

Abstract
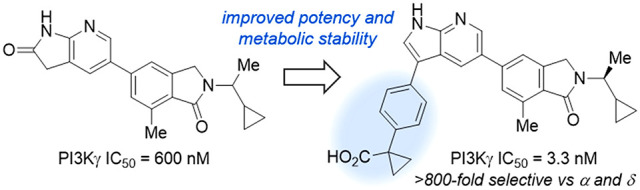
The successful application of immunotherapy in the treatment of cancer relies on effective engagement of immune cells in the tumor microenvironment. Phosphoinositide 3-kinase γ (PI3Kγ) is highly expressed in tumor-associated macrophages, and its expression levels are associated with tumor immunosuppression and growth. Selective inhibition of PI3Kγ offers a promising strategy in immuno-oncology, which has led to the development of numerous potent PI3Kγ inhibitors with variable selectivity profiles. To facilitate further investigation of the therapeutic potential of PI3Kγ inhibition, we required a potent and PI3Kγ-selective tool compound with sufficient metabolic stability for use in future in vivo studies. Herein, we describe some of our efforts to realize this goal through the systematic study of SARs within a series of 7-azaindole-based PI3Kγ inhibitors. The large volume of data generated from this study helped guide our subsequent lead optimization efforts and will inform further development of PI3Kγ-selective inhibitors for use in immunomodulation.
Keywords: PI3Kγ, inhibitor, selective, azaindole, immunomodulation, cancer
Phosphoinositide 3-kinases (PI3Ks) are a class of enzymes responsible for the regulation of a wide variety of cellular activities such as signaling, survival, metabolism, and transport. A common substrate of these kinases is the glycerophospholipid phosphatidylinositol (PIP): PI3K-mediated phosphorylation of PIP initiates a signaling cascade via downstream proteins (e.g., protein kinase B (PKB), commonly referred to as Akt, and 3-phosphoinositide-dependent protein kinase-1 (PDPK1)) and ultimately mediates critical cellular functions, such as motility and proliferation.1,2 The class I PI3Ks, including PI3Kα, PI3Kβ, PI3Kγ, and PI3Kδ, phosphorylate the C3-hydroxyl group of the inositol ring in phosphatidylinositol 4,5-bisphosphate.3
Although PI3Ks are involved in a variety of functions and are expressed throughout the body, PI3Kγ and PI3Kδ are of particular interest due to their expression in leukocytes as effectors of immune responses.4−7 In oncology, abnormalities in PI3K expression have been linked to cancer development, corresponding to high levels of PI3Kγ expression and activity within immunosuppressive myeloid cells.8−18 Due to the significance of PI3Kγ and PI3Kδ in immunomodulation, many inhibitors with varying degrees of selectivity have been developed for the treatment of cancers and other pathologies of the immune system.19,20 Structures of several potent and γ-selective inhibitors have been published, selected examples of which are shown in Figure 1a.21−27 Among these, 1 (IPI-549, developed by Infinity Pharmaceuticals) is the only γ-selective inhibitor currently in clinical trials (Ph. 1/2).28−32 Aryl sulfonamide 2 (developed by Exelixis), an advanced lead compound that resulted from optimization of a high-throughput screening hit, exhibits moderate PI3K isoform selectivity but also inhibits the mammalian target of rapamycin (mTOR) and other off-target kinases.25 Recently, researchers at AstraZeneca reported a series of aminothiazole-based PI3Kγ inhibitors such as 3, which was characterized by X-ray crystallography in complex with mouse PI3Kδ (PDB ID 6FTN).26,27 The high isoform selectivity of 3 and its analogues was attributed to the ability of the cyclopropylethyl group to displace the DFG motif of PI3Kγ, which causes a unique γ isoform-specific conformational change.27
Figure 1.
