

Abstract

Investigation of the trans-fluorine effect on the hydrolysis rate of diethyl 2-fluorocyclopropane-1,1-dicarboxylate provides synthetic access to both diastereomers of the fluorocyclopropyl analog of cabozantinib, a c-Met and VEGFR-2 inhibitor used as a first-line treatment for thyroid cancer and as a second-line treatment for renal cell carcinoma. Despite some known potent examples, there are only a few drug molecules that contain fluorocyclopropane moieties. Herein, we present a case study in which the monofluoro analog of a known cyclopropane-containing drug molecule displays an improved in vitro profile compared to the parent nonfluorinated structure. The fluorocyclopropane moiety may offer valuable fine-tuning options for lead optimization in drug discovery.
Keywords: c-Met kinase inhibitors, VEGFR-2 kinase inhibitors, cabozantinib, fluorocyclopropane, anticancer
A significant fraction of drugs newly approved by the FDA in 20181 and 20192 contain at least one fluorine atom in their chemical structure.3,4 The cyclopropane moiety can also be found in many active compounds with various indications.5,6 According to a SciFinder search, the combination of both structural motifs (fluorine-in-cyclopropane analogs) for most of the known cyclopropane-containing drug molecules has never been reported synthetically.7 Thus, despite the common approach to test fluorinated analogs during lead optimization,8 the incorporation of fluorocyclopropane9−13 is often overlooked, apparently due to the expected synthetic difficulties in obtaining such functionalities. However, there are several examples where such a structural modification has a beneficial effect on the performance of the drug molecule, for example, sitafloxacin14 or Tyk2 JH2 inhibitors.15
A prominent example of a cyclopropane-containing drug is cabozantinib16−19 (Figure 1), which is an inhibitor of the tyrosine kinases c-Met (hepatocyte growth factor receptor) and VEGFR-2 (vascular endothelial growth factor receptor 2), that has been approved for the treatment of medullary thyroid cancer20 and is used as a second-line treatment for renal cell carcinoma.21 The toxicity and resulting adverse effects of cabozantinib influence the quality of life of patients receiving this medication;22 thus, searching for more selective drug analogs is a high-priority task.
Figure 1.

trans-Fluorine effect gives access to both F-cyclopropyl cabozantinib analog diastereomers.
To the best of our knowledge, analogs of cabozantinib monofluorinated in the cyclopropane ring have never been studied. However, incorporation of an additional stereocenter always poses synthetic challenges, and if possible, isolated diastereomers should be accessed. For 2-substituted cyclopropane-1,1-diesters, the trans ester group hydrolysis rate is faster than that of the cis ester,23,24 where sterics are considered to play a major role. The fluorine atom is a relatively small substituent; however, it has a significant electronic contribution to the conformation, properties, and reactivity of the molecule.25−27 We were intrigued on whether a fluorine substituent on the cyclopropane ring can affect the hydrolysis rate of ester groups. Application of our own fluoromethylene transfer technology28−30 from diarylfluoromethylsulfonium salts allowed straightforward access to diethyl 2-fluorocyclopropane-1,1-dicarboxylate. Our DFT calculations predicted that an ester group trans relative to the fluorine should be more easily hydrolyzed to a carboxylic acid (Figure 1). These findings facilitated the development of a straightforward synthesis of both diastereomers of fluorocyclopropyl analogs of cabozantinib by a reverse hydrolysis/amide coupling strategy utilizing the same starting materials. An in vitro biological activity comparison identified a more selective fluoro analog compared to the parent structure. With this case study on cabozantinib, we would like to shed light on the fluorocyclopropane9−13 functionality for more frequent consideration in medicinal chemistry as an interesting and synthetically accessible moiety for the fine-tuning of a candidate drug.
