

Abstract
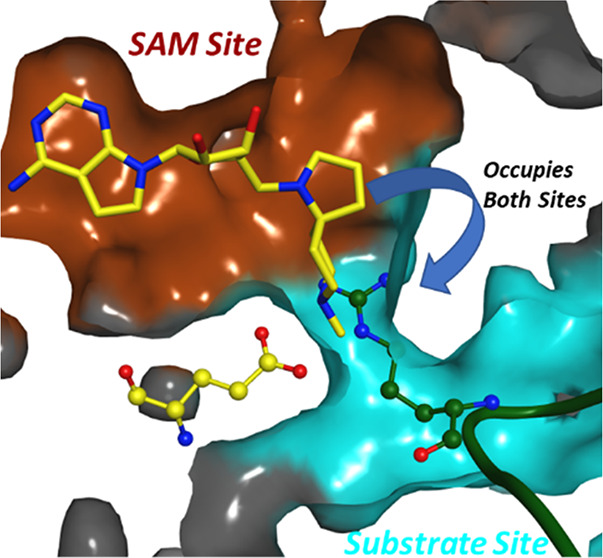
Protein arginine methyltransferase 5 (PRMT5) is an enzyme that can symmetrically dimethylate arginine residues in histones and nonhistone proteins by using S-adenosyl methionine (SAM) as the methyl donating cofactor. We have designed a library of SAM analogues and discovered potent, cell-active, and selective spiro diamines as inhibitors of the enzymatic function of PRMT5. Crystallographic studies confirmed a very interesting binding mode, involving protein flexibility, where both the cofactor pocket and part of substrate binding site are occupied by these inhibitors.
Keywords: PRMT5, Spirodiamines, Methyltransferase, Chemical Probe, Protein flexibility
In the present communication, we report the discovery of a chemical probe 15 to potently inhibit the enzymatic function of the protein arginine methyl transferase 5 (PRMT5), in vitro and in cells, and provide evidence for its molecular mechanism of action by enzyme kinetics, binding studies and structural characterization.
PRMT5 belongs to the protein family of methyltransferases that specifically methylates protein substrates on arginines.1−4 PRMT5 is a type II methyltransferase that catalyzes the formation of symmetric N,N-dimethylarginines.5 This post-translational modification can occur on histone and nonhistone proteins and is relevant for a variety of biological process, such as transcription, differentiation, and cell cycle progression.6,7 Epigenetic modification by specific methylation of histones occurs in the nucleus.5 Methylation by PRMT5 of nonhistone proteins occurs in the cytoplasm,8 an important example of which is the regulation of spliceosome assembly/activity by methylation of the ribonucleoprotein SmD3 resulting in RNA splicing.9−11 Mechanistically, the methylation of arginines by PRMT5 is accompanied by the conversion of the cofactor S-adenosylmethionine (SAM) to S-adenosylhomocysteine (SAH).2,4 This post-translational modification is a long-lasting mark, determined by the half-life of the corresponding protein substrate as no demethylase has been identified so far as a counterpart of PRMT5 activity. Stability, substrate recognition, and enzymatic activity of PRMT5 is regulated by the assembly factor methylosome protein 50 (MEP50) that forms in vitro a hetero-octameric complex (PRMT5)4/(MEP50)4.12,13
Inhibition of PRMT5 may be of therapeutic value to treat hematological cancers like AML14,15 and nonsmall cell lung cancer (NSCLC). Further, elevated expression of PRMT5 and its cofactor, MEP50, in NSCLC is highly correlated with poor survival.16 Therapeutic rationales for PRMT5 inhibition based on genetic evidence are starting to emerge. For instance, several cancer cell lines missing the common tumor suppressor CDKN2A and another metabolic enzyme, 5-methylthioadenosine phosphorylase (MTAP), became fully dependent on PRMT5 for survival, evidenced by genetic knockdown experiments.17 Recently, a few small molecule inhibitors of PRMT5 have been reported including SAM analogues such as A9145C (dehydrosinefungin)18 and LLY-28319 (see Supporting Information (SI), Section S1 for a full list).
