

Abstract
Silk fibroin (SF) was enzymatically crosslinked with tyramine-substituted silk fibroin (SF-TA) or gelatin (G-TA) to fabricate hybrid hydrogels with tunable gelation kinetics, mechanical properties and bioactivity. Horseradish peroxidase (HRP)/hydrogen peroxide (H2O2) mediated crosslinking of SF in physiological buffers results in slow gelation and limited mechanical properties. Moreover, SF lacks cell attachment sequences, leading to poor cell-material interactions. These shortcomings can limit the uses of enzymatically crosslinked silk hydrogels in injectable tissue fillings, 3D bioprinting or cell microencapsulation, where rapid gelation and high bioactivity are desired. Here SF/SF-TA and SF/G-TA composite hydrogels were characterized for hydrogel properties and the influence of conjugated cyclic arginine-glycine-aspartic acid (RGD) peptide or G-TA content on bioactivity was explored. Both SF-TA and G-TA significantly increased gelation kinetics, improved mechanical properties and delayed enzymatic degradation in a concentration-dependent manner. β-sheet formation and hydrogel stiffening were accelerated by SF-TA content but delayed by G-TA. Both cyclic RGD and G-TA significantly improved morphology and metabolic activity of human mesenchymal stem cells (hMSCs) cultured on or encapsulated in composite hydrogels. The hydrogel formulations introduced in this study provide improved control of gel formation and properties, along with biocompatible systems that can be utilized in tissue engineering and cell delivery applications.
Keywords: silk hydrogels, HRP-mediated crosslinking, tyramine substitution, gelatin composite hydrogels, cyclic RGD peptide, cell encapsulation
Graphical Abstract
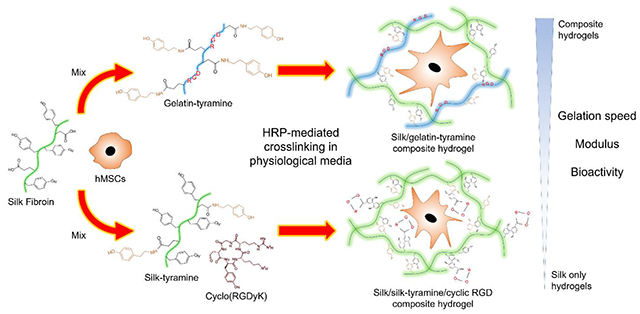
1. Introduction
Hydrogels are porous three-dimensional (3D) polymeric networks that can retain a large amount of water. Tunable physical, chemical and mechanical properties of hydrogel materials make them excellent candidates for the delivery of therapeutic agents [1, 2], tissue engineering and regenerative medicine applications [3–5]. One of the major uses of hydrogels is as injectable tissue fillers, where an empty or cell-laden pre-hydrogel solution is delivered to the defect area and cured in situ to guide cell and tissue growth [6, 7]. Besides tissue fillers, hydrogels also find use as bioinks for 3D bioprinting or as microscale particles for cell or drug delivery. Hydrogel scaffolds with a wide range of chemistry, shapes and structures have been reported for the fabrication of 3D tissue and organ systems for implantation [8, 9] or as models for in vitro drug testing [10]. Microgels fabricated by emulsification of an aqueous precursor solution in an oil phase, have been proposed for the in vivo delivery of encapsulated therapeutics and/or cells [11–15]. Silk fibroin (SF) of the domesticated silkworm Bombyx mori is a natural protein that displays excellent mechanical properties owing to hierarchical self-assembly, biocompatibility and biodegradation, and [16]. In addition to hydrogels physically crosslinked through the self-assembly of β-sheets induced by vortex [17], sonication [18], or polyalcohol treatment[19], aqueous SF solutions were also shown to form highly elastic, biocompatible hydrogels in the presence of horseradish peroxidase (HRP) and hydrogen peroxide (H2O2) through the oxidation of phenol groups in tyrosine residues into covalent crosslinks. [20] Mild crosslinking conditions in aqueous media, tunable mechanical properties, biodegradability and biocompatibility of enzymatically crosslinked silk hydrogels [21–23] make these materials excellent candidates for cell culture and encapsulation applications.
Enzymatic crosslinking of SF in physiological buffers such as phosphate buffered saline (PBS) or cell growth media was found to be much slower and result in weaker hydrogels compared to crosslinking in water [24], likely because of the salting out effect of metal ions inducing self-assembly of hydrophobic domains [25–27] which contain many tyrosine groups required for enzymatic crosslinking. Incubation of cells in water solution of SF, on the other hand, might impair cell viability due to osmotic stress [28]. This situation significantly limits the use of enzymatically crosslinked silk hydrogels in 3D bioprinting or microfluidic cell encapsulation, where rapid gelation is desired [29–31]. Another limitation of SF is that fibroin from the domesticated silkworm lacks cell attachment sequences [32], which resulted in a spherical morphology of the encapsulated cells [21, 24, 33]. The limited cell-matrix interactions may induce anoikis (homelessness), the programmed cell death of adherent cells due to missing survival signals provided by integrin-ligand interactions [34]. It is therefore useful to improve both gelation kinetics in physiological buffers and bioactivity of the enzymatically crosslinked silk hydrogels for cell encapsulation applications to preserve short or long-term cell viability.
Faster gelation of SF can be achieved by increasing the concentration of HRP [35, 36] and/or H2O2 [37]. However, high concentrations of HRP and H2O2 may cause potential immunological responses in vivo [36, 38] and apoptotic or necrotic cytotoxicity [39], respectively. Adding phenol red in silk solution was also found to increase gelation speed through bonding of phenolic hydroxyl groups to silk tyrosines [24], but the red color of the hydrogels and the risk of dye leakage at high concentrations may raise aesthetic and cytotoxicity concerns, respectively. Here we proposed faster gelation of silk by increasing the number of phenol groups available for crosslinking. Introduction of phenol groups on polymers has been reported for enzymatic crosslinking of gelatin [40–42], poly(glutamic acid) (PGA) [43], hyaluronic acid (HA) [44–48], dextran [49–51], alginate [52, 53], heparin [54], cellulose [55], chondroitin sulfate [56], poly(vinyl alcohol) (PVA) [57], poly(ethylene glycol) (PEG) [58] and their blends [59–61]. It is also possible to modify SF on the aspartic and glutamic acid residues through carbodiimide coupling [62, 63], to introduce new phenol groups via the conjugation of tyramine residues. The modified polymers can be mixed with unmodified SF at various ratios to modulate gelation kinetics and mechanical properties of resulting hydrogels. The bioactivity can also be improved through conjugation of peptides with arginine-glycine-aspartic acid (RGD) sequence, which is found in many extracellular matrix (ECM) proteins such as vitronectin, fibrinogen, collagen and laminin [64]. RGD peptides have been shown to improve cell attachment, spreading and proliferation on hydrogels of bio-inert polymers such as PEG [65–71], HA [72–75] and alginate [76], but was not conjugated into silk hydrogels before. Based on the previous reports with in situ conjugation of peptides with a tyrosine group into the hydrogels of phenol-substituted polymers during HRP-mediated crosslinking [77–79], here we proposed that silk/silk-tyramine hydrogels can be supplemented with cyclic RGD peptides with a tyrosine residue to improve cell-matrix interactions.
Another strategy to improve bioactivity of silk hydrogels would be blending with a protein that contains intrinsic RGD sequences, such as gelatin. Gelatin is a soluble protein produced by partial acid (type A) or base (type B) hydrolysis of collagen, which is the main ECM component of connective tissues including bone, cartilage and skin [80]. Gelatin is rich in RGD sequence [81], and it has been incorporated into HA, alginate and PEG to improve their bioactivity for cartilage, adipose, muscle and nerve tissue engineering applications [82]. Silk-gelatin composite hydrogels have also been reported, and the most common strategy has been the immobilization of gelatin within physically crosslinked silk network through methanol treatment [83, 84] or sonication [85]. However, significant mass losses were reported at physiological temperatures due to the sol-gel transition of gelatin at around 30°C [82, 83]. To prevent this, UV crosslinking of methacrylated gelatin prior to methanol treatment [86] or covalent crosslinking of silk and gelatin via genipin after ultrasonication [87] have been attempted, but potential cytotoxicity of methanol [88] and sonication [89] present significant issues for cell encapsulation. Silk-gelatin hydrogels directly crosslinked via genipin [90] or tyrosinase [91, 92] treatment have also been reported, but gelation times were long. It is possible to achieve rapid HRP/H2O2 mediated crosslinking of gelatin through conjugation of phenol groups [40, 41, 93–97], and hydrogel bioactivity was improved when crosslinked with other tyramine-substituted bioinert polymers such as alginate [98], HA [61, 99] and PEG [100]. This strategy, however, has not been reported for enzymatically crosslinked silk-gelatin composite hydrogels. Our group has previously demonstrated crosslinking of silk with HA-tyramine through the formation of tyrosine-tyramine bonds, resulting in significantly faster gelation compared to SF only hydrogels [101]. Using the same strategy, unmodified SF can be crosslinked with G-TA to improve both gelation kinetics in physiological buffers and bioactivity of silk hydrogels.
In this study, our aim was to improve gelation kinetics, mechanical properties and bioactivity of SF hydrogels crosslinked in physiologically relevant media through combining SF with SF-TA and cyclo(RGDyK) cyclic peptide, or only with G-TA. The influence of SF-TA and G-TA ratios on gelation kinetics, mechanical properties, β-sheet content and enzymatic degradation were explored and the effects of cyclo(RGDyK) or G-TA content on the behavior of human mesenchymal stem cells (hMSCs) on and within composite hydrogels were characterized. Our results suggest that the hydrogel formulations introduced in the present study offer biocompatible systems with tunable gelation kinetics, mechanical properties, enzymatic degradation and bioactivity that can be utilized for culture and encapsulation of mammalian cells in tissue engineering and regenerative medicine applications.
2. Materials and Methods
2.1. Synthesis of SF-TA and G-TA
Aqueous silk solutions were prepared as described before [21]. Briefly, B. mori cocoons were degummed to remove sericin protein by boiling 5 g of cut cocoons in 2 L of 0.02 M sodium carbonate (Sigma-Aldrich, St. Louis, MO) solution for 60 min and rinsing three times in deionized (DI) water. Degummed fibers were dried overnight and solubilized in 9.3 M lithium bromide (Sigma-Aldrich, St. Louis, MO) solution at a concentration of 20% (w/v) for 4 h at 60°C. The solution was then dialyzed against distilled water using regenerated cellulose dialysis tubing (3.5 kD MWCO, Spectrum Labs Inc, Rancho Dominguez, CA). Dialysis water was changed 6 times over 3 days and the resulting solution was centrifuged 2 times at 9,000 rpm at 4°C for 20 min to remove insoluble particles. The concentration of silk solution was determined by weighing a known volume of sample before and after drying overnight at 60°C.
SF-TA and G-TA derivatives were synthesized via carbodiimide-mediated reaction as described elsewhere [40]. Briefly, 2% (w/v) SF and gelatin solutions were prepared in 0.05 M 2-(N-morpholino)ethanesulfonic acid (MES) buffer (pH 6.0) and reacted with tyramine hydrochloride (Sigma-Aldrich, St. Louis, MO) (500 mg per 1 g protein) in the presence of 1-ethyl-3-(−3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) (Thermo Fisher Scientific, Rockford, IL) (184 mg per 1 g protein) and N-hydroxysuccinimide (NHS) (57 mg per 1 g protein) under stirring at room temperature (24°C) for 18 h. Solutions were dialyzed against distilled water using 3.5 MWCO tubing with 6 changes over 3 days. G-TA solution was subsequently lyophilized and stored at −20°C while SF-TA was kept as a solution at 4°C for up to a month.
2.2. Gelation Kinetics
To prepare SF/SF-TA and SF/G-TA hydrogels, aqueous SF was mixed with SF-TA or G-TA solutions at a final protein concentration of 50 mg/mL or 30 mg/mL in 40 mM hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES) (Sigma-Aldrich, St. Louis, MO) buffer (pH 7.4) or 0.5X Dulbecco’s Modified Eagle Medium (DMEM) High glucose with final SF-TA or G-TA ratios ranging between 0-100% (Table 1). Pre-hydrogel, HRP and H2O2 solutions were sterile filtered using Millex-GV 0.22 μm polyvinylidene fluoride (PVDF) syringe-driven filter units (Millipore Sigma, Darmstadt, Germany) for the rest of the study. Crosslinking kinetics of SF/SF-TA and SF/G-TA hydrogels were monitored at 37°C by measuring fluorescence emission using a SpectraMax M2 multi-mode microplate reader (Molecular Devices, Sunnyvale, CA). Gelation of 150 μL solutions was initiated with 10 U/mL HRP (type VI, Sigma-Aldrich, St. Louis, MO) and 0.01 or 0.005 wt.% H2O2 (Sigma-Aldrich, St. Louis, MO) in a black 96-well plate and fluorescence emission at 415 nm after excitation at 315 nm was monitored for 4500 s. 5% G-TA only in HEPES was omitted from material characterization due to very rapid gelation, resulting in non-homogenous hydrogels. To determine the advantage of tyramine substitution of SF over simply increasing HRP concentration, gelation kinetics of SF only and SF-TA only solutions in 0.5X DMEM with 0.01 wt% H2O2 were monitored and compared upon induction with 10 to 100 U/mL and 0.1 to 10 U/mL HRP, respectively. Results are reported after normalizing to a blank measurement taken before H2O2 was added (n=5).
Table 1.
Concentrations of each component in 5% and 3% SF/SF-TA and SF/G-TA hydrogels. Samples are denoted by the weight ratio of SF-TA or G-TA to total polymer concentration.
| SF/SF-TA | SF/G-TA | ||||
|---|---|---|---|---|---|
| Sample | SF concentration (mg/mL) | SF-TA concentration (mg/mL) | Sample | SF concentration (mg/mL) | G-TA concentration (mg/mL) |
| 5% w/v | |||||
| SF only | 50 | 0 | 5% | 47.5 | 2.5 |
| 10% | 45 | 5 | 10% | 45 | 5 |
| 20% | 40 | 10 | 15% | 42.5 | 7.5 |
| 30% | 35 | 15 | 20% | 40 | 10 |
| 50% | 25 | 25 | 25% | 37.5 | 12.5 |
| SF-TA only | 0 | 50 | 50% | 25 | 25 |
| 3% w/v | |||||
| SF only | 30 | 0 | 5% | 28.5 | 1.5 |
| 10% | 27 | 3 | 15% | 25.5 | 4.5 |
| 30% | 21 | 9 | 25% | 22.5 | 7.5 |
| 50% | 15 | 15 | G-TA only | 0 | 30 |
| SF-TA only | 0 | 30 | |||
2.3. Rheology
Rheological properties of SF/SF-TA and SF/G-TA hydrogels were measured at 37°C using an ARES-LS2 rheometer (TA Instruments, New Castle, DE) with a 25 mm stainless steel upper cone and temperature controlled Peltier bottom plate. A 420 μL aliquot of pre-hydrogel solutions with 10 U/mL HRP was loaded onto the Peltier and the cone was lowered to 47 μm. To initiate gelation, 4.2 μL of 1% or 0.5% H2O2 was injected into the gap during a 10 s precycle at a steady shear rate of 100/s. The gap was sealed with mineral oil to prevent evaporation during analysis. A dynamic time sweep was performed at 1 Hz with a 1% applied strain for 4,000 s to determine gelation kinetics and storage moduli. Dynamic frequency sweeps (0.1–100 rad/s at 1% strain) and strain sweeps (0.1%– to failure, at 1 Hz) were conducted to analyze elastic behavior of resulting hydrogels (n = 3). Rheological properties were measured in the linear viscoelastic region, where the storage modulus was independent of the applied strain.
Mesh sizes (ξ) of 3% hydrogels were estimated from the storage moduli determined by rheology using the equation below based on the rubber elasticity theory, which assumes purely elastic hydrogels without any physical interactions between the polymer chains
where G’ is the storage modulus, NA is the Avogadro constant, R is the molar gas constant, and T is the temperature [102].
2.4. Attenuated total reflectance -Fourier transform infrared spectroscopy (ATR-FTIR)
Secondary structure of the hydrogels was analyzed using a JASCO FTIR 6200 spectrometer (JASCO, Tokyo, Japan) with a MIRacle™ attenuated total reflection (ATR) with germanium crystal. Hydrogel discs (4 mm diameter, ∼2–3 mm height) were prepared by gelling 200 μL aliquots of solutions in polydimethylsiloxane (PDMS) molds incubated in 1 mL DPBS for 1, 7, 14, 21 and 28 days (n=3). After washing in deuterium oxide (Sigma-Aldrich, St. Louis, MO) three times for 30 min each, measurements were conducted by averaging 32 scans with a resolution of 4 cm−1 between 600 and 4000 cm−1. After deconvolution (Supplementary figure Fig.S1), β-sheet content was calculated as the ratio of areas of peak absorbances at 1616-1621, 1622-1627, 1628-1637 and 1697-1703 cm−1 to total area between 1580-1720 cm−1.
2.5. Unconfined compression
Unconfined compression was performed using an Instron 3366 Uniaxial Tensile Testing System (Instron, Norwood, MA) with a 10 N load cell. Hydrogel discs (4-5 mm height) prepared with 400 μL solutions in polydimethylsiloxane (PDMS) molds were incubated in 1 mL DPBS for 1, 7, 14, 21 and 28 days (n=3). After cutting with an 8 mm biopsy punch, hydrogel discs were placed between stainless steel parallel plates and stress response and elastic recovery were monitored during 30% strain at a rate of 0.667% s-1. Non-stiffened samples were objected to two pre-cycles at the same strain rate to eliminate artifacts. Tangent moduli were calculated in the linear region between 0 and 10% strain (n=5). Day 1 samples were also compressed to 80% strain to monitor the elastic behavior of the hydrogels.
2.6. In vitro enzymatic degradation
Hydrogel discs prepared from 200 μL solutions in PDMS molds were incubated in DPBS at room temperature for 4 h. Hydrogels were then transferred into 300 μL of 0.001 U/mL of protease (type XIV from Streptomyces griseus, Sigma-Aldrich, St. Louis, MO) dissolved in DPBS and incubated for 1, 2, 4, 6 or 8 days. The enzyme solution was changed every 2 days. After removal of enzyme solution, hydrogels were washed in Ultrapure™ distilled water (ThermoFisher Scientific, Waltham, MA) over night at room temperature, lyophilized and weighed. Results are reported as the mass fraction of the initial weight at day 0 (n = 4).
