

Abstract

Interleukin 1 (IL-1) receptor-associated kinases (IRAKs) are serine/threonine kinases that play critical roles in initiating the innate immune response against foreign pathogens. Additionally, dysregulation of IRAK1 signaling plays a role in neoplastic disorders. For example, IRAK1 was shown to be important for survival and proliferation in many B-cell lymphomas, including Waldenström’s macroglobulinemia (WM) and ABC subtype Diffused Large B-cell Lymphoma (DLBCL) cells. Here, we report the discovery of a highly potent and selective covalent inhibitor of IRAK1, JH-X-119-01. Intact protein MS labeling studies confirmed that JH-X-119-01 irreversibly labels IRAK1 at C302. This compound exhibited cytotoxic activity at single digit micromolar concentrations in a panel of WM, DLBCL, and lymphoma cell lines expressing MYD88. Cotreatment of JH-X-119-01 with the BTK inhibitor ibrutinib resulted in synergistic killing effects in these systems. Taken together, JH-X-119-01 represents a highly selective probe of IRAK1 for further development.
Keywords: IRAK1, MYD88, Waldenström’s macroglobulinemia, Diffused Large B-cell Lymphoma, Kinase inhibitor
Interleukin 1 (IL-1) receptor-associated kinases (IRAKs) are serine/threonine kinases that play critical roles in initiating the innate immune response against foreign pathogens. IRAKs are involved in Toll-like receptors (TLRs) and IL-1R signaling pathways and are recruited by activated MYD88 to the receptor-signaling complex upon receptors activation. Altogether there are four IRAK kinases: IRAK1 and IRAK4, which are catalytically active kinases, and IRAK2 and IRAK3, which are believed to be catalytically inactive and are hence classified as “pseudokinases.”1 IRAK1 is ubiquitously expressed with its highest expression observed in blood and immune tissues (for example, bone marrow, lymph nodes, thymus, and peripheral blood) and in hematological malignancies.2 IRAK signaling contributes to multiple signaling pathways downstream of the Toll-interleukin receptors (TIRs) that ultimately regulate NF-κB and IFN regulatory factors (IRFs).3 In the case of NF-κB, IRAK1 mediates the downstream signals of TIRs through an interaction with MYD88 which is rapidly recruited to the receptor upon ligand binding to either IL-1R or a TLR. Subsequent phosphorylation on IRAK1 by upstream signals or through autophosphorylation is the key post-translational modification and hallmark of its activation, which allows IRAK1 to bind to TRAF6 resulting in release of the IRAK1 homodimer from MYD88 and downstream NF-κB activation.4 The participation of IRAK1 in signaling networks of the innate immune response makes it a critical regulator of inflammation,5 antiviral response,6 and subsequent activation of the adaptive immune response.7 Consequently, an extensive investigation into physiological and pathological functions of IRAK1 in regulating these processes has been performed. In particular, these studies have implicated IRAK1 inhibition as potential treatment for myocardial contractile dysfunction following burn,8 autoimmune conditions associated with hyperinflammation,9,10 myocardial dysfunction,11 microbial septic response,12 human myelodysplastic syndrome (MDS),13 and acute myeloid leukemia (AML).14 IRAK1 is also overexpressed and hyperphosphorylated in a subset of breast cancers: in particular, triple-negative breast cancer (TNBC).15 Furthermore, in many B-cell lymphomas, including Waldenström’s macroglobulinemia (WM) and ABC subtype Diffused Large B-cell Lymphoma (DLBCL) cells, the MYD88 L265P somatic mutation is highly prevalent and responsible for malignant growth through activation of nuclear factor NF-κB. Genetic knockdown of either BTK or IRAK1 leads to modest cell killing,15 while co-inhibition of both BTK and IRAK1/4 results in synergistic tumor cell growth suppression.16 A number of groups, including Amgen,17 Astellas,18 AstraZeneca,19 Genentech,20 Merck,21 Pfizer,22 and UCB,23 have reported potent dual inhibitors of IRAK1/4. In addition, several more dual IRAK1/4 inhibitors have recently been reviewed.24 Notably, a highly selective IRAK4 inhibitor developed by Nimbus with over 1000-fold selectivity for IRAK4 over IRAK1 also showed synergistic killing of ABC DLBCL cells with the BTK inhibitor ibrutinib.25 However, medicinal chemistry efforts directed at the development of selective inhibitors of IRAK1 have not been reported. Herein, we describe the development of JH-X-119-01, a highly potent and selective inhibitor of IRAK1.