The fluorocyclopropanation of diethyl methylydene malonate (1) utilizing the fluoromethylsulfonium reagent 2(31) in the fluoro-Johnson–Corey–Chaykovsky30 reaction (Scheme 1) afforded 2-fluorocyclopropane-1,1-dicarboxylate (3), which was further partially hydrolyzed with LiOH to predominantly afford trans hydrolysis product monoester 4.
Scheme 1. Preparation of Monoacid 4.

Depending on whether the cis or trans diastereomer was approached (Scheme 2), key intermediate 4 was sequentially subjected to amide coupling/hydrolysis sequences first using 4-(6,7-dimethoxyquinolin-4-yloxy)-phenylamine (5) and then using 4-fluoroaniline (6) for the synthesis of the cis diastereomer JV-982; conversely, the reverse sequence—aniline 6 followed by 5—was used if the trans diastereomer JV-976 was approached. Cabozantinib F-cyclopropyl analogs JV-982 and JV-976 were obtained as racemates. Based on evaluation of the enzymatic activities against c-Met (vide infra) the chiral separation of (rac)-JV-976 by HPLC was performed, giving access to both isolated enantiomers.
Scheme 2. Synthesis of Both Diastereomers of the F-Cyclopropyl Cabozantinib Analog.
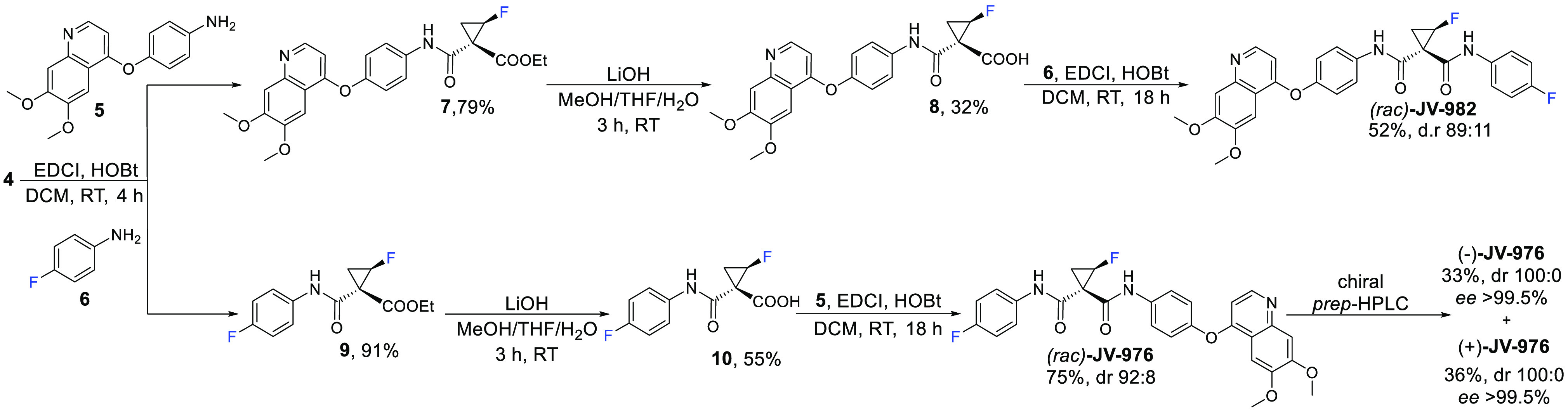
The corresponding stereochemical outcome was anticipated by investigating the trans-fluorine effect in the cyclopropane system. DFT calculations were performed using Gaussian 09 (see Supporting Information for computational details). HOMO/LUMO analysis of dimethyl 2-fluorocyclopropane-1,1-dicarboxylate (for calculations, the methyl ester was chosen for simplicity) showed that the LUMO orbitals are mostly located on the carbonyl group trans to fluorine, facilitating nucleophilic attack of the hydroxide (Figure 2). In turn, the ester group located on the same side as the fluorine is slightly turned away to avoid steric interactions with the fluorine, making OH attack less accessible.
Figure 2.
LUMO of fluorocyclopropyl diester.