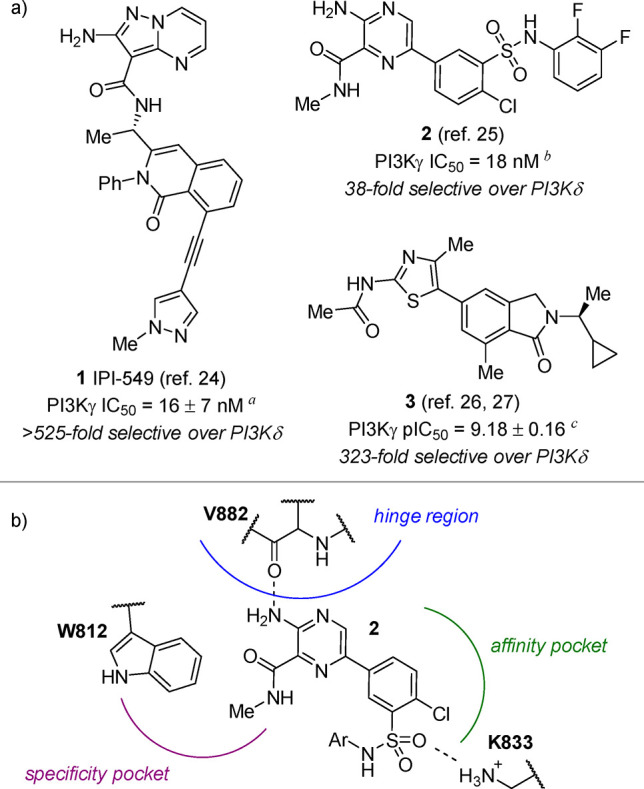
(a) Biochemical potency and selectivity values of PI3Kγ-selective inhibitors, as reported in the literature (see Supporting Information for a comparison of literature and in-house assay protocols). Inhibition determined using the ADP-Glo assay (Promega). Reported ATP Km = 29 ± 3.7 μM.34aAssay conditions: 3 mM ATP, 500 μM dioctanoyl-phosphatidylinositol-4,5-bisphosphate (diC8PIP2) as substrate, and 40 nM PI3Kγ. bAssay conditions: 1 μM ATP, 10 μM PIP2, and 30 nM PI3Kγ. cAssay conditions: 20 μM ATP, 80 μM diC8PIP2, and 1.2 nM PI3Kγ. (b) Schematic of typical inhibitor binding modes within PI3Kγ, exemplified by 2.25
As shown in Figure 1b using aryl sulfonamide 2 as a prototypical example, three regions exist within the ATP binding pocket of PI3Kγ, which are typically considered during inhibitor design.33 Interaction with the hinge region is considered essential for inhibitor activity; most known inhibitors engage in at least one hydrogen bond to V882. Inhibitor binding interactions within the affinity pocket are known to considerably influence both potency and selectivity against the other PI3K isoforms, as communicated in the discovery of 3.26,27 Most inhibitors that bind within the affinity pocket possess a functional group that engages K833 via hydrogen bonding or polar interactions. Finally, inhibitor binding within the specificity pocket also influences both potency and selectivity, as demonstrated in the discovery of 1.24 The W812 side chain in this region of the enzyme–inhibitor complex is well-positioned for an edge-to-face interaction with the inhibitor’s planar motif, as confirmed by the recently reported X-ray co-crystal structure of 1 in complex with human PI3Kγ (PDB ID 6XRL).35
Considering the promise of selective PI3Kγ inhibition for cancer immunotherapy, we pursued the design of novel inhibitor structures, utilizing the extensive body of medicinal chemistry knowledge as a source of inspiration. Our primary objective was to design a molecule possessing (1) similar or greater PI3Kγ inhibitory potency and isoform selectivity compared to known inhibitors, such as 1, as measured in both biochemical and cell-based PI3Kγ inhibition assays, and (2) suitable ADME properties for use in future in vivo studies. A molecule fulfilling these criteria would enable the eventual deconvolution and appreciation of the role of PI3Kγ inhibition in the context of our broader immunomodulatory work. To realize these goals, we embarked upon the discovery of novel inhibitors, initially through a pharmacophore mapping approach based on visual comparison of known literature inhibitors followed by a systematic SAR investigation of promising lead molecules, some of which have recently been described.35
Due to the high isoform selectivity and potency of 3 (IC50 = 4 ± 1 nM, as determined in our in-house biochemical assays, which measure a compound’s ability to inhibit the kinase activity of PI3K; see Supporting Information for details) and its analogues,26,27 we designed an initial set of compounds by attempting to mimic the hinge-binding acylaminothiazole moiety with a variety of functionally similar heterocycles (Table 1). Most of the compounds investigated contained both a hydrogen bond donor and a hydrogen bond acceptor motif, which could potentially create a stronger interaction with V882. We initiated our studies with racemic compounds 4 and 5, which bear a structural resemblance to 3. Although potency was substantially reduced for these compounds (IC50 = 0.6 and 0.53 μM, respectively), we were encouraged by the fact that replacement of the aminothiazole motif of 3 did not eliminate activity. Although triazolopyridine 6 showed decreased potency (IC50 = 1.6 μM), a less acidic analogue, (±)-7, was significantly more active (IC50 = 0.09 μM). In contrast, an isomeric analogue, 8, in which the pyrazole NH group is not properly positioned for interaction with V882, was significantly less potent (IC50 = 0.49 μM), confirming the importance of a hydrogen bond donor and acceptor pair for interaction with the hinge region. The stereochemical configuration of these compounds was also shown to be important, as 7, the (S) enantiomer of (±)-7, showed improved activity (IC50 = 0.035 μM) compared to the racemic compound. Although enantiopure pyrazolopyrazine 9 showed a 2-fold reduction in potency (IC50 = 0.08 μM) relative to 7, azaindole 10 (IC50 = 0.05 μM) had similar inhibitory activity to 7.
Table 1. Modification of the Hinge-Binding Region.


IC50 values measured using the ADP-Glo Lipid Kinase Assay (Promega) with 25 μM ATP and 50 μM phosphatidylinositol 4,5-bisphosphate as substrate.
Compound was tested once.
Based on these results and earlier studies by the Meng and Zhou laboratories,36 we evaluated the incorporation of a 4-pyridyl moiety at the C3 position of the two most promising scaffolds (i.e., azaindazole 7 and azaindole 10), which we envisioned would provide favorable interactions with the specificity pocket of PI3Kγ. Consistent with our hypothesis, azaindazole 11 (IC50 = 8 nM) was 4-fold more potent than 7, while azaindole 12 (IC50 = 3.4 nM) showed an even greater improvement in potency (approximately 15-fold) relative to the unsubstituted analogue, 10. (R)-12 was 3-fold less potent than 12, consistent with the previously reported superiority of the (S) enantiomer of the isoindolinone moiety of 3.26,27 These initial studies established 12 as a suitable starting point for further optimization.
