

Abstract
FK506-binding protein 35, FKBP35, has been implicated as an essential malarial enzyme. Rapamycin and FK506 exhibit antiplasmodium activity in cultured parasites. However, due to the highly conserved nature of the binding pockets of FKBPs and the immunosuppressive properties of these drugs, there is a need for compounds that selectively inhibit FKBP35 and lack the undesired side effects. In contrast to human FKBPs, FKBP35 contains a cysteine, C106, adjacent to the rapamycin binding pocket, providing an opportunity to develop targeted covalent inhibitors of Plasmodium FKBP35. Here, we synthesize inhibitors of FKBP35, show that they directly bind FKBP35 in a model cellular setting, selectively covalently modify C106, and exhibit antiplasmodium activity in blood-stage cultured parasites.
Keywords: FK506-binding protein, FKBP35, plasmodium, antimalarial, targeted covalent inhibition
Malaria is caused by parasites of the genus Plasmodium. Worldwide, there are over 200 million cases every year. Despite progress in antimalarial therapies and in mosquito-control, over 400,000 people die annually, the majority resulting from Plasmodium falciparum (Pf).1 Additionally, Plasmodium vivax (Pv) causes relapsing infection due to latent liver-stage parasites which remains a challenge for malaria eradication.1,2 Finally, resistance to the frontline antimalarials is emerging, accelerating the need for new therapeutics with novel mechanisms of action.3−5
FK506-binding proteins (FKBP) are conserved throughout evolution and found across the Plasmodium genus.6−8 Immunosuppression mediated by inhibitors of calcineurin and mTOR requires recruitment of FKBPs.9−11 Specifically, FK506 and rapamycin (Figure 1) induce heterodimers between human FKBP12 and calcineurin or mTOR, respectively. As a result, FKBPs became targets in drug-discovery.12,13
Figure 1.

Structures of FKBP ligands with IC50s in blood-stage antiplasmodium assays (SLF is untested against cultured parasites). The pipecolate binding motif common to the natural product ligands is in red.
In contrast to the 14 human FKPB isoforms, Plasmodium species have a single FKBP, FKBP35.7,8 Interestingly, FK506 and rapamycin have antiplasmodium activity in blood-stage assays6,14,15 and it has been hypothesized that inhibition of FKBP35 alone might suffice for antiparasitic activity.6,14 Several groups have synthesized FKBP35-targeting compounds (Figure 1). Wandless et al. developed chimeric molecules consisting of the synthetic ligand for FKBP (SLF) conjugated to methotrexate to deliver methotrexate to parasites through FKBP12 binding but did not report the antiplasmodium activity.16 Compounds SRA and D44 exhibited good antiplasmodium and PPIase activity despite lacking the pipecolate core common to many FKBP-binders (Figure 1, in red).17,18 More recently, a library of novel [4.3.1]-aza-bicyclic sulfonamides ([4.3.1]-ABS) exhibited broad antimicrobial activity, including activity against Plasmodium.19
To date, all reported inhibitors show higher binding affinity for human FKBP12. This lack of selectivity is amplified by the cellular abundance of FKBP12 (4–5 μM in erythrocytes) versus FKBP35 (50–100 nM in Plasmodium).16 A difficulty in achieving selectivity among different FKBPs is the highly conserved proline binding pocket where 13 out of 16 residues within 5 Å of rapamycin are identical between FKBP12 and PfFKBP35. Interestingly, just outside of the binding pocket, PfFKBP35 has a cysteine (C106) whereas FKBP12 has a histidine at this position. This cysteine is conserved across the human-infecting Plasmodium species and thus provides the opportunity to develop FKBP35 covalent inhibitors.20 Such therapeutics might provide improved selectivity and prolonged duration of action and could be interesting for targeting the liver hypnozoite stage of Plasmodium vivax.
We began by modifying D44 (Figure 1) to generate three analogues (Figure 2) and modeled binding of these analogues using the D44-FKBP35 structure (PDB 4J4N).18,21D44a, in which the terminal ethyl is substituted with a benzylic acrylamide, was predicted to occupy the same position as D44 and bind favorably to C106. D44b replaces the acrylamide with a propionamide to serve as a noncovalent control, and in D44c the acrylamide is placed in an unfavorable position as a control for nonspecific cysteine-reactivity. By differential scanning calorimetry (DSC), none of these compounds showed a thermal shift against full-length FKBP12 or the PfFKBP35 binding domain (FBD35) indicating absent binding compared to rapamycin. Additionally, we observed only trace, nonspecific covalent binding to C106 by mass spectrometry (Table S1). Together these data suggest that D44 does not bind to FKBP35.
Figure 2.

Structures of D44 derivatives from pilot study.