2.7. hMSCs culture on and in hydrogels
Human bone marrow mesenchymal stem cells (hMSCs) (ATCC, Manassas, VA) were cultured in DMEM High glucose supplemented with 10% fetal bovine serum (FBS), 1% Penicillin-Streptomycin (Life Technologies, Carlsbad, CA), 1% non-essential amino acids (Sigma-Aldrich, St. Louis, MO) and 1 ng/mL of fibroblast growth factor-2 (FGF-2) (Invitrogen, Carlsbad, CA). 5% SF/20% SF-TA solutions supplemented with 0, 0.1, 0.25, 0.5 or 1 mM cyclo(RGDyK) and 5% SF/G-TA solutions with G-TA weight ratios of 0, 5, 10, 15, 20, 25% were prepared in 40 mM HEPES and 300 μL aliquots with 10 U/mL HRP and 0.01% H2O2 were allowed to cure in 24-well plates for 1 h at 37°C in an incubator. 5% G-TA only hydrogels were prepared using 0.1 U/mL HRP due to very rapid gelation at 1 U/mL or 10 U/mL resulting in non-homogenous gels. Cells at passage 3 were seeded onto tissue culture plastic (TCP) controls and hydrogel surfaces at a density of 4,000 cells/cm2 in 1 mL of growth medium without FBS. At day 1, seeding media was replaced with regular growth media with FBS and it was changed every 3 days. For cell encapsulation, 300 uL pre-hydrogel solutions with 10 U/mL HRP were prepared in 40 mM HEPES or 0.5X DMEM and mixed with cells at passage 3 at a concentration of 2x105 cells/mL. Cell-laden precursor solutions with 0.01 or 0.005 wt. % H2O2 were then allowed to cure in 24 well-plates for 1 h at 37°C in an incubator. The wells were then flooded with 1 mL of culture media and media was changed every 3 days.
2.8. Fluorescence and confocal laser scanning microscopy (CLSM) imaging
To monitor the viability and morphology of hMSCs cultured on and encapsulated in hydrogels, cells were stained with Live/Dead viability kit (Invitrogen, Carlsbad, CA) and imaged with a BZ-X700 Fluorescence Microscope (Keyence Corp., Itasca, IL) or TCS SP8 microscope from Leica Microsystems (Wetzlar, Germany), respectively. After incubating the cells on hydrogel surface with calcein AM and ethidium homodimer-1 (EthD-1) for 20 min or the encapsulated cells for 1 h, samples were washed with DPBS and imaged. Day 1 fluorescence images of the cells cultured on the hydrogels were analyzed using ImageJ (1.48v, NIH, USA) to quantify cell spread areas (n ≥ 200).
2.9. Metabolic activity
Metabolic activity of hMSCs cultured on and within hydrogels was determined at days 1, 3, 7, 14, 21 and 28 by alamarBlue viability assay (Invitrogen, Carlsbad, CA) according to the manufacturer’s directions. After rinsing with DPBS, cells on hydrogel surfaces or encapsulated in hydrogels were incubated in 500 μL of 10% alamarBlue reagent in DMEM high glucose without phenol red (Sigma-Aldrich, St. Louis, MO) supplemented with 1% non-essential amino acids for 1 h or 3 h, respectively, at 37°C with 5% CO2. Following incubation, 130 μL aliquots were transferred into 96 well plates and absorbance at 570 and 595 nm were measured using a microplate reader. Results are reported as the fold change in % dye reduction at day 3, 7, 14, 21 and 28 after normalization to initial measurement at day 1 (n=4).
2.10. In vivo animal tests
Hydrogel discs of 8 mm diameter and 4 mm height were fabricated in autoclaved PDMS molds using 200 μL aliquots of 3% SF-TA only or SF/25% G-TA solutions using 10 U/mL HRP and 0.01 wt% H2O2 and implanted subcutaneously into 12-week-old female Friend Virus B NIH Jackson (FVB/NJ) mice (The Jackson Laboratory, Bar Harbor, ME) under the protocol approved by the Tufts Institutional Animal Care and Use Committee (M2019-121) (n = 3, 2 hydrogel discs per animal). The mice were anesthetized initially at 3% isoflurane and maintained at 2% for the duration of the surgery. Prior to making the incision, the area was shaved, sterilized using disinfectant and ethanol swabs (3x) and a subcutaneous injection of the sustained-release buprenorphine (0.5 mg/mL) analgesic was administered at a dose of 1 mg/kg. Two independent incisions (6-10 mm) were made on the back of each mouse to create a subcutaneous pocket using scissors and a blunt probe. The SF-TA gels were inserted into the upper back pocket (Supplementary Figure S2A) and the SF/25% G-TA discs were inserted in the lower back pocket (Supplementary Figure S2B). Incisions were closed with 1 or 2 stainless steel wound clips. The mice were monitored daily and euthanized by CO2 asphyxiation after 3 days. After resection and fixation in 4% paraformaldehyde for 48 hours, the hydrogel discs submerged in increasing concentrations of sucrose (15-30% w/v) in PBS for 48 hours, flash frozen using dry ice in optimal cutting temperature (OCT) media and stored at −80°C freezer until cryo-sectioning. The samples were sliced into 15 μm cross-sections using a cryostat and collected on Fisherbrand tissue path superfrost plus gold slides (Fisher Scientific, Hampton, NH). The samples were stained with hematoxylin and eosin (H&E) and imaged using fluorescence microscope.
2.11. Statistical analyses
All data are expres sed as means ± standard deviations for n ≥ 3. GraphPad Prism (GraphPad Software, La Jolla, CA) was used to perform One- or Two-way analysis of variance (ANOVA) with Tukey’s post hoc multiple comparison test to determine statistical significance (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001).
3. Results
3.1. Influence of buffer composition on hydrogel properties and cell encapsulation
The influence of the buffer used for hydrogel crosslinking on the shear mechanical properties of 5% SF hydrogels was assessed through rheology. It is known that salt solutions with higher ionic strength than distilled water induce self-assembly of hydrophobic domains of SF that contains many tyrosine residues into crystalline β-sheets (Fig.S3A). The 5% hydrogels crosslinked in 40 mM HEPES, 0.5X DMEM and particularly 1X PBS started gelling later (Fig.S3B) and had significantly lower storage moduli at t=4000 s (1.36, 0.74 and 0.27 kPa, respectively) compared to the hydrogels crosslinked in distilled water (3.1 kPa) (Fig.S3C). To assess the time required for hydrogels to reach mechanical stability, gel point (defined as the time point when the ratio of loss modulus to storage modulus (G”/G’) permanently drops below 0.05) of the hydrogels crosslinked in various buffers were compared (Fig.S3D). The 5% SF solutions crosslinked in HEPES, DMEM and PBS reached the gel point at ~6, 8 and 23 min, respectively, indicating a significant delay in gelation, particularly in PBS, compared to crosslinking in distilled water (~4 min).
Before encapsulation in the hydrogels, cells were incubated in 5% SF solutions prepared in distilled water, 1X PBS, 40 mM HEPES or 0.5X DMEM for 1 h at room temperature and then seeded on tissue culture plastic (TCP) to analyze the influence of buffer composition on cell viability. Almost no cells incubated in distilled water solution survived as expected due to osmotic stress. The metabolic activity of the cells incubated in HEPES buffer was reduced by half, while no significant reduction was recorded for 1X PBS or 0.5X DMEM compared to the untreated controls (Fig.S3E). Interestingly, when the cells were encapsulated in 5% hydrogels using the same buffer compositions, their viability in 1X PBS group was ~2-fold lower compared to the untreated control cells just like the HEPES group, while no significant reduction was found for 0.5X DMEM group (Fig.S3F). Distilled water and 1X PBS were eliminated from the rest of the study due to inferior cell viability and hydrogel properties, respectively.
3.2. Characterization of SF-TA and G-TA
Tyramine-conjugated biopolymers were obtained by carbodiimide coupling of tyramine groups on SF and gelatin (Fig.1A). After conjugation, UV absorbance between 250 – 300 nm and absorbance peak at 275 nm significantly increased for gelatin (Fig.S4A), while no significant change was recorded for SF due to high background UV absorbance by tyrosine residues. Liquid chromatography tandem mass spectroscopy (LC-MS/MS) analysis (Fig.S4B) was employed to demonstrate the presence of tyramine groups on SF-TA and G-TA, and SF-TA was found to have a significantly larger peak area for tyrosine but a lower peak area for tyramine compared to G-TA. To further confirm tyramine conjugation on SF, 1HNMR spectra of SF-TA and SF were compared (Fig.S4C) and alkyl proton peaks of tyramine were observed on SF-TA spectrum next to Tyr β proton peaks between 2.4-3.2 ppm. Molecular weight distributions of gelatin and silk before and after tyramine substitution were digitally quantified on polyacrylamide gel images after electrophoresis (Fig.S5A). Using the calibration curve of log molecular weight versus distance from the wells in pixels (Fig.S5B), frequency of each molecular weight in the protein smears were plotted (Fig.S5C). Interestingly, molecular weight with the highest frequency was found to decrease for SF upon tyramine substitution (122 to 57 kDa), while no significant change was observed in the mean molecular weight (SF: 85.5 kDa, SF-TA: 86.7 kDa) (Fig.S5D). For gelatin, on the other hand, molecular weight with the highest frequency shifted from 170 to 246 kDa upon tyramine substitution, while mean molecular weight slightly decreased from 131 to 124 kDa.
Figure 1.
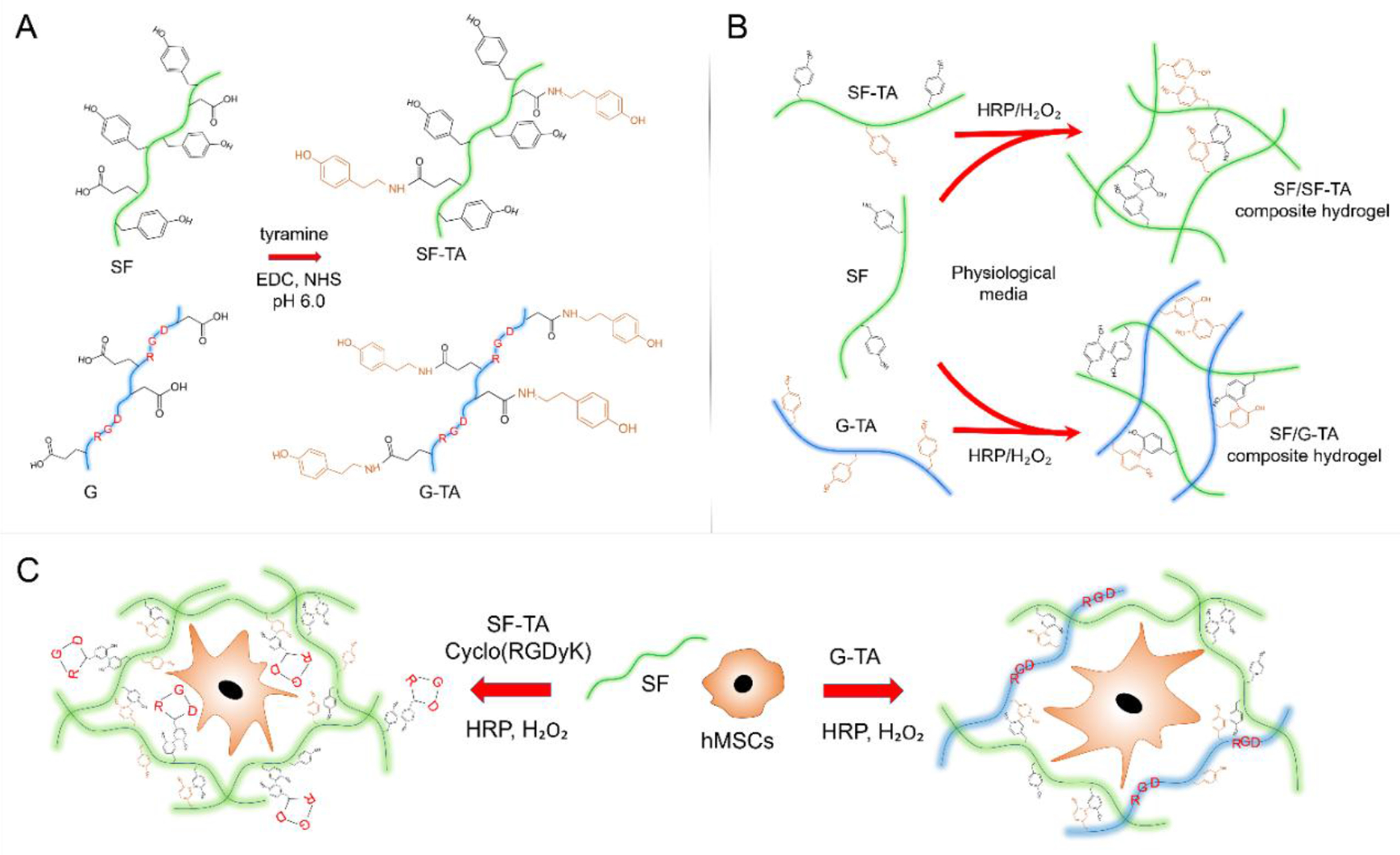
Schematic representation of the SF/SF-TA and SF/G-TA composite hydrogel systems for cell encapsulation. (A) Carbodiimide coupling of tyramine (TA) groups to carboxylic acid residues on silk fibroin (SF) and gelatin (G) chains. (B) HRP-mediated covalent crosslinking of SF with SF-TA or G-TA. (C) Encapsulation of hMSCs within enzymatically crosslinked SF/SF-TA hydrogels with in situ conjugated cyclo(RGDyK) or in SF/G-TA composite hydrogels. SF: silk fibroin, G: gelatin, SF-TA: tyramine-substituted silk fibroin, G-TA: tyramine-substituted gelatin.
3.3. Gelation of SF/SF-TA and SF/G-TA composite hydrogels
Covalent crosslinking of SF with SF-TA and G-TA was expected in the presence of HRP and H2O2 through the formation of tyrosine-tyramine linkages as well as dityrosine and dityramine crosslinks (Fig.1B). The sol-gel transition of SF/SF-TA and SF/G-TA composite hydrogels occurred upon mixing with HRP and H2O2. Unlike transparent SF only hydrogels, hybrid hydrogels were opaque, the degree of which increased with increasing SF-TA or G-TA content (Fig. 2A). Crosslinking kinetics of the hybrid hydrogels were monitored by quantifying the fluorescence emission by biphenyl-2,2′-diol groups of dityrosine, tyrosine-tyramine and dityramine at 415 nm after excitation at 315 nm. Curves of 5% SF/SF-TA (Fig.2B–i) and SF/G-TA (Fig.2B–ii) composite hydrogels crosslinked in 40 mM HEPES buffer were hyperbolic-like and reached to a plateau faster with increasing SF-TA or G-TA content, while SF only hydrogels displayed a sigmoidal shaped curve and did not reach a plateau at t = 4500 s, indicating that crosslinking was not completed. Relative intensity of fluorescence emission was found to increase significantly with increasing SF-TA content from 0% to 50% and a slight increase was observed in SF with 25% and 50% G-TA hydrogels compared to SF only group (Fig.2B–iii). For 3% hydrogels crosslinked in 0.5X DMEM buffer, all gelation curves including the SF only group had a hyperbolic shape and increasing SF-TA (Fig.2C–i) or G-TA (Fig.2C–ii) content accelerated crosslinking compared to SF only hydrogel. This increase in crosslinking speed, however, did not result in a significant increase in fluorescence intensity (Fig.2C–iii). Despite almost instant gelation, fluorescence emission by G-TA only hydrogels was significantly lower compared to the other groups. LC-MS/MS analysis revealed that the content of dityrosine bonds and unreacted tyrosines decreased while the abundance of tyrosine-tyramine and dityramine crosslinks increased with increasing SF-TA or G-TA ratio in 3% hydrogels (Fig.S6).
Figure 2.

Gelation of silk hydrogels supplemented with tyramine-modified silk or gelatin. (A) Photograph showing 5% SF/SF-TA and SF/G-TA solutions gelled in the bottom of Eppendorf tubes. Influence of (i) SF-TA or (ii) G-TA weight ratios on crosslinking kinetics and (iii) maximum fluorescence intensities of (B) 5% hydrogels prepared in 40 mM HEPES buffer and (C) 3% hydrogels prepared in 0.5X DMEM buffer (n=5, *p < 0.05, **p < 0.01 and ***p < 0.001, asterisks above the bars represent significance compared to SF only gels).
The advantage of tyramine substitution on SF over increasing HRP concentration in terms of gelation kinetics was assessed by monitoring gelation of SF only (Fig.S7A–i) and SF-TA only (Fig.S7B–i) solutions in 0.5X DMEM upon induction with HRP concentrations of 10 to 100 U/mL and 0.1 to 10 U/mL, respectively. By increasing the HRP concentration used for gelation of 3% unmodified SF solutions in 0.5X DMEM from 10 to 40 U/mL, the time required for reaching 90% of the maximum fluorescence intensity decreased from ~2400 s to ~1600 s, but it did not change significantly from 40 to 100 U/mL HRP (Fig.S7A–ii) . SF-TA solution, on the other hand, was found to reach 90% of maximum fluorescence in only 690 s at 10 U/mL, and it increased up to 2400 s when HRP concentration was dropped to 0.1 U/mL (Fig.S7B–ii), which was comparable with unmodified SF solution with 10 U/mL HRP. Maximum fluorescence intensities of SF gels at 20 or 40 U/mL HRP were significantly higher than those at 10 or 100 U/mL (Fig.S7A–iii), while a statistically significant sharp and mild increases were recorded for 0.1 to 1 U/mL and 1 to 10 U/mL for SF-TA gels (Fig.S7B–iii).
As a proof of concept, 3% SF-TA solutions prepared in 0.5X DMEM were used for printing of hydrogel patterns and fabrication of hydrogel microspheres using co-flow microfluidics approach. Upon mixing of the streams with HRP and H2O2 right before extrusion (Fig.S8A, B) through a 3D bioprinter-compatible (Fig.S8C) 2-inlet needle, the aqueous solution was observed to gel rapidly, resulting in opaque hydrogel patterns on the printing surface (Fig.S8D). Similarly, when extruded through a co-axial needle into a continuous oil phase (Fig.S9A), droplets of SF-TA pre-hydrogel solution with HRP rapidly gelled into individual hydrogel microspheres (Fig.S9B) upon diffusion of H2O2 dispersed in the oil phase. Droplets of unmodified SF solution, however, were found to fuse to each other due to slow gelation (data not shown).
Residual H2O2 in SF hydrogels supplemented with 30% w/w SF-TA or 25% w/w G-TA was quantified using the tetramethylbenzidine (TMB) colorimetric assay. A calibration curve of H2O2 concentration versus optical density at 450 nm (OD450) with a linear trend was obtained using a concentration range of 10−5 (~3.2 μM) to 5x10−4 wt.% (~163 μM) (Fig.S10A). The concentration of residual H2O2 collected by diffusing out by shaking the hydrogel discs or homogenizing them in ultrapure water was found to be below the detection limit of the assay as no significant difference was recorded between the OD450 of the blank ultrapure water or the experimental groups (Fig.S10B).
