

Abstract
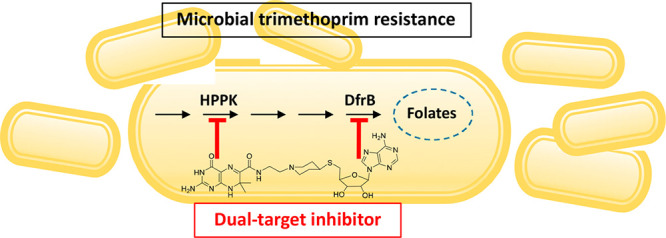
Trimethoprim (TMP) is widely used to treat infections in humans and in livestock, accelerating the incidence of TMP resistance. The emergent and largely untracked type II dihydrofolate reductases (DfrBs) are intrinsically TMP-resistant plasmid-borne Dfrs that are structurally and evolutionarily unrelated to chromosomal Dfrs. We report kinetic characterization of the known DfrB family members. Their kinetic constants are conserved and all are poorly inhibited by TMP, consistent with TMP resistance. We investigate their inhibition with known and novel bisubstrate inhibitors of 6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase (HPPK). Importantly, all are inhibited by the HPPK inhibitors, making these molecules dual-target inhibitors of two folate pathway enzymes that are strictly microbial.
Keywords: Dual-target inhibitor, type II dihydrofolate reductase, hydroxymethyl-dihydropterin pyrophosphokinase, trimethoprim resistance, bisubstrate inhibitor
The folate metabolic pathway has long been the focus of dual-target therapy as a result of its essential nature.1 Dihydrofolate reductases (Dfr), in particular, have been at the center stage in development of dual-target inhibitors, with the Dfr-thymidylate synthase pair and the Dfr-dihydropteroate synthase (DHPS) pair being among those that are topics of intense research.2−4 Trimethoprim (TMP) is an antibiotic that selectively inhibits bacterial chromosomal Dfr with little effect on mammalian Dfrs. TMP is often used in combination with sulfamethoxazole (SUL) as a front-line treatment against known or unknown infections from aerobic bacteria and occasionally against protozoa.5 SUL acts synergistically with TMP, targeting a second enzyme in the microbial folate pathway, DHPS, that is lacking in humans.6 The TMP–SUL combination is categorized by the World Health Organization as highly important due to its effectiveness in treatment of human urinary and respiratory tract infections5,7 and to reduce mortality in people living with HIV/AIDS.8
Nonetheless, the spread of resistance to TMP in humans, in livestock,5,9,10 and from livestock to humans5,11 is accelerating,12 whether in the context of TMP administered alone or with SUL. Microbial resistance to TMP may result either from mutations in their chromosomal Dfr or as a result of acquiring the evolutionarily and structurally unrelated DfrBs.13−15 DfrBs are plasmid-borne and have been identified in TMP-resistant Gram-negative bacteria, although there is little knowledge of their incidence in clinical samples.10,14,16−18 DfrB genes were first identified in TMP-resistant wastewater or clinical samples.10,14,16−18 The high TMP resistance of four DfrBs (DfrB1 to DfrB4) has been ascertained in vitro.19−21
Thus, even successful inhibition with TMP of the chromosomal Dfr of a microbe can be overcome by the presence of an evolutionarily distinct DfrB. It confers very high resistance to TMP, allowing microbial proliferation.22,23 Similarly, DfrB is highly resistant to methotrexate (KIMTX > 0.5 mM),24 highlighting the evolutionary and structural distinction between the DfrBs and chromosomal Dfrs (KIMTX = 10 nM).25
To this day, the DfrB family of dihydrofolate reductases remains poorly studied. DfrB1, the only well-characterized DfrB, is a homotetramer constituted of four β-barrel (SH3 domain) protomers. The four protomers contribute equally to the unique, symmetrical active-site tunnel (Figure 1A).13,20,21,24,26−28 DfrBs are thus evolutionarily unrelated to the monomeric chromosomal Dfrs.13
Figure 1.

The homotetrameric DfrBs are evolutionarily distinct from the chromosomal Dfrs. (A) The homotetrameric DfrB1 (PDB: 2RK1) is intrinsically TMP-resistant: its large, symmetrical active-site tunnel does not bind TMP. Each protomer is colored differently. The 20 N-terminal residues of each protomer are unstructured and are not represented. For clarity, only I68 of the V66-Q67-I68-Y69 (VQIY) region and K32 are represented as sticks on each protomer. (B) The chromosomal Dfrs are evolutionarily and structurally unrelated. The E. coli Dfr (PDB: 1DDR), shown at scale, is strongly inhibited by TMP. (C) Sequence alignment of the DfrB family; there is no DfrB8.15 The weakly conserved N-termini and the highly conserved β-barrel core including the VQIY residues that line the active site tunnel 4-fold are identified.
DfrBs share 74% to 98% sequence identity (Table S1). The β-barrel core is highly conserved (89% to 98%) whereas the unstructured N-termini are weakly conserved (Figure 1C).
