

Abstract
Biotransformation has a huge impact on the efficacy and safety of drugs. Ultimately the effects of metabolism can be the lynchpin in the discovery and development cycle of a new drug. This article discusses the impact and application of biotransformation of drugs by mammalian systems, microorganisms, and recombinant enzymes, covering active and reactive metabolites, the impact of the gut microbiome on metabolism, and how insights gained from biotransformation studies can influence drug design from the combined perspectives of a CRO specializing in a range of biotransformation techniques and pharma biotransformation scientists. We include a commentary on how biology-driven approaches can complement medicinal chemistry strategies in drug optimization and the in vitro and surrogate systems available to explore and exploit biotransformation.
Keywords: Drug metabolism, biocatalysis, late-stage oxidation, active metabolites, reactive metabolites, gut microbiome
The metabolism of a xenobiotic or drug has the potential of modifying the properties that will enhance or diminish its prospects of advancing through the discovery and development process and ultimately becoming a useful therapeutic. On one hand there are drugs on the market today whose efficacy and/or safety are contingent upon their metabolism, while others owe their failure to reach approval—or worse, their withdrawal following commercialization—to a metabolism-related liability.
Metabolism is typically understood to render a given drug more polar and thus enhance its elimination from the body. While the majority of biotransformations result in inactive metabolites, on some occasions metabolites possess activity against the intended pharmacological target, sometimes with altered specificity for a related receptor or one that is off-target. Furthermore, the pharmacokinetics of an active metabolite may result in improved efficacy, and on rare occasions a metabolite may be the major circulating active agent. On the other hand, a metabolite may be responsible for undesirable outcomes such as idiosyncratic toxicities, for example, which are hypothesized to be directed by the formation of reactive metabolite intermediates. These intermediates are known to covalently modify proteins in the body, resulting in impaired enzyme function or even eliciting an immune response.
Biotransformation in today’s integrated drug discovery environment offers opportunities for application of the insights gained in early stage metabolism studies before selection of the final candidate for development, specifically to inform drug design and expansion of SAR. A recent perspectives paper outlines the key areas that need to be considered: understanding clearance mechanisms that mediate disposition of the compound, modulation or avoidance of metabolic liabilities, identification of any bioactivation to reactive metabolites, and any impact relating to metabolites in safety testing (MIST). This is supported by relevant case studies illustrating Pfizer’s strategy in these key areas.1
The FDA’s MIST guidance, updated in March 2020, provides recommendations to industry on when and how to identify and characterize drug metabolites whose nonclinical toxicity needs to be evaluated, particularly where disproportionate drug metabolites exist.2 Disproportionate metabolites are those identified only in humans or present at higher plasma concentrations in humans than in any of the animal species used during standard nonclinical toxicology testing since metabolic profiles may vary across animal species. The FDA encourages the identification of any differences in drug metabolism between animals used in nonclinical safety assessments and humans as early as possible during the drug development process since the discovery of differences in metabolites late in drug development can cause delays. Human metabolites that can raise a safety concern are those present at greater than 10% of total drug-related exposure measured as area under the curve at steady state.
Notwithstanding the potential toxicity of some metabolites, an early exploration of biotransformation affords an opportunity to ascertain any therapeutic significance of metabolites before the structures of the parent drugs reach the public domain, thereby strengthening protection of intellectual property (IP), and creating backup/second generation development candidates. Where difficult-to-synthesize metabolites are encountered, the efforts and cost of accessing these needs to be balanced against metabolite-related liabilities that may manifest as the compound advances through clinical development. Identification, potency, and metabolic stability testing of metabolites during lead optimization could lead to the discovery of active, more polar entities opening up new chemical space, or derivatives that possess greater metabolic stability, and therefore reduce the risk of drug–drug interactions or dosing challenges due to the action of polymorphic xenobiotic-metabolizing enzymes.
Most medicinal chemistry strategies aim to reduce metabolic intrinsic clearance within chemical series, which has led to a number of challenges, not least the increase in clearance by other mechanisms such as hepatobiliary transport or alternative metabolic pathways.3 The prevalence of low-turnover drugs has also increased, where these strategies have been successful in producing drug scaffolds that are less susceptible to metabolism. It has therefore become necessary to evolve methods for characterizing biotransformation and clearance of these drugs,4 a factor that may be important in long-term therapy where metabolites may accumulate and become relevant in vivo. A recent example in the paper by Gu et al., 2018, highlighted three unusual and late-occurring CYP-derived metabolites of AZD7325 (Figure 1) that were observed as major metabolites only after observation of the profiles at steady state,5 as recommended for MIST analysis by the FDA. In fact, these three metabolites circulated at higher abundance than the parent compound at 48 h after the final dose.
Figure 1.
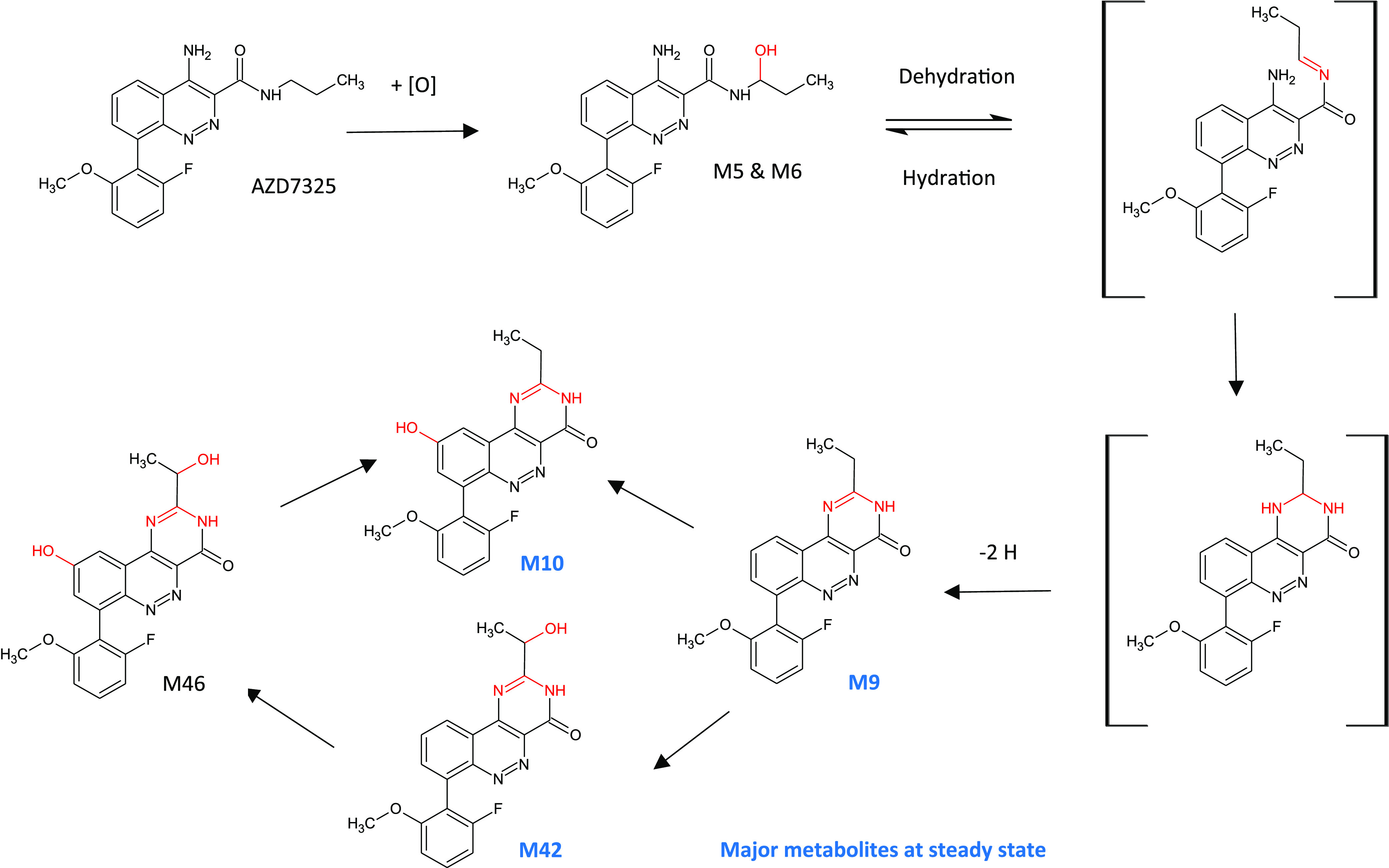
Late-occurring and long-circulating CYP-derived plasma metabolites of AZD7325 formed via metabolic cyclization and aromatization following repeat dosing in humans and preclinical animals. Adapted from Gu et al., 2018,5 under the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/).
Several drugs are known to be directly metabolized by bacteria in the human gut microbiome; however, this phenomenon and the impact thereof has only just begun to be appreciated. Recent publications such as that by Boer et al. on biotransformation and disposition of epacadostat have demonstrated that human gut bacteria can have an impact on the metabolism of drugs.6 In fact, Zimmerman et al. examined the ability of 76 strains of human gut bacteria to metabolize 271 orally administered drugs and found that two-thirds were metabolized by at least one strain. Further, they demonstrated that 30 microbiome-encoded enzymes collectively converted 20 drugs to 59 potential metabolites.7
Active Metabolites
Active metabolites arise from biotransformation of a parent drug compound by one or more xenobiotic-processing enzymes to generate (a) metabolite(s) with relevant pharmacological activity at the original target. The pharmacological effects of many existing drugs owe their effectiveness to their metabolism, as highlighted in the review paper by Fura et al.8 Generally, but not always, it is metabolism by phase I enzymes such as cytochrome P450s (CYPs) that results in active metabolites. However, there are a few examples where glucuronides of drugs also possess pharmacological activity,1,9 such as AMPK activation by acyl glucuronide conjugates of indole-3-carboxylic acid derivatives, where off- and on-target pharmacology studies are warranted.10 Glucuronide metabolites should also be considered as perpetrators of drug–drug interactions through inhibition of efflux transporters or inhibition of CYP enzymes, such as the time-dependent inhibition of CYP2C8 by the acyl glucuronides of clopidogrel and gemfibrozil.11,12
CYPs are adept at oxidizing a wide variety of organic molecules, and as such, their action results in late-stage oxidation of drug scaffolds, a property that can be exploited to generate drug leads that may have superior properties to the parent molecule. In particular, where drug leads suffer from liabilities such as lack of oral bioavailability or exclusive clearance by a polymorphic CYP, metabolism studies may reveal a better candidate. Sometimes biotransformation by CYPs results in a metabolite with an altered specificity at the target. This is possible due to the exquisite selectivity afforded from the stereochemistry created through hydroxylation, or other CYP-mediated mechanisms such as demethylation. Demethylation by CYPs can result in metabolites with improved selectivity and potency such as N-demethylated metabolites of the tricyclic antidepressants including amitriptyline and imipramine.13
Retrospective examination of metabolites can result in some costly missed opportunities in the clinic such as the classic examples of tolterodine and terfenadine, drugs superseded by their active metabolites. Tolterodine was a drug in use for urinary incontinence but suffered from CNS-related side effects and high PK variability due to exclusive metabolism by CYP2D6. Subsequently, fesoterodine, a pro-drug of the active metabolite 5-hydroxymethyltolterodine, was developed to replace tolterodine, which circumvented metabolism by a polymorphic CYP and eliminated the undesirable side effects of the original drug.14 As discussed later in this article, metabolites can be straightforward to access using in vitro biotransformation tools, illustrated in Figure 2 by the production of 5-hydroxymethyltolterodine and three other monohydroxylated derivatives by whole cell microbial biotransformation of tolterodine. In the case of the antihistamine terfenadine, the parent drug was replaced by its active carboxylated metabolite, which possessed none of the cardiotoxicity of the original drug.15
Figure 2.
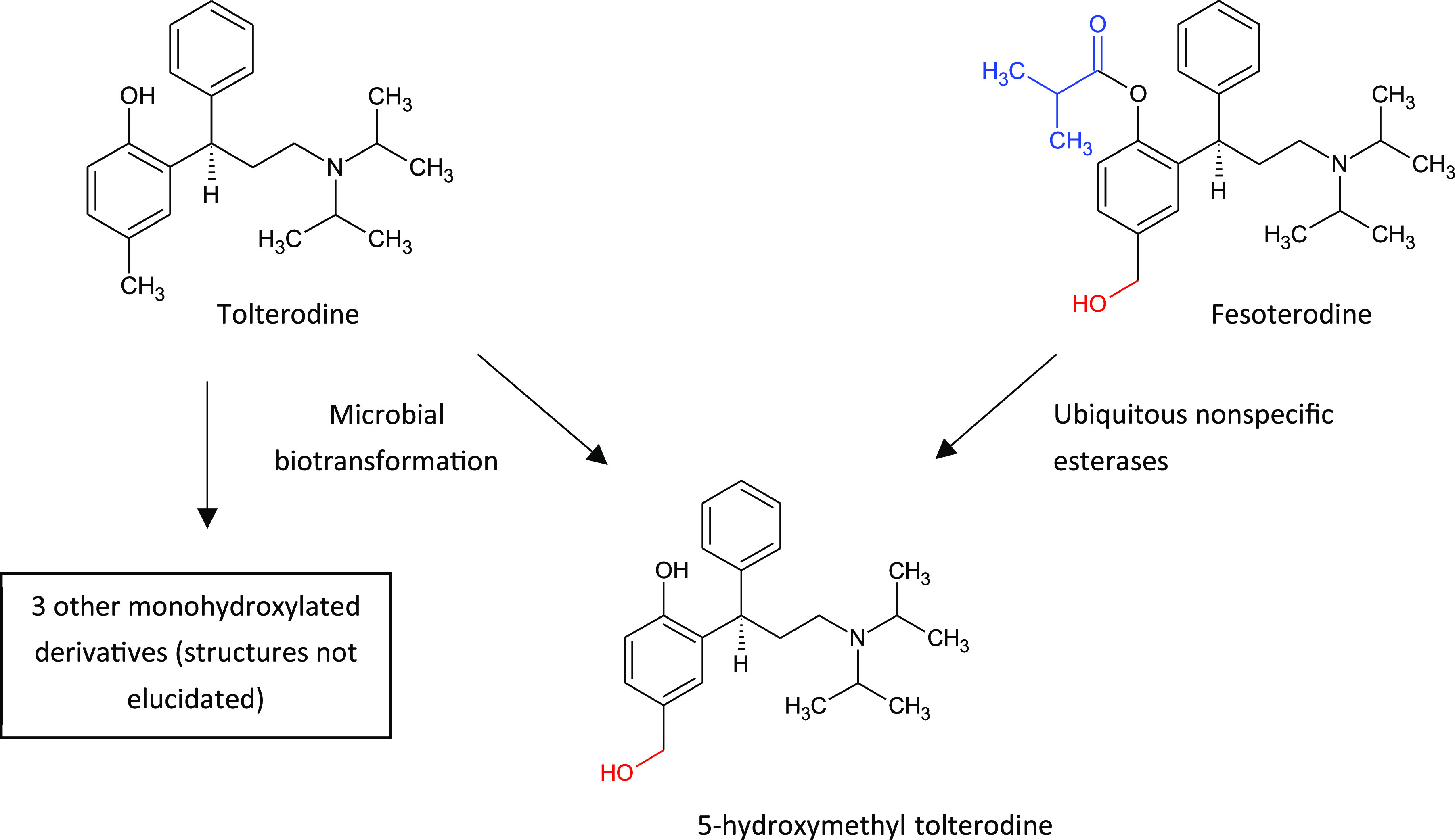
Microbial biotransformation of tolterodine to form the active metabolite 5-hydroxymethyl tolterodine, a pro-drug of which, fesoterodine, replaced the original drug to circumvent metabolism by the polymorphic CYP2D6 and eliminate undesirable side effects.
Other CYP-mediated oxidation reactions can also result in the generation of active metabolites, such as the major pyridine N-oxide (M2) of regorafenib (Stivarga), accumulation of which has a positive impact on cancer survival,16 but where concurrent N-demethylation of M2 to another major metabolite M5 is associated with adverse skin toxicity.17 Regorafenib is also metabolized by glucuronidation, and metabolites may be reduced or hydrolyzed in the gastrointestinal tract by microbial flora, allowing reabsorption of the unconjugated active substance and metabolites by enterohepatic circulation and thus increasing the complexity of its disposition (see Figure 3).18
Figure 3.