3.4. Shear mechanical properties
The influence of SF-TA and G-TA on shear mechanical properties of composite hydrogels was studied through rheology. For 5% hydrogels prepared in 40 mM HEPES buffer, time point that gelation started and the gel point (G”/G’ < 0.05) decreased with increasing SF-TA (Fig.3A–i, iii) and G-TA (Fig.3B–i, iii) ratio. Shear storage moduli of the hydrogels at t=4000 s were found to increase up to a certain point, reach a plateau and then start decreasing with increasing SF-TA (Fig.3A–ii) or G-TA (Fig.3B–ii). The maximum storage moduli recorded were ~2.2 kPa for SF with 30% SF-TA and ~ 2 kPa for 25% G-TA in comparison to 1.4 kPa for SF only hydrogels. SF with 25% unmodified gelatin started gelling much later and resulted in a significantly lower storage modulus compared to SF only or SF/25% G-TA hydrogels. The same trend of faster gelation and increase in storage modulus up to a certain point with increasing SF-TA or G-TA ratios was also observed in 3% hydrogels crosslinked in 0.5X DMEM (Fig.3C, Fig.S11), but they had lower storage moduli (0.1-0.24 kPa) compared to 5% hydrogels crosslinked in HEPES buffer (1.3-2.1 kPa). The 3% G-TA only hydrogels gelled instantly and displayed the highest storage modulus, but this measurement might not be very accurate as the solution was rapidly crosslinked and the gel was ruptured during 10 s of precycle shearing at high speed. All gels tested regardless of SF-TA or G-TA content, protein concentration or crosslinking media were frequency independent and withstood at least 100% shear strain, except for the 3% G-TA only hydrogels, which started failing at around 60% strain (Fig.S12A, B).
Figure 3.
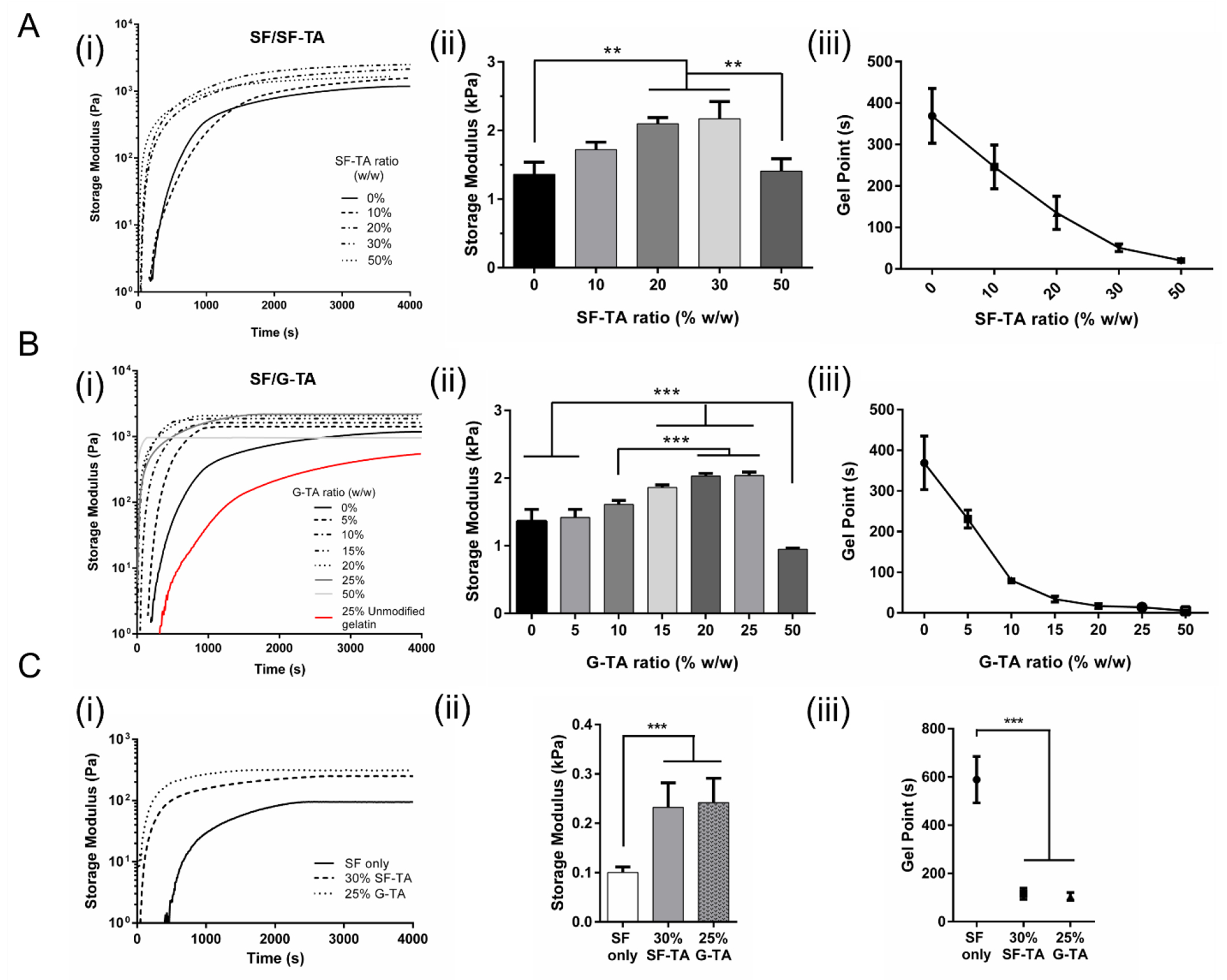
Rheological properties of composite hydrogels. (i) Representative time sweeps, (ii) shear storage moduli at t = 4000 s and (iii) gel (G”/G’ < 0.05) points of 5% (A) SF/SF-TA and (B) SF/G-TA hydrogels prepared in 40 mM HEPES buffer and (C) 3% hydrogels prepared in 0.5X DMEM. (n = 3, *p < 0.05, **p < 0.01 and ***p < 0.001).
Rheological properties of the 3% hydrogels crosslinked in 0.5X DMEM (Fig.S13A) also depended on H2O2 concentration used for crosslinking. At 0.005 wt.% H2O2, G”/G’ ratio of SF only hydrogels (Fig.S13B) did not permanently drop below 0.05 and storage modulus fluctuated with increasing frequency. It took much longer for SF/30% SF-TA hydrogels to reach gel point (~400 s) compared to 0.01 wt.% H2O2 (~120 s), while it did not change significantly for SF/25% G-TA hydrogels (Fig.S13B). Storage moduli of all 3% hydrogels crosslinked using 0.005% H2O2 were lower more than 2-fold compared to 0.01 wt.% H2O2 condition (Fig.S13C). Although no significant difference was found between SF/30% SF-TA (0.23 kPa) or 25% G-TA (0.24 kPa) at 0.01% H2O2, SF/25% G-TA (0.1 kPa) was significantly stiffer than SF/30% SF-TA (0.03 kPa) and SF only (0.02 kPa) hydrogels at 0.005% H2O2. LC-MS/MS analysis showed that the dityrosine, tyrosine-tyramine, and dityramine content gradually decreased when H2O2 concentration was lowered from 0.02 wt. % to 0.005 wt. % (Fig.S14). Cyclo(RGDyK) peptides included in 3% SF/30% SF-TA hydrogels were expected to be conjugated to silk chains during crosslinking (Fig.S15A). The hydrogels with 0.25 mM or 0.5 mM cyclo(RGDyK) started to gel (Fig.S15B) and reached the gel point (Fig.S15C) much later, and resulted in significantly lower storage modulus (Fig.S15D) compared to no peptide control.
Mesh sizes of 3% hydrogels were calculated based on rubber elasticity theory. Mesh sizes of the hydrogels crosslinked using 0.01% H2O2 were determined to range from 20 to 35 nm, which decreased from 35.1 ± 1.3 nm of SF only to 26.7 ± 2.1 of SF/30% SF-TA but then increased to 32.7 ± 1.7 nm in SF/50% SF-TA; and decreased to 26.3 ± 1.6 nm in SF/25%G-TA (Fig.S16A). G-TA only hydrogels had the lowest mesh size, which was 22.2 ± 0.5 nm. Both decreasing the H2O2 concentration to 0.005 wt% and increasing cyclic RGD peptide concentration increased mesh size (Fig.S16B), confirming that crosslinking density was decreased.
3.5. Influence of SF-TA and G-TA content on β-sheet content
IR absorption spectra of hydrogels between 1580-1720 nm (Fig.4A & D, Fig.S17) were analyzed using ATR-FTIR to quantify β-sheet content over 4 weeks of incubation in PBS at 37°C. β-sheet content of 5% hydrogels at day 1 was in the range of 20-35% and it was found to decrease with increasing SF-TA (Fig.4B) or G-TA (Fig.4C) concentration except SF-TA only hydrogels, which had the highest initial content of 32%. At day 7, SF only and SF/SF-TA hydrogels reached a β-sheet content of approximately 40% and remained approximately the same over 4 weeks, while that of SF-TA only hydrogels increased up to 48%. β-sheet content of SF with 10 to 25% G-TA, on the other hand, remained at ~25% for at least 2 weeks and then increased to 35-40% at day 21 (Fig.4C). For the 3% hydrogels prepared in 0.5X DMEM, the increase in β-sheet content over time was slower compared to 5% hydrogels. At day 1, β-sheet content of SF-TA only hydrogels was significantly lower than those of SF only or SF/30%SF-TA hydrogels. The crystalline content remained at around 20% for 3% SF only hydrogels during the first week and gradually increased to ~40% over 4 weeks, while those of SF/30% SF-TA and SF-TA only hydrogels reached above 35% and 40%, respectively (Fig.4E). After 4 weeks, SF-TA only hydrogels reached the highest crystalline ratio of 46% among the other 3% hydrogels. For SF/25% G-TA, however, no significant change was recorded over 28 days and β-sheet content remained significantly lower (~20%) compared to SF only hydrogels.
Figure 4.
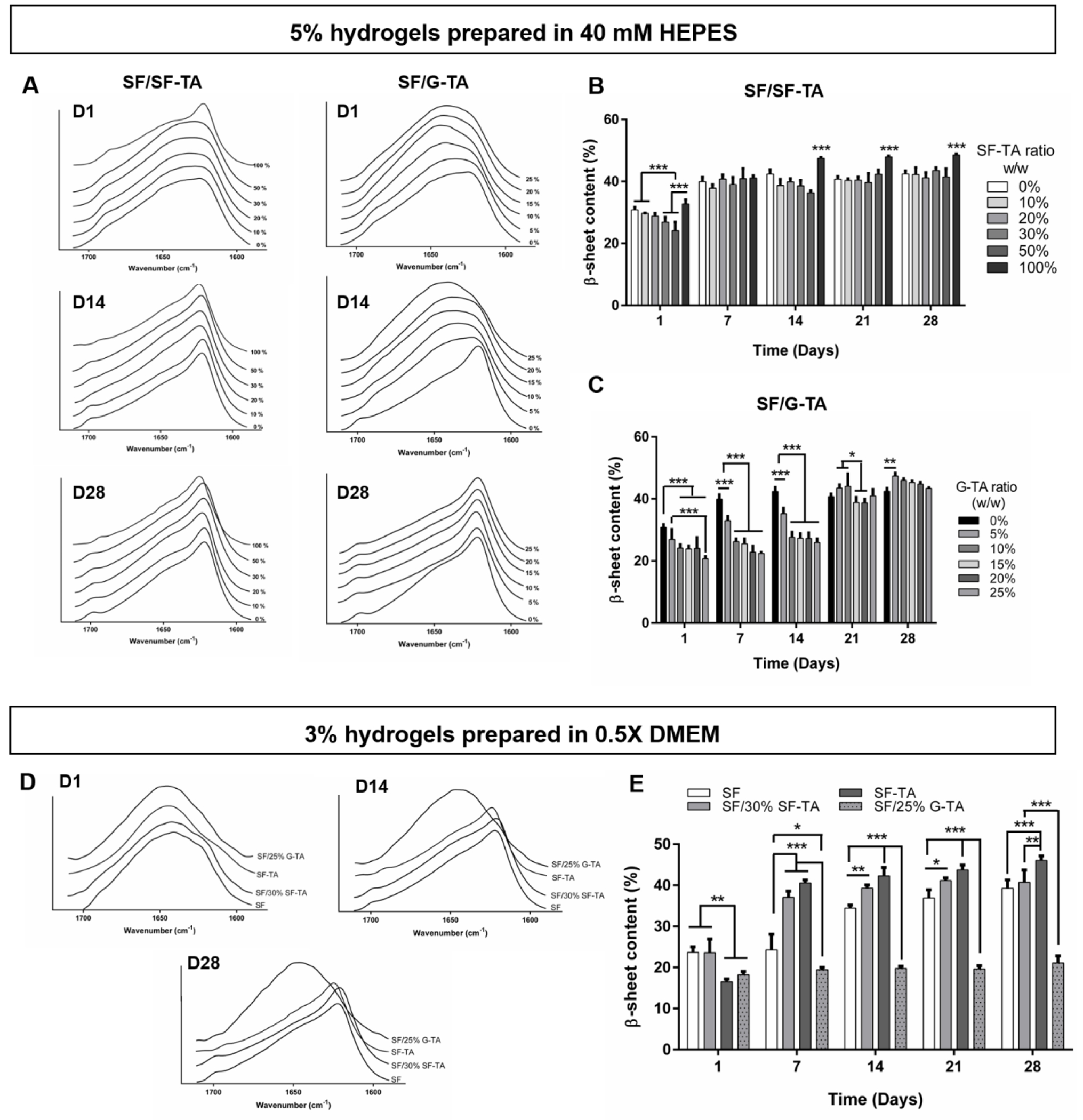
Secondary structure of composite hydrogels determined by FTIR absorbance.ATR-FTIR absorbance spectra of (A) 5% hydrogels prepared in 40 mM HEPES and (D) 3% hydrogels prepared in 0.5X DMEM. β-sheet contents of 5% (B) SF/SF-TA, (C) SF/G-TA and (E) 3% composite hydrogels over 4 weeks of culture in PBS at 37°C. (n = 3, *p < 0.05, **p < 0.01 and ***p < 0.001).
3.6. Hydrogel morphology and compressive mechanical properties
The 5% and 3% composite hydrogels prepared in 40 mM HEPES and 0.5X DMEM, respectively, were incubated in PBS at 37°C and their morphologies and compressive properties were analyzed over 4 weeks. The SF only, SF/SF-TA and SF-TA only hydrogel discs shrank by day 14 compared to day 1 (Fig.S18). The 5% SF/10% or 25% G-TA hydrogels, however, roughly preserved their initial diameters (Fig.S18, marked with red arrows) by day 14, but they also shrank significantly till day 28. Similar to G-TA only gels, no significant change was observed in the dimensions of 3% SF/25% G-TA hydrogels (Fig.S18, marked with yellow arrows), from day 1 to day 28. Unconfined compression results were parallel with the hydrogel morphologies. Stress-strain curves of all 5% (Fig.5A) and 3% (Fig.5C) hydrogels except 5% SF-TA only indicated elastic behavior with full recovery at day 1, and their compressive moduli ranged between 1.5-3 kPa and 0.3-0.6 kPa, respectively (Fig.5B, D). Initial modulus decreased with increasing SF-TA concentration but was not changed significantly by the G-TA content. 5% SF-TA only hydrogels, however, were already stiff with an average compressive modulus of 21.8 ± 2.7 kPa and displayed a large hysteresis even at day 1. All 5% hydrogels gradually stiffened over 4 weeks with an increase in hysteresis (Fig.5A, Fig.S19), and this stiffening was quicker in SF with 30% or 50% SF-TA (Fig. 5B). At day 7, elastic moduli of SF only and with 10% SF-TA hydrogels were around 4 kPa, while those with 30%, 50% or 100% SF-TA had moduli around 45, 60 and 80 kPa, respectively. At day 14, average moduli of SF only and all SF/SF-TA hydrogels were above 45 kPa and reached 60-100 kPa at day 28, while that of SF-TA only hydrogels was above 300 kPa, which could not be compressed more than 20% strain due to exceeding the detection limit of the load cell. Despite having the highest stiffness, stress-strain curves of 5% SF-TA hydrogels did not display mechanical failure over 4 weeks while 5% SF only or all SF/SF-TA hydrogels did from day 7 to day 28. Moduli of SF with 10% or 25% G-TA were maintained at around 3 kPa at day 14 and increased above 65 kPa at day 21. Among the 3% hydrogels, G-TA only (1.39 kPa) and SF-TA only (0.76 kPa) hydrogels had the highest average moduli at day 1, which were followed by SF with 30% SF-TA (0.59 kPa) or 25% G-TA (0.53 kPa) and SF only (0.34 kPa) hydrogels (Fig.5D). Compressive moduli of SF/30% SF-TA and SF-TA only increased to ~11 and ~26 kPa, respectively, at day 7 and to ~90 kPa at day 28, while it was 3.8 kPa for SF only hydrogels at day 7 and gradually increased to 39 kPa at day 28. After a slight increase from day 1 (0.53 kPa) to day 7 (1.47 kPa), compressive modulus of SF/25% G-TA hydrogels did not change significantly over 4 weeks and was measured as 1.94 kPa at day 28, where hydrogels were still able to recover. No significant change was observed in the compressive properties of G-TA only hydrogels over 28 days of incubation in PBS. At day 1, all 5% and 3% hydrogels except 5% SF-TA only gels were able to withstand 80% compressive strain and displayed elastic behavior to varying degrees (Fig.S20). 5% SF-TA only hydrogels could not be compressed above 70% strain due to the detection limit of the load cell.
Figure 5.
