

Abstract
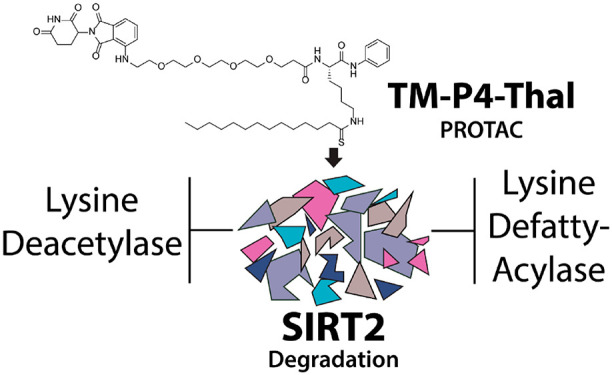
As a member of the sirtuin family of enzymes, SIRT2 promotes tumor growth and regulates various biological pathways through lysine deacetylation and defatty-acylation. In the past few years, many SIRT2-selective small molecule inhibitors have been developed, but none have demonstrated simultaneous inhibition of both SIRT2 activities in cells. To further scrutinize the physiological importance and significance of SIRT2 deacetylase and defatty-acylase activities, small molecules that can selectively inhibit both activities of SIRT2 in living cells are needed. Here, we have applied the Proteolysis Targeting Chimera (PROTAC) strategy and synthesized a new SIRT2 inhibitor (TM-P4-Thal) to degrade SIRT2 selectively, which led to simultaneous inhibition of its deacetylase and defatty-acylase activities in living cells. Additionally, this compound exemplifies the advantage of the PROTAC strategy that allows complete eradication of an enzyme and its activity in biological settings.
Keywords: SIRT2, sirtuin inhibitor, PROTAC, cancer treatment, lysine defatty-acylase, lysine deacetylase
Members of the mammalian sirtuin family of enzymes remove various acyl groups from protein lysine residues, using nicotinamide adenine dinucleotide (NAD+) as a cosubstrate.1−4 They are involved in many biological processes, including metabolism, DNA damage repair, and cell growth.5−9 Among the seven sirtuins, SIRT2 is the only one that is primarily localized in the cytosol and can remove both acetyl and other acyl groups from protein lysine residues.10−12 SIRT2 was initially reported to deacetylate protein substrates, including hypoxia-inducible factor 1 α (HIF1α) and α-tubulin.13,14 Through deacetylation of various substrates, SIRT2 promotes tumorigenesis. For example, SIRT2 deacetylates Slug to increase its stability and promotes breast cancer progression.15 By deacetylating lactate dehydrogenase A (LDH-A), which is often overexpressed in cancer cells and responsible for lactate production, SIRT2 promotes pancreatic cancer growth.16 In many cancer cell lines, the inhibition of SIRT2 leads to degradation of C-Myc, impeding cancer cell growth in vitro and tumor growth in mice.17 Besides the deacetylation activity of SIRT2, SIRT2 can also efficiently remove long-chain fatty acyl groups from lysine.10,18 SIRT2 can defatty-acylate K-Ras4a and promote K-Ras-mediated transformation.19 Recently, SIRT2 is also reported to defatty-acylate RalB, another small GTPase in the Ras subfamily.20 Through both deacetylation and defatty-acylation, SIRT2 plays significant roles in cancer growth and metabolism, which made SIRT2 an attractive target for cancer treatment.
Many SIRT2-selective inhibitors have been developed and shown to impede cancer growth. From a high throughput screening effort, AGK2 was found to be a SIRT2-selective inhibitor that binds to the “C-pocket” of SIRT2.21 Later, another SIRT2-selective inhibitor, SirReal2, was synthesized, which decreases migration and invasion of gastric cancer cells.22,23 NCO-90/140 slowed down cellular growth of several leukemia cells through induction of apoptosis and autophagy.24 Compound 53, based on NCO-90, demonstrated enhanced antiproliferative effect in breast cancer cells and promoted neurite outgrowth.25 NPD11033 effectively delayed the cell proliferation of pancreatic cancer cells.26 TM, a mechanism-based SIRT2-selective inhibitor, portrayed broad anticancer effects by promoting the degradation of c-Myc (Figure 1).17
Figure 1.

Examples of previously reported SIRT2 inhibitors.
Interestingly, recent studies that directly compared these inhibitors in various assays revealed that AGK2, SirReal2, and TM inhibit SIRT2 deacetylation activity but not the defatty-acylation activity.27 In contrast, a peptide-based SIRT2 inhibitor, S2DMi-7, could efficiently inhibit the defatty-acylase activities of SIRT1, SIRT2, and SIRT3.28 Nevertheless, being a peptide, S2DMi-7 is not SIRT2-selective and may have limited utility for cellular and in vivo studies. We recently reported that JH-T4 and NH-TM can hinder both activities of SIRT2 (Figure 1).29 However, these two compounds also inhibit SIRT1 and SIRT3, making it difficult to study SIRT2’s specific roles in cells.
Given the two different physiological catalytic activities of SIRT2, one interesting question is whether small molecules that can inhibit both activities of SIRT2 would produce different biological effects. To address this question, new inhibitors that can inhibit both the deacetylase and defatty-acylase activities of SIRT2 in live cells but are specific for SIRT2 are needed.