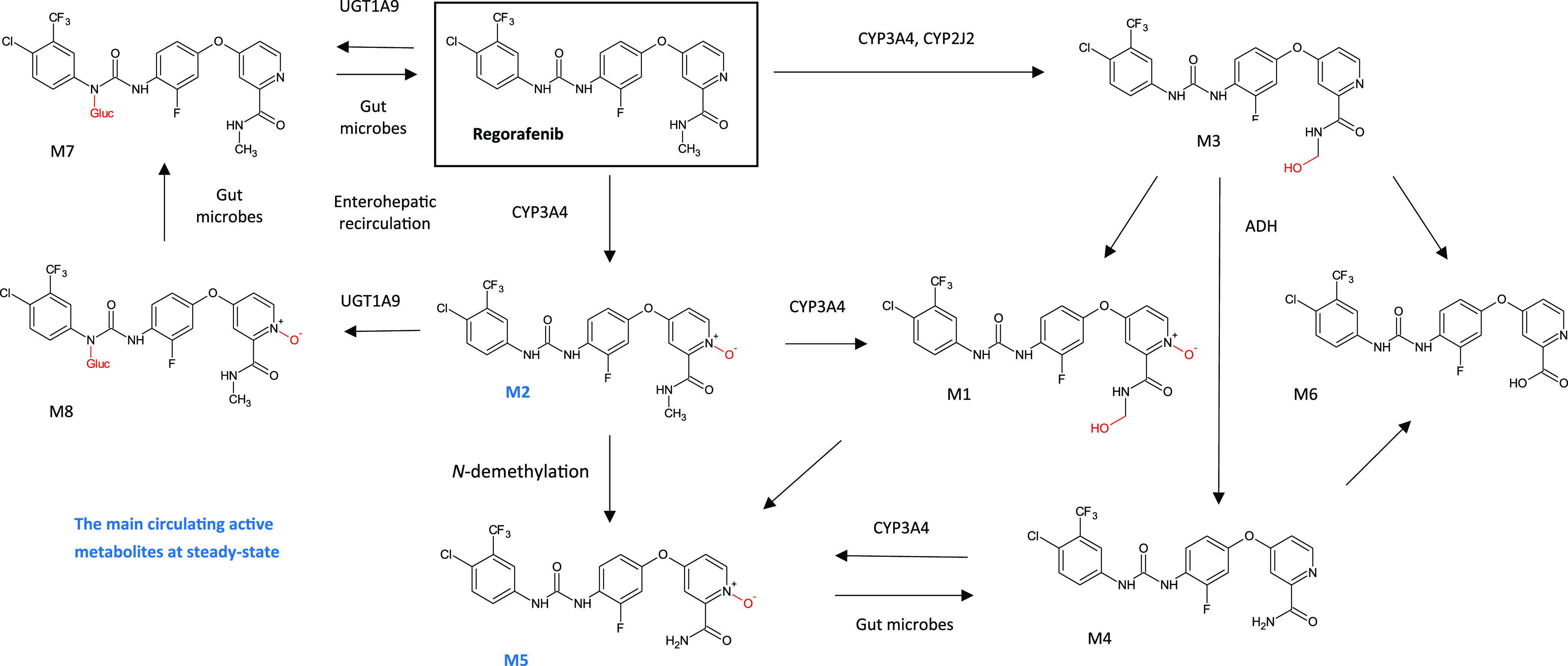
Biotransformation of regorafenib to multiple phase I and phase II metabolites, resulting in an increase in complexity of its disposition. The main circulating active metabolites at steady-state in human plasma are M2 (N-oxide) and M5 (demethylated N-oxide). Adapted from Figure 4 in Gerisch et al., 2018,19 under the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/).
Of course, N-oxide and S-oxide metabolites produced by non-CYP enzymes such as flavin-containing monooxygenases (FMOs) can also have pharmacological activity, and it has been reported that some FMO metabolites can be neurotoxic and hepatotoxic.20
The following case studies highlight some of the interesting consequences of biotransformation in drug discovery where active metabolites are involved. Scientists in several pharma companies advocate a proactive approach to enable identification of pharmacologically active metabolites early on, since a later reactive approach can lead to significant costs and delays at the drug development stage, as highlighted in the recent paper published by scientists at Pfizer.1 In this paper, they also highlight the opportunity to generate both positive and negative SAR from biotransformation of a chemical series.
Differential Selectivity of Major Active Metabolites: The Midostaurin Story
The path to approval of Novartis’ acute myeloid leukemia (AML) drug midostaurin (Rydapt) is a particularly interesting example. Midostaurin is a semisynthetic natural product which inhibits multiple receptor tyrosine kinases, including FLT3 and KIT kinases, preventing the activation of cell-growth signaling pathways. It is approved by the FDA and EMA for both newly diagnosed FLT3-mutated AML and advanced systemic mastocytosis.
The drug is metabolized mainly by CYP3A4 to active metabolites, as illustrated in Figure 4. In plasma, O-demethylation of midostaurin to CGP62221 and hydroxylation to generate epimers of CGP52421 account for 28% and 38% (epimer 1:5.3%, epimer 2:32.7%), respectively, of total exposure of the drug at 96 h. Additionally, these two metabolites attain greater steady-state plasma levels than that of midostaurin itself. It is now apparent that midostaurin and its metabolites target a variety of kinases; however, differential effects are apparent on inhibition of subtypes within each group of kinases,21 resulting in a complex interplay likely to contribute to the efficacy and distinctive effects of the drug.
Figure 4.
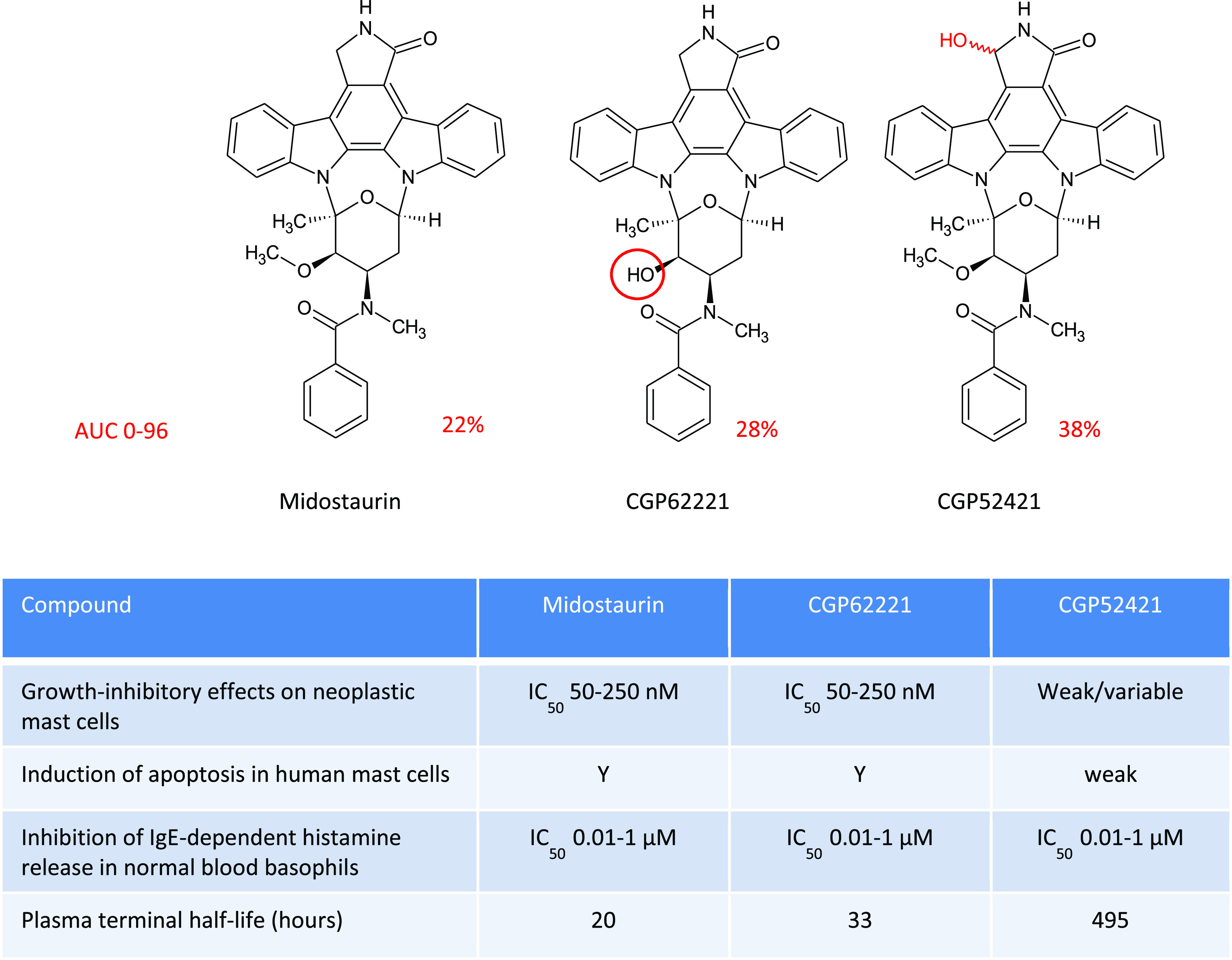
Comparison of the different pharmacological properties of midostaurin and active metabolites CGP62221 and CGP52421 produced by CYP3A4.22,23
Such effects on selectivity can offer opportunities that are likely missed for metabolites that are less significant and thus not investigated at an early stage. The information can be used to identify where metabolic blocking could be advantageous and where it would be useful to exploit the products of biotransformation, as well as add to SAR data to broaden and protect IP.
Exploiting and Protecting Metabolites of Drugs: The Valsartan Story
Novartis’ angiotensin II receptor blocker (ARB) drug valsartan (Diovan) is used to treat hypertension and heart failure. Valsartan is minimally metabolized by CYP2C9 in human liver microsomes with one main metabolite detected, 4-hydroxyvaleryl-valsartan, and which accounts for 9% of the circulating dose in humans. This metabolite was found to have no significant ARB activity; however, further investigation revealed potent inhibition of platelet aggregation. This is significant given platelet aggregation plays a key role in the pathogenesis of coronary and cerebrovascular occlusions. The finding was considered of sufficient impact for Novartis to patent use of 4-hydroxyvaleryl-valsartan for treatment of coronary disease mediated by platelet aggregation.24 Its inhibition of platelet aggregation is exerted through a different mechanism and was an unexpected discovery,25 only made possible through interrogation of the activities of 4-hydroxyvaleryl-valsartan, despite it not being a major metabolite. Metabolites and other derivatives can be accessed via several biological means including mammalian microsomes, microbial biotransformation, and through recombinant enzymes such as illustrated in Figure 5.
Figure 5.
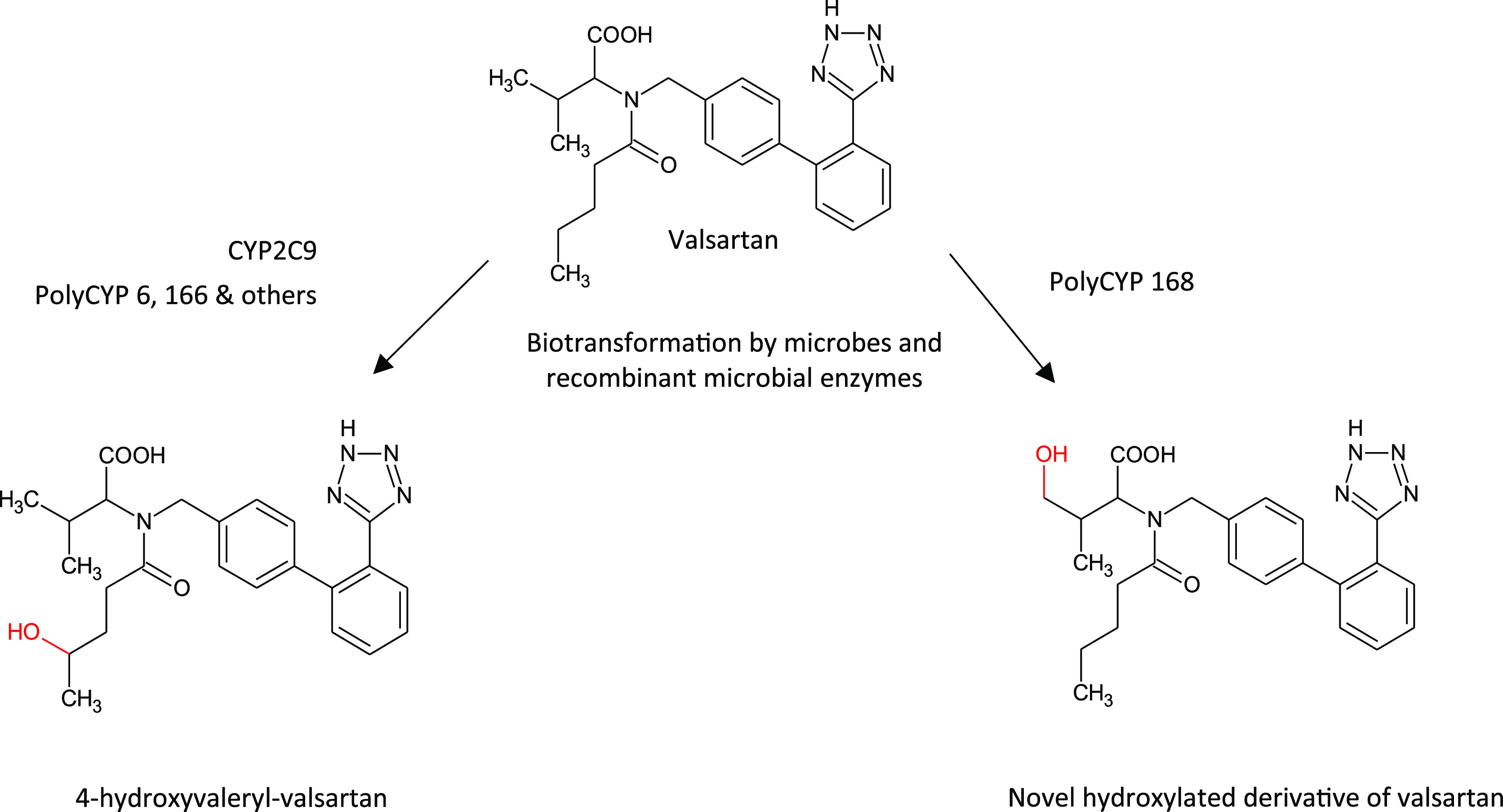
Biotransformation of the drug valsartan by microbial whole cell biotransformation and recombinant microbial enzymes to yield hydroxylated metabolites, one of which is the product of human liver microsomal biotransformation of valsartan by CYP2C9.
Learning from Metabolism: The Ezetimibe Story
Learning from biotransformation studies provides insight into which metabolic hot spots should be blocked or the metabolism redirected, and which should be harnessed for maximum benefit.
Merck’s ezetimibe (Zetia) is a textbook example of using metabolite-informed drug design where structural modifications to the drug led to blocking of “unprofitable sites” and premetabolizing “profitable” sites,26 as illustrated in Figure 6. Interestingly, ezetimibe is a good example of a drug metabolized to a pharmacologically active O-glucuronide, which is as potent an inhibitor of intestinal cholesterol absorption as the parent compound.27 The glucuronide is subject to transporter-mediated enterohepatic cycling extending the residence time in the gut.
Figure 6.
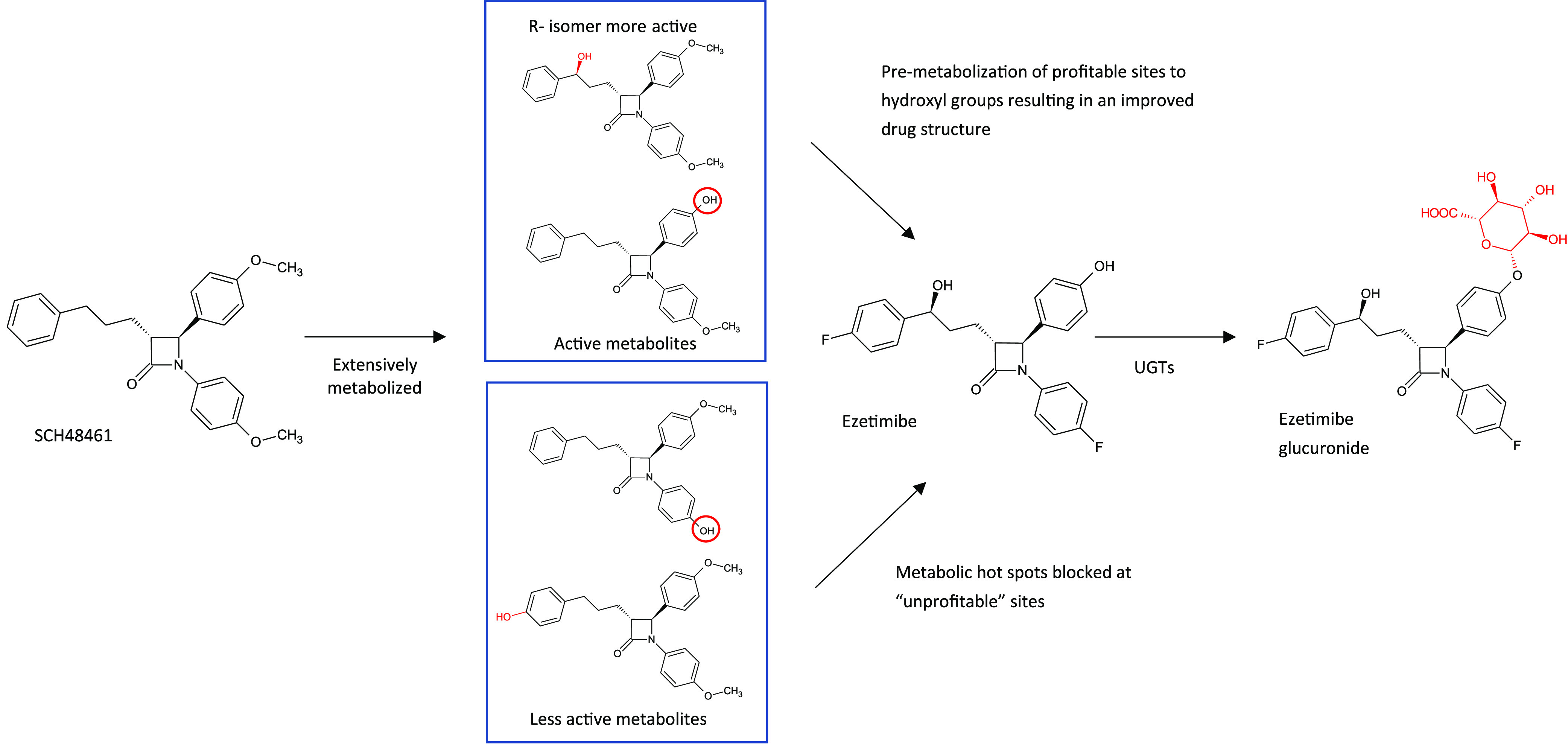
Metabolism inspired design of ezetimibe from SCH48461, and ezetimibe’s major metabolic route to a major O-glucuronide by UGTs 1A1, 1A3, and 2B15.26
Exploiting Biotransformation through Pro-drugs and Deuteration: The Development of Valbenazine and Deuterotetrabenazine
A more recent illustration of the use of insights gained from biotransformation studies is the development of the tardive dyskinesia (TD) drugs, Neurocrine’s valbenazine (Ingrezza) and Teva’s deuterotetrabenazine (Austedo), shown in Figure 7. Valbenazine and deuterotetrabenazine provide a unique mechanism of action for the treatment of TD by inhibition of vesicular monoamine transporter Type 2 (VMAT-2).28 They are the first treatments to be FDA-approved for this condition, and their efficacy relies on the presence of active metabolites observed during biotransformation of the drug tetrabenazine, use of which results in adverse side effects.
