

Abstract
Background
Critically ill coronavirus disease 2019 (COVID-19) patients may suffer persistent systemic inflammation and multiple organ failure, leading to a poor prognosis.
Research question
To examine the relevance of the novel inflammatory factor heparin-binding protein (HBP) in critically ill COVID-19 patients, and evaluate the correlation of the biomarker with disease progression.
Study design and methods
18 critically ill COVID-19 patients who suffered from respiratory failure and sepsis, including 12 cases who experienced a rapidly deteriorating clinical condition and six cases without deterioration, were investigated. They were compared with 15 age- and sex- matched COVID-19-negative patients with respiratory failure. Clinical data were collected and HBP levels were investigated.
Results
HBP was significantly increased in critically ill COVID-19 patients following disease aggravation and tracked with disease progression. HBP elevation preceded the clinical manifestations for up to 5 days and was closely correlated with patients’ pulmonary ventilation and perfusion status.
Interpretation
HBP levels are associated with COVID-19 disease progression in critically ill patients. As a potential mediator of disease aggravation and multiple organ injuries that are triggered by continuing inflammation and oxygen deficits, HBP warrants further study as a disease biomarker and potential therapeutic target.
Short abstract
For the first time, this study observed that heparin-binding protein (HBP) was significantly increased in critically ill COVID-19 patients during disease aggravation, which highlights HBP as a disease mediator and a potential therapeutic target https://bit.ly/35dz88C
Introduction
The global pandemic of coronavirus disease 2019 (COVID-19) starting in December 2019 was caused by a novel coronavirus named as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) by the International Committee on Taxonomy of Viruses [1]. Sequencing analysis of the virus genome indicates 79.5% gene sequence homology with SARS-CoV from 2002, implying that the pathogenic processes underlying severe pneumonia may share similarities between both viruses [1–3]. The pathogenesis of COVID-19 has been unfolding, and clinicians are starting to focus on rehabilitation and treatment of the associated inflammatory sequelae [4]. Consequently, several studies have reported that some SARS-CoV-2-positive patients with stable clinical conditions and nucleic acid tests converted to negative may suddenly worsen or even die during the post-viral infection stage [5–7]. While false negative testing as well as underlying chronic diseases exacerbated by the stress of infection could be contributing factors, an inflammatory condition caused by SARS-CoV-2 may persist after viral RNA is no longer detectable. A secondary inflammatory storm may occur during this stage of infection, causing multiorgan failures [5, 8–12]. The aggravation of such inflammatory processes during an otherwise clinically stable state was reported in 2002 during the SARS outbreak as well as in some cases of severe community-acquired pneumonia [13–15]. Therefore, new knowledge about the mechanisms of the worsening of COVID-19 patients is warranted to establish interventions that may significantly improve the prognosis [9, 16]. Biomarkers associated with disease aggravation as well as potential therapeutic targets would be extremely valuable in this respect.
Heparin-binding protein (HBP), also known as azurocidin or cationic antimicrobial protein of 37 kDa [17] is stored in azurophilic granules and secretory vesicles of polymorphonuclear neutrophils (PMN), at the cell membrane and in the azurophilic granules [18]. Endothelial cells are the primary targets of HBP released in the circulation and this protein plays a major role in the mechanism of capillary leakage, while it amplifies inflammatory effects by inducing cytoskeletal rearrangement and resulting in increased permeability of the vascular endothelial barrier [17, 19]. Antigens, inflammatory factors, cytokines and enzymes can stimulate PMN to release HBP by exocytosis [20, 21]. Studies in sepsis have indicated that the sensitivity and specificity of HBP are varied, compared to C-reactive protein (CRP) or procalcitonin (PCT), as an independent predictor of clinical outcome in severe infection [22, 23]. Our recent study highlighted the role of IgA deposition and its mediated vasculitis in severe COVID-19 [24]. Considering the major role of HBP in endothelial dysfunction and systemic vasculitis in severe infections, we hypothesised that elevated HBP might mediate the pathogenesis of severe COVID-19 aggravation. The goal of this study was to establish whether HBP is a useful biomarker and is associated with disease aggravation for those patients eventually requiring advanced treatment options. HBP could serve as a potential therapeutic target for patients who are at risk of developing systemic multiple organ failures, with improvement in the patient prognosis and to prevent death in severely ill patients.
Materials and methods
Participants
According to the latest international diagnostic criteria for COVID-19 [25], 18 patients with critical respiratory failure and clinical presentation of a viral-like sepsis were included in this study conducted at the First Affiliated Hospital of Guangzhou Medical University. Exclusion criteria were a history of chronic liver or kidney disease.
Among the 18 patients, two major clinical patterns were seen, i.e. 12 patients met the characteristics of a clinical stable period before exacerbations of COVID-19 rather than secondary nosocomial infections in the clinical stable period, while six other patients had been without aggravation. We referred to the studies by Blum et al. [26] and Paul et al. [27] for the definition of clinical stable period of severe infections, characterised by vital signs stable for >24 h (<37.5°C, heart rate <100 beats·min−1, respiratory rate 24 breaths·min−1), no shock, no respiratory symptoms such as dyspnoea and chest tightness. The definition of the clinical deterioration of the disease was based on the clinical studies by Vultaggio et al. [28] and Yang et al. [29], i.e. meeting one of the following criteria after a clinical stable period: 1) death during hospitalisation [28]; 2) transferred to the intensive care unit due to severe exacerbation of respiratory failure requiring invasive support measures to correct hypoxaemia [28]; 3) aggravated respiratory failure without requiring invasive ventilation support [28]; 4) respiratory distress (respiratory rate ≥30 breaths·min−1) [29]; 5) resting oxygen saturation ≤93% [29]; 6) arterial oxygen partial pressure/inhaled oxygen fraction ≤300 mmHg [29]. Considering the possibility of false negative nucleic acid tests in COVID-19 patients and differences across different sites, we assessed disease progression by a comprehensive evaluation involving three clinicians and three investigators.
According to the previous studies, most COVID-19 patients with sepsis involve nosocomial infections [30, 31]. In line with the purpose of this study on HBP and COVID-19 aggravation by persistent inflammation, it is of great importance to exclude patients aggravated by nosocomial infection. Therefore, we emphasised that no new pathogens were detected in sputum and midstream urine to exclude patients with nosocomial infection.
15 age- and sex-matched COVID-19 negative patients with respiratory failure were included for comparison. Clinical information was extensively documented, and the level of HBP was assessed in each participant. This study lasted for 125 days. Two time points were set: admission point and clinically stable point. The different clinical status/periods in each patient were comprehensively assessed and distinguished.