Proteolysis Targeting Chimera (PROTAC) uses a heterodimeric compound consisting of a ligand interacting with the protein-of-interest and another ligand recruiting E3 ligase to degrade the protein of interest.30−32 This strategy gained attention for its ability to enhance the effects of small molecule inhibitors. Traditionally, most inhibitors bind to an enzyme to inhibit its activity. PROTAC compounds, instead, catalytically remove the target proteins and thus not only inhibit the catalytic activity but also eliminate nonenzymatic functions of target proteins through degradation.30,31 Here we set out to test another possibility that could make the PROTAC strategy more effective: when an inhibitor of SIRT2 could only inhibit the deacetylation activity of SIRT2, we could enable it to inhibit all the catalytic activities of SIRT2 using the PROTAC strategy. In other words, applying the PROTAC strategy to specifically degrade SIRT2 would inhibit both catalytic activities of SIRT2, deacetylation and defatty-acylation, which currently existing small molecule inhibitors could not achieve. PROTAC utilizing SirReal2 as the ligand was synthesized but was not tested to show the advantage of the PROTAC strategy.33 Here we introduce two new PROTAC compounds, TM-P2-Thal and TM-P4-Thal, which can degrade SIRT2 efficiently in various cancer cells. Through SIRT2 degradation, we demonstrate that these compounds inhibit both activities of SIRT2 in living cells and achieve enhanced antiproliferative effect in cancer cells.
Results and Discussion
PROTAC Design and Synthesis
TM is a thiomyristoyl lysine-based SIRT2 selective inhibitor. Its thiomyristoyl lysine is important for SIRT2 inhibition, as this portion forms a stalled covalent intermediate with NAD+ to inhibit the activity of SIRT2.17 The C- and N-termini of the thiomyristoyl lysine serves as the potential modification sites to attach the linker and thalidomide, the ligand that recruits the E3 ubiquitin ligase cereblon (CRBN). According to the previously reported cocrystal structures of SIRT2 in complex with two inhibitors, BHJH-TM1 and Glucose-TM (PDB: 4R8M, 6NR0), the thiomyristoyl lysine points inward toward the enzyme pocket, while both the N- and C-terminal of the lysine are located outside of the pocket.18,34 The introduction of a hydroxyl group to the C-terminal phenyl group of TM, resulting in a new inhibitor, JH-T4, disrupted the selectivity of TM for SIRT2, as JH-T4 inhibits SIRT1, SIRT2, and SIRT3.29 From this finding, we hypothesized that altering the C-terminal phenyl ring for thalidomide attachment could potentially ruin SIRT2 selectivity. Thus, we decided to attach thalidomide to the N-terminus of TM (Scheme 1).
Scheme 1. Synthesis of TM-P2-Thal and TM-P4-Thal.

TM contains the carboxybenzoyl group (Cbz) on the N-terminal of the lysine backbone. We replaced this Cbz group with thalidomide via 2- or 4-repeating units of the polyethylene glycol (PEG) linker (Scheme 1). The PEG linker was chosen because of its aqueous solubility.35 In addition, being flexible on its own, the PEG linker could allow the thalidomide–CRBN complex to reach SIRT2’s ubiquitination site more easily. PEG linkers with two different lengths were used because the linker may affect the ubiquitination efficiency. If the linker is too short, SIRT2 and CRBN could clash with each other and not be able to form the PROTAC complex. However, if the linker is too long, CRBN and SIRT2 could be too far apart and decrease ubiquitination.
The synthesis of TM-P2-Thal or TM-P4-Thal required a total of 10 steps (Scheme 1). PEG linkers with two different lengths were attached to thalidomide. tert-Butyloxycarbonyl (Boc)-l-lysine was used to synthesize the TM analog with a free N-terminus. Then, in the last step, thalidomide with the PEG linkers was coupled to the TM analog with a free N-terminus using isobutyl chloroformate to produce TM-P2-Thal and TM-P4-Thal as the final products (Scheme 1).
TM-P2-Thal and TM-P4-Thal Maintain SIRT2 Selectivity
In vitro IC50 values of TM-P2-Thal and TM-P4-Thal on the sirtuin family members, SIRT1, SIRT2, SIRT3, SIRT5, and SIRT6, were measured to see if the selectivity toward SIRT2 had been preserved. Each compound at different concentrations was added to the sirtuin enzymatic reaction mixture to measure its inhibitory activities.17,27 Because the tested compounds inhibit sirtuin activities through forming a stalled covalent intermediate with NAD+, we preincubated these compounds with NAD+ and the corresponding sirtuin enzyme, prior to the addition of the substrate peptide. With preincubation, the IC50 values of TM-P2-Thal and TM-P4-Thal for SIRT2 deacetylase were 0.069 and 0.078 μM, respectively. Meanwhile, the IC50 values of TM-P2-Thal and TM-P4-Thal for SIRT1 deacetylase were 37 and 41 μM, respectively. The selectivity for SIRT2 over SIRT1 (IC50 SIRT1/IC50 SIRT2) was about 500 times for both TM-P2-Thal and TM-P4-Thal. In addition, both compounds did not show inhibitory activity on SIRT3, SIRT5, or SIRT6 (Table 1). Compound 8, thalidomide with PEG4 linker, could not inhibit any of the sirtuin activities (Supporting Information Table 1). Overall, even with the modifications on the N-terminus, both TM-P2-Thal and TM-P4-Thal retained SIRT2 selectivity.