The dual IRAK1/JNK inhibitor THZ-2-118 (Figure 1) served as a lead compound for development of selective IRAK1 inhibitors. THZ-2-118 potently inhibits IRAK1 with an apparent IC50 of 14.2 nM in a fixed time point assay by covalently labeling cysteine residue C302. THZ-2-118 is selective for IRAK1 over IRAK4; however, its potent inhibition of JNK1/2/3 kinases limits its utility as an IRAK1 probe. We sought to modify THZ-2-118 to remove JNK inhibition and improve the overall kinome selectivity. In order to achieve improved selectivity, we merged features of THZ series, which selects IRAK1 over IRAK4, with the excellent kinome selectivity of the dual IRAK1/4 inhibitor JH-1-2523 (Figure 1). Our resulting hybrid compound JH-X-119-01 (Figure 1) inhibited IRAK1 biochemically with an apparent IC50 of 9 nM while exhibiting no inhibition of IRAK4 at concentrations up to 10 μM. The compound showed exceptional kinome selectivity, as measured by a KINOMEScan selectivity score of S(10) = 0.01 at a concentration of 1 μM (Figure 1). JH-X-119-01 exhibited off-target inhibition of only two additional kinases, YSK4 and MEK3. Dose response analysis revealed an IC50 of 57 nM for YSK4. Biochemical assays for MEK3 were not commercially available at the time of writing this manuscript. Based on COBALT26 sequence alignment, there are no cysteine residues in YSK4 or MEK3 in a similar location to C302 or C307 of IRAK1 indicating that the binding of JH-X-119-01 to YSK4 and MEK3 is likely reversible. LC-MS analysis of labeled IRAK1 demonstrated JH-X-119-01 formed a covalent bond with the protein. Subsequent digestion and nanoflow LC-MS/MS27,28 confirmed this hybrid compound irreversibly labeled IRAK1 preferentially at C302 (95%) vs C307 (5%) (Figure 2).
Figure 1.
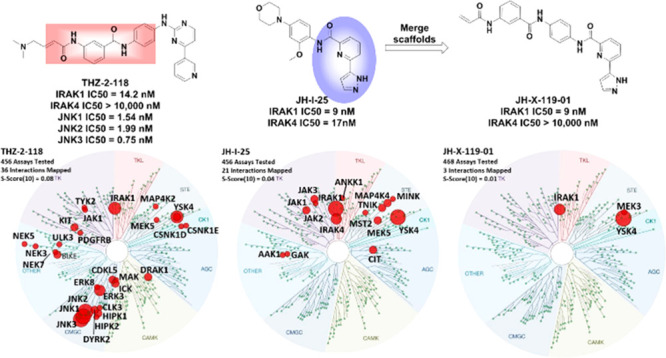
Evolution of JH-X-119-01 starting from THZ-2-118 and JH-I-25 and KinomeScan characterization of all three compounds at 1 μM showing the remarkable improvement in kinome selecvitivty for JH-X-119-01. The portions of THZ-2-118 and JH-I-25 that were fused are highlighted by pink and blue areas.
Figure 2.

JH-X-119-01 (1) forms a covalent bond with IRAK1 preferentially at C302. (A) Mass spectra (left) and zero-charge mass spectra (right) for IRAK1 protein treated with DMSO (top) or JH-X-119-01 (1). Unlabeled protein peaks are labeled with orange glyphs, while labeled protein peaks are indicated with green ones. After treatment with JH-X-119-01, the protein shows an increase in mass consistent with covalent labeling. (B) MS/MS spectrum acquired during nanoflow LC-MS/MS analysis of trypsin digested, JH-X-119-01 (1) treated IRAK1. Ions of type b and y are designated with blue and red glyphs, respectively. Inhibitor specific ions are labeled with green glyphs. Both y and b ion series localize the modification site to C302. A small amount of of C307 (∼5%) was also detectable
The initial SAR of this series focused changing the trajectory of the covalent warhead to target either C302 or C307 (Figure 4A). To accomplish this, we prepared a series of regioisomers of the benzamide linkers shown in Table 1.