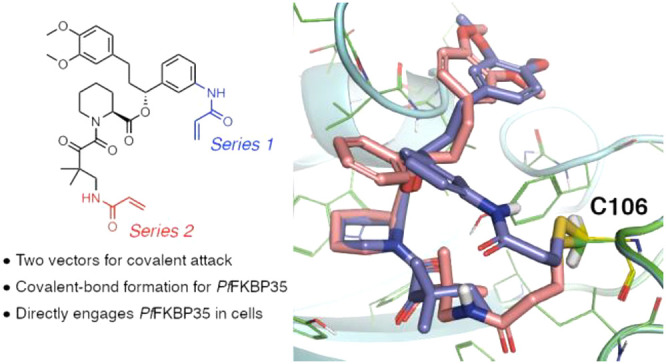
Next, we turned to SLF as a starting point.16 As there are no structures of SLF-bound FKBP35 we used the structure of the related compound SLFb bound to human FKBP51(PDB 4DRK)22 aligned with rapamycin-bound PfFKBP35 (PDB 4QT2)23 as the basis for docking. We noted two groups proximal to C106: the aryl ring of the benzylic ester (for Series 1) and the tert-pentyl group adjacent to the ketoamide (for Series 2, Figure 3a), and compounds from both series were modeled (Table S2). As a prototypical compound from Series 1, the model predicts covalently bound 1a has good overlap with SLFb in the FKBP35 binding pocket (Figure 3b). Interestingly, covalently bound compounds from Series 2 show a greater degree of overlap with SLFb (Figure 3c) resulting in better docking scores than Series 1 compounds (Table S2). Next, we investigated both series to probe the reactivity of the cysteine and determine how the vector of the covalent bond between the ligand and FKBP35 influences compound binding.
Figure 3.

Docking studies of covalent SLF analogues. (A) Structures of representative compounds from Series 1 and Series 2. (B) Model structure of 1a (purple) covalently bound to C106 (indicated by arrow) and docked in FBD35 with SLFb (gray) as a reference. (C) Model structure of 2a (yellow) covalently bound to C106 (indicated by arrow) and docked in FBD35 with SLFb (gray) as a reference. Docking model based on alignment of SLFb-bound FKBP51 binding domain (PDB 4DRK) and rapamycin-bound FKBP35 binding domain (PDB 4QT2).
For Series 1 (Scheme 1), a convergent synthesis was employed between pipecolic acid 3 or 4 with chiral benzylic alcohols 6a–d. Compounds 5a–d were synthesized via Claisen–Schmidt condensation between methylvanillin and 2′-, 3′-, or 4′-nitroacetophenone followed by hydrogenation. meta-Aniline 5b was Boc-protected to generate compound 6b. Coupling of 3 and 6b followed by Boc-deprotection furnished SLF, which was acylated with acryloyl chloride to produce 1a or propionyl chloride to produce 1b. Compounds 1c and 1d required an alternate synthetic pathway since the free anilines resulted in rapid decomposition. In order to avoid this decomposition pathway, intermediates 5a and 5c were acylated with 3-chloropropionyl chloride and then coupled with 3. Elimination of the β-chlorine with NEt3 generated compounds 1c and 1d. SLFb was synthesized by coupling benzylic alcohol 6d with 3 followed by deprotection of the acid. Compounds 1e and 1f were synthesized similarly to 1a and 1c using 4 as the coupling partner with 5b and 5a, respectively.
Scheme 1. Synthesis of Series 1 Compounds.

Reagents and conditions: (a) Methyl oxalyl chloride, DIPEA, DCM, 22 °C, 6 h; (b) 1,1-dimethylpropylmagnesium chloride, THF, −78 °C, 2 h; (c) LiOH, MeOH, 0–25 °C, 12 h; (d) SeO2, pyridine, 110 °C, 3 h; (e) oxalyl chloride, DMF, DCM, 0 °C, 30 m then: pipecolic acid methyl ester, NEt3, DCM, 0 °C, 2 h; (f) KOH, EtOH, 0 °C, 12 h; (g) H2, Pd/C, EtOAc, 2–24 h; (h) 3-chloropropionoyl chloride OR tert-butyl bromoacetate, K2CO3, acetone, 22 °C, 16 h; (i) (+)-DIP-chloride, THF, −45 °C, 16 h then: diethanolamine, Et2O, 22 °C, 2 h; (j) Boc2O, 1,4-dioxane, 150 °C, 4 h; (k) DCC, DMAP, DCM, 22 °C, 2 h; (l) TFA, DCM, 22 °C, 2 h; (m) acryloyl chloride OR propionyl chloride, NEt3, DCM, 0 °C, 2 h; (n) NEt3, MeCN, 80 °C, 6–18 h.
We investigated the binding of these compounds in two orthogonal assays: DSC and fluorescence polarization (FP, Table 1). All compounds were screened against recombinant FBD35 and FKBP12 to investigate selectivity.
Table 1. Binding of Series 1 Ligands to FBD35 and FKBP12.


Determined by fluorescence polarization and fitting the data to a competitive inhibition model (nM).
Determined by differential scanning calorimetry (°C).
Incomplete displacement of fluorescent probe.
Previous studies have shown that SLF has >10-fold lower affinity for FKBP35 than FKBP12;16 however, we observe a less profound difference in affinities. The addition of the covalent warhead to this compound (1a) does not drastically alter the IC50 as measured by FP. However, we observe a large thermal shift of +13.4 °C by DSC. Saturation of the acrylamide to the propionamide (1b) improves the binding by FP slightly but reduces the thermal shift to +2.9 °C, suggesting that the ability to form a covalent bond makes for a more thermodynamically stable complex. Moving the acrylamide warhead to the ortho position (1c) showed >10-fold weaker binding by FP but retained a large thermal shift of +9.6 °C. To rule out the possibility of nonspecific covalent bond formation, compound 1d was synthesized where the acrylamide is predicted to angle away from C106. 1d maintains good binding by FP and a thermal shift close to other noncovalent controls, suggesting that the large thermal shifts we observe result from covalent bond formation.