DfrBs are intrinsically TMP-resistant and are emergent sources of antibiotic resistance. Their anticipated health-threatening activity calls for the development of new inhibitors. We reported the first selective inhibitors of the hexahistidine-tagged DfrB1 (His6-DfrB1). These bisbenzimidazoles offer Ki of 1.7–12.0 μM.29,30
Here, we report the inhibition of DfrBs with inhibitors of E. coli 6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase (HPPK), an enzyme that catalyzes an earlier step of the bacterial folate pathway. Whereas the mammalian folate pathway starts with the intake of dietary folate, bacteria are folate autotrophs.31 Bacterial folate biosynthesis depends on HPPK, which constitutes a potential antibiotic target as it has no mammalian homologue (Figure 2).32 No antibiotic that targets HPPK has yet been reported but progress has been made in development of HPPK inhibitors.32,33
Figure 2.

Microbial pathway for biosynthesis of tetrahydrofolate (H4folate). Enzymes, shown in italics, are GTPCH, guanosine triphosphate (GTP) cyclohydrolase; PPase, phosphatase; DHNA, dihydroneopterin aldolase; HPPK, 6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase; DHPS, dihydropteroate synthase; FPGS, folylpolyglutamate synthase; DHFR, dihydrofolate reductase. The target enzymes in this study, HPPK and DHFR, are highlighted in red.
Inhibition of both DfrB1 and HPPK thus establishes these molecules as dual-target inhibitors of the folate pathway. They are based on substructures of folic acid and NADPH, qualifying them as bisubstrate inhibitors. Furthermore, we demonstrate the broad inhibitory effect on the DfrB family. To that end, we determined for the first time that DfrB members representing the entire extent of sequence diversity exhibit Dfr activity and low sensitivity to TMP, comparable to the prototypical DfrB1. The potency of the bisubstrate inhibitors toward DfrBs was comparable to that of the previously reported bisbenzimidazole inhibitors of DfrB1,29,30 confirming the interest in further developing these dual-target inhibitors to combat TMP resistance.
Results and Discussion
Dual-target inhibitors are designed to simultaneously inhibit two individual biological targets rather than requiring administration of distinct compounds.34 Also named hybrid molecules, dual-target inhibitors often consist of two linked pharmacophores, which should each bind to a target.35,36 In contrast to dual-target inhibitors built from pharmacophores, here we examine dual-target inhibitors that mimic both substrates of the target enzymes HPPK and DfrB. Both enzymes bind a pterin derivative 6-hydroxymethyl-7,8-dihydropterin and dihydrofolate (DHF), respectively—as well as an adenosine-based cofactor—ATP and NADPH, respectively (Figure 2).4
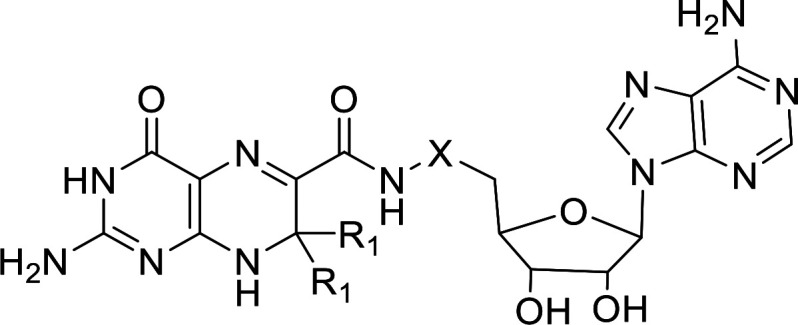
Bisubstrate molecules 1 and 2 were previously designed to mimic the adenosine portion of ATP and the pterin substrate of HPPK (Scheme 1). They inhibit HPPK with Ki of 1.1 and 3.1 μM, respectively (Table 1).32,33 Since the substrates of DfrB are DHF and NADPH, the bisubstrate molecules 1 and 2 have the potential to also inhibit DfrB, thereby acting as dual-target inhibitors of HPPK and DfrB. Neither HPPK nor DfrB is the target of an existing antibiotic; both enzymes are strictly microbial, potentially minimizing undesired side-effects.
Scheme 1. Bisubstrate Molecules Include the Pterin Moiety of DHF (Green) and the Adenosyl Moiety of NAD(P)H (Purple).
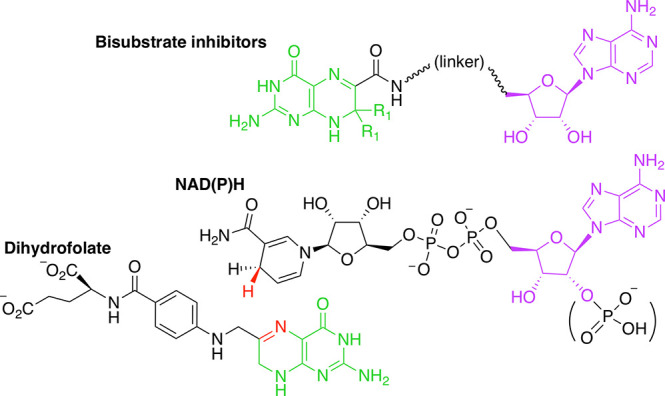
Atoms involved in hydride transfer catalyzed by DfrB1 are in red. The linker is variable.
Table 1. Inhibition of His6-DfrB1 and HPPK with Bisubstrate Inhibitors 1–5.