Our own strategy to design structurally novel and cell active PRMT5 inhibitors started from the notion that adenosine is a weak PRMT5 enzyme inhibitor (unpublished results). On the basis of principles of chemical similarity, we hypothesized that adenosine derivatives would bind to PRMT5 in a way analogous to SAM, SAH, or A9145C.18Figure 1a depicts this as a general basis of our library design concept. On the basis of the electrostatic features of the binding site, we hypothesized that the introduction of a basic nitrogen atom in the lower-right proximity of the 5′ position of adenosine (Figure 1a) would provide complementarity to the protein surface, which is predominantly rendered electronegative by the GLU444 side chain at the interface of cofactor and substrate binding sites. Taking this hypothesis into account, along with the potential flexibility of PRMT5, we designed a small library of cyclic amines and spirocyclic diamines to minimize conformational flexibility in the ligand while diversifying the topology (i.e., utilizing different vectors). We determined the enzymatic inhibition by measuring the decrease of SAM → SAH conversion in the presence of PRMT5–MEP50 complex and a recombinant human histone H2A protein by using a RapidFire high-throughput mass spectrometry assay (SI, Section S4). A cell-based target engagement assay was developed, which measures in A549 cells the inhibition of symmetric arginine dimethylation of the SmD1 and SmD3 proteins (SmD1/3), components of the survival of motor neuron (SMN) complex,20 as detected by immune histochemistry (SI, Section S5), as well as cell viability. It was shown previously20 that SmD1/3 methylation is dependent on PRMT5 catalytic activity. An example of the concentration–response curves obtained for the key compounds 14 and 15, as well as a negative control 11 is depicted in Figure 3.
Figure 1.
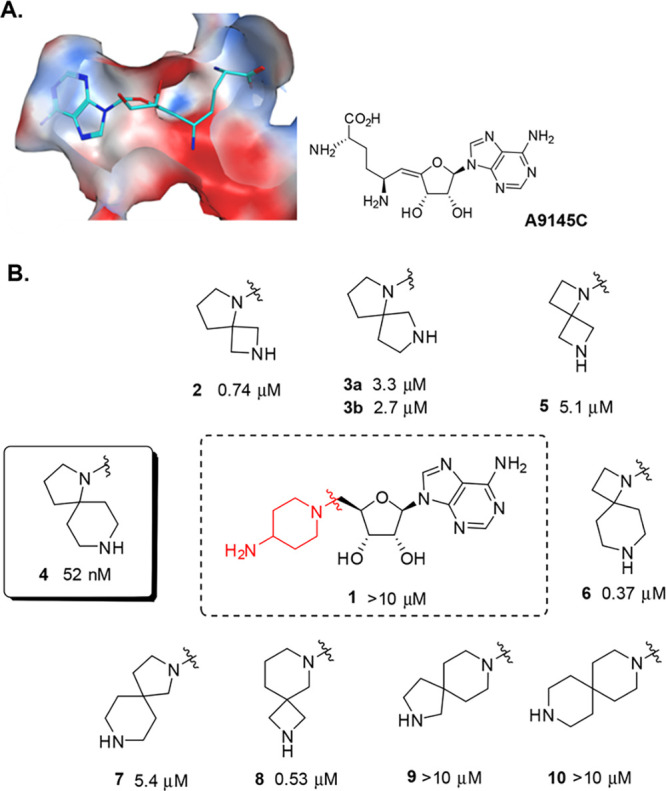
(A) Molecular surface representation of PRMT5 bound to SAM analogue A9145C (cyan sticks; PDB 4GQB). The surface (clipped for clarity, front view is semitransparent) is colored by Poisson–Boltzmann electrostatics map ranging from −40 (red) through 0 (white) to +40 (blue) as modeled in Chemical Computing Group’s MOE software version 2016.0801. The histone H4 peptide is hidden to show pocket features near the SAM binding site. (B) Initial PRMT5 hits. Activities are IC50values in the enzyme assay. The absolute stereochemistry at the spiro diamine group in 3a/b is undefined, both diastereomers were prepared separately. For details, see SI, Section S2.
Figure 3.
