

Abstract
Ribonucleotide reductase (RNR) is a central enzyme for the synthesis of DNA building blocks. Most aerobic organisms, including nearly all eukaryotes, have class I RNRs consisting of R1 and R2 subunits. The catalytic R1 subunit contains an overall activity site that can allosterically turn the enzyme on or off by the binding of ATP or dATP, respectively. The mechanism behind the ability to turn the enzyme off via the R1 subunit involves the formation of different types of R1 oligomers in most studied species and R1–R2 octamers in Escherichia coli. To better understand the distribution of different oligomerization mechanisms, we characterized the enzyme from Clostridium botulinum, which belongs to a subclass of class I RNRs not studied before. The recombinantly expressed enzyme was analyzed by size-exclusion chromatography, gas-phase electrophoretic mobility macromolecular analysis, EM, X-ray crystallography, and enzyme assays. Interestingly, it shares the ability of the E. coli RNR to form inhibited R1–R2 octamers in the presence of dATP but, unlike the E. coli enzyme, cannot be turned off by combinations of ATP and dGTP/dTTP. A phylogenetic analysis of class I RNRs suggests that activity regulation is not ancestral but was gained after the first subclasses diverged and that RNR subclasses with inhibition mechanisms involving R1 oligomerization belong to a clade separated from the two subclasses forming R1–R2 octamers. These results give further insight into activity regulation in class I RNRs as an evolutionarily dynamic process.
Keywords: Clostridium botulinum, ribonucleotide reductase, allosteric regulation, overall activity regulation, inhibition mechanism, a-site, oligomerization, phylogenetics, evolution, structure–function
Ribonucleotide reductase (RNR) catalyzes the formation of deoxyribonucleotides needed as building blocks for DNA synthesis and repair (1). Nearly all living organisms, as well as many DNA viruses, are therefore dependent on this enzyme for DNA replication. The reaction catalyzed by RNR is a free radical–dependent reaction where the 2′-OH group of ribonucleoside di- or triphosphates (NDPs/NTPs) is replaced by 2′-H to form the corresponding deoxyribonucleotides (dNDPs/dNTPs). RNRs are divided into three classes, mainly based on the mechanism to form the essential radical cofactor. Class I RNRs are found in nearly all eukaryotes, as well as in many bacteria, some archaea, and DNA viruses/bacteriophages. RNRs from this class consist of two subunits, R1 (α) and R2 (β), which are both needed for enzyme activity. The R2 subunit contains a stable free radical. It is usually a tyrosyl radical stabilized by an iron center, but the nature of the radical and the metal center varies between species (2–4). Oxygen is needed for the formation of the free radical, and class I is therefore referred to as the aerobic type of RNR. The R1 subunit contains the active site and two types of allosteric sites to regulate the activity. During the enzymatic reaction, electrons are shuttled between the active site in the R1 subunit and the free radical in the R2 subunit. Class II RNRs are one-subunit enzymes that use 5′-deoxyadenosylcobalamin as radical generator, and class III RNRs are strictly anaerobic enzymes with an oxygen-sensitive free glycyl radical. The first RNR studied was the class I enzyme from Escherichia coli, and the two genes encoding the R1 and R2 subunits were called nrdA and nrdB, respectively. Later it was discovered that E. coli also has a second class I RNR encoded by the nrdE and nrdF genes. The two RNRs belong to the two subclasses Ia and Ib in a classification system based on metal content and presence or absence of a tyrosyl radical that have recently been extended to Ic, Id, and Ie (2–4). Class I RNR proteins have also been subclassified based on phylogeny into nine NrdA/E subclasses (NrdAe/g/h/i/k/n/z/q and NrdE) and corresponding NrdB/F subclasses (5). Because it is better suited for studies of the evolution of class I RNR characteristics, we will here use the phylogenetic classification.
Initial characterization of the E. coli and mammalian class I RNRs (NrdAB) showed that they have a very advanced allosteric regulation using two types of allosteric sites (1, 6, 7), the specificity site (s-site) and the overall activity site (a-site). The s-site determines which substrate to reduce with ATP (or low concentrations of dATP) stimulating the reduction of pyrimidines (CDP/UDP), whereas dTTP and dGTP direct the substrate specificity toward GDP and ADP, respectively. At higher concentrations, dATP will also bind the a-site to turn off the enzyme, and in this way, it functions as a safeguard to prevent the total dNTP levels from getting too high in the cell. ATP can also bind to the a-site and has an opposing role to dATP by being able to activate the enzyme. The a-site is generally an ATP-cone (8) with the notable exception of the Bacillus subtilis NrdEF enzyme, which has an ATP-cone independent overall activity regulation (9, 10). The binding of ATP and dATP to the ATP-cone causes the eukaryotic R1 subunit to form two types of hexamers (11, 12). The dATP-induced hexamer can only bind the R2 dimer in a nonproductive way in which electrons cannot be transported between the two subunits, whereas the ATP-induced hexamer gives increased activity. Other structural mechanisms have been found among different RNRs, and it has been hypothesized that the evolution of new mechanisms is driven by gains and losses of ATP-cones (13). Some RNRs lack ATP-cones completely, whereas others have one, two, or three ATP-cones per R1 polypeptide. In the Pseudomonas aeruginosa class I RNR (NrdAz), which has two ATP-cones, the R1 forms a tetramer in the presence of dATP but also in this case the mechanism is that dATP induces the R1 protein to form oligomers to which the R2 dimer binds in a nonproductive way (13, 14).
The active form of the class I RNRs is generally an α2β2 complex, but in the eukaryotic subclass (NrdAe), this complex might not be a major form because ATP, which is always present in the cell at millimolar concentrations, instead promotes the formation of the active hexamer described above. In contrast, ATP has a more passive role in most other species where it mainly prevents dATP to bind but does not cause the formation of any new complex or dramatically changes the enzyme activity in any other means via the a-site. The E. coli enzyme (NrdAg), which has for a long time been considered a prototype for class I RNRs, is quite different from the other members (1, 6, 15–17). In contrast to all other examples mentioned above, dATP has no effect on the oligomerization of the individual subunits. Instead, both subunits need to be present for the formation of the inhibited form, which is an α4β4 ring composed of consecutive R1 and R2 dimers. The role of ATP in the a-site is also different from all other RNRs studied so far, and in the presence of high concentrations of dTTP or dGTP, ATP can actually turn off the enzyme via the a-site. It is the only example where it is the s-site that determines what role ATP should have in the a-site. Today it is not known whether the E. coli RNR, which once was considered a prototype of class I RNRs, is a rare exception or whether the regulation mechanism can be found in other subclasses as well.
To better understand the distribution of different oligomerization mechanisms in class I RNRs, we have studied the RNR from Clostridium botulinum, which belongs to the NrdAh subclass. This bacterium is well-known for the ability to produce the extremely potent botulinum neurotoxin that causes the life-threatening condition botulism (18). The toxin also has a wide range of therapeutic applications and is sold under the trade names Botox and Dysport. Studies of the C. botulinum RNR with X-ray crystallography, size-exclusion chromatography, gas-phase electrophoretic mobility macromolecular analysis (GEMMA), EM, and enzyme activity assays showed that it formed dATP-inhibited α4β4 octamers just as the RNR from E. coli, highlighting that this complex is found in more than one subclass. Phylogenetic analysis suggested that the original RNRs lacked this regulation, and an evolutionary hypothesis of how the different types of regulations appeared is described in the discussion.
Results
R2 structure
We have solved the crystal structure of the C. botulinum R2 subunit at 2.0 Å resolution, which crystallized in space group C2, containing two dimers per asymmetric unit. The four monomers align with an overall RMSD of 0.26 Å. Each dimer buries an area of 6800 Å2. The statistics for data collection and refinement are presented in Table 1.
Table 1.
Data collection and refinement statistics
| Data collection | |
| Space group | C 1 2 1 |
| Cell dimensions | |
| a, b, c (Å) | 210.827, 61.8538, 154.342 |
| α, β, γ (°) | 90, 127.46, 90 |
| Resolution (Å) | 2.0 |
| Rmerge | 0.073 (1.657) |
| I/σI | 10.71 (0.84) |
| Completeness (%) | 99.6 (98.2) |
| Multiplicity | 6.6 (6.6) |
| Refinement | |
| Resolution (Å) | 76.25–2.0 |
| No. reflections | 106,640 (10,493) |
| Rwork/Rfree | 0.2218/0.2367 |
| No. atoms | 10,892 |
| Protein | 10,667 |
| Ligand/ion | 32 |
| Water | 193 |
| B-factors | 54.69 |
| Protein | 54.76 |
| Ligand/ion | 52.04 |
| Water | 50.84 |
| RMSDs (Å) | |
| Bond lengths | 0.019 |
| Bond angles | 1.44 |
The residues coordinating the ferric ions in the C. botulinum R2 metal-binding site are functionally conserved. The two iron ions are coordinated by four carboxylates and two histidines. The electron density is less defined for the metal sites compared with the core of the protein, indicative of a contribution of additional structural states caused by partial metal occupancy and/or X-ray photoreduction of the metal site. A higher occupancy is observed for Fe2 compared with Fe1 in all four monomers in the asymmetric unit. Fe2 is coordinated by monodentate contacts with Glu115, Glu216, Glu182, and His219. Fe1 is coordinated by monodentate contacts with Asp85, Glu115, and His118 (Fig. 1A). In addition, the two metal ions are bridged by a monoatomic ligand. These features are displayed in all four monomers of the asymmetric unit, but the coordination distances refine to slightly different values. Notably, the metal-binding site in chain B also shows weak density for a water molecule coordinated by Fe1. A water ligand in this position is commonly observed in the oxidized (FeIII/FeIII) state of R2 (NrdB) proteins. Overall, the geometry of the metal-binding site and the distance between the ferric ions (∼3.2 Å) closely resemble the geometry in the oxidized E. coli R2 site (19, 20), suggesting that the main structural species is the oxidized (FeIII/FeIII) state. The phenolic oxygen of the presumed radical harboring tyrosine (Tyr122) is located ∼5.5 Å from Fe1. A detailed view of the metal-binding site is shown in Fig. 1A.
Figure 1.
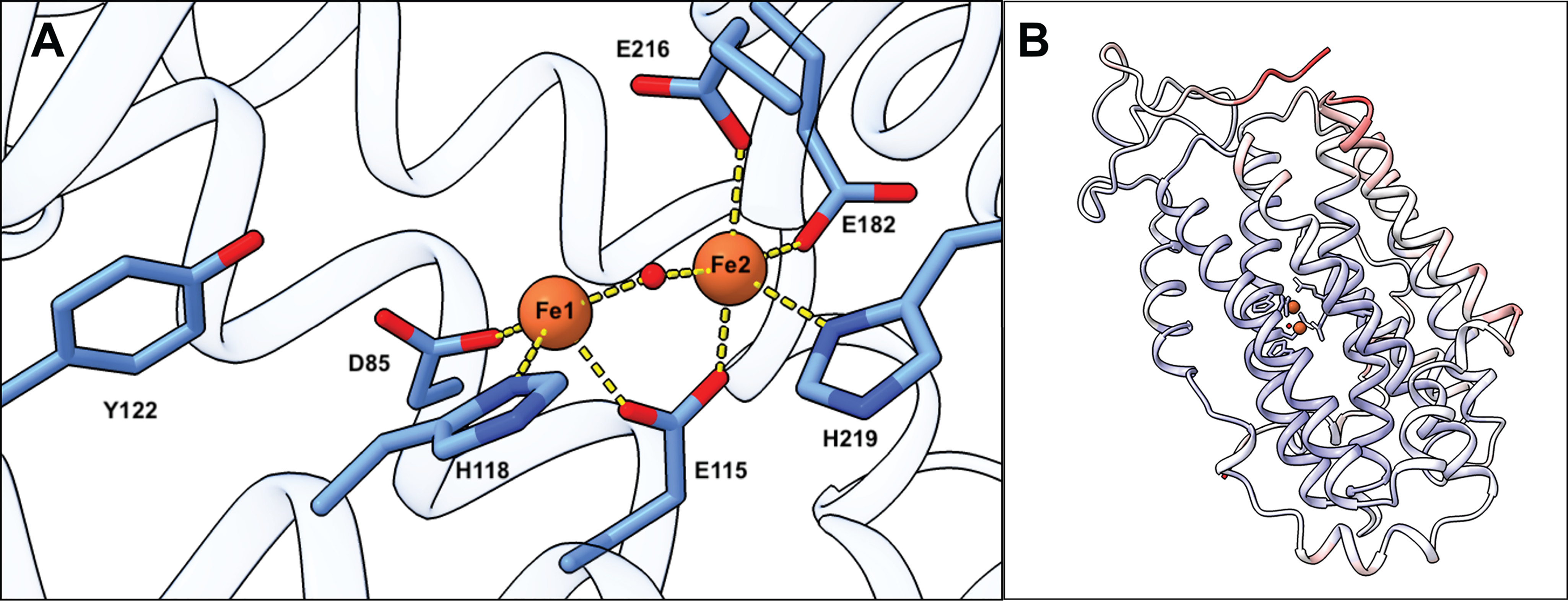
Crystal structure of the C. botulinum R2 protein (NrdB). A, coordination of the diferric site. Chain A was used to show the metal coordination. B, chain A from the crystal structure of C. botulinum R2 protein colored by B-factor (lower in blue and higher in red).
The structure is modeled up to residue 316, whereas the C-terminal region of the structure (known as the β-tail) is disordered. The C terminus is generally known to act as an interface to transfer a radical located on a tyrosine in the center of the R2 subunit to an active site cysteine in the R1 subunit, and the conserved Tyr330 residue in this interface region is part of the proposed proton-coupled electron transfer between the two subunits (21). The overall structure of the C. botulinum R2 protein colored by b-factor is presented in Fig. 1B, where the higher average b-factors of the residues close to the C terminus are shown.
A structural alignment search in the PDBeFOLD server (22, 23) revealed the R2 protein from Bacillus halodurans to have the most similar known structure (PDB ID 2RCC; RMSD, 1.21 Å over 305 residues), followed by the E. coli variant in the recently published RNR holocomplex (PDB ID 6W4X; RMSD, 1.59 Å over 289 residues) (21). The B. halodurans and C. botulinum R2 proteins both belong to the NrdBh subclass.
Radical cofactor site in the C. botulinum R2 protein
The UV-visible spectrum of C. botulinum R2 protein is characterized by a prominent band at 407 nm corresponding to a tyrosyl radical, plus additional bands at 325 and 370 nm corresponding to a diferric metal center (Fig. 2A). The tyrosyl radical content was estimated to 0.91 radical/dimeric R2 protein. The C. botulinum R2 preparation used for UV-visible and EPR spectra had a specific activity of 700 nmol/min/mg assayed with CDP as substrate and ATP as positive allosteric effector and an excess of R1 over R2 protein. The EPR spectrum of the C. botulinum R2 protein features major hyperfine coupling of 1.9–2.0 mT relating to a coupling to one of the β protons in the tyrosyl residue and smaller coupling of ∼0.7 mT from the two 3,5-ring protons (Fig. 2B). This is similar to the E. coli spectrum (Fig. 2B, inset). However, additional splitting was observed, likely emanating from a weaker coupling to the second β proton in the tyrosyl residue. This has been observed for the bacteriophage T4 R2 protein (Fig. 2B, inset) and reflects that the geometry of the ring versus the methylene group have different (locked) positions. From the EPR spectrum it was also clear that the magnetic interaction between the tyrosyl radical and the iron center is weaker in the C. botulinum R2 protein than it is in the E. coli R2 protein (Fig. S1). This can be due to the longer distance to the iron center, different geometrical arrangement, or different magnetic properties of the di-iron center itself. Although the structure of the radical state is unknown for the C. botulinum R2 protein, it can be noted that the radical harboring tyrosine is located ∼1 Å further from Fe1 compared with the E. coli FeIII/FeIII R2 protein structure (20).
Figure 2.

Spectral characterization of the C. botulinum R2 protein. A, UV-visible spectrum of 5.5 μm C. botulinum R2 protein highlighting the prominent tyrosyl radical absorption band at 407 nm and diferric bands in the 325–370 nm region. B, EPR spectrum at 77K of C. botulinum R2 protein (51.5 μm) recorded at 0.25 mW and 0.2 mT modulation amplitude; the sample is only slightly saturated during these conditions (see Fig. S1). Inset, EPR spectra at 77K of the bacteriophage T4 and E. coli R2 proteins (NrdB) (39); the width of the EPR signals have been scaled to that in Fig. 2B.
Size-exclusion chromatography shows that the C. botulinum R2 protein is a dimer and the R1 protein is in a monomer–dimer equilibrium
Size-exclusion chromatography performed with 0.15 m KCl in the mobile phase indicated that the R2 protein is a dimer, whereas the R1 protein showed a very low signal under these conditions (Fig. 3A). To increase the signal, the KCl concentration was raised to 0.3 m, and under these conditions, the R1 protein could be readily observed (Fig. 3B). The R1 protein was in a monomer–dimer equilibrium, and dimerization was promoted by dATP and to a lesser extent by ATP. The R1 signal could be enhanced further by increasing the KCl concentration to 1 m, and under these high-salt conditions, the R1 protein started to form dimers also without effectors (Fig. 3C). Similarly to the experiments in Fig. 3B, the R1 dimerization was further stimulated with dATP also in this case.
Figure 3.

Size-exclusion chromatography of the C. botulinum R1 and R2 proteins. A, R2 protein (0.025 and 0.1 mg/ml) and R1 protein (0.1 mg/ml) analyzed with a mobile phase containing 0.15 m KCl. B, 0.05 mg/ml R1 protein analyzed in the presence of 100 μm dATP, 3 mm ATP or without effector in a mobile phase containing 0.3 m KCl. C, experiments performed with 1 m KCl in the mobile phase. The top two traces show R1 protein (0.05 and 0.1 mg/ml) in the absence of allosteric effectors, and the bottom trace shows 0.05 mg/ml R1 protein in the presence of 100 μm dATP. In addition, the mobile phase contained 50 mm Tris-HCl, pH 7.6, and for the nucleotide-containing experiments, 10 mm MgCl2 was also included. The different chromatograms are distributed vertically to fit many chromatograms in each panel. The theoretical molecular masses of the R1 and R2 proteins are 88 and 41 kDa, respectively, and the molecular mass on top of each peak is derived by comparing the retention times to a set of globular standard proteins (see “Experimental procedures”).
GEMMA of the C. botulinum RNR indicates that dATP induces a stable α4β4 complex that dissociates in the presence of ATP
The size-exclusion chromatography experiments in Fig. 3 were informative for the study of the individual subunits, but because of difficulties analyzing R1–R2 complexes (explained in Fig. S2), we switched to GEMMA for these analyses. Two R1 preparations were used for the GEMMA studies: preparation 1 used in Fig. 4 (A and B) contained traces of dATP (0.3 dATP/polypeptide), whereas preparation 2 used in Fig. 4 (C and D) was dATP-free (see details under “Experimental procedures”). The reason why a dATP-containing preparation was used in the first experiments is that the GEMMA technique only allows volatile salts/buffers, and dATP helped to stabilize the protein under these conditions. Analysis of the individual subunits supported our conclusions from the size-exclusion chromatography that the R2 protein is a dimer (theoretically 82 kDa), whereas the R1 protein is mainly monomeric (theoretically 88 kDa) but becomes dimeric in the presence of allosteric effectors (Fig. 4A). Both dATP and the s-site effector dTTP promoted dimerization. Analysis of the two subunits together showed that they formed a large complex (Fig. 4B). The dependence on both subunits and the size of the complex indicated that it most likely is an α4β4 complex, similarly to what has been observed with the E. coli RNR. The complex formation was most strongly promoted by dATP but was also formed in the absence of allosteric effectors or in the presence of the s-site effectors dTTP and dGTP (Fig. 4B and Fig. S3). However, these first experiments were performed with the R1 preparation containing traces of dATP. In the presence of 300 μm ATP the complex was dissociated, and this effect is most likely via the a-site because it occurred regardless of whether 200 μm dTTP was present or not (Fig. 4B). When combined with dTTP, the concentration of ATP was lowered to 100 μm to prevent it from competing with dTTP for the s-site. The lower concentration of ATP can also explain why the large complex did not completely disappear in this case. Next, complementary analyses were performed with dATP-free enzyme. In this case, it was not possible to make buffer exchange because the dATP-free R1 protein was not stable enough for long exposures to ammonium acetate. Instead, we diluted the protein directly into ammonium acetate prior to the experiment. The analysis was then performed with higher concentrations of ammonium acetate and DTT (see figure legends) to stabilize the protein, and the concentration of allosteric effector was decreased to reduce the total amount of nonvolatile components in the mixture. The same strategy was used for both the new dATP-free preparation (preparation 2) and the earlier dATP-containing preparation (preparation 1) for comparison. With preparation 1, the α4β4 complex could readily be observed in the presence of dTTP, whereas it was not visible with the dATP-free preparation (Fig. 4C). In the presence of 50 μm dATP, both preparations showed equal levels of the complex, indicating that there was no obvious difference in protein quality between the two batches (Fig. 4C). Further studies showed that under these conditions the R1 protein was predominantly in dimer form also without effector and that the α4β4 complex could not be formed without effector or with dGTP when using the dATP-free preparation (Fig. 4D). The conclusion was that the α4β4 complex requires dATP for its formation and that the smaller α4β4 peak in Fig. 4B observed also when dATP was not added was due to traces of dATP present in the R1 preparation. The view that dATP in the a-site is crucial for α4β4 formation is supported by that ATP was the only effector that could compete with dATP and dissociate the complex in Fig. 4B.
Figure 4.

GEMMA analysis of C. botulinum RNR. A, analysis of the R2 or R1 protein in the absence or presence of 200 μm of the indicated allosteric effectors in a solution containing magnesium acetate (equimolar to the nucleotides in all experiments). B, R1 and R2 proteins analyzed together in the absence or presence of 200 μm dATP, 200 μm dTTP, 300 μm ATP, or a combination of 100 μm ATP and 200 μm dTTP. C, R1 and R2 proteins analyzed together in the presence of 50 μm dTTP or dATP. Two different R1 batches were used. The first preparation contained traces of dATP (0.3 dATP/polypeptide) and had the advantage of being stable in the GEMMA buffer, whereas the second preparation was dATP-free (<0.01 dATP/polypeptide) but less stable. The experiments in A and B were performed with 0.05 mg/ml R1 (preparation 1) and 0.025 mg/ml R2 in a solution containing 0.025 mm DTT, 0.005% (v/v) Tween 20, and 100 mm ammonium acetate, pH 7.8. The experiments in C and D were performed with 0.1 mg/ml R1 protein (both preparations in C and preparation 2 in D), 0.05 mg/ml R2 protein, 1 mm DTT, 0.005% (v/v) Tween 20, and 200 mm ammonium acetate, pH 7.8. The increased protein concentration and altered buffer conditions in D were adjustments to shorten the analysis time (fewer scans) and keep the protein stable enough during the measuring period.
EM showed that the dATP-induced C. botulinum RNR complex has an α4β4 stoichiometry with a similar overall structure as the inhibited form of the E. coli class I RNR (NrdAB)
The GEMMA experiments indicated that the C. botulinum dATP-induced complex depends on both subunits for its formation and had a similar size as the E. coli α4β4 complex. To analyze the structure of the C. botulinum complex further, we used negative-stain EM. The obtained structure was strikingly similar to the published structure of the E. coli α4β4 complex (Fig. 5).
Figure 5.

Negative-stain EM of the C. botulinum α4β4 complex. A, the most populated 2D class average of C. botulinum RNR obtained from negative-stain EM of an R1–R2 protein mixture in the presence of 200 μm dATP. The scale bar is 100 Å. B, the E. coli α4β4 complex (PDB code 5CNS) overlaid to the most populated 2D class average (40). The R1 protein is shown in green, and the R2 protein is in red. The E. coli R2 protein in the complex has been replaced with the C. botulinum R2 structure described above.
Enzyme assays showed that C. botulinum RNR has a true on/off switch and is inhibited by dATP and activated by ATP
Enzyme assays with the C. botulinum RNR were performed to elucidate the connection between oligomerization and enzyme activity. The enzyme activity with CDP as substrate and increasing concentrations of ATP (Fig. 6A) showed that the effector had an apparent Kd value of 0.70 ± 0.05 mm for ATP (kcat = 0.43 ± 0.01 s−1 per R1 polypeptide). This affinity represents a combined effect of both allosteric sites. To specifically study the a-site, an experiment was set up in which the enzyme was incubated with 0.5 mm GDP as substrate, 0.5 mm dTTP as s-site effector, and an increasing concentration ATP (Fig. 6B). Two interesting observations were then made: (i) ATP had a stimulatory role via the a-site, and (ii) the apparent affinity of ATP to this site was very high (Kd < 50 μm). The ability of s-site dNTPs to stimulate the affinity of ATP for the a-site has also been observed in E. coli, but in that case ATP inhibits the activity instead (16). The C. botulinum enzyme had instead a true on/off switch, where ATP activated the enzyme (Fig. 6B) and dATP was an inhibitor (Fig. 6C). A consequence of the high affinity for ATP to the a-site is that dATP has difficulty to compete for the a-site, as shown by the high IC50 value for dATP (0.16 ± 0.03 mm) in an assay where CDP was substrate and 1 mm ATP was allosteric activator (Fig. 6C). This means that the discrimination between dATP and ATP is only ∼6-fold (i.e. 0.16 versus 1 mm). To get more insight into the regulation, a four-substrate assay was performed with different allosteric effectors (Fig. 6D). From these studies, we could conclude that the ATP activation via the a-site was general because ATP could stimulate both dTTP-induced GDP reduction and dGTP-induced ADP reduction. The ATP stimulation must be via the a-site because ATP binding to the s-site would lead to CDP reduction, as shown when ATP was used as sole effector. The substrate specificity was similar to RNRs of most other species; ATP and low concentrations of dATP stimulated CDP reduction, whereas dTTP and dGTP stimulated GDP and ADP reduction, respectively (Fig. 6D). The enzyme activity was very low without allosteric effectors or with dCTP, which is also in line with most other RNRs. UDP was not a good substrate, and the major source of dTTP in these cells is therefore probably via deamination of dCTP or dCMP rather than from UDP reduction. The ability to use UDP as substrate varies between RNRs in different species, and the main allosteric effector for the UDP reduction is generally ATP/dATP. With the C. botulinum enzyme, a small amount of UDP reduction could be observed with ATP as effector (Fig. 6D).
Figure 6.

Enzyme assays with the C. botulinum RNR. A, assay with 0.5 mm CDP as substrate and increasing concentrations of ATP as allosteric effector. B, assay with 0.5 mm GDP as substrate and a combination of 2 mm dTTP and an increasing concentrations of ATP as allosteric effectors. C, assay with 0.5 mm CDP as substrate and a combination of 1 mm ATP and increasing concentrations of dATP as allosteric effectors. D, assay with a combination of CDP, UDP, GDP, and ADP as substrates used at 0.5 mm each. The allosteric effectors used are indicated on the abscissa. The effectors were generally used at 100 μm each, except for ATPhigh and dATPlow, which were used at 4 mm and 1 μm, respectively. All assays were performed in a buffer containing 10 mm magnesium acetate, 10 mm DTT, and 20 mm Tris-HCl, pH 7.5, with 1 µg of R1- and 2 µg of R2 proteins. Each plot represents the average of at least three independent experiments with standard errors indicated.
Phylogenetic analysis
To investigate the evolutionary relationships between different modes of activity regulation in class I RNRs, we mapped the presence of ATP-cones, as well as the five currently known forms of ATP-cone–dependent activity regulation in class I RNRs, including the recently discovered ATP-cones in the R2 protein (5, 24), on a Bayesian phylogeny of R1 (NrdA/E) sequences. (Fig. 7). The subclasses where the α4β4 mode of regulation has been found, NrdAh (C. botulinum) and NrdAg (E. coli), were found to be part of an unresolved polytomy that also includes the NrdAi/NrdAk clade, in which no NrdA sequences with ATP-cones have been found. The α4 and α6 modes were found in neighboring clades in a different part of the tree.
Figure 7.

Bayesian phylogeny of NrdA sequences with ATP-cone dependent modes of activity regulation indicated. Sequences were selected by clustering all available sequences at a cutoff of 60% identity to select sequences representing the full diversity of NrdA sequences available. The tree was rooted with a selection of class II (NrdJ) sequences. Branch colors indicate phylogenetically defined subclasses—all found to be well-supported (green circles)—NrdAe to NrdE, and colored diamonds (NrdA) and stars (NrdB) indicate the number of ATP-cones discovered in each sequence. Species in which activity regulation have been studied are labeled: Hs NrdAe, H. sapiens NrdAe; Dd NrdAe, D. discoideum NrdAe; Sc NrdAe, S. cerevisiae NrdAe; Pa NrdAz, P. aeruginosa NrdAz; Ec NrdAg, E. coli NrdAg; Cb NrdAh, C. botulinum NrdAh; Lb NrdAi, Leeuwenhoekiella blandensis NrdAi (the catalytic component of the enzyme in which activity regulation is conferred via an ATP-cone fused to the radical generating subunit NrdBi). For the full tree, with labels for all sequences and posterior probabilities for all splits, see Figshare (https://doi.org/10.17045/sthlmuni.11558187.v1).
Discussion
From the studies of the C. botulinum RNR from the NrdAh subclass, we now know that the regulation dependent on R1–R2 octamers (α4β4) that prior to this work only was found in the NrdAg subclass (E. coli) is not a rare exception in a limited number of closely related RNRs but is found in NrdAh as well. The cryo-EM structure of the inactive C. botulinum NrdAh/Bh enzyme shows a striking similarity with the E. coli NrdAg/Bg inactive complex, indicating that their inactivation mechanisms have a common origin rather than being the result of convergent evolution. With this information in hand, it is for the first time possible to propose a hypothesis of how the overall activity has evolved in the NrdA class. Although it is not possible to completely dismiss other scenarios, our phylogenetic tree is consistent with a single origin of ATP-cone–driven activity regulation in NrdA after the NrdE subclass diverged from other NrdAs (Fig. 7). This is supported by the fact that the NrdE subclass branches off first in the class I tree and that ATP-cones are rarely found in class II RNRs from which class I RNRs evolved (25). After NrdE, there is a division into four branches with unresolved order of divergence between them. One of the branches in this group contains both NrdAi and NrdAk, which lack ATP-cones in the R1 subunit and thereby are functionally similar to NrdE. A single origin of ATP-cone–driven activity regulation in class I RNR would suggest that the NrdAi and NrdAk phylogenetic subclasses diverged before the ATP-cone was gained by class I RNR or that it was lost at an early point in these subclasses because none of them contain ATP-cones in the R1 subunit today. Instead, a few of them have gained an ATP-cone in the R2 subunit and evolved R2-dependent oligomerization to achieve overall activity regulation. This is most likely a recent event because it is only found in a few related sequences. These conclusions are fairly similar to what has been made before from a phylogenetic alignment of the R2 subunit (5). Our main interest here lies rather in the three remaining branches containing RNRs with ATP-cones in the R1 subunit. Interestingly, the two entirely different ways to achieve R1-dependent oligomerization, found in NrdAe and NrdAz, respectively, are found in enzymes from the same part of the tree, indicating that R1-oligomerization–dependent mechanisms originated in the common ancestor of NrdAe and NrdAz. The NrdAz subclass is dominated by multiple cones (two or three per R1 polypeptide), of which the second cannot bind nucleotides, suggesting that it is a remnant of an ancestral cone that was functionally replaced by the later addition of another ATP-cone. The remaining two branches of the phylogeny that have been studied, NrdAh and NrdAg, both represent the R1–R2 octamer-dependent regulation. The phylogenetic tree is therefore consistent with a scenario in which overall activity regulation was first split into regulations dependent on R1–R2 octamers and R1 oligomers before the different ways of R1 oligomerizations appeared.
The distribution of different dATP inhibition mechanisms raises the question of how many regulatory mechanisms we can expect to find. Two of the subclasses are still not studied, NrdAn and NrdAq, and in particular the first subclass to branch off, NrdAq, could give more details into the evolution of the R1-oligomerization branch (Fig. 7). Another source of new oligomerization mechanisms could potentially be the enzymes with a deviating number of cones in each subclass where the gain of an extra cone can have dramatic effects on the subunit interaction surfaces. It should also be remembered that only a few members of each subclass have been studied so far. The experience from NrdAe, in which the mammalian (Bos taurus, Mus musculus, and Homo sapiens), Dictyostelium discoideum, and Saccharomyces cerevisiae enzymes have been studied, is that the regulation mechanism is coherent within the subclass with the notable exception of the ones that have lost the regulation altogether. The loss of regulation seems to happen frequently, and each subclass has several members that lack the ATP-cone. In addition to regulation disappearing because of the loss of cones, there are also smaller amino acid substitutions that can have a similar effect. The NrdAe and NrdAg enzymes from Trypanosoma brucei and bacteriophage T4, respectively, have ATP-cones that are able to bind dATP but have lost the inhibiting effect of dATP on enzyme activity (26–29), presumably because of a lack of selection for activity regulation of the enzyme.
The ATP-cone–driven regulation is similar but not identical in the NrdAh and NrdAg subclasses. In both subclasses, dATP induces an inhibited R1–R2 octamer, but the response to ATP is entirely different. In E. coli, the ATP-cone itself seems not to be able to mediate the different effects of ATP and dATP, and it is rather the combined effect of the two allosteric sites that mediates the overall activity regulation (16). ATP can also induce the formation of the inhibited R1–R2 octamer as long as the specificity allosteric site is occupied with a dNTP. The positive effect of ATP on enzyme activity is rather through the s-site; if the ATP concentration is high enough to partially compete with dNTPs for the s-site, the inhibited R1–R2 octamer is not readily formed. On the contrary, ATP was always a positive regulator in the C. botulinum NrdAh enzyme, regardless of whether dTTP or dGTP was there. Interestingly, the activity increased with the addition of ATP as compared with dTTP/dGTP used alone. This is similar to the mammalian enzyme, but in that case, ATP induces the formation of an entirely new R1 hexamer, which is structurally different from the inhibited hexamer and requires other amino acids in the interfaces between the three dimers that build up the hexamer (12). In the case of C. botulinum NrdAh, ATP activation is only leading to the formation of the same type of active complex (α2β2), which is also formed in the presence of dGTP or dTTP alone. On the other hand, the activation is also less dramatic with a less than 2-fold activation when ATP is added to a dGTP/dTTP-stimulated enzyme. For comparison, the mammalian enzyme can increase its activity 4–5-fold in this situation. The conclusion is that modest ATP activation through the a-site in class I RNRs can occur with small changes in the enzyme structure, whereas stronger effects seem to be mediated by oligomerization.
The identification of the R1–R2 octamer in two unrelated subclasses is relevant from a drug discovery viewpoint. The newly studied NrdAh subclass is not only found in C. botulinum but also in other pathogens, including Treponema pallidum causing syphilis. Drugs can potentially be developed that stabilize the R1–R2 octamer and thereby lock the enzyme in an inhibited state, and it is then important whether there are any other pathogens that can also be treated with the drugs and what effect it has on commensal bacteria. NrdAg is encoded by many pathogens, as well as commensals. In contrast, drugs developed to stabilize the R1 tetramer complex will most likely only affect species with NrdAz such as P. aeruginosa. The advantages with drugs that specifically target certain pathogens rather than being broad-spectrum antibiotics are that interspecies spread of drug resistance and side effects on the normal intestinal flora are prevented.
Experimental procedures
Protein expression and purification
The R2 cDNA construct from C. botulinum Loch Maree/Type A3 strain was synthesized and inserted into pET28TEV vectors by GenScript Inc. (New Brunswick, NJ), codon-optimized for expression in E. coli. The R1 construct was cloned from C. botulinum Loch Maree/Type A3 strain genomic DNA. 5′-CCCAAACATATGAATATTAAAATAAAAAAGAGAGATGGAC-3′ was used as forward primer, and 5′-CCCTTTCTCGAGTTAGCTTGAGCAACTTGTACATTC-3′ was used as reverse primer, introducing NdeI and XhoI restriction sites that were used for insertion into a pET28TEV vector. The construct was verified by DNA sequencing.
C. botulinum R2 protein was expressed in E. coli BL21(DE3) T1R pRARE2 in TB medium supplemented with 8 g/liter glycerol, 50 μg/ml kanamycin, and 34 μg/ml chloramphenicol (see the Acknowledgments). The cells were grown in a LEX bioreactor at 37 °C until they reached an A600 of 2. The temperature was then decreased to 18 °C, and expression was induced with 0.5 mm isopropyl β-d-1-thiogalactopyranoside and continued overnight until cells were harvested by centrifugation (10 min at 4 °C, 4500 × g). The cell pellet was resuspended in lysis buffer containing 100 mm HEPES, pH 8.0, 500 mm NaCl, 10% (v/v) glycerol, 10 mm imidazole, 0.5 mm tris(2-carboxyethyl)phosphine (TCEP) and stock solution (1 ml/1.5 liter culture, containing 1 tablet of Complete EDTA-free (protease inhibitor mixture, Roche) and 5 μl of benzonase nuclease (250 units; Sigma) per 1 ml).
The R2 protein was purified with a 2-ml HisTrap HP column (GE Healthcare) using wash buffer 1 (containing 20 mm HEPES, pH 7.5, 500 mm NaCl, 10% glycerol, 10 mm imidazole, and 0.5 mm TCEP), wash buffer 2 (containing 20 mm HEPES, pH 7.5, 500 mm NaCl, 10% glycerol, 50 mm imidazole, and 0.5 mm TCEP), and elution buffer (containing 20 mm HEPES, pH 7.5, 500 mm NaCl, 10% glycerol, 500 mm imidazole, and 0.5 mm TCEP). The eluate was then loaded onto a HiLoad 16/60 Superdex 75 column (GE Healthcare) equilibrated in SEC buffer (containing 20 mm HEPES, pH 7.5, 300 mm NaCl, 10% glycerol, and 0.5 mm TCEP). Fractions were examined by SDS-PAGE, pooled and concentrated, and fresh TCEP was added up to 2 mm.
C. botulinum R1 protein was expressed in E. coli BL21(DE3) cells grown in TB medium containing 3 g/liter glycerol and 25 μg/ml kanamycin at 37 °C. Expression was induced using 0.5 mm isopropyl β-d-1-thiogalactopyranoside at 20 °C when the cell cultures reached an A600 of 1 and continued for 16 h. The cells were harvested by centrifugation and resuspended in lysis buffer (100 mm HEPES, pH 8.0, 500 mm NaCl, 10% glycerol, 10 mm imidazole, and 0.5 mm TCEP for R1) supplemented with 1 tablet Complete EDTA-free (protease inhibitor mixture, Roche)/150 ml. Cell lysis was carried out via sonication on ice for three 5-min rounds of 10 s on/ 10 s off cycles at 80% amplitude.
The R1 protein was purified on a gravity-flow column by the addition of 100 μl protein/ml nickel–nitrilotriacetic acid, using a series of wash steps. 10 CVs of wash buffer 1 (containing 20 mm HEPES, pH 7.5, 500 mm NaCl, 10% glycerol, 10 mm imidazole, and 0.5 mm TCEP) were run through the column first and then 10 CVs of wash buffer 2 (containing 20 mm HEPES, pH 7.5, 500 mm NaCl, 10% glycerol, 50 mm imidazole, and 0.5 mm TCEP). Elution was carried out with 1 CV of elution buffer (containing 20 mm HEPES, pH 7.5, 500 mm NaCl, 10% glycerol, 500 mm imidazole, and 0.5 mm TCEP). The elution fraction was loaded onto a HiLoad 16/60 Superdex 200 column (GE Healthcare) equilibrated with a solution consisting of 20 mm HEPES, pH 7.5, 300 mm NaCl, 10% glycerol, and 0.5 mm TCEP. The fractions were collected and concentrated. This R1 preparation contained traces of dATP (0.31 ± 0.07 dATP/polypeptide, S.E. n = 2) as measured by HPLC on the soluble fraction of TCA-precipitated protein. The procedure to analyze dATP content was performed by mixing 75 μl of 6.8 μm R1 polypeptide with 5 μl of 100 mm MgCl2 and 20 μl of 50% (w/v) TCA followed by an incubation on ice for 5 min. The solution was centrifuged at 21,000 × g for 5 min at 4°C, and the supernatant was transferred to a second tube. 150 μl of Freon-trioctylamine was added (from a mixture of 500 μl of 1,1,2-trichloro-1,1,2-trifluoroethane and 140 μl of trioctylamine), vortexed for 30 s, and recentrifuged for 1 min. The upper phase was saved, mixed 1:1 with mobile phase, and analyzed with HPLC using a 50 × 2.1-mm ACE Excel C18-PFP column (Advanced Chromatography Technologies Ltd., Aberdeen, UK) at 0.4 ml/min with a mobile phase containing 7% (v/v) methanol, 0.71 g/liter tetrabutylammonium bromide, and 13.8 g/liter KH2PO4 phosphate adjusted to pH 5.6 with 4.2 ml KOH/liter. The UV-2075 detector (Jasco International Co. Ltd, Hachioji, Japan) was set to 260 nm using the standard (STD) measuring frequency of the instrument.
To obtain dATP-free R1, a second preparation was purified similarly, with the addition of an ATP wash step (5 CVs of 20 mm HEPES, pH 7.5, 500 mm NaCl, 10 mm ATP, 10% glycerol, 10 mm imidazole, and 0.5 mm TCEP) in between wash 1 and wash 2. After SDS-PAGE analysis, a significant amount of R1 protein was found in the ATP wash fraction, which was then concentrated and loaded onto the size-exclusion chromatography column. This was probably due to the proximity of the His tag to the binding site of ATP, which could have caused the detachment of R1 from the nickel–nitrilotriacetic acid resin upon binding of ATP. The preparation was free from dATP (<0.01 dATP/polypeptide).
X-ray crystallography
R2 was diluted to 20 mg/ml and used to prepare vapor-diffusion hanging-drop plates of several commercial crystallization screens. Crystals were found within a week in the PACT premierTM F2 condition (Molecular Dimensions), containing 0.2 m NaBr, 0.1 m Bis-Tris propane, pH 6.5, and 20% (w/v) PEG 3350. The crystals were frozen after equilibration in cryoprotectant solution composed of mother liquor including 20% glycerol. Diffraction data at 2.00 Å resolution of R2 were collected at Beamline I04-1 of Diamond Light Source (Oxford, UK). The data were processed using DIALS (30). Molecular replacement was carried out using Phaser (31), with the crystal structure of B. halodurans R2 as a model (PDB code 2RCC). The structure was built in Phenix (32) and Coot (33) and refined with Phenix refine (32). Relevant data processing and refinement statistics can be found in Table 1. The coordinates and structure factors of the R2 structure were deposited in the PDB with accession code 6ZJK.
EPR and UV-vis spectroscopy
Measurements were performed on a Bruker ELEXYS E500 spectrometer equipped with cold finger Dewar filled with liquid nitrogen (77K). The Xepr software package (Bruker) was used for data acquisition and processing of spectra. UV-visible spectra were measured at 25 °C on a PerkinElmer Lambda 35 spectrophotometer.
Size-exclusion chromatography
The R1 and R2 proteins were analyzed on a Superdex 200 column (GE Healthcare) run at 0.5 ml/min using a mobile phase containing 150–1000 mm KCl, 10 mm magnesium acetate, and 50 mm Tris-HCl, pH 7.6. Allosteric effectors were also included in the mobile phase, and the samples were mixed with at least 75% mobile phase and preincubated for 5 min before loading. The UV detector was set at 290 nm to minimize UV absorption from the allosteric effectors, and the sample loop was 100 μl. Molecular masses of protein complexes were derived from the retention times compared with that of a molecular mass standard composed of thyroglobulin (690 kDa), ferritin (440 kDa), IgG (150 kDa), transferrin (78 kDa), ovalbumin (45 kDa), and myoglobin (17 kDa). The logarithms of the standard protein molecular masses as a function of the retention time of the standard was plotted, and the equation derived from linear regression was used for calculations of molecular masses in the R1–R2 samples.
GEMMA
The GEMMA instrument consisted of a 3480 electrospray aerosol generator, a 3080 electrostatic classifier, a 3086 differential mobility analyzer, and a 3025A ultrafine condensation particle counter from TSI Inc. (Shoreview, MN, USA). R1 and R2 proteins were equilibrated to a solution containing 0.5 mm TCEP and 300 mm ammonium acetate, pH 7.8, by Sephadex G-50 chromatography. The R2 protein was stable in ammonium acetate and worked well in GEMMA. When analyzing the R1 protein, the instrument needed first to be equilibrated with a solution of 0.1 mg/ml solution of the protein in 400 mm ammonium acetate and 0.005% Tween 20 until the signal became stable. Because of the ability of the R1 protein to adhere to the GEMMA capillary, it usually took 30 min before the R1 signal appeared and a few minutes more before the signal was stable. At this point, the capillary walls were saturated with R1 protein, and the ammonium acetate concentration could be decreased to the working solution that was 100 or 200 mm ammonium acetate and 0.005% Tween 20. Two R1 preparations were used in the GEMMA analyses (as indicated in the figure legends). Most experiments were performed with the R1 preparation containing traces of dATP (see above). This dATP-containing preparation was more stable in ammonium acetate than dATP-free protein. The dATP-free preparation needed to be used directly without prior Sephadex-G50 chromatography by diluting a concentrated stock solution (5 mg/ml) into ammonium acetate. The nonvolatile salts (NaCl and HEPES-KOH) were then diluted enough to be able to analyze the samples with GEMMA. Generally, each experiment represents an average of three to five scans using a particle density of 0.58 g/cm3 for diameter to mass conversion.
Negative-stain EM
A mixture of the R1 and R2 proteins were incubated in a solution containing 50 mm KCl, 20 mm MgCl2, 2 mm DTT, and 50 mm Tris-HCl, pH 7.5. The protein was diluted to a final concentration of 150 nm of each protein (R1 and R2) in a buffer containing 0.2 mm dATP. After application of the protein to continuous carbon Formvar EM grids, it was stained three times for 10 s with drops of 1.5% uranyl acetate, blotted, and allowed to air dry. Imaging was performed at a Thermo Fisher Talos L120C operated at 120 kV, equipped with a 4096- × 4096-pixel Ceta camera. The nominal magnification was 57,000×, leading to an object pixel size of 2.5 Å. The predominant particle type was ring-shaped. These particles were selected from the micrographs, and two-dimensional class averages were calculated using Relion 2.0.