The tetrameric tunnel of DfrB1 is lined with the conserved VQIY sequence that is 4-fold represented, forming a single symmetrical active site. The VQIY region interacts sterically and through hydrogen bonds with the pterin moiety of the substrate, DHF.37 The adenosine moiety contributes to binding of NADPH with DfrB1 at the mouth of the active site tunnel.24,37 As a result, the pterin and adenosine moieties possess distinct binding sites.37 The design of the HPPK bisubstrate inhibitors is thus consistent with potential to inhibit DfrB1.
HPPK bisubstrate inhibitors 1 to 2 were tested for inhibition of His6-DfrB1. Inhibitors 1 and 2 inhibit His6-DfrB1 with Ki of 20 μM and 18 μM, respectively (Table 1). They are HPPK inhibitors by design.4 Their activity against His6-DfrB1 prompted us to modulate the linker length and the position of the piperidine ring for better inhibition. Novel bisubstrate molecules 3 to 5 were synthesized (Schemes S1 to S3) and validated as inhibitors of HPPK: Ki ranged between 5.5 and 12 μM, slightly less potent toward HPPK than 1 and 2. Gratifyingly, 3 to 5 also inhibit His6-DfrB1 with Ki of 10–17 μM, similar or better than inhibitors 1 and 2. The potency of inhibitors 1 to 5 is comparable to the known bisbenzimidazole inhibitors of His6-DfrB1, 6, and 7 (Scheme 2; Ki = 3.5 and 7.4 μM, respectively).30 These results confirm that this class of bisubstrate molecules acts as dual-target inhibitors of HPPK and His6-DfrB1.
Scheme 2. Bisbenzimidazole Inhibitors 6 and 7(30).

The high sequence identity of DfrBs (Table S1) suggested that His6-DfrB1 inhibitors may inhibit the broader DfrB family. To verify this hypothesis, we selected six among the eight members of the DfrB family to represent the greatest pairwise sequence differences: DfrB1 to DfrB5, and DfrB7. Variation is essentially confined to the termini and to the turns in the otherwise highly conserved β-barrel active site core (Figure 1A, C). Prior to producing these proteins, we verified the potential impact of an N-terminal His6-tag on inhibition, turning to bisbenzimidazole inhibitors 6 and 7. They were previously assayed against the N-terminally His6-tagged DfrB1 harboring additional non-native C-terminal residues (Ki6 = 3.5 and Ki7 = 7.4 μM).30 Inhibition of DfrB1 bearing its native termini yielded ≈8-fold weaker inhibition, with Ki6 = 18 μM and Ki7 = 62 μM (Table 2), demonstrating the contribution of the terminal additions to inhibitor binding. A similar 3- to 8.4-fold decrease in inhibition in the absence of non-native termini was further verified with representative bisubstrate inhibitors 1 and 3 (Table 2). This observation was unexpected since the symmetrical bisbenzimidazole inhibitors bear little resemblance to the bisubstrate inhibitors. To eliminate any contribution of the His6-tag or the non-native C-terminal residues to inhibition, all variants were produced in their nontagged, native form for further investigation.
Table 2. Inhibition of DfrBs (Non-His-tagged) with TMP and Inhibitors 1, 3, 6, and 7.
| Ki (mM)a | Ki (μM) |
||||
|---|---|---|---|---|---|
| Inhibitor: | TMP | 1 | 3 | 6 | 7 |
| DfrB1 | 0.38 ± 0.09 | 130 ± 98 | 46 ± 5 | 18 ± 6 | 62 ± 6 |
| DfrB2 | 0.81 ± 0.08 | 49 ± 3 | 52 ± 14 | 10 ± 2 | 29 ± 1 |
| DfrB3 | 0.45 ± 0.10 | 12 ± 2 | 17 ± 2 | 2.4 ± 0.4 | 6.4 ± 2.1 |
| DfrB4 | 0.50 ± 0.07 | 35 ± 4 | 51 ± 1 | 13 ± 1 | 20 ± 2 |
| DfrB5 | 1.3 ± 0.3 | 49 ± 9 | 42 ± 1 | 7.4 ± 0.1 | 29 ± 3 |
| DfrB7 | 0.66 ± 0.11 | 52 ± 32 | 37 ± 10 | 7.1 ± 2.2 | 19 ± 7 |
To access highly purified DfrB variants, we capitalized on the remarkable thermostability of DfrB1 and DfrB2, vividly illustrated by maintenance of activity upon boiling for 20 min.38−40 We purified the DfrBs to ≥95% using heat-precipitation of crude lysates at 65 to 75 °C (Table S4; Figure S1). This one-day purification method procured 6.2 to 12.4 mg (3.7 to 8.5 U) of all DfrBs from 200 mL-scale expressions in E. coli (Table S4, Table S5).
Prior to addressing their inhibition, it was essential to verify that the DfrB family members do indeed have Dfr activity. To our knowledge, Dfr activity has been reported only for DfrB1 through DfrB419−21 and kinetic parameters kcat and KM reported only for DfrB1 and DfrB4.22,30 Nonetheless, the TMP-resistant sample origin and high sequence conservation of residues VQIY and K32 implicated in ligand binding and catalysis37(Figure 1) strongly suggest that the more recent DfrB5 to DfrB9 exhibit TMP-resistant Dfr activity.