Concerned about the lipophilicity and solubility of these compounds (1a cLogP = 5.0, PBS pH 7.4 solubility = 0.31 μM; 1c cLogP = 4.4, PBS pH 7.4 solubility = 3.5 μM), we investigated swapping the tert-pentyl moiety for a 3,4,5-trimethoxybenzyl as ligands with this modification retain good affinity for human FKBPs.24 As predicted, this change resulted in a decrease in the cLogP (3.6 for 1e and 3.0 for 1f) and a concomitant increase in PBS solubility (3.1 μM and 23 μM, respectively). However, no binding to FBD35 was detected by FP, and only 1e displayed weak binding by DSC. Compound 1f resulted in a negative DSC shift indicating a destabilizing interaction with FBD35. Both 1e and 1f were able to bind to FKBP12 albeit with lower affinity when compared to 1a or 1c. These data indicate that despite the high homology between the binding pockets, FKBP35 cannot accommodate large aryl rings.
For Series 2, we examined both alkyl (2a–2e, 2h) and aryl (2f, 2g) acrylamides and acrylates. The synthesis of Series 2 required a change in strategy compared to Series 1 (Scheme 2). Compounds 2c–2g were synthesized from common intermediate 8a, which was generated from the coupling of 6e with Boc-protected pipecolic acid. 2h was similarly synthesized from intermediate 8b. Following the strategy of Holt et al.,25 4,4-dimethyldihydrofuran-2,3-dione was heated with 8a or 8b in the presence of DMAP to form intermediates 9a and 9b. Acylation of the resultant alcohol with acryloyl chloride or propionyl chloride generated esters 2c, 2d, and 2h. Inspired by this strategy, aryl acrylamides 2f and 2g were synthesized by the reaction between 8a and a Boc-protected isatin derivative followed by Boc-deprotection and acylation of the resultant aniline. Attempts to convert the alcohol of 9a into an amine in order to access 2a and 2b were unsuccessful. In order to place an acrylamide in this position, 8a was coupled with Boc-protected gamma-aminobutyric acid to synthesize 2e, which lacks the ketone and geminal dimethyl groups that were previously shown to enhance FKBP-binding affinity.26
Scheme 2. Synthesis of Series 2 Compounds.

Reagents and conditions: (a) Pd(OAc)2, NEt3, DMF, 80 °C, 16 h; (b) (+)-DIP-chloride, THF, −45 °C, 6 h then: diethanolamine, Et2O, 22 °C, 2 h; (c) DCC, Boc-Pip-OH, DMAP, DCM, 0 °C, 2 h; (d) TFA, DCM, 22 °C, 2 h; (e) 4,4-dimethyldihydrofuran-2,3-dione, DMAP, toluene, 80 °C, 16 h; (f) acryloyl chloride OR propionyl chloride, NEt3, DCM, 0 °C, 2–18 h; (g) DCC, tBuOH, DMAP, DCM, 0 °C, 2 h; (h) H2, Pd/C, EtOH, 1 h; (i) DCC, 3-methyl-2-oxobutanoic acid, DMAP, DCM, 0 °C, 2 h; (j) formaldehyde, para-methoxybenzylamine, MeOH, H2O, 50 °C, 44 h; (k) CAN, MeCN/H2O, 16 h; (l) EDC, DMAP, DCM, 0 °C, 12 h; (m) Boc2O, DMAP, THF, 6 h; (n) 8a, DMAP, toluene, 80 °C, 2 h.
Recognizing that the precursor to 2a and 2b was a beta-amino carbonyl, we envisioned that a Mannich reaction could provide access to these compounds. To test this, intermediate 11 was generated by tert-butyl protection of Cbz-protected pipecolic acid, Cbz-deprotection, and subsequent coupling with 3-methyl-2-oxobutanoic acid. 11 was treated with para-methoxybenzylamine and aqueous formaldehyde to form the Mannich adduct, which was acylated with either 3-chloropropionoyl chloride or propionyl chloride to generate 12a and 12b. Oxidative deprotection of the benzylic amide followed by TFA treatment gave intermediates 13a and 13b which were then coupled with 6e to complete the syntheses of 2a and 2b.
In our covalent docking model, 2a and 2c were predicted to form more stable covalent complexes than 1a or 1c (Table S2). While the IC50s overall did not change significantly between the two series, we observed larger thermal shifts in the DSC. As observed in Series 1, this large shift was dramatically reduced upon removal of the Michael acceptor (2b and 2d) indicating that Series 2 compounds can likely form a covalent bond with FBD35. Removal of the ketone and geminal dimethyl from 2a (2e) significantly lowered binding affinity but maintained the large thermal shift. Despite the predictions of robust binding with 2f or 2g in the docking model (Table 2), we saw no binding in the FP or DSC assays for either protein providing additional evidence that FKBP35 cannot accommodate aryl rings within the binding pocket. Similar to the solubility issues in Series 1, several of the Series 2 compounds have poor solubility and high cLogP values. In particular, 2c has a cLogP of 4.4 and a PBS solubility of <0.1 μM. By contrast, SLFb has a similar cLogP of 4.1 but a PBS solubility of 59 μM. Adding this free carboxylate to 2c to generate 2h reduced the cLogP to 3.5 while PBS solubility improved to 70 μM. Further, this compound exhibited a very good IC50 and the largest thermal shift recorded.