Our subsequent efforts centered on evaluation of various C3-substituted derivatives of 12 (Table 2). To better distinguish between compounds that generally displayed low-nanomolar potency in the biochemical kinase activity assay, we further characterized these inhibitors under more physiologically relevant conditions using a functional cellular inhibition assay, which measures the extent of PI3K-mediated phosphorylation of Akt. To assess PI3K isoform selectivity, we employed routine enzymatic kinase activity assays for the α and δ PI3K isoforms only, as most of the compounds in this series were inactive against the β isoform at concentrations up to 10 μM (>1000-fold selectivity). We were pleased to find that compound 12 exhibited submicromolar activity in THP-1 cells (THP-1 IC50 = 0.22 μM), with excellent isoform selectivity (379-fold vs PI3Kα and 290-fold vs PI3Kδ). The observed difference between biochemical and cellular potency was not surprising, as cellular potency is frequently impacted by a compound’s permeability, protein binding, and off-target activity as well as differences in the concentration of ATP present under the assay conditions.
Table 2. Evaluation of Azaindole C3 Substituents.


Enzymatic kinase activity assay.
Ratio of PI3Kα or PI3Kδ enzymatic inhibition over PI3Kγ inhibition.
Determined using the AlphaLISA SureFire Ultra Akt assay (PerkinElmer) with THP-1 cells.
Racemate.
Compound was tested once.
Although the 3-pyridyl analogue 13 (IC50 = 7 nM, THP-1 IC50 = 0.4 μM) showed comparable potency to 12, the 2-pyridyl analogue (14) was substantially less active (IC50 = 33 nM, THP-1 IC50 = 3.5 μM), indicating a hydrogen bond acceptor extending outward from the core is advantageous.37 Other modifications preserving this paradigm (e.g., 15-19) were either tolerated or had reduced activity, with biochemical potency values ranging from 2.6 to 42 nM, and THP-1 cell potencies ranging from 0.20 to 3.5 μM. Pyrazole 20 (IC50 = 3.26 nM, THP-1 IC50 = 0.2 μM) showed comparable potency to 12, but incorporation of a saturated, basic amine substituent (e.g., 21, IC50 = 281 nM, THP-1 IC50 = 3.8 μM) proved to be detrimental to activity. Interestingly, carboxylic acid 22 and its amide derivatives, 23 and 24, were moderately active in the biochemical assay (IC50 = 13, 12, and 4.5 nM, respectively) despite being structurally distinct, although cellular potencies were more variable (THP-1 IC50 = 11, 1.3, and 0.32 μM, respectively).
In addition to 5- and 6-membered aromatic heterocycles, benzoic acids were identified as suitable C3 substituents. para-Benzoic acid 25 (IC50 = 2.5 nM, THP-1 IC50 = 0.14 μM), as well as several substituted benzoic acid analogues (26–30 and 33–35), exhibited improved cellular potency compared to 20. The ortho-methyl, -isopropyl, and -cyclopropyl analogues of 25 showed an encouraging increase in cellular potency (THP-1 IC50 = 0.078, 0.059, and 0.040 μM, respectively). Disubstituted analogues of 25 (e.g., 29 and 30, THP-1 IC50 = 0.09 and 0.07 μM, respectively), for which steric effects limit the conformational freedom of the carboxylate group and may attenuate productive interactions within the affinity pocket, showed a minimal reduction in potency relative to the monosubstituted counterparts. Electron-poor benzoic acid derivatives (e.g., 31 and 32, THP-1 IC50 = 0.7 and 0.35 μM, respectively) showed a substantial decrease in potency, while analogues bearing electron-donating methoxy or pyrrolidine ortho-substituents (e.g., 33 and 34, THP-1 IC50 = 0.17 μM for both) showed similar potency to unsubstituted benzoic acid 25.
Interestingly, while meta-benzoic acid 35 (THP-1 IC50 = 0.11 μM) exhibited similar potency to the para-analogue (25), introduction of an ortho-methyl substituent to 35 resulted in a 2-fold reduction in potency (36, THP-1 IC50 = 0.26 μM). This observation further corroborates the potential effect of the conformation of the carboxylate group on compound potency. In contrast to 25 and 35, ortho-benzoic acid derivative 37 exhibited a dramatic reduction in potency (THP-1 IC50 = 15 μM), consistent with the trends observed for isomeric pyridine analogues 12–14. Amide and sulfonamide analogues of 25 generally exhibited reduced potency (38 and 39, THP-1 IC50 = 0.23 and 0.40 μM, respectively). For the compounds shown in Table 2, cellular potencies generally correlated with biochemical potency. Apart from compounds with a particularly unique geometry (21, 22, and 37), a modest correlation was observed between cellular potency and biochemical assay-derived lipophilic efficiency values (see Supporting Information for details). Although there was substantial variation of isoform selectivity among the assayed compounds, most of the active compounds exhibited >100-fold selectivity against both PI3Kα and δ. The highest isoform selectivity was observed for amide 24 and benzoic acid analogues 29 and 36.