HBP quantification
Fresh venous blood was collected from the participants and placed in an upright tube for 1–2 h, then centrifuged at 1500×g for 15 min. Plasma samples with sodium citrate anticoagulation (1:9) were tested. When separating plasma, care was taken not to aspirate the white blood cell layer to avoid the releasing of high levels of HBP. HBP is easily degraded (half-life is 1 h); therefore, all samples in the study were detected immediately after collection within 1 h. Jet-iStar 3000 automatic analyser (Joinstar Biomedical Technology, Zhejiang, China) was used to assess 50 µL of sample, which was incubated for 18 min, and the HBP level detected using dry fluorescent immunoassay methodology.
Imaging
Lungs were imaged to acquire posteroanterior oblique (PA) and lateral (LAT) views and quantitative high-resolution computed tomography (HRCT) analysis was undertaken. Care was taken for chest PA and LAT examinations to ensure that the patient's respiratory cycle did not blur the image by rapid exposure at inspiration (0.08 s). The centreline of the PA plate was aligned with the fifth dorsal thoracic vertebra, while the centreline of the LAT plate to the lateral thoracic wall at the fifth thoracic vertebra. The cassette was placed at 3 cm above the shoulder on the opposite side. The field covered the costophrenic angle. Major evaluations covered pulmonary markings, substantive area, cardiac shadow area and the costophrenic angle. The percentage of total lung area affected by the infection was adopted to evaluate the area of pulmonary involvement. HRCT was set at a slice thickness of 1.0 mm. The quantitative analysis system was used to evaluate the area of solid shadow, patch shadow and fibrous strip shadow. Additionally, we also acquired abdominal HRCT images to evaluate organ function during the study period (i.e. with abnormally low or high density).
Data analysis and statistics
Continuous variables were expressed as median and interquartile range. Nonparametric testing (Mann–Whitney test) was used to analyse differences between groups. Correlation was assessed using Spearman's correlation coefficient. A value of p<0.05 was considered statistically significant. Cross-correlation function (CCF) testing was applied to explore correlations between different indexes and HBP levels [32]. The phase relationship between different index trends and HBP trends was examined by determining the time lag at which the value of the CCF was maximal [32]. Standardised method was adopted as 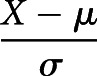 , i.e. the value of the variable minus the mean divided by the standard deviation. R (version 4.0.0; Bell Laboratories, Murray Hill, NJ, USA), GraphPad Prism 8.0.2 (GraphPad Software, San Diego, CA, USA) and IBM SPSS (Statistics for Windows version 22.0; IBM, Chicago, IL, USA) were used to analyse data and draw figures.
, i.e. the value of the variable minus the mean divided by the standard deviation. R (version 4.0.0; Bell Laboratories, Murray Hill, NJ, USA), GraphPad Prism 8.0.2 (GraphPad Software, San Diego, CA, USA) and IBM SPSS (Statistics for Windows version 22.0; IBM, Chicago, IL, USA) were used to analyse data and draw figures.
Results
Clinical characteristics
As shown in table 1, HBP levels were significantly higher in 18 critical COVID-19 patients compared to patients with other respiratory failure. CRP and PCT were also higher in COVID-19 patients. Although the white blood cell and neutrophil counts were higher in COVID-19, the lymphocyte count was lower. D-dimer level was significantly higher in COVID-19, suggesting extensive disturbances to the microcirculation despite the absence of hypotensive shock and diagnostic criteria for disseminated intravascular coagulation not being met. This finding of D-dimer change has also been observed by Zhou et al. [33]. In table 2, we show the dynamic changes of various indicators in the 12 patients with aggravation in the process of the disease. Continuous high levels of HBP were seen in these patients with further increase observed with the disease aggravation. In contrast, the six patients without aggravation had decreasing levels of HBP with the stabilisation of the disease (table 3).
TABLE 1.
Clinical characteristics of the participants#
| Respiratory failure | Critical COVID-19 | p-value | |
| Subjects | 15 | 18 | |
| Age years | 56.00 (37.00–65.00) | 59.50 (49.00–72.00) | 0.164 |
| Male/female | 9/6 | 12/6 | 0.731 |
| HBP ng·mL−1 | 12.58 (5.90–29.85) | 113.20 (50.57–186.00) | <0.001 |
| ALT U·L−1 | 23.77 (8.19–36.47) | 21.25 (13.00–34.50) | 0.064 |
| AST U·L−1 | 31.60 (30.10–40.30) | 35.74 (26.40–65.35) | <0.001 |
| BUN mg·dL−1 | 6.18 (5.43–7.20) | 10.80 (5.30–15.40) | 0.002 |
| Creatinine μmol·mL−1 | 67.10 (54.39–91.01) | 76.40 (55.60–103.50) | 0.038 |
| CRP mg·L−1 | 0.13 (0.05–0.24) | 10.37 (5.70–16.10) | <0.001 |
| D-dimer μg·L−1 | 811 (159.8–3500.00) | 3655 (1800–6353) | <0.001 |
| PCT μg·L−1 | 0.11 (0.05–0.17) | 0.30 (0.15–0.98) | <0.001 |
| K+ mmol·L−1 | 4.27 (4.10–4.62) | 4.17 (3.93–4.51) | 0.086 |
| Na+ mmol·L−1 | 135.50 (135.20–140.10) | 135.50 (131.20–143.60) | 0.305 |
| Cl− mmol·L−1 | 97.10 (94.33–104.13 | 94.50 (81.10–115.30) | 0.327 |
| Ca2+ mmol·L−1 | 2.24 (2.20–2.51) | 2.24 (2.06–2.45) | 0.394 |
| White blood cells ×109 cells·L−1 | 5.13 (4.77–7.31) | 8.46 (7.20–11.40) | <0.001 |
| Neutrophils ×109 cells·L−1 | 2.31 (1.94–4.00) | 6.80 (5.20–8.60) | <0.001 |
| Lymphocytes ×109 cells·L−1 | 1.53 (0.97–1.66) | 0.95 (0.62–1.50) | 0.021 |
| Red blood cells ×1012 cells·L−1 | 4.31 (3.44–5.19) | 2.90 (2.65–3.41) | <0.001 |
| Platelets ×109 cells·L−1 | 154.00 (117.00–203.00) | 160.00 (109.00–221.00) | 0.527 |
Data are presented as n or median (interquartile range), unless otherwise stated. COVID-19: coronavirus disease 2019; HBP: heparin-binding protein; ALT: alanine transaminase; AST: aspartate transaminase; BUN: blood urea nitrogen; CRP: C-reactive protein; PCT: procalcitonin; K+: potassium; Na+: sodium; Cl−: chloride; Ca2+: calcium. #: day 1 data, except HBP, which was the average of multiple time points.
TABLE 2.