Additionally, DFT calculations on the hydrolysis mechanism showed that the hydroxide attack on the carbonyl group is the rate determining step. Trans attack (Figure 3, Path A) is 3.2 kcal/mol more accessible than cis attack (Path B).
Figure 3.

DFT calculations on fluorocyclopropane-1,1-dicarboxylatehydrolysis. For simplicity, the Me ester was chosen for calculations. Structural minimization b3lyp/6-31g(d) integral = grid = superfine. Energies calculated b3lyp/6-311++g(2df,2p) scrf = (solvent = methanol) integral = grid = superfine.
An interesting observation of the trans-fluorine effect on carbonyl group properties arises from the comparison of N–H signals for JV-976, JV-982, and cabozantinib in the 1H NMR spectra (Figure 4A and B). The amide N–H signals positioned trans to fluorine are shifted significantly downfield compared to the cis-N–H signals or the N–H signals of nonfluorinated cabozantinib, suggesting decreased electron density of the corresponding trans-carbonyl groups.
Figure 4.
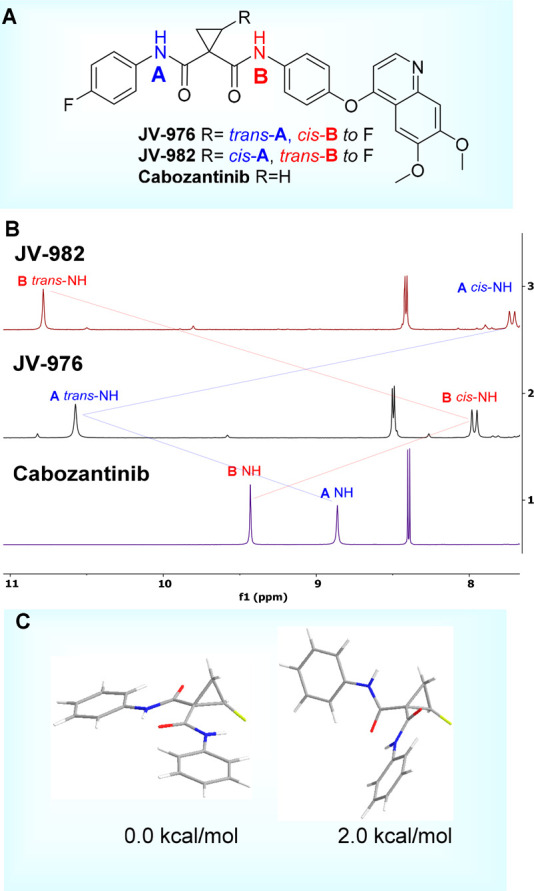
(A) Cabozantinib and its F-containing analogs. (B) Selected amide NH region of the 1H NMR spectra of JV-982, JV-976, and cabozantinib. (C) DFT-calculated conformations of the F-cyclopropane amide (Ph-amide selected for simplicity). b3lyp/6-31g(d) integral = grid = superfine.
DFT calculations on the 2-fluorocyclopropane-1,1-diamide system predicted the possible formation of intramolecular hydrogen bonds between the NH of the trans carbonyl and the O= of the cis carbonyl group, accounting for the observed strong downfield shift, whereas the experimentally observed 1H NMR JH–F = 14 Hz scalar coupling through the space between the cis amide NH and F supports close contact of the NH and fluorine atoms. This is in good agreement with the predicted diamide conformation (Figure 4C) in the fluorocyclopropane system. All of these results show that despite the small size of fluorine, it has a significant contribution to the reactivity, conformation, and properties of the substituents positioned cis or trans around the cyclopropane ring.