During the course of these investigations into the SAR of the C3 substituent, we also concurrently investigated the SAR of the N2′ isoindolinone substituent of 12 (selected due to its combination of potency, selectivity, and structural simplicity), which we envisioned would be situated within the affinity pocket of the PI3Kγ–inhibitor complex (Table 3). Replacement of the methyl group of the cyclopropylethyl moiety with either an ethyl group (40, IC50 = 6 nM, THP-1 IC50 = 0.8 μM) or a cyclopropyl group (41, IC50 = 6.8 nM, THP-1 IC50 = 0.8 μM) resulted in similar biochemical potency and selectivity but approximately 4-fold reduction in cellular potency. On the other hand, replacement of the cyclopropyl group with a methoxymethylene group (see 42, IC50 = 9 nM, THP-1 IC50 = 0.6 μM) resulted in a 3-fold reduction in both biochemical and cellular potency and a substantial reduction in isoform selectivity, confirming the important contribution of the cyclopropyl group to isoform selectivity. An even greater reduction in potency and selectivity was observed for tetrahydropyran 43 (IC50 = 50 nM, THP-1 IC50 = 1.0 μM), which reinforces the importance of the specific size and shape of the N-alkyl substituent. Analogues bearing a sterically bulky N-alkyl substituent, such as a t-butyl group (44, IC50 = 26 nM, THP-1 IC50 = 0.72 μM), showed diminished potency and isoform selectivity. Similarly, reduced potency and selectivity were observed for analogue 45 (IC50 = 28 nM), which bears an additional methyl group relative to 12 and is achiral. Despite its large size, arylethane 46 retained modest activity in the biochemical assay (IC50 = 40 nM), but its potency in cells was significantly decreased (THP-1 IC50 = 2.1 μM) relative to 12. Compound 47 (THP-1 IC50 = 1.6 μM), which bears an unbranched trifluoroethyl group, exhibited a significant reduction in both potency and selectivity compared to 12. Taken together, these results indicated that proper substitution of the isoindolinone nitrogen is critical to achieving high potency and selectivity. Moreover, these studies further supported the importance of the cyclopropylethyl group to achieving the optimal balance of potency and isoform selectivity exemplified by 12.
Table 3. Modification of the N2′ Substituent.


Enzymatic kinase activity assay.
Ratio of PI3Kα or PI3Kδ enzymatic inhibition over PI3Kγ inhibition.
Cellular potency in THP-1 cell line.
Compound was tested once.
Further characterization of 12 revealed that it was a potent inhibitor of the major CYP isoforms (IC50 values for the 1A2, 2D6, 2C9, 2C19, and 3A4 CYP isoforms were 0.7, 2.0, 0.3, <0.1, and 0.3 μM, respectively), possibly due to pyridine–iron coordination. On the other hand, pyrazole 20, which lacks the pyridine moiety, showed an improved CYP inhibition profile (IC50 values for the 1A2, 2D6, 2C9, 2C19, and 3A4 CYP isoforms were 2.9, 26, 1.4, 0.2, and 0.8 μM, respectively). Based on its decreased CYP inhibition and comparable cellular potency to 12, 20 was selected for further exploration of the isoindolinone substituent.
As shown in Table 4, improvement of cellular potency could be realized while maintaining isoform selectivity through various steric and electronic modifications of the C7′ group of 20. Incorporation of a sterically bulky isopropyl group (48, THP-1 IC50 = 0.3 μM) had little impact on potency or selectivity. Similarly, incorporation of an electron-donating methoxy group (49, THP-1 IC50 = 0.17 μM) or electron-withdrawing chloro- or nitrile groups (e.g., 50 and 51, THP-1 IC50 = 0.17 μM for both) also had little effect on potency or selectivity. Importantly, trifluoromethyl analogue 52 (THP-1 IC50 = 0.09 μM exhibited a 2-fold improvement in potency over 20 while maintaining high isoform selectivity. While both acetamide isomers 53 and 54 had reduced activity (THP-1 IC50 = 0.73 and 0.5 μM, respectively) relative to 20, C7′-sulfonamide and -sulfone analogues exhibited up to a 4-fold improvement in potency (55-58, THP-1 IC50 = 0.05–0.15 μM). Despite the promising potency of 55 and 58, these compounds exhibited extrahepatic clearance in rat models (6.0 and 4.5 L/h/kg, respectively), which precluded their further progression.
Table 4. Modification of the C7′ Substituent of 20.


Enzymatic kinase activity assay.
Ratio of PI3Kα or PI3Kδ enzymatic inhibition over PI3Kγ inhibition.
Cellular potency in THP-1 cell line.
Faced with the CYP inhibition liabilities of C3-pyridine analogues and the poor metabolic stability of the C3-pyrazole series, we turned our attention to the C3-benzoic acid series, exemplified by analogues 25, 26, and 29 (Table 5). Benzoic acid 25 and the corresponding ortho-substituted analogue, 26, both exhibited poor in vitro metabolic stability, as measured in human and rat hepatocytes, with rat intrinsic clearance (CLint) values ≥40 μL/min/106 cells. While hepatocyte stability could be improved through the incorporation of an additional ortho substituent on the benzoic acid moiety (29, rat CLint 2.0 μL/min/106 cells), this compound exhibited extrahepatic clearance in rat models (e.g., CL = 6.9 L/h/kg). While the C7′-CF3 analogues of 26 and 29 (see 59 and 60, THP-1 IC50 = 0.04 and 0.03 μM, respectively) demonstrated improved potency—consistent with our previous observations in the C3-pyrazole series—their metabolic stability was comparable to the C7′-Me analogues. The poor in vitro/in vivo correlation for this series of compounds may be due to active efflux, as Caco-2 permeability studies revealed parent compound 25 to have low permeability (Papp = 1.07 × 10–6 cm/sec) and an efflux ratio of 46.6.
