7.9, 1.4 Hz, 1H), 7.42 (td, J = 7.5, 1.4 Hz, 1H), 7.37–7.16 (m, 3H), 6.93–6.79 (m, 2H), 6.36 (d, J = 8.0 Hz, 1H), 5.98 (dd, J = 10.0, 2.2 Hz, 1H), 5.90 (ddd, J = 16.3, 10.7, 5.4 Hz, 1H), 5.37–5.15 (m, 2H), 5.10 (t, J = 4.4 Hz, 1H), 5.03–4.97 (m, 1H), 4.75–4.45 (m, 5H), 4.08 (q, J = 6.6 Hz, 1H), 4.03–3.95 (m, 2H), 3.90 (ddt, J = 12.4, 7.4, 4.1 Hz, 1H), 3.85–3.78 (m, 2H), 3.76 (s, 3H), 3.49 (s, 1H), 2.60 (ddd, J = 15.0, 8.8, 5.7 Hz, 1H), 2.42 (ddd, J = 15.7, 7.7, 5.4 Hz, 1H), 2.31 (ddt, J = 13.9, 8.8, 5.2 Hz, 1H), 2.24–2.15 (m, 2H), 2.10 (tt, J = 10.4, 5.5 Hz, 2H), 1.81 (td, J = 12.3, 9.9 Hz, 1H), 1.50 (tt, J = 7.8, 5.4 Hz, 1H), 1.36–1.27 (m, 6H), 0.97 (d, J = 6.7 Hz, 3H), 0.87 (dd, J = 7.6, 5.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 211.1, 164.3, 159.3, 155.8, 134.3, 132.9, 132.0, 130.3, 129.1, 127.0, 125.2, 117.7, 113.9, 101.8, 99.8, 98.0, 93.2, 80.3, 75.1, 74.5, 72.9, 72.4, 71.9, 70.3, 68.7, 65.7, 55.4, 50.0, 34.0, 32.2, 31.1, 29.5, 17.4, 14.8, 9.0, 0.8. HRMS: [M + Na]+ calcd for C42H51NO12Na 784.3309; found 784.3322.
7-[2,3-Dideoxy-4-ulo-α-l-fucopyranosyl-2-deoxy-3-p-methoxybenzyl-α-l-fucopyranosyl-(1 → 4)-3-amino-2,3-dideoxy-α-l-fucopyranoside]-14-O-tert-butyldimethylsilyl-doxorubicinone (31)
Prepared according to General Procedure C from donor 30 (422 mg, 0.552 mmol) and doxorubicinone acceptor 16(35) (1.5 equiv) to give after column chromatography (20:80–100:0 EtOAc/pentane) the crude anthracycline trisaccharide. To a solution of the above trisaccharide in DCM (93 mL) and phosphate buffer (9.3 mL, pH = 7) was added DDQ (1.25 g, 5.52 mmol, 10 equiv) at 0 °C, after which the mixture was stirred at that temperature for 45 min. It was then stirred at room temperature for an additional 2.5 h, after which it was diluted with DCM and washed with H2O four times. The organic layer was then dried over Na2SO4 and concentrated in vacuo. Column chromatography (5:95–12:88 acetone/toluene) gave the free 3″-hydroxyl anthracycline trisaccharide as a red solid (310 mg, 0.315 mmol, 57% over two steps). 1H NMR (400 MHz, chloroform-d) δ 13.93 (s, 1H), 13.24 (s, 1H), 8.03 (dd, J = 7.8, 1.0 Hz, 1H), 7.78 (t, J = 8.1 Hz, 1H), 7.39 (dd, J = 8.6, 1.1 Hz, 1H), 6.02 (d, J = 7.9 Hz, 1H), 5.84 (ddt, J = 16.2, 10.8, 5.5 Hz, 1H), 5.51 (d, J = 3.7 Hz, 1H), 5.26 (td, J = 3.4, 1.7 Hz, 1H), 5.23–5.05 (m, 2H), 4.99–4.93 (m, 1H), 4.90 (d, J = 2.8 Hz, 2H), 4.58–4.41 (m, 4H), 4.19–4.10 (m, 3H), 4.09 (s, 3H), 3.93–3.82 (m, 1H), 3.78–3.70 (m, 2H), 3.58 (s, 1H), 3.20 (dd, J = 18.7, 1.8 Hz, 1H), 2.97 (d, J = 18.9 Hz, 1H), 2.55–2.39 (m, 3H), 2.29 (d, J = 14.8 Hz, 1H), 2.24–2.02 (m, 4H), 1.92 (ddd, J = 14.0, 10.0, 3.8 Hz, 2H), 1.83–1.72 (m, 1H), 1.37–1.22 (m, 10H), 0.96 (s, 9H), 0.14 (d, J = 2.2 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 211.5, 209.9, 187.2, 186.8, 161.1, 156.5, 156.0, 155.6, 135.8, 135.6, 134.2, 134.0, 133.0, 121.0, 119.9, 118.5, 117.6, 111.6, 111.4, 101.6, 100.9, 100.3, 82.2, 81.1, 72.0, 69.8, 67.9, 66.8, 65.6, 65.0, 56.8, 46.6, 35.8, 34.4, 34.2, 33.5, 31.4, 27.6, 26.0, 18.7, 17.5, 16.9, 14.9. HRMS: [M + Na]+ calcd for C49H65NO18SiNa 1006.3869; found 1006.3876.