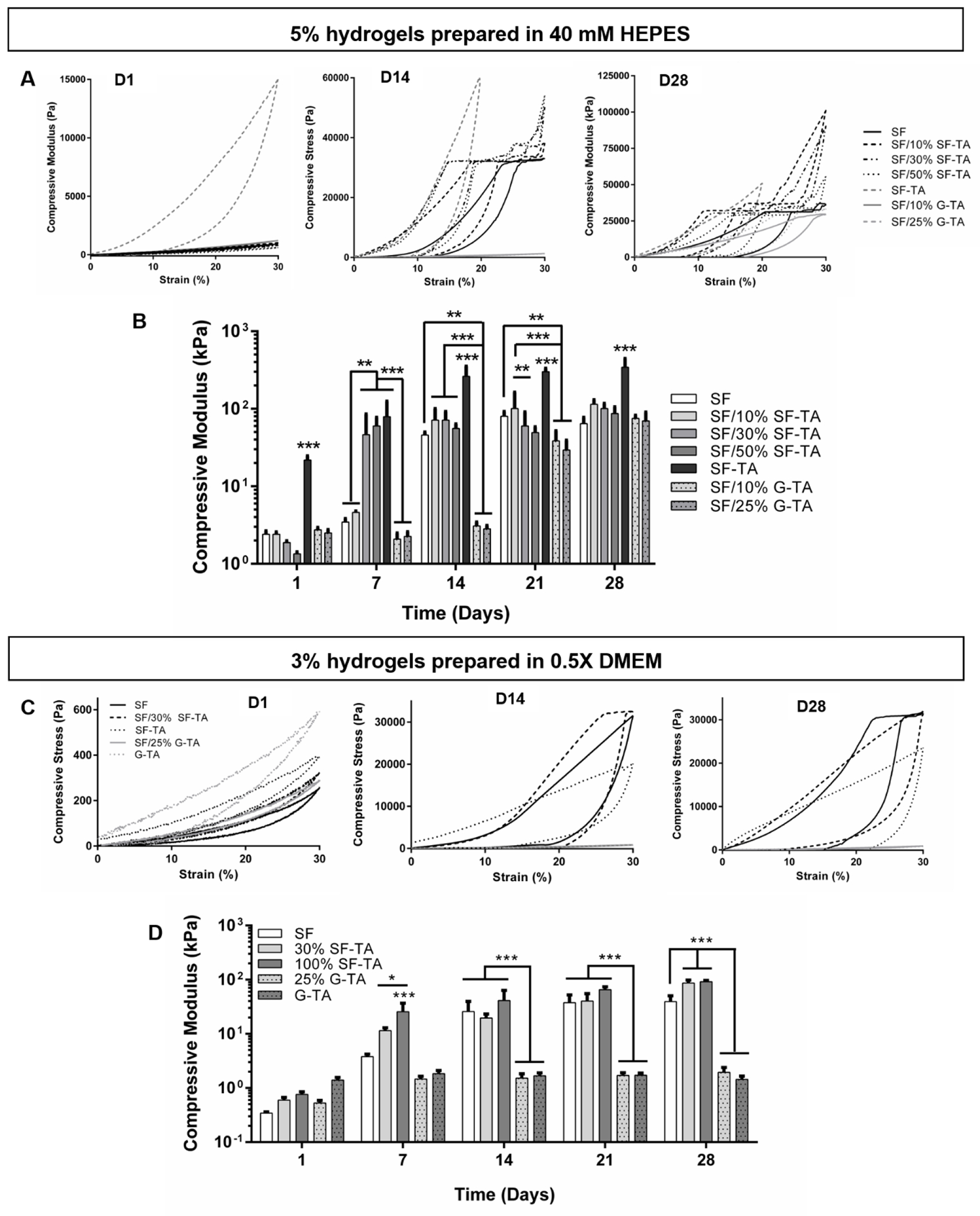
Unconfined compression tests of SF/SF-TA and SF/G-TA composite hydrogels. Representative stress-strain curves of (A) 5% hydrogels prepared in 40 mM HEPES and (C) 3% hydrogels prepared in 0.5X DMEM. Influence of SF-TA and G-TA on compressive moduli of (B) 5% and (D) 3% silk hydrogels over 4 weeks of incubation in PBS at 37 °C. (n = 5, compressive moduli are on log scale, *p < 0.05, **p < 0.01 and ***p < 0.001).
3.7. Influence of SF-TA and G-TA content on proteolytic degradation
The influence of SF-TA and G-TA content on enzymatic degradation of composite hydrogels was determined by monitoring the percent initial mass over 8 days of incubation in protease XIV solution (Fig.6). Increasing SF-TA or G-TA content was found to delay the degradation of 5% hydrogels crosslinked in HEPES buffer. Less than 5% of the initial mass of SF only hydrogels remained at day 4 while those of SF/50% SF-TA and SF/25% G-TA were around 60% and 50% their original weights at day 4, and 30% and 15% at day 8, respectively (Fig.6A). SF-TA only hydrogels degraded significantly slower and maintained ~65% of its initial weight at day 8. A similar trend was also recorded for the 3% hydrogels crosslinked in 0.5X DMEM buffer, but their degradation was slower compared to 5% hydrogels (Fig.6B). Less than 40% and 5% of the SF only hydrogels were left at day 4 and 8, respectively, while SF/30% SF-TA and SF/25% G-TA were approximately 60% and 30% of their initial weights at days 4 and 8, respectively. SF-TA only hydrogels, on the other hand, were ~48% of their original weight at day 8, while G-TA only hydrogels displayed the fastest degradation and completely dissolved away by day 6.
Figure 6.
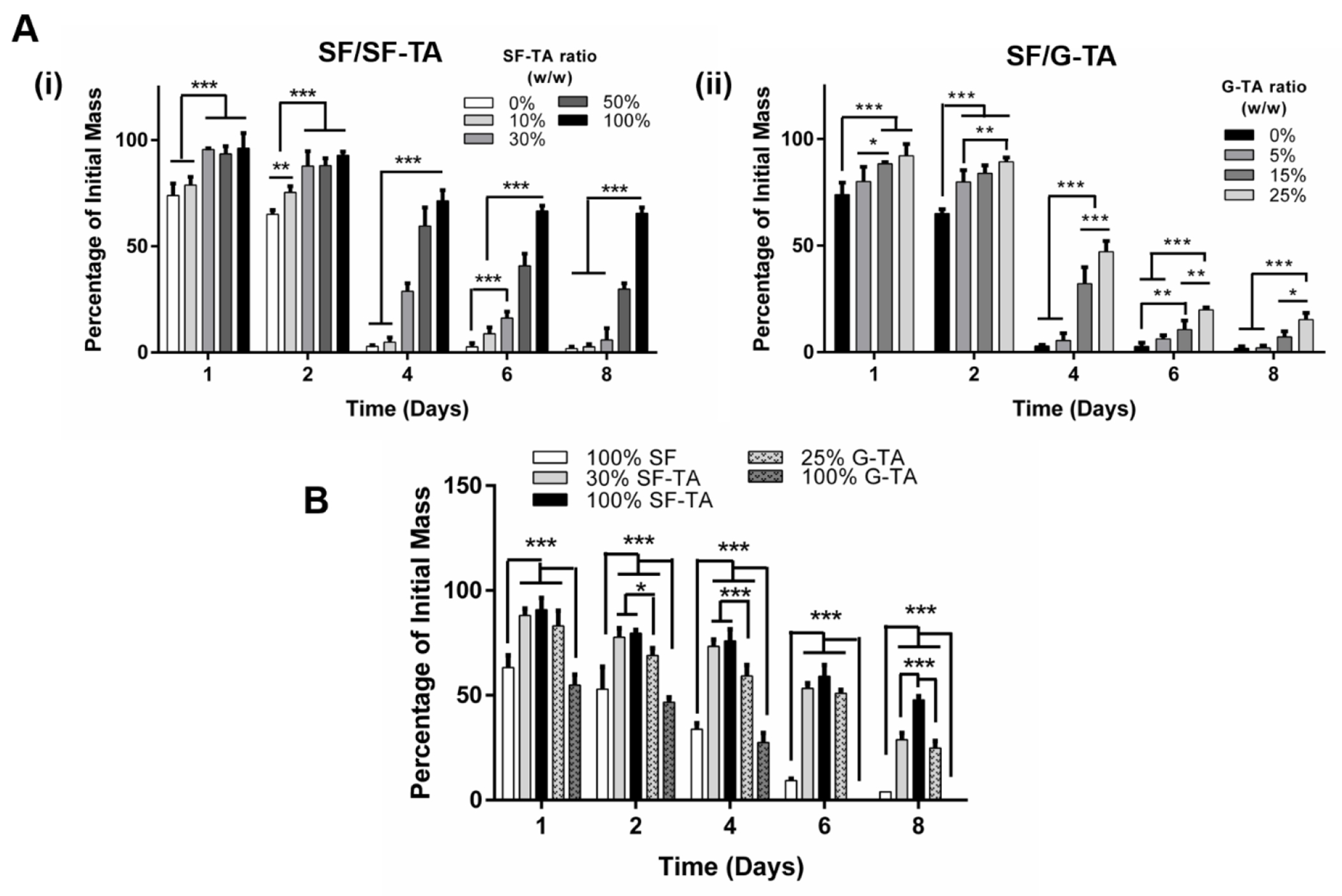
In vitro degradation of hydrogels over 8 days of incubation in PBS with 0.001 U/mL protease XIV. (A) Degradation of 5% silk hydrogels prepared in 40 mM HEPES buffer and supplemented with varying weight ratios of (i) SF-TA or (ii) G-TA. (B) Degradation of 3% silk only, with 30% SF-TA or 25% G-TA hydrogels prepared in 0.5X DMEM (n = 4, *p < 0.05, **p < 0.01 and ***p < 0.001).
3.8. Effects of cyclo(RGDyK) and G-TA on response of hMSCs cultured on hydrogel surfaces
To determine the effects of cyclo(RGDyK) (0.1-1 mM) and G-TA (5-25%) on cell behavior, the area of hMSC spreading at day 1 and metabolic activity over 4 weeks of culture were analyzed. Cells were seeded onto hydrogels in FBS free medium immediately after crosslinking to evaluate the cell-surface interactions without adsorption of any serum proteins onto the surfaces as they guide cell adhesion and fate of adherent cells [103]. Many cells on no RGD hydrogels (controls) displayed a round morphology at day 1 while those on the hydrogels supplemented with cyclic RGD or G-TA flattened and spread well (Fig.7A). The highest and lowest cell spreading were observed on G-TA only and SF only hydrogels, respectively, and the area of cell spreading increased and reached a plateau with increasing cyclo(RGDyK) or G-TA concentrations in composite hydrogels (Fig.7B). The maximum spread area on SF/G-TA hydrogels was much higher than that on SF/SF-TA with cyclic RGD. At day 14, cells on all hydrogels spread well and reached confluency at day 28 except the ones on G-TA only gels (Fig.S21). Metabolic activity at day 1 decreased (Fig.S22A) but the fold change over 4 weeks increased significantly with increasing cyclo(RGDyK) concentration. A slight decrease in metabolic activity was also recorded with increasing G-TA ratio at day 1 (Fig.S22B), but the fold change over 4 weeks increased significantly even though a plateau was reached after 15% G-TA (Fig.7C). The fold change in metabolic activity on G-TA only gels was significantly higher than the other groups during the first two weeks but leveled with others at day 14. At day 28, the highest average fold change was observed on G-TA only hydrogels (4.88 ± 0.96-fold) followed by SF/20% G-TA (4.63 ± 0.13-fold), which were higher than that recorded on SF/SF-TA with 1 mM cyclo(RGyK) (3.92 ± 0.09-fold).
Figure 7.
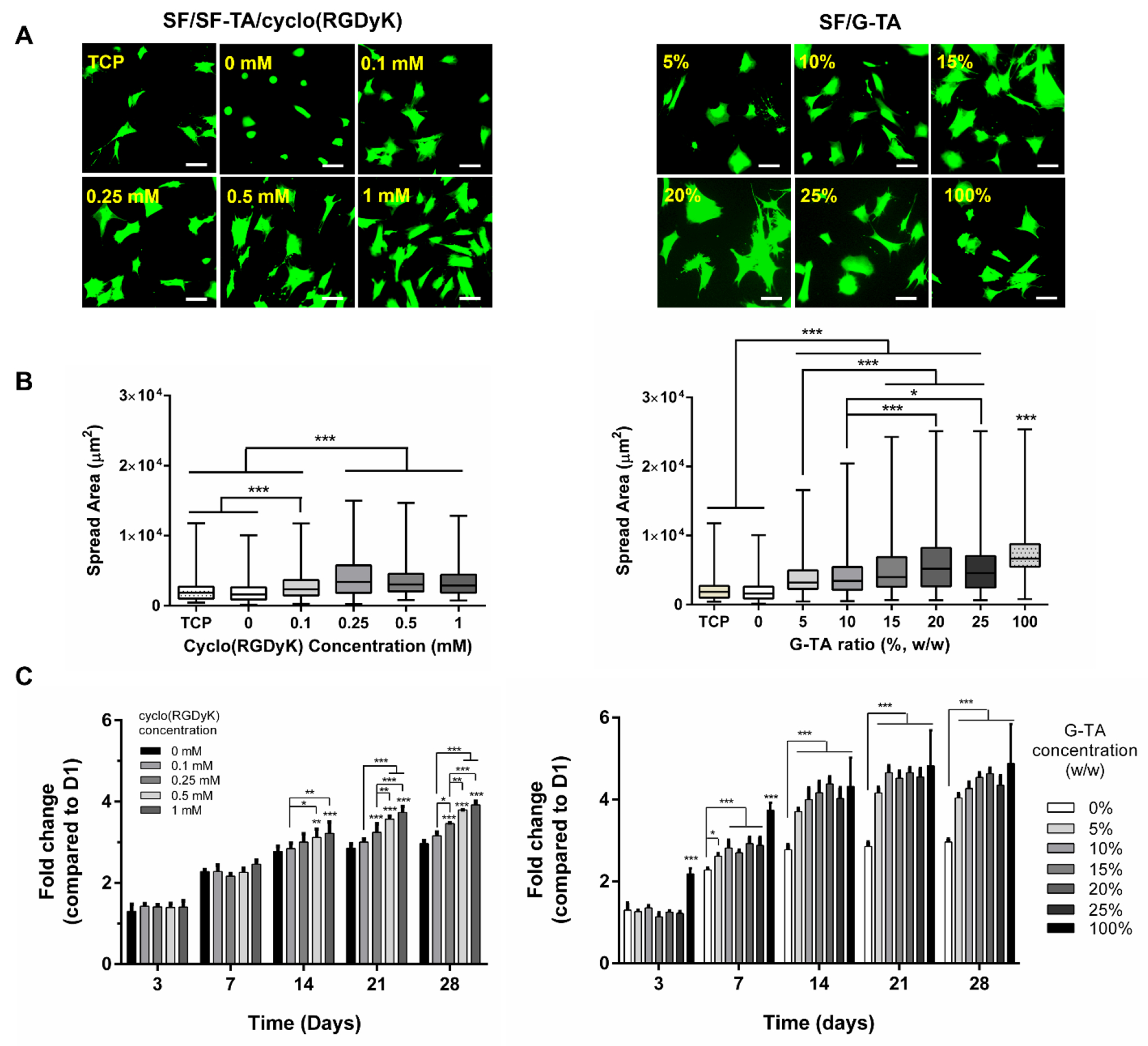
hMSCs behavior on 5% silk hydrogels supplemented with cyclo(RGDyK) or G-TA. (A) Fluorescence images of hMSCs 24 h after seeding on hydrogel surfaces (green: calcein, scale bars: 100 μm). (B) Box-whisker distribution graphs showing the effects of cyclo(RGDyK) or G-TA content on spread area of hMSCs on silk hydrogels at day 1 (spread areas of individual cells were quantified on fluorescent micrographs using ImageJ software, n ≥ 200). (C) Fold changes in metabolic activity of hMSCs on 5% silk hydrogels supplemented with cyclo(RGDyK) or G-TA over 4 weeks of culture compared to day 1. (n = 4, *p < 0.05, **p < 0.01 and ***p < 0.001, asterisks above the bars represent significance compared to no cycloRGD control).
3.9. In vitro response of encapsulated hMSCs
hMSCs were encapsulated in SF/SF-TA with or without cyclo(RGDyK) and SF/G-TA composite hydrogels and their viability and morphologies were monitored over 4 weeks. First, cells were encapsulated in 5% hydrogels crosslinked in HEPES buffer to determine whether accelerated gelation with increasing SF-TA ratio improved initial viability. Indeed, fewer dead cells were observed in the hydrogels (Fig.8A–i) and % dye reduction at day 1 increased significantly with increasing SF-TA concentration (Fig.8A–ii). At day 7, however, metabolic activity decreased in all groups compared to day 1 and the fold change in metabolic activity for SF/30%SF-TA was 0.18 at day 28 compared to day 1 (Fig.8A–iii). The same decrease over time was also observed with the SF/25% G-TA hydrogels, but the fold change over 28 days (0.41-fold) was higher than those in SF only (0.21-fold) SF/30% SF-TA hydrogels. When hydrogels were crosslinked in 0.5X DMEM instead of 40 mM HEPES (Fig.8A–iv), the fold change was still below 1 in SF only (0.64-fold) and even lower in SF/30% SF-TA hydrogels (0.41), while a 1.4-fold increase was recorded in SF /25% G-TA hydrogels at day 28. 0.5X DMEM was used for preparation of hydrogel precursor solutions for the rest of the study.
Figure 8.
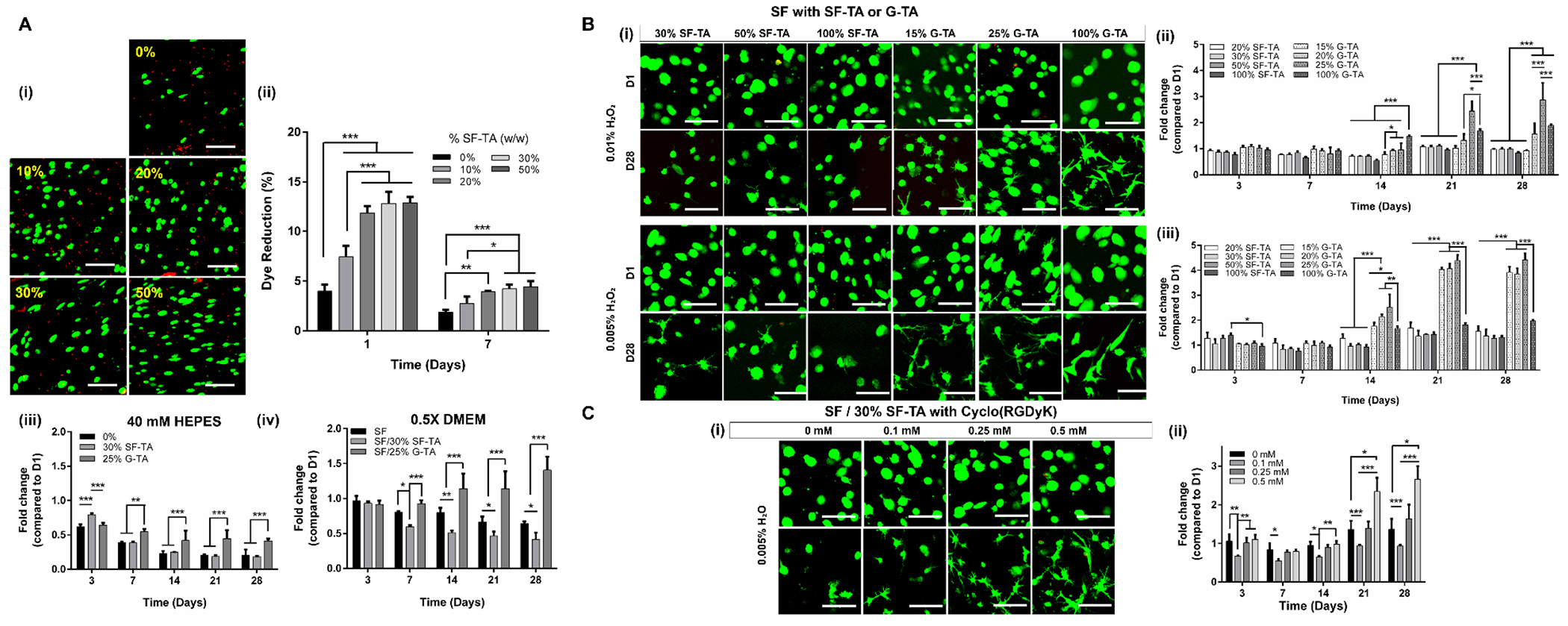
Viability of hMSCs encapsulated in silk hydrogels. (A) (i) Day 1 fluorescent micrographs of hMSCs encapsulated in 5% silk hydrogels (prepared in 40 mM HEPES) with different SF-TA weight ratios (green: live (calcein), red: dead (EthD-1), scale bars: 100 μm). (ii) % dye reduction by the cells encapsulated in 5% SF/SF-TA composite hydrogels. Fold changes in metabolic activity of hMSCs encapsulated in 5% SF, SF/SF-TA or SF/G-TA hydrogels prepared in (iii) HEPES or (iv) 0.5X DMEM buffers. (B) (i) Day 1 and 28 fluorescent micrographs of hMSCs encapsulated in 3% SF/SF-TA or SF/G-TA composite hydrogels prepared in 0.5X DMEM buffer using 0.01% or 0.005% H2O2 (scale bars: 100 μm). Fold changes in metabolic activities of hMSCs compared to day 1 in composite hydrogels prepared using (ii) 0.01% or (iii) 0.005% H2O2. (C) (i) Day 1 and 28 fluorescent micrographs and (ii) fold changes in metabolic activities of hMSCs encapsulated in 3% SF/30%SF-TA hydrogels supplemented with various concentrations of cyclo(RGDyK) and crosslinked using 0.005% H2O2. (n = 4 for metabolic activity assays, *p < 0.05, **p < 0.01 and ***p < 0.001).
