

Abstract

Bruton’s tyrosine kinase (BTK) has been shown to play a key role in the pathogenesis of autoimmunity. Therefore, the inhibition of the kinase activity of BTK with a small molecule inhibitor could offer a breakthrough in the clinical treatment of many autoimmune diseases. This Letter describes the discovery of BMS-986143 through systematic structure–activity relationship (SAR) development. This compound benefits from defined chirality derived from two rotationally stable atropisomeric axes, providing a potent and selective single atropisomer with desirable efficacy and tolerability profiles.
Keywords: BTK, atropisomer, CD69, autoimmune disease
Protein kinases have been linked, directly and indirectly, to the pathophysiology of a large number of diseases.1 Inhibition of kinase activity has the potential to interfere with critical signaling cascades, thus making kinases an attractive target for a wide variety of therapeutic areas.1 One such protein kinase, Bruton’s Tyrosine Kinase (BTK), is a nonreceptor kinase expressed in all hematopoietic cells including B cells, mast calls, and macrophages, but not in T cells or differentiated plasma cells. BTK, one of the five Tec family kinases, plays a crucial role in B cell receptor mediated signaling in B cells and Fcγ receptor (e.g., FcγRlla and FcγRllla) and Fcε receptor mediated signaling in myeloid cells.2−4 Autoimmune disease development in humans, including rheumatoid arthritis (RA) and lupus, is reliant on many of the BTK regulated signaling pathways.5−10 Consequently, the inhibition of the kinase activity of BTK has emerged as a clinical strategy for the treatment of many autoimmune diseases, without depleting B cells or inducing B cell immune deficiency.11 This has led to a significant effort across the pharmaceutical industry to identify both irreversible and reversible small molecule inhibitors of BTK as clinical therapeutic agents to treat autoimmunity.12−26
We previously disclosed a potent, reversible carbazole inhibitor of BTK (1, BTK IC50 = 3 nM; human whole blood IC50 = 550 nM measuring the expression of CD69). A notable characteristic was that 1 existed as a mixture of four interconverting atropisomers. Although carbazole 1 demonstrated desirable efficacy in mouse models of RA, undesired side effects were noted during tolerability studies in multiple species. More recently, we reported on a strategy to improve the intrinsic activity, selectivity, and ultimately the tolerability profile through the identification of a single, stable atropisomer by rotationally locking two atropisomeric axes into the preferred bioactive conformation to inhibit BTK.28 Removing the less target relevant atropisomers could potentially mitigate undesired off-target interactions that could be contributing to the toxicity observed with 1. This was accomplished by replacing the quinazolinone with a quinazolinedione to lock the lower atropisomeric axis and by adding small substituents at C3 to rotationally lock the carbazole C4 atropisomeric axis. This effort resulted in the identification of a single, rotationally stable atropisomer, carbazole 2, which provided a 6-fold improvement in human whole blood potency when compared to 1 (IC50 = 90 vs 550 nM), as well as improved kinase selectivity.28 As previously disclosed, demethylation of the quinazolinedione occurred in vivo in both mouse (66%) and rat (80%), resulting in the formation of metabolite 3.28 Further structure–activity relationship (SAR) evolution, focused on exploring carbazole C3 substitution as well as other structural variations, led to the identification of clinical asset BMS-986142 (4).28,29 In this letter, we outline a parallel strategy to overcome the stability issue through the replacement of the quinazolinedione with pyridopyrimidinedione bicyclic systems 5 and 6 (Figure 1).
Figure 1.
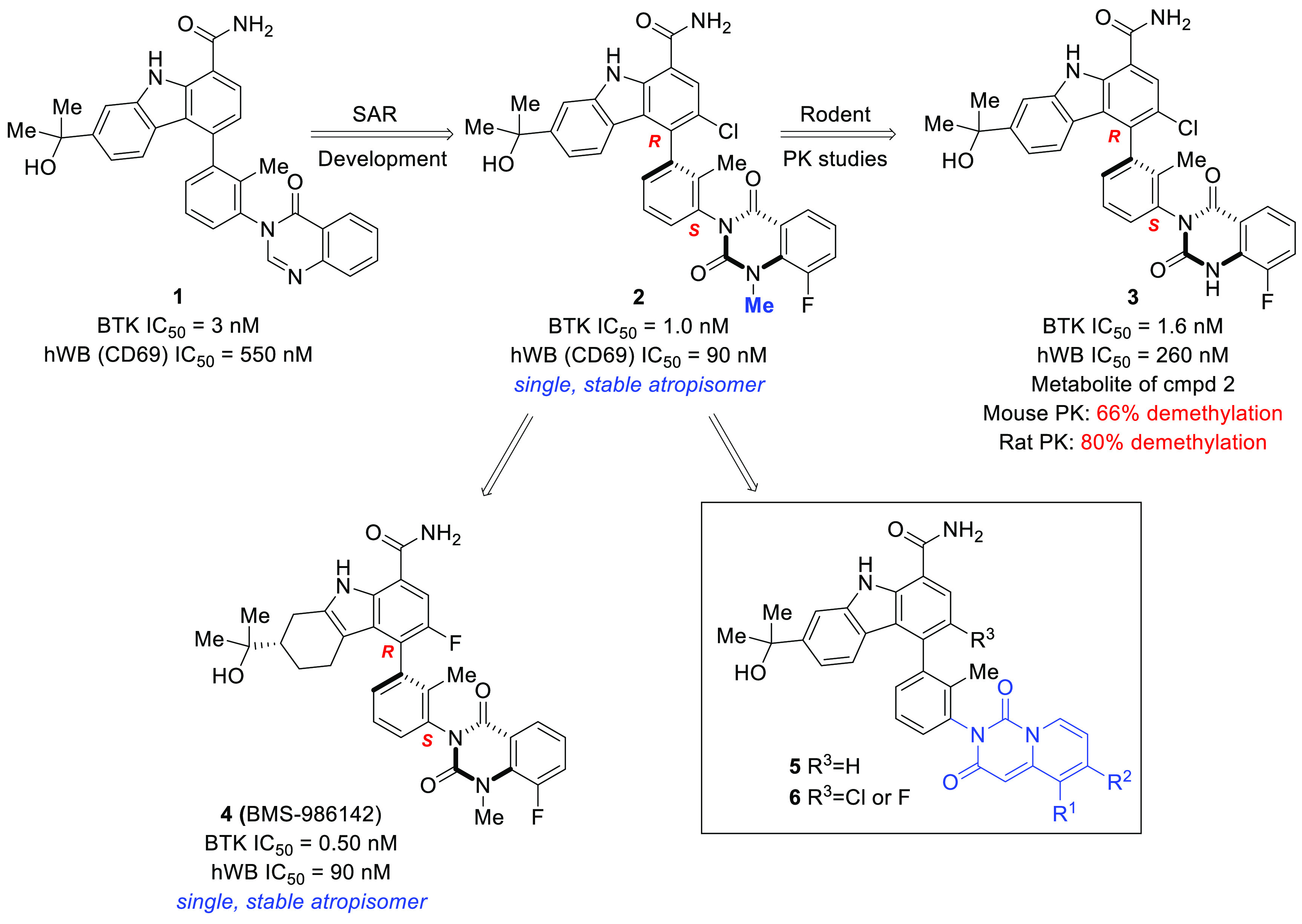
Identification of pyridopyrimidinedione inhibitors.