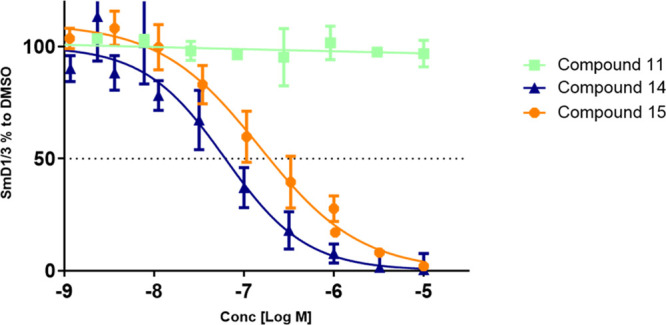
A549 cells were treated at indicated concentrations for 48 h and cellular arginine, dimethylation of SmD1/3 proteins was determined by immune fluorescence microscopy. Dose response curves of representatives are shown. The graphs represent ratios of nuclear to cytoplasmic symmetric dimethylation intensities normalized to the DMSO control. The results are mean ± SE of at least three replicates.
As depicted in Figure 1B, several of our initial diamines displayed only weak to modest enzyme inhibition. In contrast, spiro diamine 4 stood out as an inhibitor with nanomolar potency, which translated into cellular activity (Table 1). The isomeric diamines 7 and 9 showed much reduced activity. The monocyclic diamine 1 showed no measurable inhibition. Few other spiro diamines, such as 6 and 8, showed submicromolar inhibition. We used rigid protein docking to rationalize these structure–activity relationships (SARs) without any success and hypothesized the role of protein flexibility in modeling the putative binding modes of this class of molecules. Eventually, an induced-fit docking (IFD) model was developed (SI, Section S3). With the IFD approach, a semiquantitative structure-based model justifying the SARs emerged, where most of the active compounds (SI, Section S3.2) formed a hydrogen-bond via their distal amines with Glu444 residue at the boundary of the substrate site, while all the weakly active compounds did not form such an interaction.
Table 1. Enzymatic and Cellular Inhibition Data of Compounds to Evaluate the SAR.


PRMT5-MEP50 enzyme inhibition (5.0 nM enzyme concentration in assay), mean for n ≥ 2.
Inhibition of symmetric arginine dimethylation of SmD1/3 proteins in A549 cancer cells after 2 days incubation, detected by immuno histochemistry, mean for n ≥ 3.
We sought to prove the importance of the distal basic nitrogen by SAR expansion (Table 1). Removing the piperidine ring (compound 11) led to loss of most of the activity, while the cyclohexyl analogue 16 and the acetyl piperidine 17 showed diminished activity by 3 orders of magnitude. Interestingly, the enzymatic inhibition of 4 in the biochemical assay did translate to cell-based inhibition (Table 1), indicating sufficient cellular permeability, without significant effects on cell viability (SI, Section S5). Replacing the adenine by 7-deaza adenine (compound 14) and methylating the piperidine to a tertiary amine (compound 15) was well tolerated and enhanced the cellular activity to 58 nM and 200 nM, respectively. Ring-opening of the piperidine in 4 to obtain amino ethyl pyrolidines 12 and 13 maintained enzymatic activity which is stereodependent, with the S stereochemistry as in 13 preferred. Taking these results together, we have identified spirocyclic diamine analogues of SAM (14 and 15), in which the piperidine spirocyle significantly contributes to the inhibition of PRMT5 in vitro and in cells.