Table 1. In Vitro IC50 values of TM-P2-Thal and TM-P4-Thal.
| IC50 (μM) | TM-P2-Thal | TM-P4-Thal | TM |
|---|---|---|---|
| SIRT1 | 37 ± 3.5 | 41 ± 5.1 | >83 |
| SIRT2 Deacetylase (with preincubation) | 0.069 ± 0.032 | 0.078 ± 0.018 | 0.093 ± 0.012 |
| SIRT2 Deacetylase (without preincubation) | 0.12 ± 0.020 | 0.093 ± 0.025 | 0.15 ± 0.036 |
| SIRT2 Defatty-acylase (with preincubation) | 0.17 ± 0.044 | 0.11 ± 0.063 | 0.37 ± 0.031 |
| SIRT2 Defatty-acylase (without preincubation) | >83 | >83 | >83 |
| SIRT3 | >83 | >83 | >83 |
| SIRT5 | >83 | >83 | >83 |
| SIRT6 | >83 | >83 | >83 |
We have further tested TM, TM-P2-Thal, and TM-P4-Thal against SIRT2 deacetylase and defatty-acylase activities with and without preincubation. Even without preincubation, these three compounds could inhibit SIRT2 deacetylase efficiently. However, without preincubation, they could not inhibit SIRT2 defatty-acylase activity even at 83 μM. With preincubation, the IC50 values for TM, TM-P2-Thal, and TM-P4-Thal were 0.37, 0.17, and 0.11 μM, respectively. This IC50 difference between the cases with and without preincubation may explain why TM could not inhibit SIRT2 defatty-acylase activity in cells from previous studies.29
TM-P2-Thal and TM-P4-Thal Degrade SIRT2 Selectively in Cells
We next set out to check the effects of TM, TM-P2-Thal, and TM-P4-Thal on SIRT2 protein levels in two breast cancer cell lines, MCF7 and BT549. After 48 h of treatment at various concentrations, TM-P2-Thal and TM-P4-Thal degraded SIRT2 selectively without changing the levels of SIRT1 and SIRT3 (Figure 2A). As expected, TM did not decrease the level of SIRT2 in the two cell lines. In MCF7 cells, 5 μM of the PROTAC compounds degraded SIRT2 more effectively than 25 μM, which is likely due to the Hook effect of the PROTAC strategy.36 High concentrations of PROTAC primarily form a binary complex with either the E3 ligase or the protein target, thereby preventing the ternary complex formation that is required for the efficient degradation of target protein. Similarly, SIRT2 selective degradations were observed in MDA-MB-231 and MDA-MB-468 cell lines (Supporting Information Figure 1A and 1B) in the presence of TM-P2-Thal and TM-P4-Thal.
Figure 2.

TM-P2-Thal and TM-P4-Thal degrade SIRT2 selectively in cells. (A) Immunoblots for SIRT1, SIRT2, and SIRT3 after treating with indicated concentrations of inhibitors for 48 h in MCF7 and BT-549 cells. (B) Immunoblot for SIRT2 after MCF7 cells were treated with indicated concentrations of TM-P4-Thal for 48 h. (C) Immunoblot for SIRT2 after MCF7 cells were treated with 10 μM of TM-P4-Thal for the indicated incubation times.
In MCF7 cells, we then further tested SIRT2-selective degradation with lower than 5 μM TM-P4-Thal for 48 h. Even at 0.5 μM, the SIRT2 level was significantly decreased compared to that of a vehicle-treated sample. Furthermore, the decreased levels of SIRT2 at both 0.5 and 10 μM were similar, suggesting the high efficiency of TM-P4-Thal (Figure 2B). Again, at 50 μM TM-P4-Thal, the SIRT2 level did not change much likely due to the hook effect.36
Next, we examined the time course of TM-P4-Thal-induced SIRT2 degradation in MCF7 cells. As the incubation time increased, 10 μM of TM-P4-Thal lowered SIRT2 even further. Thus, SIRT2 was degraded by TM-P4-Thal in a time-dependent manner (Figure 2C). However, with 25 μM of TM-P4-Thal, the SIRT2 level decreased slightly at 24 h and no significant SIRT2 degradation was observed at later time points (Supporting Information Figure 1C). The decreased degradation at 25 μM of TM-P4-Thal was again due to the hook effect. We also treated cells with 10 and 25 μM of TM-P2-Thal at different time points. A similar result was observed, but the overall efficiency in the degradation of SIRT2 was lower compared to that with TM-P4-Thal (Supporting Information Figure 1C). Thus, we mainly used TM-P4-Thal for the rest of the study. Furthermore, we treated MCF7 cells with compound 8, thalidomide with a PEG4 linker, and detected the SIRT2 level through immunoblotting. As expected, without the TM moiety, Compound 8 could not degrade SIRT2 (Supporting Information Figure 2A). Also, we synthesized TM-P4-Thal-CH3 with a methyl group on thalidomide, which disrupts the overall binding to CRBN and hinders PROTAC degradation.37 Like compound 8, TM-P4-Thal-CH3 could not degrade SIRT2 in cells (Supporting Information Figure 2B).