The pyridopyrimidinedione-carbazole compounds presented in this Letter were evaluated in both biochemical and cellular assays to determine their intrinsic activity against BTK (see Supporting Information S2). Enzymatic activity was established in a human recombinant BTK enzymatic assay.28 Cellular activity was determined in a B cell receptor (BCR) stimulated calcium flux assay in Ramos B cells.28 Potent analogues were then triaged with a human BCR stimulated whole blood assay (hWB), measuring the expression of CD69.28 Additionally, compounds were evaluated against a subset of kinases with the goal of identifying a highly selective inhibitor. In this Letter, selectivity for BTK over JAK2 is shown in Tables 1 and 2, highlighting improved kinase selectivity relative to 1. Clinically, JAK2 inhibition has been associated with anemia,30 a potentially undesirable side effect when considering the treatment of autoimmune disease. Select compounds, with desirable potency, selectivity, and in vitro liability profiles were subsequently evaluated in vivo.
Table 1. In Vitro Potency of Carbazoles 5.

| in vitro
activity |
mouse
PK (10 mg/kg) |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| compd | dione chirality | R1 | R2 | BTK IC50 (nM)a | JAK2/BTK selectivity | Ramos IC50 (nM)a | hWB IC50 (nM)a | Cma× (μM) | AUC (μM·h) |
| 1 | NA | NA | NA | 3.0 ± 0.10 | 94× | 26 ± 15 | 550 ± 100 | 8.9 | 80 |
| 5a | peak 1 | H | H | 1.7 (n = 1) | 1200× | 19 ± 21 | ND | 0.54 | 2.5 |
| 5b | peak 2 | H | H | 1.8 (n = 1) | 530× | 8.5 ± 3.0 | 410 (n = 1) | ||
| 5c | racemate | H | OMe | 0.63 (n = 1) | 1600× | 19 (n = 2) | 140 (n = 1) | 1.3 | 3.9 |
| 5d | racemate | OMe | H | 0.52 (n = 1) | 1900× | 19 ± 10 | 140 ± 73 | 1.3 | 4.0 |
| 5e | racemate | H | Cl | 0.49 ± 0.20 | 1500× | 3.1 ± 2.7 | 3,000 (n = 3) | ||
| 5f | racemate | Cl | H | 0.94 (n = 1) | 1400× | 12 (n = 2) | 120 (n = 2) | 3.5 | 23 |
| 5g | R | Cl | H | 1.0 (n = 1) | 1100× | 18 (n = 1) | 78 (n = 1) | ||
| 5h | S | Cl | H | 0.41 (n = 2) | 2500× | 13 ± 7 | 69 (n = 2) | ||
| 5i | racemate | F | H | 0.86 (n = 2) | 1100× | 19 (n = 2) | 75 (n = 2) | 5.2 | 28 |
| 5j | peak 1 | F | H | 1.7 (n = 2) | 500× | 18 (n = 2) | 91 (n = 1) | ||
| 5k | peak 2 | F | H | 1.2 ± 0.8 | 870× | 14 ± 1 | 79 ± 20 | ||
IC50 values are shown as mean values of at least three determinations unless specified otherwise; ND = not determined.
Table 2. In Vitro Potency of Carbazoles 6.

| in vitro
activity |
||||||
|---|---|---|---|---|---|---|
| compd | R3 | chirality | BTK IC50 (nM)a | JAK2/BTK selectivity | Ramos IC50 (nM)a | hWB IC50 (nM)a |
| 6a | Cl | homochiral | 180 (n = 1) | 11× | >300 | ND |
| 6b | Cl | homochiral | 0.55 ± 0.16 | 2600× | 10 ± 2 | 162 (n = 1) |
| 6c | Cl | homochiral | 17 (n = 1) | 60× | 87 (n = 1) | ND |
| 6d | Cl | homochiral | 0.26 ± 0.12 | 3800× | 6.9 ± 3.4 | 25 ± 19 |
| 6e | F | homochiral | 6.3 (n = 1) | 330× | 600 (n = 1) | ND |
| 6f | F | homochiral | 0.22 ± 0.07 | 6000× | 6.6 ± 0.9 | 64 (n = 2) |
| 6g | F | homochiral | 7.2 (n = 1) | 140× | 170 (n = 2) | ND |
| 6h | F | homochiral | 0.19 ± 0.02 | 7200× | 7.6 ± 2.4 | 37 (n = 2) |
IC50 values are shown as mean values of at least three determinations unless specified otherwise; ND = Not determined.
