

Abstract
Much of our understanding of the spatial organization of and interactions between cellular organelles and macromolecular complexes has been the result of imaging studies utilizing either light- or electron-based microscopic analyses. These classical approaches, while insightful, are nonetheless limited either by restrictions in resolution or by the sheer complexity of generating multidimensional data. Recent advances in the use and application of X-rays to acquire micro- and nanotomographic data sets offer an alternative methodology to visualize cellular architecture at the nanoscale. These new approaches allow for the subcellular analyses of unstained vitrified cells and three-dimensional localization of specific protein targets and have served as an essential tool in bridging light and electron correlative microscopy experiments. Here, we review the theory, instrumentation details, acquisition principles, and applications of both soft X-ray tomography and X-ray microscopy and how the use of these techniques offers a succinct means of analyzing three-dimensional cellular architecture. We discuss some of the recent work that has taken advantage of these approaches and detail how they have become integral in correlative microscopy workflows.
Keywords: X-ray microscopy, tomography, subcellular organelle, yeast, S. cerevisiae, development, correlative microscopy, microscopy, Schizosaccharomyces pombe, hard and soft X-rays, water window
Understanding the three-dimensional spatial organization of inter- and intracellular components is paramount in elucidating their function in both healthy and diseased states. Modern microscopy has evolved from a disparate array of isolated techniques to a cohesive toolbox of imaging approaches that routinely allow the visualization of proteins of interest, dynamic events, and how changes in cellular morphology and macromolecular structure affect physiology. However, despite the unique insights enabled by these approaches, limitations in each imaging modality, along with the increasing desire to generate data from a near-native state with limited perturbation, have led to the need for and development of new approaches and correlative techniques.
Light microscopy approaches are widely available and relatively simple and have enabled significant insights into cellular organization. This vast array of imaging modalities, spanning from brightfield transmitted light microscopy to superresolution fluorescence nanoscopy, provide tremendous flexibility in the analyses of cellular and subcellular morphology. Three-dimensional light microscopy is routinely performed through the use of confocal (1–3), light-sheet (4), and multiphoton (5, 6) approaches. These techniques are, by necessity, dependent on either fluorescent reporter probes or the presence of intrinsic signals, such as autofluorescence or harmonic signal generation (7). As such, signals obtained from these imaging experiments are limited to visualizing structures that are either “tagged” or generate the intrinsic signals being measured, thus rendering the remainder of the cell invisible.
EM is routinely used to explore the ultrastructural organization of cells and tissues at nanometer or less resolution. As the electron beam has a limited depth of penetration, volumetric analyses have often proven to be complex, difficult, and time-consuming. Typically, preparation of samples for transmission EM (one of the most widely used ultrastructural imaging techniques) involves a primary aldehyde fixation with a secondary heavy metal fixation, infiltration into a resin support matrix, and thin sections taken (50–100 nm), which can then be visualized under the microscope. This methodology can be extended to three dimensions using tilt-series tomography (8, 9) and serial sectioning (10, 11), with such approaches being utilized to study a wide array of tissues and cell types. These tomographic techniques, however, are limited by sectioning precision and the arduous alignment and processing steps, which can be very time-consuming. More recently, scanning EM (SEM) has been used to generate volumetric data. Specifically, serial block-face-SEM (SBF-SEM) (12), array tomography (13), and the use of a focused ion beam in conjunction with SEM (FIB-SEM) (14) have all been used in nanotomography studies (15–18). Despite improved acquisition speeds and automation options, SBF-SEM and array tomography are limited by nonisotropic voxels, having different dimensions in the x-y and z planes and thus obfuscating volumetric analysis, whereas FIB-SEM is limited by the small sample volumes that may be acquired (10–40 μm in the z-plane).
In contrast, tomographic techniques that utilize X-rays, including soft X-ray tomography (SXT) (19) and X-ray microscopy (XRM) have recently been shown to be an invaluable tool in studying the spatial architecture of individual cells, as well as the localization of specific proteins of interest within cells and tissues. These approaches offer significant gains when compared with EM, with hard X-rays capable of penetrating through the entire organism and soft X-rays allowing single-cell visualization of minimally perturbed native specimens, without the need for contrast-enhancing agents. X-rays, with their substantially shorter wavelengths, offer almost an order of magnitude better resolution as compared with light microscopy, with more recent advances resolving objects down to 5 nm (20). Naturally, these resolutions are atypical of the vast part of the work that has been done with the approach, with much more objective values shown in Table 1. Nonetheless, the use of X-rays on vitrified samples offers a nanotomographic technique with preparation ease only matched by cryo-FIB systems (21–23), but with substantially higher acquisition speed. Analyses of the mesoscale architecture and the ability to target specific regions of whole tissues offer a bridge from the micro- to the nanoscale, and X-ray–based microscopy has become a vital step in correlative studies, linking multifluorophore microscopy with ultrastructural analyses. The cryopreservation requirements of soft X-ray microscopy have expanded to include cryo-light microscopy (24), whereas XRM has been employed as a mapping tool to facilitate localization of regions of interest in 3D nanotomography studies (25).
Table 1.
Comparison of imaging modalities
This comparison table shows the divergent approaches of generating tomographic data, focusing on resolution, speed of image acquisition, and average cost of instruments (in thousands (k) and millions (M) of US dollars).
| Technique | Resolution (limits) | Imaging time | Fixation method | Staining methods | Protein labeling | Instrument cost range | References |
|---|---|---|---|---|---|---|---|
| Confocal microscopy | ∼200 nm | Minutes | Chemical/None | Fluorescence | Immunofluorescence | $300k–500k | 1, 2 |
| Superresolution microscopy | ∼20 nm | Minutes | Chemical/None | Fluorescence | Immunofluorescence | $750k–1M | 124, 125 |
| TEM | <1 nm | Minutes | Chemical/Vitrification | Heavy metals | Immunogold | $750k–2M | 8, 9, 126 |
| SEM | <5 nm | Minutes | Chemical | Heavy metals | Immunogold | $500k–1M | 127 |
| FIB-SEM | ∼5 nm | Hours | Chemical/Vitrification | Heavy metals | Ni-DAB | $2M–4M | 14, 128, 129 |
| Cryo-TEM | ∼2 Å | Days | Vitrification | None | None | $5M–10M+ | 101 |
| