

Abstract

The excitatory amino acid transporters (EAATs) mediate uptake of the major excitatory neurotransmitter l-glutamate (Glu). The essential functions governed by these transporters in regulating the central Glu level make them interesting therapeutic targets in a wide range of neurodegenerative and psychiatric disorders. l-Aspartate (Asp), another EAAT substrate, has served as a privileged scaffold for the development of EAAT inhibitors. In this study, we designed and synthesized the first β-indolyloxy Asp analogs 15a–d with the aim to probe a hitherto unexplored adjacent pocket to the substrate binding site. The pharmacological properties of 15a–d were characterized at hEAAT1-3 and rEAAT4 in a conventional [3H]-d-Asp uptake assay. Notably, thiophene analog 15b and the para-trifluoromethyl phenyl analog 15d were found to be hEAAT1,2-preferring inhibitors exhibiting IC50 values in the high nanomolar range (0.21–0.71 μM) at these two transporters versus IC50 values in the low micromolar range at EAAT3,4 (1.6–8.9 μM). In summary, the results presented herein open up for further structure–activity relationship studies of this new scaffold.
Keywords: EAAT inhibitors, TBOA analogs, aspartate analogs, glutamate transporters
In the central nervous system (CNS), the excitatory amino acid transporters (EAATs) mediate uptake of the major excitatory neurotransmitter l-glutamate (Glu) from the synaptic cleft and its close surroundings into glial cells and neurons.1,2 Thus, the EAATs play a vital role in regulation of glutamatergic neurotransmission including maintaining synaptic and extrasynaptic Glu concentrations below the neurotoxic level.2−4 To date, five EAAT subtypes have been identified, termed EAAT1–5 in humans, which correspond to GLAST, GLT-1, EAAC1, EAAT4, and EAAT5, respectively, in rodents. The two glial transporters EAAT1,2 and the neuronal transporter EAAT3 are the primary EAATs involved in synaptic Glu homeostasis in the brain, whereas EAAT4 and EAAT5 almost exclusively are expressed in the cerebellum and retina, respectively, where they mediate substrate-activated chloride conductance uncoupled from substrate translocation.2,5−7 EAAT dysfunction in the glutamatergic system has been associated with several neurological diseases, including Huntington’s disease, Alzheimer’s disease, Parkinson’s disease, and epilepsy, but also with psychiatric disorders such as schizophrenia and depression.8−11 Moreover, a series of studies suggest that EAAT-mediated Glu homeostasis fails dramatically in ischemia, where EAAT-mediated uptake is converted into transporter-mediated Glu release adding to the neurotoxicity observed in this condition.12 However, the exact contributions of EAATs to mechanisms underlying these physiological functions and pathophysiological conditions are still quite unclear, with conflicting observations having been reported in different studies. Development of subtype-selective small-molecule EAAT activators and inhibitors is thus an important objective, since such ligands constitute valuable pharmacological tools for study of the neurological processes underlying the aforementioned diseases and since new insight therein may promote development of novel drugs. In light of the different abundance and distribution of the EAAT subtypes in the CNS and their distinct roles governed here, selective modulation of the Glu uptake through inhibition of each of them may impact glutamatergic neurotransmission very differently, both when it comes to the characteristics and the levels of the induced effects but also in terms of the different CNS regions affected.
While only a few noncompetitive EAAT ligands have been disclosed,13−17 a large number of competitive EAAT ligands (substrates and nonsubstrate inhibitors) have been developed using Glu and dl-aspartate (Asp) but also the natural product kainic acid (KA) as leads (Figure 1).18−21 Early on in this field, the Asp analog dl-threo-β-hydroxyaspartate (dl-THA, Figure 1) was shown to inhibit Glu uptake in synaptosomes,22,23 and a series of l-THA analogs were reported by Shimamoto and co-workers, with the pinnacle analog being l-threo-3-benzyloxyaspartate (l-TBOA, Figure 1).24,25 Further incorporation of substituents onto the phenyl ring led to the identification of the nanomolar potent EAAT inhibitor (l-threo)-3-[3-[4-(trifluoromethyl)benzoylamino]benzyloxy]aspartate (TFB-TBOA, Figure 1).26,27 Although TFB-TBOA exhibited low nanomolar inhibitory activity at EAAT1,2 and submicromolar inhibitory activity at EAAT3,4, none of the 3-aryloxy substituted Asp analogs exhibited substantial subtype-selectivity across the EAATs. Elimination of the tethering ether oxygen in l-TBOA resulted in l-β-threo-benzyl-aspartate (l-β-BA, Figure 1), which surprisingly induced a 10-fold preference for EAAT3 over EAAT1 and EAAT2.28 On this basis, a series of β-arylsulfonamide functionalized Asp analogs were explored in our group, and the [3-(trifluoromethyl)phenyl]-sulfonamide-l-Asp analog (1a, Figure 1) was shown to be a EAAT2-selective inhibitor displaying low-micromolar potency at this transporter and 30- and 50-fold selectivity over EAAT1 and EAAT3, respectively.29 Another group has directed its efforts on exploring the conversion of the 4-carboxylate functionality (in Asp) to an amide, for further substitution. This work led to the identification of N-[4-(2-bromo-4,5-difluorophenoxy)phenyl]-l-asparagine (WAY-213613, Figure 1) as a midnanomolar potency EAAT2-selective inhibitor with 10- to 50-fold selectivity over other EAATs.30 Finally, upon hybridization of nonselective EAATs inhibitor l-TBOA with EAAT2-selective inhibitor WAY-213613, the EAAT inhibitor (l-threo)-2-amino-3-(benzyloxy)-4-[[4-(2-bromo-4,5-difluorophenoxy)phenyl]amino]-4-oxobutanoic acid (1b, Figure 1) was obtained. However, the strategy failed to preserve the EAAT2-selectivity of WAY-213613.31
Figure 1.

Chemical structures: (A) Endogenous EAAT substrates Glu and Asp; (B) EAAT inhibitors with l-Asp scaffold and pharmacological properties at EAAT1–4.29,31
In 2017, several X-ray crystal structures of thermostable EAAT1 variants (EAAT1cryst and EAAT1cryst-II) in complex with a substrate (l-Asp) or a nonsubstrate inhibitor (TFB-TBOA) with/without the allosteric inhibitor UCPH-101 were reported by Reyes group.32 The structure of EAAT1cryst displays a symmetric homotrimer, and each subunit is made up of two domains (Supporting Information): a scaffold domain (ScaD), including transmembrane helices TM1–TM2 and TM4–TM5, and a transport domain (TranD), including TM3, TM6–TM8, and re-entrant helical loops 1–2 (HP1–HP2).31,32 In this work, we chose to compare the TFB-TBOA/UCPH-101-bound EAAT1cryst crystal structure (PDB: 5mju) with the crystal structure of EAAT1cryst in complex with l-Asp and UCPH-101 (PDB: 5llm).
Superimposition of the two EAAT1cryst structures reveals that the binding modes of l-Asp and TFB-TBOA are very similar with respect to the amino acid and distal carboxylic acid functionalities (Figure 2a). Furthermore, the 3-benzyloxy group of TFB-TBOA is embedded in the re-entrant HP2 and is moved as much as 12 Å from its position in the Asp-bound complex (Figure 2a). The trifluoromethyl benzyl amide group occupies a hydrophobic cavity formed by residues of HP1b and TM7a and possibly residues in TM2/4c. Together, the observed ∼400-fold increase in EAAT1 inhibitory potency of TFB-TBOA compared with TBOA originates from these differences.32 From the X-ray structure it can also been seen that an additional subpocket is defined by HP2 and TM2/4c/7a (Figure 2a–b). In fact, no structure–activity relationship (SAR) study has been reported for ligands that project substituents in this direction. This motivated us to revisit the l-Asp scaffold and grow C-3 substituents in this direction with the aim to identify EAAT inhibitors with novel subtype-selectivity profiles.
Figure 2.
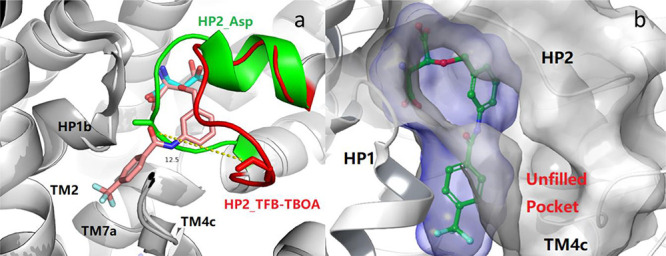
EAAT1 substrate binding pocket. (a) Overlay of crystal structures of EAAT1cryst (white cartoon) in complex with l-Asp (PDB: 5llm, cyan stick) and EAAT1cryst (gray cartoon) in complex with TFB-TBOA (PDB: 5mju, pink stick). Ile423 (red stick) of HP2 (red) in TFB-TBOA-bound complex is moved ∼12 Å away from its closed position (green stick) in the l-Asp-bound complex (green). (b) Excess space of TFB-TBOA binding pocket (PDB: 5mju). TFB-TBOA depicted as green and the ligand surface depicted as blue. Protein and protein surface depicted as gray (truncated at 7.0 Å from ligand).
In summary, a bulky substituent in the C-3 position of l-Asp dictates EAAT inhibition,25 which is further observed from comparing X-ray structures of EAAT1cryst in complex with l-Asp (PDB: 5llm) and with TFB-TBOA (PDB: 5mju).
To explore the identified unfilled subpocket in the EAAT substrate binding pocket, we designed a small series of l-threo-β-indolyloxy-Asp analogs (Figure 3). This scaffold jump (from an acyl aniline to an acyl indole) allows for introduction of substituents on the newly introduced C′-3 position, which are directed into this previously unexplored area of the EAAT substrate binding pocket. However, taking the hydrolytic instability of the N′-1 amide into consideration, the flipping of the indole ring was applied (Strategy A). Unfortunately, all attempts to synthesize analogs 16a–b failed in our hands (Supporting Information). At this point we concluded that exchange of the N-acyl group for an N-tosyl group (Strategy B) would solve our synthesis challenges and at the same time resolve the potential hydrolytic instability problem. However, the tetrahedral geometry of the sulfonamide is very different from the planar amide.33 Moreover, the slightly hydrophobic pocket environment of the binding pocket might favor compounds with lipophilic aromatic substituents at the C′-3 position.34 The following four analogs were included in a modeling study to address the applicability of a sulfonamide in the design: No substituent (15a) at the C′-3 position and analogs with (hetero)aromatic substitutions of differentiated size, including thiophene group (15b), phenyl group (15c), and para-trifluoromethyl phenyl group (15d).
Figure 3.
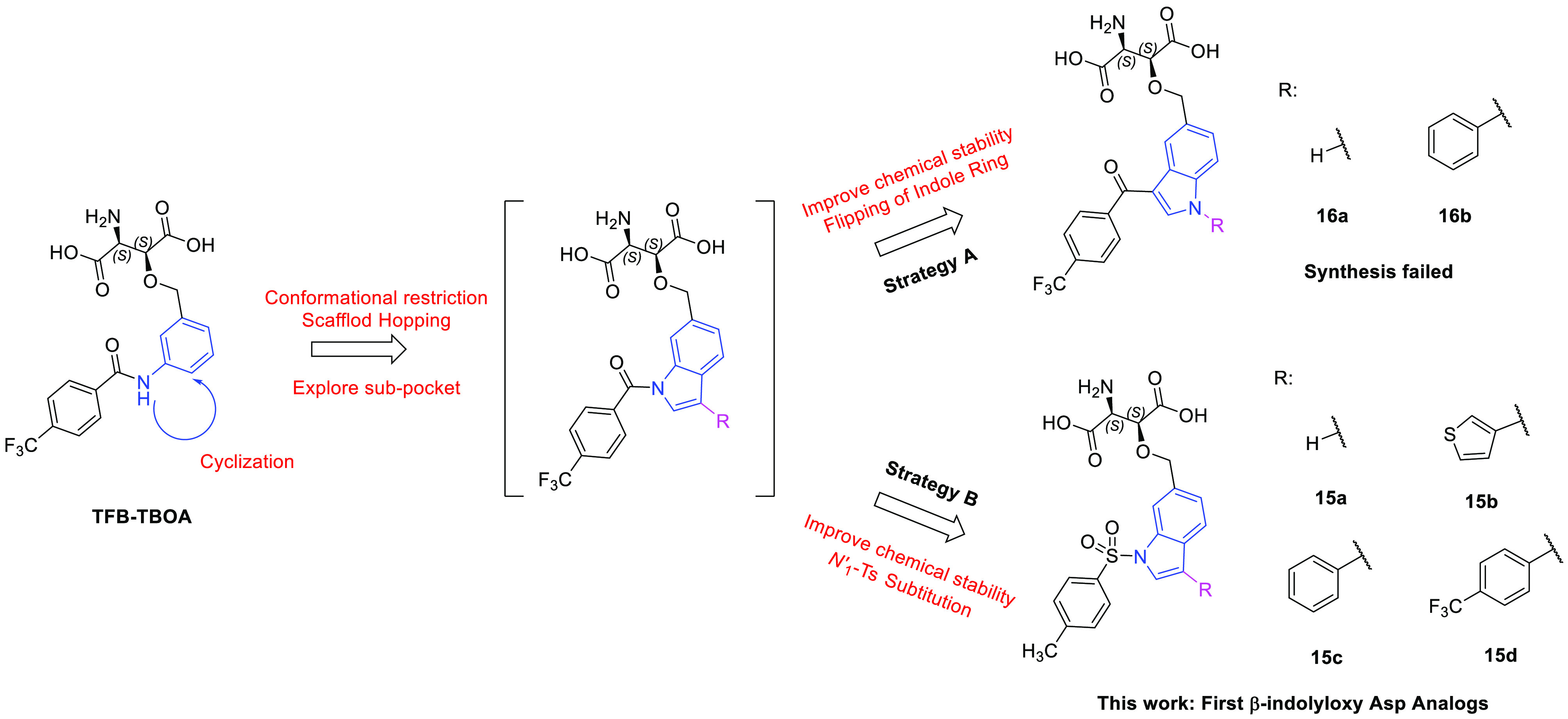
Rational design of the first β-indolyloxy Asp analogs 15a–d.
We first constructed a homology model of EAAT1 (from here on referred to as hmEAAT1) using the crystal structure of EAAT1cryst in complex with competitive inhibitor TFB-TBOA and allosteric inhibitor UCPH-101 (PDB: 5mju).32 Induced-fit docking of 15a–d, l-TBOA, and TFB-TBOA into hmEAAT1 was performed to estimate binding affinities of these inhibitors (Figure 4). The pose which showed good overlay with TFB-TBOA of EAAT1cryst complex with respect to the Asp part and lowest induced-fit docking score was selected (IFDScore) (Figure 4a). For the ligands investigated, the IFDScore ranking was determined to be 15d ≪ 15c ∼ 15b ∼ TFB-TBOA ∼ 15a ≪ l-TBOA (Table 1). The modeling study confirmed that TFB-TBOA binds significantly better to the EAAT1 protein compared to l-TBOA, and the newly designed analogs 15a–d were predicted to do that as well.
Figure 4.
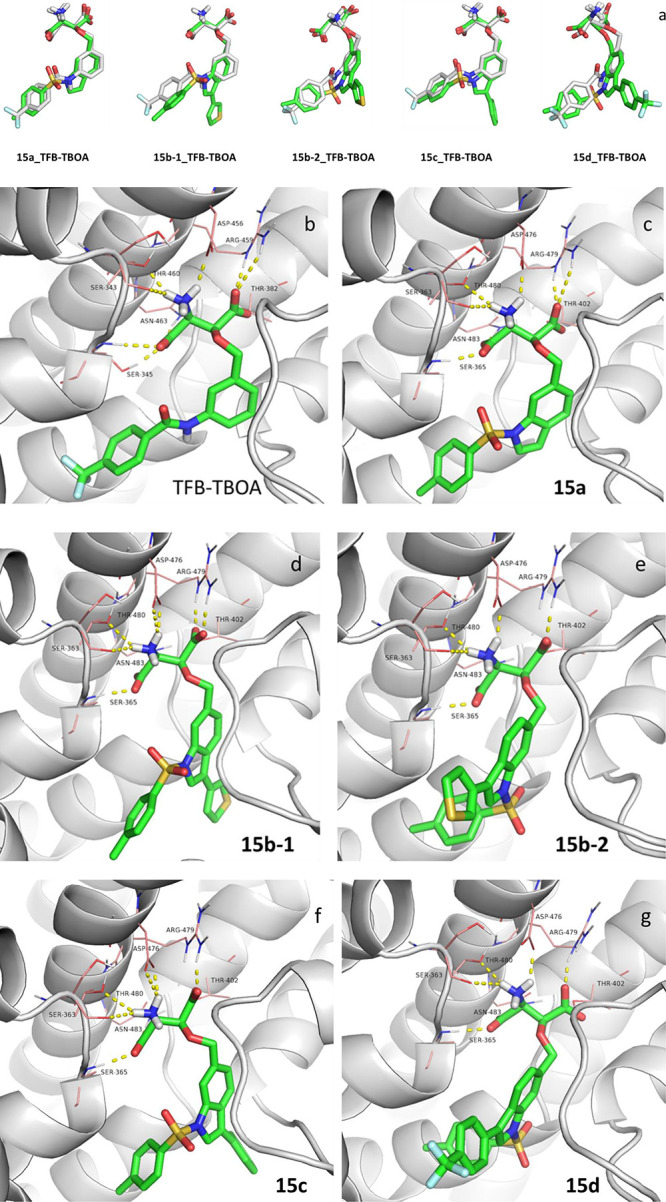
Modeling of ligands into EAAT1cryst (green stick) or into hmEAAT1 (white stick), and outline of the key residues (pink line) involved in binding the ligands and hydrogen bonds (yellow dash). (a) Overlay of binding pose of ligands (green) into hmEAAT1 and TFB-TBOA (PDB: 5mju, white) in EAAT1cryst. (b) Binding mode of TFB-TBOA with EAAT1cryst (PDB: 5mju). (c–g) Binding mode of compound 15a–d from induced-fit docking into hmEAAT1. Compound numbers are labeled on the respective figures.