Figure 7.
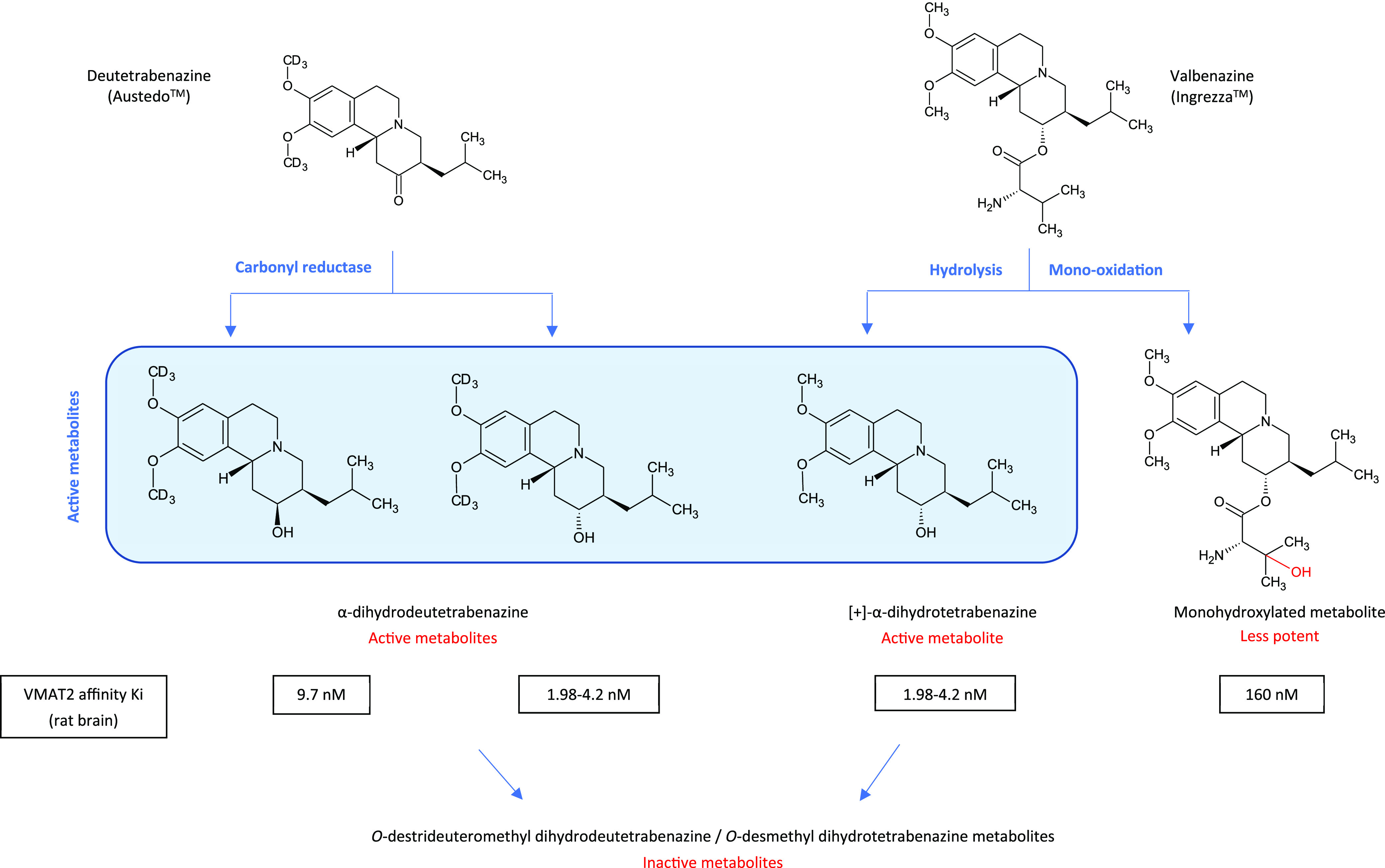
Metabolism of drugs approved to treat tardive dyskinesia through inhibition of VMAT2 (vesicular monoamine 2 transporter). Austedo uses a deuterated form of tetrabenazine (3R, 11bR enantiomer shown) to improve exposure of hydroxylated active metabolites. In contrast Ingrezza uses a prodrug approach involving hydrolysis of the valine ester to a specific hydroxylated active metabolite.28,29
Each enantiomer of tetrabenazine gives rise to two isomers of dihydrotetrabenazine, resulting in four isomeric metabolites. Of these, only those metabolites derived from α-tetrabenazine are active VMAT-2 inhibitors and contribute to the therapeutic effects of the drug. The two dihydrotetrabenazine derivatives of β-tetrabenazine show high-affinity binding to other CNS-relevant targets and may be responsible for the off-target side effects such as sedation and parkinsonism observed during chronic tetrabenazine intake.
The development of deuterotetrabenazine successfully utilizes deuteration to cause a slowdown in metabolism of the methoxy groups on the aryl ring. It is prolongation of the active alpha dihydrodeutetrabenazine metabolites that are formed following the action of carbonyl reductase on the parent drug that give the deuterated drug a dosing advantage.
The application of deuteration to influence metabolism is a technique that needs to be considered carefully, since Cerny et al. reported that it has little effect on slowing or preventing metabolism via CYP enzymes due to breakage of the C–H bond not being the rate limiting step in the CYP catalytic cycle. Deuteration can however result in the alteration of the metabolic profile without a decrease in metabolic rate through metabolic switching.1
In contrast, valbenazine is a pro-drug, metabolized by hydrolysis of the valine ester to the most active metabolite, [+]-alpha-dihydrotetrabenazine ([+]-α-HTBZ) and a mono-oxidized metabolite (NBI-136110), the two most abundant circulating metabolites of valbenazine.29 [+]-α-HTBZ is then further metabolized to the inactive O-desmethyl metabolite by CYP2D6. The prodrug is more slowly hydrolyzed to the specific active metabolite.
Biotransformation Tales Still Unfolding: The Ketamine Story
Ketamine has been introduced as a fast-acting drug for treatment-resistant depression through Janssen’s launch of esketamine (Spravato), the S enantiomer of ketamine. While ketamine is a known antagonist of the N-methyl-d-aspartate receptor (NMDAR), its precise mechanism of action is unknown due to the numerous effects at other receptor sites.
Ketamine is extensively metabolized to a number of major circulating active metabolites, major pathways of which are illustrated in Figure 8. Intriguingly, some of the metabolites of ketamine, produced by hydroxylation of the cyclohexyl ring at the C6 position, exert NMDAR inhibition-independent antidepressant actions.30 In particular, treatment of rodents with (2R,6R)-hydroxynorketamine (HNK) resulted in an antidepressant effect without binding to the NMDAR and was devoid of any of the sensory dissociative side-effects. Recent findings reveal that (2R,6R)-HNK acts in an mGlu2 (group II metabotropic glutamate receptors subtype 2) receptor-dependent manner,31 which is interesting given that inhibitors of group II mGlu receptors have gained interest for their actions to exert rapid antidepressant effects. Of particular interest is the possible use of these metabolites in the treatment of depression and the potential for opening new paths for investigating their role in other brain disorders.
Figure 8.
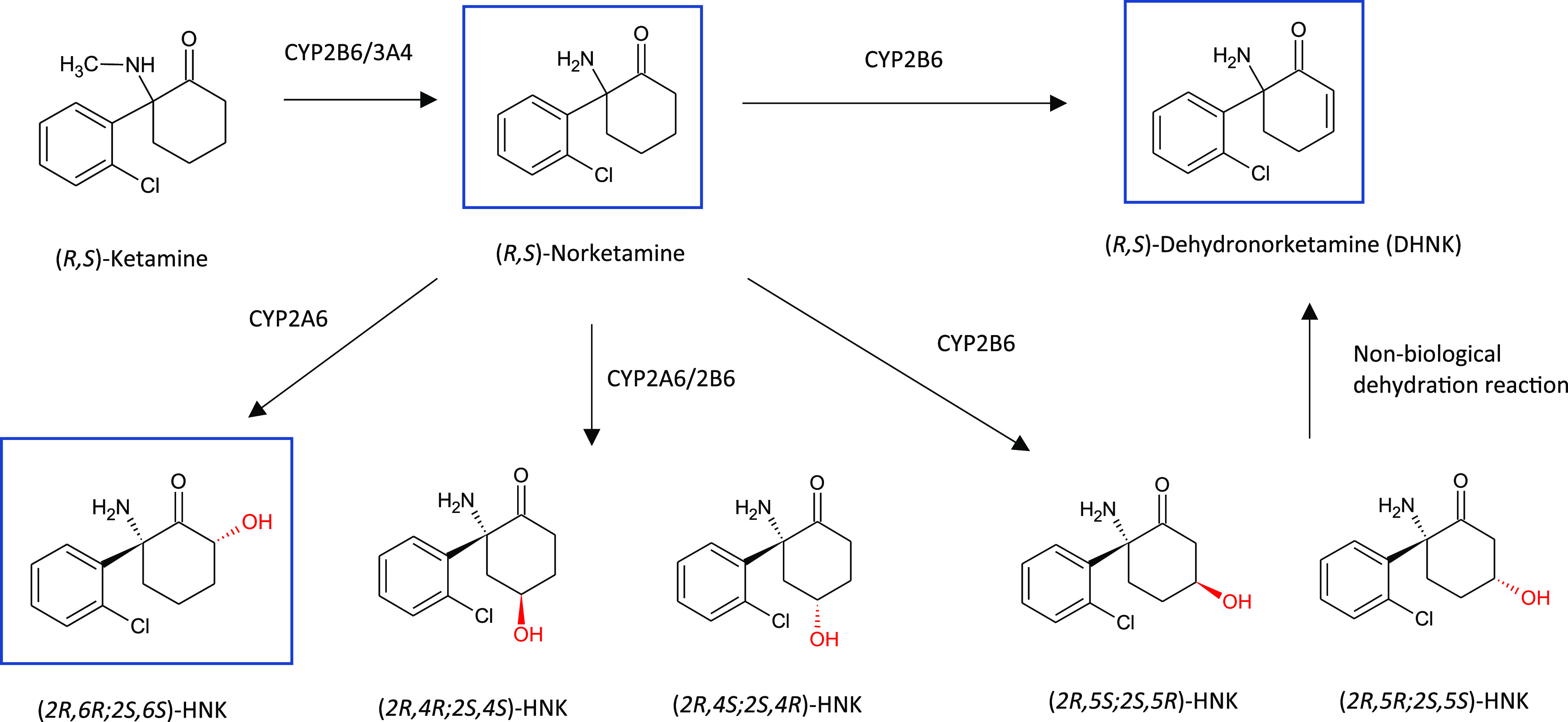
Major metabolic pathways of ketamine (HNK= Hydroxynorketamine). Major circulating metabolites of ketamine in plasma are boxed in blue. Adapted from Figure 1 with permission from Zanos P. et al. Ketamine and ketamine metabolite pharmacology. Pharmacol. Rev.2018, 70 (3), 630. Copyright 2018 ASPET.30
Drug Design and Metabolic Stability
One consequence of the current practice of selection of molecules during the discovery phase that have high metabolic stability in liver microsome systems is that alternative non-CYP pathways are now becoming more prevalent as clearance mechanisms, such as those mediated by aldehyde oxidase, and which can result in some unwelcome surprises in the clinic due to unexpectedly high clearance of the drug.32,33 Aldehyde oxidase (AO) is not only present in the liver but is extensively expressed extrahepatically with wide differences in expression across animal species. To alleviate concerns about differences in AO enzyme expression between species used for preclinical assessment and humans, one recent study examining two AO-mediated metabolites of a methylquinoline-containing drug candidate used early in vitro and in vivo models to ensure adequate animal species coverage to predict relatively high exposure of one of the AO metabolites in human plasma.34
AO has broad substrate specificity and is particularly adept at oxidizing azaheterocycles, which are now commonly used in medicinal chemistry programs to lower lipophilicity. However, scientists at UCB propose that AO should not be perceived as an enemy of medicinal chemistry, as oxidized metabolites formed by AO are reported as metabolically stable and, if pharmacologically active, may provide valuable inspiration for novel scaffolds and improved drug design. Their recommendation is that AO-mediated metabolism is best approached at the drug design stage to control it, by either mitigation (i.e., stop, decrease, diversify) or utilization (e.g., design of a prodrug or a modified scaffold) of the enzymatic reaction.35
Another consequence of the increasing use of nitrogen-containing heterocycles in drugs (including nitrogen containing fused ring systems) is the potential rise in N-glucuronidation as a clearance mechanism.36 It has been our observation from projects we have conducted for various pharma companies over recent years that the need to access major N-glucuronide metabolites is a rising trend. Accessing N-glucuronide metabolites is not always straightforward; however, a number of examples exist of successful biotransformations of drugs to the corresponding N-glucuronide using microbes. A recent example involving biotransformation of molidustat using Streptomyces griseochromogenes was successfully scaled up in fermenters to generate 1.2 g of the analytically pure N-glucuronide, illustrated in Figure 9.37
Figure 9.

Production of the major N-glucuronide metabolite of moludistat by microbial fermentation.37
Other biotransformation pathways can also become more prevalent when drug design programs steer away from CYP-based mechanisms, such as the hydrolytic ring opening of substituted oxetane rings to diols by epoxide hydrolase (EH), the extent of which is influenced by even slight structural changes.38 Oxetane-containing ring systems are increasingly used in drug design to modulate drug-like properties, and thus the recommendation is that metabolism by EH should be assessed early in the design process to understand the complete metabolic fate of these compounds. An oxetane ring has also been incorporated in the design of γ-secretase inhibitors where a cycloalkane moiety was ultimately replaced with the smaller oxetane ring structure to reduce the extent of oxidative metabolism by CYPs.39
Biology-Driven Late-Stage Oxidation and Application to Medicinal Chemistry
As we have seen in the previous examples, biotransformation of drugs has led to intriguing findings. In particular, oxidation of drug leads by hydroxylation, subsequent oxidation to carboxyls, or dealkylation can lead to derivatives that have superior properties to the parent molecule. These derivatives may or may not be the major metabolites observed in the in vitro/in vivo system, as sometimes the minor metabolites or derivatives that might otherwise be overlooked, or not made in the specific in vitro system employed, may possess the improved properties.
Addition of a hydroxyl group can have a significant impact on binding affinity and selectivity and influence pharmacokinetic (PK) properties. Pfizer’s late stage oxidation program looks to explore the SAR around lead compounds, dialing in increased solubility and improving PK properties where needed. This biologically driven approach has been used to identify oxidized derivatives that not only possess reduced lipophilicity but also address liabilities possessed by some lead molecules such as metabolic stability. In the work published by Stepan et al., one of Pfizer’s PDE2 (phosphodiesterase 2) inhibitor lead compounds suffered from exclusive clearance by CYP3A4.40 Through screening of the parent compound with liver microsomes sourced from multiple species, several hydroxylated derivatives were produced, all of which maintained potency and also possessed higher metabolic stability, illustrated in Figure 10. Production of hundreds of milligrams of one of these hydroxylated derivatives was scaled up for ADME characterization using a whole cell biotransformation reaction utilizing a streptomycete bacterium, a process subsequently replaced by an 8-step chemical synthesis in order to provide multigram amounts for in vivo studies. The hydroxylated candidate was found to possess exquisite drug-like properties including a low oxidative turnover, increased renal excretion, and good selectivity against other phosphodiesterases.
Figure 10.
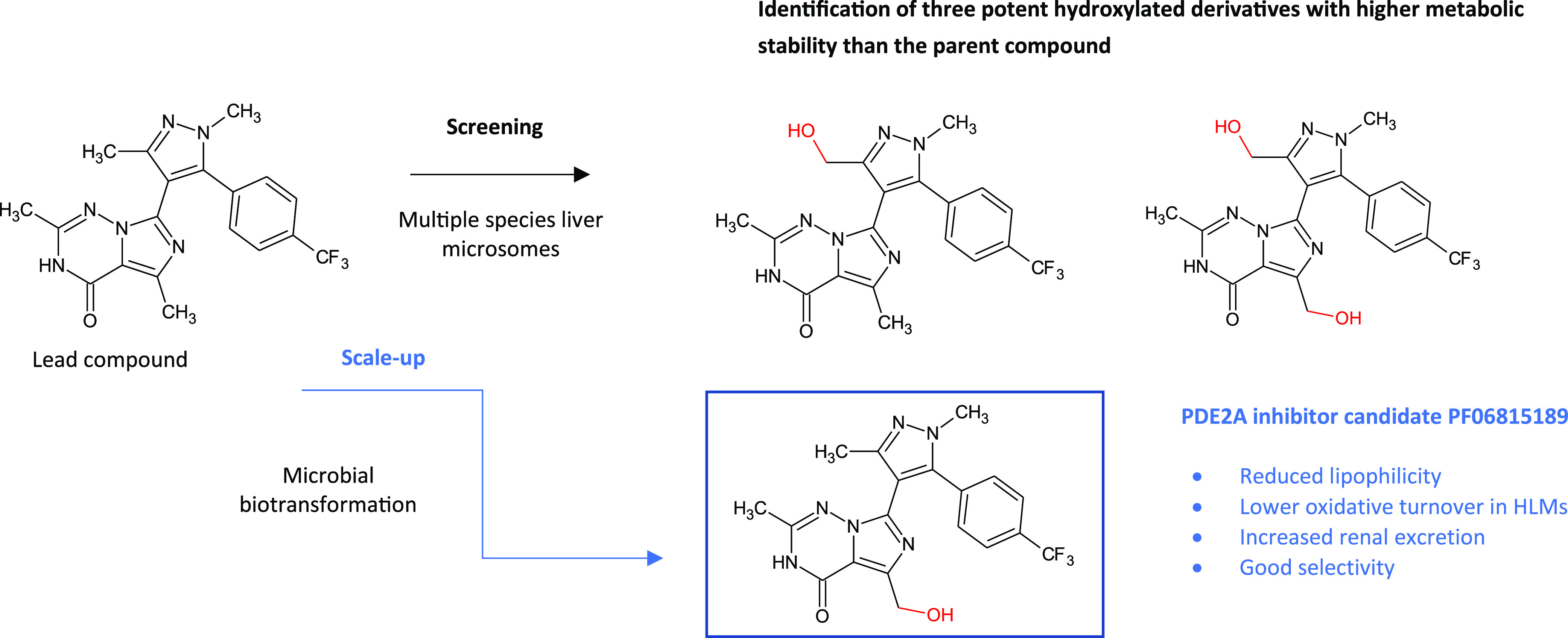
Late stage oxidation of a phosphodiesterase 2A (PDE2) inhibitor lead compound using a biotransformation approach to generate lead candidate PF06815189, which possessed “exquisite drug-like attributes”.40
Similarly, introduction of a hydroxyl group at the tert-butyl position of a Gilead kinase inhibitor using microbial biotransformation resulted in not only increased polarity but also an unanticipated 20-fold boost in potency such that the ligand-lipophilicity efficiency (LLE) was increased by 2.6 units to a more drug-like 6.0 (see Figure 11).41 This oxidized derivative was not a compound that would ordinarily have been synthesized, thus biotransformation provided an unexpected and valuable insight.