7-[2,3-Dideoxy-4-ulo-α-l-fucopyranosyl-2-deoxy-α-l-fucopyranosyl-(1 → 4)-3-amino-2,3-dideoxy-α-l-fucopyranoside]-doxorubicinone (9)
A solution of 31 (159 mg, 0.162 mmol) and N,N-dimethylbarbituric acid (115 mg, 0.729 mmol, 4.5 equiv) in DCM (16.3 mL) was degassed for 5 min. Then, Pd(PPh3)4 (9.0 mg, 81 μmol, 0.05 equiv) was added, and the mixture was allowed to stir for 20 min. It was then directly subjected to column chromatography on neutral silica (0:100–3:97 MeOH/DCM) to give the free amine as a red solid (118 mg, 0.131 mmol, 81%). 1H NMR (500 MHz, chloroform-d) δ 13.90 (s, 1H), 7.97 (dd, J = 7.7, 1.1 Hz, 1H), 7.75 (t, J = 8.1 Hz, 1H), 7.38 (dd, J = 8.7, 1.1 Hz, 1H), 5.48 (d, J = 3.7 Hz, 1H), 5.23 (dd, J = 4.1, 2.2 Hz, 1H), 5.10 (t, J = 6.1 Hz, 1H), 5.01 (d, J = 3.6 Hz, 1H), 4.94–4.81 (m, 2H), 4.50 (q, J = 6.7 Hz, 1H), 4.25 (q, J = 6.6 Hz, 1H), 4.13 (ddd, J = 12.2, 4.7, 2.7 Hz, 1H), 4.08 (s, 3H), 4.03 (q, J = 6.4 Hz, 1H), 3.73 (s, 1H), 3.52 (d, J = 2.5 Hz, 1H), 3.13 (dd, J = 18.8, 1.9 Hz, 1H), 3.00 (ddd, J = 12.4, 4.7, 2.4 Hz, 1H), 2.89 (d, J = 18.7 Hz, 1H), 2.56–2.38 (m, 3H), 2.30 (dt, J = 14.8, 2.1 Hz, 1H), 2.23–2.00 (m, 3H), 1.89 (td, J = 12.4, 3.8 Hz, 1H), 1.75 (td, J = 12.9, 3.9 Hz, 1H), 1.68 (dd, J = 13.1, 4.5 Hz, 1H), 1.33 (d, J = 6.8 Hz, 3H), 1.28 (d, J = 6.5 Hz, 3H), 1.22 (d, J = 6.5 Hz, 3H), 0.96 (s, 9H), 0.14 (d, J = 1.2 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 211.2, 210.0, 186.9, 186.6, 161.1, 156.4, 155.8, 135.7, 135.5, 134.1, 120.8, 119.8, 118.5, 111.4, 101.4, 100.8, 100.2, 82.3, 81.7, 71.9, 69.6, 68.4, 67.4, 66.6, 65.2, 56.7, 46.8, 35.6, 34.4, 33.9, 33.5, 27.7, 26.0, 18.7, 17.7, 17.2, 14.8. HRMS: [M + H]+ calcd for C45H62NO16Si 900.3838; found 900.3836.
To a solution of the above compound (19.7 mg, 21.9 μmol) in pyridine (0.7 mL) and THF (1.4 mL) in a PTFE tube was added HF·pyr complex (70 wt % HF, 86 μL) at 0 °C. After 3 h, an additional such portion of HF·pyr complex was added. After stirring one more hour, solid NaHCO3 was added to quench, and the mixture was stirred until cessation of effervescence. It was then filtered off, and the filtrate was poured into DCM/H2O. The organic layer was dried over Na2SO4 and concentrated in vacuo. Column chromatography on neutral silica (DCM; 4:96 MeOH/DCM) gave the title compound as a red solid (12.7 mg, 16.2 μmol, 74%). 1H NMR (500 MHz, chloroform-d) δ 13.94 (s, 1H), 8.13–7.89 (m, 1H), 7.78 (t, J = 8.1 Hz, 1H), 7.52–7.31 (m, 1H), 5.51 (d, J = 3.8 Hz, 1H), 5.36–5.27 (m, 1H), 5.09 (t, J = 6.1 Hz, 1H), 5.01 (d, J = 3.7 Hz, 1H), 4.81–4.68 (m, 2H), 4.49 (q, J = 6.6 Hz, 1H), 4.23 (q, J = 6.4 Hz, 1H), 4.16–4.05 (m, 4H), 4.01 (q, J = 6.5 Hz, 1H), 3.72 (s, 1H), 3.52 (s, 1H), 3.25 (dd, J = 18.9, 2.0 Hz, 1H), 3.08–2.96 (m, 2H), 2.46 (dtt, J = 17.8, 10.3, 5.8 Hz, 4H), 2.32 (dt, J = 14.5, 2.1 Hz, 1H), 2.25 (t, J = 7.6 Hz, 1H), 2.22–2.05 (m, 4H), 1.89 (td, J = 12.3, 3.7 Hz, 1H), 1.76 (td, J = 12.9, 3.9 Hz, 1H), 1.70 (d, J = 4.5 Hz, 1H), 1.33 (d, J = 6.5 Hz, 3H), 1.28 (d, J = 6.4 Hz, 3H), 1.22 (d, J = 6.7 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 213.9, 210.0, 187.2, 186.8, 161.2, 156.4, 155.8, 135.9, 135.6, 134.0, 133.7, 121.0, 120.0, 118.6, 111.7, 111.5, 101.3, 100.9, 100.3, 82.4, 81.7, 72.0, 69.2, 68.5, 67.5, 65.6, 65.3, 56.8, 46.8, 35.6, 34.5, 34.1, 33.6, 27.7, 17.8, 17.2, 14.9. HRMS: [M + H]+ calcd for C39H48NO16: 786.2973; found 786.2982.
7-[2,3-Dideoxy-4-ulo-α-l-fucopyranosyl-2-deoxy-α-l-fucopyranosyl-(1 → 4)-3-dimethylamino-2,3-dideoxy-α-l-fucopyranoside]-doxorubicinone (11)
A solution of 31 (159 mg, 0.162 mmol) and N,N-dimethylbarbituric acid (115 mg, 0.729 mmol, 4.5 equiv) in DCM (16.3 mL) was degassed for 5 min. Then, Pd(PPh3)4 (9.0 mg, 81 μmol, 0.05 equiv) was added, and the mixture was allowed to stir for 20 min. It was then directly subjected to column chromatography on neutral silica (0:100–3:97 MeOH/DCM) to give the free amine as a red solid (118 mg, 0.131 mmol, 81%).