The dynamic changes of various clinical indicators in the 12 patients with aggravation
| Admission time days | |||||||||||
| 1 | 5 | 10 | 15 | 20 | 25 | 30 | 35 | 40 | 45 | 50 | |
| PSI score | 147.20 (133.00–172.00) | 149.50 (117.30–172.00) | 118.50 (104.30–141.00) | 109.50 (88.00–130.50) | 103.50 (89.00–151.00) | 108.50 (89.00–151.00) | 115.50 (101.30–144.00) | 102.00 (91.00–122.00) | 104.00 (85.00–1156.00) | 100.00 (72.50–115.00) | 85.00 (61.00–116.00) |
| HBP mg·mL−1 | 116.20 (47.65–230.00) | 85.39 (35.98–184.00) | 92.96 (54.83–162.30) | 89.12 (45.10–145.30) | 72.22 (35.26–108.60) | 106.30 (37.39–142.00) | 92.31 (35.00–183.40) | 162.80 (91.94–288.60) | 179.90 (109.50–248.80) | 165.40 (94.52–266.90) | 129.20 (43.12–238.80) |
| IgA AU·mL−1 | 17.97 (7.98–155.50) | 15.96 (7.30–67.68) | 18.70 (9.65–34.73) | 22.28 (13.70–70.89) | 18.88 (11.40–92.46) | 20.43 (8.41–32.82) | 9.18 (5.02–22.42) | 7.66 (5.21–11.43) | 20.69 (6.42–35.74) | 23.56 (8.82–67.11) | 22.62 (5.31–51.25) |
| PCT ng·mL−1 | 0.38 (0.17–1.18) | 0.38 (0.10–1.08) | 0.24 (0.10–0.71) | 0.30 (0.15–0.75) | 0.73 (0.20–2.09) | 0.80 (0.19–1.29) | 0.88 (0.44–1.32) | 0.48 (0.19–1.38) | 1.01 (0.33–1.15) | 0.48 (0.24–0.63) | 0.42 (0.14–1.08) |
| CRP ng·mL−1 | 8.64 (6.93–9.96) | 10.51 (7.91–12.32) | 6.05 (2.08–13.37) | 4.09 (1.07–13.71) | 15.44 (7.71–19.15) | 10.17 (6.77–17.83) | 7.45 (5.87–11.98) | 8.98 (5.76–11.34) | 15.93 (11.42–19.95) | 18.77 (16.12–23.74) | 4.18 (3.09–9.27) |
| Neutrophils ×109 cells·L−1 | 5.70 (3.80–7.40) | 5.45 (3.30–7.45) | 5.70 (4.43–7.50) | 6.10 (5.28–7.08) | 6.10 (5.10–7.10) | 6.30 (4.58–6.80) | 6.35 (5.05–7.15) | 6.50 (5.40–8.20) | 6.15 (5.48–7.73) | 5.60 (5.10–5.95) | 4.90 (4.20–5.50) |
| D-dimer μg·L−1 | 2939.00 (869.80–5477.00) | 2972.00 (896.00–4897.00) | 3186.00 (1738.00–6927.00) | 2320.00 (1994.00–3453.00) | 2455.00 (1509.00–2936.00) | 2060.00 (1271.00–3596.00) | 1320.00 (615.80–5552.00) | 4530.00 (2131.00–4792.00) | 7576.00 (3810.00–8252.00) | 6390.00 (5607.00–7420.00) | 4308.00 (1094.00–6163.00) |
| HRCT % | 35 | 35 | 35 | 30 | 30 | 27 | 30 | 30 | 50 | 60 | 58 |
| Chest PA and LAT % | 45 | 40 | 35 | 25 | 23 | 35 | 35 | 35 | 55 | 65 | 45 |
Data are presented as median (interquartile range), unless otherwise stated. PSI: pneumonia severity index; HBP: heparin-binding protein; AU: arbitrary unit; PCT: procalcitonin; CRP: C-reactive protein; HRCT: high-resolution computed tomography; PA: posteroanterior oblique; LAT: lateral.
TABLE 3.
The dynamic changes of various clinical indicators in the six patients without aggravation
| Admission time days | |||||||||||
| 1 | 5 | 10 | 15 | 20 | 25 | 30 | 35 | 40 | 45 | 50 | |
| PSI score | 104.00 (89.25–115.00) | 98.00 (79.50–115.0) | 96.00 (64.50–109.30) | 85.50 (59.00–98.75) | 85.50 (59.00–98.75) | 75.50 (54.50–87.50) | 71.00 (54.50–86.75) | 66.00 (50.25–86.75) | 53.00 (43.00–80.00) | 59.00 (44.50–65.50) | 50.00 (37.50–57.00) |
| HBP mg·mL−1 | 103.30 (52.22–106.80) | 107.60 (77.31–160.60) | 78.22 (60.10–147.70) | 69.15 (63.14–195.40) | 77.75 (24.75–135.40) | 35.45 (29.45–41.45) | 30.75 (16.46–98.79) | 23.66 (9.89–30.64) | 15.39 (5.77–26.75) | 21.39 (6.41–39.19) | 27.16 (14.77–35.14) |
| IgA AU·mL−1 | 43.40 (11.37–221.80) | 77.65 (23.47–182.00) | 47.34 (15.96–232.80) | 34.91 (23.86–174.80) | 59.86 (22.61–177.10) | 64.94 (16.48–157.90) | 37.18 (15.46–126.30) | 37.38 (7.72–86.80) | 8.78 (5.95–47.73) | 8.55 (5.75–39.14) | 7.77 (5.58–40.47) |
| PCT ng·mL−1 | 0.26 (0.19–1.35) | 0.15 (0.06–1.23) | 0.13 (0.06–1.61) | 0.18 (0.09–1.23) | 0.24 (0.11–1.41) | 0.80 (0.19–1.29) | 0.54 (0.20–0.97) | 0.31 (0.04–1.05) | 0.30 (0.04–0.93) | 0.15 (0.03–0.88) | 0.09 (0.02–0.56) |
| CRP ng·mL−1 | 2.71 (0.81–11.34) | 2.71 (0.40–9.73) | 9.27 (0.16–13.06) | 4.83 (1.30–10.63) | 3.24 (0.93–12.18) | 2.26 (0.56–10.21) | 1.60 (0.33–9.03) | 1.35 (1.08–8.99) | 2.19 (0.60–3.83) | 2.22 (1.09–4.09) | 1.73 (1.00–2.81) |
| Neutrophil ×109 cells·L−1 | 5.40 (3.35–11.18) | 5.50 (3.10–7.70) | 5.40 (3.35–9.18) | 5.10 (4.00–8.80) | 5.60 (3.60–6.40) | 4.60 (3.23–6.00) | 4.65 (2.58–6.48) | 5.60 (3.10–6.75) | 4.00 (2.93–5.78) | 4.40 (3.55–5.65) | 4.60 (4.00–4.85) |
| D-dimer μg·L−1 | 751.50 (656.30–3770.00) | 2101.00 (741.50–5586.00) | 1799.00 (482.00–5210.00) | 1296.00 (567.00–7045.00) | 689.00 (237.00–9545.00) |
358.00 (266.50–2212.00) | 482.00 (267.30–2204.00) | 270.00 (166.00–806.00) | 259.00 (147.00–350.00) | 489.00 (457.50–545.50) | 475.00 (255.00–730.50) |
| HRCT % | 45 | 43 | 50 | 40 | 42 | 27 | 30 | 27 | 10 | 5 | 5 |
| Chest PA and LAT % | 50 | 39 | 35 | 31 | 26 | 20 | 15 | 20 | 15 | 10 | 5 |
Data are presented as median (interquartile range), unless otherwise stated. PSI: pneumonia severity index; HBP: heparin-binding protein; AU: arbitrary unit; PCT: procalcitonin; CRP: C-reactive protein; HRCT: high-resolution computed tomography; PA: posteroanterior oblique; LAT: lateral.