Table 1. Functional Properties Displayed by l-Glu, dl-TBOA, and 15a–d at hEAAT1-HEK293, hEAAT2-HEK293, hEAAT3-HEK293, and rEAAT4-tsA201 Cell Lines in the [3H]-d-Aspartate Uptake Assaye.


IFDScore of the compound pose shown in the table was the one with the highest overlay quality like the reference ligand TFB-TBOA of EAAT1cryst complex (PDB: 5mju).
IC50 values were estimate from fitted concentration-inhibition curves that were not fully completed in the concentration ranges tested (up to 300 μM).
The purity of compound 15c tested was 80%.
IC50 values are given in μM with pIC50 ± S.E.M. values in brackets, and all data are based on 3 independent experiments (n = 3).
In the substrate-binding site of EAAT1cryst, the Asp fragment of TFB-TBOA is involved in a network of hydrogen-bond interactions with the nearby polar residues (Figure 4b), including Ser343 and Ser345 in HP1, Thr460, Asp456, and Arg459 in TM8b, and Thr382 in TM7b. The additional benzoylamino and trifluoromethyl groups of TFB-TBOA only formed van der Waals interactions with surrounding residues Ile96 and Ile100 in TM2 and with Pro372 and Thr376 in TM7a. According to the induced-fit docking results (Figure 4c–g), the key interactions with the Asp fragment were conserved among analogs 15a–d, and the residues at equivalent positions in hmEAAT1 were Ser363, Ser365, Thr402, Asp476, Arg479, and Thr480. No hydrogen bonds could be identified for the β-indolyloxy part. However, for the binding modes identified for 15b–d in hmEAAT1, residues of differentiation to EAAT2,3 could lead to subtype selectivity (Supporting Information). Furthermore, the incorporated groups on the C′-3 position of 15c and one binding mode of 15b oriented to the unfilled back subpocket constructed by TM7a/4c and HP2 (Figure 4d, 4f). For 15d and one different binding mode of 15b, the C′-3 substituents are directed into the hitherto unexplored front solvent exposure region shaped by TM2/4c and HP2 (Figure 4e, 4g). In summary, the modeling study substantiated our design and motivated us to continue with the synthesis and pharmacological evaluation of 15a–d.
Asymmetric synthesis of C-3 substituted Asp analogs is challenging due to the required l-threo configuration at vicinal chiral centers. This is evidenced by the published asymmetric syntheses of l-TBOA and TFB-TBOA which are both lengthy.27,35 Alternatively, the Lochner group has reported an asymmetric aminohydroxlyation strategy to access the l-threo isomer, however, with low enantiomeric selectivity.36 An efficient chemoenzymatic methodology by use of MAL-L384A as biocatalyst was reported by Poelarends group. However, the drawback is the narrow substrate specificity of enzyme together with its limited commercial availability.31,37,38
We thus turned to develop a new strategy. Our retrosynthetic analysis suggests that 15a–d could be derived from O-alkylation of dimethyl (l-threo)-N-Boc-3-hydroxyaspartate (7, Asp fragment) with C′-3 appropriately substituted (R group) 6-(bromomethyl)-1-tosyl-1H-indole (13a–d, indole part) followed by deprotection (Scheme 1). The key precursor 7 is readily obtained from commercially available d-tartaric acid (2) through the reported synthesis route for dimethyl (3S)-2-azido-3-hydroxysuccinate (5a/5b), followed by azide reduction and protection.39 We predicted the indole part 13a–d is obtained from commercially available methyl 1H-indole-6-carboxylate (8) via bromination, N′-1 position substitution, Suzuki–Miyaura reaction, reduction, and nucleophilic substitution.
Scheme 1. Retrosynthetic Analysis of Newly Designed EAAT Inhibitors 15a–d.

The synthesis of Asp fragment 7 (Scheme 2) commenced with esterification of commercially available d-tartaric acid (2) under standard conditions to give dimethyl ester 3.40 After treatment with HBr in AcOH and acetate hydrolysis in acidic methanol, the corresponding bromo alcohol 4 was obtained in 74% yield over two steps. Nucleophilic substitution with sodium azide gave 5 in 68% yield as a mixture of l-threo5a and d-erythro5b diastereomers (threo/erythro = 5.6:1). Hereafter the diastereomeric azides 5a/5b were reduced to amine 6a/6b by H2 and 10% Pd/C, and the diastereomeric mixture was separated by column chromatography to give the l-threo isomer 6a isolated in 36%. The absolute configuration of 6a was confirmed by comparison with reported 1H NMR data for the analogous diethyl ester analog.41 After Boc-protection, optically pure Asp fragment 7 was ready for the following O-alkylation. Overall, key intermediate 7 was prepared in only five steps with an 11% overall yield.
Scheme 2. Synthesis of Asp Fragment 7 from d-Tartaric Acid 2.

Reagents and conditions: (a) SOCl2, MeOH, 0 °C to reflux, 4 h, quant.; (b) (i) HBr/AcOH, 0 °C to rt, overnight, (ii) AcCl, MeOH, 0 °C to reflux, 4 h, 74%; (c) NaN3, DMF, rt, overnight, 68%; (d) H2,10% Pd/C, MeOH, 45 °C, 3 h, 36%; (e) (Boc)2O, Et3N, DCM, 10 h, 62%.
For the synthesis of indole part 13a–d (Scheme 3), the route began with selective bromination at the C′-3 position of 6-methoxycarbonylindole with N-bromosuccinimide (NBS) at low temperature (−78 °C) to give bromine 9 in 80% yield. The strong base NaH was used to deprotonate the indole ring of compound 9 (or 8), which was then reacted with tosyl chloride to give N′-1 tosylamides 10 (or 11a) in over 90% yield. The appropriate boronic acid was used in the subsequent palladium coupling with 10 to give the series of analogs 11b–d in 69%–79% yield. To introduce the alkyl bromide functionality, esters 11a–d were first reduced to their corresponding alcohols 12a–d by LiAlH4. After some experimentation, we established that excess equivalents of MsCl (3.0 equiv), LiBr (8.0 equiv), and Et3N (4.0 equiv) in THF provided the desired alkyl bromides 13a–d in 68%–79% yield.42
Scheme 3. Synthesis of Compounds 15a–d from Methyl 6-Indolecarboxylate 8.