Figure 4.

(A) Molecular model of 1 in complex with IRAK1. (B) Molecular model of 1 in complex with IRAK1 with only the interacting residues shown. Interactions are shown with dashed lines. Distances are in Ångstroms.
Table 1. SAR of the JH-X-119-01 Series.

| compd | IRAK1 IC50 (nM) | IRAK4 IC50 (nM) | labeling Site |
|---|---|---|---|
| JH-X-119-01 (1) | 9 | >10 000 | Cys 302 |
| 2 | 47 | >10 000 | Cys 307 |
| 3 | 55 | >10 000 | Cys 302 and Cys 307 (3:1) |
| 4 | 25 | >10 000 | Cys 302 |
| 5 | 259 | >10 000 | N/A |
| 6 | 52 | >10 000 | N/A |
The representative preparation of JH-X-119-01 is presented in Scheme 1. The 1,4–1,4 regioisomer (2) was less potent against IRAK1 with an apparent enzymatic IC50 of 47 nM. Interestingly, nanoflow LC-MS/MS analysis revealed this compound was selective for C307. MS labeling studies revealed (3) targeted both C302 and C307 (∼3:1 ratio). The 1,3–1,3 regioisomer (4) was more potent than 2 and 3 in the IRAK1 biochemical assay with an IC50 of 25 nM but was still less potent than JH-X-119-01, and showed a strong preference for C302 by nanoflow LC-MS/MS experiments. We also prepared 5, the reversible version of JH-X-119-01 with a propyl amide in place of the acrylamide warhead, which showed a 25-fold loss in potency, supporting the importance of the covalent bond formation for potency. In addition, compound 6 with a dimethylamino group attached to the warhead was prepared to improve the physicochemical properties of JH-X-119-01, however, compound 6 showed a 5-fold loss in potency.
Scheme 1. Preparation of JH-X-119-01.

Reagents and conditions: (a) HATU, DIEA, DCM; (b) TFA, DCM; (c) oxalyl chloride, 3-((tert-butoxycarbonyl)amino)benzoic acid, DCM, DMF, pyridine; (d) acryloyl chloride, THF, NaHCO3, H2O; (e) Pd(dppf)Cl2, t-BuXPhos, 1-(tetrahydro-2H-pyran-2-yl)-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole, Na2CO3, 1,4-dioxane, H2O, 100 °C.
We then tested compounds 1–6 in a panel of WM cells, DLBCL cells, and lymphoma cells expressing mutant MYD88. As seen in Figure 3A, JH-X-119-01 was the most potent, showing moderate cell killing effects in these cells with EC50’s ranging from 0.59 to 9.72 μM. Interestingly, despite having apparent enzyme IC50’s that are 2–5-fold less than that of JH-X-119-01, compounds 2 and 3 were much less potent in many of these cells. In many cases, cellular EC50’s could not be generated due to the lack of potency as indicated by N/A (>1000 μM) in Figure 3A. EC50 curves for all compounds are available in the Supporting Information (SI; Figure S1). However, compound 6 was within 2–5-fold potency in these cells as JH-X-119-01 despite having an apparent enzyme IC50 that is 5-fold less. This suggests that 6 might have better cell permeabilility due to the dimethylamino solubilizing group. Additionally, the reversible version of JH-X-119-01, compound 5 was much less potent highlighting the importance of the covalent bond formation. Overall, these results are in agreement with what has previously been observed with IRAK1 genetic knockdowns, where IRAK1 knockdown had moderate cell killing effects.15 We were initially concerned that some of the cell killing effects could be due to potency against YSK4. Therefore, we checked our gene expression profiling data for primary tumor cells from patients with WM and for healthy donor peripheral blood B-cells and confirmed that YSK4 expression levels were very low in both cells. We also searched the public protein atlas database for this kinase expression and found the similar results that YSK4 expression are barely detectable in general in B-cells or other hematologic cells except dendritic cells. In addition, in a study using shRNA library to screen multiple Defuse Large B-cell Lymphoma cell lines, YSK4 was not on the list of genes that have impact on cell survival.29 Based on this data, we do not believe YSK4 inhibition contributes to the efficacy of JH-X-119-01 in MYD88 mutated B-cell lymphoma cells.