To a solution of the above amine (48.0 mg, 53.3 μmol) in EtOH (10.8 mL) and 37% aq CH2O (132 μL, 30 equiv) was added NaBH(OAc)3 (21.5 mg, 0.101 mmol, 1.9 equiv). The mixture was stirred for 1.5 h before being poured into sat. aq NaHCO3. This was repetitively extracted with DCM, dried over Na2SO4, and concentrated in vacuo. Column chromatography on neutral silica (10:90–40:60 acetone/toluene) followed by size-exclusion chromatography (Sephadex LH-20, 1:1 DCM/MeOH v/v) gave the dimethylamine as a red solid (25.8 mg, 27.8 μmol, 52%). 1H NMR (500 MHz, chloroform-d) δ 13.93 (s, 1H), 13.24 (s, 1H), 8.01 (dt, J = 7.7, 1.5 Hz, 1H), 7.83–7.70 (m, 1H), 7.45–7.36 (m, 1H), 5.53 (d, J = 3.8 Hz, 1H), 5.26 (dd, J = 4.1, 2.1 Hz, 1H), 5.10–5.06 (m, 1H), 5.03 (d, J = 3.4 Hz, 1H), 4.97–4.82 (m, 2H), 4.77 (s, 1H), 4.55 (q, J = 6.4 Hz, 1H), 4.50 (q, J = 6.7 Hz, 1H), 4.09 (d, J = 3.3 Hz, 4H), 3.92 (q, J = 6.6 Hz, 1H), 3.75 (s, 1H), 3.72–3.58 (m, 2H), 3.18 (dd, J = 18.9, 2.0 Hz, 1H), 2.98 (d, J = 18.8 Hz, 1H), 2.53–2.38 (m, 3H), 2.32 (dt, J = 14.6, 2.2 Hz, 1H), 2.26–2.01 (m, 10H), 1.94–1.73 (m, 4H), 1.33 (d, J = 6.8 Hz, 3H), 1.31–1.20 (m, 7H), 1.17 (d, J = 6.4 Hz, 3H), 0.96 (s, 9H), 0.14 (d, J = 2.8 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 211.4, 210.3, 187.1, 186.7, 161.1, 156.6, 155.9, 135.8, 135.6, 134.3, 134.1, 124.9, 121.0, 119.9, 118.5, 111.5, 111.4, 101.5, 100.3, 99.6, 83.1, 74.1, 71.9, 69.7, 68.6, 66.7, 65.4, 61.7, 56.8, 43.4, 35.6, 34.4, 34.0, 33.6, 30.4, 29.8, 27.7, 26.0, 18.1, 17.1, 14.9. HRMS: [M + H]+ calcd for C47H66NO16Si: 928.4151; found 928.4157.
To a solution of the above compound (20.6 mg, 22.2 μmol) in pyridine (1.4 mL) and THF (1.4 mL) in a PTFE tube was added HF·pyr complex (70 wt % HF, 87 μL) at 0 °C. Four more additional such amounts of HF·pyr complex were added over the course of 4.5 h. Then, solid NaHCO3 was added to quench, and the mixture was stirred until cessation of effervescence. It was then filtered off, and the filtrate was poured into DCM/H2O. The organic layer was dried over Na2SO4 and concentrated in vacuo. Column chromatography on neutral silica (DCM – 10:90 MeOH/DCM) gave the title compound as a red solid (13.3 mg, 16.3 μmol, 73%). 1H NMR (500 MHz, chloroform-d) δ 13.95 (s, 1H), 13.26 (s, 1H), 8.03 (dd, J = 7.7, 1.0 Hz, 1H), 7.79 (dd, J = 8.5, 7.7 Hz, 1H), 7.40 (dd, J = 8.7, 1.1 Hz, 1H), 5.55 (d, J = 3.8 Hz, 1H), 5.32–5.28 (m, 1H), 5.08 (dd, J = 7.0, 5.6 Hz, 1H), 5.03 (s, 1H), 4.92 (s, 1H), 4.76 (d, J = 1.0 Hz, 2H), 4.54 (d, J = 6.6 Hz, 1H), 4.49 (q, J = 6.7 Hz, 1H), 4.16–4.03 (m, 4H), 3.91 (q, J = 6.5 Hz, 1H), 3.76 (s, 1H), 3.71–3.60 (m, 2H), 3.26 (dd, J = 18.8, 2.0 Hz, 1H), 3.03 (d, J = 18.8 Hz, 1H), 2.54–2.40 (m, 3H), 2.34 (dt, J = 14.6, 2.2 Hz, 1H), 2.24–2.12 (m, 7H), 2.10 (dd, J = 12.1, 4.6 Hz, 1H), 2.03 (d, J = 15.0 Hz, 1H), 1.83 (td, J = 12.2, 3.8 Hz, 3H), 1.33 (d, J = 6.7 Hz, 3H), 1.27 (d, J = 6.6 Hz, 3H), 1.17 (d, J = 6.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 213.9, 210.3, 187.3, 186.9, 161.2, 156.5, 155.9, 135.9, 135.6, 134.2, 133.7, 121.1, 119.9, 118.5, 111.7, 111.5, 101.4, 100.3, 99.6, 83.1, 74.1, 71.9, 69.3, 68.8, 66.9, 65.6, 65.4, 61.8, 56.8, 43.5, 35.6, 34.4, 34.1, 33.7, 27.8, 18.2, 17.1, 14.9. HRMS: [M + H]+ calcd for C41H52NO16: 814.3286; found 814.3301.
7-[2,3-Dideoxy-4-ulo-α-l-fucopyranosyl-2-deoxy-3-O-p-methoxybenzyl-α-l-fucopyranosyl-(1 → 4)-3-N-allyloxycarbonyl-2,3-dideoxy-α-l-fucopyranoside]-aklavinone (32)
Prepared according to General Procedure C from donor 30 (211 mg, 0.276 mmol) and aklavinone 14(34) (2 equiv) at −20 °C to give after column chromatography (10:90 EtOAc/pentane and then 2:98–20:80 acetone/toluene) the title compound as a yellow solid (210 mg, 0.213 mmol, 77%). 1H NMR (400 MHz, chloroform-d) δ 12.66 (s, 1H), 12.01 (s, 1H), 7.82 (dd, J = 7.5, 1.1 Hz, 1H), 7.72–7.61 (m, 2H), 7.34–7.21 (m, 2H), 6.93–6.82 (m, 2H), 6.07 (d, J = 7.8 Hz, 1H), 5.83 (ddt, J = 16.0, 10.8, 5.6 Hz, 1H), 5.46 (d, J = 3.8 Hz, 1H), 5.30–5.06 (m, 4H), 4.98 (s, 1H), 4.71–4.62 (m, 1H), 4.62–4.49 (m, 2H), 4.46 (ddt, J = 6.9, 5.5, 1.5 Hz, 2H), 4.22 (s, 2H), 4.12 (s, 1H), 4.09–3.90 (m, 3H), 3.87 (d, J = 7.1 Hz, 1H), 3.82 (s, 3H), 3.70 (s, 3H), 3.55 (s, 1H), 2.66–2.47 (m, 2H), 2.42 (ddd, J = 15.7, 7.6, 5.5 Hz, 1H), 2.36–2.25 (m, 2H), 2.25–2.04 (m, 3H), 2.00 (dd, J = 12.9, 4.5 Hz, 1H), 1.74 (dq, J = 13.5, 6.0, 4.3 Hz, 2H), 1.50 (dq, J = 14.6, 7.1 Hz, 1H), 1.30 (d, J = 6.6 Hz, 3H), 1.28–1.24 (m, 3H), 1.08 (t, J = 7.3 Hz, 3H), 0.98 (d, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 211.1, 192.8, 181.4, 171.5, 162.6, 162.2, 159.3, 155.5, 142.7, 137.4, 133.5, 133.0, 133.0, 131.1, 130.3, 129.1, 124.8, 121.0, 120.3, 117.5, 115.8, 114.8, 113.9, 101.6, 101.5, 98.0, 80.9, 75.0, 72.5, 71.8, 71.4, 70.3, 68.5, 67.7, 65.5, 57.1, 55.4, 52.6, 46.5, 34.0, 32.2, 31.6, 31.1, 29.5, 17.4, 17.3, 14.8, 6.8. HRMS: [M + Na]+ calcd for C52H61NO18Na 1010.3786; found 1010.3796.