Table 5. Further Characterization and SAR of C3 Benzoic Acid and Phenylacetic Acid Azaindole Derivatives.


Enzymatic kinase activity assay.
Ratio of PI3Kα or PI3Kδ enzymatic inhibition over PI3Kγ inhibition.
Cellular potency in THP-1 cell line.
Compound was tested once.
n.d. = value not determined.
Finally, a breakthrough came with the synthesis of C3 phenylacetic acid derivatives 61 (THP-1 IC50 = 0.16 μM) and 62 (THP-1 IC50 = 0.15 μM). Although these molecules showed somewhat diminished inhibitory activity relative to the benzoic acids, they retained high isoform selectivity and displayed an encouraging improvement in hepatocyte stability (rat CLint = 17 and 23 μL/min/106 cells for 61 and 62, respectively). Moreover, compound 62 exhibited significantly lower in vivo clearance (CL = 1.8 L/h/kg) relative to the benzoic acid analogues, which lack a methylene spacer. Gratifyingly, substitution of the α-methylene group of 61 (see 63–65, THP-1 IC50 = 0.11–0.18 μM) resulted in further improvements in human hepatocyte stability without substantially altering the potency or selectivity of these inhibitors. While this optimization campaign did not ultimately deliver a compound suitable for further development, compound 64 emerged as the most promising inhibitor due to its high cellular potency, excellent isoform selectivity, and acceptable pharmacokinetic profile.
The general synthetic routes used to access our azaindole/azaindazole inhibitors are shown in Scheme 1. The overall sequence is highly modular and allows for the introduction of diversity in several independent steps. Depending on the specific substrate, conditions such as the coupling partner, palladium catalyst, base, or solvent may vary (see Supporting Information for details). In general, the respective coupling steps can be done in any order to facilitate SAR exploration.
Scheme 1. General Synthetic Routes to Azaindole-Based Inhibitors.

Reagents and conditions: (a) NBS, (PhCO2)2, CCl4, 80 °C; (b) RNH2, B(OH)3, K2CO3, ACN, r.t.; (c) B2pin2, (dppf)PdCl2, KOAc, dioxane, 100 °C; (d) (dppf)PdCl2, Na2CO3, dioxane/H2O, 100 °C; then deprotection (conditions vary, see Supporting Information); (e) (dppf)PdCl2, Na2CO3, dioxane/H2O, 80 °C; (f) RNH2, HATU, i-Pr2NEt, DMF, 40 °C; (g) NBS, CH2Cl2, r.t.; (h) ArB(OR)2, (dppf)PdCl2, Na2CO3, dioxane/H2O, 100 °C; (i) TFA, r.t.; N,N-DMEDA, MeOH, 45 °C.
As shown in Scheme 1a, isoindolinone intermediates A and B are generated from methyl benzoate and primary amine precursors. These can be coupled to a variety of heteroaryl bromides to yield various simple inhibitors (4–10, 22). Alternatively, A or B can be coupled with C3-substituted azaindole/azaindazole intermediates, as shown in Scheme 1b. This sequence proved particularly useful during SAR evaluation of isoindolinone substituents. Finally, the sequence shown in Scheme 1c enabled rapid evaluation of C3 aryl substituents in combination with the most promising isoindolinone scaffolds.
Traditionally, ligand interactions within the affinity and specificity pockets of PI3Kγ have been thought to independently affect potency and selectivity, respectively. In contrast, our SAR investigations demonstrate that improvements in both metrics can be realized through careful design of inhibitors that are capable of occupying both regions of the enzyme active site. Through systematic SAR analysis, we have discovered a series of novel, potent, and selective azaindole-based PI3Kγ inhibitors. These compounds exhibit cellular IC50 values as low as 0.040 μM (28), while maintaining >300-fold selectivity against all other class I PI3K isoforms. In further inhibitor design iterations, C3 phenylacetic acid derivatives (e.g., 64) exhibited significantly improved pharmacokinetic properties, helping pave the way for future biological studies. The volume of data generated in this study will help expedite future campaigns toward potent and orally bioavailable PI3Kγ inhibitors for potential clinical applications.
Glossary
Abbreviations
- SAR
structure–activity relationship
- ATP
adenosine triphosphate
- ADME
absorption, distribution, metabolism, and excretion
- CYP
cytochrome P450
- NBS
N-bromosuccinimide
- ACN
acetonitrile
- r.t.
room temperature (23 °C)
- pin
pinacolato
- dppf
1,1′-bis(diphenylphosphino)ferrocene
- HATU
(1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate
- DMF
N,N-dimethylformamide
- SEM
2-(trimethylsilyl)ethoxymethyl
- TFA
trifluoroacetic acid
- N,N-DMEDA
N,N-dimethylethane-1,2-diamine
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00387.