Figure 3.

(A) Cellular EC50’s for compounds 1–6. Cells were incubated with inhibitors for 72 h and monitored via CellTiter-Glo assay. (B) Design of desthiobiotin probe JH-XIII-39-1. (C) Western blots for IRAK1 following pulldown assay by JH-XIII-39-01 at 5 μM after the pretreatment of BCWM.1 WM cells with 1, 4, or 6 at the indicated concentrations.
To understand how JH-X-119-01 binds to IRAK1, we initiated a molecular modeling study (Figure 4) using the previously reported cocrystal structure of JH-I-25 with IRAK1.30 See the SI for details regarding construction of the molecular modeling study. Our molecular modeling study predicted that JH-X-119-01 forms the same single hinge contact with the carbonyl oxygen of L291. The pyridine ring participates in a π–π stacking interation with Y288. In addition, the central phenyl ring participates in a π–π stacking interation with F290. As previously reported,30 Y288 participates in a polar interaction network with the catalytic lysine K239, E259, and D358 from the Asp-Phe-Gly (DFG) motif to stabilize the DFG-inactive conformation. The pyrazole moiety of 1 interacts with this network by forming a hydrogen bond with the carboxylate group of D358. We also observed an additional hydrogen bonding interaction with the amide linker of 1 and D298. Consistent with our MS study, the acrylamide warhead of 1 is in closer to C302 than C307 and is therefore predicted to form a covalent bond with C302 instead of C307.
To understand the selectivity of JH-X-119-01 for IRAK1 over IRAK4, we superimposed the cocrystal structure of JH-I-25 in complex with IRAK1 with the cocrystal structure of JH-I-25 in complex with IRAK4 (Figure 5). JH-I-25 is able to bind both IRAK1 and IRAK4 due to the conserved ATP pocket of IRAK1 and IRAK4. However, IRAK4 has a smaller ATP front pocket (Figure 5 inset). Due to the smaller ATP front pocket, JH-X-119-01 likely clashes with D278 of IRAK4. Therefore, IRAK4 cannot accommodate JH-X-119-01.
Figure 5.

Structural analysis of JH-X-119-01 docked into the overlay of the structure of IRAK1 and IRAK4.
Nevertheless, to confirm cellular target engagement, we designed the desthiobiotinylated version of JH-X-119-01, JH-XIII-39-01 (7), by appending a linker from the solvent-directed acrylamide moeity (Figure 3B). To ensure the viability of 7 as a useful probe, we tested it in an enzyme assay and found that it maintained inhibition of IRAK1 enzyme with an apparent enzyme IC50 of 70 nM. We then conducted pull-down experiments (see the SI for experimental details) using BCWM.1 WM cells (Figure 3C). All three compounds were able to block IRAK1 pull-down by the desthiobiotin compound 7, thereby confirming cellular target engagement. However, our lead compound, JH-X-119-01 (1), as well as compound 4 significantly blocked IRAK1 pull-down at 10 μM and less so at 5 μM, as compared to 6, which effectively blocked IRAK1 pull-down at 5 μM. These differences are most likely due to better cell permeability of 6, resulting from the presence of the dimethylamino solubilizing group or faster covalent bond formation of 6 with IRAK1. As BTK inhibition has been previously shown to synergize with IRAK inhibition, we cotreated JH-X-119-01 with ibrutinib, a covalent BTK inhibitor in BCWM.1 and TMD8 cells, which resulted in increased killing compared to JH-X-119-01 alone (Figure 6).
Figure 6.

Combination index (CI) values for the determination of synergistic (CI < 1.0) or additive (CI = 1.0) effects when combining ibrutinib and JH-X-119-01 at indicated concentrations in (A) MYD88 mutated BCWM.1 WM and (B) TMD8 ABC DLBCL cells.
In summary, we have discovered that (1) is a potent and remarkably selective inhibitor of IRAK1 kinase activity. Compound 1 is a covalent inhibitor of IRAK1 and labels C302 as confirmed by mass spectrometry. Characterization of 1 in a panel of WM cells, DLBCL cells, and lymphoma cells expressing MYD88 revealed that 1 exhibited moderate cell killing effects in these cells. Compound 1 exhibits excellent kinase selectivity as assessed by KinomeScan (468 kinases) with only two off targets YSK4 and MEK3. Further optimization of this chemotype especially in regard to in vivo pharmacokinetics will be reported in due course.
Acknowledgments
The authors wish to thank The David and Janet Bingham Research Fund and Yang Family Fund of the International Waldenstrom’s Macroglobulinemia Foundation, the Leukemia and Lymphoma Society (Grant: R6507-18), the NIH SPORE in Multiple Myeloma (Grant: 2P50CA100707-16A1), and Peter S. Bing M.D. The authors also acknolwedge generous financial support from the NIH: R01 CA233800, R01 CA222218 (to J.A.M.), and R03 TR002933; R37 AI050872 (to H.W.).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00378.
Full Ambit profiling data for JH-X-119-01, experimental details, EC50 curves, and MS labeling data for compounds 2–4 (PDF)
Author Contributions
Conceptualization: J.M.H., S.B., and N.S.G. Compound synthesis: J.M.H. Cellular experiments: G.Y. MS studies: S.B.F. and L.W. All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): N.S.G. is a founder, science advisory board member (SAB), and equity holder in Gatekeeper, Syros, Petra, C4, B2S, Aduro, Inception, Allorion, Jengu and Soltego (board member). The Gray lab receives or has received research funding from Novartis, Takeda, Astellas, Taiho, Janssen, Kinogen, Voronoi, Her2llc, Deerfield, and Sanofi. S.P.T. lab receives or has received funding from Pharmacyclics/Abbvie, Janssen Pharmaceuticals, Beigene, and Bristol Myers Squibb. J.A.M. serves on the SAB of 908 Devices. The Marto lab receives sponsored research support from Vertex and Astra-Zeneca. S.B. is a scientific advisory board member of Adenoid Cystic Carcinoma Foundation. H.W. is a co-founder, SAB member, and equity holder of Ventus Therapeutics.
Supplementary Material
References
- Flannery S.; Bowie A. G. The interleukin-1 receptor-associated kinases: Critical regulators of innate immune signalling. Biochem. Pharmacol. 2010, 80 (12), 1981–1991. 10.1016/j.bcp.2010.06.020. [DOI] [PubMed] [Google Scholar]
- Cao Z. D.; Henzel W. J.; Gao X. O. IRAK: A kinase associated with the interleukin-1 receptor. Science 1996, 271 (5252), 1128–1131. 10.1126/science.271.5252.1128. [DOI] [PubMed] [Google Scholar]