HBP and multisystem indices
HBP changes and their correlation with inflammatory markers
As shown, there is significant correlation between HBP and PMN in the aggravation group (p<0.05, r=0.35); however, in the remission group there was no significant correlation (figure 1a). Although the two mechanisms are closely related, they do not look completely parallel from the data. The HBP level in the case group with aggravation showed a significant upward trend since the clinical stable period, which peaked on the 10th day after the aggravation period, and it began to decline at the 20th day as the disease got under control. As disease progression of patients with severe COVID-19 who developed aggravation occurred at varied time points, we used time ranges to indicate aggravation. PMN showed a similar trend, but the increase was not as significant as for HBP and the decrease at later time points was greater. In contrast, the HBP level of the patients with remission of the disease showed a significant downward trend. In these patients, the HBP levels were significantly correlated with CRP and PCT (r=0.463 and 0.497, respectively; both p<0.001).
FIGURE 1.

Changes in heparin-binding protein (HBP) levels over time and correlation with the level of inflammation. a) Comparison of longitudinal trends of HBP changes and polymorphonuclear neutrophils (PMN) during hospitalisation. On the graph, severe acute respiratory syndrome coronavirus 2 infection represents the remission stage of coronavirus disease 2019 in the presence of a positive RNA test. The period of remission/recurrence represents the period of viral negativity during remission during which the patient's condition nonetheless worsened again. It should be noted that the trend curve of PMN in the figure is for patients with aggravation. b) Further analysis of the remission/recurrence period. Six inflammatory markers were compared after normalisation. The larger the sphere, the higher the level. IL: interleukin; TNF: tumour necrosis factor; IFN: interferon.
Comparing HBP levels with an inflammatory index composed of interleukin (IL)-2, IL-4, IL-6, IL-10, tumour necrosis factor-α and interferon-γ (figure 1b and supplementary table 1) revealed that IL-6 showed the most significant increase over the progress of disease. We further projected these variables onto the plane of the inflammatory indices and HBP to enable more intuitive visualisation of the linear relationship, and found that IL-6 had a strikingly positive correlation with HBP (r=0.693, p<0.001), and a significant correlation with disease duration (p<0.001).
Coagulation index analysis
Activated partial thromboplastin time (APTT), which closely tracks endogenous coagulation, is longer in the aggravation period than the clinical stable period (supplementary table 2). D-dimer was markedly increased in the aggravation period in clinical stable period and correlated with HBP. However, patients’ clinical manifestations did not meet the diagnostic criteria for disseminated intravascular coagulation.
Correlation of blood gas index and lung exudation level
We compared data on the changes of pulmonary effusion from HRCT and chest PA and LAT at the admission time point, with the subsequent trajectory of HBP in different clinical periods (figure 2a). The aggravation period after the clinical stable period shows that the increase of HBP occurs earlier, at ∼35–40 days, while HRCT and chest PA and LAT peak at ∼45 days. After calculating the cross-correlation function of HRCT and chest PA and LAT (figure 2b and c), we found that the strongest correlation with HBP was when HRCT and chest PA and LAT lagged by 5 days. Thus, HBP rises and reaches a peak ∼5 days prior to HRCT and chest PA and LAT. Our further analysis showed that HBP was correlated with the arterial–alveolar oxygen differential, intrapulmonary shunt volume, respiratory index and oxygenation index (supplementary figure S2).
FIGURE 2.

Comparison between the trends of high-resolution computed tomography (HRCT) and chest posteroanterior oblique (PA) and lateral (LAT) views in critically ill coronavirus disease 2019 patients. a) The y-axis is the percentage of lung area with exudative lesions in HRCT and chest PA and LAT. The figure shows the time when the three indicators peak. The time line was from the start of admission to the end of the study for a total of 125 days. b) Cross-correlation function (CCF) analysis between HRCT and heparin-binding protein (HBP). The correlation of HRCT and HBP is highest with a 5-day lag for HRCT (r=0.92, p<0.05). c) CCF analysis between chest PA and LAT and HBP. The correlation between chest PA and LAT and HBP is highest at 5 days (r=0.804, p<0.05).
Evaluation of extrapulmonary organ function in patients with COVID-19
The overall levels of aspartate transaminase (AST), creatine kinase (CK), CK isoenzymes, cardiac troponin I and myoglobin increased at 35–45 days at the aggravation period after clinical stable period (supplementary table 3, supplementary figure S3A). During this period, the pro-brain natriuretic peptide of the patients was 2479 (95% CI 603.4–3379) pg·mL−1, which was significantly higher than the healthy level (<50 pg·mL−1). No patients had a significant decrease in urine output. Imaging indicated that one patient had a pericardial effusion, but no enlargement in cardiac shadow or abnormalities in cardiovascular performance. Compared with the changes of HBP levels (peaking at 35–40 mg·mL−1), there was a time lag in the changes of the above five markers. After calculating the cross-correlation (supplementary figure S3B), it was again found that a 5-day lag yielded the strongest correlation with HBP, peaking ∼5 days earlier than AST, CK, CK isoenzymes, cardiac troponin I and myoglobin.
We selected blood urea nitrogen (BUN), creatinine and potassium (K+) as indicators to reflect kidney function. In the aggravation stage, BUN and creatinine, but not K+ showed a significant upward trend (supplementary table 4 and supplementary figure S4A). During this period, abdominal computed tomography (CT) showed no obvious abnormalities in the kidneys. CCF analysis (supplementary figure S4B) showed a strong positive correlation between BUN, creatinine and HBP, indicating that changes in BUN and creatinine mirror those of HBP, again with a 5-day time lag. For K+, values held steady without an obvious rise or peak, so the correlation between K+ and HBP is very weak.
The AST/alanine transaminase (ALT) ratio, as an indicator reflecting liver injury, showed an upward trend starting at 35 days, and reached the peak of >3 at 40 days (supplementary figure S4A), indicating more severe hepatocyte destruction. However, during this period, abdominal CT indicated no new abnormal low-density lesions in the liver.
SARS-CoV-2 RNA positivity
In the severe COVID-19 patients, seven patients exhibited a change from negative to positive during the stable period. We summarised the nucleic acid test results at different times and at different sites in the patient population in the study (supplementary table 5). Unsurprisingly, we did not find a correlation between HBP and these test results, and thus concluded that the increase in HBP was unrelated to the reversion of RNA positivity.