As we initiated work in this series, compounds 5a and 5b (Table 1) were prepared from enantiomerically pure pyridopyrimidinedione atropisomeric intermediates, isolated from supercritical fluid chromatography (SFC) chiral resolution (see Supporting Information S1). Both compounds demonstrated comparable activity and selectivity when evaluated in the in vitro assays. Further SAR development focused on exploring substitution at the R1 and R2 positions to enhance potency. Since chiral resolution of the lower stable atropisomeric center did not result in significant differentiation, subsequent early compounds were evaluated as a racemic mixture of atropisomers at the dione and as a mixture of interconverting atropisomers at carbazole C4. The addition of a donating methoxy group at either the R1 or R2 carbons (5d and 5c) respectively, was tolerated, both providing very similar activity (BTK biochemical assay, the Ramos cellular assay, and the human whole blood assay) and similar oral plasma blood levels in mouse PK studies. Interestingly, both compounds were ∼4-fold more potent in the whole blood assay when compared to compound 1 (140 vs 550 nM, respectively). Replacing the R2 methoxy with a chloro (5e) maintained intrinsic activity; however, the compound was found to be inactive in the whole blood assay (IC50 > 3 μM). Further analysis revealed that 5e was unstable in whole blood when incubated at a concentration of 0.5 μM, showing >85% degradation within 10 min and complete degradation by the 2 h time point. To further understand the source of the instability, 5e was incubated in human liver microsomes with glutathione (10 μM, 60 min). The major metabolite identified corresponded to direct glutathione replacement of the pyridopyrimidinedione chloro substituent, indicating that the R2 chloro is highly reactive. This is not surprising considering the δ-chloroenone motif. The R1 chloro derivative (5f), on the other hand, was found to be stable in the same assay, and as a result was stable in the whole blood assay providing 120 nM activity. Enantiomerically pure chloro diones were prepared as stable atropisomers, derived from chiral boronic esters with confirmed absolute chirality. Both 5g (R) and 5h (S) provided ∼7–8 fold improvement in whole blood potency relative to 1 and 13–27 fold improvement in the selectivity for BTK over JAK2. The R1 fluoro substitution (5i–k) provided similar results. Although the systemic oral exposures in mouse PK for the compounds shown in Table 1 were lower than those seen with 1, we were encouraged by the significant improvement in human whole blood potency observed in this series.
The addition of either a chloro or a fluoro at the carbazole C3 position (R3) of compound 5i rotationally restricted the C3 atropiosmeric center, allowing for the isolation of four single, stable atropisomeric compounds, as shown in Table 2. Although chloro versus fluoro substitution provided little differentiation in intrinsic potency, the chirality of the C4 individual atropisomers had a profound effect on potency and selectivity, with compound 6d providing a human whole blood IC50 of 25 nM (22-fold more potent than 1) and selectivity for BTK over JAK2 of 3,800-fold (40-fold more selective than 1). Single crystal X-ray crystallographic analysis of compound 6d confirmed that the carbazole C4 atropisomer is the R configuration (Figure 2; CDCC # 1501157), consistent with the preferred bioactive conformation observed in the cocrystal structure of clinical asset BMS-986142 bound in the active site of BTK.28 The lower dione atropisomer was confirmed to be the S configuration. When comparing the desired, bioactive conformation (6d) with the undesired conformation (6c), the observed increase in biochemical, cellular, and human whole blood potencies, as well as the significant differences in the selectivity for BTK over JAK2, clearly demonstrates the potential benefit of preparing and isolating a single, rotationally stable atropisomeric compound. This effect is consistent with the benefits observed with traditional chiral center resolution. It is worth noting that both atropisomers 6c and 6d showed similar inhibition of JAK2 with an IC50 of ∼1 μM, so the improvement in selectivity observed is derived from the increased affinity of 6d for BTK. With highly desirable whole blood potency and selectivity, 6d was advanced for further evaluation.
Figure 2.

Single crystal X-ray structure of 6d confirming the absolute atropisomeric stereochemistry (CDCC # 1501157).
A more in depth in vitro activity profile for 6d is presented in Table 3. In multiple assays aimed at establishing the effectiveness of the compound in inhibiting critical B cell functions derived through BCR stimulation, including proliferation, antibody production, and costimulatory molecule expression, 6d potently inhibited signaling and functional end points with single digit nanomolar activity. Consistent with the inhibition observed in BCR stimulated pathways, 6d provided potent inhibition of end points derived from IgG-containing immune complex low affinity activating Fcγ receptor signaling in peripheral blood mononuclear cells (PBMC) (IC50 = 2 nM). Of particular interest, 6d inhibited the expression of CD63 on the surface of basophils in human whole blood, driven by FcεRI signaling (IC50 of 54 nM). This is similar to the previously stated human whole blood activity when measuring the BCR-stimulated expression of CD69 on the surface of B cells (IC50 = 25 nM). Compared to our early lead compound 1 (Table 3), 6d provided significantly enhanced cellular and whole blood potencies.
Table 3. Partial In Vitro Cell Activity Data and Whole Blood Data for 6d.

| IC50 (nM)a |
|||
|---|---|---|---|
| assay | receptor/stimulation | 6d | 1 |
| cellular assays | |||
| calcium flux in Ramos B Cells | BCR/anti-IgM | 7 ± 3 | 26 ± 15 |
| proliferation of human peripheral B Cells | BCR/anti-IgM/IgG | 1 ± 0.4 | 8b |
| CD86 surface expression in peripheral B Cells | BCR/anti-IgM/IgG | 1 ± 0.5 | 40 ± 30 |
| CD86 surface expression in peripheral B Cells | CD40/CD40L | >10 000 | >10 000 |
| TNFα from human PBMC Cells | FCλR/immune complex | 2b | 14b |
| human whole blood assays | |||
| human whole blood CD69 surface expression in peripheral B Cells | BCR/anti-IgM | 25 ± 10 | 550 ± 100 |
| mouse whole blood CD69 surface expression in peripheral B Cells | BCR/anti-IgM/IgG | 130 | 2,100 ± 200 |
| human whole blood CD63 surface expression in basophils | FceRI/anti-IgE | 54 ± 20 | ND |
IC50 values are shown as mean values of at least three determinations.
IC50 values are shown as a single determination; PBMC = peripheral blood mononuclear cells; ND = not determined.
Carbazole 6d was evaluated against 384 kinases, inhibiting only seven kinases with less than 100-fold selectivity relative to BTK (Table 4). Four of the seven were Tec family members, TEC, BMX, TXK, and ITK, and only three kinases, TEC, BLK, and BMX, were inhibited with less than 30-fold selectivity relative to BTK.