Biological assay procedures, synthetic procedures and characterization of PI3Kγ inhibitors (PDF)
Author Contributions
D.H.M. and J.L.J. wrote the manuscript; D.H.M., J.L.J., E.U.S., G.M., K.V.L., M.R.L., and J.P.P. edited the manuscript; J.L.J., X.Y., R.T.-T., J.F., E.U.S., D.H.M., G.M., S.D., K.V.L., M.R.L., and J.P.P. contributed to inhibitor design; J.L.J., X.Y., R.T.-T., J.F., E.U.S., D.H.M., G.M., S.D., and K.V.L. synthesized the inhibitors; E.G., K.W., D.S., P.D., T.P., D.S., J.C., and L.J. provided ADME support; S.G.S., C.M., A.C., X.Z., S.W.Y. provided biological assay support; A.P., U.S., and N.P.W. provided protein support. All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): All authors are current or former employees of Arcus Biosciences.
Supplementary Material
References
- Wymann M. P.; Pirola L. Structure and Function of Phosphoinositide 3-Kinases. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 1998, 1436, 127–150. 10.1016/S0005-2760(98)00139-8. [DOI] [PubMed] [Google Scholar]
- Amzel L. M.; Huang C.-H.; Mandelker D.; Lengauer C.; Gabelli S. B.; Vogelstein B. Structural Comparisons of Class I Phosphoinositide 3-Kinases. Nat. Rev. Cancer 2008, 8, 665–669. 10.1038/nrc2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toker A.; Cantley L. C. Signalling through the Lipid Products of Phosphoinositide-3-OH Kinase. Nature 1997, 387, 673–676. 10.1038/42648. [DOI] [PubMed] [Google Scholar]
- Cahill C. M.; Rogers J. T.; Walker W. A. The Role of Phosphoinositide 3-Kinase Signaling in Intestinal Inflammation. J. Signal Transduction 2012, 2012, 1–13. 10.1155/2012/358476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T.; Irie-Sasaki J.; Jones R. G.; Oliveira-Dos-Santos A. J.; Stanford W. L.; Bolon B.; Wakeham A.; Itie A.; Bouchard D.; Kozieradzki I.; Joza N.; Mak T. W.; Ohashi P. S.; Suzuki A.; Penninger J. M. Function of PI3Kγ in Thymocyte Development, T Cell Activation, and Neutrophil Migration. Science 2000, 287, 1040–4046. 10.1126/science.287.5455.1040. [DOI] [PubMed] [Google Scholar]
- Kaneda M. M.; Messer K. S.; Ralainirina N.; Li H.; Leem C. J.; Gorjestani S.; Woo G.; Nguyen A. V.; Figueiredo C. C.; Foubert P.; Schmid M. C.; Pink M.; Winkler D. G.; Rausch M.; Palombella V. J.; Kutok J.; McGovern K.; Frazer K. A.; Wu X.; Karin M.; Sasik R.; Cohen E. E. W.; Varner J. A. PI3Kγ Is a Molecular Switch That Controls Immune Suppression. Nature 2016, 539, 437–442. 10.1038/nature19834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-García A.; Sánchez-Ruiz J.; Flores J. M.; Carrera A. C. Phosphatidylinositol 3-Kinase γ Inhibition Ameliorates Inflammation and Tumor Growth in a Model of Colitis-Associated Cancer. Gastroenterology 2010, 138, 1374–1383. 10.1053/j.gastro.2009.12.001. [DOI] [PubMed] [Google Scholar]
- Fruman D. A.; Rommel C. PI3K and Cancer: Lessons, Challenges and Opportunities. Nat. Rev. Drug Discovery 2014, 13, 140–156. 10.1038/nrd4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark A. K.; Sriskantharajah S.; Hessel E. M.; Okkenhaug K. PI3K Inhibitors in Inflammation, Autoimmunity and Cancer. Curr. Opin. Pharmacol. 2015, 23, 82–91. 10.1016/j.coph.2015.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorpe L. M.; Yuzugullu H.; Zhao J. J. PI3K in Cancer: Divergent Roles of Isoforms, Modes of Activation and Therapeutic Targeting. Nat. Rev. Cancer 2015, 15, 7–24. 10.1038/nrc3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneda M. M.; Cappello P.; Nguyen A. V.; Ralainirina N.; Hardamon C. R.; Foubert P.; Schmid M. C.; Sun P.; Mose E.; Bouvet M.; Lowy A. M.; Valasek M. A.; Sasik R.; Novelli F.; Hirsch E.; Varner J. A. Macrophage PI3Kγ Drives Pancreatic Ductal Adenocarcinoma Progression. Cancer Discovery 2016, 6, 870–885. 10.1158/2159-8290.CD-15-1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janku F. Phosphoinositide 3-Kinase (PI3K) Pathway Inhibitors in Solid Tumors: From Laboratory to Patients. Cancer Treat. Rev. 2017, 59, 93–101. 10.1016/j.ctrv.2017.07.005. [DOI] [PubMed] [Google Scholar]