To validate that SIRT2 degradation occurs through CRBN and the ubiquitin-proteasome system, we tried to rescue the degradation through cotreatment of a proteasome inhibitor, MG132. MCF7 cells were pretreated with MG132 (5 μM) for 2 h, followed by cotreatment of TM-P4-Thal (5 μM) and MG132 (5 μM) for an additional 16 h. While the treatment with TM-P4-Thal decreased the SIRT2 level, the cotreatment with TM-P4-Thal and MG132 did not decrease the SIRT2 level, suggesting that TM-P4-Thal promotes proteasome-dependent degradation of SIRT2 (Supporting Information Figure 3).
To further validate SIRT2 degradation by TM-P4-Thal, we performed Tandem Mass Tag (TMT) global proteomic with HEK 293T cells treated with 5 μM TM-P4-Thal for 16 h. Consistent with the immunoblots, the SIRT2 abundance ratio between TM-P4-Thal and DMSO treated cells was about 0.36, suggesting substantial degradation of SIRT2. Meanwhile, abundance ratios of other sirtuins, including SIRT1, 3, 5, and 6, did not alter significantly after TM-P4-Thal treatment. Overall, the TMT global proteomic result further confirms selective SIRT2 degradation by TM-P4-Thal (Supporting Information Figure 4, Supporting Information Table 3).
TM-P4-Thal Efficiently Inhibits Both SIRT2 Deacetylation and Defatty-Acylation in Cells
To assess the inhibition of SIRT2 deacetylation by TM-P4-Thal in cells, acetylation of α-tubulin, a previously reported SIRT2 deacetylation target, was examined by immunofluorescence after treating MCF7 cells with 5 and 10 μM TM-P4-Thal for 12 h.14 If SIRT2 deacetylation is inhibited, the level of acetylation would be increased by the treatments. TM-P4-Thal or TM at 5 and 10 μM concentrations significantly increased the acetylation of α-tubulin (Figure 3A). Furthermore, quantification of the fluorescence signals indicated that at 5 μM, TM-P4-Thal increased approximately 2.9-fold while TM increased about 2.6-fold compared to the vehicle control (Figure 3B), suggesting both compounds inhibit SIRT2 deacetylase with similar efficiency.
Figure 3.

TM-P4-Thal inhibits both SIRT2 deacetylase and defatty-acylase activities in cells. (A) Immunofluorescence images of acetyl α-tubulin (K40) in MCF7 cells treated with DMSO (control), TM-P4-Thal (5, 10 μM), and TM (5, 10 μM) for 12 h. (B) Normalized Ac-α-tubulin fluorescence intensity level of 5 μM TM-P4-Thal and TM for 12 h, using ImageJ software for quantification. (C) Immunoblot with pan-Ras antibody to show the stable overexpression of Flag-tagged K-Ras4a-G12V in HEK 293T cells. (D) Immunoblots for SIRT1, SIRT2, and SIRT3 after treating HEK 293T cells with 1 μM of TM-P4-Thal or 10 μM of TM in the presence of 50 μM of Alk-14 for 24 h. (E) Alk14 labeling and in-gel fluorescence to detect lysine fatty acylation of K-Ras4a in HEK 293T cells. Cells were treated with 1 μM TM-P4-Thal or 10 μM TM in the presence of 50 μM Alk-14. (F) Quantified relative lysine fatty acylation level of K-Ras4a from part E, using ImageJ for quantification.
Previous work demonstrated that SIRT2 can efficiently remove long chain fatty acyl groups from K182, 184, and 185 of K-Ras4a. SIRT2 knockdown consequently increased K-Ras4a lysine fatty acylation.19 Furthermore, we also found that TM could not inhibit SIRT2’s activity on K-Ras4a lysine fatty acylation.29 Because TM-P4-Thal degrades SIRT2, we expected that TM-P4-Thal could effectively inhibit not only the deacetylase but also the defatty-acylase activity of SIRT2.
To test this, we generated HEK 293T stably expressing Flag-tagged K-Ras4a-G12V (Figure 3C). In these cells, 1 μM of TM-P4-Thal treatment for 48 h decreased the SIRT2 level. Meanwhile, both SIRT1 and SIRT3 levels remained unchanged upon treatment (Figure 3D). Next, to visualize K-Ras4a fatty acylation levels, the cells were treated with 1 μM TM-P4-Thal or 10 μM TM for 42 h and then incubated with Alkyne-14 (Alk-14) for an additional 6 h to label fatty acylated proteins. After immunoprecipitating the Flag-tagged K-Ras4a-G12V, attaching a fluorescent dye through click chemistry and treating with NH2OH to remove cysteine fatty acylation, we detected the lysine fatty acylation level by in-gel fluorescence (Figure 3E, F). TM-P4-Thal increased fluorescence significantly compared to the DMSO control, suggesting efficient inhibition of SIRT2 defatty-acylation by TM-P4-Thal. In contrast, TM did not significantly increase the fluorescence level of K-Ras4a (Figure 3E, F). This suggests that while TM could not inhibit SIRT2 defatty-acylation, its PROTAC-version induced degradation of SIRT2 efficiently and inhibited the defatty-acylation activity in live cells.