3′,3′-Didesmethyl-aclarubicin (10)
To a biphasic mixture of 32 (210 mg, 0.213 mmol) in DCM (36 mL) and phosphate buffer (3.6 mL, pH = 7) was added DDQ (484 mg, 2.13 mmol, 10 equiv) at 0 °C after which the mixture was stirred at that temperature for 90 min. It was diluted with DCM and washed with H2O four times, after which the organic layer was dried over Na2SO4 and concentrated in vacuo. Column chromatography (5:95–10:90 acetone/toluene) gave the intermediate free 3″-hydroxyl as a yellow solid (155 mg, 0.179 mmol, 84%). 1H NMR (400 MHz, chloroform-d) δ 12.65 (s, 1H), 12.00 (s, 1H), 7.81 (dd, J = 7.5, 1.2 Hz, 1H), 7.75–7.60 (m, 2H), 7.32–7.25 (m, 1H), 6.05 (d, J = 7.8 Hz, 1H), 5.83 (ddt, J = 16.3, 10.7, 5.5 Hz, 1H), 5.46 (d, J = 3.8 Hz, 1H), 5.27–5.06 (m, 4H), 4.95 (d, J = 3.5 Hz, 1H), 4.53–4.38 (m, 3H), 4.28–4.18 (m, 2H), 4.18–4.06 (m, 3H), 3.86 (dd, J = 12.2, 6.5 Hz, 1H), 3.81–3.72 (m, 2H), 3.70 (s, 3H), 3.55 (s, 1H), 2.59–2.38 (m, 4H), 2.31 (d, J = 15.0 Hz, 1H), 2.24–2.06 (m, 2H), 2.01 (dd, J = 12.9, 4.6 Hz, 1H), 1.92 (td, J = 12.4, 3.8 Hz, 1H), 1.83–1.68 (m, 2H), 1.49 (dq, J = 14.7, 7.2 Hz, 1H), 1.36–1.24 (m, 9H), 1.08 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 209.9, 192.8, 181.4, 171.5, 162.6, 162.2, 155.5, 142.7, 137.4, 133.6, 133.0, 133.0, 131.1, 124.8, 121.0, 120.3, 117.5, 115.9, 114.8, 101.6, 101.6, 100.3, 82.1, 81.2, 71.9, 71.5, 71.4, 67.9, 67.7, 65.5, 65.0, 57.1, 52.6, 46.6, 34.4, 34.0, 33.5, 32.2, 31.6, 27.6, 17.3, 16.9, 14.8, 6.8. HRMS: [M + Na]+ calcd for C44H53NO17Na 890.3211; found 890.3220.
A solution of the above compound (155 mg, 0.179 mmol) and N,N-dimethylbarbituric acid (125 mg, 0.806 mmol, 4.5 equiv) in DCM (18 mL) was degassed for 5 min. Then, Pd(PPh3)4 (10.0 mg, 0.0090 mmol, 0.05 equiv) was added, and the mixture was allowed to stir for 15 min. It was then directly subjected to column chromatography on neutral silica (0:100–3:97 MeOH/DCM), followed by size-exclusion chromatography (Sephadex LH-20; eluent, 1:1 DCM/MeOH) twice and finally column chromatography on neutral silica (3:97 MeOH/DCM) to give the title compound as a yellow solid (86 mg, 0.11 mmol, 61%). 1H NMR (500 MHz, chloroform-d + MeOD) δ 7.81 (dt, J = 7.4, 2.0 Hz, 1H), 7.74–7.62 (m, 2H), 7.30 (d, J = 1.2 Hz, 1H), 5.47 (t, J = 2.5 Hz, 1H), 5.26 (dd, J = 4.4, 1.8 Hz, 1H), 5.10 (t, J = 6.2 Hz, 1H), 4.99 (d, J = 3.6 Hz, 1H), 4.52 (q, J = 6.7 Hz, 1H), 4.19 (q, J = 6.7 Hz, 1H), 4.17–4.04 (m, 3H), 3.74 (s, 1H), 3.70 (s, 3H), 3.50 (d, J = 2.4 Hz, 1H), 2.99 (ddd, J = 10.9, 6.3, 2.4 Hz, 1H), 2.56–2.37 (m, 4H), 2.30 (dt, J = 14.9, 1.8 Hz, 2H), 2.21–2.12 (m, 1H), 2.09 (dd, J = 12.4, 4.6 Hz, 1H), 1.91 (td, J = 12.4, 3.8 Hz, 1H), 1.75 (ddd, J = 14.1, 9.4, 5.7 Hz, 3H), 1.50 (dp, J = 13.8, 7.0 Hz, 1H), 1.32 (d, J = 6.8 Hz, 3H), 1.29 (d, J = 6.4 Hz, 3H), 1.23 (d, J = 6.5 Hz, 3H), 1.08 (t, J = 7.3 Hz, 3H). 13C NMR (126 MHz, CDCl3 + MeOD) δ 210.3, 192.7, 181.4, 171.4, 162.4, 162.0, 142.6, 137.4, 133.5, 133.0, 131.2, 124.8, 120.9, 120.2, 115.8, 114.7, 101.6, 100.9, 100.0, 81.9, 81.8, 71.9, 71.6, 70.9, 68.1, 67.5, 65.1, 57.1, 52.6, 46.6, 34.2, 34.2, 33.8, 33.5, 32.1, 27.6, 17.4, 17.0, 14.7, 6.6. HRMS: [M + H]+ calcd for C40H50NO15 784.3181; found 784.3196.