Further, the biological activities of the fluorocyclopropyl analogs of cabozantinib were studied. The abilities of the newly synthesized compounds JV-982 and JV-976 to inhibit c-Met kinase were compared to the inhibitory potency of cabozantinib. As shown in Table 1, both fluorocyclopropyl analogs are highly potent c-Met kinase inhibitors with activity in the low nanomolar range. cis-F diastereomer JV-982 showed the same activity as cabozantinib, while the trans-F diastereomer JV-976 indicated slightly higher inhibitory activity than cabozantinib. These encouraging results prompted us to further investigate the cytotoxic activity of newly synthesized compounds in cancerous and noncancerous cell lines. For this purpose, racemic JV-976 was separated into the pure enantiomers (+)-JV-976 and (−)-JV-976.
Table 1. IC50 Values of the Tested Compounds against c-Met Kinasea.
| Compound | (rac)-JV-976 | (rac)-JV-982 | Cabozantinib |
|---|---|---|---|
| IC50, nM | 18.6 ± 6.2 | 29.9 ± 7.2 | 29.9 ± 11.9 |
The values of two independent replicates are calculated.
The compounds JV-982, (+)-JV-976, (−)-JV-976, and cabozantinib were evaluated for their cytotoxic activity against Hep G2 liver cancer cells.32 The results are summarized in Table 2. All tested compounds displayed similar cytotoxicities in the low micromolar range in Hep G2 cells. Cabozantinib and JV-982 were slightly more active than (+)-JV-976 and (−)-JV-976. However, the difference in cytotoxicity was quite pronounced against noncancerous HEK293 cells, which express a low level of c-Met, and the results were compared to the reference compound cabozantinib. As a result, the selectivity in toxicity for HEK293/Hep G2 was more than 10-fold for (+)-JV-976 compared to an ∼3-fold selectivity margin for (−)-JV-976, JV-982, and the parent compound cabozantinib. Additionally, we performed an ex vivo blood stability assay using Wistar rat blood for the compounds under discussion. The assay was performed for up to 60 min at 37 °C, and we found that the fluoro-containing compounds had good metabolic stability in rat blood ex vivo (see the Supporting Information). This example shows that fine-tuning of the candidate drug by incorporating a fluorocyclopropane moiety can adjust the properties of the drug candidate; as a result, (+)-JV-976 was identified as an active and potentially less toxic alternative to cabozantinib.
Table 2. IC50 Values of the Tested Compounds against Noncancerous HEK293 and Hep G2 Liver Carcinoma Cells.
| IC50, μM |
|||
|---|---|---|---|
| Compound | Hep G2 | HEK293 | Selectivity HEK293/Hep G2 |
| (+)-JV-976 | 11.7 ± 3.2 | >100 | >10 |
| (−)-JV-976 | 17.4 ± 8.4 | 53.3 ± 24 | 3.1 |
| JV-982 | 9.4 ± 2.0 | 25.6 ± 3.1 | 2.7 |
| Cabozantinib | 8.3 ± 2.7 | 25.8 ± 13.3 | 3.1 |
Molecular docking simulations for JV-982 and JV-976 were performed using crystal structures of c-Met kinase (PDB ID: 3LQ8)17 and VEGFR-2 kinase (PDB ID: 3U6J).33
The most promising compound, JV-976 (red), was selected to dock into the active site of the kinases c-Met and VEGFR-2 and overlaid with cabozantinib (yellow). As shown in the binding models (Figure 5), cabozantinib and JV-976 occupy nearly identical conformations in both receptors. We performed additional MD (molecular dynamics) simulations of the synthesized compounds in complex with both enzymes discussed, c-Met kinase (PDB ID: 3LQ8) and VEGFR-2 kinase (PDB ID: 3U6J). As expected, no substantial difference in the binding mode of these compounds was observed. The RMSD values of all synthesized compounds were comparable and fluctuated at ∼0.7 Å2. This shows that fluorocyclopropane can act as a good bioisosteric replacement of the fluorocyclopropane moiety and does not disrupt any of the existing interactions.
Figure 5.