All DfrBs displayed clear Dfr activity (Table 3). The DHF productive affinities were nearly identical for all DfrBs (KMDHF within 1.4-fold variation) and strongly conserved for NADPH (KMNADPH: 1 to 4.5-fold variation). Those values are similar to the E. coli chromosomal Dfr (EcDfr) (KMDHF = 1.2 μM and KMNADPH = 0.94 μM),41 as befits enzymes that compete for the same cellular resources.
Table 3. Kinetic Constants for the Dihydrofolate Reductase Activity of DfrBs (Non-His-tagged).
| Dfr | KMDHF (μM) | KMNADPH (μM) | kcatDHF (s–1) | kcatDHF/kMDHF (μM–1·s–1) |
|---|---|---|---|---|
| EcDfr | 1.2a | 0.94a | 12.0b,c | 10 |
| DfrB1 | 4.4 ± 0.5 | 5.8 ± 0.8 | 0.32 ± 0.006 | 0.073 |
| DfrB2 | 2.3 ± 0.2 | 3.3 ± 0.3 | 0.41 ± 0.01 | 0.18 |
| DfrB3 | 3.0 ± 0.5 | 1.3 ± 0.3 | 0.20 ± 0.01 | 0.067 |
| DfrB4 | 4.6 ± 0.7 | 3.5 ± 0.5 | 0.28 ± 0.01 | 0.061 |
| DfrB5 | 3.2 ± 0.4 | 3.1 ± 0.2 | 0.32 ± 0.01 | 0.10 |
| DfrB7 | 3.4 ± 0.4 | 2.8 ± 0.3 | 0.24 ± 0.007 | 0.071 |
The kcatDHF determined by varying DHF (kcatDHF = 0.20 to 0.41 s–1; Table 3) showed excellent agreement with the kcatNADPH, determined by varying NADPH (Table S6). DfrBs are poor catalysts relative to EcDfr (kcat = 12.0 s–1).42 Their catalytic efficiency (kcatDHF /KMDHF) is nearly 2 orders of magnitude lower than that of EcDfr (Table 3). The high sequence similarity of DfrB6 and DfrB9 with their closest homologue among the assayed DfrBs (91% and 85%, respectively; Table S1) suggests that their catalytic properties, and those of closely related homologues that may be identified in the future, lie within this range.
The Ki for TMP for the chromosomal EcDfr is 20 pM, demonstrating that TMP procures strong inhibition of EcDfr43 (Figure 1). E. coli cannot overcome TMP inhibition by increasing the copy number of EcDfr since its overexpression to the required concentration would result in cellular toxicity by folate sequestration.44 The lesser-studied dfrB genes are plasmid-borne; their copy level in clinically relevant strains is unknown. KiTMP has been reported only for DfrB219. We determined that the KiTMP values for the purified DfrBs (0.38 mM to 1.3 mM) are 107–108-fold higher than for EcDfr, confirming that the DfrB family displays high TMP resistance. Our data demonstrate that the DfrBs possess the Dfr activity and tolerance to TMP to support bacterial proliferation in the presence of high concentrations of TMP, providing a survival advantage to bacteria.13,14,21
Considering the kinetic similarities between all DfrBs, we hypothesized that they should be similarly inhibited. To verify this, we tested bisubstrate inhibitor 1 against the purified DfrBs (Table 2). Bisubstrates 1 and 3 inhibit all DfrBs with Ki1 = 12–130 μM and Ki3 = 17–52 μM, demonstrating that they are dual inhibitors of HPPK and of the DfrB family. We further verified that bisbenzimidazole inhibitors 6 and 7 inhibit all DfrBs with Ki in the low μM range (Ki6 = 2.4–17.5 μM and Ki7 = 6.4–61.7 μM; Table 2). Overall, bisbenzimidazole inhibitors 6 and 7 offer slightly better affinity than bisubstrate inhibitors 1 and 3 (Ki values differ 2–7-fold for any given DfrB). Our results demonstrate the promise held by the bisubstrate inhibitors, given their dual-target advantage; further development as leads for antibiotics should address microbial permeation.
Short molecular dynamic studies of molecules 6 and 7 with DfrB1 previously revealed three binding hotspots: the K32 network (K32, G35, and A36), the YTT cluster (Y46, T48, and T51), and the VQIY region (Figure 1).30 These regions are conserved among DfrBs. The substrates DHF and NADPH, and the bisbenzimidazole inhibitors 6 and 7, share two among those binding hotspots: the K32 network and the VQIY region. Despite interacting with the same residues, crystal structures and simulations determined that the substrates and the bisbenzimidazole inhibitors bind to different faces of the active-site tunnel and are therefore mutually exclusive.30 Bisubstrate inhibitors 1 and 3 mimic DHF and NADPH, suggesting that these inhibitors should bind to those same hotspots and tunnel faces as DHF and NADPH.