Table 4. Partial In Vitro Selectivity Data for 6d.
| kinase | biochemical IC50 (nM) | kinase/BTK fold selectivity |
|---|---|---|
| BTK (Tec family) | 0.26 | |
| TEC (Tec family) | 3 | 10× |
| BLK | 5 | 17× |
| BMX (Tec family) | 7 | 23× |
| TXK (Tec family) | 10 | 33× |
| FGR | 15 | 50× |
| YES1 | 19 | 63× |
| ITK (Tec family) | 21 | 70× |
In pharmacokinetic (PK) studies in mice and dogs (Table 5), carbazole 6d was highly absorbed with bioavailability of 100% and 82%, respectively. The compound had a low rate of plasma clearance with a large steady-state volume of distribution in both species. On the basis of the PK and liability profiles (Table 6), coupled with demonstrated potency and selectivity, 6d was further evaluated in vivo in models of human RA.
Table 5. Pharmacokinetic Parameters for 6d.
| parameter | mouseb | doga |
|---|---|---|
| po dose (mg/kg) | 6 | 2 |
| iv dose (mg/kg) | 3 | 1 |
| Cmax (μM), PO | 4.3 | 1.2 ± 0.4 |
| Tmax (μM), PO | 1.0 | 3.7 ± 1.2 |
| AUC (μM·h), PO | 20 | 13 ± 6 |
| T1/2 (h), iv | 3.6 | 7.9 ± 0.6 |
| MRT (h), iv | 3.5 | 10.1 ± 1.5 |
| CL (mL/min/kg), iv | 8.6 | 4.4 ± 0.7 |
| Vss (L/kg), iv | 1.8 | 2.6 ± 0.3 |
| Fpo (%) | 100 | 82 ± 31 |
Average of three animals.
Average of two animals. Vehicle: (po) 80% PEG400, 20% water; (iv) 10% DMAC, 30% PEG300, 60% water; (iv dog) 10% EtOH, 70% PEG400, 20% water.
Table 6. Partial In Vitro Profiling Data for 6d.
| parameter | result |
|---|---|
| protein binding (bound) | 99.7% human |
| 99.4% mouse | |
| 99.5% rat | |
| 98.7% dog | |
| 98.2% monkey | |
| mutagenicity | Ames negative |
| hERG (patch clamp) | IC50 > 30 μM |
| Na+ (patch clamp) | 13% @ 10 μM (1 and 4 Hz) |
| Ca+ (patch clamp) | 19% @ 10 μM |
| CYP inhibition (IC50)a | >12 μM 1A2, 2B6, 2D6, 2C19 |
| 3.2 μM 2C8 | |
| 5.7 μM 2C9 | |
| 11 μM 3A4 | |
| PAMPA permeability | 1836/1302 nm/s (pH 5.5/7.4) |
| Caco2 Permeability: | ND due to insufficient recovery |
| Aqueous solubility | <0.001 mg/mL |
| FaSSIF solubilityb | 14 μg/mL |
| FeSSIF solubilityc | 551 μg/mL |
| log D at pH 7.0 (HPLC) | 3.84 |
CYP = Cytochrome P450.
FaSSIF = Fasted state simulated intestinal fluid.
FeSSIF = Fed state simulated intestinal fluid; ND = not determined.
In order to understand the compounds impact on in vivo efficacy in models of human RA, 6d was evaluated in two mouse models, a collagen-induced arthritis (CIA) model,19 dependent on both BCR-signaling and Fcγ receptor signaling, and an anticollagen antibody-induced model,28 dependent solely on Fcγ receptor signaling. In the CIA study (Figure 3), 6d was dosed orally at 15 and 45 mg/kg BID and provided dose-dependent inhibition of observed clinical disease progression (63% and 80%, respectively), representing 17 and 19 h coverage of the mouse whole blood IC50 (130 nM). In the anticollagen antibody-induced arthritis (CAIA) study (Figure 4), 6d when dosed orally at 10 and 25 mg/kg BID resulted in 78% and 100% suppression of clinically evident paw swelling, respectively. The observed efficacy corresponded to 17 h (10 mg/kg BID) and 23 h (25 mg/kg BID) coverage of the mouse whole blood IC50 (130 nM). In summary, PK/PD relationships for 6d in preclinical models of arthritis suggested that coverage of the whole blood IC50 for 18 h duration is needed to achieve robust efficacy of >70% reduction in clinical scores. Doses providing close to 24 h duration of whole blood IC50 coverage resulted in maximal efficacy (100% reductions in clinical scores).
Figure 3.

Efficacy of 6d in mouse model of human collagen-induced arthritis. A 15 mg/kg BID dose provided 17 h coverage of the mouse WB IC50 (130 nM) while a 45 mg/kg BID dose provided 19 h coverage. Error bars represent the mean ± SEM. *P < 0.05 versus vehicle control group.
Figure 4.

Efficacy of 6d in FcgR-dependent collagen Ab-induced arthritis (CAIA). Robust efficacy was observed with a 10 mg/kg BID dose providing 18 h coverage of the mouse WB IC50 (130 nM), and a 25 mg/kg BID dose provided 23 h coverage. Error bars represent the mean ± SEM. *P < 0.05 versus vehicle control group.
Compounds represented by pyridopyrimidinediones 5(31) and 6(32) were prepared as outlined in Schemes 1–3. The final compounds were synthesized as shown in Scheme 1. Carbazole 7(27,28) was coupled with the appropriate pyridopyrimidinedione (8) under standard Suzuki coupling conditions33 to give the racemic compounds 5 and 9. If R3 is Cl or F, subsequent SFC chiral resolution provided each of the four rotationally stable atropisomers 6a–h. Alternatively, compounds 5g, 5h, and 6d were prepared starting with the appropriate chiral boronic ester 10 or 16, as depicted in Scheme 2. The absolute chiral configuration of 6d was established through single crystal X-ray structural elucidation (Figure 2; CDCC # 1501157)
Scheme 1. Preparation of Carbazoles 5, 6, and 9.