Figure 11.

Late-stage oxidation of a protein kinase inhibitor through microbial biotransformation resulting in a hydroxylated derivative with improved potency, as well as achieving the desired reduction in lipophilicity.
Interactions of hydroxyl groups are particularly interesting as they can mimic water in the binding site, resulting in dramatic changes in binding affinity, such as that seen with a hydroxylated derivative of the drug Arbidol (umifenovir). Arbidol is an “old” broad spectrum antiviral drug licensed in China and Russia with activity against several influenza strains. It binds in a hydrophobic cavity at the interface of two protomers of hemeagglutinin (HA), primarily making hydrophobic interactions but also inducing some conformational rearrangements to form a network of salt bridges. The drug functions as a “molecular glue”, stabilizing the prefusion conformation of a HA and thereby inhibiting virus-host cell fusion.42 At the bottom of this binding pocket sits an ordered water molecule. Recently, scientists displaced this ordered water molecule with a hydroxyl group added at the meta position of the thiophenol ring of Arbidol, and in so doing increased the binding affinity over a thousand-fold for some influenza strains.43
Addition of hydroxyl groups can also dramatically impact PK properties as exemplified by the type 2 diabetes drug saxagliptin.44 Addition of a hydroxyl group at the bridgehead position of a precursor to saxagliptin had a huge effective on PK properties, resulting in a derivative that was orally bioavailable and where any inhibition of CYP3A4 had been abolished. Interestingly the inspiration for the hydroxylation derived from some metabolism studies in which hydroxylation of a related series of compounds had also resulted in an improvement in oral bioavailability and where an active metabolite had been implicated.45 Accordingly, it is not always the metabolites themselves which result in an improved candidate, but rather the learning from metabolism studies that can provide an inspiration for a chemical change that results in profound benefits.
Similarly, oxidative biotransformation by non-CYP enzymes can provide inspiration in drug design, as illustrated in the paper by Ouvry et al., where minor structural changes were found to have a dramatic impact on properties of a series of mTOR inhibitors.46 Low systemic exposure was required for the topical drug candidate, which was particularly susceptible to phase II metabolism. However, a proposed aldehyde oxidase metabolite of one derivative turned out to be active and possessed increased selectivity, providing inspiration for a new direction to identify improved derivatives.
Late-stage hydroxylation is a key tactic highlighted by Bostrom et al. where addition of hydroxyl groups to a drug lead can result in improved activity, selectivity, solubility, and lipophilicity through replacing or mimicking water in binding site interactions.41 Fortunately, the location of water molecules in binding sites can be predicted by water prediction software, where good crystallographic data exists. However, it can be difficult to predict which water-mediated protein–ligand interactions are going to be beneficial, since Nittinger et al. reported that predicted binding free energies correlate poorly with the observed changes in inhibitor potency when solvent atoms were displaced by chemical changes in closely related compounds.47 Thus, empirical methods are needed to fully evaluate the effect of displacement and mimicking of water by hydroxyl and carboxyl groups, relevant also for targets where there is no or only poor-quality crystallographic data available.
In Vitro and Surrogate Biological Systems for Biotransformation
Many drug metabolites have complex structures, and synthesis of phase 1 metabolites needs to address regioisomeric and stereoisomeric considerations, with that of phase 2 metabolites requiring additional considerations for conjugation reactions. The most versatile systems for metabolite generation therefore involve biocatalysis by subcellular fractions such as recombinant enzymes, microbial whole cell biotransformation, and liver/S9 microsome systems.8 These approaches all have advantages and disadvantages, and pragmatic choices need to be made. If the site of metabolism is known, appropriate subcellular fractions such as liver S9 or microsome preparations can be used, but if they do produce the desired metabolite, scalability for identification and evaluation can be an issue, not least because of the expense of the cofactors required. If appropriate, whole cell microbial biotransformation is readily scalable to produce milligram to gram quantities at reasonable cost because the microbes utilize their own cofactors, but the time to deliver the desired metabolite can be longer (several weeks) and not all mammalian metabolites are easily accessible via the microbial route. Recombinant enzymes of mammalian or microbial origin offer a potentially quicker alternative, but extraneous cofactors need to be addressed as a cost-consideration. All of these biological approaches require an empirical screening step to see whether they can produce the target metabolite in the required time/quantity. If time is of the essence then a parallel approach using multiple technologies is recommended. It should be noted that significant advances are being made in chemical synthetic alternatives for oxidative production of drug metabolites such as the heme-mimetic systems for late stage aliphatic hydroxylations of highly functionalized molecules,48 and aromatic hydroxylations that can subsequently allow deoxyfluorination to block metabolic sites,49 but these are outside the scope of this article.
Recombinant Enzymes
Recombinant cytochrome P450 enzyme systems, derived from either mammalian or microbial origins, are convenient tools for producing scalable quantities of the products of biotransformation for evaluation, catalyzing a variety of metabolic reactions including hydroxylation, epoxidation, N- and O-dealkylation and Baeyer–Villiger oxidation.
Recombinant human CYPs have been successfully employed for generating metabolites by several pharmaceutical companies.50 Although some have reportedly shown low catalytic activity and poor stability for industrial use51 and thus may be less suited to larger scale preparation, a review by Winkler et al. describes the synthesis of drug metabolites in hundreds of milligram to gram scale using recombinant CYPs expressed in E. coli, Schizosaccharomyces pombe, and Sf21 insect cells, including the production of the main human metabolite of sagopilone in gram amounts by a genetically engineered E. coli expressing human CYP2C19 and cytochrome P450 reductase (see Figure 12a).52 In many cases microbial oxygenases have high turnover and good operational stability53 and bacterial CYPs usually have much higher specific activities50 and thus are more suitable biocatalysts for obtaining scalable quantities of derivatives. Recombinant microbial-derived CYP kits are convenient for determining susceptibility of lead compounds to biotransformation by oxidation, such as PolyCYPs enzymes, composed of a diverse set of recombinant CYPs cloned from actinomycete strains with known oxidizing ability, and expressed in E. coli together with the appropriate redox partners,54 and MicroCyps, derived from a series of mutants of the fatty acid hydroxylase CYP from Bacillus megaterium (BM3). Both systems enable scalable synthesis of CYP-derived human metabolites for definitive metabolite identification and pharmacological testing. A selection of human CYP derived metabolites produced by a small number of PolyCYPs enzymes is illustrated in Figure 12b to illustrate some of the oxidations possible with microbial CYPs. The ability of microbial CYPs to mimic human CYPs and capture nearly all of the mammalian P450 scope of reactivity is exemplified through the production of 12 of 13 known metabolites of three drug compounds and 7 new metabolites by a 120 member BM3 mutant panel, demonstrating considerable versatility and activating C–H bonds of varying strength.55
Figure 12.
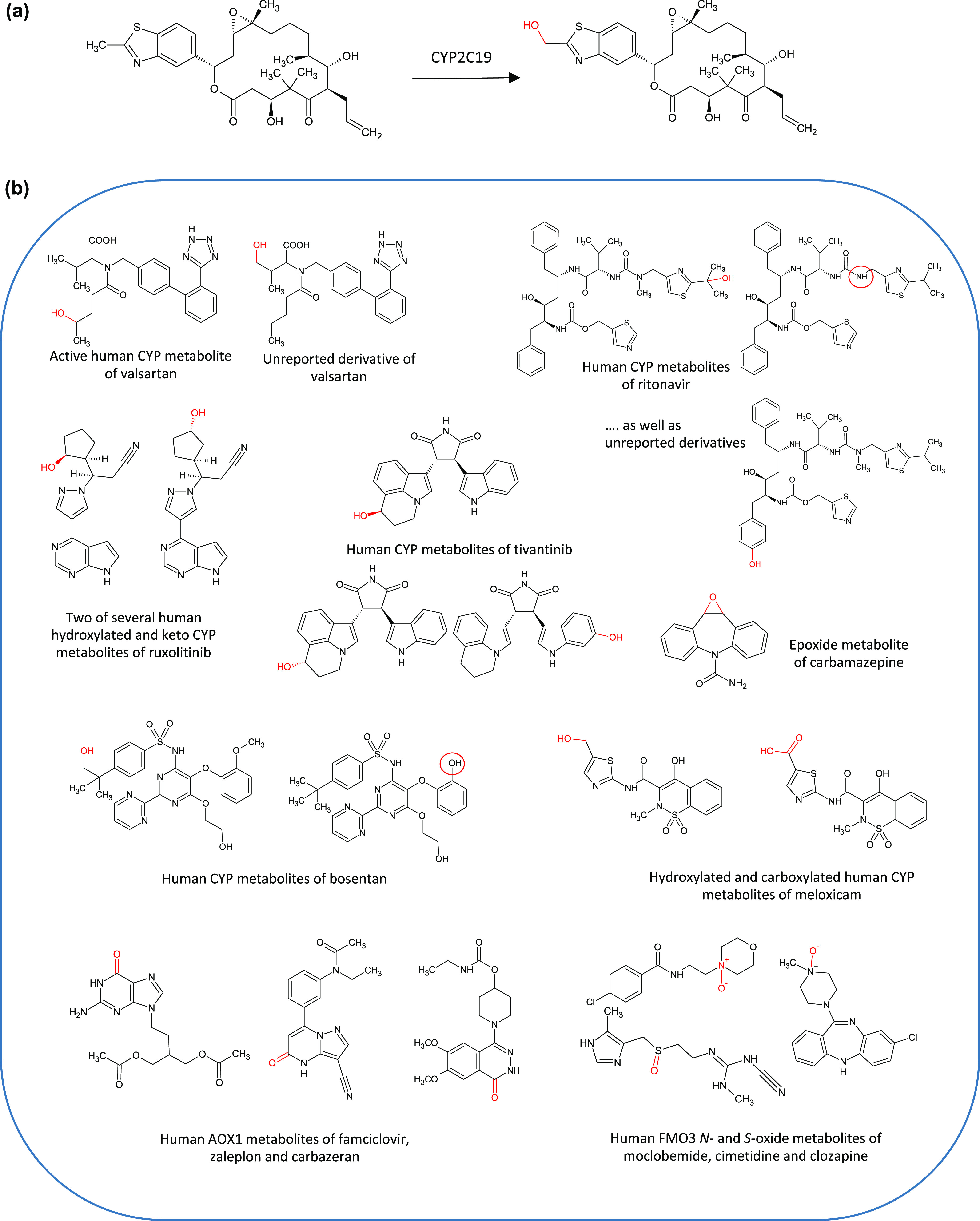
(a) Gram scale production of the main human metabolite of the epothilone analogue, sagopilone, by human CYP2C19 expressed in E. coli. A 100 L scale biotransformation of sagopilone resulted in 5 g of metabolite after 23 h at an isolated yield of 54%. (b) Selection of human CYP, AO, and FMO3 derived metabolites and other oxidized analogues synthesized by recombinant enzymes in the PolyCYPs+ screening kit. Reactions catalyzed by recombinant microbial PolyCYPs isoforms include aliphatic and aromatic hydroxylation, carboxylation, epoxidation, and dealkylation.
A wide range in catalytic ability is achieved by PolyCYPs through their possession of a high degree of native diversity in their CYP domains. Substrate promiscuity has been broadened by engineering point mutations in some PolyCYP isoforms, with the potential for further property optimization for specific reactions of interest.56 In addition, CYPs can be engineered to improve regio- and stereospecificity of hydroxylation reactions such as that undertaken with BM3 mutants using directed evolution.57
In addition to their utility in making human metabolites, bacterial CYP enzymes are also employed as a complement to organic chemical synthesis in the optimization of discovery-stage compounds. Sets of enzymes are used to create oxidized derivatives, providing an empirical approach to access multiple derivatives in parallel. A process has thus been designed to screen compounds directly from compound libraries at relevant concentrations, to generate products of interest as determined by LC-MS analysis, such as the PolarExplorer platform.58 Fractions generated from these reactions can be screened in the relevant bioassays to prioritize work up of products that retain bioactivity. Selected reactions can then be readily scaled up using the specific enzyme isoforms proven to produce oxidized derivatives of interest. Subsequently, large quantities of specific hydroxylated derivatives of interest may be chemically synthesized for in vivo work. In the case of oxidation of the antiviral drug ritonavir by PolyCYPs enzymes, several oxidized derivatives are produced by different PolyCYPs isoforms including the main metabolite of ritonavir, ε-hydroxy-N-methylisopropylthiazoloylmethyl-ritonavir (M2), as illustrated in Figure 13.
Figure 13.

Biotransformation of the HIV-1 protease inhibitor ritonavir to oxidized metabolites by recombinant microbial CYP enzymes. In humans the main metabolite formed is ε-HO-N-methylisopropylthiazoloylmethyl-ritonavir (M2) by the action of CYP3A4 and CYP2D6.
Microbial Biotransformation
Microbial biotransformation is a technique that is effective both for metabolite production and for late-stage diversification. It was first reported in 1974 as a method to produce human metabolites59 and its use was commonplace in many larger pharmaceutical companies, but in-house capabilities have waned with the decline in natural products expertise since the late 1990s. Microorganisms possess enzymes that are homologous to mammalian phase I and II xenobiotic-metabolizing enzymes, such as cytochrome P450 monooxygenases, UDP-glucuronosyltransferases, aryl sulfotransferases, and glutathione S-transferases, and they are also capable of producing metabolites that are formed by other oxidative mechanisms in humans such as aldehyde oxidase and flavin-containing monooxygenases.60
The fact that microbes do not require extraneous cofactors to generate metabolites is advantageous, since these are generated by the microorganism itself in whole cell culture. Hence microbial biotransformation is often a cost-effective way to access metabolites that cannot be easily synthesized by chemical means. In addition, the presence of multiple enzymes during the biotransformation reaction permits the possibility of formation of metabolites resulting from more than one xenobiotic pathway, such as the glucuronide, CYP metabolite, and gut metabolite of epacadostat produced in “one pot” by a single micro-organism.6 In addition, secondary metabolites arising from sequential biotransformation steps can be accessed. In the example below, application of microbial biotransformation enabled production of the aldehyde oxidase mediated hydroxylated metabolite of samotolisib, as well as the N-oxide.61
Identification of AO Metabolism during Discovery: The Story of Samotolisib (LY3023414)
Samotolisib (LY3023414), a dual PI3k/mTOR kinase inhibitor, was recently discontinued during phase 2 studies for metastatic prostate cancer for reasons unrelated to its metabolism. Biotransformation results in six metabolites formed by aldehyde oxidase and CYP enzymes through hydroxylation, N-demethylation, and N-oxidation (see Figure 14). Aldehyde oxidase was implicated and understood early on and found to contribute approximately half the clearance of the drug, facilitated by the production and structure elucidation of the AO-mediated hydroxylated metabolite (M2) and the N-oxide (M4) by the action of a single microbe on the parent molecule. The CYP-mediated metabolite had already been chemically synthesized separately.61
Figure 14.

Provision of human CYP and non-CYP metabolites M2 (20.1 mg), M4 (66.2 mg), and M12 (18.4 mg) of Samatolisib at multi-milligram scale utilizing a mixed approach of microbial biotransformation, chemical synthesis, and liver S9 incubations.61
In contrast to accessing metabolites by chemical synthesis, it is also not necessary to know the structure of the target metabolite; however, it is important to perform thorough LC-MS/MS comparative analysis of the output from a microbial biotransformation “screen” with a biological reference (e.g., a plasma sample containing the target metabolite), particularly where multiple, closely eluting stereo- or regioisomeric possibilities are likely.
These attributes, along with the ability to readily scale-up production, make microbial biotransformation a very powerful technique to produce milligram or gram amounts of a target metabolite, which at the very least can be used to elucidate the metabolite’s structure after it has been made. One example of how microbial biotransformation was deployed in this way was the production of a disproportionate oxidized metabolite of LEO Pharma’s ingenol disoxate (see Figure 15).62 The position of the oxidation was unknown, and the complexity of the ingenol portion of the molecule meant that even had the structure of the metabolite been known, chemical synthesis would not have been facile. Once the best microbial system for the production of the target metabolite had been identified, an initial 86 mg yield of 99% pure M27 was obtained from an incubation with 400 mg of parent compound, subsequently followed by a further 415 mg supply to facilitate pharmacological profiling.
Figure 15.