We investigated the binding mode of bisubstrate inhibitor 1 on DfrB1 as a model for these interactions. Because the promiscuous active-site tunnel of DfrB1 can bind either DHF and NADPH or two molecules of either24 and binds two molecules of the bisbenzimidazole inhibitors,30 we attempted to determine the Hill coefficient for 1 as we previously did for 6 and 7.29,30 However, absorbance of 1 interfered with the activity assay. Nonetheless, in the absence of the substrate DHF, a low but clearly detectable change in the absorbance of NADPH was observed (data not shown), suggesting slow reduction of 1 by NADPH. This suggested that binding of 1 may be facilitated when its DHF-like pterin moiety is juxtaposed with the nicotinamide ring of NADPH.
Consistent with that hypothesis, we undertook molecular docking of 1 + NADPH into DfrB1 (refer to Methods for details of all simulations). NADPH was modeled based on cocrystallized NADP+ and remained fixed. The pterin moiety of cocrystallized DHF served as a template for initial placement of 1 at the VQIY binding hotspot; the remainder of 1 was free for conformational exploration. Following conformational refinement, analysis of the 25 top-scored poses showed that the adenosine moiety of 1 is compatible with binding at the K32 network hotspot at the tunnel entrance, mimicking the adenosine of NADPH and thus achieving the design objective of the bisubstrate inhibitors (Figure 3; Figure S2). Nonetheless, because 1 does not include the 2′-phosphate of NADPH, its adenosine moiety was not exclusively bound by the K32 network as are NADPH and inhibitors 6 and 7.30 Instead, it also explored a diversity of other configurations, consistent with its modest affinity (Figure S3). This was facilitated by the lack of consistent binding events between the linker of 1 and DfrB1 and suggests high entropy of the adenosine moiety of 1 at the tunnel entrance (Figure S4).
Figure 3.
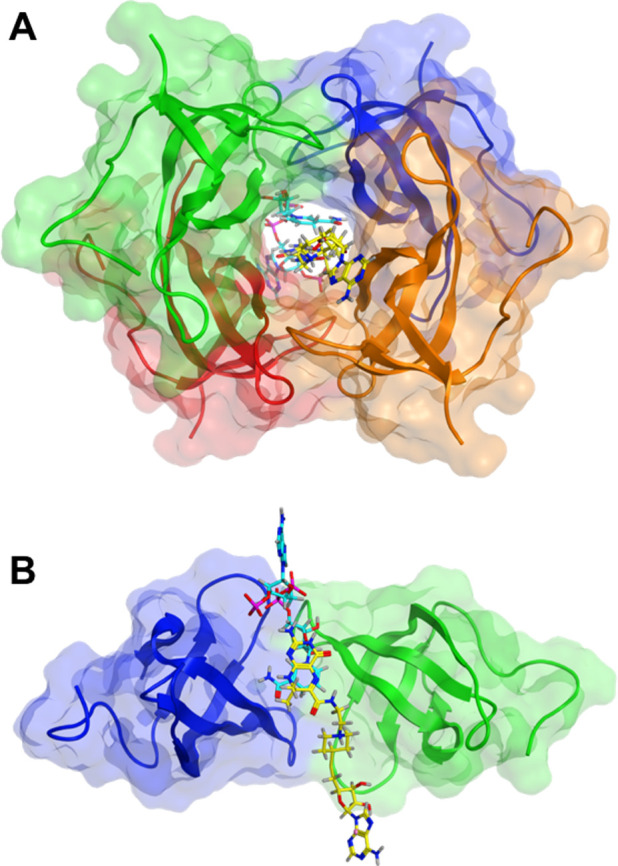
Docking of 1 (yellow) into the DfrB1 tunnel with NADPH (cyan) (PDB: 2RK1). A pose with the adenosine moiety of 1 overlaying best with that of NADPH is shown (Figure S3). In the top 25 poses, inhibitor 1 forms contacts most frequently with K32, V66, and I68 (Figure 1; Figure S4). Contacts are rarely or not established with G35 and A36 that participate in binding the 2′-phosphate of NADPH, and with the YTT cluster.30 (A) Front view. (B) Side view; two subunits are represented.
We also docked either one or two molecules of 1 into DfrB1, without NADPH; this is plausible because DfrB1 can bind two molecules of DHF.24 Similar to docking of 1 + NADPH, the adenosine moiety of 1 established diverse contacts at the tunnel entrance that included, but were not exclusive to, the K32 network.
The docking results served to initiate conformational exploration by LowModeMD as previously described for the bisbenzimidazole inhibitors 6 and 7;30 the ligands were free, and nearby protein atoms were flexible. During the initial stages of conformational exploration, inhibitor 1 established or maintained contacts with the VQIY and K32 network hotspots, confirming that binding of 1 mimics binding of NADPH and DHF. However, in all cases, inhibitor 1 diffused out of the tunnel (Figure S5), reflecting its modest affinity (Table 2). When NADPH was included, it remained at its binding site, confirming the coherence of the simulations.
The diffusion of bisubstrate inhibitor 1 out of DfrB1 during the course of simulations contrasts with bisbenzimidazole inhibitors 6 and 7: although 6 and 7 display affinity within the same order of magnitude (Table 2), they did not diffuse out during the same procedure.30 As mentioned above, 6 and 7 interacted not only with the VQIY region and the K32 network, as do NADPH and DHF, but also with the YTT cluster. Here, the linker of 1 did not establish interactions with the YTT cluster. This is reminiscent of our previous report where the lack of stabilizing interactions at the YTT cluster characterized the weakest bisbenzimidazole inhibitors.30 The linker of bisubstrates 1 and 3 thus constitutes a target for optimization.