Cell Culture
K562 cells (B. Pang, Stanford University), HCT116 cells (T. van Hall, LUMC, The Netherlands), and PC3 and DU145 cells (C. Robson, Newcastle University, U.K.) were maintained in Roswell Park Memorial Institute (RPMI)-1640 medium supplemented with 8% fetal bovine serum (FCS). Wild-type MelJuSo cells were maintained in IMDM (IMDM = Iscove’s Modified Dulbecco’s Medium) supplemented with 8% FCS. MelJuSo cells stably expressing PAGFP-H2A were maintained in IMDM supplemented with 8% FCS and G-418, as described.17 U87 cells (ATCC HTB-14) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 8% FCS. Cell lines were maintained in a humidified atmosphere of 5% CO2 at 37 °C and regularly tested for the absence of mycoplasma.
Western Blot and Constant-Field Gel Electrophoresis (CFGE)
Cells were treated with drugs at indicated doses for 2 h. These concentrations and treatment times correspond to physiological serum peak concentrations in cancer patients under standard treatment.17,50 Subsequently, drugs were removed by extensive washing and cells were collected at indicated time points after drug removal and processed immediately for the assay. Cells were lysed directly in sodium dodecyl sulfate (SDS) sample buffer (2% SDS, 10% glycerol, 5% β-mercaptoethanol, 60 mM Tris–HCl pH 6.8, and 0.01% bromophenol blue). Lysates were resolved by SDS-polyacrylamide gel electrophoresis (PAGE) followed by Western blotting. Primary antibodies used for blotting were γH2AX (1:1000, 05-036, Millipore) and β-actin (1:10000, A5441, Sigma). DNA double-strand breaks were visualized by constant-field gel electrophoresis, as described.51 Images were quantified with ImageJ.
Microscopy
PAGFP-H2A photoactivation and time-lapse confocal imaging were performed as described17 on a Leica SP8 confocal microscope system, 63× lens, equipped with a climate chamber. Loss of fluorescence after different treatments was quantified using ImageJ software. For TopoIIα live cell imaging, MelJuSo cells were transiently transfected with a construct encoding TopoIIα-GFP.17 Fractional distance calculations for the TopoIIα relocalization were done using LAS X software (Leica).
Cell Viability Assay
Cells were seeded into 96-well plates. Twenty-four hours after seeding, the cells were treated with indicated drugs for 2 h. Subsequently, drugs were removed and cells were left to grow for an additional 72 h. Cell viability was measured using the CellTiter-Blue viability assay (Promega). Relative survival was normalized to the untreated control and corrected for background signal.
Flow Cytometry for Measuring Drug Uptake in Cells
Cells were treated with 1 μM of the indicated drugs for 2 h. The samples were washed, collected, and fixed with paraformaldehyde. The samples were analyzed by flow cytometry using a BD FACS Aria II, with a 561 nm laser and a 610/20 nm detector.
Glossary
Abbreviations
- AML
acute myeloid leukemia
- TopoIIα
topoisomerase IIα
- PAGFP-H2A
photoactivated GFP-H2A
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c01191.
Detailed synthetic procedures of the monosaccharide building blocks 13, 17, 18, 25, and 28; NMR and other analytical data of all new compounds including compounds 3–11; evaluation of DNA break capacity of compounds 1–12; confocal microscopy of PAGFP-H2A-tagged histones to visualize chromatin damage by compounds 1–12; confocal microscopy of TopoIIα relocalization by compounds 1–12; and figures on correlation of anthracycline structural features versus cytotoxicity of compounds 1–12 (PDF)
Molecular formula strings and tabulated biological assays data (CSV)
Author Contributions
D.P.A.W. and S.Y.v.d.Z. contributed equally to this work. D.P.A.W., S.Y.v.d.Z., H.S.O., J.N., and J.D.C.C. conceived the experiments. D.P.A.W. under supervision of H.S.O., G.A.v.d.M., and J.D.C.C. performed the synthesis. S.Y.v.d.Z. under supervision of JN performed all biochemical and cellular experiments. The manuscript was written by D.P.A.W. and S.Y.v.d.Z. with input of all authors.
This work was supported by grants from the Dutch Cancer Society KWF (J.N.) and by the Institute for Chemical Immunology, an NWO Gravitation project funded by the Ministry of Education, Culture and Science of the Netherlands to H.S.O. and J.N.
The authors declare the following competing financial interest(s): J.N. is a shareholder in NIHM that aims to produce aclarubicin for clinical use.