To determine whether lowering protein and/or H2O2 concentration improves long term cell response, morphologies and metabolic activity of hMSCs encapsulated in 3% composite hydrogels crosslinked with 0.01 wt.% or 0.005 wt.% H2O2 were monitored over 28 days. The SF only group was omitted since the cells settled to the bottom of the plate and proliferated there due to the slow gelation. The majority of the cells encapsulated in the SF/SF-TA hydrogels crosslinked using 0.01% H2O2 remained spherical over 4 weeks, while some cells in SF/G-TA hydrogels and almost all cells in G-TA only gels had cytoplasmic extensions branching from their bodies at day 28 (Fig.8B–i, Fig.S23). No significant change was found in metabolic activities in any SF/SF-TA or SF/15% G-TA hydrogels over 4 weeks, while there was a statistically insignificant 1.5 fold increase in SF/20% G-TA hydrogels and statistically significant 1.9-fold and 2.9-fold increase in G-TA only and SF/25% G-TA hydrogels, respectively (Fig.8B–ii). When the H2O2 concentration was lowered to 0.005 wt.%, however, more cells exhibited elongated or branched morphologies, particularly in SF/G-TA hydrogels. An increase in % dye reduction up to 4-fold compared to day 1 was recorded in SF hydrogels with 15-25% G-TA, while the increase in G-TA only gels was still around 2-fold. Again, no significant change was observed in SF/SF-TA hydrogels (Fig.8B–iii).
Cell response to SF/SF-TA hydrogels with cyclic RGD peptides was also determined by monitoring cell morphology and metabolic activity over 28 days. At both 0.01 and 0.005 wt.%, more and more cells displayed cytoplasmic extensions with an increase in cyclo(RGDyK) concentration (Fig.8C–i, Fig.S24A). No significant change was found in metabolic activity over 28 days in any group at 0.01 wt.% H2O2 (Fig.S24B), while 1.6 and 2.7-fold increases were recorded in the hydrogels supplemented with 0.25 and 0.5 mM cyclo(RGDyK), respectively at 0.005 wt. % H2O2 (Fig.8C–ii).
3.10. In vivo response to SF-TA only and SF/G-TA composite hydrogels
Preformed hydrogel discs of 3% w/v SF-TA only and SF/25% G-TA were implanted subcutaneously in mice and no adverse effects from the implants were observed over 3 days. The samples were extracted at 3 days post-implantation (Fig.9A, B) and examined histologically. H&E staining showed formation of a thin inflammatory capsule surrounding the gels (pointed with black arrows) indicating a typical foreign body response without excessive inflammation or cell infiltration into the bulk of the gels (Fig.9C, Fig.S25). SF/25% G-TA discs mostly remained intact (Fig.9C–i) while SF-TA only hydrogel discs broke up into several pieces (Fig.9C–ii).
Figure 9.
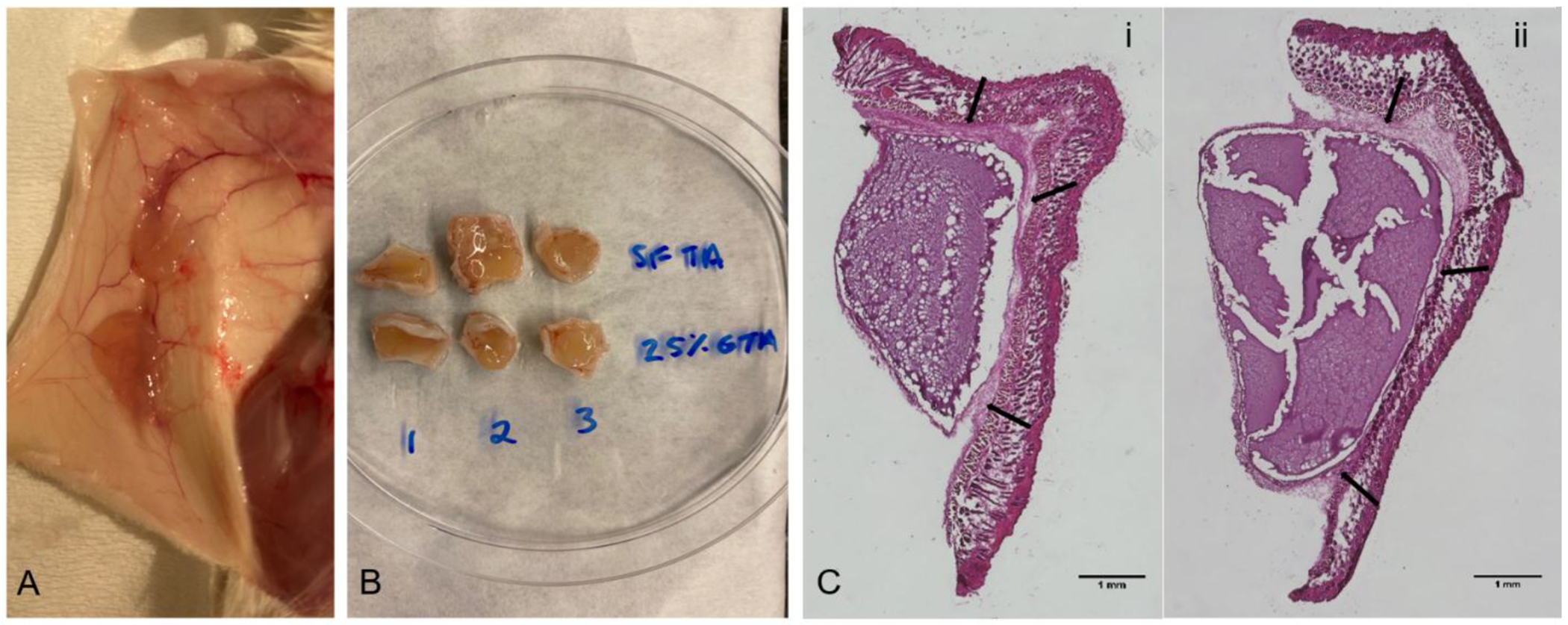
In vivo animal tests. Day 3 photographs of subcutaneously implanted 3% SF-TA only and SF/25% G-TA composite hydrogels crosslinked in 0.5X DMEM using 0.01 wt% H2O2 and 10 U/mL HRP (A) before and (B) after removal from mice. (C) Hematoxylin and eosin stained slice of cutaneous tissues containing (i) SF/25% G-TA and (ii) SF-TA only hydrogel discs. Scale bars: 1 mm.
4. Discussion
Enzymatically crosslinked SF/SF-TA and SF/G-TA hydrogels were characterized for gelation kinetics, mechanical properties, secondary structure and enzymatic degradation. In addition, the influence of cyclic RGD or G-TA on the bioactivity of composite hydrogels were explored for the culture and encapsulation of hMSCs. In the presence of HRP and H2O2, aqueous solutions of SF formed highly elastic and biodegradable hydrogels through oxidation of tyrosine residues into dityrosine crosslinks; complete gelation took 10 to more than 30 min in distilled water depending on the average molecular weight of the regenerated SF chains [21, 23]. Even though cell encapsulation in distilled water was cytocompatible when the cells were added shortly before gelation was complete [21], this approach is impractical for 3D bioprinting or microencapsulation, where cells may need to be incubated in the pre-hydrogel solution for extended periods of time, thus, the ionic strength of the solution must be controlled carefully [104, 105]. Crosslinking of SF in physiological buffers, however, takes much longer for gelation start and completion, and results in mechanically weaker hydrogels likely because of the high ionic strength. Solutions of the salts of Na+ and K+ have been shown to induce the self-assembly of the hydrophobic domains in SF sequence into β-sheets through salting out effect [25, 27], which has been utilized before for the fabrication of physically crosslinked silk microparticles [106] or aqueous-derived silk scaffolds [26] using high concentration salt solutions. In the case of enzymatic crosslinking, however, physiological buffers with relatively high ionic strength could be expected to reduce the accessibility of tyrosine residues available in these hydrophobic domains, impairing the gelation speed as well as the overall crosslinking density. Slow gelation is an issue for tissue fillers, bioinks for 3D bioprinting, and cell encapsulating microgels, where rapidly gelling hydrogel materials with high structural integrity are desired [14, 104, 107]. One of the most commonly used rapidly gelling hydrogel system in these applications has been the ionically crosslinked polysaccharide alginate [108–110], which have very limited stability at physiological conditions due to the ongoing ion exchange mechanisms and their structural and mechanical integrities have been shown to deteriorate very quickly [111, 112]. Slow degradation and long-term structural stability [113] of silk hold promise as an alternative hydrogel system and therefore the gelation kinetics of enzymatically crosslinked silk hydrogels in physiological buffers should be improved for widespread biomedical applications.
Our first approach to improve gelation kinetics was to conjugate tyramine residues onto SF to increase the total phenol content. Moreover, we hypothesized that tyramine conjugation on aspartic and glutamic acid residues that are located in the amorphous regions of SF [114] would provide more accessible phenol groups for crosslinking at high ionic strength. Despite the low theoretical increase in the number of phenol groups upon 100% conversion of all carboxylic acid residues (1.1 mol% [115]), even 20 wt. % SF-TA significantly decreased the time required for the initiation of gelation, from 5-10 min to tens of seconds. Gelation kinetics of aqueous silk solutions could also be modulated by changing the concentrations of H2O2 and/or HRP. Besides potential cytotoxicity [39], high concentrations of H2O2 could also impair gelation as it was shown that increasing it above 2.4 mM (~0.007 wt%) gradually decreased crosslinking density of 2.5% w/v SF hydrogels, likely because of the inhibition of the enzymatic activity of HRP [37]. Using high concentrations of HRP, on the other hand, may raise concerns of immune response since HRP is a plant-derived enzyme and both its protein core and N-glycans were shown to induce immune reaction in rodents [36]. Moreover, we showed that tyramine-substitution enables gelation speeds at a moderate concentration (10 U/mL) of HRP that cannot be achieved with unmodified silk using a high (100 U/mL) concentration of HRP. An important advantage of SF-TA over other tyramine-substituted polymers is the control over gelation kinetics through modulation of SF and SF-TA ratios. This control is not possible with other polymers such as gelatin, HA or PEG, which have few or no phenol residues, therefore unmodified polymers cannot be crosslinked with tyramine-modified chains. SF, on the other hand, is already rich in phenol residues because of its high tyrosine content, which enables crosslinking of unmodified SF with SF-TA at any weight ratio to modulate gelation kinetics.
As an alternative strategy, we crosslinked SF with tyramine-substituted gelatin (G-TA) to improve both bioactivity and gelation kinetics of SF hydrogels. Dityrosine crosslinking of SF with gelatin was demonstrated before through tyrosinase treatment, but complete gelation took more than 40 min [91]. Here we also found that unmodified gelatin increases the gelation time and deteriorates shear mechanical properties of enzymatically crosslinked silk hydrogels due to relatively low tyrosine content (0.26 mol%) of porcine skin gelatin [116] compared to SF. After chemical modification, G-TA had a significantly higher tyramine content than SF-TA because of higher aspartic and glutamic acid content of porcine skin gelatin (12.4 mol% [116]) available for carbodiimide coupling compared to SF (1.1 mol% [115]). Since both SF and gelatin have free amine and carboxylic acid residues in their primary structure, crosslinking of polymer chains during carbodiimide coupling of tyramine groups could be an issue. To prevent this, tyramine was used at molar excess of aspartic and glutamic acid residues on SF (~15X) and gelatin (~3X), and it was added to protein solutions before EDC and NHS to ensure carboxylic acids would already be saturated by amine groups of the tyramine residues upon activation by EDC and NHS. Still, gel electrophoresis showed that although mean molecular weight did not significantly changed for neither groups, molecular weight with highest frequency increased significantly for gelatin, suggesting an increase in chain length likely because of crosslinking. Interestingly, molecular weight with highest frequency in the distribution of SF-TA chains was remarkably lower compared to unmodified silk. This could be explained with precipitation and removal of the crosslinked large chains due to low solubility of SF. G-TA increased the gelation speed of silk hydrogels to a higher degree than SF-TA at the same weight ratio, as expected. A similar increase in gelation rate was also reported before for SF crosslinked with HA-TA, but high viscosity and hydrophilicity of HA were stated as issues for effective mixing of the two polymers at high HA-TA ratios [101]. Unlike HA-TA, G-TA had a low viscosity at 37°C and was completely miscible with SF-TA at any ratio. At room temperature, on the other hand, solutions with final G-TA solutions above 1% w/v undergoes thermal gelation similar to unmodified gelatin. Our results indicate that oxidative crosslinking of silk with G-TA through tyrosine-tyramine bridges is more advantageous over other strategies such as genipin-mediated crosslinking of primary amine residues, completion of which was found to increase up to 48 h with increasing SF ratio due to low basic amino acid content of SF [90]. Oxidative crosslinking of SF with SF-TA or G-TA introduced in this study is also compatible with enzyme-free fabrication strategies such as using syringes packed with HRP-immobilized beads [117], riboflavin-mediated photocrosslinking [118] or Fe(III) ion-mediated Fenton reaction [119].
Besides improved gelation kinetics, one of the most significant effects of the SF-TA and G-TA content on SF hydrogels was the improved shear mechanical properties. Both tyramine-substituted polymers decreased the time required for gels to reach mechanical stability and increased their storage modulus up to a point, suggesting an increase in crosslinking density [101, 120]. This was confirmed by LC-MS/MS analysis revealing the presence tyrosine-tyramine and dityramine crosslinks in addition to dityrosine bonds. Interestingly, the increase in storage modulus was not linearly correlated with polymer-TA concentration, as it reached a plateau at ~20 wt. % SF-TA or G-TA and then sharply dropped at higher ratios. This is likely because of very rapid local gelation that prevents homogenous diffusion of H2O2 and limits chain mobility required for collision of phenolic radicals to form new crosslinks. Decreasing H2O2 concentration by half decreased crosslinking density and resulted in higher gelation times and lower storage moduli. The same trend was reported before for tyramine-substituted polymers such as PGA-TA [43] or HA-TA [121] as well as SF [101], which was explained with a decrease in H2O2/phenol mol ratio and the number of phenolic radicals available for crosslinking. SF-TA and particularly G-TA significantly improved shear mechanical properties at low H2O2 concentration as well, indicating that improved gelation was independent of H2O2 concentration. Another parameter that influenced the shear mechanical properties of SF/SF-TA hydrogels was the cyclic RGD content, which gradually increased gelation time and decreased storage moduli at increasing concentrations. This was an expected outcome since some of the phenolic radicals generated on the polymer chains are occupied by the tyrosine residues of the peptides, resulting in a decrease in crosslinking density. The same observation was also made by others for in situ conjugation of peptides into hydrogels of acrylate-crosslinked HA [74] and dithiol crosslinked PGA [71] as well as dityramine crosslinked tetronic-tyramine [77], gelatin-PEG-tyramine [122] and HA-tyramine hydrogels [78, 79]. In the SF/SF-TA system, the decrease in crosslinking density with increasing peptide concentration can be compensated by increasing the weight ratio of SF-TA to obtain desired gelation kinetics and mechanical properties.
Another notable effect of the polymer-tyramine content was the β-sheet content and gradual stiffening of SF hydrogels incubated in PBS. Stiffness of hydrogel matrices is an important parameter as it is sensed by the cells through mechanotransduction, which alters gene expression and influences morphology, proliferation and lineage commitment [123–125]. Modulation of the mechanical properties of enzymatically crosslinked silk hydrogels has been demonstrated before through changing the average molecular weight (boiling time), protein concentration [21], or the amount of H2O2 used for crosslinking [37]. Here we report modulation of secondary structure and stiffness of SF hydrogels by altering SF-TA or G-TA content without changing any other parameter. Initially, all 3% and 5% hydrogels except 5% SF-TA only were very elastic at day 1 with maximum compressive moduli of 0.6 and 3 kPa, respectively, and could withstand 80% of compressive strain without failure. SF-TA ratio in SF/SF-TA composites was negatively correlated with the β-sheet content and compressive moduli of 5% hydrogels at day 1, likely because of increased crosslinking density that was proposed to restrict movement of SF chains in the network required for the formation of β-sheet domains [126]. Both SF only and SF/SF-TA composite hydrogels shrank and stiffened over 4 weeks with an increase in compressive modulus up to 100 kPa as a result of a gradual increase in β-sheet ratio. Stiffening of enzymatically crosslinked SF hydrogels has been reported before as a result of self-assembly of hydrophobic silk domains into crystalline β-sheet structures [101, 127, 128], which can be explained with SF chains in the gel network being still partly movable and capable of rearranging into crystalline domains [126]. Despite initially providing lower β-sheet content and stiffness, increasing SF-TA content accelerated β-sheet formation and stiffening compared to SF only control likely because of an increase in hydrophobicity of SF chains upon conversion of the charged carboxylic acid residues into hydrophobic phenol groups. 5% SF-TA only hydrogels, for instance, were found to stiffen rapidly even in a single day. Higher hydrophobicity might have favored water removal and formation of β-sheets through a similar mechanism with silk self-assembly induced by protonation of carboxylic acid residues at acidic pH [129, 130]. Moreover, the increase in the number of phenol residues on SF-TA chains might have enhanced π–π and π–OH group interactions, which were proposed to provide templating effects and foster silk self-assembly [131]. Unlike SF-TA, G-TA significantly delayed crystallization and stiffening of silk hydrogels. Hindered self-assembly of silk in physically crosslinked composite hydrogels has been reported before as a result of the topological constraints caused by entanglements with gelatin molecules [83, 86, 87]. Moreover, higher hydrophilicity of gelatin because of its significantly larger content of charged amino acids (26.2 mol%) [116] compared to SF (1.7 mol%) [115] might have provided higher water retention, which was shown to delay β-sheet formation in SF crosslinked with HA-TA that has a large negative charge [101]. Our findings indicate that a wide range of hydrogel stiffness with compressive moduli ranging between 1-100 kPa could be achieved simply by altering SF-TA or G-TA content. These hydrogels can be utilized as scaffold materials that favor stem cell differentiation towards a wide range of lineages including neurogenic (0.1-1 kPa), adipogenic (1-5 kPa), myogenic (10-20 kPa), and osteogenic (>25 kPa) cells [123, 132].