Biotransformation of ingenol disoxate to its main human metabolite (M27), using a microbial strain. M27 is a metabolite that was more prevalent in humans than in preclinical species, i.e. a disproportionate metabolite, and thus subject to the FDA’s MIST guidelines. Yield of M27 was optimized by early harvest of the reaction to prevent onward oxidation to dihydroxylated derivatives, resulting in the purification of 100s of milligrams of the metabolite for further evaluation. M27 was shown to have similar pharmacological effects to the parent compound but was less potent.62
Although chemical synthesis is often the first strategy used to attempt to access metabolites, the risk of failure can be mitigated by evaluating microbial biotransformation in parallel. This approach was taken by Janssen, who were confronted with the challenge of producing a secondary (hydroxylated des-methyl) metabolite of (S)-ketamine, which was in clinical development for the treatment of resistant depression. At the time, there was little prior art relating to a synthetic route to the putative structure detected in human plasma; hence, Janssen elected to commission both chemical and microbial synthesis in parallel to meet the urgent timeline to produce the metabolite for analysis. Both routes were successful within approximately two months; in the case of microbial biotransformation, a 600 mg dose of the commercially available (S)-norketamine parent substrate in a culture volume of 6 L yielded 83 mg of (2S,6S)-6-hydroxynorketamine at >95% purity by LC-UV-ELSD.60
Wild-type microbes have broad oxidation capabilities due to the high number of CYP enzymes that can be expressed (25–50 for bacterial species, 100+ for fungi). Hydroxylated derivatives can be produced, including aliphatic and aromatic hydroxylated derivatives, with the former usually of greater interest to medicinal chemists. The process is more resource-intensive than for enzymatic diversification, with an incubation duration of 3–5 days compared to overnight reaction for enzymes. Consideration must be given to the choice of parent compound to biotransform with respect to unwanted side reactions that are known to be performed by the microbes to be used, such as compounds containing easily reducible functional groups, or those that are susceptible to hydrolytic enzymes or N-acetylation of primary amines. Once a derivative has been deemed to be of interest, microbial biotransformation has the added benefit of scalability, with any derivative produced in reasonable yield being amenable to production at larger scale in multiple shaken flasks or stirred tank reactors to yield 10s–100s of milligrams for further evaluation in a matter of a few weeks.
A final example illustrating the utility of microbial biotransformation both for human metabolite production and for late-stage diversification is the production of metabolites of ruxolitinib. While it was possible for low milligram amounts of metabolites to be made using chemical synthesis, larger amounts were required for absolute stereochemical assignment by X-ray crystallography and for further biological testing. Many microbes were able to produce some of the desired metabolites; however, one in particular was able to produce all the cyclopentyl hydroxyl derivatives and so was scaled up to enable the purification of 50–120 mg of each metabolite (see Figure 16). Several of these metabolites were found to possess pharmacological activity, including two metabolites resulting from hydroxylation at the 2- and 3-position on the cyclopentyl moiety, and the third a ketone resulting from further oxidation at the 3-position. Based on the IC50 values, the contribution of the metabolites to pharmacodynamic activity relative to parent was estimated to be 15–18% in healthy subject studies.63
Figure 16.
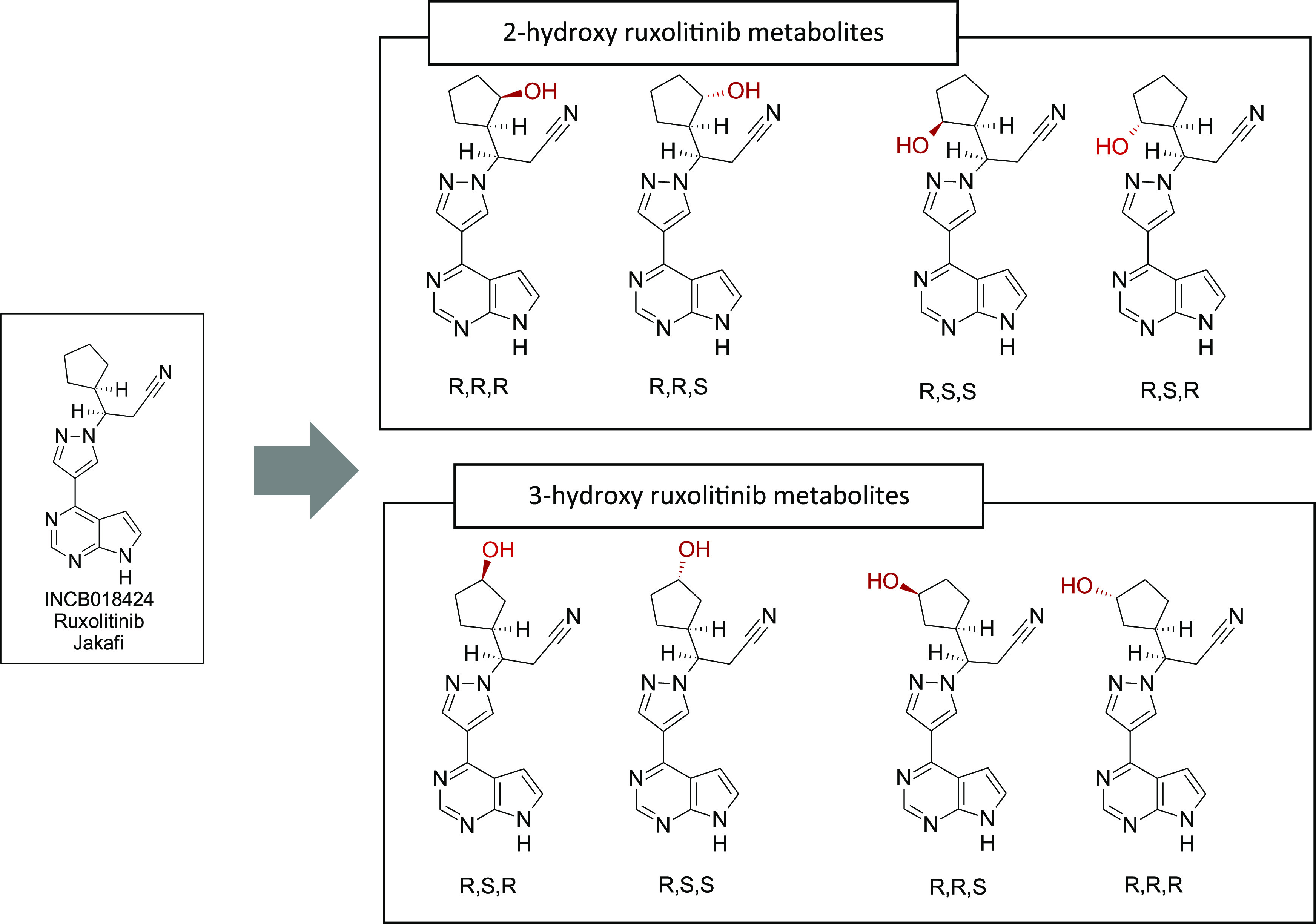
Hydroxylated metabolites of ruxolitinib produced by a single bacterial species, following a panel screen consisting of multiple microbial species. Keto derivatives were also produced and identified.
In Vitro Animal Tissue Derived Systems (Liver S9/Microsomes/Hepatocytes)
Drug metabolism investigators have a variety of animal and human derived in vitro tools at their disposal to aid their discovery chemistry partners in, inter alia, derisking potential toxicophores, optimizing metabolic stability, and informing on potential liabilities associated with the MIST guidelines. In addition to the more widely used liver, intestine, kidney, and lung subcellular fractions (microsomes, S9, and cytosol), whole liver cells (hepatocytes), and recombinant cytochromes P450 (CYPs), newer tools have emerged including hepatoma cell lines (HepG2 and HepaRG), intestinal whole cells, and membrane immobilized liver cell cocultures (liver on a chip). Each in vitro system can be used in combination with appropriate cofactors, specific or broad enzyme inhibitors, positive control substrates, and trapping reagents to answer specific questions regarding a drug’s metabolic disposition.
A recent example of the judicious use of liver subcellular fractions, hepatocytes, and recombinant CYP450s to investigate a human disproportionate circulating metabolite, M21 (morpholino lactam), of momelotinib (MMB) was described by Zheng et al.64 (see Figure 17). Hepatocyte incubations in the presence of 1-aminobenzotriazole (ABT), a pan-CYP inhibitor, demonstrated a significantly increased proportion of MMB remaining. In contrast, addition of the aldehyde oxidase (AOX) inhibitor hydralazine to hepatocyte incubations had little effect on MMB turnover. Both inhibitors, however, significantly reduced the amount of M21 formed over the incubation time course, indicating a multistep metabolism pathway involving CYPs and AOX. Reaction phenotyping experiments with CYP450 isoenzymes revealed that MMB was efficiently metabolized (% MMB remaining) by CYP3A4, 2C8, 2C19, 2C9, and 1A2. The involvement of multiple CYPs in MMB metabolism was corroborated using specific CYP450 chemical inhibitors in human hepatocyte incubations with MMB; furthermore, each inhibitor appeared to diminish M21 formation to a similar extent with the exception of CYP2C9. M21 was proposed to be formed via a multioxidation pathway, first to a carbinolamine intermediate (CYP450) and then by AOX to M21. Further, liver microsome incubations with MMB in the presence of β-NADPH cofactor and potassium cyanide produced a cyano adduct, indicating involvement of an iminium ion intermediate. No circulating M21 was observed in rat or dog toxicokinetic plasma thus necessitating additional safety studies in which MMB was coadministered with M21 to satisfy the MIST guidance.
Figure 17.
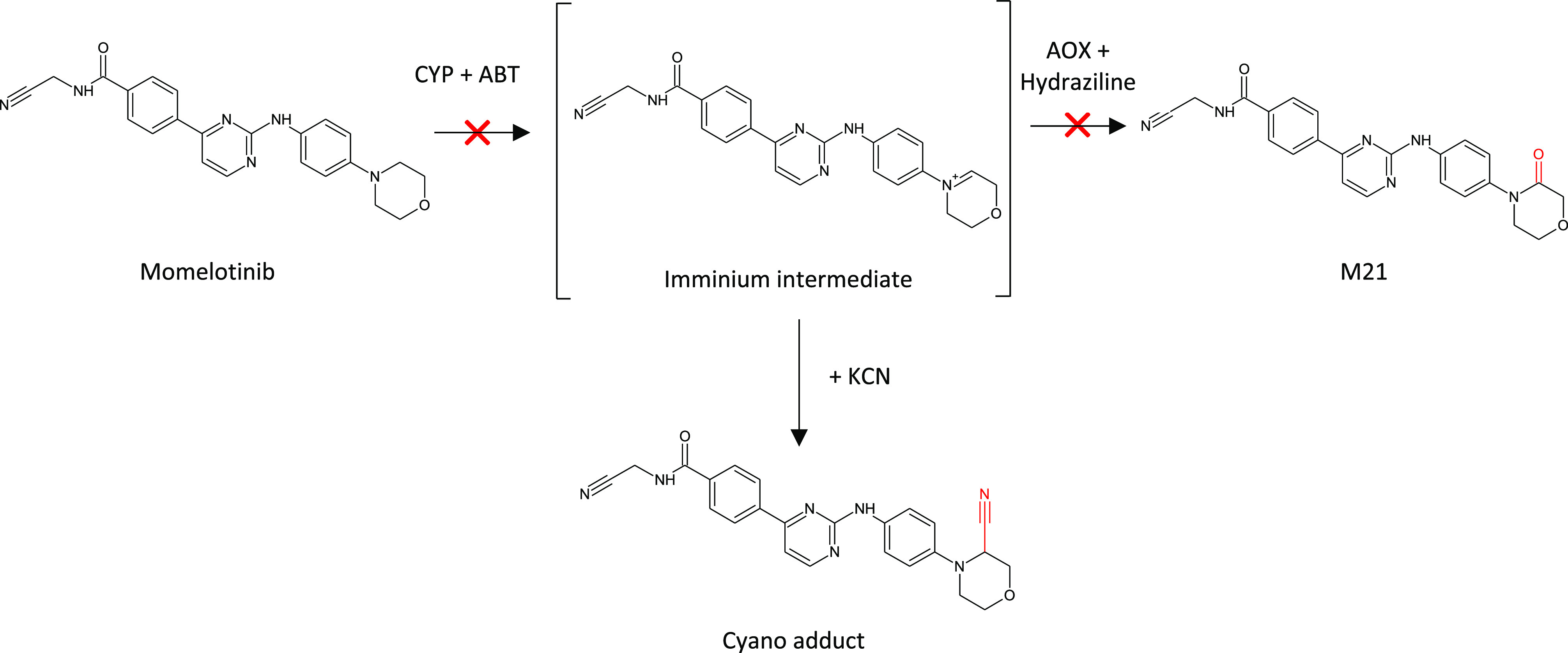
Proposed pathway for the CYP/AOX dependent metabolism of momelotinib. Reprinted in part with permission from Zheng, J.; et al. Drug Metab. Dispos., 2018, 46, 237–247. Copyright 2018, ASPET.
Polymorphisms among the various CYP450 enzymes are well-known and are ideally avoided as a major route of drug clearance.65,66 The epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor 2 (HER2) inhibitor lapatinib is metabolized by CYP3A4 and CYP3A5 (in combination referred to as CYP3A), in the presence of β-NADPH, to a p-hydroxy aniline (M1, O-debenzylation) which in turn is proposed to be further oxidized by CYP3A to a quinone imine reactive metabolite intermediate67 (see Figure 18). M1 forms adducts in incubations supplemented with glutathione (GSH) and has been implicated in the idiosyncratic liver toxicity of lapatinib observed in the clinic. Bissada et al. recently reported on investigations with CYP3A5 genotyped liver microsomes and hepatocytes from individual donors.68 CYP3A5 has several functional variants (polymorphs) which confer high (*1/*1), low (*3/*3), and medium (*1/*3) enzyme activity.68 Each lot of genotyped microsomes was characterized with regard to the ability to metabolize midazolam (a marker for CYP3A activity) and T-5, a selective substrate for CYP3A5. Only T-5 N-oxide formation demonstrated a positive relationship with CYP3A5 genotype. No such relationship could be established for the formation of lapatinib M1 or its GSH adduct (M1-GSH) and CYP3A5 genotype. M1 and M1-GSH levels, however, correlated well with 1′-hydroxymidazolam, but not T-5 N-oxide formation. A strong correlation also existed for formation of M1-GSH and its precursor, M1. Inhibitors of CYP3A4 (CYP3cide, a CYP3A4 specific inactivator) and CYP3A (ketoconazole, a CYP3A pan-inhibitor) were then investigated to assess the contribution of CYP3A5 to lapatinib M1 formation in a 150-donor pooled lot of human liver microsomes and the CYP3A5 genotyped individual donors. The investigators concluded that the CYP3A5 contribution to M1 product formation was greatest in microsomes of *1/*1 donors (26%), followed by the multidonor pool (16%), and *3/*3 donors (6%). Incubations in 3A5 genotyped pooled and individual donor hepatocytes confirmed the positive relationship with regard to 1′-hydroxymidazolam, T-5 N-oxide, and lapatinib M1 formation observed in genotyped microsomes. Formation of both M1 and M1-GSH correlated well with 1′-hydroxymidazolam but not T-5 N-oxide formation in hepatocytes regardless of genotype. Finally, CYP3cide and ketoconazole inhibition experiments in hepatocytes from a low (*3/*3) and a high CYP3A5 (*1/*1) expresser demonstrated a greater contribution of CYP3A5 to lapatinib M1 formation in the high expresser lot. Ultimately, while CYP3A5 contributed significantly to lapatinib O-debenzylation (M1), the total CYP3A phenotype, and not CYP3A5 alone, provided the most compelling marker of lapatinib bioactivation.
Figure 18.
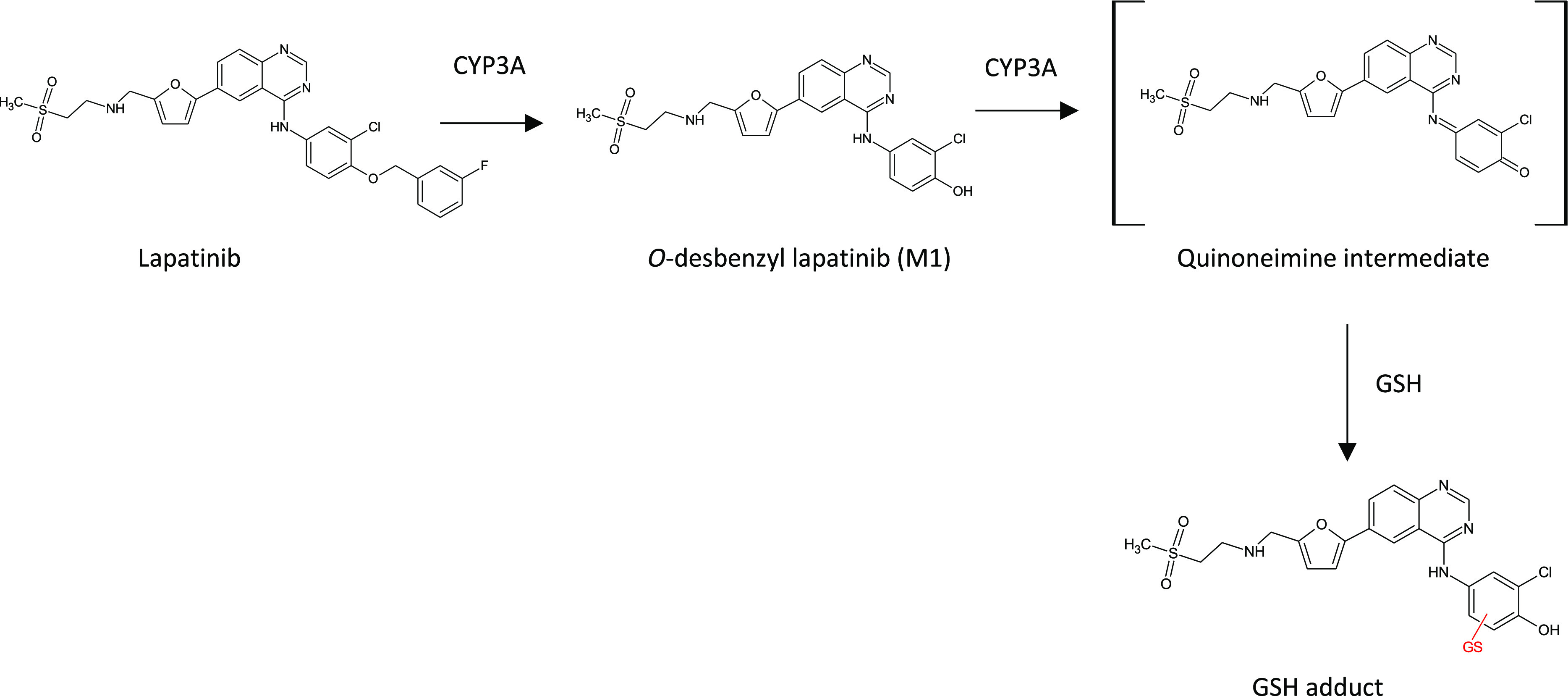
Proposed pathway for the CYP3A dependent metabolism and bioactivation of lapatinib. Reprinted in part with permission from Bissada, J.E. et al. Drug Metab. Dispos., 2019, 47, 1257–1269. Copyright 2019, ASPET.