Table 2. Binding of Series 2 Ligands to FBD35 and FKBP12.


Determined by fluorescence polarization and fitting the data to a competitive inhibition model (nM).
Determined by differential scanning calorimetry (°C).
We predict that this large shift in Tm is a result of a covalent bond between these ligands and C106. In support of this, all of the covalent compounds that were able to bind to FBD35 by FP also exhibited much larger thermal shifts (>9 °C vs 2–4 °C, generally) relative to their noncovalent controls (1a vs 1b, 2a vs 2b, 2c vs 2d, and 1c, 2e, and 2h). Notably, we observed two distinct melting points in the DSC traces for putative covalent compounds that were recorded after short incubations with FBD35. To highlight this phenomenon, compound 1c was measured by DSC after a short incubation time with FBD35 and FKBP12. When incubated with FBD35, we observe two overlapping melting curves with distinct ΔTm at 1.6 and 9.6 °C relative to the apoprotein (Figure 4a). Repeating the same experiment with FKBP12 results in a single ΔTm of 3.2 °C (Figure 4b). The first ΔTm is likely indicative of the overall weaker affinity of 1c for FBD35, which is corroborated by our FP results. This ΔTm is within the same range of all the noncovalent controls, which we interpret as resulting from a reversible ligand–protein interaction. With extended incubation times (>24 h), we observe only the second, larger thermal shift with the covalent compounds whereas the noncovalent controls remain unchanged (data not shown). All compounds showed only a single thermal shift when tested against human FKBP12, regardless of incubation time.
Figure 4.
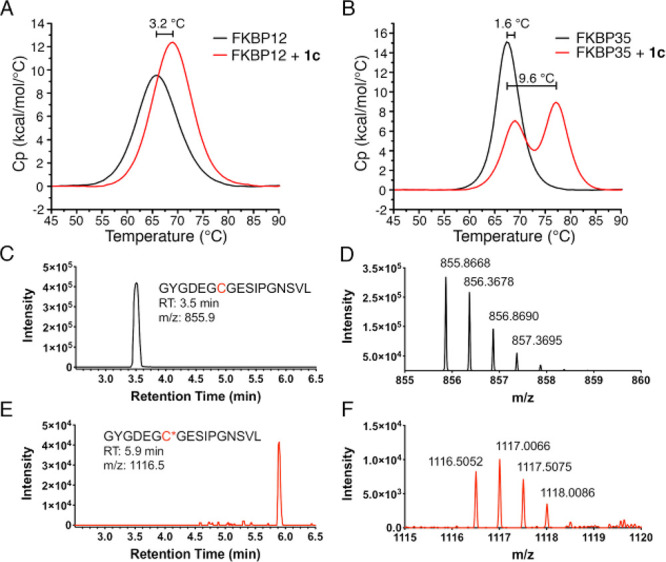
Evidence for covalent bond formation between 1c and FBD35 observed in DSC and confirmed by LC-MS. (A) DSC trace of 1c measured on FBD showing two distinct melting curves at Δ = 1.6 °C and Δ = 9.6 °C. (B) DSC trace of 1c on FKBP12 showing a single melting curve shift at Δ = 3.2 °C. (C) Extracted ion chromatogram of iodoacetamide-treated, chymotrypsin-digested FBD35 found the expected carbamidomethylated peptide (GYGDEGCGESIPGN, m/z = 855.9, 2nd charge state) at RT = 3.5 min. (D) Mass spectrum of the peak at 3.5 min in panel C. (E) Extracted ion chromatogram of 1c-treated, chymotrypsin-digested FBD35 found the expected 1c-modified peptide (GYGDEGC*GESIPGN, m/z = 1116.5, 2nd charge state) at RT = 5.9 min. (F) Mass spectrum of the peak at 5.9 min in panel E.
To confirm the formation of a covalent adduct, we analyzed FBD35 by LC-MS alone or after 24 h of incubation with 1c (Figure S1). After extended incubation with 1c, we observed a nearly 1 min shift in the retention time of the protein complex vs the apo protein with an increase of mass to 14.5 kDa, consistent with the expected mass of the apo protein (13.9 kDa) plus the ligand (579 Da). Additionally, we subjected both the FBD35 apoprotein and the 1c-FBD35 adduct to enzymatic digestion to generate the peptide containing C106 (GYGDEGCGESIPGN, Figure 4c–f, Figure S1). In the extracted ion chromatogram of the apoprotein, the carbamidomethylated peptide was found at 3.5 min (Figure 4c) with the correct mass (m/z = 855.9, second charge state, Figure 4d). When incubated with 1c, there was some residual unmodified peptide found at 3.5 min (Figure S1c–d) as well as the predicted 1c-inclusion peptide at RT = 5.9 (Figure 4e) with the correct mass (m/z = 1116.5, second charge state, Figure 4f). This mass was not found in the untreated protein (Figure S1e–f).
We next determined the relative rates of covalent adduct formation and FKBP35 consumption (Figure S2a,b) for compounds 1a–c, 2a, and 2c via LC-MS. Measuring the reaction half-life reveals a rank order of 2c > 1c > 2a > 1a and no binding by 1b (Figure S2c). Given their highly conserved structures, the stark difference in reaction half-lives between 2c (t1/2 = 0.78 h) and 2a (t1/2 = 11.7 h) is likely a result of the difference in reactivities between acrylates and acrylamides,27 highlighting the importance of fine-tuning warhead electrophilicity.