SF-TA and G-TA content modulated the enzymatic degradation of the composite hydrogels. Spatial and temporal control of biodegradation is an important parameter in terms of tissue regeneration and potential release of biochemical agents or encapsulated cells. Tissue engineering scaffolds should be able to guide cell growth and allow new ECM deposition while gradually degrading in the body until the original tissue architecture is restored and delivery vehicles should sustain desired release kinetics [133]. High crystalline content of silk-based hydrogels has been shown to provide slower degradation compared to other common biopolymers such as collagen/gelatin, elastin or HA due to its high crystalline content 102]. To determine the degradation behavior of composite hydrogels we incubated them in a solution of protease XIV, an enzyme that was shown to favor the degradation of non-β-sheet regions in silk hydrogels [134], and found that both SF-TA and G-TA reduced degradation rates, likely because of higher crosslinking densities of the composite hydrogels than SF only control. Reduced enzymatic degradation rate with increased crosslinking density was reported before for enzymatically crosslinked silk hydrogels [135] as well as many other covalently crosslinked polymer networks including gelatin [40], HA [35], and PEG [136]. This can be explained with a decrease in average mesh size with increased crosslinking density [137], leading to reduced enzyme diffusivity into the gel. Interestingly, 3% hydrogels degraded more slowly compared to their 5% counterparts, which might seem to contradict the faster enzymatic degradation of physically crosslinked silk hydrogels at lower protein concentrations [33]. However, crosslinking density of enzymatically crosslinked silk hydrogels decreases with increasing protein concentration above 1.25% (w/v) as a result of lower chain mobility and limited collisions of phenolic radicals. Diffusivity of enzymatically crosslinked silk hydrogels was therefore found to increase above 1.25% silk [37]. Despite having the lowest mesh size, 3% G-TA only hydrogels completely degraded in 6 days, while SF-TA only hydrogels had a larger calculated mesh size but degraded the slowest. This observation can be explained with both the lack of protease sensitive sequences in SF primary structure and its high crystalline content that limits water retention and diffusivity. It should also be noted that the mesh sizes were calculated for day 0 samples based on rubber elasticity theory, which assumes no physical interactions between the polymer chains [102]. Therefore, it would not be accurate to apply it to silk hydrogels at later time points due to physical crosslinking through β-sheet formation.
To test the cell-matrix interactions of hydrogels supplemented with cyclic RGD peptides or with G-TA, hMSCs were first cultured on the hydrogel surfaces and their viability, spreading and metabolic activity were analyzed. RGD peptides regulate integrin mediated focal adhesion formation followed by cell adhesion, spreading and organization of actin cytoskeleton, which further modulate survival, proliferation, and differentiation via mechanotransduction [64]. We preferred a cyclic RGD peptide rather than a linear peptide because cyclic RGD peptides were found to improve hydrogel-matrix interactions to a higher degree compared to linear counterparts [67, 138] as they bind selectively to αvβ3 and αvβ5 integrins with higher affinity and display lower susceptibility to enzymatic degradation [139]. Even though tethering of linear RGD peptides improved cell-matrix interactions on silk films [63, 140, 141], fibers [142], and nanofibrous mats [143], conjugation of linear or cyclic RGD peptides into physically or enzymatically crosslinked silk hydrogels has not been reported before. The only example to our knowledge is the ionically crosslinked silk-alginate hydrogels supplemented with cyclic RGD peptides, which was shown to provide better adhesion of mouse embryonic stem cells compared to a no RGD control [144]. Here we showed that cyclic RGD peptides significantly improved initial cell spreading and the metabolic activity over 4 weeks on SF/SF-TA hydrogels. This is parallel with earlier studies reporting improved adhesion of murine osteoblasts on elastin-mimetic polypeptide hydrogels [138], better adhesion, spreading, and proliferation of endothelial cells on PEG diacrylate (PEGDA) hydrogels [67], and higher adhesion and proliferation of murine fibroblasts on PEGDA [70] and PGA [71] hydrogels after covalent conjugation of cyclic RGD peptides. Similarly, HRP-mediated conjugation of cyclo(RGDyK) on surface deposited resilin-like peptides was shown to improve fibroblast attachment significantly through the binding of integrin αV chains [145]. Although we observed no significant cytotoxicity with live/dead staining, initial metabolic activity decreased gradually with increasing cyclic RGD concentration, which might be due to the unconjugated peptides that diffused out and reduced cell attachment by blocking the surface integrins [69]. Crosslinking SF with G-TA improved cell spreading and metabolic activity of hMSCs on hydrogels to a higher extent than cyclic RGD peptides. Improved cell-hydrogel interactions upon blending silk with gelatin was also reported by others before. For example, Das et al. demonstrated that goat chondrocytes displayed prominent adhesion with many filopodial extensions and reach confluency earlier on physically crosslinked SF-gelatin hydrogels compared to SF only controls [75]. Similarly, attachment, spreading, and proliferation of human MG63 osteosarcoma cells increased with increasing gelatin content in silk-gelatin blend hydrogels [87]. Increasing the concentration of tyramine-substituted gelatin also improved adhesion, spreading, and proliferation of human aortic endothelial cells and murine embryo fibroblast on gelatin-alginate composite hydrogels [98]. The increase in cell spreading and metabolic activity recorded on SF/G-TA hydrogels, however, was not linearly correlated with G-TA concentration; it reached a plateau and then started declining after 15% G-TA, probably due to the increase in modulus, which was reported to reduce spread area and proliferation of hMSCs on hydrogel surfaces [124].
Encapsulating cells in hydrogel scaffolds has been an attractive approach for tissue engineering applications by providing a biodegradable and highly hydrated tissue-like environment for the growth of cells and tissues. Since the cells to be encapsulated are present in the pre-gel solution during the crosslinking process, pre-gel formulations and crosslinking chemistries should be cytocompatible [6]. Here we initially used 40 mM HEPES solution, a non-toxic tissue culture buffer [146], to maintain a constant physiological pH of SF solution without significantly increasing the ionic strength. SF solution in HEPES buffer, however, turned out to be hypotonic and could not maintain high viability of suspended or encapsulated cells. Interestingly, increasing SF-TA content significantly improved initial viability of hMSCs encapsulated in 5% hydrogels, likely because of faster gelation that lowered the duration of exposure to osmotic stress. However, cell viability gradually decreased in SF only or composite hydrogels from day 1 to 7, suggesting a long-term damage caused by initial osmotic stress. When the buffer was replaced with 0.5X DMEM to provide better physiological conditions [21, 24], there was still a decrease in metabolic activity in SF only and particularly in SF/SF-TA composite hydrogels as opposed to a slight increase observed in SF/G-TA hydrogels. This trend suggests that β-sheet formation and stiffening, which were enhanced by SF-TA but delayed by G-TA, impaired growth of encapsulated hMSCs. This was not unexpected as it was reported before that β-sheet transition in enzymatically crosslinked SF hydrogels led to an increase in hydrogel stiffness and induced apoptosis of encapsulated cancer cells [124] or limited the growth of encapsulated cardiac fibroblasts [117]. Decreasing protein concentration to 3% improved metabolic activity of encapsulated cells, likely because of a decrease in hydrogel stiffness [21] as well as an increase in hydrogel porosity [127], which was shown to enhance cell proliferation in hydrogel matrices [147]. The most remarkable improvement in the metabolic activity of encapsulated cells, however, was obtained when H2O2 concentration was reduced by half. It is known that H2O2 may induce apoptotic cell death even at low (0.1-10 mM) concentrations [39], but its cytotoxicity was shown to be minimized if rapidly consumed by the HRP mediated oxidative crosslinking reaction [36]. The concentration of residual H2O2 in hydrogels crosslinked using 0.01 wt% H2O2 could not be detected using TMB assay, suggesting that it was rapidly consumed by HRP and was lower than 3.2 μM, which was the detection limit we recorded. Indeed, cytotoxicity of the initial H2O2 concentration was not an issue as the metabolic activity at day 1 was not improved when H2O2 concentration was reduced to 0.005 wt%. The increase in fold change in metabolic activity over time could be attributed to lower crosslinking density at lower H2O2 concentrations [37] that results in a decrease in hydrogel modulus and an increase in mesh size [121], which were reported to improve proliferation and spreading of encapsulated cells [148, 149]. The fold change in metabolic activity in SF hydrogels was not affected significantly by SF-TA content, but it increased significantly when pre-gel solution was supplemented with 0.5 mM cyclic RGD peptide, indicating signs of cell growth. With increasing cyclo (RGDyK) peptide concentration, encapsulated cells also started to spread and displayed long cytoplasmic extensions. These observations are parallel with previous studies, where encapsulated MSCs were shown to display higher viability [66, 68] and spreading [74] in RGD-tethered PEG and HA hydrogels compared to no RGD controls, which could be explained with higher expression of integrins [57, 65]. It is likely that the decrease in crosslinking density with increasing cyclic peptide that is expected to result in larger mesh size also enhanced cell growth and spreading further. Despite increasing the crosslinking density and storage modulus, G-TA improved metabolic activity even to a higher degree than cyclic RGD peptides and provided cell spreading to some degree, particularly at low H2O2 concentration. An increase in spreading and proliferation upon crosslinking with gelatin-tyramine was also reported for myoblasts [61] and dermal fibroblasts [100] encapsulated in HA-tyramine and PEG-tyramine hydrogels, respectively, indicating that the intrinsic RGD peptides in gelatin sequence improve cell-matrix interactions and cell growth. In another study, MG-63 osteoblast-like cells were shown to spread and proliferate better in gelatin-alginate composite hydrogels compared to alginate only or RGD-conjugated alginate hydrogels [150], which is parallel with our observations. Besides providing cell-adhesive sequences, G-TA also inhibited crystallization and stiffening of 3% composite hydrogels and maintained a well-hydrated structure over 4 weeks of culture, which probably contributed to better growth of hMSCs that were shown to proliferate faster in softer hydrogel matrices [124, 151]. Interestingly, the fold change in metabolic activity of encapsulated hMSCs over 4 weeks of culture was significantly higher in SF/25% G-TA composite hydrogels than in G-TA control, likely because of the highest crosslinking density and therefore smallest mesh size of G-TA only gels among all groups, which might have limited cellular activity within the polymer network. Still, the cells in G-TA only gels were observed to spread earlier and more than SF/25% G-TA at both 0.01 and 0.005 wt% H2O2, suggesting that the primary sequence of protein network played a more important role in the morphology of encapsulated cells than the mesh size.
Overall, our results indicate that SF/SF-TA with cyclo(RGDyK) and SF/G-TA composite hydrogels have several advantages over G-TA only hydrogels. Firstly, gelation kinetics, mechanical properties and bioactivity of SF/SF-TA/cyclo(RGDyK) and SF/G-TA composites can be tuned by changing the weight ratios of individual components, while G-TA only solutions gel very rapidly even at very low HRP concentrations, making it hard to obtain homogenous hydrogels due to improper mixing. Secondly, unlike aqueous G-TA solutions, SF/SF-TA formulations or SF/G-TA blends with a G-TA weight ratio of 10 to 25% do not display thermal gelation. This enables storage and handling of aqueous solutions at room temperature without a significant increase in solution viscosity, which could otherwise cause issues during filter sterilization as well as 3D printing and microfluidic applications, such as a decrease in cell viability as a result of higher shear stress [152] or a drop in the efficiency of microfluidic fabrication of monodisperse droplets [153], respectively. Finally, SF/SF-TA and SF/G-TA composite hydrogels were found to be more resistant to enzymatic degradation, and SF/G-TA composite hydrogels allowed for a significantly higher increase in the metabolic activity of the encapsulated hMSCs compared to G-TA only hydrogels, suggesting that they could be a better alternative as tissue engineering scaffolds with longer-term stability to guide cell growth and tissue organization until native tissue is regenerated. SF/SF-TA/cyclo(RGDyK) composite gels also led to a higher fold change in metabolic activity compared to G-TA only controls and they could be utilized in hard-tissue engineering applications [154] or disease models [155] considering the self-assembly and gradual stiffening of SF/SF-TA or SF-TA only gels in physiological buffers, a pattern that is not observed with G-TA-based hydrogels.
5. Conclusions
Gelation kinetics, mechanical properties, enzymatic degradation and bioactivity of silk hydrogels were successfully modulated by combining SF with SF-TA and cyclic RGD, or only with G-TA. Both SF-TA and G-TA increased gelation speed and mechanical stability and slowed the enzymatic degradation of silk hydrogels crosslinked in physiological buffers. Temporal crystallization and stiffening of silk hydrogels were enhanced by SF-TA but delayed by G-TA, enabling modulation of mechanical properties within a wide range suitable for both soft and hard tissue engineering applications. We were able to modulate the bioactivity of the silk hydrogels by changing the cyclo(RGDyK) or G-TA content, which enables modification of cell microenvironment for specific scaffold or cell delivery applications. Our future studies will focus on utilizing these composite hydrogel formulations as bioinks for 3D bioprinting of cell-laden scaffolds and as mammalian cell-coating sheets using microfluidic encapsulation.
Supplementary Material
Statement of Significance.
HRP-mediated covalent crosslinking of aqueous silk solutions allows for fabrication of highly elastic, mechanically tunable, degradable and biocompatible hydrogels. However, slow gelation and impaired mechanical properties of silk hydrogels enzymatically crosslinked in physiological media as well as the lack of cell-recognition sequences in silk fibroin limit potential cell encapsulation applications. In the present study, crosslinking of regenerated silk with tyramine-substituted silk or gelatin was performed for to improve gelation kinetics and mechanical properties of silk hydrogels by increasing the number of phenol groups available for enzymatic crosslinking. Moreover, in situ conjugation of cyclic RGD peptides into silk hydrogels was employed to enhance cell-matrix interactions. Our findings indicate significant improvement of gelation kinetics and modulus, as well as modulation of silk secondary structure and enzymatic degradation of composite hydrogels in addition to enhanced bioactivity provided by cyclic RGD peptides or tyramine-substituted gelatin. Silk hydrogel formulations introduced here hold great promise as injectable cell-laden tissue fillers and as bioinks for cell encapsulation through 3D bioprinting or droplet-based microgel fabrication.
Acknowledgements
We thank the NIH (P41EB002520, R01EB021264, R01NS094218), the Air Force Office of Scientific Research (FA9550-17-1-0333), and the Army Research Office (W911NF-17-1-0384) for support of this work. We also acknowledge the Turkish Fulbright Commission for PhD fellowship of O.H. The authors would like to thank Morgan Hawker, Nicole R. Raia, and Meghan McGill for valuable experimental advice.