- Rao N.; Nguyen S.; Ngo K.; Fung-Leung W. P. A novel splice variant of interleukin-1 receptor (IL-1R)-associated kinase 1 plays a negative regulatory role in toll/IL-1R-induced inflammatory signaling. Mol. Cell. Biol. 2005, 25 (15), 6521–6532. 10.1128/MCB.25.15.6521-6532.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A.; Kaczanowska S.; Davila E. IL-1 receptor-associated kinase signaling and its role in inflammation, cancer progression, and therapy resistance. Front. Immunol. 2014, 5, 553. 10.3389/fimmu.2014.00553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringwood L.; Li L. W. The involvement of the interleukin-1 receptor-associated kinases (IRAKs) in cellular signaling networks controlling inflammation. Cytokine+ 2008, 42 (1), 1–7. 10.1016/j.cyto.2007.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong W. UnPINning IRAK1. Sci. Signaling 2011, 4, ec349. 10.1126/scisignal.4204ec349. [DOI] [Google Scholar]
- Gottipati S.; Rao N. L.; Fung-Leung W. P. IRAK1: A critical signaling mediator of innate immunity. Cell. Signalling 2008, 20 (2), 269–276. 10.1016/j.cellsig.2007.08.009. [DOI] [PubMed] [Google Scholar]
- Thomas J. A.; Tsen M. F.; White D. J.; Horton J. W. IRAK contributes to burn-triggered myocardial contractile dysfunction. American Journal of Physiology-Heart and Circulatory Physiology 2002, 283 (2), H829–H836. 10.1152/ajpheart.00416.2001. [DOI] [PubMed] [Google Scholar]
- Deng C.; Radu C.; Diab A.; Tsen M. F.; Hussain R.; Cowdery J. S.; Racke M. K.; Thomas J. A. IL-1 receptor-associated kinase 1 regulates susceptibility to organ-specific autoimmunity. J. Immunol. 2003, 170 (6), 2833–2842. 10.4049/jimmunol.170.6.2833. [DOI] [PubMed] [Google Scholar]
- Jacob C. O.; Zhu J. K.; Armstrong D. L.; Yan M.; Han J.; Zhou X. J.; Thomas J. A.; Reiff A.; Myones B. L.; Ojwang J. O.; Kaufman K. M.; Klein-Gitelman M.; McCurdy D.; Wagner-Weiner L.; Silverman E.; Ziegler J.; Kelly J. A.; Merrill J. T.; Harley J. B.; Ramsey-Goldman R.; Vila L. M.; Bae S. C.; Vyse T. J.; Gilkeson G. S.; Gaffney P. M.; Moser K. L.; Langefeld C. D.; Zidovetzki R.; Mohan C. Identification of IRAK1 as a risk gene with critical role in the pathogenesis of systemic lupus erythematosus. Proc. Natl. Acad. Sci. U. S. A. 2009, 106 (15), 6256–6261. 10.1073/pnas.0901181106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas J. A.; Haudek S. B.; Koroglu T.; Tsen M. F.; Bryant D. D.; White D. J.; Kusewitt D. F.; Horton J. W.; Giroir B. P. IRAK1 deletion disrupts cardiac Toll/IL-1 signaling and protects against contractile dysfunction. American Journal of Physiology-Heart and Circulatory Physiology 2003, 285 (2), H597–H606. 10.1152/ajpheart.0655.2001. [DOI] [PubMed] [Google Scholar]
- Chandra R.; Federici S.; Bishwas T.; Nemeth Z. H.; Deitch E. A.; Thomas J. A.; Spolarics Z. IRAK1-Dependent Signaling Mediates Mortality in Polymicrobial Sepsis. Inflammation 2013, 36 (6), 1503–1512. 10.1007/s10753-013-9692-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhyasen G. W.; Bolanos L.; Fang J.; Jerez A.; Wunderlich M.; Rigolino C.; Mathews L.; Ferrer M.; Southall N.; Guha R.; Keller J.; Thomas C.; Beverly L. J.; Cortelezzi A.; Oliva E. N.; Cuzzola M.; Maciejewski J. P.; Mulloy J. C.; Starczynowski D. T. Targeting IRAK1 as a Therapeutic Approach for Myelodysplastic Syndrome. Cancer Cell 2013, 24 (1), 90–104. 10.1016/j.ccr.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhyasen G.; Bolanos L.; Fang J.; Rigolino C.; Cortelezzi A.; Oliva E. N.; Cuzzola M.; Starczynowski D. T. Inhibition of IRAK1 As a Novel Therapeutic Strategy in Acute Myeloid Leukemia and Myelodysplastic Syndrome. Blood 2011, 118 (21), 612. 10.1182/blood.V118.21.612.612. [DOI] [Google Scholar]