The Glu444 interaction model and the role of protein flexibility in this series was further validated by a cocrystal structure of compound 14 as a representative, bound to PRMT5 (Figure 2). Compound 14 binds to the SAM binding site of PRMT5 (Figure 2a) with its deazapurine substructure making interactions analogous to the adenosine portion of A9145C. The spirodiamine moiety, however, interacts partially with the substrate binding site, via a hydrogen-bonding interaction with the catalytic E444 residue. In comparison to A9145C bound cocrystal structure (Figure 2b), compound 14 bound PRMT5 structure shows a conformational change in the F327 side chain to accommodate and interact with the spiro diamine moiety. Further, the side chain of the second catalytic residue E435 moves upward to accommodate the spiro moiety and interestingly occupies the equivalent space overlapping with the putative acidic moiety of A9145C (and in analogy, the acidic moeity of SAM and SAH). The mechanism of action of compound 14 as well as compound 15 was determined (SI, Section S4.4) and confirmed that both molecules are competitive with SAM and noncompetitive with histone H2A. This observation is in agreement with the crystal structure where the SAM pocket is fully occupied by these compounds. The noncompetitive inhibition mechanism with the protein substrate is however not directly obvious by observing the crystallographic binding mode where part of the protein substrate site is simultaneously occupied by these inhibitors. Previous crystallographic studies on apo and SAH bound Caenorhabditis elegans PRMT5 structures have revealed that a segment including a N-terminal loop (L0) and a following helix (αA) became ordered upon SAH binding.21 A superimposition of these C. elegans structures with highly conserved human PRMT5 structure bound to a histone H4 tail peptide (SI, Section S7.2) shows that SAM/SAH binding is necessary to stabilize and create the substrate binding site. The spiroamines reported here can indeed stabilize and create the substrate binding site in an analogous manner. They would, however, sterically hinder specific binding of the substrate arginine side chain, where the arginine side chain also needs to engage in an interaction with Glu444. It is possible that in solution and as observed by our MOA studies, the substrate could still bind nonspecifically to the remainder of the substrate site. Regarding experimentally observed features of compound binding modes, it is interesting to note that there are two classes of (representatives in SI, Section S1) well-characterized PRMT5 inhibitors published (and structures reported in the Protein Data Bank), either SAM site binders or substrate site binders, the latter having a requirement of SAM or a SAM mimetic cobound. Structurally, compound 14 and by analogy, compound 15 bind in the SAM site as well as part of the substrate site, simultaneously, which makes their binding mode a novel one, e.g., when compared to recently reported compounds like LLY-283 (SI, Section S7.3). In addition, the binding of compound 15, as a representative was also studied by surface plasmon resonance (SPR) (SI, Section S6) using immobilized PRMT5:MEP50 complex. A Kd value of 7.6 nM was calculated based on the SPR analysis.
Figure 2.

(A) Interactions of compound 14 (yellow sticks) with PRMT5 (gray sticks). Hydrogen bonds are depicted as dotted lines. Molecular surface of the binding pocket is colored as brown and cyan to indicate the putative SAM and substrate binding sites, respectively. (B) Superimposition of cocrystal structure from this study (PDB 6RLL; yellow) with SAM analogue A9145C and histone H4 peptide bound PRMT5 (PDB 4GQB; cyan).
Finally, the selectivity of compound 15 was assessed against 21 purified recombinant human arginine (Arg) and lysine (Lys) methyltransferases (Figure 4 and SI, Section S8).22,23 At 10 μM compound 15, only the PRMT5/MEP50 enzyme was inhibited by >90%, while another closely related Arg methyltransferase, such as PRMT7, was minimally inhibited (<25% inhibition). No Lys methyltransferases were inhibited at >25%. Taken together, these data show the high degree of PRMT5 selectivity of this molecule 15.
Figure 4.

Selectivity of compound 15, tested at 10 μM concentration in a lysine and arginine methyl transferase panel, phylogenetic tree representation as modified from Richon et al.23 Small gray circles represent <25% inhibition, large red circle >90% inhibition (PRMT5-MEP50 only).
In conclusion, structure-based design was used to discover a novel chemical class of potent, selective and cell-active spirodiamines as PRMT5 inhibitors with a novel binding mode, occupying parts of both the SAM and the substrate sites, as demonstrated by compounds 14 and 15. While the physicochemical properties of compound 15 are in the right range to study its behavior in cells, the high degree of hydrophilicity and in particular the presence of two basic centers precludes the assessment of 15 in vivo (mouse) following the oral route due to limited absorption (not shown). A full account of the optimization of the probe molecule 15 to obtain a better drug-like molecule with robust tumor growth inhibiting properties will be reported in due course.
Glossary
Abbreviations
- PRMT5
protein arginine methyl transferase 5
- SmD1/3
small nuclear riboprotein D 1/3
- SAM
S-adenosyl methionine
- SAH
S-adenosyl homocysteine
- MEP50
methylosome protein 50
- NSCLC
nonsmall cell lung cancer
- CDKN2A
cyclin dependent kinase inhibitor 2A
- MTAP
5-methylthioadenosine phosphorylase
- H2A
histone 2A
- SMN
survival of motor neuron
- IFD
induced-fit docking
- SPR
surface plasmon resonance
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00355.