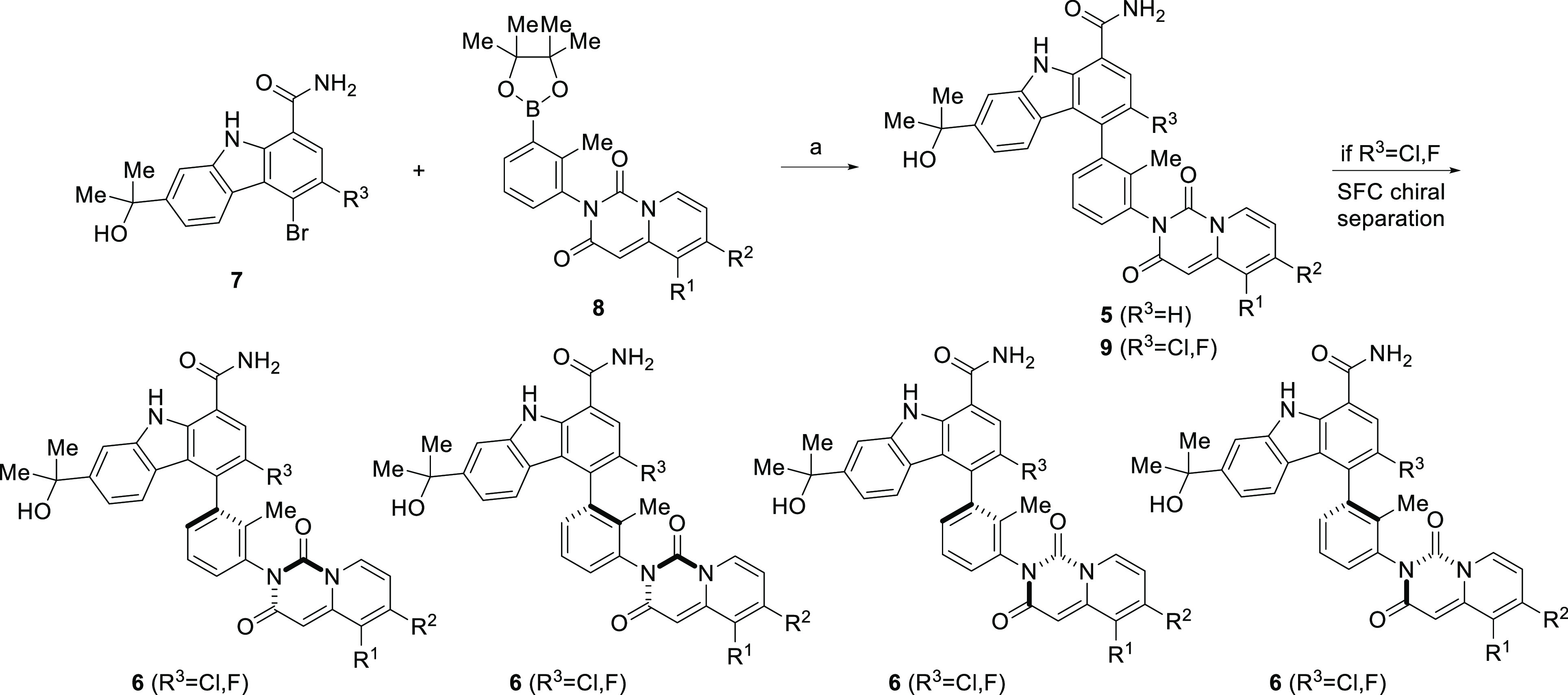
Reagents and conditions: (a). Dichloro 1,1′-bis(diphenylphosphino)ferrocene palladium(II)–CH2Cl2 adduct, Cs2CO3, THF–water, 45 °C, 60–65% yield.
Scheme 3. Preparation of Boronic Esters Intermediates 8, 10, and 17.

Reagents and conditions: (a) Diethyl malonate, Cs2CO3, DMSO, 100 °C; (b) NaCl, H2O, DMSO, 145 °C; (c) 3N NaOH, THF, rt.; (d) DIEA, HATU, DMF, rt., 89% yield over 4 steps; (e) Diborane, dichloro 1,1′-bis(diphenylphosphino)ferrocene palladium(II)–CH2Cl2 adduct, potassium acetate, DMSO, 90 °C, 85%; (f) CDI, Toluene, 110 °C, 65% yield; (g) SFC chiral separation.
Scheme 2. Preparation of Atropisomers 5g and 5h and Preparation of Single, Rotationally Stable Atropisomer 6d.

Reagents and conditions: (a). Dichloro 1,1′-bis(diphenylphosphino)ferrocene palladium(II)–CH2Cl2 adduct, Cs2CO3, THF–water, 45 °C; 60–68% yield.
The synthesis of boronic ester intermediates 8, 10, and 16 is shown in Scheme 3. Commercially available phenyl acetic acids or sodium salts prepared as outlined in Scheme 3 were coupled with 3-bromo-2-methylaniline in the presence of HATU and diisopropylamine in DMF to give 14. Intermediate 14 was converted to boronic ester 15, which was subsequently heated with carbondiimidazole in toluene at 100 °C to provide 8. SFC chiral resolution afforded single, rotationally stable atropisomeric intermediates 10 and 16. The absolute configuration of boronic ester 10 was confirmed to be S through single crystal X-ray structural elucidation (CDCC # 1501156; refer to the supplemental section for structural details).
The inhibition of the kinase activity of BTK with a small molecule inhibitor has emerged as a clinical strategy for the treatment of many autoimmune diseases. Pyridopyrimidinedione-carbazoles were envisioned to resolve a metabolic stability issue observed in the quinazolinedione series. An iterative SAR effort established the viability of the pyridopyrimidinediones as a progressable series. This effort resulted in the identification of a single atropisomer 6d, conformationally stable under physiological conditions, which demonstrated significant improvements in human whole blood potency (25 versus 550 nM, respectively) and overall selectivity relative to earlier lead 1. Importantly, in multiple species, carbazole 6d had a desirable safety and tolerability profile. This clearly demonstrates the potential benefit of preparing and isolating a single, rotationally stable atropisomeric compound to enhance potency, selectivity, and safety, similar to the benefits observed with traditional chiral center resolution. With a desirable in vitro and in vivo profile, 3-chloro-4-(R)-(3-(S)-(5-chloro-1,3-dioxo-1H-pyrido[1,2-c]pyrimidin-2(3H)-yl)-2-methylphenyl)-7-(2-hydroxypropan-2-yl)-9H-carbazole-1-carboxamide (6d, BMS-986143) was selected as a development candidate for further evaluation as a potential agent for the treatment of autoimmune diseases.