We next were interested in comparing the interaction of compounds with FKBP35 and human FKBP12 in a consistent cellular setting. To this end we employed a nanoluciferase bioluminescent resonant energy transfer (nanoBRET) assay to measure target engagement.28 We generated HEK293T cells stably expressing either nanoluciferase-tagged (nLuc) full length PfFKBP35 or nLuc-tagged FKBP12 (Figure S3). Using a BODIPY-conjugated rapamycin tracer (Rap-Gly-BDP), we were able to measure the interaction with both FKBPs and measure its displacement in cells with unlabeled compounds to infer direct target engagement (Figure S4).
As a positive control we measured displacement of the tracer with unmodified rapamycin (Figure 5a,b black squares) and found robust tracer displacement with IC50s of 34 nM (FKBP35) and 20 nM (FKBP12), respectively (Table S3). As a negative control, we measured tracer displacement using GPI-1046 (Figure S5), an early FKBP12 ligand29 of similar structure to SLF whose ability to bind FKBPs was subsequently called into question.22,30 We proceeded to measure matched pairs 1a and 1b, 2a and 2b, and 2c and 2d in the assay. After a 2 h incubation all compounds displace the fluorescent tracer, providing evidence that these compounds not only engage with FKBPs but are also cell permeable. Despite submicromolar IC50 values against both FKBPs in the FP assay SLFb displayed markedly reduced probe displacement likely due to reduced cell permeability as a result of the free carboxylate. Similar to the FP assay, there were no significant differences between covalent and noncovalent compounds in this assay. This suggests the intrinsic binding of the compounds remains a major driver of target affinity at 2 h. Nonetheless, the NanoBRET assay confirms that these compounds are not only cell-permeable but can also directly engage the target FKBP in a cellular setting.
Figure 5.
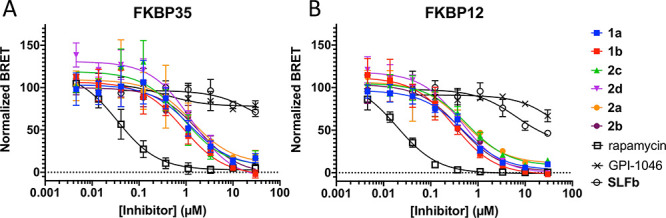
Intracellular binding of compounds to FKBP35 and FKBP12 in a NanoBRET cellular target engagement assay. (A) Normalized BRET ratio of covalent and noncovalent matched pairs in HEK-293T cells expressing nLuc-tagged FKBP35. (B) Normalized BRET ratio of covalent and noncovalent matched pairs in HEK-293T cells expressing nLuc-tagged FKBP12.
Given the success of our biochemical assays, we investigated the activity of compounds 1a and 1b on live, cultured parasites (Figure 6). Using a luciferase reporter NF54 P. falciparum parasite line, both 1a (IC50 = 1.4 μM) and 1b (IC50 = 1.9 μM) showed dose-dependent growth-inhibition (Figure 6a). Gratifyingly, these compounds performed similarly to unmodified rapamycin (IC50 = 1.1 μM). Despite displaying poorer binding in the FP assay, 1c shows similar antiplasmodium activity as 1a (IC50 = 1.9 μM), possibly linked to faster adduct formation with FKBP35 (Figure S2c). As we observed in the NanoBRET assay, SLFb (IC50 = 33.7 μM) performs poorly compared to 1a and 1b (Figure 6b). Despite similar affinity and higher warhead reactivity, 2c also performs poorly in this assay (IC50 = 13 μM, Figure S6a). This discrepancy is likely due to differences in plasma stability as 2c is rapidly degraded by plasma where 1a–c are not (Figure S6b). Additionally, these compounds were not cytotoxic in HEK293T cells across the relevant concentration range indicating that the Plasmodium assay data are not a result of general cytotoxicity (Figure S7).
Figure 6.
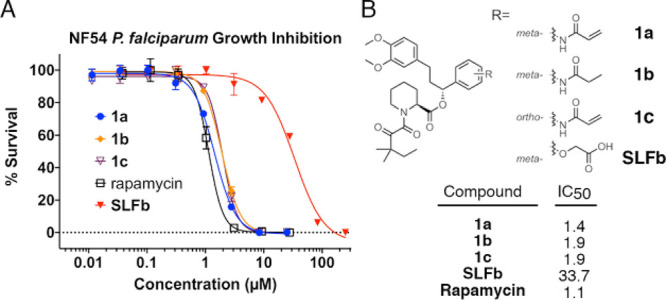
P. falciparum strain NF54 growth-inhibition assay. (A) NF54 parasites were incubated with 1a, 1b, SLFb, or rapamycin in a dose-dependent manner. Proliferation was measured by luminescence after a complete life-cycle and normalized to DMSO (100%) and chloroquine (100 nM, 0%). (B) Structural differences between synthetic ligands and compound IC50s (μM).