- Wee Z. N.; Yatim S.; Kohlbauer V. K.; Feng M.; Goh J. Y.; Yi B.; Lee P. L.; Zhang S. J.; Wang P. P.; Lim E.; Tam W. L.; Cai Y.; Ditzel H. J.; Hoon D. S. B.; Tan E. Y.; Yu Q. IRAK1 is a therapeutic target that drives breast cancer metastasis and resistance to paclitaxel. Nat. Commun. 2015, 6, 8746. 10.1038/ncomms9746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G.; Zhou Y. S.; Liu X.; Xu L.; Cao Y.; Manning R. J.; Patterson C. J.; Buhrlage S. J.; Gray N.; Tai Y. T.; Anderson K. C.; Hunter Z. R.; Treon S. P. A mutation in MYD88 (L265P) supports the survival of lymphoplasmacytic cells by activation of Bruton tyrosine kinase in Waldenstrom macroglobulinemia. Blood 2013, 122 (7), 1222–1232. 10.1182/blood-2012-12-475111. [DOI] [PubMed] [Google Scholar]
- Powers J. P.; Li S. Y.; Jaen J. C.; Liu J. Q.; Walker N. P. C.; Wang Z. L.; Wesche H. Discovery and initial SAR of inhibitors of interleukin-1 receptor-associated kinase-4. Bioorg. Med. Chem. Lett. 2006, 16 (11), 2842–2845. 10.1016/j.bmcl.2006.03.020. [DOI] [PubMed] [Google Scholar]
- Kondo M.; Tahara A.; Hayashi K.; Abe M.; Inami H.; Ishikawa T.; Ito H.; Tomura Y. Renoprotective effects of novel interleukin-1 receptor-associated kinase 4 inhibitor AS2444697 through anti-inflammatory action in 5/6 nephrectomized rats. Naunyn-Schmiedeberg's Arch. Pharmacol. 2014, 387 (10), 909–919. 10.1007/s00210-014-1023-z. [DOI] [PubMed] [Google Scholar]
- Scott J. S.; Degorce S. L.; Anjum R.; Culshaw J.; Davies R. D. M.; Davies N. L.; Dillman K. S.; Dowling J. E.; Drew L.; Ferguson A. D.; Groombridge S. D.; Halsall C. T.; Hudson J. A.; Lamont S.; Lindsay N. A.; Marden S. K.; Mayo M. F.; Pease J. E.; Perkins D. R.; Pink J. H.; Robb G. R.; Rosen A.; Shen M. H.; McWhirter C.; Wu D. D. Discovery and Optimization of Pyrrolopyrimidine Inhibitors of Interleukin-1 Receptor Associated Kinase 4 (IRAK4) for the Treatment of Mutant MYD88(L265P) Diffuse Large B-Cell Lymphoma. J. Med. Chem. 2017, 60 (24), 10071–10091. 10.1021/acs.jmedchem.7b01290. [DOI] [PubMed] [Google Scholar]
- Bryan M. C.; Drobnick J.; Gobbi A.; Kolesnikov A.; Chen Y. S.; Rajapaksa N.; Ndubaku C.; Feng J. W.; Chang W.; Francis R.; Yu C.; Choo E. F.; DeMent K.; Ran Y. Q.; An L.; Emson C.; Huang Z. Y.; Sujatha-Bhaskar S.; Brightbill H.; DiPasquale A.; Maher J.; Wai J.; McKenzie B. S.; Lupardus P. J.; Zarrin A. A.; Kiefer J. R. Development of Potent and Selective Pyrazolopyrimidine IRAK4 Inhibitors. J. Med. Chem. 2019, 62 (13), 6223–6240. 10.1021/acs.jmedchem.9b00439. [DOI] [PubMed] [Google Scholar]
- Lim J.; Altman M. D.; Baker J.; Brubaker J. D.; Chen H.; Chen Y.; Fischmann T.; Gibeau C.; Kleinschek M. A.; Leccese E.; Lesburg C.; Maclean J. K. F.; Moy L. Y.; Mulrooney E. F.; Presland J.; Rakhilina L.; Smith G. F.; Steinhuebel D.; Yang R. Discovery of 5-Amino-N-(1H-pyrazol-4-yl)pyrazolo 1,5-a pyrimidine-3-carboxamide Inhibitors of IRAK4. ACS Med. Chem. Lett. 2015, 6 (6), 683–688. 10.1021/acsmedchemlett.5b00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumey L. N.; Boschelli D. H.; Bhagirath N.; Shim J.; Murphy E. A.; Goodwin D.; Bennett E. M.; Wang M. M.; Lin L. L.; Press B.; Shen M.; Frisbie R. K.; Morgan P.; Mohan S.; Shin J.; Rao V. R. Identification and optimization of indolo 2,3-c quinoline inhibitors of IRAK4. Bioorg. Med. Chem. Lett. 2014, 24 (9), 2066–2072. 10.1016/j.bmcl.2014.03.056. [DOI] [PubMed] [Google Scholar]