The authors declare no competing financial interest.
Supplementary Material
References
- Bedford M. T.; Clarke S. G. Protein arginine methylation in mammals: who, what, and why. Mol. Cell 2009, 33, 1–13. 10.1016/j.molcel.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaniskan H. U.; Jin J. Recent progress in developing selective inhibitors of protein methyltransferases. Curr. Opin. Chem. Biol. 2017, 39, 100–108. 10.1016/j.cbpa.2017.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf S. S. The protein arginine methyltransferase family: an update about function, new perspectives and the physiological role in humans. Cell. Mol. Life Sci. 2009, 66, 2109–2121. 10.1007/s00018-009-0010-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.; Bedford M. T. Protein arginine methyltransferases and cancer. Nat. Rev. Cancer 2013, 13, 37–50. 10.1038/nrc3409. [DOI] [PubMed] [Google Scholar]
- Karkhanis V.; Hu Y. J.; Baiocchi R. A.; Imbalzano A. N.; Sif S. Versatility of PRMT5-induced methylation in growth control and development. Trends Biochem. Sci. 2011, 36, 633–641. 10.1016/j.tibs.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedford M. T.; Richard S. Arginine methylation an emerging regulator of protein function. Mol. Cell 2005, 18, 263–272. 10.1016/j.molcel.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Jansson M.; Durant S. T.; Cho E. C.; Sheahan S.; Edelmann M.; Kessler B.; La Thangue N. B. Arginine methylation regulates the p53 response. Nat. Cell Biol. 2008, 10, 1431–1439. 10.1038/ncb1802. [DOI] [PubMed] [Google Scholar]
- Stopa N.; Krebs J. E.; Shechter D. The PRMT5 arginine methyltransferase: many roles in development, cancer and beyond. Cell. Mol. Life Sci. 2015, 72, 2041–2059. 10.1007/s00018-015-1847-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahms H.; Meheus L.; de Brabandere V.; Fischer U.; Luhrmann R. Symmetrical dimethylation of arginine residues in spliceosomal Sm protein B/B’ and the Sm-like protein LSm4, and their interaction with the SMN protein. RNA 2001, 7, 1531–1542. 10.1017/S135583820101442X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang T. W.; Peng P. J.; Tarn W. Y. The exon junction complex component Y14 modulates the activity of the methylosome in biogenesis of spliceosomal small nuclear ribonucleoproteins. J. Biol. Chem. 2011, 286, 8722–8728. 10.1074/jbc.M110.190587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesen W. J.; Paushkin S.; Wyce A.; Massenet S.; Pesiridis G. S.; Van Duyne G.; Rappsilber J.; Mann M.; Dreyfuss G. The methylosome, a 20S complex containing JBP1 and pICln, produces dimethylarginine-modified Sm proteins. Mol. Cell. Biol. 2001, 21, 8289–8300. 10.1128/MCB.21.24.8289-8300.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho M. C.; Wilczek C.; Bonanno J. B.; Xing L.; Seznec J.; Matsui T.; Carter L. G.; Onikubo T.; Kumar P. R.; Chan M. K.; Brenowitz M.; Cheng R. H.; Reimer U.; Almo S. C.; Shechter D. Structure of the arginine methyltransferase PRMT5-MEP50 reveals a mechanism for substrate specificity. PLoS One 2013, 8, e57008 10.1371/journal.pone.0057008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng X.; Wang Z. Protein arginine methyltransferase 5 regulates multiple signaling pathways to promote lung cancer cell proliferation. BMC Cancer 2016, 16, 567. 10.1186/s12885-016-2632-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik S.; Liu F.; Veazey K. J.; Gao G.; Das P.; Neves L. F.; Lin K.; Zhong Y.; Lu Y.; Giuliani V.; Bedford M. T.; Nimer S. D.; Santos M. A. Genetic deletion or small-molecule inhibition of the arginine methyltransferase PRMT5 exhibit anti-tumoral activity in mouse models of MLL-rearranged AML. Leukemia 2018, 32, 499–509. 