In summary, heat-precipitation rapidly procured highly purified DfrBs, allowing confirmation of their dihydrofolate reductase activity and their low sensitivity to TMP. We demonstrate, for the first time, that known and new HPPK inhibitors broadly inhibit all of the DfrBs assayed. The features revealed by our kinetic and computational study provide many insights toward design of dual-target antibiotics targeting HPPK and the DfrBs. Further optimization of affinity and additional properties of bisubstrate inhibitors 1 and 3 as HPPK inhibitors would result in a dual-target antibiotic analogous to the SUL–TMP combination, to slow the emergence of TMP resistance without the inconvenience of coadministering two antibiotics.
Acknowledgments
This work was supported by NSERC grants 227853 and 2018-04686 awarded to J.N.P., CFI grant 11510 to J.N.P., the Fondation Marcel et Rolande Gosselin to A.M., and the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research to X.J. J.L.T. received scholarships from FQRNT, Hydro-Québec, Université de Montréal, and PROTEO. C.L.-St-D. held a Graham-Bell scholarship from NSERC.
Glossary
Abbreviations
- Dfr
dihydrofolate reductase
- DfrB
type II Dfr
- DHF
dihydrofolate
- EcDfr
E. coli chromosomal Dfr
- H2
dihydro
- H4
tetrahydro
- His6-DfrB1
hexahistidine-tagged DfrB1
- HPPK
6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase
- K32 network
Lys32, Gly35, and Ala36
- SUL
sulfomethoxazole
- TMP
trimethoprim
- VQIY
Val66-Gln67-Ile68-Tyr69
- YTT cluster
Tyr46, Thr48, and Thr51
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00393.
Additional enzyme kinetics and inhibition data, DfrB purification data, modeling data, synthetic schemes, and materials and methods (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Bourne C. R. Utility of the Biosynthetic Folate Pathway for Targets in Antimicrobial Discovery. Antibiotics 2014, 3 (1), 1–28. 10.3390/antibiotics3010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arooj M.; Sakkiah S.; Cao G.; Lee K. W. An innovative strategy for dual inhibitor design and its application in dual inhibition of human thymidylate synthase and dihydrofolate reductase enzymes. PLoS One 2013, 8 (4), e60470 10.1371/journal.pone.0060470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzam R. A.; Elsayed R. E.; Elgemeie G. H. Design, Synthesis, and Antimicrobial Evaluation of a New Series of N-Sulfonamide 2-Pyridones as Dual Inhibitors of DHPS and DHFR Enzymes. ACS Omega 2020, 5 (18), 10401–10414. 10.1021/acsomega.0c00280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw G. X.; Li Y.; Shi G.; Wu Y.; Cherry S.; Needle D.; Zhang D.; Tropea J. E.; Waugh D. S.; Yan H.; Ji X. Structural enzymology and inhibition of the bi-functional folate pathway enzyme HPPK-DHPS from the biowarfare agent Francisella tularensis. FEBS J. 2014, 281 (18), 4123–4137. 10.1111/febs.12896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giguère S.; Prescott J. F.; Dowling P. M.. Antimicrobial therapy in veterinary medicine; John Wiley & Sons Inc, 2013; Vol. 47, pp 256–257. [Google Scholar]
- Debabov D. Antibiotic resistance: Origins, mechanisms, approaches to counter. Appl. Biochem. Microbiol. 2013, 49, 665–671. 10.1134/S0003683813080024. [DOI] [Google Scholar]
- WHO . WHO list of critically important antimicrobials for human medicine, 6th ed.; World Health Organization, 2019. [Google Scholar]
- Suthar A. B.; Vitoria M. A.; Nagata J. M.; Anglaret X.; Mbori-Ngacha D.; Sued O.; Kaplan J. E.; Doherty M. C. Co-trimoxazole prophylaxis in adults, including pregnant women, with HIV: a systematic review and meta-analysis. Lancet HIV 2015, 2 (4), e137–150. 10.1016/S2352-3018(15)00005-3. [DOI] [PubMed] [Google Scholar]
- Yassin A. K.; Gong J.; Kelly P.; Lu G.; Guardabassi L.; Wei L.; Han X.; Qiu H.; Price S.; Cheng D.; Wang C. Antimicrobial resistance in clinical Escherichia coli isolates from poultry and livestock, China. PLoS One 2017, 12 (9), e0185326 10.1371/journal.pone.0185326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunde M. Prevalence and characterization of class 1 and class 2 integrons in Escherichia coli isolated from meat and meat products of Norwegian origin. J. Antimicrob. Chemother. 2005, 56, 1019–1024. 10.1093/jac/dki377. [DOI] [PubMed] [Google Scholar]
- Skold O. Resistance to trimethoprim and sulfonamides. Vet. Res. 2001, 32, 261–273. 10.1051/vetres:2001123. [DOI] [PubMed] [Google Scholar]