Supplementary Material
References
- Arcamone F.; Franceschi G.; Penco S.; Selva A. Adriamycin (14-Hydroxydaunomycin), a Novel Antitumor Antibiotic. Tetrahedron Lett. 1969, 10, 1007–1010. 10.1016/S0040-4039(01)97723-8. [DOI] [PubMed] [Google Scholar]
- Röthig H. J.; Kraemer H. P.; Sedlacek H. H. Aclarubicin: Experimental and Clinical Experience. Drugs Exp. Clin. Res. 1985, 11, 123–125. [PubMed] [Google Scholar]
- Weiss R. B. The Anthracyclines: Will We Ever Find a Better Doxorubicin?. Semin. Oncol. 1992, 19, 670–686. [PubMed] [Google Scholar]
- Rizvi S. F. A.; Tariq S.; Mehdi M. Anthracyclines: Mechanism of Action, Classification, Pharmacokinetics and Future – A Mini Review. Int. J. Biotechnol. Bioeng. 2018, 4, 81–85. [Google Scholar]
- Rayson D.; Richel D.; Chia S.; Jackisch C.; van der Vegt S.; Suter T. Anthracycline–Trastuzumab Regimens for HER2/Neu-Overexpressing Breast Cancer: Current Experience and Future Strategies. Ann. Oncol. 2008, 19, 1530–1539. 10.1093/annonc/mdn292. [DOI] [PubMed] [Google Scholar]
- Sadurska E. Current Views on Anthracycline Cardiotoxicity in Childhood Cancer Survivors. Pediatr. Cardiol. 2015, 36, 1112–1119. 10.1007/s00246-015-1176-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotrionte M.; Biondi-Zoccai G.; Abbate A.; Lanzetta G.; D’Ascenzo F.; Malavasi V.; Peruzzi M.; Frati G.; Palazzoni G. Review and Meta-Analysis of Incidence and Clinical Predictors of Anthracycline Cardiotoxicity. Am. J. Cardiol. 2013, 112, 1980–1984. 10.1016/j.amjcard.2013.08.026. [DOI] [PubMed] [Google Scholar]
- Felix C. A. Secondary Leukemias Induced by Topoisomerase-Targeted Drugs. Biochim. Biophys. Acta, Gene Struct. Expression 1998, 1400, 233–255. 10.1016/S0167-4781(98)00139-0. [DOI] [PubMed] [Google Scholar]
- Mistry A. R.; Felix C. A.; Whitmarsh R. J.; Mason A.; Reiter A.; Cassinat B.; Parry A.; Walz C.; Wiemels J. L.; Segal M. R.; Adès L.; Blair I. A.; Osheroff N.; Peniket A. J.; Lafage-Pochitaloff M.; Cross N. C. P.; Chomienne C.; Solomon E.; Fenaux P.; Grimwade D. DNA Topoisomerase II in Therapy-Related Acute Promyelocytic Leukemia. N. Engl. J. Med. 2005, 352, 1529–1538. 10.1056/NEJMoa042715. [DOI] [PubMed] [Google Scholar]
- Lefrak E. A.; Pit’ha J.; Rosenheim S.; Gottlieb J. A. A Clinicopathologic Analysis of Adriamycin Cardiotoxicity. Cancer 1973, 32, 302–314. . [DOI] [PubMed] [Google Scholar]
- Jones R. L.; Swanton C.; Ewer M. S. Anthracycline Cardiotoxicity. Expert Opin. Drug Saf. 2006, 5, 791–809. 10.1517/14740338.5.6.791. [DOI] [PubMed] [Google Scholar]
- Cappetta D.; De Angelis A.; Sapio L.; Prezioso L.; Illiano M.; Quaini F.; Rossi F.; Berrino L.; Naviglio S.; Urbanek K. Oxidative Stress and Cellular Response to Doxorubicin: A Common Factor in the Complex Milieu of Anthracycline Cardiotoxicity. Oxid. Med. Cell. Longevity 2017, 2017, 1–13. 10.1155/2017/1521020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Songbo M.; Lang H.; Xinyong C.; Bin X.; Ping Z.; Liang S. Oxidative Stress Injury in Doxorubicin-Induced Cardiotoxicity. Toxicol. Lett. 2019, 307, 41–48. 10.1016/j.toxlet.2019.02.013. [DOI] [PubMed] [Google Scholar]
- Hashimoto K.; Ito K.; Ishimori Y. Novel DNA Sensor for Electrochemical Gene Detection. Anal. Chim. Acta 1994, 286, 219–224. 10.1016/0003-2670(94)80163-0. [DOI] [Google Scholar]
- Qiao X.; van der Zanden S. Y.; Wander D. P. A.; Borràs D. M.; Song J.-Y.; Li X.; van Duikeren S.; van Gils N.; Rutten A.; van Herwaarden T.; van Tellingen O.; Giacomelli E.; Bellin M.; Orlova V.; Tertoolen L. G. J.; Gerhardt S.; Akkermans J. J.; Bakker J. M.; Zuur C. L.; Pang B.; Smits A. M.; Mummery C. L.; Smit L.; Arens R.; Li J.; Overkleeft H. S.; Neefjes J. Uncoupling DNA Damage from Chromatin Damage to Detoxify Doxorubicin. Proc. Natl. Acad. Sci. U.S.A. 2020, 117, 15182–15192. 10.1073/pnas.1922072117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitiss J. L. Targeting DNA Topoisomerase II in Cancer Chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. 10.1038/nrc2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang B.; Qiao X.; Janssen L.; Velds A.; Groothuis T.; Kerkhoven R.; Nieuwland M.; Ovaa H.; Rottenberg S.; van Tellingen O.; Janssen J.; Huijgens P.; Zwart W.; Neefjes J. Drug-Induced Histone Eviction from Open Chromatin Contributes to the Chemotherapeutic Effects of Doxorubicin. Nat. Commun. 2013, 4, 1908 10.1038/ncomms2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang B.; de Jong J.; Qiao X.; Wessels L. F. A.; Neefjes J. Chemical Profiling of the Genome with Anti-Cancer Drugs Defines Target Specificities. Nat. Chem. Biol. 2015, 11, 472–480. 10.1038/nchembio.1811. [DOI] [PubMed] [Google Scholar]
- Yang F.; Kemp C. J.; Henikoff S. Doxorubicin Enhances Nucleosome Turnover around Promoters. Curr. Biol. 2013, 23, 782–787. 10.1016/j.cub.2013.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iihoshi H.; Ishihara T.; Kuroda S.; Ishihara N.; Saitoh H. Aclarubicin, an Anthracycline Anti-Cancer Drug, Fluorescently Contrasts Mitochondria and Reduces the Oxygen Consumption Rate in Living Human Cells. Toxicol. Lett. 2017, 277, 109–114. 10.1016/j.toxlet.2017.06.006. [DOI] [PubMed] [Google Scholar]