References
- [1].Nguyen QV, Huynh DP, Park JH, Lee DS, Injectable polymeric hydrogels for the delivery of therapeutic agents: A review, Eur. Polym. J 72 (2015) 602–619. [Google Scholar]
- [2].Li, Mooney DJ, Designing hydrogels for controlled drug delivery, Nat. Rev. Mater 1 1(12) (2016) 16071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Vlierberghe SV, Dubruel P, Schacht E, Biopolymer-based hydrogels as scaffolds for tissue engineering applications: A review, Biomacromolecules. 12 (2011) 1387–1408. [DOI] [PubMed] [Google Scholar]
- [4].Wu Y, Wang L, Guo B, Ma PX, Interwoven aligned conductive nanofiber yarn/hydrogel composite scaffolds for engineered 3D cardiac anisotropy. ACS Nano, 11(6) (2017), 5646–5659. [DOI] [PubMed] [Google Scholar]
- [5].Wang L, Wu Y, Guo B, Ma PX, Nanofiber yarn/hydrogel core–shell scaffolds mimicking native skeletal muscle tissue for guiding 3D myoblast alignment, elongation, and differentiation. ACS Nano, 9(9) (2015), 9167–9179. [DOI] [PubMed] [Google Scholar]
- [6].Nicodemus GD, Bryant SJ, Cell Encapsulation in Biodegradable Hydrogels for Tissue Engineering Applications, Tissue Eng. Part B Rev 14 (2008) 149–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Guo B, Qu J, Zhao X, Zhang M, Degradable conductive self-healing hydrogels based on dextran-graft-tetraaniline and N-carboxyethyl chitosan as injectable carriers for myoblast cell therapy and muscle regeneration. Acta Biomater. 84 (2019) 180–193. [DOI] [PubMed] [Google Scholar]
- [8].Tasoglu S, Demirci U, Bioprinting for stem cell research, Trends Biotechnol. 31(1) (2013) 10–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zhang YS, Yue K, Aleman J, Mollazadeh-Moghaddam K, Bakht SM, Yang J, Jia W, Dell’Erba V, Assawes P, Shin SR, Dokmeci MR, Oklu R, Khademhosseini A 3D bioprinting for tissue and organ fabrication. Ann. Biomed. Eng, 45(1) (2017) 148–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gudapati H, Dey M, Ozbolat I, A comprehensive review on droplet-based bioprinting: Past, present and future, Biomaterials. 102 (2016) 20–42 [DOI] [PubMed] [Google Scholar]
- [11].Wan J, Microfluidic-based synthesis of hydrogel particles for cell microencapsulation and cell-based drug delivery, Polymers 4(2) (2012) 1084–1108. [Google Scholar]
- [12].Kang AR, Park JS, Ju J, Jeong GS, Lee SH, Cell encapsulation via microtechnologies, Biomaterials 35(9) (2014) 2651–2663. [DOI] [PubMed] [Google Scholar]
- [13].Rossow T, Lienemann PS, Mooney DJ, Cell Microencapsulation by Droplet Microfluidic Templating, Macromol. Chem. Phys 218(2) (2017) 1600380. [Google Scholar]
- [14].Hasturk O, Kaplan DL, Cell armor for protection against environmental stress: Advances, challenges and applications in micro-and nanoencapsulation of mammalian cells. Acta Biomater. 95 (2018) 3–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wu Y, Wang L, Guo B, Ma PX, Injectable biodegradable hydrogels and microgels based on methacrylated poly (ethylene glycol)-co-poly (glycerol sebacate) multi-block copolymers: synthesis, characterization, and cell encapsulation. J. Mater. Chem. B 2(23) (2014), 3674–3685. [DOI] [PubMed] [Google Scholar]
- [16].Vepari C, Kaplan DL, Silk as a biomaterial. Prog. Polym. Sci 32(8-9) (2007) 991–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Yucel T, Cebe P, Kaplan DL, Vortex-induced injectable silk fibroin hydrogels. Biophys. J 97(7) (2009) 2044–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wang X, Kluge JA, Leisk GG, Kaplan DL, Sonication-induced gelation of silk fibroin for cell encapsulation. Biomaterials, 29(8) (2008) 1054–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zhao H, Xiong S, Li M, Zhang Q, Liu G, Comparison of gelation time and polyalcohol effect on hydrogels from domestic and wild silk fibroins. Adv. Mater. Sci. Eng 2012 (2012). [Google Scholar]
- [20].Aeschbach R, Amadò R, Neukom H, Formation of dityrosine cross-links in proteins by oxidation of tyrosine residues. Biochim. Biophys. Acta 439(2) (1976) 292–301. [DOI] [PubMed] [Google Scholar]
- [21].Partlow BP, Hanna CW, Rnjak-Kovacina J, Moreau JE, Applegate MB, Burke KA, Kaplan DL, Highly tunable elastomeric silk biomaterials. Adv. Funct. Mater 24(29) (2014) 4615–4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Bradner SA, Partlow BP, Cebe P, Omenetto FG, Kaplan DL, Fabrication of elastomeric silk fibers. Biopolymers, 107(9) (2017) e23030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tabatabai AP, Partlow BP, Raia NR, Kaplan DL, Blair DL, Silk Molecular Weight Influences the Kinetics of Enzymatically Cross-linked Silk Hydrogel Formation. Langmuir, 34(50) (2018) 15383–15387. [DOI] [PubMed] [Google Scholar]
- [24].Sundarakrishnan A, Acero EH, Coburn J, Chwalek K, Partlow B, Kaplan DL, Phenol red-silk tyrosine cross-linked hydrogels. Acta Biomater. 42 (2016) 102–113. [DOI] [PubMed] [Google Scholar]
- [25].Zhou L, Chen X, Shao Z, Huang Y, Knight DP, Effect of metallic ions on silk formation in the mulberry silkworm, Bombyx mori. J. Phys. Chem. B 109(35) (2005), 16937–16945. [DOI] [PubMed] [Google Scholar]
- [26].Kim UJ, Park J, Kim HJ, Wada M, Kaplan DL, Three-dimensional aqueous-derived biomaterial scaffolds from silk fibroin, Biomaterials, 26(15) (2005), 2775–2785. [DOI] [PubMed] [Google Scholar]
- [27].Ruan QX, Zhou P, Sodium ion effect on silk fibroin conformation characterized by solid-state NMR and generalized 2D NMR–NMR correlation, J. Mol. Struct 883 (2008), 85–90. [Google Scholar]
- [28].Ho SN, Intracellular water homeostasis and the mammalian cellular osmotic stress response. J. Cell. Physiol 206(1) (2006) 9–15. [DOI] [PubMed] [Google Scholar]
- [29].Chung BG, Lee KH, Khademhosseini A, Lee SH, Microfluidic fabrication of microengineered hydrogels and their application in tissue engineering. Lab Chip, 12(1) (2012) 45–59. [DOI] [PubMed] [Google Scholar]
- [30].Kirchmajer DM, Gorkin R, An overview of the suitability of hydrogel-forming polymers for extrusion-based 3D-printing. J. Mater. Chem. B 3(20) (2015) 4105–4117. [DOI] [PubMed] [Google Scholar]
- [31].Chimene D, Lennox KK, Kaunas RR, Gaharwar AK, Advanced bioinks for 3D printing: a materials science perspective. Ann. Biomed. Eng, 44(6) (2016) 2090–2102. [DOI] [PubMed] [Google Scholar]
- [32].Wang Y, Kim HJ, Vunjak-Novakovic G, Kaplan DL, Stem cell-based tissue engineering with silk biomaterials. Biomaterials, 27(36) (2006) 6064–6082. [DOI] [PubMed] [Google Scholar]
- [33].Wang X, Kluge JA, Leisk GG, Kaplan DL, Sonication-induced gelation of silk fibroin for cell encapsulation. Biomaterials, 29(8) (2008) 1054–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Chiarugi P, Giannoni E, Anoikis: a necessary death program for anchorage-dependent cells. Biochem. Pharmacol 76(11) (2008) 1352–1364. [DOI] [PubMed] [Google Scholar]
- [35].Lee F, Chung JE, Kurisawa M, An injectable enzymatically crosslinked hyaluronic acid–tyramine hydrogel system with independent tuning of mechanical strength and gelation rate. Soft Matter. 4(4) (2008) 880–887. [DOI] [PubMed] [Google Scholar]
- [36].Lee F, Bae KH, Kurisawa M, Injectable hydrogel systems crosslinked by horseradish peroxidase. Biomed. Mater 11(1) (2015) 014101. [DOI] [PubMed] [Google Scholar]
- [37].McGill M, Coburn JM, Partlow BP, Mu X, Kaplan DL, Molecular and macro-scale analysis of enzyme-crosslinked silk hydrogels for rational biomaterial design. Acta Biomater. 63 (2017) 76–84. [DOI] [PubMed] [Google Scholar]
- [38].Sminia T, Delemarre F, Janse EM, Histological observations on the intestinal immune response towards horseradish peroxidase in rats. Immunology 50(1) (1983) 53. [PMC free article] [PubMed] [Google Scholar]
- [39].Gardner AM, Xu FH, Fady C, Jacoby FJ, Duffey DC, Tu Y, Lichtenstein A, Apoptotic vs. nonapoptotic cytotoxicity induced by hydrogen peroxide. Free Radic. Biol. Med 22(1-2) (1997) 73–83. [DOI] [PubMed] [Google Scholar]
- [40].Sakai S, Hirose K, Taguchi K, Ogushi Y, Kawakami K, An injectable, in situ enzymatically gellable, gelatin derivative for drug delivery and tissue engineering. Biomaterials 30(20) (2009) 3371–3377. [DOI] [PubMed] [Google Scholar]
- [41].Amini AA, Nair LS, Enzymatically cross-linked injectable gelatin gel as osteoblast delivery vehicle. J. Bioact. Compat. Polym 27(4) (2012) 342–355. [Google Scholar]
- [42].Liang Y, Zhao X, Hu T, Han Y, Guo B, Mussel-inspired, antibacterial, conductive, antioxidant, injectable composite hydrogel wound dressing to promote the regeneration of infected skin. J. Colloid Interface Sci 556 (2019), 514–528. [DOI] [PubMed] [Google Scholar]
- [43].Peng Z, She Y, Chen L, Synthesis of poly (glutamic acid)-tyramine hydrogel by enzyme-mediated gelation for controlled release of proteins. J. Biomater. Sci. Polym. Ed 26(2) (2015) 111–127. [DOI] [PubMed] [Google Scholar]
- [44].Kurisawa M, Chung JE, Yang YY, Gao SJ, Uyama H, Injectable biodegradable hydrogels composed of hyaluronic acid–tyramine conjugates for drug delivery and tissue engineering. ChemComm (34) (2005) 4312–4314. [DOI] [PubMed] [Google Scholar]
- [45].Kim KS, Park SJ, Yang JA, Jeon JH, Bhang SH, Kim BS, Hahn SK, Injectable hyaluronic acid–tyramine hydrogels for the treatment of rheumatoid arthritis. Acta Biomater. 7(2) (2011) 666–674. [DOI] [PubMed] [Google Scholar]
- [46].Toh WS, Lim TC, Kurisawa M, Spector M, Modulation of mesenchymal stem cell chondrogenesis in a tunable hyaluronic acid hydrogel microenvironment. Biomaterials, 33(15) (2012) 3835–3845. [DOI] [PubMed] [Google Scholar]
- [47].Xu K, Narayanan K, Lee F, Bae KH, Gao S, Kurisawa M, Enzyme-mediated hyaluronic acid–tyramine hydrogels for the propagation of human embryonic stem cells in 3D. Acta Biomater. 24 (2015) 159–171. [DOI] [PubMed] [Google Scholar]
- [48].Liang Y, Zhao X, Hu T, Chen B, Yin Z, Ma PX, Guo B, Adhesive hemostatic conducting injectable composite hydrogels with sustained drug release and photothermal antibacterial activity to promote full-thickness skin regeneration during wound healing. Small, 15(12) (2019), 1900046. [DOI] [PubMed] [Google Scholar]
- [49].Jin R, Hiemstra C, Zhong Z, Feijen J, Enzyme-mediated fast in situ formation of hydrogels from dextran–tyramine conjugates. Biomaterials, 28(18) (2007) 2791–2800. [DOI] [PubMed] [Google Scholar]
- [50].Teixeira LSM, Bijl S, Pully VV, Otto C, Jin R, Feijen J, van Blitterswijk CA, Dijkstra PJ, Karperien M, Self-attaching and cell-attracting in-situ forming dextran-tyramine conjugates hydrogels for arthroscopic cartilage repair. Biomaterials, 33(11) (2012) 3164–3174. [DOI] [PubMed] [Google Scholar]
- [51].Wei H, Xie J, Jiang X, Ye T, Chang A, Wu W, Synthesis and Characterization of Dextran–Tyramine-Based H2O2-Sensitive Microgels. Macromolecules, 47(17) (2014) 6067–6076. [Google Scholar]
- [52].Sakai S, Kawakami K, Synthesis and characterization of both ionically and enzymatically cross-linkable alginate. Acta Biomater. 3(4) (2007) 495–501. [DOI] [PubMed] [Google Scholar]
- [53].Sakai S, Hirose K, Moriyama K, Kawakami K, Control of cellular adhesiveness in an alginate-based hydrogel by varying peroxidase and H2O2 concentrations during gelation. Acta Biomater. 6(4) (2010) 1446–1452. [DOI] [PubMed] [Google Scholar]
- [54].Joung YK, You SS, Park KM, Go DH, Park KD, In situ forming, metal-adhesive heparin hydrogel surfaces for blood-compatible coating. Colloids Surf. B Biointerfaces, 99 (2012) 102–107. [DOI] [PubMed] [Google Scholar]
- [55].Sakai S, Ogushi Y, Kawakami K, Enzymatically crosslinked carboxymethylcellulose–tyramine conjugate hydrogel: cellular adhesiveness and feasibility for cell sheet technology. Acta Biomater, 5(2) (2009) 554–559. [DOI] [PubMed] [Google Scholar]
- [56].Jin R, Lou B, Lin C, Tyrosinase-mediated in situ forming hydrogels from biodegradable chondroitin sulfate–tyramine conjugates. Polym. Int 62(3) (2013) 353–361. [Google Scholar]
- [57].Lim KS, Ramaswamy Y, Roberts JJ, Alves MH, Poole-Warren LA, Martens PJ, Promoting cell survival and proliferation in degradable Poly (vinyl alcohol)–Tyramine Hydrogels. Macromol. Biosci 15(10) (2015) 1423–1432. [DOI] [PubMed] [Google Scholar]
- [58].Kamperman T, Henke S, Zoetebier B, Ruiterkamp N, Wang R, Pouran B, Weinans H, Karperien M, Leijten J, Nanoemulsion-induced enzymatic crosslinking of tyramine-functionalized polymer droplets. J. Mater. Chem. B, 5(25) (2017) 4835–4844. [DOI] [PubMed] [Google Scholar]
- [59].Wennink JW, Niederer K, Bochyńska AI, Moreira Teixeira LS, Karperien M, Feijen J, Dijkstra PJ, Injectable Hydrogels by Enzymatic Co-Crosslinking of Dextran and Hyaluronic Acid Tyramine Conjugates. Macromol Symp. 309(1) (2011) 213–221). [Google Scholar]
- [60].Jin R, Teixeira LSM, Dijkstra PJ, van Blitterswijk CA, Karperien M, Feijen J, Chondrogenesis in injectable enzymatically crosslinked heparin/dextran hydrogels. J. Control. Release 152(1) (2011) 186–195. [DOI] [PubMed] [Google Scholar]
- [61].Poveda-Reyes S, Moulisova V, Sanmartín-Masiá E, Quintanilla-Sierra L, Salmerón-Sánchez M, Ferrer GG, Gelatin—hyaluronic acid hydrogels with tuned stiffness to counterbalance cellular forces and promote cell differentiation. Macromol. Biosci, 16(9) (2016) 1311–1324. [DOI] [PubMed] [Google Scholar]
- [62].Bai L, Zhu L, Min S, Liu L, Cai Y, Yao J, Surface modification and properties of Bombyx mori silk fibroin films by antimicrobial peptide. Appl. Surf. Sci 254(10) (2008) 2988–2995. [Google Scholar]
- [63].Gil ES, Mandal BB, Park SH, Marchant JK, Omenetto FG, Kaplan DL, Helicoidal multi-lamellar features of RGD-functionalized silk biomaterials for corneal tissue engineering. Biomaterials, 31(34) (2010) 8953–8963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Hersel U, Dahmen C, Kessler H, RGD modified polymers: biomaterials for stimulated cell adhesion and beyond. Biomaterials, 24(24) (2003) 4385–4415. [DOI] [PubMed] [Google Scholar]
- [65].Yang F, Williams CG, Wang DA, Lee H, Manson PN, Elisseeff J, The effect of incorporating RGD adhesive peptide in polyethylene glycol diacrylate hydrogel on osteogenesis of bone marrow stromal cells. Biomaterials, 26(30) (2005) 5991–5998. [DOI] [PubMed] [Google Scholar]
- [66].Salinas CN, Anseth KS, The influence of the RGD peptide motif and its contextual presentation in PEG gels on human mesenchymal stem cell viability. J. Tissue Eng. Regen. Med 2(5) (2008) 296–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Zhu J, Tang C, Kottke-Marchant K, Marchant RE, Design and synthesis of biomimetic hydrogel scaffolds with controlled organization of cyclic RGD peptides. Bioconjugate Chem. 20(2) (2009) 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Liu SQ, Tian Q, Wang L, Hedrick JL, Hui JHP, Yang YY, Ee PLR, Injectable biodegradable poly (ethylene glycol)/RGD peptide hybrid hydrogels for in vitro chondrogenesis of human mesenchymal stem cells. Macromol. Rapid Commun 31(13) (2010) 1148–1154. [DOI] [PubMed] [Google Scholar]
- [69].Wilson MJ, Liliensiek SJ, Murphy CJ, Murphy WL, Nealey PF, Hydrogels with well-defined peptide-hydrogel spacing and concentration: impact on epithelial cell behavior. Soft Matter 8(2) (2012) 390–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Beria L, Gevrek TN, Erdog A, Sanyal R, Pasini D, Sanyal A, ‘Clickable’hydrogels for all: facile fabrication and functionalization. Biomater. Sci 2(1) (2014) 67–75. [DOI] [PubMed] [Google Scholar]
- [71].Xu Q, Zhang Z, Xiao C, He C, Chen X, Injectable polypeptide hydrogel as biomimetic scaffolds with tunable bioactivity and controllable cell adhesion. Biomacromolecules, 18(4) (2017) 1411–1418. [DOI] [PubMed] [Google Scholar]
- [72].Shu XZ, Ghosh K, Liu Y, Palumbo FS, Luo Y, Clark RA, Prestwich GD, Attachment and spreading of fibroblasts on an RGD peptide–modified injectable hyaluronan hydrogel. J. Biomed. Mater. Res. A 68(2) (2004) 365–375. [DOI] [PubMed] [Google Scholar]
- [73].Cui FZ, Tian WM, Hou SP, Xu QY, Lee IS, Hyaluronic acid hydrogel immobilized with RGD peptides for brain tissue engineering. J. Mater. Sci.: Mater. Med 17(12) (2006) 1393–1401. [DOI] [PubMed] [Google Scholar]
- [74].Lam J, Segura T, The modulation of MSC integrin expression by RGD presentation. Biomaterials, 34(16) (2013) 3938–3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Zhang D, Sun MB, Lee J, Abdeen AA, Kilian KA, Cell shape and the presentation of adhesion ligands guide smooth muscle myogenesis. J. Biomed. Mater. Res. A 104(5) (2016) 1212–1220. [DOI] [PubMed] [Google Scholar]
- [76].Kang SW, Cha BH, Park H, Park KS, Lee KY, Lee SH, The effect of conjugating RGD into 3D alginate hydrogels on adipogenic differentiation of human adipose-derived stromal cells. Macromol. Biosci 11(5) (2011) 673–679. [DOI] [PubMed] [Google Scholar]
- [77].Park KM, Jun I, Joung YK, Shin H, Park KD, In situ hydrogelation and RGD conjugation of tyramine-conjugated 4-arm PPO–PEO block copolymer for injectable bio-mimetic scaffolds. Soft Matter 7(3) (2011) 986–992. [Google Scholar]
- [78].Wang LS, Lee F, Lim J, Du C, Wan AC, Lee SS, Kurisawa M, Enzymatic conjugation of a bioactive peptide into an injectable hyaluronic acid–tyramine hydrogel system to promote the formation of functional vasculature. Acta Biomater. 10(6) (2014) 2539–2550. [DOI] [PubMed] [Google Scholar]
- [79].Loebel C, Szczesny SE, Cosgrove BD, Alini M, Zenobi-Wong M, Mauck RL, Eglin D, Cross-linking chemistry of tyramine-modified hyaluronan hydrogels alters mesenchymal stem cell early attachment and behavior. Biomacromolecules 18(3) (2017) 855–864. [DOI] [PubMed] [Google Scholar]