Discussion
Severe COVID-19 and its aggravation
Respiratory symptoms are the main manifestation of COVID-19, and severe diffuse alveolar epithelial injury is one of the major pathological features [34]. In severely ill patients, pulmonary exudation is significantly increased and abnormal dilatation of the pulmonary vessels at the sites of lesions is commonly observed [35]. However, extrapulmonary organs such as the liver, kidney and heart may also suffer direct or secondary damage [36]. SARS-CoV-2 can trigger inflammatory storms affecting perfusion, resulting in multiorgan dysfunction, which has been reported in imaging, biopsy and autopsy studies, despite there being no direct evidence of viral infection in these extrapulmonary organs [36–38]. At the clinical stable stage of severe COVID-19 with respiratory failure combined with sepsis, some patients aggravated and suffered multiple organ injuries. Studies have reported that systemic inflammatory responses are stronger in the elderly, and those with weakened adaptive immunity and malnutrition may have more severe clinical manifestations and are more likely to relapse without again testing positive for the virus, due to non-infection-related secondary inflammation. This is one of the leading causes of death during the stable period and is comparable to the risk of sudden cardiac death from COVID-19 [39–42]. We speculated that it may be caused by persistent inflammation triggered by SARS-CoV-2. Pathogens and their fragments released after tissue destruction are recognised as foreign bodies. Immune dysfunction may lead to clinical deterioration. Excessive activation of innate immunity aggravates the damage to tissues and organs, reinforcing a vicious cycle [43].
HBP: a biomarker and a critical mediator of COVID-19 aggravation
In this study, we found that the novel inflammatory factor HBP was closely correlated with the aggravation of severe COVID-19 with respiratory failure, as well as its correlation with levels of inflammatory markers, coagulation abnormalities and pulmonary injuries.
HBP is stored in vesicles in PMN near the cell membrane and is released when they are activated. In the present study, correlations of the changes of HBP levels and PMN with the exacerbation of disease in the stable stage were consistent; both began to rise at 30 days and peaked at 35 days. Correlation analysis of the six inflammatory factors measured in this study showed that IL-6 levels were significantly increased and related to HBP, and the two factors paralleled disease progression over time. The level of IL-6 can be elevated in the early stages of an inflammatory storm, after which CRP, PCT and amyloid protein increase, all positively correlated with the progression of inflammation and inhibition of PMN apoptosis [44]. Studies have shown that IL-6 amplifies the inflammatory response and enhances PMN production, and activates them to release HBP [45–47].
Pulmonary exudation is significantly increased in COVID-19 patients, which is closely related to the mechanism of pulmonary vascular leakage activated by HBP (supplementary figure S5). Vascularity and neutrophil exosmosis and aggregation can further aggravate systemic inflammatory responses, and finally exacerbate sepsis. Moreover, Albarello et al. [48] found local abnormal dilatation of blood vessels in the lung exudation area (not caused by microthrombi). Pulmonary blood flow perfusion imbalance is also one of the causes of hypoxaemia, but the mechanism is unknown. We made speculated further, according to the research from Morsing et al. [49] and Lauritzen et al. [50], that the local release of vasodilatory substances such as histamines may be caused by leukocyte extravasation which is activated by endothelial cytoskeletal reorganisation, in turn activated by HBP, leading to pulmonary vascular dilatation and tortuosity when under the inflammatory environment. Song et al. [51] found that increasing HBP damages surface polysaccharides of epithelial cells, and activates endogenous coagulation mechanisms, causing APTT prolongation and D-dimer increases.
The cytoskeleton consists of microfilaments, microtubules and intermediate fibres: the microfilaments are composed of actin, myosin and actin-binding proteins [52]. HBP binds to and internalises the surface polysaccharide receptor of endothelial cells to activate protein kinase C (PKC) and Pho kinase [53, 54]. PKC in turn activates voltage-gated calcium (Ca) channels, facilitating Ca2+ influx, increasing concentrations of which result in altered actin alignment [54]. In addition, PKC phosphorylates cytoskeletal proteins directly and causes contraction. Rho can promote myosin light chain phosphorylation and initiate cytoskeletal rearrangement [55]. Eventually, gap formation between otherwise tightly adhering endothelial cells causes increased permeability and thus capillary leakage, allowing neutrophil migration (supplementary figure S5).
Additionally, we identified a dynamic correlation of pulmonary effusion lesions with HBP, HRCT and chest PA and LAT. It was found that HBP elevation preceded the progression of pulmonary lesions by 5 days, and was significantly correlated with changes in pulmonary effusion area. Therefore, from the imaging perspective, the elevation of HBP precedes the progression of pulmonary lesions. Compared with the commonly used inflammatory factors CRP, PCT and amyloid, HBP may not only reflect the level of inflammation as a biomarker in evaluating severe patients, but may also be an important mediator in the mechanism of inflammation progression.
Severe hypoxaemia is prevalent in patients with severe COVID-19, and acute diffuse respiratory distress syndrome is a major factor contributing to a poor prognosis [56]. In the present study, HBP levels were found to be significantly correlated with intrapulmonary shunt volume, arterial alveolar oxygen differential, respiratory index and oxygenation index. Under the influence of capillary leakage syndrome and vascular endothelial cell destruction caused by pathological factors including HBP, the respiratory membrane and pulmonary blood perfusion are affected, eventually leading to the decline of pulmonary ventilation function [57, 58]. Hence, we found that HBP is closely related to pulmonary ventilation in COVID-19, parallel to the degree of hypoxia in this study. This close correlation highlights the association between HBP and abnormal intrapulmonary shunting.
Extrapulmonary organ injuries and negative testing in COVID-19 aggravation
Extrapulmonary organ injuries were commonly seen in this study. Markers of myocardial cell damage increased in the present study, with pericardial effusion seen in one patient. Inciardi et al. [59] and Zheng et al. [60] both reported myocarditis as manifestations that may be evidence of extrapulmonary organ involvement in COVID-19, accompanied by an increase in myocardial indicators. Here, we found peak values of AST/ALT ratio >3. Zhang et al. [36] found that liver injury was more common in patients with severe COVID-19 than those with mild disease and reported a significant increase in AST in severely ill patients, consistent with the results of this study. At this stage of severe COVID-19, hepatocyte mitochondria were damaged, with a large amount of AST released and the AST/ALT ratio remarkably increased. A ratio >3 indicates serious liver tissue damage. Additionally, BUN and creatinine were increased, and urine occult blood and protein were positive; two severely ill patients were diagnosed with acute kidney damage. Renal perfusion is affected by hypoxia and systemic inflammation, resulting in glomerular filtration dysfunction. Zheng et al. [60] reported that severe COVID-19 patients can exhibit secondary renal dysfunction with accompanying fluid retention resulting in some developing AKI. However, our study did not find a significant decrease in urine volume or electrolyte imbalance such as for K+. Correlation between HBP levels and the extrapulmonary organ injuries seen in this study might be due to the consequences of COVID-19 aggravation. Inflammation and hypoxia of lung injuries may be the direct causes.