Reagents and conditions: (a) NBS, THF, −78 °C, 2 h, 80%; (b) TsCl, NaH, THF, 0 °C to rt, 30 min, 98% (11a), 90% (10); (c) RB(OH)2, Pd(dppf)Cl2·CH2Cl2, Cs2CO3, dioxane/H2O (4:1), 110 °C (M.W.), 15 min, 73% (11b), 69% (11c), 79% (11d); (d) LiAlH4, THF, 0 °C to rt, 30 min, 86% (12a), 84% (12b), 80% (12c), 87% (12d); (e) MsCl, Et3N, LiBr, THF, 0 °C to rt, 4 h, 68% (13a), 73% (13b), 69% (13c), 79% (13d); (f) compound 7, NaH, DMF, −15 °C, 4 h; (g) (i) TFA, DCM, 0 °C to rt, 2 h, (ii) LiOH·H2O, THF/H2O (1:1), 5 h, prep. HPLC, 8% (15a), 6% (15b), 5% (15c), 3% (15d) over 3 steps.
The synthesis of target molecules 15a–d (Scheme 3) was finalized by treatment of Asp fragment 7 with NaH and appropriate bromines 13a–d at low temperature (−15 °C) for 4 h to minimize the risk of epimerization to give the desired ethers 14a–d. Deprotection of 14a–d was conducted by first treatment with TFA then hydrolysis with LiOH, to give the desired targets 15a as a HCl salt, and 15b–d as TFA salts in yields of 3%–8%, over three steps.
The pharmacological properties of newly synthesized β-indolyloxy Asp analogs 15a–d were determined at stable hEAAT1-HEK293, hEAAT2-HEK293, hEAAT3-HEK293, and rEAAT4-tsA201 cell lines in a conventional [3H]-d-Asp uptake assay (Table 1 and Figure 5). dl-TBOA and Glu were assayed in parallel as reference ligands.
Figure 5.

Concentration-inhibition curves for dl-TBOA, l-Glu, and compounds 15a–d at stable hEAAT1-HEK293, hEAAT2-HEK293, hEAAT3-HEK293, and rEAAT4-tsA201 cell lines in the [3H]-d-Asp uptake assay. Data are from representative experiments (out of a total of 3 independent experiments) and given as mean ± SD values in counts per min (CPM) based on duplicate determinations.
The synthesized β-indolyloxy Asp analogs 15a–d were all found to be inhibitors at EAAT1–4 with preference ranging from 3- to 42-fold for hEAAT1,2 over hEAAT3 and rEAAT4. In detais, analog 15a (R = H) was slightly less potent compared to dl-TBOA at the EAAT1–4 subtypes. The phenyl analog 15c displayed comparable inhibitory potencies at hEAAT1,2 with those of dl-TBOA, but was a 6-fold weaker inhibitor of hEAAT3 compared to dl-TBOA, thus exhibiting a ∼30-fold preference for hEAAT1,2 over hEAAT3. Notably, both 15b and 15d displayed high nanomolar potencies as hEAAT1,2 inhibitors and low micromolar inhibitory potencies at hEAAT3 and rEAAT4. The para-trifluoromethyl phenyl analog 15d displayed ∼25-fold and ∼5-fold preference for hEAAT1,2 over hEAAT3 and rEAAT4, respectively. The thiophene analog 15b displayed 3-, 42-, and 10-fold higher inhibitor potency at hEAAT2 than at hEAAT1, hEAAT3, and rEAAT4, respectively. Thus, the increase in inhibitory potency at the EAATs seemed to correlate with increasing size of substitutions compared to dl-TBOA. Furthermore, from the SAR studies of 15a and 15b–d, the substitutions on the C′-3 position of the indole ring generally were observed to increase the preference for hEAAT1,2 over hEAAT3 and rEAAT4.
On the comparison of functional data (EAAT1 potencies) with calculated IFDScores (EAAT1 binding affinities) of 15a–d (Table 1) it is evident that the calculated binding affinities do not translate well into experimentally determined inhibitory potencies. For 15a–d, the calculated IFDScores lie in the order of that for TFB-TBOA, but the experimentally determined potencies are generally closer to that of l-TBOA. This discrepancy lies at the fundamental problem between binding affinities and functional potencies, calculated and/or experimentally determined, which is an ongoing research topic.43−45 However, the calculated IFDscores did predict 15d to be the analog with the highest EAAT1 affinity, and this analog was shown to be the most potent analog in the series (EAAT1 data).
In conclusion, we have explored four β-indolyloxy Asp analogs as inhibitors of the EAATs. A new synthesis of Asp derivatives with aryloxy substituents at the C-3 position was developed, and a SAR study probing the effects of the different substitutions at the C′-3 position of the indole ring was carried out. The thiophene analog 15b and the para-trifluoromethyl phenyl analog 15d were found to be potent hEAAT1,2-preferring inhibitors, displaying IC50 values in the high nanomolar range (0.21–0.71 μM) at EAAT1,2 and low micromolar range at (1.6–8.9 μM) EAAT3,4. Our in silico study failed to shed substantial light on the molecular bases for the observed differences in pharmacological profiles of the analogs at EAAT1–4. However, it did elucidate the existence of the extra area adjacent to the substrate-binding pocket constructed by TM2/4c or TM7a/4c from the ScaD in the transporter when binding bulky ligands like TFB-TBOA. Occupancy of the extra pocket by substituents in analogs could benefit the EAAT activity and subtype-selectivity/preference, as evidenced by the functional profiles by 15a versus 15b–d. Therefore, this extra pocket could constitute an interesting target region for analogs in the future search for subtype-selective EAAT ligands. Furthermore, considering the relatively limited SAR work done focused on the C′-3 position of the indole ring in the present work, it remains to be seen whether future SAR studies based on this novel scaffold could yield lead analogs with further increased inhibitory potencies and interesting selectivity profiles.
Acknowledgments
The authors gratefully acknowledge the financial support from the Lundbeck Foundation and the China Scholarship Council (CSC). Peter Kovermann and Christoph Fahlke are thanked for their generous gift of the stable rEAAT4-tsA201 cell line.