Acknowledgments
We would like to thank our colleagues in the Department of Discovery Synthesis at the Biocon-BMS Center (Bengaluru, India) for their contributions toward intermediate preparation.
Glossary
Abbreviations
- BTK
Bruton’s tyrosine kinase
- PK
pharmacokinetic
- SAR
structure–activity relationship
- hERG
human ether-a-go-go-related gene.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00335.
Experimental details and compound characterization data for key compounds as well as biological protocols (PDF)
The authors declare no competing financial interest.
This paper was originally published ASAP on September 22, 2020, with an error in the TOC/Abstract graphic. The corrected version was reposted on September 23, 2020.
Supplementary Material
References
- Zhang J.; Yang P. L.; Gray N. S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. 10.1038/nrc2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed A. J.; Yu L.; Bäckesjö C.-M.; Vargas L.; Faryal R.; Aints A.; Christensson B.; Berglöf A.; Vihinen M.; Nore B. F.; Smith C. I. E. Bruton’s tyrosine kinase (Btk): function, regulation, and transformation with special emphasis on the PH domain. Immunol. Rev. 2009, 228, 58–73. 10.1111/j.1600-065X.2008.00741.x. [DOI] [PubMed] [Google Scholar]
- Mohamed A. J.; Nore B. F.; Christensson B.; Smith C. I. E. Signaling of Bruton’s tyrosine kinase, Btk. Scand. Scand. J. Immunol. 1999, 49, 113–118. 10.1046/j.1365-3083.1999.00504.x. [DOI] [PubMed] [Google Scholar]
- Takata M.; Kurosaki T. A role for Bruton’s tyrosine kinase in B cell antigen receptor-mediated activation of phospholipase C-γ2. J. Exp. Med. 1996, 184, 31–40. 10.1084/jem.184.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson L.; Holmdahl R. Genes on the X chromosome affect development of collagen-induced arthritis in mice. Clin. Exp. Immunol. 1993, 94, 459–465. 10.1111/j.1365-2249.1993.tb08218.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg B. J.; Smathers P. A.; Frederiksen K.; Steinberg A. D. Ability of the xid gene to prevent autoimmunity in (NZBXNZW)F1 mice during the course of their natural history, after polyclonal stimulation, or following immunization with DNA. J. Clin. Invest. 1982, 70, 587–597. 10.1172/JCI110651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D.; Kim Y.; Postelnek J.; Vu M. D.; Hu D.-Q.; Liao C.; Bradshaw M.; Hsu J.; Zhang J.; Pashine A.; Srinivasan D.; Woods J.; Levin A.; O’Mahony A.; Owens T. D.; Lou Y.; Hill R. J.; Narula S.; DeMartino J.; Fine J. S. RN486, a selective Bruton’s tyrosine kinase inhibitor, abrogates immune hypersensitivity responses and arthritis in rodents. J. Pharmacol. Exp. Ther. 2012, 341, 90–103. 10.1124/jpet.111.187740. [DOI] [PubMed] [Google Scholar]
- Di Paolo J. A.; Huang T.; Balazs M.; Barbosa J.; Barck K. H.; Bravo B. J.; Carano R. A. D.; Darrow J.; Davies D. R.; DeForge L. E.; Diehl L.; Ferrando R.; Gallion S. L.; Giannetti A. M.; Gribling P. P.; Hurez V.; Hymowitz S. G.; Jones R.; Kropf J. E.; Lee W. P.; Maciejewski P. M.; Mitchell S. A.; Rong H.; Staker B. L.; Whitney J. A.; Yeh S.; Young W. B.; Yu C.; Zhang J.; Reif K.; Currie K. S. Specific Btk inhibition suppresses B cell and myeloid cell-mediated arthritis. Nat. Chem. Biol. 2011, 7, 41–50. 10.1038/nchembio.481. [DOI] [PubMed] [Google Scholar]
- Rankin A. L.; Seth N.; Keegan S.; Andreyeva T.; Cook T. A.; Edmonds J.; Mathialagan N.; Benson M. J.; Syed J.; Zhan Y.; Benoit S. E.; Miyashiro J. S.; Wood N.; Mohan S.; Peeva E.; Ramaiah S. K.; Messing D.; Homer B. L.; Dunussi-Joannopoulos K.; Nickerson- Nutter C. L.; Schnute M. E.; Douhan J. III. Selective inhibition of BTK prevents murine lupus and antibody-mediated glomerulonephritis. J. Immunol. 2013, 191, 4540–4550. 10.4049/jimmunol.1301553. [DOI] [PubMed] [Google Scholar]
- Mease P. J. B cell-targeted therapy in autoimmune disease: rationale, mechanisms, and clinical application. J. Rheumatol. 2008, 35, 1245–1255. [PubMed] [Google Scholar]
- Crofford L. J.; Nyhoff L. E.; Sheehan J. H.; Kendall P. L. The role of Bruton’s tryorsine kinase in autoimmunity and implications for therapy. Expert Rev. Clin. Immunol. 2016, 12, 763–773. 10.1586/1744666X.2016.1152888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford J. J.; Johnson A. R.; Misner D. L.; Belmont L. D.; Castanedo G.; Choy R.; Coraggio M.; Dong L.; Eigenbrot C.; Erickson R.; Ghilardi N.; Hau J.; Katewa A.; Kohli P. B.; Lee W.; Lubach J. W.; McKenzie B. S.; Ortwine D. F.; Schutt L.; Tay S.; Wei B.-Q.; Reif K.; Liu L.; Wong H.; Young W. B. Discovery of GDC-0853: A potent, selective, and noncovalent Bruton’s tyrosine kinase inhibitor in early development. J. Med. Chem. 2018, 61, 2227–2245. 10.1021/acs.jmedchem.7b01712. [DOI] [PubMed] [Google Scholar]