TM-P4-Thal Shows Improved Cytotoxicity in Breast Cancer Cells at Lower Concentrations
After demonstrating that TM-P4-Thal can inhibit both activities of SIRT2, we evaluated its effect on the proliferation of MCF7 and MDA-MB-231 cells (Figure 4). In both breast cancer cell lines, we observed selective SIRT2 degradation after treatment of TM-P4-Thal (Figure 2 and Supporting Information Figure 1). In MCF7 cells, after 72 h incubation, TM-P4-Thal showed much stronger cytotoxicity than TM at lower concentrations. However, toward higher concentrations, the difference between TM and TM-P4-Thal became smaller. A similar trend was observed in MDA-MB-231. However, in MDA-MB-231 cells, at higher concentrations, the cytotoxicity of TM exceeded that of TM-P4-Thal (Figure 4). This is likely because of inefficient SIRT2 degradation at high concentrations of TM-P4-Thal due to the hook effect.36
Figure 4.
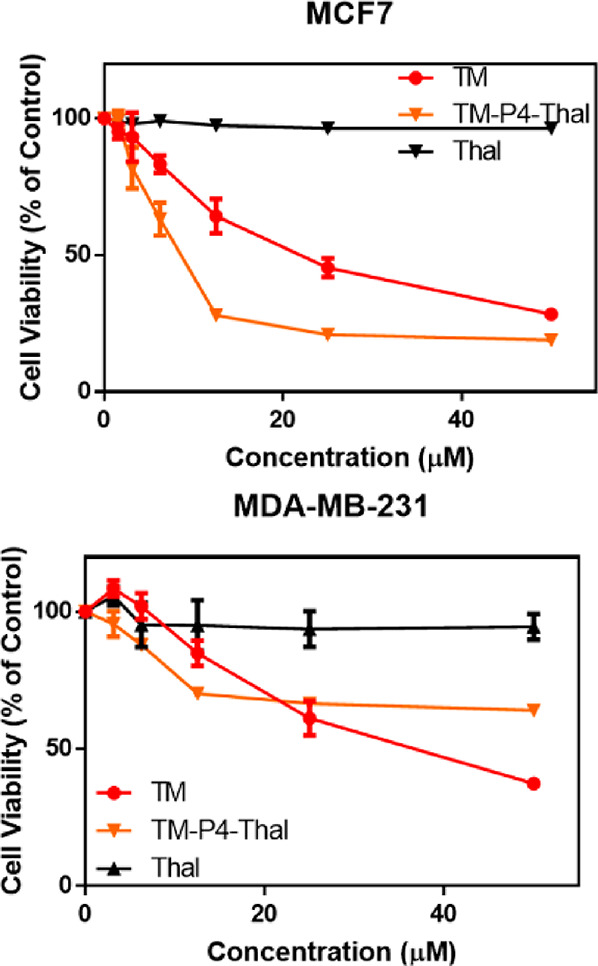
TM-P4-Thal shows improved cellular cytotoxicity in breast cancer cell lines at lower concentrations. Cell viability of MCF7 and MDA-MB-231 cells after treatment of TM, TM-P4-Thal, and thalidomide for 72 h.
There was some discrepancy between the SIRT2 level and the cytotoxicity. For instance, 1 μM TM-P4-Thal induced SIRT2 degradation but did not exert significant cytotoxicity in the cellular proliferation assay. SIRT2 may need to be completely inhibited/degraded for a longer period to see a stronger impact. Also, there could be an undiscovered reparative biological mechanism for the deceased protein level. Further studies are needed to understand this difference.
To test the cellular permeabilities, after treatment of TM-P4-Thal and TM (50 μM) for 6 h, MCF7 cells were washed with cold PBS three times, and the small molecules were extracted using methanol. The extracted samples were analyzed using liquid chromatography–mass spectrometry (LC-MS). Because these compounds with poor aqueous solubility are not suitable for traditional PAMPA assays, we have tried to measure the cellular permeability in this way. Overall, TM-P4-Thal showed less cell permeability, which could explain the lower cytotoxicity of TM-P4-Thal than that of TM at high concentrations (Supporting Information Table 2). Thalidomide did not affect cellular proliferation, suggesting that the cytotoxicity of TM-P4-Thal did not originate from the thalidomide moiety (Figure 4).
The stronger cytotoxicity of TM-P4-Thal may come from the inhibition of SIRT2 defatty-acylase activity. As shown earlier, at the same concentrations (5 and 10 μM), TM and TM-P4-Thal inhibited SIRT2 deacetylase similarly (Figure 3A and B), while only TM-P4-Thal could inhibit SIRT2 defatty-acylase. As such, the inhibition of SIRT2 defatty-acylase activity may have led to the stronger cytotoxicity of TM-P4-Thal, suggesting that inhibiting the defatty-acylation activity of SIRT2 could enhance the anticancer activity of SIRT2 inhibitors. We note that it remains possible that the stronger cytotoxicity of TM-P4-Thal may also come from the inhibition of the nonenzymatic function of SIRT2. However, given no such nonenzymatic function has been reported for SIRT2, we think the stronger cytotoxicity of TM-P4-Thal is likely due to the inhibition of SIRT2 defatty-acylase activity.