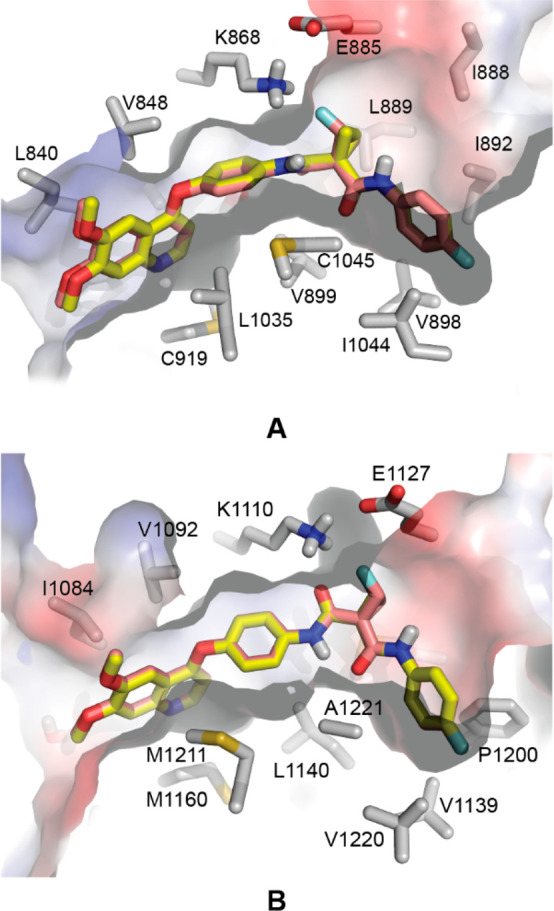
Molecular docking of JV-976 and cabozantinib into the (A) c-Met and (B) VEGFR-2 enzyme active sites.
In conclusion, investigation of the fluorine effect on hydrolysis selectivity for diethyl 2-fluorocyclopropane-1,1-dicarboxylate resulted in diastereoselective access to the cis and trans fluorocyclopropyl analogs of cabozantinib. The obtained trans-fluoro analog JV-976 displayed an improved inhibitory effect against c-Met enzymatic activity. The subsequent chiral separation of JV-976 allowed us to identify that enantiomer (+)-JV-976 has more than 3-fold higher selectivity toward liver cancer Hep G2 cells than noncancerous HEK293 cells when compared to the parent drug cabozantinib (selectivity of >10 vs 3.1), offering a potentially less toxic alternative for further investigation of liver cancer treatment. In combination with recent synthetic progress toward the synthesis of fluorocyclopropanes using fluoromethylsulfonium reagents along with the present case study on cabozantinib, we would like to shed light on the fluorocyclopropane functionality for more frequent consideration in medicinal chemistry, allowing for fine-tuning of the chemical and biological properties of a drug candidate.
Acknowledgments
Financial support from the ERDF project Nr.1.1.1.5/17/A/003. The authors wish to thank Aigars Jirgensons and Maija Dambrova for scientific discussions; Renate Melngaile, Armands Kazia, and Arturs Sperga for the synthesis of reagent 2; Andulis Smidlers for technical assistance; and LIOS analytical service for NMR (Juris Popelis and Marina Petrova) and MS (Solveiga Grinberga, Dace Hartmane, Valerija Krizanovska, and Baiba Gukalova).
Glossary
Abbreviations
- DCM
dichloromethane
- DFT
density functional theory
- EDCI
1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide
- FDA
United States Food and Drug Administration
- HOBt
hydroxybenzotriazole
- IC50
50% inhibitory concentration
- PDB ID
Protein Data Bank identification code
- RMDS
root-mean-square deviation of atomic positions
- Tyk2 JH2
tyrosine kinase 2 JH2
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00220.