- Zhao H.-F.; Wang J.; Shao W.; Wu C.-P.; Chen Z.-P.; To S.-S. T.; Li W.-P. Recent Advances in the Use of PI3K Inhibitors for Glioblastoma Multiforme: Current Preclinical and Clinical Development. Mol. Cancer 2017, 16, 1–16. 10.1186/s12943-017-0670-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W.; Qiu Y.; Kong D. Class I Phosphatidylinositol 3-Kinase Inhibitors for Cancer Therapy. Acta Pharm. Sin. B 2017, 7, 27–37. 10.1016/j.apsb.2016.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi S.; Durden D. L. Combinatorial Approach to Improve Cancer Immunotherapy: Rational Drug Design Strategy to Simultaneously Hit Multiple Targets to Kill Tumor Cells and to Activate the Immune System. J. Oncol. 2019, 2019, 1–18. 10.1155/2019/5245034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruman D. A. Targeting PI3K-Gamma in Non-Hodgkin Lymphoma. J. Clin. Oncol. 2019, 37, 932–934. 10.1200/JCO.19.00215. [DOI] [PubMed] [Google Scholar]
- De Henau O.; Rausch M.; Winkler D.; Campesato L. F.; Liu C.; Cymerman D. H.; Budhu S.; Ghosh A.; Pink M.; Tchaicha J.; Douglas M.; Tibbitts T.; Sharma S.; Proctor J.; Kosmider N.; White K.; Stern H.; Soglia J.; Adams J.; Palombella V. J.; McGovern K.; Kutok J. L.; Wolchok J. D.; Merghoub T. Overcoming Resistance to Checkpoint Blockade Therapy by Targeting PI3Kγ in Myeloid Cells. Nature 2016, 539, 443–447. 10.1038/nature20554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassetta L.; Kitamura T. Targeting Tumor-Associated Macrophages as a Potential Strategy to Enhance the Response to Immune Checkpoint Inhibitors. Front. Cell Dev. Biol. 2018, 6, 1–6. 10.3389/fcell.2018.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry M. W. D.; Abdulai R.; Mogemark M.; Petersen J.; Thomas M. J.; Valastro B.; Westin Eriksson A. Evolution of PI3Kγ and δ Inhibitors for Inflammatory and Autoimmune Diseases. J. Med. Chem. 2019, 62, 4783–4814. 10.1021/acs.jmedchem.8b01298. [DOI] [PubMed] [Google Scholar]
- Garces A. E.; Stocks M. J. Class 1 PI3K Clinical Candidates and Recent Inhibitor Design Strategies: A Medicinal Chemistry Perspective. J. Med. Chem. 2019, 62, 4815–4850. 10.1021/acs.jmedchem.8b01492. [DOI] [PubMed] [Google Scholar]
- Come J. H.; Collier P. N.; Henderson J. A.; Pierce A. C.; Davies R. J.; Le Tiran A.; O’Dowd H.; Bandarage U. K.; Cao J.; Deininger D.; Grey R.; Krueger E. B.; Lowe D. B.; Liang J.; Liao Y.; Messersmith D.; Nanthakumar S.; Sizensky E.; Wang J.; Xu J.; Chin E. Y.; Damagnez V.; Doran J. D.; Dworakowski W.; Griffith J. P.; Jacobs M. D.; Khare-Pandit S.; Mahajan S.; Moody C. S.; Aronov A. M. Design and Synthesis of a Novel Series of Orally Bioavailable, CNS-Penetrant, Isoform Selective Phosphoinositide 3-Kinase γ (PI3Kγ) Inhibitors with Potential for the Treatment of Multiple Sclerosis (MS). J. Med. Chem. 2018, 61, 5245–5256. 10.1021/acs.jmedchem.8b00085. [DOI] [PubMed] [Google Scholar]
- Shvartsbart A.; Combs A. P.; Falahatpisheh N.; Polam P.; Shao L.; Shepard S.. 3-(5-Amino-pyrazin-2-yl)-benzenesulfonamide Derivatives and Related Compounds as PI3K-gamma Kinase Inhibitors for Treating Cancer. WIPO 2019, WO 2019/126505 A1.
- Bellenie B. R.; Bloomfield G. C.; Bruce I.; Culshaw A. J.; Hall E. C.; Hollingworth G.; Neef J.; Spendiff M.; Watson S. J.. Amino Pyrazine Derivatives as Phosphatidylinositol 3-Kinase Inhibitors. WIPO 2015, WO 2015/162459 A1.