Primary aliphatic alcohols are featured in many marketed and investigational drugs spanning a range of therapeutic targets. Abacavir (HIV), dasatinib (oncology), and ozanimod (MS), to name a few, bear primary alcohol groups which are all metabolized to their carboxylic acid analogues by alcohol dehydrogenase (ADH), with or without the contribution of aldehyde dehydrogenase (ALDH). ADH and ALDH each comprise a family of enzymes found primarily in the liver. Both enzymes are found in the cytosolic fraction and require NAD+ cofactor to catalyze their reactions. The pH optima for xenobiotic substrates of ADH/ALDH may be substantially different than physiological pH in in vitro incubations, and enzymes may be active in vitro for extended periods of time.69 Diao et al. identified a major carboxylic acid metabolite of 3-n-butylphthalide (NBP), M5-2, in human plasma following a 200 mg oral dose.70 NBP itself does not feature a primary alcohol, but is efficiently converted to 11-hydroxy-NBP (ω oxidation of the butyl side chain) by CYP mediated oxidation, and then to the corresponding carboxylic acid M5-2 (see Figure 19). M5-2 was observed as only a minor metabolite in human liver microsomes (HLM), belying its relative abundance in human plasma. The investigators demonstrated the near quantitative conversion of 11-hydroxy-NBP to M52 in human liver cytosol (HLC) incubations supplemented with NAD+ at pH 7.4. When 4-methylpyrazole or disulfiram, specific inhibitors of ADH and ALDH, respectively, was included in HLC incubations with 11-hydroxy-NBP, M5-2 formation was significantly reduced. Combined, these results supported the role of both ADH and ALDH in the formation of the major human circulating metabolite of NBP, M5-2. M5-2 was also formed in HLM incubations with 11-hydroxy-NBP supplemented with NADPH, but to a lesser extent than observed in HLC incubations. Xenobiotics featuring primary aliphatic alcohols should thus be considered as potential substrates for ADH/ALDH, as should drug candidates which can be converted to primary aliphatic alcohols by oxidation or O-dealkylation.
Figure 19.
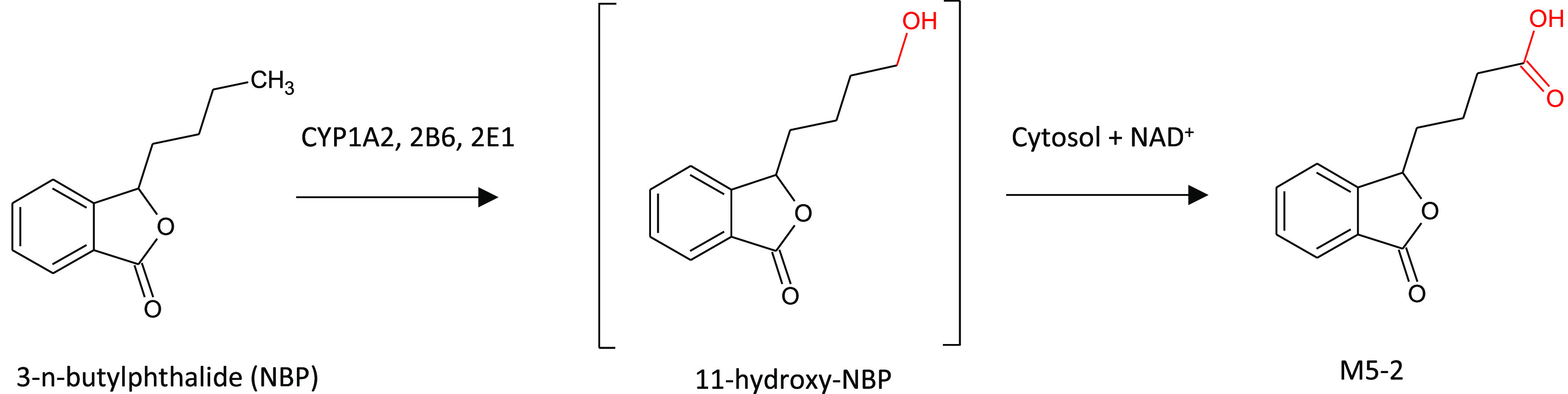
Proposed pathway for the CYP and ADH/ALDH dependent metabolism of NBP to its major circulating metabolite, M5-2. Reprinted in part with permission from Diao, X. et al. Drug Metab. Dispos., 2013, 41, 430–444. Copyright 2013, ASPET.
Biotransformation capabilities of in vitro liver systems can also be exploited by chemists at the discovery stage where an appreciation of metabolites produced can lead to novel chemical matter. This technique has been exploited by scientists at Pfizer for late stage functionalization of several unique scaffolds, as discussed earlier. The use of multiple species of liver microsomes to generate oxidized derivatives of six compounds in their M1 positive allosteric modulator (PAM) program was instrumental in providing rapid SAR exploration and feedback to the medicinal chemistry design cycle of where polarity could and could not be tolerated.71 Nanomole quantities of 21 oxidized derivatives were purified across the six templates. NMR spectroscopy was used to elucidate structures and provide quantitation to enable calculation of in vitro potency of each derivative relative to the parent compound in the relevant biological assay. Biotransformation thus proved a useful alternative where de novo synthesis of multiple derivatives would be time-consuming and costly.
Reactive Metabolites
Bioactivation is the metabolic process whereby a compound is converted to an electrophilic reactive metabolite. Bioactivation may result in the covalent modification of cellular macromolecules and is believed to be associated with idiosyncratic adverse drug reactions (IADRs), including drug-induced liver injury (DILI), through an immune-mediated response.72 IADRs are difficult to predict and usually manifest only after a great number of patients have received the drug, either late in drug development or after it is marketed, resulting in drug discontinuation or black box warnings.73 Accordingly, the concept of structural alerts, functional groups often associated with reactive metabolite formation, has been incorporated into the drug discovery paradigm in an attempt to avoid or mitigate bioactivation and the potentially associated IADRs. A recent review thoroughly discusses the development and implementation of the structural alerts concept.73
Examples of retrospective analysis of drug bioactivation following IADR in the clinic are plentiful. Lapatinib, as discussed earlier, is bioactivated to a reactive quinone imine metabolite by CYPs 3A4 and 3A5; this bioactivation is thought to be responsible for lapatinib-associated hepatotoxicity, resulting in a black box warning shortly after its launch.74 Bromfenac, a nonsteroidal anti-inflammatory drug (NSAID), was discontinued following reports of acute hepatotoxicity in the clinic.75 Again, bioactivation to a reactive metabolite is thought causative in bromfenac-associated DILI. A series of publications on bromfenac bioactivation illustrates the complex and unusual nature of its bioactivation involving initial acyl glucuronide formation, which then forms an indolinone metabolite through intramolecular nucleophilic substitution75,76 (see Figure 20). It is this indolinone that undergoes further CYP-mediated oxidation resulting in a quinone imine and/or quinone methide reactive intermediate. Lastly, the clinical development of MK-8666, a GPR40 agonist for the treatment of type 2 diabetes mellitus, was halted due to DILI.77 MK-8666 was shown to form both acyl glucuronide and CoA thioester reactive metabolites resulting in protein modification in vitro.77 Scalable quantities of acyl glucuronides are often required by pharma companies to undertake evaluation of their potential to form reactive species; these can often be accessed using microbial biotransformation, if chemical synthesis is not facile.60
Figure 20.
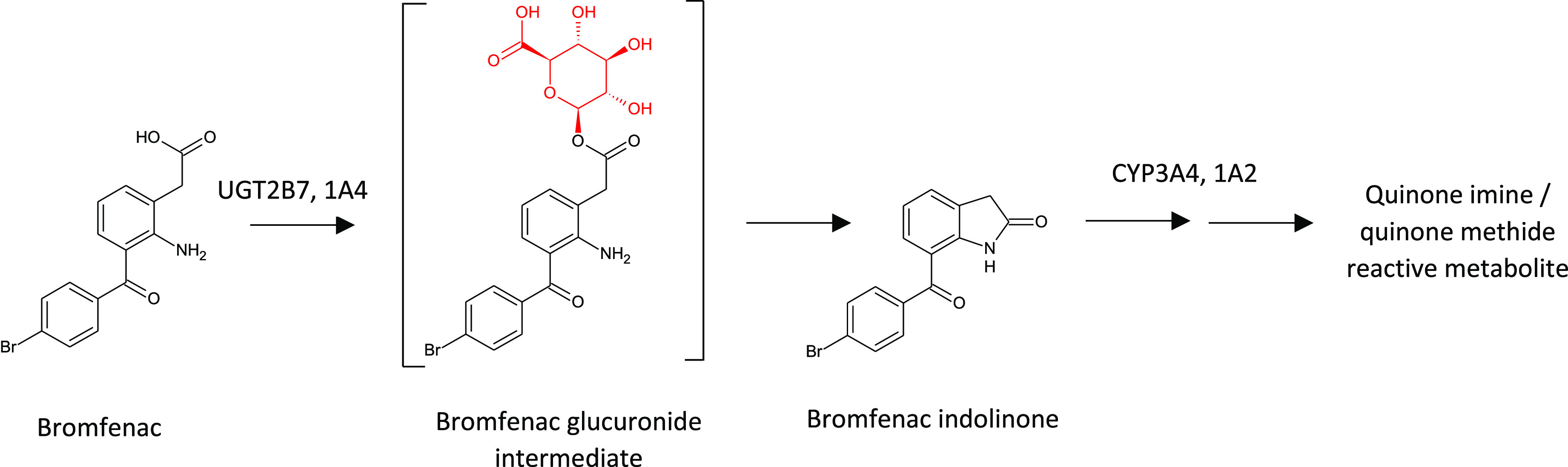
Primary route of bromfenac bioactivation is initiated by glucuronidation and culminates in CYP-dependent quinone imine/methide reactive metabolite formation.
When characterizing the metabolism of a new compound or chemical series, it has become standard practice to assess molecules for their propensity to form reactive metabolites using in vitro incubations, supplemented with appropriate cofactors, in the presence of exogenous nucleophiles such as GSH for soft electrophilic reactive metabolites and potassium cyanide (KCN) for hard electrophilic reactive metabolites (e.g., imines). While not as commonplace because of the required resource investment, covalent binding assays with radiolabeled compounds can provide a measure of the extent of covalent modification of proteins and, by extension, reactive metabolite formation. Again, the reader is directed to the review article by Kalgutkar for a comprehensive discussion of case studies illustrating the use of reactive metabolite trapping and biotransformation analysis to design out bioactivation.73 In an additional example, Lou et al. recently discussed the characterization and efforts to mitigate bioactivation en route to the discovery of RN941, a Bruton’s tyrosine kinase (BTK) inhibitor for the treatment of inflammatory diseases and B cell malignancies.78 The authors used a combination of 14C covalent binding, GSH trapping, KCN trapping, and semicarbazide trapping (for aldehydes) to characterize the bioactivation of a series of structural analogs. NMR analysis of an isolated GSH adduct of one analogue lead to the proposal of a common bioactivation pathway involving epoxidation of the aminopyridone core and discovery of RN941, which has a pyridazinone core and is devoid of bioactivation.
Impact of the Gut Microbiome on Metabolism
Interest in the gut microbiome’s contribution to xenobiotic metabolism has increased dramatically in recent years, yet only ∼1% of approved drugs are known substrates of gut microbiota.79,80 The actual number is likely significantly higher but unappreciated and uncharacterized because microbiota-derived metabolites are often not absorbed and therefore not of toxicological concern in terms of regulatory guidance regarding systemically circulating metabolites.2 Often, gut microbiome-dependent metabolism is elucidated only when attempting to source unexpected major circulating metabolites (≥10% of total circulating compound-related material), which have implications in the design of clinical drug–drug interaction studies. Of note to the discovery scientist, in contrast to the host’s mostly oxidative and conjugative metabolic capabilities, gut microbiota-associated metabolism is reductive/hydrolytic in nature,81 such that certain functional groups may predispose a compound to metabolism by gut bacteria. This is especially true with sustained-release formulations or poorly absorbed compounds.82 Multiple reviews have been written discussing examples of the impact of gut microbiota metabolism on drug efficacy and toxicity over the past several decades.80,83,84
Epacadostat (EPAC) presents an unusual case where, in addition to gut microbiome-facilitated enterohepatic circulation of parent, major circulating metabolites derive from extensive Phase II metabolism, reductive metabolism by gut microbiota, and secondary systemic Phase I metabolism of the absorbed gut metabolite.6 Epacadostat, an investigational compound targeting the enzyme indoleamine 2,3-dioxygenase 1 (IDO1), is metabolized to three major circulating metabolites in humans and preclinical species: M9, a direct O-glucuronide of the EPAC amidoxime; M11, a reduction of the amidoxime of EPAC to an amidine; and M12, N-dealkylated M11 (see Figure 21). In vitro experiments with recombinant human UDP-glucuronosyltransferases (UGTs) showed that M9 is a product of UGT1A9. However, only trace amounts of M11 and M12 could be formed from EPAC in any of the typically evaluated in vitro systems, including human microsomes from multiple tissues, hepatocytes, recombinant human cytochromes P450, and non-P450 enzymatic systems. The negative results with the typical metabolic systems in conjunction with the amidine (reduced amidoxime) functional group in M11 and M12 suggested the involvement of gut microbiota. Incubation of EPAC in feces homogenates, with and without antibiotic pretreatment, confirmed the role of gut bacteria in the formation of M11, but not M12. Further, incubation of a synthetic standard of M11 with recombinant P450s showed that M12 is formed via N-dealkylation of M11 by CYP3A4, CYP2C19, and CYP1A2. Lastly, incubation of a synthetic standard of M9 in feces homogenates showed that gut microbiota microorganisms are capable of efficient deconjugation of M9 back to parent EPAC, consistent with observed enterohepatic circulation in the clinic.85 All three major metabolites of epacadostat could be produced by microbial biotransformation. While M12 could also be accessed by chemical synthesis, microbial biotransformation enabled tens of milligrams of M9 and M11 to be produced for further evaluation of the metabolites.85
Figure 21.

Major metabolites of epacadostat produced by microbial biotransformation via mixed metabolic pathways.6 Scale-up of the most productive biotransforming microbes for M9 and M11 enabled the supply of 112 mg of the glucuronide (M9) and 69 mg of the gut metabolite (M11).
The metabolism of BILR 355 illustrates the complex interplay between host metabolic processes (CYP and non-CYP) and gut microbiota-mediated metabolism86,87 (see Figure 22). BILR 355, a second-generation non-nucleoside reverse transcriptase inhibitor (NNRTI) developed for the treatment of HIV-1 infection, is extensively metabolized by CYP3A4 resulting in unfavorable pharmacokinetics in the clinic. Co-dosing with ritonavir, a known CYP3A4 inhibitor, dramatically increased the systemic exposure and t1/2 of BILR 355 but also resulted in the appearance of a disproportionate human metabolite, BILR 516. BILR 516 is a quinolone secondary metabolite of BILR 355 requiring initial reduction of the parent molecule’s quinoline N-oxide to a quinoline (BILR 402) by gut bacteria. Oxidation of BILR 402 to BILR 516 was shown to be mediated by aldehyde oxidase (AO). Parent BILR 355 is readily metabolized by CYP3A4 to multiple metabolites, as is the gut metabolite BILR 402, which can also be oxidized back to parent. When BILR 355 is dosed alone, the metabolism by CYP3A4 dominates the metabolic processes, dispersing the metabolite burden to numerous minor metabolites. With inhibition of CYP3A4 by coadministration of ritonavir, BILR 355 plasma levels increase, but BILR 402 is predominantly metabolized by AO to BILR 516. Complicating matters further, human and monkey AO activity is much higher than the rat, while dog has little to none. Accordingly, BILR 516 is observed at low levels in the rat and dog toxicological studies, resulting in BILR 516 being observed as a disproportionate human metabolite.
Figure 22.
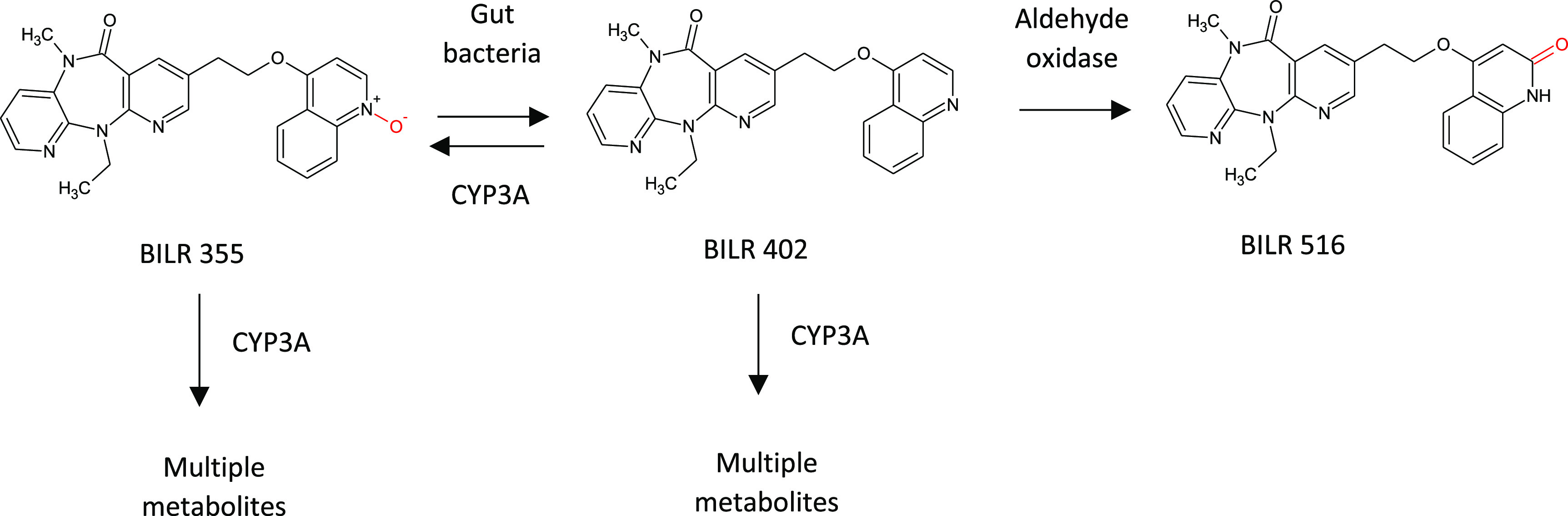
Metabolism of BILR 355 involves a complex interplay between CYP-, AO-, and gut microbiota-mediated processes.