Experimental details and data on synthetic procedures, biological activity studies, molecular docking, NMR spectra of synthesized products, and DFT calculations (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Mei H.; Han J.; Fustero S.; Medio-Simon M.; Sedgwick D. M.; Santi C.; Ruzziconi R.; Soloshonok V. A. Fluorine-Containing Drugs Approved by the FDA in 2018. Chem. - Eur. J. 2019, 25 (51), 11797–11819. 10.1002/chem.201901840. [DOI] [PubMed] [Google Scholar]
- Mei H.; Remete A. M.; Zou Y.; Moriwaki H.; Fustero S.; Kiss L.; Soloshonok V. A.; Han J.. Fluorine-Containing Drugs Approved by the FDA in 2019. Chin. Chem. Lett. 2020, 10.1016/j.cclet.2020.03.050. [DOI] [PubMed] [Google Scholar]
- Wang J.; Sánchez-Roselló M.; Aceña J. L.; Pozo C. D.; Sorochinsky A. E.; Fustero S.; Soloshonok V. A.; Liu H. Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011). Chem. Rev. 2014, 114 (4), 2432–2506. 10.1021/cr4002879. [DOI] [PubMed] [Google Scholar]
- Purser S.; Moore P. R.; Swallow S.; Gouverneur V. Fluorine in Medicinal Chemistry. Chem. Soc. Rev. 2008, 37 (2), 320–330. 10.1039/B610213C. [DOI] [PubMed] [Google Scholar]
- Talele T. T. The “Cyclopropyl Fragment” Is a Versatile Player That Frequently Appears in Preclinical/Clinical Drug Molecules. J. Med. Chem. 2016, 59 (19), 8712–8756. 10.1021/acs.jmedchem.6b00472. [DOI] [PubMed] [Google Scholar]
- Chawner S. J.; Cases-Thomas M. J.; Bull J. A. Divergent Synthesis of Cyclopropane-Containing Lead-Like Compounds, Fragments and Building Blocks through a Cobalt Catalyzed Cyclopropanation of Phenyl Vinyl Sulfide. Eur. J. Org. Chem. 2017, 2017 (34), 5015–5024. 10.1002/ejoc.201701030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SciFinder and DrugBank search on fluorocyclopropyl analogues of cyclopropane containing drug molecules reviewed in ref (5).
- Meanwell N. A. Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem. 2018, 61 (14), 5822–5880. 10.1021/acs.jmedchem.7b01788. [DOI] [PubMed] [Google Scholar]
- Jubault P.; Pons A.; Poisson T.; Pannecoucke X.; Charette A. Synthesis and Applications of Fluorocyclopropanes. Synthesis 2016, 48 (23), 4060–4071. 10.1055/s-0035-1560557. [DOI] [Google Scholar]
- David E.; Milanole G.; Ivashkin P.; Couve-Bonnaire S.; Jubault P.; Pannecoucke X. Syntheses and Applications of Monofluorinated Cyclopropanes. Chem. - Eur. J. 2012, 18 (47), 14904–14917. Selected recent synthetic advances toward the synthesis of fluorocyclopropanes: 10.1002/chem.201202831. [DOI] [PubMed] [Google Scholar]
- Pons A.; Tognetti V.; Joubert L.; Poisson T.; Pannecoucke X.; Charette A. B.; Jubault P. Catalytic Enantioselective Cyclopropanation of α-Fluoroacrylates: An Experimental and Theoretical Study. ACS Catal. 2019, 9 (3), 2594–2598. 10.1021/acscatal.9b00354. [DOI] [Google Scholar]