Glossary
Abbreviations
- CNS
central nervous system
- ddpf
1,1′-bis(diphenylphosphino)ferrocene
- EAAT
excitatory amino acid transporter
- l-TBOA
l-threo-benzyloxyaspartate
- IFDScore
induced-fit docking score
- MsCl
methanesulfonyl chloride
- NBS
N-bromosuccinimide
- TFB-TBOA
(l-threo)-3-[3-[4-(trifluoromethyl)benzoylamino]benzyloxy]aspartate
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- DCM
dichloromethane
- Boc
tert-butoxycarbonyl.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00342.
HPLC purity of compounds 15a–d and solvent used for dissolving the compounds (ZIP)
Homology model of EAAT1 built on EAAT1cryst (PDB: 5mju) (PDB)
Experimental section describing the synthesis procedures, pharmacological characterization, and modeling. Overview of crystal structure of EAAT1cryst and domain organization (PDB: 5mju), sequence alignment, and structure overlay of hEAAT1 with EAAT1cryst (PDB: 5mju), comparison of residues difference within hmEAAT1–3 for the binding model of TFB-TBOA and 15a–d into hmEAAT1, induced-fit docking score for l-TBOA, TFB-TBOA, and 15a–d and synthesis route for analog 16b (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Seal R. P.; Amara S. G. Excitatory Amino Acid Transporters: A Family in Flux. Annu. Rev. Pharmacol. Toxicol. 1999, 39 (1), 431–456. 10.1146/annurev.pharmtox.39.1.431. [DOI] [PubMed] [Google Scholar]
- Danbolt N. Glutamate Uptake. Prog. Neurobiol. 2001, 65 (1), 1–105. 10.1016/S0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Frandsen A.; Schousboe A. Development of Excitatory Amino Acid Induced Cytotoxicity in Cultured Neurons. Int. J. Dev. Neurosci. 1990, 8 (2), 209–216. 10.1016/0736-5748(90)90013-R. [DOI] [PubMed] [Google Scholar]
- Rosenberg P.; Amin S.; Leitner M. Glutamate Uptake Disguises Neurotoxic Potency of Glutamate Agonists in Cerebral Cortex in Dissociated Cell Culture. J. Neurosci. 1992, 12 (1), 56–61. 10.1523/JNEUROSCI.12-01-00056.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen A. A.; Fahlke C.; Bjørn-Yoshimoto W. E.; Bunch L. Excitatory Amino Acid Transporters: Recent Insights into Molecular Mechanisms, Novel Modes of Modulation and New Therapeutic Possibilities. Curr. Opin. Pharmacol. 2015, 20, 116–123. 10.1016/j.coph.2014.10.008. [DOI] [PubMed] [Google Scholar]
- Vandenberg R. J.; Ryan R. M. Mechanisms of Glutamate Transport. Physiol. Rev. 2013, 93 (4), 1621–1657. 10.1152/physrev.00007.2013. [DOI] [PubMed] [Google Scholar]
- Arriza J.; Fairman W.; Wadiche J.; Murdoch G.; Kavanaugh M.; Amara S. Functional Comparisons of Three Glutamate Transporter Subtypes Cloned from Human Motor Cortex. J. Neurosci. 1994, 14 (9), 5559–5569. 10.1523/JNEUROSCI.14-09-05559.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges R. J.; Esslinger C. S. The Excitatory Amino Acid Transporters: Pharmacological Insights on Substrate and Inhibitor Specificity of the EAAT Subtypes. Pharmacol. Ther. 2005, 107 (3), 271–285. 10.1016/j.pharmthera.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Benarroch E. E. Glutamate Transporters: Diversity, Function, and Involvement in Neurologic Disease. Neurology 2010, 74 (3), 259–264. 10.1212/WNL.0b013e3181cc89e3. [DOI] [PubMed] [Google Scholar]
- Horiuchi Y.; Iida S.; Koga M.; Ishiguro H.; Iijima Y.; Inada T.; Watanabe Y.; Someya T.; Ujike H.; Iwata N.; Ozaki N.; Kunugi H.; Tochigi M.; Itokawa M.; Arai M.; Niizato K.; Iritani S.; Kakita A.; Takahashi H.; Nawa H.; Arinami T. Association of SNPs Linked to Increased Expression of SLC1A1 with Schizophrenia. Am. J. Med. Genet., Part B 2012, 159B (1), 30–37. 10.1002/ajmg.b.31249. [DOI] [PubMed] [Google Scholar]
- Tanaka K.; Watase K.; Manabe T.; Yamada K.; Watanabe M.; Takahashi K.; Iwama H.; Nishikawa T.; Ichihara N.; Kikuchi T.; Okuyama S.; Kawashima N.; Hori S.; Takimoto M.; Wada K. Epilepsy and Exacerbation of Brain Injury in Mice Lacking the Glutamate Transporter GLT-1. Science 1997, 276 (5319), 1699–1702. 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- Rossi D. J.; Oshima T.; Attwell D. Glutamate Release in Severe Brain Ischaemia is Mainly by Reversed Uptake. Nature 2000, 403, 316–321. 10.1038/35002090. [DOI] [PubMed] [Google Scholar]