- Buckley G. M.; Gowers L.; Higueruelo A. P.; Jenkins K.; Mack S. R.; Morgan T.; Parry D. M.; Pitt W. R.; Rausch O.; Richard M. D.; Sabin V.; Fraser J. L. IRAK-4 inhibitors. Part 1: A series of amides. Bioorg. Med. Chem. Lett. 2008, 18 (11), 3211–3214. 10.1016/j.bmcl.2008.04.058. [DOI] [PubMed] [Google Scholar]
- Genung N. E.; Guckian K. M. Small Molecule Inhibition of Interleukin-1 Receptor-Associated Kinase 4 (IRAK4). Prog. Med. Chem. 2017, 56, 117–163. 10.1016/bs.pmch.2016.11.004. [DOI] [PubMed] [Google Scholar]
- Kelly P. N.; Romero D. L.; Yang Y.; Shaffer A. L. III; Chaudhary D.; Robinson S.; Miao W.; Rui L.; Westlin W. F.; Kapeller R.; Staudt L. M. Selective interleukin-1 receptor-associated kinase 4 inhibitors for the treatment of autoimmune disorders and lymphoid malignancy. J. Exp. Med. 2015, 212 (13), 2189–2201. 10.1084/jem.20151074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos J. S.; Agarwala R. COBALT: constraint-based alignment tool for multiple protein sequences. Bioinformatics 2007, 23 (9), 1073–1079. 10.1093/bioinformatics/btm076. [DOI] [PubMed] [Google Scholar]
- Ficarro S. B.; Zhang Y.; Lu Y.; Moghimi A. R.; Askenazi M.; Hyatt E.; Smith E. D.; Boyer L.; Schlaeger T. M.; Luckey C. J.; Marto J. A. Improved Electrospray Ionization Efficiency Compensates for Diminished Chromatographic Resolution and Enables Proteomics Analysis of Tyrosine Signaling in Embryonic Stem Cells. Anal. Chem. 2009, 81 (9), 3440–3447. 10.1021/ac802720e. [DOI] [PubMed] [Google Scholar]
- Ficarro S. B.; Browne C. M.; Card J. D.; Alexander W. M.; Zhang T.; Park E.; McNally R.; Dhe-Paganon S.; Seo H.-S.; Lamberto I.; Eck M. J.; Buhrlage S. J.; Gray N. S.; Marto J. A. Leveraging Gas-Phase Fragmentation Pathways for Improved Identification and Selective Detection of Targets Modified by Covalent Probes. Anal. Chem. 2016, 88 (24), 12248–12254. 10.1021/acs.analchem.6b03394. [DOI] [PubMed] [Google Scholar]
- Ngo V. N.; Young R. M.; Schmitz R.; Jhavar S.; Xiao W.; Lim K.-H.; Kohlhammer H.; Xu W.; Yang Y.; Zhao H.; Shaffer A. L.; Romesser P.; Wright G.; Powell J.; Rosenwald A.; Muller-Hermelink H. K.; Ott G.; Gascoyne R. D.; Connors J. M.; Rimsza L. M.; Campo E.; Jaffe E. S.; Delabie J.; Smeland E. B.; Fisher R. I.; Braziel R. M.; Tubbs R. R.; Cook J. R.; Weisenburger D. D.; Chan W. C.; Staudt L. M. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011, 470 (7332), 115–U133. 10.1038/nature09671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Qiao Q.; Ferrao R.; Shen C.; Hatcher J. M.; Buhrlage S. J.; Gray N. S.; Wu H. Crystal structure of human IRAK1. Proc. Natl. Acad. Sci. U. S. A. 2017, 114 (51), 13507–13512. 10.1073/pnas.1714386114. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.