- Beaulieu L.-P. B.; Schneider J. F.; Charette A. B. Highly Enantioselective Simmons–Smith Fluorocyclopropanation of Allylic Alcohols via the Halogen Scrambling Strategy of Zinc Carbenoids. J. Am. Chem. Soc. 2013, 135 (21), 7819–7822. 10.1021/ja402393w. [DOI] [PubMed] [Google Scholar]
- Ye S.; Yoshida S.; Fröhlich R.; Haufe G.; Kirk K. L. Fluorinated Phenylcyclopropylamines. Part 4: Effects of Aryl Substituents and Stereochemistry on the Inhibition of Monoamine Oxidases by 1-Aryl-2-Fluoro-Cyclopropylamines. Bioorg. Med. Chem. 2005, 13 (7), 2489–2499. 10.1016/j.bmc.2005.01.043. [DOI] [PubMed] [Google Scholar]
- Kimura Y.; Atarashi S.; Kawakami K.; Sato K.; Hayakawa I. (Fluorocyclopropyl)Quinolones. 2. Synthesis and Stereochemical Structure-Activity Relationships of Chiral 7-(7-Amino-5-Azaspiro[2.4]Heptan-5-Yl)-1-(2-Fluorocyclopropyl)Quinolone Antibacterial Agents. J. Med. Chem. 1994, 37 (20), 3344–3352. 10.1021/jm00046a019. [DOI] [PubMed] [Google Scholar]
- Liu C.; Lin J.; Moslin R.; Tokarski J. S.; Muckelbauer J.; Chang C.; Tredup J.; Xie D.; Park H.; Li P.; et al. Identification of Imidazo[1,2-b]Pyridazine Derivatives as Potent, Selective, and Orally Active Tyk2 JH2 Inhibitors. ACS Med. Chem. Lett. 2019, 10 (3), 383–388. 10.1021/acsmedchemlett.9b00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakes F. M.; Chen J.; Tan J.; Yamaguchi K.; Shi Y.; Yu P.; Qian F.; Chu F.; Bentzien F.; Cancilla B.; et al. Cabozantinib (XL184), a Novel MET and VEGFR2 Inhibitor, Simultaneously Suppresses Metastasis, Angiogenesis, and Tumor Growth. Mol. Cancer Ther. 2011, 10 (12), 2298–2308. 10.1158/1535-7163.MCT-11-0264. [DOI] [PubMed] [Google Scholar]
- Qian F.; Engst S.; Yamaguchi K.; Yu P.; Won K.-A.; Mock L.; Lou T.; Tan J.; Li C.; Tam D.; et al. Inhibition of Tumor Cell Growth, Invasion, and Metastasis by EXEL-2880 (XL880, GSK1363089), a Novel Inhibitor of HGF and VEGF Receptor Tyrosine Kinases. Cancer Res. 2009, 69 (20), 8009–8016. 10.1158/0008-5472.CAN-08-4889. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Jiang X.; Jiang Y.; Guo M.; Zhang S.; Li J.; He J.; Liu J.; Wang J.; Ouyang L. Recent Advances in the Development of Dual VEGFR and c-Met Small Molecule Inhibitors as Anticancer Drugs. Eur. J. Med. Chem. 2016, 108, 495–504. 10.1016/j.ejmech.2015.12.016. [DOI] [PubMed] [Google Scholar]
- Wei D.; Fan H.; Zheng K.; Qin X.; Yang L.; Yang Y.; Duan Y.; Zhang Q.; Zeng C.; Hu L. Synthesis and Anti-Tumor Activity of [1,4] Dioxino [2,3-f] Quinazoline Derivatives as Dual Inhibitors of c-Met and VEGFR-2. Bioorg. Chem. 2019, 88, 102916. 10.1016/j.bioorg.2019.04.010. [DOI] [PubMed] [Google Scholar]
- Bannen L. C.; Sze-Ming Chan D.; Chen J.; Dalrymple L. E.; Forsyth T. P.; Huynh T. P.; Jammalamadaka V.; Khoury R. G.; Leahy J. W.; Mac M. B.; Mann G.; Mann L. W.; Nuss J. M.; Parks J. J.; Takeuchi C. S.; Wang Y.; Xu W.. C-met Modulators and Methods of Use. WO2005030140A2, 2004.