- Jensen A. A.; Erichsen M. N.; Nielsen C. W.; Stensbøl T. B.; Bunch L. Discovery of the First Selective Inhibitor of Excitatory Amino Acid Transporter Subtype 1. J. Med. Chem. 2009, 52 (4), 912–915. 10.1021/jm8013458. [DOI] [PubMed] [Google Scholar]
- Erichsen M. N.; Huynh T. H.; Abrahamsen B.; Bastlund J. F.; Bundgaard C.; Monrad O.; Bekker-Jensen A.; Nielsen C. W.; Frydenvang K.; Jensen A. A.; Bunch L. Structure-Activity Relationship Study of First Selective Inhibitor of Excitatory Amino Acid Transporter Subtype 1:2-Amino-4-(4-methoxyphenyl)-7-(naphthalen-1-yl)-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (UCPH-101). J. Med. Chem. 2010, 53 (19), 7180–91. 10.1021/jm1009154. [DOI] [PubMed] [Google Scholar]
- Hansen S. W.; Erichsen M. N.; Fu B.; Bjørn-Yoshimoto W. E.; Abrahamsen B.; Hansen J. C.; Jensen A. A.; Bunch L. Identification of a New Class of Selective Excitatory Amino Acid Transporter Subtype 1 (EAAT1) Inhibitors Followed by a Structure-Activity Relationship Study. J. Med. Chem. 2016, 59 (19), 8757–8770. 10.1021/acs.jmedchem.6b01058. [DOI] [PubMed] [Google Scholar]
- Kortagere S.; Mortensen O. V.; Xia J.; Lester W.; Fang Y.; Srikanth Y.; Salvino J. M.; Fontana A. C. K. Identification of Novel Allosteric Modulators of Glutamate Transporter EAAT2. ACS Chem. Neurosci. 2018, 9 (3), 522–534. 10.1021/acschemneuro.7b00308. [DOI] [PubMed] [Google Scholar]
- Wu P.; Bjørn-Yoshimoto W. E.; Staudt M.; Jensen A. A.; Bunch L. Identification and Structure-Activity Relationship Study of Imidazo[1,2-a]pyridine-3-amines as First Selective Inhibitors of Excitatory Amino Acid Transporter Subtype 3 (EAAT3). ACS Chem. Neurosci. 2019, 10 (10), 4414–4429. 10.1021/acschemneuro.9b00447. [DOI] [PubMed] [Google Scholar]
- Bunch L.; Erichsen M. N; Jensen A. A Excitatory Amino Acid Transporters as Potential Drug Targets. Expert Opin. Ther. Targets 2009, 13 (6), 719–731. 10.1517/14728220902926127. [DOI] [PubMed] [Google Scholar]
- Dunlop J.; Butera J. Ligands Targeting the Excitatory Amino Acid Transporters (EAATs). Curr. Top. Med. Chem. 2006, 6 (17), 1897–1906. 10.2174/156802606778249829. [DOI] [PubMed] [Google Scholar]
- Faure S.; Jensen A. A.; Maurat V.; Xin G.; Sagot E.; Aitken D. J.; Bolte J.; Gefflaut T.; Bunch L. Stereoselective Chemo-Enzymatic Synthesis of the Four Stereoisomers of l-2-(2-Carboxycyclobutyl)-glycine and Pharmacological Characterization at Human Excitatory Amino Acid Transporter Subtypes 1, 2 and 3. J. Med. Chem. 2006, 49 (22), 6532–8. 10.1021/jm060822s. [DOI] [PubMed] [Google Scholar]
- Jensen A. A.; Brøuner-Osborne H. Pharmacological Characterization of Human Excitatory Amino Acid Transporters EAAT1, EAAT2 and EAAT3 in a Fluorescence-Based Membrane Potential Assay. Biochem. Pharmacol. 2004, 67 (11), 2115–2127. 10.1016/j.bcp.2004.02.013. [DOI] [PubMed] [Google Scholar]
- Balcar V. J.; Johnston G. A. R. The Structural Specificity of the High Affinity Uptake of l-Glutamate and l-Asparate by Rat Brain Slices. J. Neurochem. 1972, 19 (11), 2657–2666. 10.1111/j.1471-4159.1972.tb01325.x. [DOI] [PubMed] [Google Scholar]
- Barbour B.; Brew H.; Attwell D. Electrogenic Uptake of Glutamate and Aspartate into Glial Cells Isolated from the Salamander (Ambystoma) Retina. J. Physiol. 1991, 436 (1), 169–193. 10.1113/jphysiol.1991.sp018545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamoto K.; Lebrun B.; Yasudakamatani Y.; Sakaitani M.; Shigeri Y.; Yumoto N.; Nakajima T. dl-threo-β-Benzyloxyaspartate, a Potent Blocker of Excitatory Amino Acid. Mol. Pharmacol. 1998, 53 (2), 195–201. 10.1124/mol.53.2.195. [DOI] [PubMed] [Google Scholar]
- Lebrun B.; Sakaitani M.; Shimamoto K.; Yasuda-Kamatani Y.; Nakajima T. New β-Hydroxyaspartate Derivatives are Competitive Blockers for the Bovine Glutamate/Aspartate Transporter. J. Biol. Chem. 1997, 272 (33), 20336–9. 10.1074/jbc.272.33.20336. [DOI] [PubMed] [Google Scholar]
- Shimamoto K.; Sakai R.; Takaoka K.; Yumoto N.; Shigeri Y. Characterization of Novel l-threo-Benzyloxyaspartate Derivatives, Potent Blockers of the Glutamate Transporters. Mol. Pharmacol. 2004, 65 (4), 1008–1015. 10.1124/mol.65.4.1008. [DOI] [PubMed] [Google Scholar]
- Shimamoto K.β-Benzyloxyaspartic Acid Derivatives with Amino Group on Benzene Ring. WO03000698A1, 2003.