Conclusion
Here, we introduced two new PROTAC compounds, TM-P2-Thal and TM-P4-Thal, which can degrade SIRT2 efficiently and selectively in living cells. In addition, degradation of SIRT2 by TM-P4-Thal allowed inhibition of both the deacetylation and defatty-acylation activities of SIRT2. As a comparison, previously reported traditional SIRT2 inhibitors could not inhibit the defatty-acylation activity of SIRT2.27 As such, this case portrays an additional advantage of PROTAC strategy over traditional inhibition strategy. Furthermore, degradation of SIRT2 led to stronger cytotoxicity, suggesting the importance of defatty-acylation activity of SIRT2 in cancer cells. Since not much has been revealed about SIRT2’s defatty-acylation activity, TM-P4-Thal, together with TM, can be utilized as useful chemical tools to further scrutinize and understand the biological significance of SIRT2’s defatty-acylation activity.
Acknowledgments
Imaging data was acquired through the Cornell University Biotechnology Resource Center, with NYSTEM (CO29155) and NIH (S10OD018516) funding for the shared Zeiss LSM880 confocal/multiphoton microscope. This work had use of the Cornell University NMR facility, which is supported, in part, by the NSF through MRI award CHE-1531632. Proteomic data was acquired by the Proteomic and Metabolomics Facility of Cornell University and was supported by HHMI Transformative Technology 2019 program.
Glossary
Abbreviations
- PROTAC
Proteolysis-Targeting Chimera
- NAD+
nicotinamide adenine dinucleotide
- HIF1α
Hypoxia-Inducible Factor 1 α
- LDH-A
lactate dehydrogenase A
- CRBN
Cereblon
- PEG
polyethylene glycol
- UPS
ubiquitin-proteasome system
- TMT
tandem mass tag
- Boc
tert-butyloxycarbonyl
- Cbz
carboxybenzoyl
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00423.
Materials and methods, Supplementary Figures 1, 2, 3, and 4, Supplementary Tables 1, 2, and 3, and Supplementary Scheme 1 (PDF)
Author Contributions
J.Y.H. and H.L. wrote the manuscript. All the authors have given approval to the final version of the manuscript.
The work is support in part by NIH grant R01DK107868.
The authors declare no competing financial interest.
Supplementary Material
References
- Feldman J. L.; Dittenhafer-Reed K. E.; Denu J. M. Sirtuin Catalysis and Regulation. J. Biol. Chem. 2012, 287 (51), 42419–42427. 10.1074/jbc.R112.378877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauve A. A.; Celic I.; Avalos J.; Deng H.; Boeke J. D.; Schramm V. L. Chemistry of gene silencing: the mechanism of NAD+-dependent deacetylation reactions. Biochemistry 2001, 40 (51), 15456–15463. 10.1021/bi011858j. [DOI] [PubMed] [Google Scholar]
- Sauve A. A.; Wolberger C.; Schramm V. L.; Boeke J. D. The biochemistry of sirtuins. Annu. Rev. Biochem. 2006, 75, 435–465. 10.1146/annurev.biochem.74.082803.133500. [DOI] [PubMed] [Google Scholar]
- Imai S.-i.; Armstrong C. M.; Kaeberlein M.; Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 2000, 403 (6771), 795–800. 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- Hu J.; Jing H.; Lin H. Sirtuin inhibitors as anticancer agents. Future Med. Chem. 2014, 6 (8), 945–966. 10.4155/fmc.14.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao R.; Xiong X.; DePinho R. A.; Deng C. X.; Dong X. C. FoxO3 transcription factor and Sirt6 deacetylase regulate low density lipoprotein (LDL)-cholesterol homeostasis via control of the proprotein convertase subtilisin/kexin type 9 (Pcsk9) gene expression. J. Biol. Chem. 2013, 288 (41), 29252–9. 10.1074/jbc.M113.481473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du C.; Lin X.; Xu W.; Zheng F.; Cai J.; Yang J.; Cui Q.; Tang C.; Cai J.; Xu G.; Geng B. Sulfhydrated Sirtuin-1 Increasing Its Deacetylation Activity Is an Essential Epigenetics Mechanism of Anti-Atherogenesis by Hydrogen Sulfide. Antioxid. Redox Signaling 2019, 30 (2), 184–197. 10.1089/ars.2017.7195. [DOI] [PubMed] [Google Scholar]