- [80].Gómez-Guillén MC, Giménez B, López-Caballero MA, Montero MP, Functional and bioactive properties of collagen and gelatin from alternative sources: A review. Food Hydrocoll. 25(8) (2011) 1813–1827. [Google Scholar]
- [81].Heino J, The collagen family members as cell adhesion proteins. Bioessays 29(10) (2007) 1001–1010. [DOI] [PubMed] [Google Scholar]
- [82].Van Vlierberghe S, Dubruel P, Schacht E, Biopolymer-based hydrogels as scaffolds for tissue engineering applications: a review. Biomacromolecules 12(5) (2011) 1387–1408. [DOI] [PubMed] [Google Scholar]
- [83].Gil ES, Frankowski DJ, Spontak RJ, Hudson SM, Swelling behavior and morphological evolution of mixed gelatin/silk fibroin hydrogels. Biomacromolecules 6(6) (2005) 3079–3087. [DOI] [PubMed] [Google Scholar]
- [84].Das S, Pati F, Chameettachal S, Pahwa S, Ray AR, Dhara S, Ghosh S, Enhanced redifferentiation of chondrocytes on microperiodic silk/gelatin scaffolds: toward tailor-made tissue engineering. Biomacromolecules 14(2) (2013) 311–321. [DOI] [PubMed] [Google Scholar]
- [85].Bragg JC, Kweon H, Jo Y, Lee KG, Lin CC, In situ formation of silk-gelatin hybrid hydrogels for affinity-based growth factor sequestration and release. RSC Advances 6(115) (2016) 114353–114360. [Google Scholar]
- [86].Xiao W, He J, Nichol JW, Wang L, Hutson CB, Wang B, Du Y, Fan H, Khademhosseini A, Synthesis and characterization of photocrosslinkable gelatin and silk fibroin interpenetrating polymer network hydrogels. Acta Biomater. 7(6) (2011) 2384–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Xiao W, Liu W, Sun J, Dan X, Wei D, Fan H, Ultrasonication and genipin cross-linking to prepare novel silk fibroin–gelatin composite hydrogel. J. Bioact. Compat. Polym 27(4) (2012) 327–341. [Google Scholar]
- [88].Liesivuori J, Savolainen AH, Methanol and formic acid toxicity: biochemical mechanisms. Pharmacol. Toxicol 69(3) (1991) 157–163. [DOI] [PubMed] [Google Scholar]
- [89].Feril LB Jr, Kondo T, Zhao QL, Ogawa R, Tachibana K, Kudo N, Fujimoto S, Nakamura S, Enhancement of ultrasound-induced apoptosis and cell lysis by echo-contrast agents. Ultrasound Med. Biol 29(2) (2003) 331–337. [DOI] [PubMed] [Google Scholar]
- [90].Sun W, Incitti T, Migliaresi C, Quattrone A, Casarosa S, Motta A, Genipin-crosslinked gelatin–silk fibroin hydrogels for modulating the behaviour of pluripotent cells. J. Tissue Eng. Regen. Med 10(10) (2016) 876–887. [DOI] [PubMed] [Google Scholar]
- [91].Das S, Pati F, Choi YJ, Rijal G, Shim JH, Kim SW, Ray AR, Cho D, Ghosh S, Bioprintable, cell-laden silk fibroin–gelatin hydrogel supporting multilineage differentiation of stem cells for fabrication of three-dimensional tissue constructs. Acta Biomater. 11 (2015) 233–246. [DOI] [PubMed] [Google Scholar]
- [92].Chameettachal S, Midha S, Ghosh S, Regulation of chondrogenesis and hypertrophy in silk fibroin-gelatin-based 3D bioprinted constructs. ACS Biomater. Sci. Eng 2(9) (2016) 1450–1463. [DOI] [PubMed] [Google Scholar]
- [93].Hu M, Kurisawa M, Deng R, Teo CM, Schumacher A, Thong YX, Wang L, Shimacher KM, Ying JY, Cell immobilization in gelatin–hydroxyphenylpropionic acid hydrogel fibers. Biomaterials, 30(21) (2009) 3523–3531. [DOI] [PubMed] [Google Scholar]
- [94].Wang LS, Boulaire J, Chan PP, Chung JE, Kurisawa M, The role of stiffness of gelatin–hydroxyphenylpropionic acid hydrogels formed by enzyme-mediated crosslinking on the differentiation of human mesenchymal stem cell. Biomaterials, 31(33) (2010) 8608–8616. [DOI] [PubMed] [Google Scholar]
- [95].Wang LS, Du C, Chung JE, Kurisawa M, Enzymatically cross-linked gelatin-phenol hydrogels with a broader stiffness range for osteogenic differentiation of human mesenchymal stem cells. Acta Biomater. 8(5) (2012) 1826–1837. [DOI] [PubMed] [Google Scholar]
- [96].Li Z, Qu T, Ding C, Ma C, Sun H, Li S, Liu X, Injectable gelatin derivative hydrogels with sustained vascular endothelial growth factor release for induced angiogenesis. Acta Biomater. 13 (2015) 88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Chuang CH, Lin RZ, Tien HW, Chu YC, Li YC, Melero-Martin JM, Chen YC, Enzymatic regulation of functional vascular networks using gelatin hydrogels. Acta Biomater. 19 (2015) 85–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Liu Y, Sakai S, Taya M, Impact of the composition of alginate and gelatin derivatives in bioconjugated hydrogels on the fabrication of cell sheets and spherical tissues with living cell sheaths. Acta Biomater. 9(5) (2013) 6616–6623. [DOI] [PubMed] [Google Scholar]
- [99].Sanmartín-Masiá E, Poveda-Reyes S, Gallego Ferrer G, Extracellular matrix–inspired gelatin/hyaluronic acid injectable hydrogels. Int. J. Polym. Mater. Po 66(6) (2017) 280–288. [Google Scholar]
- [100].Hoang Thi TT, Lee JS, Lee Y, Park KM, Park KD, Enhanced Cellular Activity in Gelatin-Poly (Ethylene Glycol) Hydrogels without Compromising Gel Stiffness. Macromol. Biosci 16(3) (2016) 334–340. [DOI] [PubMed] [Google Scholar]
- [101].Raia NR, Partlow BP, McGill M, Kimmerling EP, Ghezzi CE, Kaplan DL, Enzymatically crosslinked silk-hyaluronic acid hydrogels. Biomaterials 131 (2017) 58–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Lohmann N, Schirmer L, Atallah P, Wandel E, Ferrer RA, Werner C, Simon JC, Franz S, Freudenberg U, Glycosaminoglycan-based hydrogels capture inflammatory chemokines and rescue defective wound healing in mice, Sci. Transl. Med 9(386) (2017), eaai9044. [DOI] [PubMed] [Google Scholar]
- [103].Arima Y, Iwata H, Effect of wettability and surface functional groups on protein adsorption and cell adhesion using well-defined mixed self-assembled monolayers. Biomaterials 28(20) (2007) 3074–3082. [DOI] [PubMed] [Google Scholar]
- [104].Rutz AL, Lewis PL, Shah RN, Toward next-generation bioinks: tuning material properties pre- and post-printing to optimize cell viability. MRS Bulletin 42(8) (2017), 563–570. [Google Scholar]
- [105].Gopinathan J, Noh I, Recent trends in bioinks for 3D printing. Biomater. Res 22(1) (2018) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Lammel AS, Hu X, Park SH, Kaplan DL, Scheibel TR, Controlling silk fibroin particle features for drug delivery, Biomaterials, 31(16) (2010), 4583–4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Hilborn J, In vivo injectable gels for tissue repair. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol 3(6) (2011) 589–606. [DOI] [PubMed] [Google Scholar]
- [108].Goh CH, Heng PWS, Chan LW, Alginates as a useful natural polymer for microencapsulation and therapeutic applications. Carbohyd. Polym 88(1) (2012) 1–12. [Google Scholar]
- [109].Rzany B, Overview on injectable fillers: Efficacy and safety, in: De Maio M, Rzany B (Eds.), Injectable fillers in aesthetic medicine, Springer, Berlin, 2014, pp.1–16. [Google Scholar]
- [110].Axpe E, Oyen M, Applications of alginate-based bioinks in 3D bioprinting. Int. J. Mol. Sci 17(12) (2016) 1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Kong HJ, Kaigler D, Kim K, Mooney DJ, Controlling rigidity and degradation of alginate hydrogels via molecular weight distribution. Biomacromolecules 5(5) (2004) 1720–1727. [DOI] [PubMed] [Google Scholar]
- [112].Shahriari D, Koffler J, Lynam DA, Tuszynski MH, Sakamoto JS, Characterizing the degradation of alginate hydrogel for use in multilumen scaffolds for spinal cord repair. J. Biomed. Mater. Res. A, 104(3) (2016) 611–619. [DOI] [PubMed] [Google Scholar]
- [113].Kapoor S, Kundu SC, Silk protein-based hydrogels: promising advanced materials for biomedical applications. Acta Biomater. 31 (2016) 17–32. [DOI] [PubMed] [Google Scholar]
- [114].Shulha H, Foo CWP, Kaplan DL, Tsukruk VV, Unfolding the multi-length scale domain structure of silk fibroin protein. Polymer, 47(16) (2006) 5821–5830. [Google Scholar]
- [115].Murphy AR, Kaplan DL, Biomedical applications of chemically-modified silk fibroin. J. Mater. Chem 19(36) (2009) 6443–6450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Hafidz RMRN, Yaakob CM, Amin I, Noorfaizan A, Chemical and functional properties of bovine and porcine skin gelatin. Int. Food Res. J 18(2011) (2011) 813–817. [Google Scholar]
- [117].Bae JW, Kim BY, Lih E, Choi JH, Lee Y, Park KD, In situ formation of enzyme-free hydrogels via ferromagnetic microbead-assisted enzymatic cross-linking. ChemComm 50(89) (2014) 13710–13713. [DOI] [PubMed] [Google Scholar]
- [118].Applegate MB, Partlow BP, Coburn J, Marelli B, Pirie C, Pineda R, Kaplan DL, Omenetto FG, Photocrosslinking of silk fibroin using riboflavin for ocular prostheses. Adv. Mater 28(12) (2016) 2417–2420. [DOI] [PubMed] [Google Scholar]
- [119].Choi J, McGill M, Raia NR, Hasturk O, Kaplan DL, Silk Hydrogels Crosslinked by the Fenton Reaction. Adv. Healthcare Mater (2019) 1900644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Winter HH, Chambon F, Analysis of linear viscoelasticity of a crosslinking polymer at the gel point. J. Rheol 30(2) (1986) 367–382. [Google Scholar]
- [121].Lee F, Chung JE, Kurisawa M, An injectable hyaluronic acid–tyramine hydrogel system for protein delivery. J. Control. Release 134(3) (2009) 186–193. [DOI] [PubMed] [Google Scholar]
- [122].Park KM, Lee Y, Son JY, Bae JW, Park KD, In situ SVVYGLR peptide conjugation into injectable gelatin-poly (ethylene glycol)-tyramine hydrogel via enzyme-mediated reaction for enhancement of endothelial cell activity and neo-vascularization. Bioconjugate Chem. 23(10) (2012) 2042–2050. [DOI] [PubMed] [Google Scholar]
- [123].Engler AJ, Sen S, Sweeney HL, Discher DE, Matrix elasticity directs stem cell lineage specification. Cell 126(4) (2006) 677–689. [DOI] [PubMed] [Google Scholar]
- [124].Pek YS, Wan AC, Ying JY, The effect of matrix stiffness on mesenchymal stem cell differentiation in a 3D thixotropic gel. Biomaterials 31(3) (2010) 385–391. [DOI] [PubMed] [Google Scholar]
- [125].Olivares-Navarrete R, Lee EM, Smith K, Hyzy SL, Doroudi M, Williams JK, Schwartz Z, Substrate stiffness controls osteoblastic and chondrocytic differentiation of mesenchymal stem cells without exogenous stimuli. PloS one 12(1) (2017) e0170312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Su D, Yao M, Liu J, Zhong Y, Chen X, Shao Z, Enhancing mechanical properties of silk fibroin hydrogel through restricting the growth of β-sheet domains. ACS Appl. Mater. Interfaces. 9(20) (2017) 17489–17498. [DOI] [PubMed] [Google Scholar]
- [127].Zhao S, Chen Y, Partlow BP, Golding AS, Tseng P, Coburn J, Applegate MB, Moreau JE, Omenetto FG, Kaplan DL, Bio-functionalized silk hydrogel microfluidic systems. Biomaterials 93 (2016) 60–70. [DOI] [PubMed] [Google Scholar]
- [128].Stoppel WL, Gao AE, Greaney AM, Partlow BP, Bretherton RC, Kaplan DL, Black LD III, Elastic, silk-cardiac extracellular matrix hydrogels exhibit time-dependent stiffening that modulates cardiac fibroblast response. J. Biomed. Mater. Res. A 104(12) (2016) 3058–3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Matsumoto A, Chen J, Collette AL, Kim UJ, Altman GH, Cebe P, Kaplan DL, Mechanisms of silk fibroin sol– gel transitions. J. Phys. Chem. B 110(43) (2006) 21630–21638. [DOI] [PubMed] [Google Scholar]
- [130].He YX, Zhang NN, Li WF, Jia N, Chen BY, Zhou K, Zhang J, Chen Y, Zhou CZ, N-Terminal domain of Bombyx mori fibroin mediates the assembly of silk in response to pH decrease. J. Mol. Biol 418(3-4) (2012) 197–207. [DOI] [PubMed] [Google Scholar]
- [131].Partlow BP, Bagheri M, Harden JL, Kaplan DL, Tyrosine templating in the self-assembly and crystallization of silk fibroin. Biomacromolecules 17(11) (2016) 3570–3579. [DOI] [PubMed] [Google Scholar]
- [132].Wen JH, Vincent LG, Fuhrmann A, Choi YS, Hribar KC, Taylor-Weiner H, Chen S, Engler AJ, Interplay of matrix stiffness and protein tethering in stem cell differentiation. Nat. Mater 13(10) (2014) 979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Yildirimer L, Seifalian AM, Three-dimensional biomaterial degradation—Material choice, design and extrinsic factor considerations. Biotechnol. Adv 32(5) (2014) 984–999. [DOI] [PubMed] [Google Scholar]
- [134].Brown J, Lu CL, Coburn J, Kaplan DL, Impact of silk biomaterial structure on proteolysis. Acta Biomater. 11 (2015) 212–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Yan LP, Silva-Correia J, Ribeiro VP, Miranda-Gonçalves V, Correia C, da Silva Morais A, Sousa RA, Reis RM, Oliveira AL, Oliveira JM, Reis RL, Tumor growth suppression induced by biomimetic silk fibroin hydrogels. Sci. Rep 6 (2016) 31037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Weber LM, Lopez CG, Anseth KS, Effects of PEG hydrogel crosslinking density on protein diffusion and encapsulated islet survival and function. J. Biomed. Mater. Res. A 90(3) (2009) 720–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Peppas NA, Mikos NA, Hydrogels in Medicine and Pharmacy. Vol. 1, Taylor & Francis Group, LLC, Boca Raton, 1986. [Google Scholar]
- [138].Kaufmann D, Fiedler A, Junger A, Auernheimer J, Kessler H, Weberskirch R, Xhemical conjugation of linear and cyclic RGD moieties to a recombinant elastin-mimetic polypeptide-A versatile approach towards bioactive protein hydrogels. Macromol. Biosci 8(6) (2008), 577–588. [DOI] [PubMed] [Google Scholar]
- [139].Kantlehner M, Schaffner P, Finsinger D, Meyer J, Jonczyk A, Diefenbach B, Kessler H, Surface coating with cyclic RGD peptides stimulates osteoblast adhesion and proliferation as well as bone formation. Chembiochem. 1(2) (2000) 107–114. [DOI] [PubMed] [Google Scholar]
- [140].Kardestuncer T, McCarthy MB, Karageorgiou V, Kaplan DL, Gronowicz G, RGD-tethered silk substrate stimulates the differentiation of human tendon cells. Clin. Orthop. Relat. Res 448 (2006) 234–239. [DOI] [PubMed] [Google Scholar]
- [141].Wang H, Ma L, Yang S, Shao Z, Meng C, Duan D, Li Y, Effect of RGD-modified silk material on the adhesion and proliferation of bone marrow-derived mesenchymal stem cells. J. Huazhong U. Sci-Med 29(1) (2009), 80–83. [DOI] [PubMed] [Google Scholar]
- [142].Chen J, Altman GH, Karageorgiou V, Horan R, Collette A, Volloch V, Colabro T, Kaplan DL, Human bone marrow stromal cell and ligament fibroblast responses on RGD-modified silk fibers. J. Biomed. Mater. Res. A 67A(2) (2003) 559–570. [DOI] [PubMed] [Google Scholar]
- [143].Kim JW, Ki CS, Park YH, Kim HJ, Um IC, Effect of RGDS and KRSR peptides immobilized on silk fibroin nanofibrous mats for cell adhesion and proliferation. Macromol. Res 18(5) (2010) 442–448. [Google Scholar]
- [144].Ziv K, Nuhn H, Ben-Haim Y, Sasportas LS, Kempen PJ, Niedringhaus TP, Hrynykd M, Sinclair R, Barron AE, Gambhir SS, A tunable silk–alginate hydrogel scaffold for stem cell culture and transplantation. Biomaterials 35(12) (2014) 3736–3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Vashi AV, Ramshaw JA, Glattauer V, Elvin CM, Lyons RE, Werkmeister JA, Controlled surface modification of tissue culture polystyrene for selective cell binding using resilin-inspired polypeptides. Biofabrication 5(3) (2013) 035005. [DOI] [PubMed] [Google Scholar]
- [146].Shipman C Jr, Evaluation of 4-(2-hydroxyethyl)-1-piperazineëthanesulfonic acid (HEPES) as a tissue culture buffer. Exp. Biol. Med 130(1) (1969) 305–310. [DOI] [PubMed] [Google Scholar]
- [147].Hwang CM, Sant S, Masaeli M, Kachouie NN, Zamanian B, Lee SH, Khademhosseini A, Fabrication of three-dimensional porous cell-laden hydrogel for tissue engineering. Biofabrication 2(3) (2010) 035003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Mahoney MJ, Anseth KS, Three-dimensional growth and function of neural tissue in degradable polyethylene glycol hydrogels. Biomaterials 27(10) (2006) 2265–2274. [DOI] [PubMed] [Google Scholar]
- [149].Liao H, Munoz-Pinto D, Qu X, Hou Y, Grunlan MA, Hahn MS, Influence of hydrogel mechanical properties and mesh size on vocal fold fibroblast extracellular matrix production and phenotype. Acta Biomater. 4(5) (2008) 1161–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Grigore A, Sarker B, Fabry B, Boccaccini AR, Detsch R, Behavior of encapsulated MG-63 cells in RGD and gelatine-modified alginate hydrogels. Tissue Eng. Part A 20(15-16) (2014) 2140–2150. [DOI] [PubMed] [Google Scholar]
- [151].Guvendiren M, Burdick JA, Stiffening hydrogels to probe short-and long-term cellular responses to dynamic mechanics. Nat. Commun 3 (2012) 792. [DOI] [PubMed] [Google Scholar]
- [152].Blaeser A, Duarte Campos DF, Puster U, Richtering W, Stevens MM, Fischer H, Controlling shear stress in 3D bioprinting is a key factor to balance printing resolution and stem cell integrity. Adv. Healthc. Mater 5(3) (2016) 326–333. [DOI] [PubMed] [Google Scholar]
- [153].Choi CH, Jung JH, Rhee YW, Kim DP, Shim SE, Lee CS, Generation of monodisperse alginate microbeads and in situ encapsulation of cell in microfluidic device, Biomed. Microdevices 9(6) (2007), 855–862. [DOI] [PubMed] [Google Scholar]
- [154].Breuls RG, Jiya TU, Smit TH, Scaffold stiffness influences cell behavior: opportunities for skeletal tissue engineering, Open Orthop J. 2 (2008) 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Smithmyer ME, Sawicki LA, Kloxin AM, Hydrogel scaffolds as in vitro models to study fibroblast activation in wound healing and disease, Biomater. Sci, 2(5) (2014), 634–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.