- Cohen S.; Tuckwell K.; Zhao R.; Galanter J.; Lee C.; Rae J.; Toth B.; Ramamoorthi N.; Hackney J. A.; Chinn L. W.; Townsend M. J.; Morimoto A. M.; Katsumoto T. R.; Genovese M. C.; Berman A.; Damjanov N.; Fedkov D.; Jeka S.; et al. Fenebrutinib versus placebo or adalimumab in rheumatoid arthritis: A randomized, double-blind, phase II trial (ANDES Study). Arthritis Rheumatol. 2020, 72, 1435. 10.1002/art.41275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell R. D.; Qiu H.; Askew B. C.; Bender A. T.; Brugger N.; Camps M.; Dhanabal M.; Dutt V.; Eichhorn T.; Gardberg A. S.; Goutopoulos A.; Grenningloh R.; Head J.; Healey B.; Hodous B. L.; Huck B. R.; Johnson T. L.; Jones C.; Jones R. C.; Mochalkin I.; Morandi F.; Nguyen N.; Meyring M.; Potnick J. R.; Santos D. C.; Schmidt R.; Sherer B.; Shutes A.; Urbahns K.; Follis A. V.; Wegener A. A.; Zimmerli S. C.; Liu-Bujalski L. Discovery of evobrutinib: An oral, potent, and highly selective, covalent Bruton’s tyrosine kinase (BTK) inhibitor for the treatment of immunological diseases. J. Med. Chem. 2019, 62, 7643–7655. 10.1021/acs.jmedchem.9b00794. [DOI] [PubMed] [Google Scholar]
- Montalban X.; Arnold D. L.; Weber M. S.; Staikov I.; Piasecka-Stryczynska K.; Willmer J.; Martin E. C.; Dangond F.; Syed S.; Wolinsky J. S. Placebo-controlled trial of an oral BTK inhibitor in multiple sclerosis. N. Engl. J. Med. 2019, 380, 2406–2417. 10.1056/NEJMoa1901981. [DOI] [PubMed] [Google Scholar]
- Smith P. F.; Krishnarajah J.; Nunn P. A.; Hill R. J.; Karr D.; Tam D.; Masjedizadeh M.; Funk J. O.; Gourlay S. G. A phase I trial of PRN1008, a novel reversible covalent inhibitor of Bruton’s tyrosine kinase, in healthy volunteers. Br. J. Clin. Pharmocol. 2017, 83, 2367–2376. 10.1111/bcp.13351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill R. J.; Smith P., Krishnarajah J.; Bradshaw J. M.; Masjedizadeh M.; Bisconte A.; Karr D.; Owens T. D.; Brameld K.; Funk J. O.; Goldstein D. M.; Nunn P. A., Gourlay S. G.. Discovery of PRN1008, a novel, reversible covalent BTK inhibitor in clinical development for rheumatoid arthritis. Arthritis Rheumatol. 2015, 67 ( (suppl 10), ). https://acrabstracts.org/abstract/discovery-of-prn1008-a-novel-reversible-covalent-btk-inhibitor-in-clinical-development-for-rheumatoid-arthritis/. [Google Scholar]
- Smith P. F.; Owens T. D.; Langrish C. L.; Xing Y.; Francesco M. R.; Shu J.; Hartmann S.; Karr D.; Burns R.; Quesenberry R.; Neale A.; Gourlay S. G.; Redfern A. Discovery of PRN1008, a novel, reversible covalent BTK inhibitor in clinical development for rheumatoid arthritis. ACTRIMS 2019, 3790. [Google Scholar]
- Watterson S. H.; Liu Q.; Beaudoin Bertrand M.; Batt D. G.; Li L.; Pattoli M. A.; Skala S.; Cheng L.; Obermeier M. T.; Moore R.; Yang Z.; Vickery R.; Elzinga P. A.; Discenza L.; D’Arienzo C.; Gillooly K. M.; Taylor T. L.; Pulicicchio C.; Zhang Y.; Heimrich E.; McIntyre K. W.; Ruan Q.; Westhouse R. A.; Catlett I. M.; Zheng N.; Chaudhry C.; Dai J.; Galella M. A.; Tebben A. J.; Pokross M.; Li J.; Zhao R.; Smith D.; Rampulla R.; Allentoff A.; Wallace M. A.; Mathur A.; Salter-Cid L.; Macor J. E.; Carter P. H.; Fura A.; Burke J. R.; Tino J. A. Discovery of Branebrutinib (BMS-986195): A strategy for identifying a highly potent and selective covalent inhibitor providing rapid in vivo inactivation of Bruton’s tyrosine kinase (BTK). J. Med. Chem. 2019, 62, 3228–3250. 10.1021/acs.jmedchem.9b00167. [DOI] [PubMed] [Google Scholar]
- Catlett I. M.; Nowak M.; Kundu S.; Zheng N.; Liu A.; He B.; Girgis I. G.; Grasela D. M. Safety, pharmacokinetics and pharmacodynamics of branebrutinib (BMS-986195), a covalent, irreversible inhibitor of Bruton’s tyrosine kinase: Randomised phase I, placebo-controlled trial in healthy participants. Br. J. Clin. Pharmacol. 2020, 86, 1849. 10.1111/bcp.14290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans E. K.; Tester R.; Aslanian S.; Karp R.; Sheets M.; Labenski M. T.; Witowski S. R.; Lounsbury H.; Chaturvedi P.; Mazdiyasni H.; Zhu Z.; Nacht M.; Freed M. I.; Petter R. C.; Dubrovsky A.; Singh J.; Westlin W. F. Inhibition of BTK with CC-292 provides early pharmacodynamic assessment of activity in mice and humans. J. Pharmacol. Exp. Ther. 2013, 346, 219–228. 10.1124/jpet.113.203489. [DOI] [PubMed] [Google Scholar]