- Jing H.; Lin H. Sirtuins in Epigenetic Regulation. Chem. Rev. 2015, 115 (6), 2350–2375. 10.1021/cr500457h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosciuk T.; Wang M.; Hong J. Y.; Lin H. Updates on the epigenetic roles of sirtuins. Curr. Opin. Chem. Biol. 2019, 51, 18–29. 10.1016/j.cbpa.2019.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Fung Y. M. E.; Zhang W.; He B.; Chung M. W. H.; Jin J.; Hu J.; Lin H.; Hao Q. Deacylation Mechanism by SIRT2 Revealed in the 1’-SH-2’-O-Myristoyl Intermediate Structure. Cell Chem. Biol. 2017, 24 (3), 339–345. 10.1016/j.chembiol.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman J. L.; Dittenhafer-Reed K. E.; Kudo N.; Thelen J. N.; Ito A.; Yoshida M.; Denu J. M. Kinetic and Structural Basis for Acyl-Group Selectivity and NAD+ Dependence in Sirtuin-Catalyzed Deacylation. Biochemistry 2015, 54 (19), 3037–3050. 10.1021/acs.biochem.5b00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J.; He B.; Zhang X.; Lin H.; Wang Y. SIRT2 Reverses 4-Oxononanoyl Lysine Modification on Histones. J. Am. Chem. Soc. 2016, 138 (38), 12304–7. 10.1021/jacs.6b04977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo K. S.; Park J. H.; Heo J. Y.; Jing K.; Han J.; Min K. N.; Kim C.; Koh G. Y.; Lim K.; Kang G. Y.; Uee Lee J.; Yim Y. H.; Shong M.; Kwak T. H.; Kweon G. R. SIRT2 regulates tumour hypoxia response by promoting HIF-1α hydroxylation. Oncogene 2015, 34 (11), 1354–1362. 10.1038/onc.2014.76. [DOI] [PubMed] [Google Scholar]
- North B. J.; Marshall B. L.; Borra M. T.; Denu J. M.; Verdin E. The Human Sir2 Ortholog, SIRT2, Is an NAD+-Dependent Tubulin Deacetylase. Mol. Cell 2003, 11 (2), 437–444. 10.1016/S1097-2765(03)00038-8. [DOI] [PubMed] [Google Scholar]
- Zhou W.; Ni T. K.; Wronski A.; Glass B.; Skibinski A.; Beck A.; Kuperwasser C. The SIRT2 Deacetylase Stabilizes Slug to Control Malignancy of Basal-like Breast Cancer. Cell Rep. 2016, 17 (5), 1302–1317. 10.1016/j.celrep.2016.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao D.; Zou S. W.; Liu Y.; Zhou X.; Mo Y.; Wang P.; Xu Y. H.; Dong B.; Xiong Y.; Lei Q. Y.; Guan K. L. Lysine-5 acetylation negatively regulates lactate dehydrogenase A and is decreased in pancreatic cancer. Cancer Cell 2013, 23 (4), 464–76. 10.1016/j.ccr.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing H.; Hu J.; He B.; Negrón Abril Y. L.; Stupinski J.; Weiser K.; Carbonaro M.; Chiang Y.-L.; Southard T.; Giannakakou P.; Weiss R. S.; Lin H. A SIRT2-Selective Inhibitor Promotes c-Myc Oncoprotein Degradation and Exhibits Broad Anticancer Activity. Cancer Cell 2016, 29 (3), 297–310. 10.1016/j.ccell.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng Y.-B.; Jing H.; Aramsangtienchai P.; He B.; Khan S.; Hu J.; Lin H.; Hao Q. Efficient Demyristoylase Activity of SIRT2 Revealed by Kinetic and Structural Studies. Sci. Rep. 2015, 5, 8529. 10.1038/srep08529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing H.; Zhang X.; Wisner S. A.; Chen X.; Spiegelman N. A.; Linder M. E.; Lin H., SIRT2 and lysine fatty acylation regulate the transforming activity of K-Ras4a. eLife 2017, 6, 10.7554/eLife.32436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegelman N. A.; Zhang X.; Jing H.; Cao J.; Kotliar I. B.; Aramsangtienchai P.; Wang M.; Tong Z.; Rosch K. M.; Lin H. SIRT2 and Lysine Fatty Acylation Regulate the Activity of RalB and Cell Migration. ACS Chem. Biol. 2019, 14 (9), 2014–2023. 10.1021/acschembio.9b00492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Outeiro T. F.; Kontopoulos E.; Altmann S. M.; Kufareva I.; Strathearn K. E.; Amore A. M.; Volk C. B.; Maxwell M. M.; Rochet J.-C.; McLean P. J.; Young A. B.; Abagyan R.; Feany M. B.; Hyman B. T.; Kazantsev A. G. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science 2007, 317 (5837), 516–519. 10.1126/science.1143780. [DOI] [PubMed] [Google Scholar]
- Rumpf T.; Schiedel M.; Karaman B.; Roessler C.; North B. J.; Lehotzky A.; Olah J.; Ladwein K. I.; Schmidtkunz K.; Gajer M.; Pannek M.; Steegborn C.; Sinclair D. A.; Gerhardt S.; Ovadi J.; Schutkowski M.; Sippl W.; Einsle O.; Jung M. Selective Sirt2 inhibition by ligand-induced rearrangement of the active site. Nat. Commun. 2015, 6, 6263. 10.1038/ncomms7263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Zhang M.; Dorfman R. G.; Pan Y.; Tang D.; Xu L.; Zhao Z.; Zhou Q.; Zhou L.; Wang Y.; Yin Y.; Shen S.; Kong B.; Friess H.; Zhao S.; Wang L.; Zou X. SIRT2 Promotes the Migration and Invasion of Gastric Cancer through RAS/ERK/JNK/MMP-9 Pathway by Increasing PEPCK1-Related Metabolism. Neoplasia 2018, 20 (7), 745–756. 10.1016/j.neo.2018.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozako T.; Mellini P.; Ohsugi T.; Aikawa A.; Uchida Y. I.; Honda S. I.; Suzuki T. Novel small molecule SIRT2 inhibitors induce cell death in leukemic cell lines. BMC Cancer 2018, 18 (1), 791. 10.1186/s12885-018-4710-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellini P.; Itoh Y.; Elboray E. E.; Tsumoto H.; Li Y.; Suzuki M.; Takahashi Y.; Tojo T.; Kurohara T.; Miyake Y.; Miura Y.; Kitao Y.; Kotoku M.; Iida T.; Suzuki T. Identification of Diketopiperazine-Containing 2-Anilinobenzamides as Potent Sirtuin 2 (SIRT2)-Selective Inhibitors Targeting the ″Selectivity Pocket″, Substrate-Binding Site, and NAD(+)-Binding Site. J. Med. Chem. 2019, 62 (12), 5844–5862. 10.1021/acs.jmedchem.9b00255. [DOI] [PubMed] [Google Scholar]