- Esslinger C. S.; Agarwal S.; Gerdes J.; Wilson P. A.; Davis E. S.; Awes A. N.; O’Brien E.; Mavencamp T.; Koch H. P.; Poulsen D. J.; Rhoderick J. F.; Chamberlin A. R.; Kavanaugh M. P.; Bridges R. J. The Substituted Aspartate Analogue l-β-threo-Benzyl-Aspartate Preferentially Inhibits the Neuronal Excitatory Amino Acid Transporter EAAT3. Neuropharmacology 2005, 49 (6), 850–61. 10.1016/j.neuropharm.2005.08.009. [DOI] [PubMed] [Google Scholar]
- Hansen J. C.; Bjørn-Yoshimoto W. E.; Bisballe N.; Nielsen B.; Jensen A. A.; Bunch L. β-Sulfonamino Functionalized Aspartate Analogues as Excitatory Amino Acid Transporter Inhibitors: Distinct Subtype Selectivity Profiles Arising from Subtle Structural Differences. J. Med. Chem. 2016, 59 (19), 8771–8786. 10.1021/acs.jmedchem.6b01066. [DOI] [PubMed] [Google Scholar]
- Dunlop J.; McIlvain H. B.; Carrick T. A.; Jow B.; Lu Q.; Kowal D.; Lin S.; Greenfield A.; Grosanu C.; Fan K.; Petroski R.; Williams J.; Foster A.; Butera J. Characterization of Novel Aryl-Ether, Biaryl, and Fluorene Aspartic Acid and Diaminopropionic Acid Analogs as Potent Inhibitors of the High-Affinity Glutamate Transporter EAAT2. Mol. Pharmacol. 2005, 68 (4), 974–982. 10.1124/mol.105.012005. [DOI] [PubMed] [Google Scholar]
- Fu H.; Zhang J.; Tepper P. G.; Bunch L.; Jensen A. A.; Poelarends G. J. Chemoenzymatic Synthesis and Pharmacological Characterization of Functionalized Aspartate Analogues as Novel Excitatory Amino Acid Transporter Inhibitors. J. Med. Chem. 2018, 61 (17), 7741–7753. 10.1021/acs.jmedchem.8b00700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canul-Tec J. C.; Assal R.; Cirri E.; Legrand P.; Brier S.; Chamot-Rooke J.; Reyes N. Structure and Allosteric Inhibition of Excitatory Amino Acid Transporter 1. Nature 2017, 544 (7651), 446–451. 10.1038/nature22064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brameld K. A.; Kuhn B.; Reuter D. C.; Stahl M. Small Molecule Conformational Preferences Derived from Crystal Structure Data. A Medicinal Chemistry Focused Analysis. J. Chem. Inf. Model. 2008, 48 (1), 1–24. 10.1021/ci7002494. [DOI] [PubMed] [Google Scholar]
- Pedretti A.; De Luca L.; Sciarrillo C.; Vistoli G. Fragmental Modeling of Human Glutamate Transporter EAAT1 and Analysis of its Binding Modes by Docking and Pharmacophore Mapping. ChemMedChem 2008, 3 (1), 79–90. 10.1002/cmdc.200700197. [DOI] [PubMed] [Google Scholar]
- Shimamoto K.; Shigeri Y.; Yasuda-Kamatani Y.; Lebrun B.; Yumoto N.; Nakajima T. Syntheses of Optically Pure β-Hydroxyaspartate Derivatives as Glutamate Transporter Blockers. Bioorg. Med. Chem. Lett. 2000, 10 (21), 2407–2410. 10.1016/S0960-894X(00)00487-X. [DOI] [PubMed] [Google Scholar]
- Leuenberger M.; Ritler A.; Simonin A.; Hediger M. A.; Lochner M. Concise Asymmetric Synthesis and Pharmacological Characterization of All Stereoisomers of Glutamate Transporter Inhibitor TFB-TBOA and Synthesis of EAAT Photoaffinity Probes. ACS Chem. Neurosci. 2016, 7 (5), 534–9. 10.1021/acschemneuro.5b00311. [DOI] [PubMed] [Google Scholar]
- Fu H.; Younes S. H.; Saifuddin M.; Tepper P. G.; Zhang J.; Keller E.; Heeres A.; Szymanski W.; Poelarends G. J. Rapid Chemoenzymatic Route to Glutamate Transporter Inhibitor l-TFB-TBOA and Related Amino Acids. Org. Biomol. Chem. 2017, 15 (11), 2341–2344. 10.1039/C7OB00305F. [DOI] [PubMed] [Google Scholar]
- Raj H.; Szymański W.; de Villiers J.; Rozeboom H. J.; Veetil V. P.; Reis C. R.; de Villiers M.; Dekker F. J.; de Wildeman S.; Quax W. J.; Thunnissen A.-M. W. H.; Feringa B. L.; Janssen D. B.; Poelarends G. J. Engineering Methylaspartate Ammonia Lyase for the Asymmetric Synthesis of Unnatural Amino Acids. Nat. Chem. 2012, 4 (6), 478–484. 10.1038/nchem.1338. [DOI] [PubMed] [Google Scholar]
- Cheng B.; Shchepakin D.; Kavanaugh M. P.; Trauner D. Photoswitchable Inhibitor of a Glutamate Transporter. ACS Chem. Neurosci. 2017, 8 (8), 1668–1672. 10.1021/acschemneuro.7b00072. [DOI] [PubMed] [Google Scholar]
- Degel B.; Staib P.; Rohrer S.; Scheiber J.; Schirmeister T. Cis-Configured Aziridines are New Pseudo-Irreversible Dual-Mode Inhibitors of Candida albicans Secreted Aspartic Protease 2. ChemMedChem 2008, 3 (2), 302–315. 10.1002/cmdc.200700101. [DOI] [PubMed] [Google Scholar]
- Tang Z.; Yang Z.-H.; Chen X.-H.; Cun L.-F.; Mi A.-Q.; Jiang Y.-Z.; Gong L.-Z. A Highly Efficient Organocatalyst for Direct Aldol Reactions of Ketones with Aldedydes. J. Am. Chem. Soc. 2005, 127 (25), 9285–9289. 10.1021/ja0510156. [DOI] [PubMed] [Google Scholar]
- Kodet J. G.; Wiemer D. F. Synthesis of Indole Analogues of the Natural Schweinfurthins. J. Org. Chem. 2013, 78 (18), 9291–302. 10.1021/jo4014244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitchen D. B.; Decornez H.; Furr J. R.; Bajorath J. Docking and Scoring in Virtual Screening for Drug Discovery: Methods and Applications. Nat. Rev. Drug Discovery 2004, 3 (11), 935–49. 10.1038/nrd1549. [DOI] [PubMed] [Google Scholar]
- Edgar P. P.; Schwartz R. D. Functionally Relevant Gamma-Aminobutyric acidA Receptors: Equivalence Between Receptor Affinity (Kd) and Potency (EC50)?. Mol. Pharmacol. 1992, 41 (6), 1124–1129. [PubMed] [Google Scholar]
- Tosco P.; Ahring P. K.; Dyhring T.; Peters D.; Harpsøe K.; Liljefors T.; Balle T. Complementary Three-Dimensional Quantitative Structure-Activity Relationship Modeling of Binding Affinity and Functional Potency: A Study on α4β2 Nicotinic Ligands. J. Med. Chem. 2009, 52 (8), 2311–2316. 10.1021/jm801060h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.