Conclusions and Future Directions
Studies of metabolism are crucial in the development of new drugs, with many potentially useful candidates falling by the wayside due to various liabilities or limitations. As we have recounted, however, there are numerous examples of metabolism studies providing insights that range from understanding how the effects of some drugs are mediated by their active metabolites, to the design of chemical modifications that lead to improved drugs with greater metabolic stability and efficacy.
Historically, metabolic investigations have been conducted mostly in the development phases, potentially leading to late recognition of issues or opportunities for drug improvement, sometimes even after approval and consequently opening the door to competitors. There is therefore a current trend to bring metabolism studies forward into drug discovery at the lead optimization stage so that findings can inform and guide the selection of the most suitable candidate, thus avoiding a later reactive approach leading to costs and delays at the development stage. This is facilitated by the availability of a very broad toolkit for in vitro metabolic studies, comprising animal tissue derived systems (liver S9, microsomes, hepatocytes), microbial biotransformation, and an expanding range of recombinant enzymes. While the animal tissue-derived systems allow exquisite dissection of metabolic processes and identification of the key enzymes involved, reaction scale-up to identify the actual metabolites can prove to be very expensive and has ethical concerns; hence, we employ use of these for scale-up as the technique of last resort. Microbial biotransformation has long been used as a cost-effective alternative for reaction scale-up for metabolite ID but does also produce nonmammalian metabolites, so careful chromatographic comparison is required to pinpoint the desired products. The recombinant enzymes available include CYPs of mammalian and microbial origin, and those derived from actinomycete bacteria have a notably wide range of catalytic abilities. These are ideal for small-scale screening to identify those that produce metabolites of interest, again with chromatographic comparison to confirm those products as the mammalian targets. The microbially derived enzymes are particularly suited to reaction scale-up and for larger metabolite amounts this can utilize a whole cell biotransformation process via a recombinant strain or even the wild type producing strain to control costs.
The trend in drug design to select lead compounds that have a high metabolic stability in liver microsome systems is leading to a greater use of N-heterocyclic chemistry and an increased observation of non-CYP metabolism via enzymes such as AO. Recombinant phase I enzyme kits for metabolic studies should therefore ideally include AO and other nitrogen-focused enzymes such as flavin monooxygenases (FMOs). Another consequence of such chemistry is the likely rise in N-glucuronidation as a clearance mechanism and concomitant increase in the need to characterize N-glucuronide metabolites. Also noteworthy is a need that is likely to increase to produce and characterize metabolites formed from new candidate drugs by gut microbiota in order to evaluate their biological effects. This may become more routine as the implications of these reactions becomes more widely understood.
Acknowledgments
The Hypha authors would like to acknowledge the contribution of all staff involved in biotransformation work at Hypha.
The authors declare no competing financial interest.
This paper published ASAP on August 28, 2020. Figures 6 and 21 have been replaced and the corrected version was reposted on September 1, 2020.
References
- Cerny M. A.; Kalgutkar A. S.; Obach R. S.; Sharma R.; Spracklin D. K.; Walker G. S. The effective application of metabolite profiling in drug design and discovery. J. Med. Chem. 2020, 63, 6387–6406. 10.1021/acs.jmedchem.9b01840. [DOI] [PubMed] [Google Scholar]
- Safety Testing of Drug Metabolites Guidance for Industry. U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER). March 2020. Pharmacology/Toxicology, Revision 2. https://www.fda.gov/media/72279/download (accessed 2020-08-12). [Google Scholar]
- Smith D. A.; Beaumont K.; Maurer T. S.; Di L. Clearance in drug design. J. Med. Chem. 2019, 62 (5), 2245–2255. 10.1021/acs.jmedchem.8b01263. [DOI] [PubMed] [Google Scholar]
- Hutzler J. M.; Ring B. J.; Anderson S. R. Low turnover drug molecules: a current challenge for drug metabolism scientists. Drug Metab. Dispos. 2015, 43, 1917–1928. 10.1124/dmd.115.066431. [DOI] [PubMed] [Google Scholar]
- Gu C.; Artelsmair M.; Elmore C. S.; Lewis R. J.; Davis P.; Hall J. E.; Dembofsky B. T.; Christoph G.; Smith M. A.; Chapdelaine M.; Sunzel M. Late-occurring and long-circulating metabolites of GABAAα2,3 receptor modulator AZD7325 involving metabolic cyclization and aromatization: relevance to MIST analysis and application for patient compliance. Drug Metab. Dispos. 2018, 46 (3), 303–315. 10.1124/dmd.117.078873. [DOI] [PubMed] [Google Scholar]
- Boer J.; Young-Sciame R.; Lee F.; Bowman K. J.; Yang X.; Shi J. G.; Nedza F. M.; Frietze W.; Galya L.; Combs A. P.; Yeleswaram S.; Diamond S. Roles of UGT, P450, and gut microbiota in the metabolism of epacadostat in humans. Drug Metab. Dispos. 2016, 44, 1668–1674. 10.1124/dmd.116.070680. [DOI] [PubMed] [Google Scholar]
- Zimmermann M.; Zimmermann-Kogadeeva R.; Wegmann R.; Goodman A. L. Mapping human microbiome drug metabolism by gut bacteria and their genes. Nature 2019, 570, 462–467. 10.1038/s41586-019-1291-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fura A.; Shu Y.-Z.; Zhu M.; Hanson R. L.; Roongta V.; Humphreys W. G. Discovering drugs through biological transformation: role of pharmacologically active metabolites in drug discovery. J. Med. Chem. 2004, 47 (18), 4339–4351. 10.1021/jm040066v. [DOI] [PubMed] [Google Scholar]
- Schadt S.; Bister B.; Chowdhury S. K.; Funk C.; Hop C. E. C. A.; Humphreys W. G.; Igarashi F.; James A. D.; Kagan M.; Khojasteh S. C.; Nedderman A. N. R.; Prakash C.; Runge F.; Scheible H.; Spracklin D. K.; Swart P.; Tse S.; Yuan J.; Obach R. S. A decade in the MIST: learnings from investigations of drug metabolites in drug development under the “metabolites in safety testing” regulatory guidelines. Drug Metab. Dispos. 2018, 46 (6), 865–878. 10.1124/dmd.117.079848. [DOI] [PubMed] [Google Scholar]
- Ryder T. F.; Calabrese M. F.; Walker G. S.; Cameron K. O.; Reyes A. R.; Borzilleri K. A.; Delmore J.; Miller R.; Kurumbail R. G.; Ward J.; Kung D. W.; Brown J. A.; Edmonds D. J.; Eng H.; Wolford A. C.; Kalgutkar A. S. Acyl glucuronide metabolites of 6-chloro-5-[1-hydroxycyclobutyl)phenyl]-1H-indole-3-carboxylic acid (PF-06409577) and related indole-3-carboxylic acid derivatives are direct activators of adenosine monophosphate-activated protein kinase (AMPK). J. Med. Chem. 2018, 61 (16), 7273–7288. 10.1021/acs.jmedchem.8b00807. [DOI] [PubMed] [Google Scholar]
- Tornio A.; Filppula A. M.; Kailari O.; Neuvonen M.; Nyronen T. H.; Tapaninen T.; Neuvonen P. J.; Niemi M.; Backman J. T. Glucuronidation converts clopidogrel to a strong time-dependent inhibitor of CYP2C8: a phase II metabolite as a perpetrator of drug-drug interactions. Clin. Pharmacol. Ther. 2014, 96 (4), 498–507. 10.1038/clpt.2014.141. [DOI] [PubMed] [Google Scholar]
- Ogilvie B. W.; Zhang D.; Li W.; Rodrigues A. D.; Gipson A. E.; Holsapple J.; Toren P.; Parkinson A. Glucuronidation converts gemfibrozil to a potent, metabolism-dependent inhibitor of CYP2C8: implications for drug-drug interactions. Drug Metab. Dispos. 2006, 34 (1), 191–197. 10.1124/dmd.105.007633. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Tang W. Drug Metabolism in drug discovery and development. Acta Pharm. Sin. B 2018, 8 (5), 721–732. 10.1016/j.apsb.2018.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra B.; Gandelman K.; Sachse R.; Wood N.; Michel M. C. The design and development of fesoterodine as a prodrug of 5-hydroxymethyl tolterodine (5-HMT), the active metabolite of tolterodine. Curr. Med. Chem. 2009, 16, 4481–4489. 10.2174/092986709789712835. [DOI] [PubMed] [Google Scholar]
- Bauman J. L. The role of pharmacokinetics, drug interactions and pharmacogenomics in the acquired long QT syndrome. Eur. Heart J. Suppl. 2001, 3, K93–100. 10.1016/S1520-765X(01)90012-4. [DOI] [Google Scholar]
- European Medicines Agency , Stivarga, https://www.ema.europa.eu/en/documents/product-information/stivarga-epar-product-information_en.pdf (accessed 2020-08-12).
- Rousseau B.; Boukerma A. K.; Henriques J.; Cohen R.; Lucidarme O.; Borg C.; Tournigand C.; Kim S. C. H.; Bachet J.-B.; Mazard T.; Louvet C.; Chibaudel B.; Diaz L. A.; Vernerey D.; Andre T.; Hulin A. Accumulation of active metabolite M-2 predicts overall survival (OS) of chemorefractory metastatic colorectal cancer patients treated with regorafenib (REGO). J. Clin. Oncol. 2019, 37 (15 suppl), 3121. 10.1200/JCO.2019.37.15_suppl.3121. [DOI] [Google Scholar]
- Taguchi D.; Inoue M.; Fukuda K.; et al. Therapeutic drug monitoring of regorafenib and its metabolite M5 can predict treatment efficacy and the occurrence of skin toxicities. Int. J. Clin. Oncol., 2020, 25, 531. 10.1007/s10147-019-01593-w. [DOI] [PubMed] [Google Scholar]
- Gerisch M.; Hafner F.; Lang D.; Radtke M.; Diefenbach K.; Cleton A.; Lettieri J. Mass balance, metabolic disposition, and pharmacokinetics of a single oral dose of regorafenib in healthy human subjects. Cancer Chemother. Pharmacol. 2018, 81, 195–206. 10.1007/s00280-017-3480-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruciani G.; Valeri A.; Goracci L.; Pellegrino M. R.; Buonerba F.; Baroni M. Flavin monooxygenase metabolism: why medicinal chemists should matter. J. Med. Chem. 2014, 57 (14), 6183–6196. 10.1021/jm5007098. [DOI] [PubMed] [Google Scholar]
- Stone R. M.; Manley P. W.; Larson R. L.; Capdeville R. Midostaurin: its odyssey from discovery to approval for treating acute myeloid leukemia and advanced systemic mastocytosis. Blood Advances 2018, 2 (4), 444–453. 10.1182/bloodadvances.2017011080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter B.; Winter G. E.; Blatt K.; Bennett K. L.; Stefanzl G.; Rix U.; Eisenwort G.; Hadzijusufovic E.; Gridling M.; Dutreix C.; Hoermann G.; Schwaab J.; Radia D.; Roesel J.; Manley P. W.; Reiter A.; Superti-Furga G.; Valent P. Target interaction profiling of midostaurin and its metabolites in neoplastic mast cells predicts distinct effects on activation and growth. Leukemia 2016, 30 (2), 464–472. 10.1038/leu.2015.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He H.; Tran P.; Gu H.; Tedesco V.; Zhang J.; Lin W.; Gatlik E.; Klein K.; Heimbach T. Absorption, metabolism, and excretion of midostaurin in humans. Drug Metab. Dispos. 2017, 45 (5), 540–555. 10.1124/dmd.116.072744. [DOI] [PubMed] [Google Scholar]
- Novartis patent WO 2003094915 A1. Use of valsartan and its metabolite to inhibit platelet aggregation. https://patents.google.com/patent/WO2003094915A1/en (accessed 2020-08-12).