- Kudo N.; Ito A.; Arata M.; Nakata A.; Yoshida M.. Identification of a novel small molecule that inhibits deacetylase but not defatty-acylase reaction catalysed by SIRT2. Philos. Trans. R. Soc., B 2018, 373 ( (1748), ), 20170070. 10.1098/rstb.2017.0070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegelman N. A.; Price I. R.; Jing H.; Wang M.; Yang M.; Cao J.; Hong J. Y.; Zhang X.; Aramsangtienchai P.; Sadhukhan S.; Lin H. Direct Comparison of SIRT2 Inhibitors: Potency, Specificity, Activity-Dependent Inhibition, and On-Target Anticancer Activities. ChemMedChem 2018, 13 (18), 1890–1894. 10.1002/cmdc.201800391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi M.; Ieda N.; Nakagawa H. Development of Peptide-Based Sirtuin Defatty-Acylase Inhibitors Identified by the Fluorescence Probe, SFP3, That Can. Efficiently Measure Defatty-Acylase Activity of Sirtuin. J. Med. Chem. 2019, 62 (11), 5434–5452. 10.1021/acs.jmedchem.9b00315. [DOI] [PubMed] [Google Scholar]
- Spiegelman N. A.; Hong J. Y.; Hu J.; Jing H.; Wang M.; Price I. R.; Cao J.; Yang M.; Zhang X.; Lin H. A Small-Molecule SIRT2 Inhibitor That Promotes K-Ras4a Lysine Fatty-Acylation. ChemMedChem 2019, 14 (7), 744–748. 10.1002/cmdc.201800715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun B.; Fiskus W.; Qian Y.; Rajapakshe K.; Raina K.; Coleman K. G.; Crew A. P.; Shen A.; Saenz D. T.; Mill C. P.; Nowak A. J.; Jain N.; Zhang L.; Wang M.; Khoury J. D.; Coarfa C.; Crews C. M.; Bhalla K. N. BET protein proteolysis targeting chimera (PROTAC) exerts potent lethal activity against mantle cell lymphoma cells. Leukemia 2018, 32 (2), 343–352. 10.1038/leu.2017.207. [DOI] [PubMed] [Google Scholar]
- Raina K.; Lu J.; Qian Y.; Altieri M.; Gordon D.; Rossi A. M. K.; Wang J.; Chen X.; Dong H.; Siu K.; Winkler J. D.; Crew A. P.; Crews C. M.; Coleman K. G. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (26), 7124–7129. 10.1073/pnas.1521738113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottis P.; Crews C. M. Proteolysis-Targeting Chimeras: Induced Protein Degradation as a Therapeutic Strategy. ACS Chem. Biol. 2017, 12 (4), 892–898. 10.1021/acschembio.6b01068. [DOI] [PubMed] [Google Scholar]
- Schiedel M.; Herp D.; Hammelmann S.; Swyter S.; Lehotzky A.; Robaa D.; Olah J.; Ovadi J.; Sippl W.; Jung M. Chemically Induced Degradation of Sirtuin 2 (Sirt2) by a Proteolysis Targeting Chimera (PROTAC) Based on Sirtuin Rearranging Ligands (SirReals). J. Med. Chem. 2018, 61 (2), 482–491. 10.1021/acs.jmedchem.6b01872. [DOI] [PubMed] [Google Scholar]
- Hong J. Y.; Price I. R.; Bai J. J.; Lin H. A Glycoconjugated SIRT2 Inhibitor with Aqueous Solubility Allows Structure-Based Design of SIRT2 Inhibitors. ACS Chem. Biol. 2019, 14 (8), 1802–1810. 10.1021/acschembio.9b00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurz R. P.; Dellamaggiore K.; Dou H.; Javier N.; Lo M. C.; McCarter J. D.; Mohl D.; Sastri C.; Lipford J. R.; Cee V. J. A ″Click Chemistry Platform″ for the Rapid Synthesis of Bispecific Molecules for Inducing Protein Degradation. J. Med. Chem. 2018, 61 (2), 453–461. 10.1021/acs.jmedchem.6b01781. [DOI] [PubMed] [Google Scholar]
- Lu J.; Qian Y.; Altieri M.; Dong H.; Wang J.; Raina K.; Hines J.; Winkler J. D.; Crew A. P.; Coleman K.; Crews C. M. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol. 2015, 22 (6), 755–763. 10.1016/j.chembiol.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng M.; Xiong Y.; Safaee N.; Nowak R. P.; Donovan K. A.; Yuan C. J.; Nabet B.; Gero T. W.; Feru F.; Li L.; Gondi S.; Ombelets L. J.; Quan C.; Janne P. A.; Kostic M.; Scott D. A.; Westover K. D.; Fischer E. S.; Gray N. S. Exploring Targeted Degradation Strategy for Oncogenic KRAS(G12C). Cell Chem. Biol. 2020, 27 (1), 19–31. (e6) 10.1016/j.chembiol.2019.12.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.