In severely ill COVID-19 patients in our hospital, the viral RNA test may convert to negative. However, in these patients, sudden exacerbation caused by persistent inflammation in the stable stage may still occur and lead to multiorgan damage. The absence of viral inclusion bodies in the heart, liver and kidney suggests that direct viral infection may not be the cause, consistent with several other studies [36, 59]. In contrast, elevated HBP in these patients were highlighted as the potential culprit. Nonetheless, we also found that some indicators reflecting levels of organ injury level did not decrease, even when inflammation was controlled.
Akhmerov et al. [61] postulated that this persistent state of organ injury may last for a long time, and that especially for severe COVID-19, organ damage may not improve in the short term (by days), so early treatment interventions would significantly improve the prognosis. The critical issue is to find the ideal therapeutic targets. Considering the potential pathological roles of HBP in severe COVID-19, its value as a therapeutic target is highlighted [62], e.g. by heparin-based therapy [63]. This is especially important when considering that coagulation disorders have been suggested an important issue in COVID-19 [64]. Investigation on HBP for its therapeutic value is thus warranted.
Conclusions
The correlation of HBP with COVID-19 aggravation suggests its key role in systemic inflammation in patients with severe COVID-19. Our longitudinal analysis revealed that HBP increased ∼5 days preceding any imaging features of organ dysfunction. We therefore propose that HBP monitoring can be a useful biomarker for COVID-19 aggravation and multiple organ injuries as sequelae of SARS-CoV-2 infection. In addition, early intervention targeting HBP to improve the prognosis of severe COVID-19 patients warrants clinical investigation. However, this study has its limitations. The sample size of this study is small. Because HBP has a short half-life and is affected by white blood cells during detection, its detection needs to be undertaken in fresh serum. For these reasons, we still lack solid data about the sensitivity and specificity of HBP assay in severe COVID-19. Further study is warranted to confirm our conclusions about its clinical applications.
Supplementary material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material 00741-2020.SUPPLEMENT (418.5KB, pdf)
Supplementary figure s1 00741-2020 supplementary figure S1 (36.2KB, pdf)
Supplementary figure s2 00741-2020 supplementary figure S2 (346.9KB, pdf)
Supplementary figure s3 00741-2020 supplementary figure S3 (130.7KB, pdf)
Supplementary figure s4 00741-2020 supplementary figure S4 (156.6KB, pdf)
Supplementary figure s5 00741-2020 supplementary figure S5 (194.7KB, pdf)
Supplementary figures 00741-2020.supplement figure legends (364.7KB, pdf)
Acknowledgements
The authors would like to express their gratitude to EditSprings (https://www.editsprings.com/) for the expert linguistic services provided. The authors also appreciate the YuanGuJi Technology Co., Ltd for the technical support.
Footnotes
This article has supplementary material available from openres.ersjournals.com.
Ethics statement: The study was approved by the Ethics Committee of the First Affiliated Hospital of Guangzhou Medical University (2020-77).
Author contributions: Conception and design of the research was performed by B. Sun, N. Zhong and H. Hakonarson; drafting the manuscript by M. Xue, Y. Zeng and H-Q. Qu; samples collection and detection by N. Li; acquisition of data by H. Hu, H. Huang and P. Zheng; statistical analysis by X.D. Zhang and T. Zhang; and analysis and interpretation of data by L. Zhou, Z. Duan, L-f. Tian, Y. Zhang and H. Hakonarson.
Conflict of interest: M. Xue has nothing to disclose.
Conflict of interest: Y. Zeng has nothing to disclose.
Conflict of interest: H-Q. Qu has nothing to disclose.
Conflict of interest: T. Zhang has nothing to disclose.
Conflict of interest: N. Li has nothing to disclose.
Conflict of interest: H. Huang has nothing to disclose.
Conflict of interest: P. Zheng has nothing to disclose.
Conflict of interest: H. Hu has nothing to disclose.
Conflict of interest: L. Zhou has nothing to disclose.
Conflict of interest: Z. Duan has nothing to disclose.
Conflict of interest: Y. Zhang has nothing to disclose.
Conflict of interest: W. Bao has nothing to disclose.
Conflict of interest: L-f. Tian has nothing to disclose.
Conflict of interest: H. Hakonarson has nothing to disclose.
Conflict of interest: N. Zhong has nothing to disclose.
Conflict of interest: X.D. Zhang has nothing to disclose.
Conflict of interest: B. Sun has nothing to disclose.
Support statement: This study was supported by the Guangdong Science and Technology Fund (project number 2020B1111300001), and the Zhejiang University special scientific research fund for COVID-19 prevention and control (project numbers 2020XGZX001 and 2020XGZX025). Funding information for this article has been deposited with the Crossref Funder Registry.
References
- 1.Chan JF, Yip CC, To KK, et al. Improved molecular diagnosis of COVID-19 by the novel, highly sensitive and specific COVID-19-RdRp/Hel real-time reverse transcription-PCR assay validated in vitro and with clinical specimens. J Clin Microbiol 2020; 58: e00310-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jin Y, Yang H, Ji W, et al. Virology, epidemiology, pathogenesis, and control of COVID-19. Viruses 2020; 12: 372. doi: 10.3390/v12040372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sohrabi C, Alsafi Z, O'Neill N, et al. World Health Organization declares global emergency: a review of the 2019 novel coronavirus (COVID-19). Int J Surg 2020; 76: 71–76. doi: 10.1016/j.ijsu.2020.02.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ye Q, Wang B, Mao J. The pathogenesis and treatment of the ‘cytokine storm’ in COVID-19. J Infect 2020; 80: 607–613. doi: 10.1016/j.jinf.2020.03.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen D, Xu W, Lei Z, et al. Recurrence of positive SARS-CoV-2 RNA in COVID-19: a case report. Int J Infect Dis 2020; 93: 297–299. doi: 10.1016/j.ijid.2020.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Montali F, Palmieri G, Casali L, et al. Rapidly fatal outcome of Covid-19 after successful emergency surgery during pandemic outbreak in Northern Italy. Int J Surg Case Rep 2020; 73: 9–12. doi: 10.1016/j.ijscr.2020.06.073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bai Y, Yao L, Wei T, et al. Presumed asymptomatic carrier transmission of COVID-19. JAMA 2020; 323: 1406–1407. doi: 10.1001/jama.2020.2565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Giudicessi JR, Roden DM, Wilde AAM, et al. Genetic susceptibility for COVID-19-associated sudden cardiac death in African Americans. Heart Rhythm 2020; 17: 1487–1492. doi: 10.1016/j.hrthm.2020.04.