Enzyme assays
The R1 and R2 proteins were premixed on ice in a buffer solution containing 300 mm NaCl, 0.5 mm TCEP, and 50 mm HEPES-KOH, pH 7.5. 3.67 μl of this mixture (containing 1 μg of R1 and 2 μg of R2) was added to 46.33 μl of an ice-cold solution containing substrate and allosteric effectors, 10 mm magnesium acetate, 10 mm DTT, and 20 mm Tris-HCl, pH 7.5 (final concentrations indicated). The small amount of NaCl coming from the premix (final concentration, 22 μm) was important for full enzyme activity. The enzyme assays were incubated at 37 °C for 20 min and transferred to ice before adding 450 μl of ice-cold water (the low temperature slowed down the reaction to nondetectable levels). The samples were centrifuged through 3-kDa cutoff filters to remove the protein and mixed 1:1 with mobile phase before HPLC analysis on a 150 × 2.1 mm Sunshell C18-WP 2.6 μm column (ChromaNik Technologies Inc., Osaka, Japan). The analysis was performed at 48 °C using a mobile phase of 23% solution A, 57% solution B, and 20% solution C. Solution A contained 7% (v/v) acetonitrile and 23 g/liter KH2PO4 adjusted to pH 6.2 with KOH, solution B contained 7% (v/v) acetonitrile, and solution C contained 7% methanol and 3.56 g/liter tetrabutylammonium bromide. The size of the sample loop was 5 μl, and the flow rate was 0.4 ml/min. The NDPs and dNDPs came out within 12 min, and subsequently the mobile phase was changed to a composition of 75% A, 5% B, and 20% C to elute the allosteric effectors and re-equilibrated to original conditions before loading the next sample. The peaks were detected by their absorption at 270 nm (FAST setting) with a UV-2075 Plus detector. The enzyme assay was linear with respect to time and R1 protein concentration (the R2 protein was in molar excess). Kd and kcat values for allosteric effectors were calculated by using the one-site specific binding formula in Fig. 6A and the Agonist versus response (three parameters) formula in Fig. 6B using GraphPad Prism 7.04 software (GraphPad Software Inc., San Diego, CA, USA) on the average graph from three independent data sets, and the standard error was based on how well the average data fitted with the formula. For the IC50 value of dATP inhibition, the [Inhibitor] versus response formula was used to first calculate the individual IC50 values of the experiments and the standard error was based on the variation between the individual IC50 values. Activity measurements of the EPR sample differed from the above in that 1.6 times excess of R1 over R2 was used, and the substrate concentration was 0.8 mm CDP.
Phylogenetic analysis
Sequences from NCBI's RefSeq and GenBankTM databases (34), downloaded in March 2019, were searched with subclass specific HMMER (35) profiles for NrdA with NrdJ (encoding class II RNRs) serving as outgroup. The resulting sequences were clustered at 60% identity with UCLUST (36) to create a representative set of sequences. After manual inspection of sequences, 342 of 27,821 original NrdA sequences remained, plus 26 NrdJ sequences selected for aligning well to NrdA. The sequences were aligned with ProbCons (37), and 283 reliably aligned positions were selected with BMGE (38) using the BLOSUM30 matrix. The ATP-cones were not included in the positions used for phylogenetic reconstruction, because they potentially have a different evolutionary history than the rest of the sequences.
Data availability
The R2 protein crystal structure data are accessible in the Protein Data Bank (code 6ZJK), and a more detailed phylogenetic tree can be found at https://doi.org/10.17045/sthlmuni.11558187.v1. All other data are contained in the article.
Supplementary Material
Acknowledgments
We acknowledge the facilities and technical assistance of the Umeå Core Facility Electron Microscopy at the Chemical Biological Centre of Umeå University, which is part of the National Microscopy Infrastructure. We also thank the Protein Science Facility at Karolinska Institute for protein expression and purification.
This article contains supporting information.
Author contributions—M. M.-C., D. L., M. S., L.-A. C., and A. H. data curation; M. M.-C., V. R. J., D. L., M. S., L.-A. C., N. J., and A. H. methodology; M. M.-C., D. L., M. S., L.-A. C., M. H., B.-M. S., P. S., and A. H. writing-review and editing; V. R. J., B.-M. S., P. S., and A. H. investigation; D. L., P. S., and A. H. conceptualization; L.-A. C., M. H., B.-M. S., P. S., and A. H. funding acquisition; B.-M. S., P. S., and A. H. supervision; P. S. and A. H. project administration; A. H. writing-original draft; M. M.-C. performed the work and analysis on the R2 crystal structure; V. R. J. and A. H. performed and analyzed GEMMA, SEC and enzyme assays; D. L. performed the phylogenetic analysis; M. S. performed and analyzed the work on the R2 protein radical; L.-A. C. performed and analyzed the cryo-EM structure; N. J. performed and analyzed some of the enzyme assays.
Funding and additional information—This work was supported by Swedish Research Council Grants 2019-01242 (to A. H.), 2018-03406 (to P. S.), and 2019-01400 (to B.-M. S.); Swedish Cancer Foundation Grants 2017/716 (to P. S.) and 2018/820 (to B.-M. S.); funds from the Wallenberg Centre for Molecular Medicine, Umeå (to L.-A. C.); Human Frontier Science Program Grant CDA00047/2017-C (to L.-A. C.); and funds from the Wenner-Gren Foundation (to B.-M. S.). Some work was done with the facilities and technical assistance of the Umeå Core Facility Electron Microscopy at the Chemical Biological Centre of Umeå University, a part of the National Microscopy Infrastructure, under Grant VR-RFI 2016-00968.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- RNR
- ribonucleotide reductase
- GEMMA
- gas-phase electrophoretic mobility macromolecular analysis
- TCEP
- tris(2-carboxyethyl)phosphine
- PDB
- Protein Data Bank
- RMSD
- root-mean-square deviation
- s-site
- specificity site
- a-site
- activity site
- CV
- column volume.
References
- 1. Hofer A., Crona M., Logan D. T., and Sjöberg B.-M. (2012) DNA building blocks: keeping control of manufacture. Crit. Rev. Biochem. Mol. Biol. 47, 50–63 10.3109/10409238.2011.630372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cotruvo J. A., and Stubbe J. (2011) Class I ribonucleotide reductases: metallocofactor assembly and repair in vitro and in vivo. Annu. Rev. Biochem. 80, 733–767 10.1146/annurev-biochem-061408-095817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rose H. R., Ghosh M. K., Maggiolo A. O., Pollock C. J., Blaesi E. J., Hajj V., Wei Y., Rajakovich L. J., Chang W. C., Han Y., Hajj M., Krebs C., Silakov A., Pandelia M. E., Bollinger J. M. Jr., et al. (2018) Structural basis for superoxide activation of Flavobacterium johnsoniae class I ribonucleotide reductase and for radical initiation by its dimanganese cofactor. Biochemistry 57, 2679–2693 10.1021/acs.biochem.8b00247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Srinivas V., Lebrette H., Lundin D., Kutin Y., Sahlin M., Lerche M., Eirich J., Branca R. M. M., Cox N., Sjöberg B. M., and Högbom M. (2018) Metal-free ribonucleotide reduction powered by a DOPA radical in Mycoplasma pathogens. Nature 563, 416–420 10.1038/s41586-018-0653-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rozman Grinberg I., Lundin D., Hasan M., Crona M., Jonna V. R., Loderer C., Sahlin M., Markova N., Borovok I., Berggren G., Hofer A., Logan D. T., and Sjöberg B.-M. (2018) Novel ATP-cone-driven allosteric regulation of ribonucleotide reductase via the radical-generating subunit. eLife 7, e31529 10.7554/eLife.31529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brown N. C., and Reichard P. (1969) Role of effector binding in allosteric control of ribonucleoside diphosphate reductase. J. Mol. Biol. 46, 39–55 10.1016/0022-2836(69)90056-4 [DOI] [PubMed] [Google Scholar]
- 7. Eriksson S., Thelander L., and Akerman M. (1979) Allosteric regulation of calf thymus ribonucleoside diphosphate reductase. Biochemistry 18, 2948–2952 10.1021/bi00581a005 [DOI] [PubMed] [Google Scholar]
- 8. Aravind L., Wolf Y. I., and Koonin E. V. (2000) The ATP-cone: an evolutionarily mobile, ATP-binding regulatory domain. J. Mol. Microbiol. Biotechnol. 2, 191–194 [PubMed] [Google Scholar]