10.1038/leu.2017.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarighat S. S.; Santhanam R.; Frankhouser D.; Radomska H. S.; Lai H.; Anghelina M.; Wang H.; Huang X.; Alinari L.; Walker A.; Caligiuri M. A.; Croce C. M.; Li L.; Garzon R.; Li C.; Baiocchi R. A.; Marcucci G. The dual epigenetic role of PRMT5 in acute myeloid leukemia: gene activation and repression via histone arginine methylation. Leukemia 2016, 30, 789–799. 10.1038/leu.2015.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyorffy B.; Surowiak P.; Budczies J.; Lanczky A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS One 2013, 8, e82241 10.1371/journal.pone.0082241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavrakis K. J.; McDonald E. R. 3rd; Schlabach M. R.; Billy E.; Hoffman G. R.; deWeck A.; Ruddy D. A.; Venkatesan K.; Yu J.; McAllister G.; Stump M.; deBeaumont R.; Ho S.; Yue Y.; Liu Y.; Yan-Neale Y.; Yang G.; Lin F.; Yin H.; Gao H.; Kipp D. R.; Zhao S.; McNamara J. T.; Sprague E. R.; Zheng B.; Lin Y.; Cho Y. S.; Gu J.; Crawford K.; Ciccone D.; Vitari A. C.; Lai A.; Capka V.; Hurov K.; Porter J. A.; Tallarico J.; Mickanin C.; Lees E.; Pagliarini R.; Keen N.; Schmelzle T.; Hofmann F.; Stegmeier F.; Sellers W. R. Disordered methionine metabolism in MTAP/CDKN2A-deleted cancers leads to dependence on PRMT5. Science 2016, 351, 1208–1213. 10.1126/science.aad5944. [DOI] [PubMed] [Google Scholar]
- Antonysamy S.; Bonday Z.; Campbell R. M.; Doyle B.; Druzina Z.; Gheyi T.; Han B.; Jungheim L. N.; Qian Y.; Rauch C.; Russell M.; Sauder J. M.; Wasserman S. R.; Weichert K.; Willard F. S.; Zhang A.; Emtage S. Crystal structure of the human PRMT5:MEP50 complex. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 17960–17965. 10.1073/pnas.1209814109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonday Z. Q.; Cortez G. S.; Grogan M. J.; Antonysamy S.; Weichert K.; Bocchinfuso W. P.; Li F.; Kennedy S.; Li B.; Mader M. M.; Arrowsmith C. H.; Brown P. J.; Eram M. S.; Szewczyk M. M.; Barsyte-Lovejoy D.; Vedadi M.; Guccione E.; Campbell R. M. LLY-283, a Potent and Selective Inhibitor of Arginine Methyltransferase 5, PRMT5, with Antitumor Activity. ACS Med. Chem. Lett. 2018, 9, 612–617. 10.1021/acsmedchemlett.8b00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister G.; Eggert C.; Buhler D.; Brahms H.; Kambach C.; Fischer U. Methylation of Sm proteins by a complex containing PRMT5 and the putative U snRNP assembly factor pICln. Curr. Biol. 2001, 11, 1990–1994. 10.1016/S0960-9822(01)00592-9. [DOI] [PubMed] [Google Scholar]
- Sun L.; Wang M.; Lv Z.; Yang N.; Liu Y.; Bao S.; Gong W.; Xu R. M. Structural insights into protein arginine symmetric dimethylation by PRMT5. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 20538–20543. 10.1073/pnas.1106946108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi K. Y.; Eason M. M.; Ferry J. J.; Planck J. L.; Walsh C. P.; Smith R. F.; Howitz K. T.; Ma H. Assay development for histone methyltransferases. Assay Drug Dev. Technol. 2013, 11, 227–236. 10.1089/adt.2012.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richon V. M.; Johnston D.; Sneeringer C. J.; Jin L.; Majer C. R.; Elliston K.; Jerva L. F.; Porter Scott M.; Copeland R. A. Chemogenetic analysis of human protein methyltransferases. Chem. Biol. Drug Des. 2011, 78, 199–210. 10.1111/j.1747-0285.2011.01135.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.