- Schafer P. H.; Kivitz A. J.; Ma J.; Korish S.; Sutherland D.; Li L.; Azaryan A.; Kosek J.; Adams M.; Capone L.; Hur E. M.; Hough D. R.; Ringheim G. E. Spebrutinib (CC-292) affects markers of B cell activation, chemotaxis, and osteoclasts in patients with rheumatoid arthritis: Results from a mechanistic study. Rheumatol. Thera. 2020, 7, 101–119. 10.1007/s40744-019-00182-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J. K.; Byun J.-Y.; Park J. A.; Kim Y.-Y.; Lee Y. J.; Oh J. I.; Jang S. Y.; Kim Y. H.; Song Y. W.; Son J.; Suh K. H.; Lee Y.-M.; Lee E. B. HM71224, a novel Bruton’s tyrosine kinase inhibitor, suppresses B cell and monocyte activation and ameliorates arthritis in a mouse model: a potential drug for rheumatoid arthritis. Arthritis Res. Ther. 2016, 18, 91–1. 10.1186/s13075-016-0988-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goess C.; Harris C. M.; Murdock S.; McCarthy R. W.; Sampson E.; Twomey R.; Mathieu S.; Mario R.; Perham M.; Goedken E. R.; Long A. J. ABBV-105, a selective and irreversible inhibitor of Bruton’s tyrosine kinase, is efficacious in multiple preclinical models of inflammation. Mod. Rheumatol. 2019, 29, 510–522. 10.1080/14397595.2018.1484269. [DOI] [PubMed] [Google Scholar]
- Angst D.; Gessier F.; Janser P.; Vulpetti A.; Walchli R.; Beerli C.; Littlewood-Evans A.; Dawson J.; Nuesslein-Hildesheim B.; Wieczorek G.; Gutmann S.; Scheufler C.; Hinniger A.; Zimmerlin A.; Funhoff E. G.; Pulz R.; Cenni B. Discovery of LOU064 (remibrutinib), a potent and highly selective covalent inhibitor of Bruton’s tyrosine kinase. J. Med. Chem. 2020, 63, 5102. 10.1021/acs.jmedchem.9b01916. [DOI] [PubMed] [Google Scholar]
- Hosoi F.; Iguchi S.; Yoshiga Y.; Kaneko R.; Nakachi Y.; Akasaka D.; Yonekura K.; Iwasawa Y.; Sasaki E.; Utsugi T. OP0075 TAS5315, A novel Bruton’s tyrosine kinase (BTK) inhibitor, demonstrates potent efficacy in mouse collagen-induced arthritis model. Ann. Rheum. Dis. 2015, 74, 97.1. 10.1136/annrheumdis-2015-eular.3864. [DOI] [Google Scholar]
- De Lucca G. B.; Shi Q.; Liu Q.; Batt D. G.; Bertrand M. B.; Rampulla R.; Mathur A.; Discenza L.; D’Arienzo C.; Dai J.; Obermeier M. T.; Vickery R.; Zhang Y.; Yang Z.; Marathe P. H.; Tebben A. J.; Muckelbauer J. K.; Chang C. Y.; Zhang H.; Gillooly K.; Taylor T. L.; Pattoli M. A.; Skala S.; Kukral D. W.; McIntyre K. W.; Salter-Cid L.; Fura A.; Burke J. R.; Barrish J. C.; Carter P. H.; Tino J. A. Small molecule reversible inhibitors of Bruton’s tyrosine Kinase (BTK): Structure-activity relationships leading to the identification of 7-(2-hydroxypropan-2-yl)-4-[2- methyl-3-(4-oxo-3,4-dihydroquinazolin-3-yl)phenyl]-9H-carbazole-1-carboxamide (BMS-935177). J. Med. Chem. 2016, 59, 7915–7935. 10.1021/acs.jmedchem.6b00722. [DOI] [PubMed] [Google Scholar]
- Watterson S. H.; Liu Q.; Beaudoin Bertrand M.; Batt D. G.; Li L.; Pattoli M. A.; Skala S.; Cheng L.; Obermeier M. T.; Moore R.; Yang Z.; Vickery R.; Elzinga P. A.; Discenza L.; D’Arienzo C.; Gillooly K. M.; Taylor T. L.; Pulicicchio C.; Zhang Y.; Heimrich E.; McIntyre K. W.; Ruan Q.; Westhouse R. A.; Catlett I. M.; Zheng N.; Chaudhry C.; Dai J.; Galella M. A.; Tebben A. J.; Pokross M.; Li J.; Zhao R.; Smith D.; Rampulla R.; Allentoff A.; Wallace M. A.; Mathur A.; Salter-Cid L.; Macor J. E.; Carter P. H.; Fura A.; Burke J. R.; Tino J. A. Discovery of Branebrutinib (BMS-986195): A strategy for identifying a highly potent and selective covalent inhibitor providing rapid in vivo inactivation of Bruton’s tyrosine kinase (BTK). J. Med. Chem. 2019, 62, 3228–3250. 10.1021/acs.jmedchem.9b00167. [DOI] [PubMed] [Google Scholar]
- Lee S. K.; Xing J.; Catlett I. M.; Adamczyk R.; Griffies A.; Liu A.; Murthy B.; Nowak M. Safety, pharmacokinetics, and pharmacodynamics of BMS-986142, a novel reversible BTK inhibitor, in healthy participants. Eur. J. Clin. Pharmacol. 2017, 73, 689–698. 10.1007/s00228-017-2226-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winton E. F.; Kota V. Momelotinib in myelofibrosis: JAK1/2 inhibitor with a role in treating and understanding anemia. Future Oncol. 2017, 13, 395–407. 10.2217/fon-2016-0417. [DOI] [PubMed] [Google Scholar]
- Ko S. S.; Batt D. A.; Bertrand M. B.; Delucca G. V.; Langevine C. M.; Liu Q.; Srivastava A. S.; Watterson S. H.. Carbazole carboxamide compounds. US 9,714,234, 2017.
- Batt D. G.; Bertrand M. B.; Delucca G.; Galella M. A.; Ko S. S.; Langevine C. M.; Liu Q.; Shi Q.; Srivastava A. S.; Tino J. A.; Watterson S. H.. Substituted tetrahydrocarbazole and carbazole carboxamide compounds. US 9,334,290, 2016.
- Miyaura N.; Suzuki A. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem. Rev. 1995, 95, 2457–2483. 10.1021/cr00039a007. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.