While the in vitro data confirm these compounds covalently modify C106 and the NanoBRET assay demonstrates direct target engagement of FKBP35 in a cellular context, additional genetic studies will be necessary to fully establish whether FKBP35 inhibition is responsible for their antiplasmodium activity. Despite this limitation, our results suggest that covalent inhibition of FKBP35 could be an avenue for the development of antimalarial therapeutics. The presence of C106 provides a functional handle for high-throughput screening of covalent fragment libraries for the development of novel chemical matter that could provide improved selectivity between FKBP35 and the human FKBPs. While we predominantly examined simple acrylamides, the slow rate of covalent-bond formation indicates more electrophilic warheads might prove beneficial for additional selectivity and potency. In fact, switching to a more reactive acrylate drastically accelerated the rate of covalent bond formation but at the cost of plasma stability. Additional experiments examining warhead reactivity should be a fruitful endeavor. Alternatively, it is possible that the pipecolic acid core does not provide an optimal foundation upon which to covalently attack C106. Thus, screening efforts to uncover new high affinity FKBP-binding chemotypes optimized for the covalent modification strategy presented here have the potential to create more potent and selective compounds for this highly conserved enzyme. Such compounds will be of significant interest for testing against liver-stage disease.
Acknowledgments
The authors thank Eamon Comer for thoughtful chemistry discussions; Dale Porter for helping secure internal funding sources; and Christian Stephan Meyners and Wei Jiang for assistance with the fluorescence polarization assay.
Glossary
Abbreviations
- FKBP
FK506 binding protein
- FBD
FKBP binding domain
- SLF
synthetic ligand for FKBP
- PPIase
peptidyl prolyl isomerase
- DSC
differential scanning calorimetry
- FP
fluorescence polarization
- nLuc
nanoluciferase
- BRET
bioluminescent resonant energy transfer
Biographies
Thomas Atack received his Ph.D. degree from Indiana University Bloomington where he developed novel iron- and manganese-catalyzed cross-coupling reactions under Prof. Silas Cook. He then transitioned to the Broad Institute of MIT and Harvard where he joined the research group of Dr. William R. Sellers to study therapeutic development for diseases with unmet medical need. His research interests revolve around the design, synthesis, and characterization of novel chemical matter for therapeutic development.
William Sellers is a physician-scientist, Core Institute Member at the Broad Institute and a Professor of Medicine at the Dana-Farber Cancer Institute and Harvard Medical School. He directs a research group focused on utilizing functional genomic technologies to discover new targets and new therapeutics in cancer. He is a co-founder of Civetta Therapeutics. Previously, Dr. Sellers directed cancer drug discovery and early cancer clinical development at the Novartis, led the Cancer Cell Line Encyclopedia project, co-discovered EGFR mutations in lung cancer and served on the National Cancer Advisory Board.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00272.
Synthesis and characterization of all compounds and biophysical, biochemical, and biological assay procedures (PDF)
Author Present Address
& (C.A.B.) Vertex Pharmaceuticals, 490 Arsenal Way, Watertown, Massachusetts 02472, United States.
Author Present Address
^ (P.R.M.) Tango Therapeutics, 100 Binney Street, Suite 700, Cambridge, Massachusetts 02142, United States.
Author Contributions
T.C.A., A.I., and W.R.S. generated the concept and experiments. T.C.A. designed and synthesized all compounds, analyzed data, and prepared the manuscript. D.D.R. prepared and isolated recombinant proteins and ran DSC experiments and mass spec assays. C.A.B. performed cell culture and nanoBRET experiments. C.F.P. and L.Y.E. performed and analyzed parasite viability experiments. P.R.M. performed computational modeling. T.C.A. and H.B.B. performed FP experiments. J.M. ran and with T.C.A. analyzed mass spec experiments. F.P.R. and D.P. performed cell viability experiments. A.I. and J.C.N. facilitated the collaboration. All authors discussed the results and commented on the manuscript. All authors have given approval to the final version of the manuscript.
This research was supported by The Broad Institute (SPARC grant (T.C.A., D.D.R., W.R.S.) and Broad Next10 (J.C.N)) and The Bill and Melinda Gates Foundation (OPP1158199 and OPP1162467 (C.F.A.P., L.Y.E., J.C.N.)).
The authors declare the following competing financial interest(s): W.R.S. is a Board or SAB member and holds equity in Peloton Therapeutics, Ideaya Biosciences, Civetta Therapeutics, and Bluebird and has consulted for Array, Astex, Dynamo Therapeutics, Ipsen, PearlRiver Therapeutics, Sanofi, and Servier and receives research funding from Pfizer Pharmaceuticals and Deerfield Management.
Supplementary Material
References
- WHO . World Malaria Report 2017; 2017.