- Vecchio S. D.; Ellis R. J. Cabozantinib for the Management of Metastatic Clear Cell Renal Cell Carcinoma. J. Kidney Cancer VHL 2018, 5 (4), 1. 10.15586/jkcvhl.2018.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidinger M.; Danesi R. Management of Adverse Events Associated with Cabozantinib Therapy in Renal Cell Carcinoma. Oncologist 2018, 23 (3), 306–315. 10.1634/theoncologist.2017-0335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess K.; Li W. Practical Asymmetric Syntheses of All Four 2,3-Methanoleucine Stereoisomers. Tetrahedron Lett. 1995, 36 (16), 2725–2728. 10.1016/0040-4039(95)00382-M. [DOI] [Google Scholar]
- Jiang B.; Zhang F.; Xiong W. A Convenient Stereoselective Synthesis of Trifluoromethyl-Substituted Polyfunctionalized Cyclopropane: Synthesis of (±)-Trans-Trifluoronorcoronamic Acid. Chem. Commun. 2003, (4), 536–537. 10.1039/b210642f. [DOI] [PubMed] [Google Scholar]
- Thiehoff C.; Rey Y. P.; Gilmour R. The Fluorine Gauche Effect: A Brief History. Isr. J. Chem. 2017, 57 (1–2), 92–100. 10.1002/ijch.201600038. [DOI] [Google Scholar]
- Ni C.; Hu J. The Unique Fluorine Effects in Organic Reactions: Recent Facts and Insights into Fluoroalkylations. Chem. Soc. Rev. 2016, 45 (20), 5441–5454. 10.1039/C6CS00351F. [DOI] [PubMed] [Google Scholar]
- Díaz N.; Jiménez-Grávalos F.; Suárez D.; Francisco E.; Martín-Pendás Á. Fluorine Conformational Effects Characterized by Energy Decomposition Analysis. Phys. Chem. Chem. Phys. 2019, 21 (45), 25258–25275. 10.1039/C9CP05009D. [DOI] [PubMed] [Google Scholar]
- Veliks J.; Kazia A. Fluoromethylene Transfer from Diarylfluoromethylsulfonium Salts: Synthesis of Fluorinated Epoxides. Chem. - Eur. J. 2019, 25 (15), 3786–3789. 10.1002/chem.201900349. [DOI] [PubMed] [Google Scholar]
- Melngaile R.; Sperga A.; Baldridge K. K.; Veliks J. Diastereoselective Monofluorocyclopropanation Using Fluoromethylsulfonium Salts. Org. Lett. 2019, 21 (17), 7174–7178. 10.1021/acs.orglett.9b02867. [DOI] [PubMed] [Google Scholar]
- Kazia A.; Melngaile R.; Mishnev A.; Veliks J. Johnson–Corey–Chaykovsky Fluorocyclopropanation of Double Activated Alkenes: Scope and Limitations. Org. Biomol. Chem. 2020, 18 (7), 1384–1388. 10.1039/C9OB02712B. [DOI] [PubMed] [Google Scholar]
- Prakash G. K. S.; Ledneczki I.; Chacko S.; Olah G. A. Direct Electrophilic Monofluoromethylation. Org. Lett. 2008, 10 (4), 557–560. 10.1021/ol702500u. [DOI] [PubMed] [Google Scholar]
- Wang S. Y.; Chen B.; Zhan Y. Q.; Xu W. X.; Li C. Y.; Yang R. F.; Zheng H.; Yue P. B.; Larsen S. H.; Sun H. B.; Yang X. SU5416 Is a Potent Inhibitor of Hepatocyte Growth Factor Receptor (c-Met) and Blocks HGF-Induced Invasiveness of Human HepG2 Hepatoma Cells. J. Hepatol. 2004, 41 (2), 267–273. 10.1016/j.jhep.2004.04.013. [DOI] [PubMed] [Google Scholar]
- Norman M. H.; Liu L.; Lee M.; Xi N.; Fellows I.; D’Angelo N. D.; Dominguez C.; Rex K.; Bellon S. F.; Kim T.-S.; Dussault I. Structure-Based Design of Novel Class II c-Met Inhibitors: 1. Identification of Pyrazolone-Based Derivatives. J. Med. Chem. 2012, 55 (5), 1858–1867. 10.1021/jm201330u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.