- Evans C. A.; Liu T.; Lescarbeau A.; Nair S. J.; Grenier L.; Pradeilles J. A.; Glenadel Q.; Tibbitts T.; Rowley A. M.; Dinitto J. P.; Brophy E. E.; O’Hearn E. L.; Ali J. A.; Winkler D. G.; Goldstein S. I.; O’Hearn P.; Martin C. M.; Hoyt J. G.; Soglia J. R.; Cheung C.; Pink M. M.; Proctor J. L.; Palombella V. J.; Tremblay M. R.; Castro A. C. Discovery of a Selective Phosphoinositide-3-Kinase (PI3K)-γ Inhibitor (IPI-549) as an Immuno-Oncology Clinical Candidate. ACS Med. Chem. Lett. 2016, 7, 862–867. 10.1021/acsmedchemlett.6b00238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leahy J. W.; Buhr C. A.; Johnson H. W. B.; Kim B. G.; Baik T.; Cannoy J.; Forsyth T. P.; Jeong J. W.; Lee M. S.; Ma S.; Noson K.; Wang L.; Williams M.; Nuss J. M.; Brooks E.; Foster P.; Goon L.; Heald N.; Holst C.; Jaeger C.; Lam S.; Lougheed J.; Nguyen L.; Plonowski A.; Song J.; Stout T.; Wu X.; Yakes M. F.; Yu P.; Zhang W.; Lamb P.; Raeber O. Discovery of a Novel Series of Potent and Orally Bioavailable Phosphoinositide 3-Kinase γ Inhibitors. J. Med. Chem. 2012, 55, 5467–5482. 10.1021/jm300403a. [DOI] [PubMed] [Google Scholar]
- Pemberton N.; Mogemark M.; Arlbrandt S.; Bold P.; Cox R. J.; Gardelli C.; Holden N. S.; Karabelas K.; Karlsson J.; Lever S.; Li X.; Lindmark H.; Norberg M.; Perry M. W. D.; Petersen J.; Blomqvist S. R.; Thomas M.; Tyrchan C.; Eriksson A. W.; Zlatoidsky P.; Öster L. Discovery of Highly Isoform Selective Orally Bioavailable Phosphoinositide 3-Kinase (PI3K)-γ Inhibitors. J. Med. Chem. 2018, 61, 5435–5441. 10.1021/acs.jmedchem.8b00447. [DOI] [PubMed] [Google Scholar]
- Gangadhara G.; Dahl G.; Bohnacker T.; Rae R.; Gunnarsson J.; Blaho S.; Öster L.; Lindmark H.; Karabelas K.; Pemberton N.; Tyrchan C.; Mogemark M.; Wymann M. P.; Williams R. L.; Perry M. W. D.; Papavoine T.; Petersen J. A Class of Highly Selective Inhibitors Bind to an Active State of PI3Kγ. Nat. Chem. Biol. 2019, 15, 348–357. 10.1038/s41589-018-0215-0. [DOI] [PubMed] [Google Scholar]
- A Dose-Escalation Study to Evaluate the Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of IPI-549. Last updated May 28, 2020. https://clinicaltrials.gov/ct2/show/NCT02637531 (accessed Jul 10, 2020).
- A Study to Evaluate Safety/Tolerability of Immunotherapy Combinations in Participants With Triple-Negative Breast Cancer or Gynecologic Malignancies. Last updated Jul 8, 2020. https://clinicaltrials.gov/ct2/show/NCT03719326 (accessed Jul 10, 2020).
- Window of Opportunity Study of IPI-549 in Patients With Locally Advanced HPV+ and HPV- Head and Neck Squamous Cell Carcinoma. Last updated May 21, 2020. https://clinicaltrials.gov/ct2/show/NCT03795610 (accessed Jul 10, 2020).
- Study to Evaluate the Efficacy/Safety of IPI-549 in Combination With Nivolumab in Patients With Advanced Urothelial Carcinoma (MARIO-275). Last updated May 18, 2020. https://clinicaltrials.gov/ct2/show/NCT03980041 (accessed Jul 10, 2020).
- Evaluation of IPI-549 Combined With Front-line Treatments in Pts. With Triple-Negative Breast Cancer or Renal Cell Carcinoma (MARIO-3). Last updated May 18, 2020. https://clinicaltrials.gov/ct2/show/NCT03961698 (accessed Jul 10, 2020).
- Miller M. S.; Thompson P. E.; Gabelli S. B. Structural Determinants of Isoform Selectivity in PI3K Inhibitors. Biomolecules 2019, 9, 1–35. 10.3390/biom9030082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidugiriene J.; Zegzouti H.; Goueli S. A. Evaluating the Utility of a Bioluminescent ADP-Detecting Assay for Lipid Kinases. Assay Drug Dev. Technol. 2009, 7, 585–597. 10.1089/adt.2009.0223. [DOI] [PubMed] [Google Scholar]
- Drew S. L.; Thomas-Tran R.; Beatty J. W.; Fournier J.; Lawson K. V.; Miles D. H.; Mata G.; Sharif E. U.; Yan X.; Mailyan A. K.; Ginn E.; Chen J.; Wong K.; Soni D.; Dhanota P.; Chen P.-Y.; Shaqfeh S. G.; Meleza C.; Pham A. T.; Chen A.; Zhao X.; Banuelos J.; Jin L.; Schindler U.; Walters M. J.; Young S. W.; Walker N. P.; Leleti M. R.; Powers J. P.; Jeffrey J. L.. Discovery of Potent and Selective PI3Kγ Inhibitors. J. Med. Chem. 2020, Article ASAP, 10.1021/acs.jmedchem.0c01203. [DOI] [PubMed] [Google Scholar]
- Yang C.; Zhang X.; Wang Y.; Yang Y.; Liu X.; Deng M.; Jia Y.; Ling Y.; Meng L.-H.; Zhou Y. Discovery of a Novel Series of 7-Azaindole Scaffold Derivatives as PI3K Inhibitors with Potent Activity. ACS Med. Chem. Lett. 2017, 8, 875–880. 10.1021/acsmedchemlett.7b00222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The 2-pyridyl substituent may also differentially affect the electronics of the central core, potentially decreasing the azaindole binding affinity to the hinge region of the enzyme.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.