- Baird J. K. Real-World Therapies and the Problem of Vivax Malaria. N. Engl. J. Med. 2008, 359 (24), 2601–2603. 10.1056/NEJMe0808729. [DOI] [PubMed] [Google Scholar]
- Corey V. C.; Lukens A. K.; Istvan E. S.; Lee M. C. S.; Franco V.; Magistrado P.; Coburn-Flynn O.; Sakata-Kato T.; Fuchs O.; Gnädig N. F.; Goldgof G.; Linares M.; Gomez-Lorenzo M. G.; De Cózar C.; Lafuente-Monasterio M. J.; Prats S.; Meister S.; Tanaseichuk O.; Wree M.; Zhou Y.; Willis P. A.; Gamo F.-J.; Goldberg D. E.; Fidock D. A.; Wirth D. F.; Winzeler E. A. A Broad Analysis of Resistance Development in the Malaria Parasite. Nat. Commun. 2016, 7 (May), 11901. 10.1038/ncomms11901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trape J. F.; Pison G.; Preziosi M. P.; Enel C.; Du Loû A. D.; Delaunay V.; Samb B.; Lagarde E.; Molez J. F.; Simondon F. Impact of Chloroquine Resistance on Malaria Mortality. C. R. Acad. Sci., Ser. III 1998, 321 (8), 689–697. 10.1016/S0764-4469(98)80009-7. [DOI] [PubMed] [Google Scholar]
- Das D.; Phyo A. P.; Tarning J.; Ph D.; Lwin K. M.; Ariey F.; Hanpithakpong W.; Lee S. J.; Ringwald P.; Silamut K.; Herdman T.; An S. S.; Yeung S.; Socheat D.; White N. J. Artemisinin Resistance in Plasmodium Falciparum Malaria. N. Engl. J. Med. 2009, 361 (5), 455–467. 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaghan P.; Fardis M.; Revill W. P.; Bell A. Antimalarial Effects of Macrolactones Related to FK520 (Ascomycin) Are Independent of the Immunosuppressive Properties of the Compounds. J. Infect. Dis. 2005, 191 (July), 1342–1349. 10.1086/428454. [DOI] [PubMed] [Google Scholar]
- Monaghan P.; Bell A. A Plasmodium Falciparum FK506-Binding Protein (FKBP) with Peptidyl-Prolyl Cis-Trans Isomerase and Chaperone Activities. Mol. Biochem. Parasitol. 2005, 139 (2), 185–195. 10.1016/j.molbiopara.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Bell A.; Monaghan P.; Page A. P. Peptidyl-Prolyl Cis–Trans Isomerases (Immunophilins) and Their Roles in Parasite Biochemistry, Host–Parasite Interaction and Antiparasitic Drug Action. Int. J. Parasitol. 2006, 36 (3), 261–276. 10.1016/j.ijpara.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Van Duyne G.; Standaert R.; Karplus P.; Schreiber S.; Clardy J. Atomic Structure of FKBP-FK506, an Immunophilin-Immunosuppressant Complex. Science (Washington, DC, U. S.) 1991, 252 (5007), 839–842. 10.1126/science.1709302. [DOI] [PubMed] [Google Scholar]
- Michnick S. W.; Rosen M. K.; Wandless T. J.; Karplus M.; Schreiber S. L. Solution Structure of FKBP, a Rotamase Enzyme and Receptor for FK506 and Rapamycin. Science (Washington, DC, U. S.) 1991, 252 (5007), 836–839. 10.1126/science.1709301. [DOI] [PubMed] [Google Scholar]
- Banaszynski L. A.; Liu C. W.; Wandless T. J. Characterization of the FKBP-Rapamycin-FRB Ternary Complex. J. Am. Chem. Soc. 2005, 127 (13), 4715–4721. 10.1021/ja043277y. [DOI] [PubMed] [Google Scholar]
- Kolos J. M.; Voll A. M.; Bauder M.; Hausch F.. FKBP Ligands—Where We Are and Where to Go? Front. Pharmacol. 2018, 9 ( (December), ), 10.3389/fphar.2018.01425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunyak B. M.; Gestwicki J. E. Peptidyl-Proline Isomerases (PPIases): Targets for Natural Products and Natural Product-Inspired Compounds. J. Med. Chem. 2016, 59 (21), 9622–9644. 10.1021/acs.jmedchem.6b00411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaghan P.; Leneghan D. B.; Shaw W.; Bell A. The Antimalarial Action of FK506 and Rapamycin: Evidence for a Direct Effect on FK506-Binding Protein PfFKBP35. Parasitology 2017, 144 (7), 869–876. 10.1017/S0031182017000245. [DOI] [PubMed] [Google Scholar]
- Nsanzabana C.; Rosenthal P. J. In Vitro Activity of Antiretroviral Drugs against Plasmodium Falciparum. Antimicrob. Agents Chemother. 2011, 55 (11), 5073–5077. 10.1128/AAC.05130-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun P. D.; Barglow K. T.; Lin Y. M.; Akompong T.; Briesewitz R.; Ray G. T.; Haldar K.; Wandless T. J. A Bifunctional Molecule That Displays Context-Dependent Cellular Activity. J. Am. Chem. Soc. 2003, 125 (25), 7575–7580. 10.1021/ja035176q. [DOI] [PubMed] [Google Scholar]