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu Y, Gayle AA, Wilder-Smith A, et al. The reproductive number of COVID-19 is higher compared to SARS coronavirus. J Travel Med 2020; 27: taaa021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bos JM, Hebl VB, Oberg AL, et al. Marked up-regulation of ACE2 in hearts of patients with obstructive hypertrophic cardiomyopathy: implications for SARS-CoV-2-mediated COVID-19. Mayo Clin Proc 2020; 95: 1354–1368. doi: 10.1016/j.mayocp.2020.04.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoang VT, Dao TL, Gautret P. Recurrence of positive SARS-CoV-2 in patients recovered from COVID-19. J Med Virol 2020; 92: 2366–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Loconsole D, Passerini F, Palmieri VO, et al. Recurrence of COVID-19 after recovery: a case report from Italy. Infection 2020: 48: 965–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peiris JS, Yuen KY, Osterhaus AD, et al. The severe acute respiratory syndrome. N Engl J Med 2003; 349: 2431–2441. doi: 10.1056/NEJMra032498 [DOI] [PubMed] [Google Scholar]
- 14.Peiris JS Severe acute respiratory syndrome (SARS). J Clin Virol 2003; 28: 245–247. doi: 10.1016/j.jcv.2003.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.French Intensive Care Society, International congress – Réanimation 2016. Ann Intensive Care 2016; 6: Suppl. 1, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu J, Li W, Shi X, et al. Early antiviral treatment contributes to alleviate the severity and improve the prognosis of patients with novel coronavirus disease (COVID-19). J Intern Med 2020: 288: 128–138. doi: 10.1111/joim.13063 [DOI] [PubMed] [Google Scholar]
- 17.Fisher J, Linder A. Heparin-binding protein: a key player in the pathophysiology of organ dysfunction in sepsis. J Intern Med 2017; 281: 562–574. doi: 10.1111/joim.12604 [DOI] [PubMed] [Google Scholar]
- 18.Tapper H, Karlsson A, Mörgelin M, et al. Secretion of heparin-binding protein from human neutrophils is determined by its localization in azurophilic granules and secretory vesicles. Blood 2002; 99: 1785–1793. doi: 10.1182/blood.V99.5.1785 [DOI] [PubMed] [Google Scholar]
- 19.Gautam N, Olofsson AM, Herwald H, et al. Heparin-binding protein (HBP/CAP37): a missing link in neutrophil-evoked alteration of vascular permeability. Nat Med 2001; 7: 1123–1127. doi: 10.1038/nm1001-1123 [DOI] [PubMed] [Google Scholar]
- 20.Yang Y, Liu G, He Q, et al. A promising candidate: heparin-binding protein steps onto the stage of sepsis prediction. J Immunol Res 2019; 2019: 7515346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Soehnlein O, Kai-Larsen Y, Frithiof R, et al. Neutrophil primary granule proteins HBP and HNP1-3 boost bacterial phagocytosis by human and murine macrophages. J Clin Invest 2008; 118: 3491–3502. doi: 10.1172/JCI35740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Halldorsdottir HD, Eriksson J, Persson BP, et al. Heparin-binding protein as a biomarker of post-injury sepsis in trauma patients. Acta Anaesthesiol Scand 2018; 62: 962–973. doi: 10.1111/aas.13107 [DOI] [PubMed] [Google Scholar]
- 23.Zhou Y, Liu Z, Huang J, et al. Usefulness of the heparin-binding protein level to diagnose sepsis and septic shock according to Sepsis-3 compared with procalcitonin and C reactive protein: a prospective cohort study in China. BMJ Open 2019; 9: e026527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu HQ, Sun BQ, Fang ZF, et al. Distinct features of SARS-CoV-2-specific IgA response in COVID-19 patients. Eur Respir J 2020; 56: 2001526. doi: 10.1183/13993003.01526-2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hanson KE, Caliendo AM, Arias CA, et al. Infectious Diseases Society of America guidelines on the diagnosis of COVID-19. Clin Infect Dis 2020: ciaa760. doi: 10.1093/cid/ciaa760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blum CA, Nigro N, Briel M, et al. Adjunct prednisone therapy for patients with community-acquired pneumonia: a multicentre, double-blind, randomised, placebo-controlled trial. Lancet 2015; 385: 1511–1518. doi: 10.1016/S0140-6736(14)62447-8 [DOI] [PubMed] [Google Scholar]
- 27.Paul M, Daikos GL, Durante-Mangoni E, et al. Colistin alone versus colistin plus meropenem for treatment of severe infections caused by carbapenem-resistant Gram-negative bacteria: an open-label, randomised controlled trial. Lancet Infect Dis 2018; 18: 391–400. doi: 10.1016/S1473-3099(18)30099-9 [DOI] [PubMed] [Google Scholar]
- 28.Vultaggio A, Vivarelli E, Virgili G, et al. Prompt predicting of early clinical deterioration of moderate-to-severe COVID-19 patients: usefulness of a combined score using IL-6 in a preliminary study. J Allergy Clin Immunol Pract 2020; 8: 2575–2581. doi: 10.1016/j.jaip.2020.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang Y, Shen C, Li J, et al. Plasma IP-10 and MCP-3 levels are highly associated with disease severity and predict the progression of COVID-19. J Allergy Clin Immunol 2020; 146: 119–127. doi: 10.1016/j.jaci.2020.04.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alhazzani W, Møller MH, Arabi YM, et al. Surviving Sepsis campaign: guidelines on the management of critically ill adults with coronavirus disease 2019 (COVID-19). Intensive Care Med 2020; 46: 854–887. doi: 10.1007/s00134-020-06022-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Denstaedt SJ, Singer BH, Standiford TJ. Sepsis and nosocomial infection: patient characteristics, mechanisms, and modulation. Front Immunol 2018; 9: 2446. doi: 10.3389/fimmu.2018.02446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chang ST, Lai KL, Kuo FC, et al. Cross-correlation between spine and hip joint kinematics differs in healthy individuals and subgroups of ankylosing spondylitis patients during trunk lateral flexion. Musculoskelet Sci Pract 2018; 38: 8–14. doi: 10.1016/j.msksp.2018.09.001 [DOI] [PubMed] [Google Scholar]
- 33.Zhou F, Yu T, Du R, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 2020; 395: 1054–1062. doi: 10.1016/S0140-6736(20)30566-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Varga Z, Flammer AJ, Steiger P, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020; 395: 1417–1418. doi: 10.1016/S0140-6736(20)30937-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pan F, Ye T, Sun P, et al. Time course of lung changes at chest CT during recovery from coronavirus disease 2019 (COVID-19). Radiology 2020; 295: 715–721. doi: 10.1148/radiol.2020200370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang C, Shi L, Wang FS. Liver injury in COVID-19: management and challenges. Lancet Gastroenterol Hepatol 2020; 5: 428–430. doi: 10.1016/S2468-1253(20)30057-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hanley B, Lucas SB, Youd E, et al. Autopsy in suspected COVID-19 cases. J Clin Pathol 2020; 73: 239–242. doi: 10.1136/jclinpath-2020-206522 [DOI] [PubMed] [Google Scholar]