- Wijdeven R. H.; Pang B.; van der Zanden S. Y.; Qiao X.; Blomen V.; Hoogstraat M.; Lips E. H.; Janssen L.; Wessels L.; Brummelkamp T. R.; Neefjes J. Genome-Wide Identification and Characterization of Novel Factors Conferring Resistance to Topoisomerase II Poisons in Cancer. Cancer Res. 2015, 75, 4176–4187. 10.1158/0008-5472.CAN-15-0380. [DOI] [PubMed] [Google Scholar]
- Gottesman M. M.; Ling V. The Molecular Basis of Multidrug Resistance in Cancer: The Early Years of P-Glycoprotein Research. FEBS Lett. 2006, 580, 998–1009. 10.1016/j.febslet.2005.12.060. [DOI] [PubMed] [Google Scholar]
- Mechetner E.; Kyshtoobayeva A.; Zonis S.; Kim H.; Stroup R.; Garcia R.; Parker R. J.; Fruehauf J. P. Levels of Multidrug Resistance (MDR1) P-Glycoprotein Expression by Human Breast Cancer Correlate with in Vitro Resistance to Taxol and Doxorubicin. Clin. Cancer Res. 1998, 4, 389–398. [PubMed] [Google Scholar]
- Cox J.; Weinman S. Mechanisms of Doxorubicin Resistance in Hepatocellular Carcinoma. Hepatic Oncol. 2016, 3, 57–59. 10.2217/hep.15.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajic M.; Iyer J. K.; Kersbergen A.; Van Der Burg E.; Nygren A. O. H.; Jonkers J.; Borst P.; Rottenberg S. Moderate Increase in Mdr1a/1b Expression Causes in Vivo Resistance to Doxorubicin in a Mouse Model for Hereditary Breast Cancer. Cancer Res. 2009, 69, 6396–6404. 10.1158/0008-5472.CAN-09-0041. [DOI] [PubMed] [Google Scholar]
- Krohn K., Ed. Anthracycline Chemistry and Biology II; Topics in Current Chemistry; Springer: Berlin, Heidelberg, 2008; Vol. 283. [Google Scholar]
- Yoshimoto A.; Johdo O.; Nishida H.; Okamoto R.; Takeuchi T. Anthracycline Metabolites from Baumycin-Producing Streptomyces Sp. D788. III. New Anthracycline Metabolites Produced by Blocked Mutants 4L-660 and YDK-18. J. Antibiot. 1993, 46, 1758–1761. 10.7164/antibiotics.46.1758. [DOI] [PubMed] [Google Scholar]
- Tong G. L.; Wu H. Y.; Smith T. H.; Henry D. W. Adriamycin Analogs. 3. Synthesis of N-Alkylated Anthracyclines with Enhanced Efficacy and Reduced Cardiotoxicity. J. Med. Chem. 1979, 22, 912–918. 10.1021/jm00194a005. [DOI] [PubMed] [Google Scholar]
- Kunnari T.; Niemi J.; Ylihonko K.; Mäntsälä P.; Hakala J. Hybrid Anthracyclines by a Genetically Engineered Streptomyces Galilaeus Mutant. Bioorg. Med. Chem. Lett. 1997, 7, 725–726. 10.1016/S0960-894X(97)00092-9. [DOI] [Google Scholar]
- Lu W.; Leimkuhler C.; Oberthür M.; Kahne D.; Walsh C. T. AknK Is An L-2-Deoxyfucosyltransferase in the Biosynthesis of the Anthracycline Aclacinomycin A. Biochemistry 2004, 43, 4548–4558. 10.1021/bi035945i. [DOI] [PubMed] [Google Scholar]
- Oki T.; Kitamura I.; Matsuzawa Y.; Shibamoto N.; Ogasawara T.; Yoshimoto A.; Inui T.; Naganawa H.; Takeuchi T.; Umezawa H. Antitumor Anthracycline Antibiotics, Aclacinomycin A and Analogues. II. Structural Determination. J. Antibiot. 1979, 32, 801–819. 10.7164/antibiotics.32.801. [DOI] [PubMed] [Google Scholar]
- Tanaka H.; Yoshioka T.; Shimauchi Y.; Matsushita Y.; Matsuzawa Y.; Oki T.; Ishikura T. Chemical Modification of Anthracycline Antibiotics. IV. Synthesis of New Anthracyclines with Trisaccharide. J. Antibiot. 1982, 35, 312–320. 10.7164/antibiotics.35.312. [DOI] [PubMed] [Google Scholar]
- Yu B. Gold(I)-Catalyzed Glycosylation with Glycosyl o-Alkynylbenzoates as Donors. Acc. Chem. Res. 2018, 51, 507–516. 10.1021/acs.accounts.7b00573. [DOI] [PubMed] [Google Scholar]
- Smith A.; Toward A.. General Method for the Construction of Anthracycline Antibiotics. Ph.D. Thesis, Princeton University, 1998. [Google Scholar]
- Horton D.; Priebe W.; Valera O. Synthesis and Antitumour Activity of 3′-Deamino-3′-Hydroxydoxorubicin. J. Antibiot. 1984, 37, 853–857. 10.7164/antibiotics.37.853. [DOI] [PubMed] [Google Scholar]
- Hansen T.; Elferink H.; van Hengst J. M. A.; Houthuijs K. J.; Remmerswaal W. A.; Kromm A.; Berden G.; van der Vorm S.; Rijs A. M.; Overkleeft H. S.; Filippov D. V.; Rutjes F. P. J. T.; van der Marel G. A.; Martens J.; Oomens J.; Codée J. D. C.; Boltje T. J. Characterization of Glycosyl Dioxolenium Ions and Their Role in Glycosylation Reactions. Nat. Commun. 2020, 11, 2664 10.1038/s41467-020-16362-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komarova B. S.; Tsvetkov Y. E.; Nifantiev N. E. Design of α-Selective Glycopyranosyl Donors Relying on Remote Anchimeric Assistance. Chem. Rec. 2016, 16, 488–506. 10.1002/tcr.201500245. [DOI] [PubMed] [Google Scholar]
- Hansen T.; Lebedel L.; Remmerswaal W. A.; van der Vorm S.; Wander D. P. A.; Somers M.; Overkleeft H. S.; Filippov D. V.; Désiré J.; Mingot A.; Bleriot Y.; van der Marel G. A.; Thibaudeau S.; Codée J. D. C. Defining the SN1 Side of Glycosylation Reactions: Stereoselectivity of Glycopyranosyl Cations. ACS Cent. Sci. 2019, 5, 781–788. 10.1021/acscentsci.9b00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearlman B. A.; McNamara J. M.; Hasan I.; Hatakeyama S.; Sekizaki H.; Kishi Y. Practical Total Synthesis of (±)-Aklavinone and Total Synthesis of Aklavin. J. Am. Chem. Soc. 1981, 103, 4248–4251. 10.1021/ja00404a047. [DOI] [Google Scholar]