- Serebruany V. L.; Malinin A. I.; Lowry D. R.; Sane D. C.; Webb R. L.; Gottlieb S. O.; O’Connor C. M.; Hennekens C. H. Effects of Valsartan and Valeryl 4-Hydroxy Valsartan on Human Platelets: A Possible Additional Mechanism for Clinical Benefits. J. Cardiovasc. Pharmacol. 2004, 43 (5), 677–684. 10.1097/00005344-200405000-00010. [DOI] [PubMed] [Google Scholar]
- Clader J. W. The discovery of ezetimibe: a view from outside the receptor. J. Med. Chem. 2004, 47 (1), 1–9. 10.1021/jm030283g. [DOI] [PubMed] [Google Scholar]
- Kosoglou T.; Statkevich P.; Johnson-Levonas A. O.; Paolini J. F.; Bergman A. J.; Alton K. B. Ezetimibe: a review of its metabolism, pharmacokinetics and drug interactions. Clin. Pharmacokinet. 2005, 44 (5), 467–494. 10.2165/00003088-200544050-00002. [DOI] [PubMed] [Google Scholar]
- Touma K. T. B.; Scarff J. R. Valbenazine and deutetrabenazine for tardive dyskinesia. Innovations in Clinical Neuroscience 2018, 15 (5−6), 13–16. [PMC free article] [PubMed] [Google Scholar]
- Grigoriadis D. E.; Smith E.; Hoare S. R. J.; Madan A.; Bozigian H. Phamacologic characterization of valbenazine (NBI-98843) and its metabolites. J. Pharmacol. Exp. Ther. 2017, 361 (3), 454–461. 10.1124/jpet.116.239160. [DOI] [PubMed] [Google Scholar]
- Zanos P.; Moaddel R.; Morris P. J.; Riggs L. M.; Highland J. N.; Georgiou P.; Pereira E. F. R.; Albuquerque E. X.; Thomas C. J.; Thomas C. J.; Zarate C. A.; Gould T. D. Ketamine and ketamine metabolite pharmacology: insights into therapeutic mechanisms. Pharmacol. Rev. 2018, 70 (3), 621–660. 10.1124/pr.117.015198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanos P.; Highland J. N.; Stewart B. W.; Georgiou P.; Jenne C. E.; Lovett J.; Morris P. J.; Thomas C. J.; Moaddel R.; Zarate C. A.; Gould T. D. (2R,6R)-hydroxynorketamine exerts mGlu2 receptor-dependent antidepressant actions. Proc. Natl. Acad. Sci. U. S. A. 2019, 116 (13), 6441–6450. 10.1073/pnas.1819540116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheshmazar N.; Dastmalchi S.; Terao M.; Garattini E.; Hamzeh-Mivehroud M. Aldehyde oxidase at the crossroad of metabolism and preclinical screening. Drug Metab. Rev. 2019, 51 (4), 428–452. 10.1080/03602532.2019.1667379. [DOI] [PubMed] [Google Scholar]
- Pryde D.; Dalvie D.; Hu Q.; Jones P.; Obach R. S.; Tran T.-D. Aldehyde oxidase: an enzyme of emerging importance in drug discovery. J. Med. Chem. 2010, 53, 8441–8460. 10.1021/jm100888d. [DOI] [PubMed] [Google Scholar]
- Li A. C.; Cui D.; Yu E.; Dobson K.; Hellriegel E. T.; Robertson P. Jr. Identification and human exposure prediction of two aldehyde oxidase-mediated metabolites of a methylquinoline-containing drug candidate. Xenobiotica 2019, 49 (3), 302–312. 10.1080/00498254.2018.1444815. [DOI] [PubMed] [Google Scholar]
- Manevski N.; King L.; Pitt W. R.; Lecomte F.; Toselli F. Metabolism by aldehyde oxidase: drug design and complementary approaches to challenges in drug discovery. J. Med. Chem. 2019, 62 (24), 10955–10994. 10.1021/acs.jmedchem.9b00875. [DOI] [PubMed] [Google Scholar]
- Stachulski A. V.; Meng X. Glucuronides from metabolites to medicines: a survey of the in vivo generation, chemical synthesis and properties of glucuronides. Nat. Prod. Rep. 2013, 30, 806–848. 10.1039/c3np70003h. [DOI] [PubMed] [Google Scholar]
- Beck H.; Jeske M.; Thede K.; Stoll F.; Flamme I.; Akbaba M.; Erguden J.-K.; Karig G.; Keldenich J.; Oehme F.; Militzer H. C.; Hartung I. V.; Thuss U. Discovery of molidustat (BAY 85–3934): a small-molecule oral hif-prolyl hydroxylase (HIF-PH) inhibitor for the treatment of renal anemia. ChemMedChem 2018, 13, 988–1003. 10.1002/cmdc.201700783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toselli F.; Fredenwall M.; Svensson P.; Li X.-Q.; Johansson A.; Weidolf L.; Hayes M. A. Hip to be square: oxetanes as design elements to alter metabolic pathways. J. Med. Chem. 2019, 62 (16), 7383–7399. 10.1021/acs.jmedchem.9b00030. [DOI] [PubMed] [Google Scholar]
- Stepan A. F.; Karki K.; McDonald W. S.; Dorff P. H.; Dutra J. K.; Dirico K. J.; Won A.; Subramanyam C.; Efremov I. V.; O’Donnell C. J.; Nolan C. E.; Becker S. L.; Pustilnik L. R.; Sneed B.; Sun H.; Lu Y.; Robshaw A. E.; Riddell D.; O’Sullivan T. J.; Sibley E.; Capetta S.; Atchison K.; Hallgren A. J.; Miller E.; Wood A.; Obach R. S. Metabolism-directed design of oxetane-containing arylsulfonamide derivatives as gamma-secretase inhibitors. J. Med. Chem. 2011, 54 (22), 7772–7783. 10.1021/jm200893p. [DOI] [PubMed] [Google Scholar]
- Stepan A. F.; Tran T. P.; Helal C. J.; Brown M. S.; Chang C.; O’Connor R. E.; De Vivo M.; Doran S. D.; Fisher E. L.; Jenkinson S.; Karanian D.; Kormos B. L.; Sharma R.; Walker G. S.; Wright A. S.; Yang E. X.; Brodney M. A.; Wager T. T.; Verhoest P. R.; Obach R. S. Late-stage microsomal oxidation reduces drug-drug interaction and identifies phosphodiesterase 2A inhibitor PF-06815189. ACS Med. Chem. Lett. 2018, 9 (2), 68–72. 10.1021/acsmedchemlett.7b00343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostrom J.; Brown D. G.; Young R. J.; Kesuri G. M. Expanding the medicinal chemistry synthetic toolbox. Nat. Rev. Drug Discovery 2018, 17, 709–727. 10.1038/nrd.2018.116. [DOI] [PubMed] [Google Scholar]
- Kadam U.; Wilson I. A. Structure of Arbidol with influenza hemagglutinin. Proc. Natl. Acad. Sci. U. S. A. 2017, 114 (2), 206–214. 10.1073/pnas.1617020114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright Z. V. F.; Wu N. C.; Kadam R. U.; Wilson I. A.; Wolan D. W. Structure-based optimization and synthesis of antiviral drug Arbidol analogues with significantly improved affinity to influenza hemagglutinin. Bioorg. Med. Chem. Lett. 2017, 27 (16), 3744–3748. 10.1016/j.bmcl.2017.06.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer J.; Sager C. P.; Ernst B. Hydroxyl groups in synthetic and natural-product-derived therapeutics: a perspective on a common functional group. J. Med. Chem. 2019, 62 (20), 8915–8930. 10.1021/acs.jmedchem.9b00179. [DOI] [PubMed] [Google Scholar]
- Augeri D. J.; Robl J. A.; Betebenner D. A.; Magnin D. R.; Khanna A.; Robertson J. G.; Wang A.; Simpkins L. M.; Taunk P.; Huang Q.; Han S. P.; Abboa-Offei B.; Cap M.; Xin L.; Tao L.; Tozzo E.; Welzel G. E.; Egan D. M.; Marcinkeviciene J.; Chang S. Y.; Biller S. A.; Kirby M. S.; Parker R. A.; Hamann L. G. Discovery and preclinical profile of saxagliptin (BMS-477118): a highly potent, long-acting, orally active dipeptidy peptidase IV inhibitor for the treatment of type 2 diabetes. J. Med. Chem. 2005, 48, 5025–5037. 10.1021/jm050261p. [DOI] [PubMed] [Google Scholar]
- Ouvry G.; Clary L.; Tomas L.; Aurelly M.; Bonnary L.; Borde E.; Bouix-Peter C.; Chantalat L.; Defoin-Platel C.; Deret S.; Forissier M.; Harris C. S.; Isabet T.; Lamy L.; Luzy A. P.; Pascau J.; Soulet C.; Taddei A.; Taquet N.; Thoreau E.; Varvier E.; Vial E.; Hennequin L. F. Impact of minor structural modifications on properties of a series of mTOR inhibitors. ACS Med. Chem. Lett. 2019, 10 (11), 1561–1567. 10.1021/acsmedchemlett.9b00401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nittinger E.; Gibbons P.; Eligenbrot C.; Davies D. R.; Maurer B.; Yu C. L.; Kiefer J. R.; Kuglstatter A.; Murray J.; Ortwine D. F.; Tang Y.; Tsui V. Water molecules in protein-ligand interfaces. Evaluation of software tools and SAR comparison. J. Comput.-Aided Mol. Des. 2019, 33, 307–330. 10.1007/s10822-019-00187-y. [DOI] [PubMed] [Google Scholar]
- White M. C.; Zhao J. Aliphatic C-H oxidations for late-stage functionalization. J. Am. Chem. Soc. 2018, 140, 13988–14009. 10.1021/jacs.8b05195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Börgel J.; Tanwar L.; Berger F.; Ritter T. Late-stage aromatic C-H oxygenation. J. Am. Chem. Soc. 2018, 140, 16026–16031. 10.1021/jacs.8b09208. [DOI] [PubMed] [Google Scholar]
- Schroer K.; Kittelmann M.; Lütz S. Recombinant human cytochrome P450 monooxygenases for drug metabolite synthesis. Biotechnol. Bioeng. 2010, 106, 699–706. 10.1002/bit.22775. [DOI] [PubMed] [Google Scholar]
- Di Nardo G.; Gilardi G. Optimization of the bacterial cytochrome P450 BM3 system for the production of human drug metabolites. Int. J. Mol. Sci. 2012, 13, 15901–24. 10.3390/ijms131215901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler M.; Geier M.; Hanlon S. P.; Nidetzky B.; Glieder A. Human enzymes for organic synthesis. Angew. Chem., Int. Ed. 2018, 57, 13406–13423. 10.1002/anie.201800678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghisalba O.; Kittelmann M.. Preparation of Drug Metabolites Using Fungal and Bacterial Strains. In Modern Biooxidation: Enzymes, Reactions and Applications; Schmid R.D., Urlacher V.B., Eds.; John Wiley & Sons., 2007; Chapter 9, pp 215. [Google Scholar]
- Steele J.; de Riso A.; Williams H.; Phipps R.; Bardoni S.; Drake C.; Wrigley S.; Shanu-Wilson J.; Scheffler F.; Schulz S.; Ward J. Human metabolites of bosentan produced by enzymes in Hypha’s PolyCYPs® cytochrome P450 kit. Drug Metab. Pharmacokinet. 2018, 33, S32. 10.1016/j.dmpk.2017.11.121. [DOI] [Google Scholar]
- Sawayama A.; Chen M. Y.; Kulanthaivel P.; Kuo M.-S.; Hemmerle H.; Arnold F. H. A Panel of Cytochrome P450 BM3 Variants to Produce Drug Metabolites and Diversify Lead Compounds. Chem. - Eur. J. 2009, 15, 11723–11729. 10.1002/chem.200900643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele J. C. P.; De Riso A.; Falcioni F.; Phipps R. K.; Wrigley S. K.; Hopkins E. J.; Khan R. K.; Kokubun T.; Nytko K. L.; Poon V.; Schulz S.; Ward J. W.; Bawn M.. Biocatalytic techniques. PCT/GB2019/053337, 2019. [Google Scholar]
- Acevedo-Rocha C. G.; Gamble C. G.; Lonsdale R.; Li A.; Nett N.; Hoebenreich S.; Lingnau J. B.; Wirtz C.; Fares C.; Hinrichs H.; Deege A.; Mulholland A. J.; Nov Y.; Leys D.; McLean K. J.; Munro A. W.; Reetz M. T. P450-catalyzed regio- and diastereoselective steroid hydroxylation: efficient directed evolution enabled by mutability landscaping. ACS Catal. 2018, 8 (4), 3395–3410. 10.1021/acscatal.8b00389. [DOI] [Google Scholar]
- PolarExplorer - Late stage hydroxylation using PolyCYPs® enzymes. https://www.hyphadiscovery.co.uk/polycyps/polycyps-diversification-kits/.
- Smith R. V.; Rosazza J. P. Microbial models of mammalian metabolism. Aromatic hydroxylation. Arch. Biochem. Biophys. 1974, 161 (2), 551–558. 10.1016/0003-9861(74)90338-5. [DOI] [PubMed] [Google Scholar]
- Salter R.; Beshore D. C.; Colletti S. L.; Evans L.; Gong Y.; Helmy R.; Liu Y.; Maciolek C. M.; Martin G.; Pajkovic N.; Phipps R.; Small J.; Steele J.; de Vries R.; Williams H.; Martin I. J. Microbial biotransformation - an important tool for the study of drug metabolism. Xenobiotica 2019, 49, 877. 10.1080/00498254.2018.1512018. [DOI] [PubMed] [Google Scholar]
- Zhou L.; Satonin D.; Chappell J.; Yi P.; Moulton R.; Callies S.; Cassidy K. Elimination of [14C]-LY3023414 by aldehyde oxidase and CYP enzymes in humans following oral administration. Drug Metab. Pharmacokinet. 2019, 34 (1), S63. 10.1016/j.dmpk.2018.09.219. [DOI] [Google Scholar]
- Carlsen M.Biosynthesis, structural identification and quantification of low pg/mL levels of a major human metabolite of a dermal drug candidate. Presented at European Bioanalysis Forum, Barcelona, November 2016. http://www.e-b-f.eu/wp-content/uploads/2018/06/bcn2016-D2A3-2-Morten-Carlsen_Leo.pdf (accessed 2020-08-12). [Google Scholar]
- Center for Drug Evaluation and Research Application Number: 202192Orig1s000, Clinical Pharmacology and Biopharmaceutics Review for Ruxolitinib; Incyte Corporation, June 2011, p 23, https://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/202192Orig1s000ClinPharmR.pdf (accessed 2020-08-12). [Google Scholar]
- Zheng J.; Xin Y.; Zhang J.; Subramanian R.; Murray B. P.; Whitney J. A.; Warr M. R.; Ling J.; Moorehead L.; Kwan E.; Hemenway J.; Smith B. J.; Silverman J. A. Pharmacokinetics and disposition of momelotinib revealed a disproportionate human metabolite - resolution for clinical development. Drug Metab. Dispos. 2018, 46, 237–247. 10.1124/dmd.117.078899. [DOI] [PubMed] [Google Scholar]
- Johansson I.; Ingelman-Sundberg M. Genetic polymorphism and toxicology—with emphasis on cytochrome P450. Toxicol. Sci. 2011, 120 (1), 1–13. 10.1093/toxsci/kfq374. [DOI] [PubMed] [Google Scholar]
- Fukasawa T.; Suzuki A.; Otani K. Effects of genetic polymorphism of cytochrome P450 enzymes on the pharmacokinetics of benzodiazepines. J. Clin. Pharm. Ther. 2007, 32, 333–341. 10.1111/j.1365-2710.2007.00829.x. [DOI] [PubMed] [Google Scholar]
- Castellino S.; O’Mara M.; Koch K.; Borts D. J.; Bowers G. D.; MacLauchlin C. Human Metabolism of lapatinib, a dual kinase inhibitor: implications for hepatotoxicity. Drug Metab. Dispos. 2012, 40, 139–150. 10.1124/dmd.111.040949. [DOI] [PubMed] [Google Scholar]
- Bissada J. E.; Truong V.; Abouda A. A.; Wines K. J.; Crouch R. D.; Jackson K. D. Interindividual variation in CYP3A activity influences lapatinib bioactivation. Drug Metab. Dispos. 2019, 47, 1257–1269. 10.1124/dmd.119.088823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh J. S.; Reese M. J.; Thurmond L. M. The metabolic activation of abacavir by human liver cytosol and expressed human alcohol dehydrogenase isozymes. Chem.-Biol. Interact. 2002, 142, 135–154. 10.1016/S0009-2797(02)00059-5. [DOI] [PubMed] [Google Scholar]
- Diao X.; Deng P.; Xie C.; Li X.; Zhong D.; Zhang Y.; Chen X. Metabolism and pharmacokinetics of 3-n-butylphthalide (NBP) in humans: the role of cytochrome P450s and alcohol dehydrogenase in biotransformation. Drug Metab. Dispos. 2013, 41, 430–444. 10.1124/dmd.112.049684. [DOI] [PubMed] [Google Scholar]
- Brodney M. A.; Sharma R.; Lazzaro J. T.; Walker G. S.; Obach R. S. Harnessing biosynthesis and quantitative NMR for late stage functionalization of lead molecules: Application to the M1 positive allosteric modulator (PAM) program. Bioorg. Med. Chem. Lett. 2018, 28, 2068–2073. 10.1016/j.bmcl.2018.04.054. [DOI] [PubMed] [Google Scholar]
- Stepan A. F.; Walker D. P.; Bauman J.; Price D. A.; Baillie T. A.; Kalgutkar A. S.; Aleo M. D. Structural alert/reactive metabolite concept as applied in medicinal chemistry to mitigate the risk of idiosyncratic drug toxicity; a perspective based on the critical examination of trends in the top 200 drugs marketed in the United States. Chem. Res. Toxicol. 2011, 24, 1345–1410. 10.1021/tx200168d. [DOI] [PubMed] [Google Scholar]
- Kalgutkar A. S. Designing around structural alerts in drug discovery. J. Med. Chem. 2020, 63 (12), 6276–6302. 10.1021/acs.jmedchem.9b00917. [DOI] [PubMed] [Google Scholar]
- Towles J. K.; Clark R. N.; Wahlin M. D.; Uttamsingh V.; Rettie A. E.; Jackson K. D. Cytochrome P450 3A4 and CYP3A5-catalysed bioactivation of lapatinib. Drug Metab. Dispos. 2016, 44, 1584–1597. 10.1124/dmd.116.070839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll J. P.; Yadav A. S.; Shay N. R. Role of glucuronidation and P450 oxidation in the bioactivation of bromfenac. Chem. Res. Toxicol. 2018, 31, 223–230. 10.1021/acs.chemrestox.7b00293. [DOI] [PubMed] [Google Scholar]
- Yadav A. S.; Shah N. R.; Carlson T. J.; Driscoll J. P. Metabolite profiling and reaction phenotyping for the in vitro assessment of the bioactivation of bromfenac. Chem. Res. Toxicol. 2020, 33, 249–257. 10.1021/acs.chemrestox.9b00268. [DOI] [PubMed] [Google Scholar]
- Shang J.; Tschirret-Guth R.; Cancilla M.; Samuel K.; Chen Q.; Chobanian H. R.; Thomas A.; Tong W.; Josein H.; Buevich A. V.; Mitra K. Bioactivation of GPR40 agonist MK-8666: formation of protein adducts in vitro from reactive acyl glucuronide and acyl CoA thioester. Chem. Res. Toxicol. 2020, 33, 191–201. 10.1021/acs.chemrestox.9b00226. [DOI] [PubMed] [Google Scholar]
- Lou Y.; Lopez F.; Jiang Y.; Han X.; Brotherton C.; Billedeau R.; Gabriel S.; Gleason S.; Goldstein D. M.; Hilgenkamp R.; Kocer B.; Orzechowski L.; Tan J.; Wovkulich P.; Wen B.; Fry D.; Di Lello P.; Chen L.; Zhang F.-J.; Fretland J.; Nangia A.; Yang T.; Owens T. D. Mitigation of reactive metabolite formation for a series of 3-amino-2-pyridone inhibitors of Bruton’s tyrosine kinase (BTK). Bioorg. Med. Chem. Lett. 2017, 27, 632–635. 10.1016/j.bmcl.2016.11.092. [DOI] [PubMed] [Google Scholar]
- Yip L. Y.; Chan E. C. Y. Investigation of host-gut microbiota modulation of therapeutic outcome. Drug Metab. Dispos. 2015, 43, 1619–1631. 10.1124/dmd.115.063750. [DOI] [PubMed] [Google Scholar]
- Sousa T.; Paterson R.; Moore V.; Carlsson A.; Abrahamsson B.; Basit A. W. The gastrointestinal microbiota as a site for biotransformation of drugs. Int. J. Pharm. 2008, 363, 1–25. 10.1016/j.ijpharm.2008.07.009. [DOI] [PubMed] [Google Scholar]
- Koppel N.; Rekdal V. M.; Balskus E. P. Chemical transformation of xenobiotics by the human gut microbiota. Science 2017, 356, eaag2770 10.1126/science.aag2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke G.; Sandhu K. V.; Griffin B. T.; Dinan T. G.; Cryan J. F.; Hyland N. P. Gut reactions: breaking down xenobiotic-microbiome interactions. Pharmacol. Rev. 2019, 71, 198–224. 10.1124/pr.118.015768. [DOI] [PubMed] [Google Scholar]
- Xie Y.; Hu F.; Xiang D.; Lu H.; Li W.; Zhao A.; Huang L.; Wang R. The metabolic effect of gut microbiota on drugs. Drug Metab. Rev. 2020, 52, 139–156. 10.1080/03602532.2020.1718691. [DOI] [PubMed] [Google Scholar]
- Wilson I. D.; Nicholson J. K. Gut microbiome interactions with drug metabolism, efficacy, and toxicity. Translational Research 2017, 179, 204–222. 10.1016/j.trsl.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q.; Zhang Y.; Boer J.; Shi J. G.; Hu P.; Diamond S.; Yeleswaram S. In vitro interaction of epacadostat and its major metabolites with human efflux and uptake transporters: implications for pharmacokinetics and drug interactions. Drug Metab. Dispos. 2017, 45, 612–623. 10.1124/dmd.116.074609. [DOI] [PubMed] [Google Scholar]
- Li Y.; Lai W. G.; Whitcher-Johnstone A.; Busacca C. A.; Eriksson M. C.; Lorenz J. C.; Tweedie D. J. Metabolic switching of BILR 355 in the presence of ritonavir. I. Identifying an unexpected disproportionate human metabolite. Drug Metab. Dispos. 2012, 40, 1122–1129. 10.1124/dmd.111.044354. [DOI] [PubMed] [Google Scholar]
- Li Y.; Xu J.; Lai W. G.; Whitcher-Johnstone A.; Tweedie D. J. Metabolic switching of BILR 355 in the presence of ritonavir. II. Uncovering novel contributions by gut bacteria and aldehyde oxidase. Drug Metab. Dispos. 2012, 40, 1130–1137. 10.1124/dmd.111.044362. [DOI] [PubMed] [Google Scholar]