- Stapleton P. J.; Lundon D. J.; McWade R.; Scanlon N.; Hannan M. M.; O’Kelly F.; Lynch M. Antibiotic resistance patterns of Escherichia coli urinary isolates and comparison with antibiotic consumption data over 10 years, 2005–2014. Ir. J. Med. Sci. 2017, 186 (3), 733–741. 10.1007/s11845-016-1538-z. [DOI] [PubMed] [Google Scholar]
- Howell E. E. Searching sequence space: Two different approaches to dihydrofolate reductase catalysis. ChemBioChem 2005, 6, 590–600. 10.1002/cbic.200400237. [DOI] [PubMed] [Google Scholar]
- Huovinen P. Trimethoprim resistance. Antimicrob. Agents Chemother. 1987, 31, 1451–1456. 10.1128/AAC.31.10.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faltyn M.; Alcock B.; McArthur A. Evolution and nomenclature of the trimethoprim resistant dihydrofolate (dfr) reductases. Preprints 2019, 10.20944/preprints201905.0137.v1. [DOI] [Google Scholar]
- Masters P. A.; O’Bryan T. A.; Zurlo J.; Miller D. Q.; Joshi N. Trimethoprim-sulfamethoxazole revisited. Arch. Intern. Med. 2003, 163, 402–410. 10.1001/archinte.163.4.402. [DOI] [PubMed] [Google Scholar]
- Kadlec K.; Kehrenberg C.; Schwarz S. Molecular basis of resistance to trimethoprim, chloramphenicol and sulphonamides in Bordetella bronchiseptica. J. Antimicrob. Chemother. 2005, 56, 485–490. 10.1093/jac/dki262. [DOI] [PubMed] [Google Scholar]
- L’Abée-Lund T. M.; Sørum H. Class 1 integrons mediate antibiotic resistance in the fish pathogen Aeromonas salmonicida worldwide. Microb. Drug Resist. 2001, 7, 263–272. 10.1089/10766290152652819. [DOI] [PubMed] [Google Scholar]
- Amyes S. G.; Smith J. T. The purification and properties of the trimethoprim-resistant dihydrofolate reductase mediated by the R-factor, R388. Eur. J. Biochem. 1976, 61 (2), 597–603. 10.1111/j.1432-1033.1976.tb10055.x. [DOI] [PubMed] [Google Scholar]
- Toulouse J. L.; Edens T. J.; Alejaldre L.; Manges A. R.; Pelletier J. N. Integron-associated DfrB4, a previously uncharacterized member of the trimethoprim-resistant dihydrofolate reductase B family, is a clinically identified emergent source of antibiotic resistance. Antimicrob. Agents Chemother. 2017, 61, 1–5. 10.1128/AAC.02665-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broad D. F.; Smith J. T. Classification of trimethoprim-resistant dihydrofolate reductases mediated by R-plasmids using isoelectric focussing. Eur. J. Biochem. 1982, 125, 617–622. 10.1111/j.1432-1033.1982.tb06727.x. [DOI] [PubMed] [Google Scholar]
- Schmitzer A. R.; Lepine F.; Pelletier J. N. Combinatorial exploration of the catalytic site of a drug-resistant dihydrofolate reductase: creating alternative functional configurations. Protein Eng., Des. Sel. 2004, 17 (11), 809–19. 10.1093/protein/gzh090. [DOI] [PubMed] [Google Scholar]
- Pattishall K. H.; Acar J.; Burchall J. J.; Goldstein F. W.; Harvey R. J. Two distinct types of trimethoprim-resistant dihydrofolate reductase specified by R-plasmids of different compatibility groups. J. Biol. Chem. 1977, 252 (7), 2319–23. [PubMed] [Google Scholar]
- Jackson M.; Chopra S.; Smiley R. D.; Maynord P. O. N.; Rosowsky A.; London R. E.; Levy L.; Kalman T. I.; Howell E. E. Calorimetric studies of ligand binding in R67 dihydrofolate reductase. Biochemistry 2005, 44, 12420–12433. 10.1021/bi050881s. [DOI] [PubMed] [Google Scholar]
- Rajagopalan P. T. R.; Zhang Z.; McCourt L.; Dwyer M.; Benkovic S. J.; Hammes G. G. Interaction of dihydrofolate reductase with methotrexate: Ensemble and single-molecule kinetics. Proc. Natl. Acad. Sci. U. S. A. 2002, 99 (21), 13481–13486. 10.1073/pnas.172501499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krahn J. M.; Jackson M. R.; DeRose E. F.; Howell E. E.; London R. E. Crystal structure of a type II dihydrofolate reductase catalytic ternary complex. Biochemistry 2007, 46, 14878–14888. 10.1021/bi701532r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duff M. R.; Chopra S.; Strader M. B.; Agarwal P. K.; Howell E. E. Tales of dihydrofolate binding to R67 dihydrofolate reductase. Biochemistry 2016, 55, 133–145. 10.1021/acs.biochem.5b00981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yachnin B. J.; Colin D. Y.; Volpato J. P.; Ebert M.; Pelletier J. N.; Berghuis A. M. Novel crystallization conditions for tandem variant R67 DHFR yield a wild-type crystal structure. Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun. 2011, 67, 1316–1322. 10.1107/S1744309111030417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastien D.; Ebert M. C. C. J. C.; Forge D.; Toulouse J.; Kadnikova N.; Perron F.; Mayence A.; Huang T. L.; Vanden Eynde J. J.; Pelletier J. N. Fragment-based design of symmetrical bis-benzimidazoles as selective inhibitors of the trimethoprim-resistant, type II R67 dihydrofolate reductase. J. Med. Chem. 2012, 55, 3182–3192. 10.1021/jm201645r. [DOI] [PubMed] [Google Scholar]