- 9. Parker M. J., Zhu X., and Stubbe J. (2014) Bacillus subtilis class Ib ribonucleotide reductase: high activity and dynamic subunit interactions. Biochemistry 53, 766–776 10.1021/bi401056e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Thomas W. C., Brooks F. P. 3rd, Burnim A. A., Bacik J. P., Stubbe J., Kaelber J. T., Chen J. Z., and Ando N. (2019) Convergent allostery in ribonucleotide reductase. Nat. Commun. 10, 2653 10.1038/s41467-019-10568-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rofougaran R., Vodnala M., and Hofer A. (2006) Enzymatically active mammalian ribonucleotide reductase exists primarily as an α6β2 octamer. J. Biol. Chem. 281, 27705–27711 10.1074/jbc.M605573200 [DOI] [PubMed] [Google Scholar]
- 12. Fairman J. W., Wijerathna S. R., Ahmad M. F., Xu H., Nakano R., Jha S., Prendergast J., Welin R. M., Flodin S., Roos A., Nordlund P., Li Z., Walz T., and Dealwis C. G. (2011) Structural basis for allosteric regulation of human ribonucleotide reductase by nucleotide-induced oligomerization. Nat. Struct. Mol. Biol. 18, 316–322 10.1038/nsmb.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jonna V. R., Crona M., Rofougaran R., Lundin D., Johansson S., Brännström K., Sjöberg B.-M., and Hofer A. (2015) Diversity in overall activity regulation of ribonucleotide reductase. J. Biol. Chem. 290, 17339–17348 10.1074/jbc.M115.649624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Johansson R., Jonna V. R., Kumar R., Nayeri N., Lundin D., Sjöberg B.-M., Hofer A., and Logan D. T. (2016) Structural mechanism of allosteric activity regulation in a ribonucleotide reductase with double ATP cones. Structure 24, 906–917 10.1016/j.str.2016.03.025 [DOI] [PubMed] [Google Scholar]
- 15. Brown N. C., and Reichard P. (1969) Ribonucleoside diphosphate reductase: formation of active and inactive complexes of proteins B1 and B2. J. Mol. Biol. 46, 25–38 10.1016/0022-2836(69)90055-2 [DOI] [PubMed] [Google Scholar]
- 16. Rofougaran R., Crona M., Vodnala M., Sjöberg B.-M., and Hofer A. (2008) Oligomerization status directs overall activity regulation of the Escherichia coli class Ia ribonucleotide reductase. J. Biol. Chem. 283, 35310–35318 10.1074/jbc.M806738200 [DOI] [PubMed] [Google Scholar]
- 17. Ando N., Brignole E. J., Zimanyi C. M., Funk M. A., Yokoyama K., Asturias F. J., Stubbe J., and Drennan C. L. (2011) Structural interconversions modulate activity of Escherichia coli ribonucleotide reductase. Proc. Natl. Acad. Sci. U.S.A. 108, 21046–21051 10.1073/pnas.1112715108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dong M., Masuyer G., and Stenmark P. (2019) Botulinum and tetanus neurotoxins. Annu. Rev. Biochem. 88, 811–837 10.1146/annurev-biochem-013118-111654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nordlund P., and Eklund H. (1993) Structure and function of the Escherichia coli ribonucleotide reductase protein R2. J. Mol. Biol. 232, 123–164 10.1006/jmbi.1993.1374 [DOI] [PubMed] [Google Scholar]
- 20. Högbom M., Galander M., Andersson M., Kolberg M., Hofbauer W., Lassmann G., Nordlund P., and Lendzian F. (2003) Displacement of the tyrosyl radical cofactor in ribonucleotide reductase obtained by single-crystal high-field EPR and 1.4-A x-ray data. Proc. Natl. Acad. Sci. U.S.A. 100, 3209–3214 10.1073/pnas.0536684100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kang G., Taguchi A. T., Stubbe J., and Drennan C. L. (2020) Structure of a trapped radical transfer pathway within a ribonucleotide reductase holocomplex. Science 368, 424–427 10.1126/science.368.6486.42-d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Krissinel E., and Henrick K. (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268 10.1107/S0907444904026460 [DOI] [PubMed] [Google Scholar]
- 23. Krissinel E., and Henrick K. (2005) Multiple alignment of protein structures in three dimensions. In Computational Life Sciences (Berthold M., Glen R. C., Diederichs K., Kohlbacher O., and Fischer I., eds) vol. 3695, pp. 67–78, Springer, Berlin, Germany: 10.1007/11560500_7 [DOI] [Google Scholar]
- 24. Rozman Grinberg I., Lundin D., Sahlin M., Crona M., Berggren G., Hofer A., and Sjöberg B.-M. (2018) A glutaredoxin domain fused to the radical-generating subunit of ribonucleotide reductase (RNR) functions as an efficient RNR reductant. J. Biol. Chem. 293, 15889–15900 10.1074/jbc.RA118.004991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lundin D., Berggren G., Logan D. T., and Sjöberg B.-M. (2015) The origin and evolution of ribonucleotide reduction. Life (Basel) 5, 604–636 10.3390/life5010604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hofer A., Ekanem J. T., and Thelander L. (1998) Allosteric regulation of Trypanosoma brucei ribonucleotide reductase studied in vitro and in vivo. J. Biol. Chem. 273, 34098–34104 10.1074/jbc.273.51.34098 [DOI] [PubMed] [Google Scholar]
- 27. Hofer A., Schmidt P. P., Gräslund A., and Thelander L. (1997) Cloning and characterization of the R1 and R2 subunits of ribonucleotide reductase from Trypanosoma brucei. Proc. Natl. Acad. Sci. U.S.A. 94, 6959–6964 10.1073/pnas.94.13.6959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Berglund O., and Eckstein F. (1972) Synthesis of ATP- and dATP-substituted sepharoses and their application in the purification of phage-T4–induced ribonucleotide reductase. Eur. J. Biochem. 28, 492–496 10.1111/j.1432-1033.1972.tb01936.x [DOI] [PubMed] [Google Scholar]
- 29. Berglund O. (1972) Ribonucleoside diphosphate reductase induced by bacteriophage T4: II. Allosteric regulation of substrate sepecificity and catalytic activity. J. Biol. Chem. 247, 7276–7281 [PubMed] [Google Scholar]
- 30. Waterman D. G., Winter G., Gildea R. J., Parkhurst J. M., Brewster A. S., Sauter N. K., and Evans G. (2016) Diffraction-geometry refinement in the DIALS framework. Acta Crystallogr. D Struct. Biol. 72, 558–575 10.1107/S2059798316002187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liebschner D., Afonine P. V., Baker M. L., Bunkóczi G., Chen V. B., Croll T. I., Hintze B., Hung L. W., Jain S., McCoy A. J., Moriarty N. W., Oeffner R. D., Poon B. K., Prisant M. G., Read R. J., et al. (2019) Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D Struct. Biol. 75, 861–877 10.1107/S2059798319011471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Haft D. H., DiCuccio M., Badretdin A., Brover V., Chetvernin V., O'Neill K., Li W., Chitsaz F., Derbyshire M. K., Gonzales N. R., Gwadz M., Lu F., Marchler G. H., Song J. S., Thanki N., et al. (2018) RefSeq: an update on prokaryotic genome annotation and curation. Nucleic Acids Res. 46, D851–D860 10.1093/nar/gkx1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Eddy S. R. (2011) Accelerated profile HMM searches. PLoS Comput. Biol. 7, e1002195 10.1371/journal.pcbi.1002195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Edgar R. C. (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 10.1093/bioinformatics/btq461 [DOI] [PubMed] [Google Scholar]
- 37. Do C. B., Mahabhashyam M. S., Brudno M., and Batzoglou S. (2005) ProbCons: probabilistic consistency-based multiple sequence alignment. Genome Res. 15, 330–340 10.1101/gr.2821705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Criscuolo A., and Gribaldo S. (2010) BMGE (Block Mapping and Gathering with Entropy): a new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol. Biol. 10, 210 10.1186/1471-2148-10-210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sahlin M., Gräslund A., Ehrenberg A., and Sjöberg B.-M. (1982) Structure of the tyrosyl radical in bacteriophage T4-induced ribonucleotide reductase. J. Biol. Chem. 257, 366–369 [PubMed] [Google Scholar]
- 40. Zimanyi C. M., Chen P. Y., Kang G., Funk M. A., and Drennan C. L. (2016) Molecular basis for allosteric specificity regulation in class Ia ribonucleotide reductase from Escherichia coli. eLife 5, e07141 10.7554/eLife.07141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sahlin M., Gräslund A., and Ehrenberg A. (1986) Determination of relaxation times for a free-radical from microwave saturation studies. J. Magn. Reson. 67, 135–137 10.1016/0022-2364(86)90417-8 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The R2 protein crystal structure data are accessible in the Protein Data Bank (code 6ZJK), and a more detailed phylogenetic tree can be found at https://doi.org/10.17045/sthlmuni.11558187.v1. All other data are contained in the article.