- Garro-Helion F.; Merzouk A.; Guibé F. Mild and Selective Palladium(0)-Catalyzed Deallylation of Allylic Amines. Allylamine and Diallylamine as Very Convenient Ammonia Equivalents for the Synthesis of Primary Amines. J. Org. Chem. 1993, 58, 6109–6113. 10.1021/jo00074a044. [DOI] [Google Scholar]
- Johdo O.; Nishida H.; Okamoto R.; Yoshimoto A.; Takeuchi T. New Anthracycline Antibiotics 10-Epi-Oxaunomycin and 10-Epi-11-Deoxyoxaunomycin. J. Antibiot. 1995, 48, 1153–1158. 10.7164/antibiotics.48.1153. [DOI] [PubMed] [Google Scholar]
- Veeneman G. H.; Van Leeuwen S. H.; Zuurmond H.; Van Boom J. H. Synthesis of Carbohydrate-Antigenic Structures of Mycobacterium tuberculosis Using an Iodonium Ion Promoted Glycosidation Approach. J. Carbohydr. Chem. 1990, 9, 783–796. 10.1080/07328309008543874. [DOI] [Google Scholar]
- Cipollone A.; Berettoni M.; Bigioni M.; Binaschi M.; Cermele C.; Monteagudo E.; Olivieri L.; Palomba D.; Animati F.; Goso C.; Maggi C. Novel Anthracycline Oligosaccharides: Influence of Chemical Modifications of the Carbohydrate Moiety on Biological Activity. Bioorg. Med. Chem. 2002, 10, 1459–1470. 10.1016/S0968-0896(01)00411-4. [DOI] [PubMed] [Google Scholar]
- Craine L.; Raban M. The Chemistry of Sulfenamides. Chem. Rev. 1989, 89, 689–712. 10.1021/cr00094a001. [DOI] [Google Scholar]
- Noshita T.; Sugiyama T.; Kitazumi Y.; Oritani T. Phenolic Ferrier Reaction and Its Application to the Natural Product Synthesis. Tetrahedron Lett. 1994, 35, 8259–8262. 10.1016/0040-4039(94)88297-5. [DOI] [Google Scholar]
- Zhang X.; Zhou Y.; Zuo J.; Yu B. Total Synthesis of Periploside A, a Unique Pregnane Hexasaccharide with Potent Immunosuppressive Effects. Nat. Commun. 2015, 6, 5879 10.1038/ncomms6879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y.; Li Z.; Shi H.; Zhang J.; Yu B. Assembly of Digitoxin by Gold(I)-Catalyzed Glycosidation of Glycosyl o-Alkynylbenzoates. J. Org. Chem. 2011, 76, 9748–9756. 10.1021/jo201850z. [DOI] [PubMed] [Google Scholar]
- Soubeyrand S.; Pope L.; Haché R. J. G. Topoisomerase IIα-Dependent Induction of a Persistent DNA Damage Response in Response to Transient Etoposide Exposure. Mol. Oncol. 2010, 4, 38–51. 10.1016/j.molonc.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Maanen J. M. S.; Retel J.; de Vries J.; Pinedo H. M. Mechanism of Action of Antitumor Drug Etoposide: A Review. JNCI, J. Natl. Cancer Inst. 1988, 80, 1526–1533. 10.1093/jnci/80.19.1526. [DOI] [PubMed] [Google Scholar]
- Speth P. A. J.; van Hoesel Q. G. C. M.; Haanen C. Clinical Pharmacokinetics of Doxorubicin. Clin. Pharmacokinet. 1988, 15, 15–31. 10.2165/00003088-198815010-00002. [DOI] [PubMed] [Google Scholar]
- Olive P. L.; Wlodek D.; Banáth J. P. DNA Double-Strand Breaks Measured in Individual Cells Subjected to Gel Electrophoresis. Cancer Res. 1991, 51, 4671–4676. [PubMed] [Google Scholar]
- Dordal M. S.; Ho A. C.; Jackson-Stone M.; Fu Y. F.; Goolsby C. L.; Winter J. N. Flow Cytometric Assessment of the Cellular Pharmacokinetics of Fluorescent Drugs. Cytometry 1995, 20, 307–314. 10.1002/cyto.990200406. [DOI] [PubMed] [Google Scholar]
- Capranico G.; De Isabella P.; Penco S.; Tinelli S.; Zunino F. Role of DNA Breakage in Cytotoxicity of Doxorubicin, 9-Deoxydoxorubicin, and 4-Demethyl-6-Deoxydoxorubicin in Murine Leukemia P388 Cells. Cancer Res. 1989, 49, 2022–2027. [PubMed] [Google Scholar]
- Gate L.; Couvreur P.; Nguyen-Ba G.; Tapiero H. N-Methylation of Anthracyclines Modulates Their Cytotoxicity and Pharmacokinetic in Wild Type and Multidrug Resistant Cells. Biomed. Pharmacother. 2003, 57, 301–308. 10.1016/S0753-3322(03)00037-4. [DOI] [PubMed] [Google Scholar]
- Gianni L. Anthracycline Resistance: The Problem and Its Current Definition. Semin. Oncol. 1997, 24, S10–S17. [PubMed] [Google Scholar]
- Mansour O. C.; Evison B. J.; Sleebs B. E.; Watson K. G.; Nudelman A.; Rephaeli A.; Buck D. P.; Collins J. G.; Bilardi R. A.; Phillips D. R.; Cutts S. M. New Anthracenedione Derivatives with Improved Biological Activity by Virtue of Stable Drug-DNA Adduct Formation. J. Med. Chem. 2010, 53, 6851–6866. 10.1021/jm901894c. [DOI] [PubMed] [Google Scholar]
- Liaw Y. C.; Gao Y. G.; Robinson H.; Van der Marel G. A.; Van Boom J. H.; Wang A. H. J. Antitumor Drug Nogalamycin Binds DNA in Both Grooves Simultaneously: Molecular Structure of Nogalamycin-DNA Complex. Biochemistry 1989, 28, 9913–9918. 10.1021/bi00452a006. [DOI] [PubMed] [Google Scholar]
- Frederick C. A.; Williams L. D.; Ughetto G.; van der Marel G. A.; Van Boom J. H.; Rich A.; Wang A. H. J. Structural Comparison of Anticancer Drug-DNA Complexes: Adriamycin and Daunomycin. Biochemistry 1990, 29, 2538–2549. 10.1021/bi00462a016. [DOI] [PubMed] [Google Scholar]
- Zeman S. M.; Phillips D. R.; Crothers D. M. Characterization of Covalent Adriamycin-DNA Adducts. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 11561–11565. 10.1073/pnas.95.20.11561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloc K.; Mlochowski J.; Syper L. Synthesis of Novel Quinones with Silver (II) Dipicolinate as a New Selective Oxidant. Chem. Lett. 1980, 9, 725–728. 10.1246/cl.1980.725. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.