- Toulouse J. L.; Yachnin B. J.; Ruediger E. H.; Deon D.; Gagnon M.; Saint-Jacques K.; Ebert M. C. C. J. C.; Forge D.; Bastien D.; Colin D. Y.; Vanden Eynde J. J.; Marinier A.; Berghuis A. M.; Pelletier J. N. Structure-Based Design of Dimeric Bisbenzimidazole Inhibitors to an Emergent Trimethoprim-Resistant Type II Dihydrofolate Reductase Guides the Design of Monomeric Analogues. ACS Omega 2019, 4 (6), 10056–10069. 10.1021/acsomega.9b00640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright D. L.; Anderson A. C. Antifolate agents: a patent review (2006 - 2010). Expert Opin. Ther. Pat. 2011, 21, 1293–1308. 10.1517/13543776.2011.587804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi G.; Shaw G.; Liang Y. H.; Subburaman P.; Li Y.; Wu Y.; Yan H.; Ji X. Bisubstrate analogue inhibitors of 6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase: New design with improved properties. Bioorg. Med. Chem. 2012, 20, 47–57. 10.1016/j.bmc.2011.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi G.; Shaw G.; Li Y.; Wu Y.; Yan H.; Ji X. Bisubstrate analog inhibitors of 6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase: New lead exhibits a distinct binding mode. Bioorg. Med. Chem. 2012, 20, 4303–4309. 10.1016/j.bmc.2012.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domalaon R.; Idowu T.; Zhanel G. G.; Schweizer F.. Antibiotic Hybrids: the Next Generation of Agents and Adjuvants against Gram-Negative Pathogens? Clin. Microbiol. Rev. 2018, 31 ( (2), ), 10.1128/CMR.00077-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T.; Wan Y.; Xiao Y.; Xia C.; Duan G. Dual-Target Inhibitors Based on HDACs: Novel Antitumor Agents for Cancer Therapy. J. Med. Chem. 2020, 63 (17), 8977–9002. 10.1021/acs.jmedchem.0c00491. [DOI] [PubMed] [Google Scholar]
- Guo Z. R. Strategy of molecular drug design: dual-target drug design. Acta Pharmaceutica Sinica 2009, 44 (3), 209–18. [PubMed] [Google Scholar]
- Krahn J. M.; Jackson M. R.; DeRose E. F.; Howell E. E.; London R. E. Crystal structure of a type II dihydrofolate reductase catalytic ternary complex. Biochemistry 2007, 46, 14878–14888. 10.1021/bi701532r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert M. C.; Morley K. L.; Volpato J. P.; Schmitzer A. R.; Pelletier J. N. Asymmetric mutations in the tetrameric R67 dihydrofolate reductase reveal high tolerance to active-site substitutions. Protein Sci. 2015, 24 (4), 495–507. 10.1002/pro.2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolg J. W.; Hanggi U. J.; Zachau H. G. Isolation of a small DNA fragment carrying the gene for a dihydrofolate reductase from a trimethoprim resistance factor. Mol. Gen. Genet. 1978, 164 (1), 15–29. 10.1007/BF00267594. [DOI] [PubMed] [Google Scholar]
- Brito R. M.; Reddick R.; Bennett G. N.; Rudolph F. B.; Rosevear P. R. Characterization and stereochemistry of cofactor oxidation by a type II dihydrofolate reductase. Biochemistry 1990, 29 (42), 9825–31. 10.1021/bi00494a011. [DOI] [PubMed] [Google Scholar]
- Howell E. E.; Booth C.; Farnum M.; Kraut J.; Warren M. S. A second-site mutation at phenylalanine-137 that increases catalytic efficiency in the mutant aspartate-27 to serine Escherichia coli dihydrofolate reductase. Biochemistry 1990, 29 (37), 8561–8569. 10.1021/bi00489a009. [DOI] [PubMed] [Google Scholar]
- Fierke C. A.; Johnson K. A.; Benkovic S. J. Construction and evaluation of the kinetic scheme associated with dihydrofolate reductase from Escherichia coli. Biochemistry 1987, 26 (13), 4085–4092. 10.1021/bi00387a052. [DOI] [PubMed] [Google Scholar]
- Stone S. R.; Morrison J. F. Mechanism of inhibition of dihydrofolate reductases from bacterial and vertebrate sources by various classes of folate analogues. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 1986, 869 (3), 275–285. 10.1016/0167-4838(86)90067-1. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya S.; Bershtein S.; Yan J.; Argun T.; Gilson A. I.; Trauger S. A.; Shakhnovich E. I.. Transient protein-protein interactions perturb Escherichia coli metabolome and cause gene dosage toxicity. eLife 2016, 5, 10.7554/eLife.20309 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.