- Harikishore A.; Leow M. L.; Niang M.; Rajan S.; Pasunooti K. K.; Preiser P. R.; Liu X.; Yoon H. S. Adamantyl Derivative as a Potent Inhibitor of Plasmodium FK506 Binding Protein 35. ACS Med. Chem. Lett. 2013, 4 (11), 1097–1101. 10.1021/ml400306r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harikishore A.; Niang M.; Rajan S.; Preiser P. R.; Yoon H. S. Small Molecule Plasmodium FKBP35 Inhibitor as a Potential Antimalaria Agent. Sci. Rep. 2013, 3, 2501. 10.1038/srep02501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomplun S.; Sippel C.; Hähle A.; Tay D.; Shima K.; Klages A.; Ünal C. M.; Rieß B.; Toh H. T.; Hansen G.; Yoon H. S.; Bracher A.; Preiser P.; Rupp J.; Steinert M.; Hausch F. Chemogenomic Profiling of Human and Microbial FK506-Binding Proteins. J. Med. Chem. 2018, 61 (8), 3660–3673. 10.1021/acs.jmedchem.8b00137. [DOI] [PubMed] [Google Scholar]
- MacDonald C. A.; Boyd R. J. Molecular Docking Study of Macrocycles as Fk506-Binding Protein Inhibitors. J. Mol. Graphics Modell. 2015, 59, 117–122. 10.1016/j.jmgm.2015.04.009. [DOI] [PubMed] [Google Scholar]
- Zhu K.; Borrelli K. W.; Greenwood J. R.; Day T.; Abel R.; Farid R. S.; Harder E. Docking Covalent Inhibitors: A Parameter Free Approach to Pose Prediction and Scoring. J. Chem. Inf. Model. 2014, 54 (7), 1932–1940. 10.1021/ci500118s. [DOI] [PubMed] [Google Scholar]
- Gopalakrishnan R.; Kozany C.; Gaali S.; Kress C.; Hoogeland B.; Bracher A.; Hausch F. Evaluation of Synthetic FK506 Analogues as Ligands for the FK506-Binding Proteins 51 and 52. J. Med. Chem. 2012, 55 (9), 4114–4122. 10.1021/jm201746x. [DOI] [PubMed] [Google Scholar]
- Bianchin A.; Allemand F.; Bell A.; Chubb A. J.; Guichou J. F. Two Crystal Structures of the FK506-Binding Domain of Plasmodium Falciparum FKBP35 in Complex with Rapamycin at High Resolution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2015, 71, 1319–1327. 10.1107/S1399004715006239. [DOI] [PubMed] [Google Scholar]
- Armistead D. M.; Badia M. C.; Deininger D. D.; Duffy J. P.; Saunders J. O.; Tung R. D.; Thomson J. A.; DeCenzo M. T.; Futer O.; Livingston D. J.; Murcko M. A.; Yamashita M. M.; Navia M. A. Design, Synthesis and Structure of Non-Macrocyclic Inhibitors of FKBP12, the Major Binding Protein for the Immunosuppressant FK506. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1995, 51 (4), 522–528. 10.1107/S0907444994014502. [DOI] [PubMed] [Google Scholar]
- Holt D. A.; Luengo J. I.; Yamashita D. S.; Oh H. J.; Konialian A. L.; Yen H. K.; Rozamus L. W.; Brandt M.; Bossard M. J.; Levy M. A.; Eggleston D. S.; Liang J.; Schultz L. W.; Stout T. J.; Clardy J. Design, Synthesis, and Kinetic Evaluation of High-Affinity FKBP Ligands and the X-Ray Crystal Structures of Their Complexes with FKBP12. J. Am. Chem. Soc. 1993, 115 (22), 9925–9938. 10.1021/ja00075a008. [DOI] [Google Scholar]
- Holt D. A.; Konialian-Beck A. L.; Oh H. J.; Yen H. K.; Rozamus L. W.; Krog A. J.; Erhard K. F.; Ortiz E.; Levy M. A.; Brandt M.; Bossard M. J.; Luengo J. I. Structure-Activity Studies of Synthetic FKBP Ligands as Peptidyl-Prolyl Isomerase Inhibitors. Bioorg. Med. Chem. Lett. 1994, 4 (2), 315–320. 10.1016/S0960-894X(01)80135-9. [DOI] [Google Scholar]
- Jackson P. A.; Widen J. C.; Harki D. A.; Brummond K. M. Covalent Modifiers: A Chemical Perspective on the Reactivity of α,β-Unsaturated Carbonyls with Thiols via Hetero-Michael Addition Reactions. J. Med. Chem. 2017, 60 (3), 839–885. 10.1021/acs.jmedchem.6b00788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robers M. B.; Dart M. L.; Woodroofe C. C.; Zimprich C. A.; Kirkland T. A.; Machleidt T.; Kupcho K. R.; Levin S.; Hartnett J. R.; Zimmerman K.; Niles A. L.; Ohana R. F.; Daniels D. L.; Slater M.; Wood M. G.; Cong M.; Cheng Y.-Q.; Wood K. V. Target Engagement and Drug Residence Time Can Be Observed in Living Cells with BRET. Nat. Commun. 2015, 6 (1), 10091. 10.1038/ncomms10091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner J. P.; Hamilton G. S.; Ross D. T.; Valentine H. L.; Guo H.; Connolly M. A.; Liang S.; Ramsey C.; Li J. H. J.; Huang W.; Howorth P.; Soni R.; Fuller M.; Sauer H.; Nowotnik A. C.; Suzdak P. D. Neurotrophic Immunophilin Ligands Stimulate Structural and Functional Recovery in Neurodegenerative Animal Models. Proc. Natl. Acad. Sci. U. S. A. 1997, 94 (5), 2019–2024. 10.1073/pnas.94.5.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper S.; Bilsland J.; Young L.; Bristow L.; Boyce S.; Mason G.; Rigby M.; Hewson L.; Smith D.; O’Donnell R.; O’Connor D.; Hill R. G.; Evans D.; Swain C.; Williams B.; Hefti F. Analysis of the Neurotrophic Effects of GPI-1046 on Neuron Survival and Regeneration in Culture and in Vivo. Neuroscience 1999, 88 (1), 257–267. 10.1016/S0306-4522(98)00221-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.