- 38.Wichmann D, Sperhake JP, Lütgehetmann M, et al. Autopsy findings and venous thromboembolism in patients with COVID-19: a prospective cohort study. Ann Intern Med 2020; 173: 268–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Butler MJ, Barrientos RM. The impact of nutrition on COVID-19 susceptibility and long-term consequences. Brain Behav Immun 2020; 87: 53–54. doi: 10.1016/j.bbi.2020.04.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jawhara S Could intravenous immunoglobulin collected from recovered coronavirus patients protect against COVID-19 and strengthen the immune system of new patients? Int J Mol Sci 2020; 21: 2272. doi: 10.3390/ijms21072272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zabetakis I, Lordan R, Norton C, et al. COVID-19: the inflammation link and the role of nutrition in potential mitigation. Nutrients 2020; 12: 1466. doi: 10.3390/nu12051466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sullivan RJ, Johnson DB, Rini BI, et al. COVID-19 and immune checkpoint inhibitors: initial considerations. J Immunother Cancer 2020; 8: e000933. doi: 10.1136/jitc-2020-000933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Agrati C, Sacchi A, Bordoni V, et al. Expansion of myeloid-derived suppressor cells in patients with severe coronavirus disease (COVID-19). Cell Death Differ 2020; 27: 3196–3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kishimoto T IL-6: from its discovery to clinical applications. Int Immunol 2010; 22: 347–352. doi: 10.1093/intimm/dxq030 [DOI] [PubMed] [Google Scholar]
- 45.Meng ZH, Dyer K, Billiar TR, et al. Essential role for IL-6 in postresuscitation inflammation in hemorrhagic shock. Am J Physiol Cell Physiol 2001; 280: C343–C351. doi: 10.1152/ajpcell.2001.280.2.C343 [DOI] [PubMed] [Google Scholar]
- 46.Choy EH, De Benedetti F, Takeuchi T, et al. Translating IL-6 biology into effective treatments. Nat Rev Rheumatol 2020; 16: 335–345. doi: 10.1038/s41584-020-0419-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gubernatorova EO, Gorshkova EA, Polinova AI, et al. IL-6: relevance for immunopathology of SARS-CoV-2. Cytokine Growth Factor Rev 2020; 53: 13–24. doi: 10.1016/j.cytogfr.2020.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Albarello F, Pianura E, Di Stefano F, et al. 2019-novel coronavirus severe adult respiratory distress syndrome in two cases in Italy: an uncommon radiological presentation. Int J Infect Dis 2020; 93: 192–197. doi: 10.1016/j.ijid.2020.02.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morsing SKH, Al-Mardini C, van Stalborch AD, et al. Double-hit-induced leukocyte extravasation driven by endothelial adherens junction destabilization. J Immunol 2020; 205: 511–520. doi: 10.4049/jimmunol.1900816 [DOI] [PubMed] [Google Scholar]
- 50.Lauritzen B, Lykkesfeldt J, Djurup R, et al. Effects of heparin-binding protein (CAP37/azurocidin) in a porcine model of Actinobacillus pleuropneumoniae-induced pneumonia. Pharmacol Res 2005; 51: 509–514. doi: 10.1016/j.phrs.2005.01.004 [DOI] [PubMed] [Google Scholar]
- 51.Song M, Liu L, Qi X, et al. [Mechanism of heparin-binding protein increasing vascular permeability in early burn]. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue 2020; 32: 330–335. [DOI] [PubMed] [Google Scholar]
- 52.Kounakis K, Tavernarakis N. The cytoskeleton as a modulator of aging and neurodegeneration. Adv Exp Med Biol 2019; 1178: 227–245. doi: 10.1007/978-3-030-25650-0_12 [DOI] [PubMed] [Google Scholar]
- 53.Bentzer P, Fisher J, Kong HJ, et al. Heparin-binding protein is important for vascular leak in sepsis. Intensive Care Med Exp 2016; 4: 33. doi: 10.1186/s40635-016-0104-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu Z, Khalil RA. Evolving mechanisms of vascular smooth muscle contraction highlight key targets in vascular disease. Biochem Pharmacol 2018; 153: 91–122. doi: 10.1016/j.bcp.2018.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ringvold HC, Khalil RA. Protein kinase C as regulator of vascular smooth muscle function and potential target in vascular disorders. Adv Pharmacol 2017; 78: 203–301. doi: 10.1016/bs.apha.2016.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xie J, Covassin N, Fan Z, et al. Association between hypoxemia and mortality in patients with COVID-19. Mayo Clin Proc 2020; 95: 1138–1147. doi: 10.1016/j.mayocp.2020.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Azkur AK, Akdis M, Azkur D, et al. Immune response to SARS-CoV-2 and mechanisms of immunopathological changes in COVID-19. Allergy 2020; 75: 1564–1581. doi: 10.1111/all.14364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Teuwen L-A, Geldhof V, Pasut A, et al. COVID-19: the vasculature unleashed. Nat Rev Immunol 2020; 20: 389–391. doi: 10.1038/s41577-020-0343-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Inciardi RM, Lupi L, Zaccone G, et al. Cardiac involvement in a patient with coronavirus disease 2019 (COVID-19). JAMA Cardiol 2020; 5: 819–824. doi: 10.1001/jamacardio.2020.1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zheng YY, Ma YT, Zhang JY, et al. COVID-19 and the cardiovascular system. Nat Rev Cardiol 2020; 17: 259–260. doi: 10.1038/s41569-020-0360-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Akhmerov A, Marbán E. COVID-19 and the heart. Circ Res 2020; 126: 1443–1455. doi: 10.1161/CIRCRESAHA.120.317055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bentzer P, Fisher J, Kong HJ, et al. Heparin-binding protein is important for vascular leak in sepsis. Intensive Care Med Exp 2016; 4: 33. doi: 10.1186/s40635-016-0104-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fisher J, Russell JA, Bentzer P, et al. Heparin-binding protein (HBP): a causative marker and potential target for heparin treatment of human sepsis-induced acute kidney injury. Shock 2017; 48: 313–320. doi: 10.1097/SHK.0000000000000862 [DOI] [PubMed] [Google Scholar]
- 64.Giannis D, Ziogas IA, Gianni P. Coagulation disorders in coronavirus infected patients: COVID-19, SARS-CoV-1, MERS-CoV and lessons from the past. J Clin Virol 2020; 127: 104362. doi: 10.1016/j.jcv.2020.104362 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material 00741-2020.SUPPLEMENT (418.5KB, pdf)
Supplementary figure s1 00741-2020 supplementary figure S1 (36.2KB, pdf)
Supplementary figure s2 00741-2020 supplementary figure S2 (346.9KB, pdf)
Supplementary figure s3 00741-2020 supplementary figure S3 (130.7KB, pdf)
Supplementary figure s4 00741-2020 supplementary figure S4 (156.6KB, pdf)
Supplementary figure s5 00741-2020 supplementary figure S5 (194.7KB, pdf)
Supplementary figures 00741-2020.supplement figure legends (364.7KB, pdf)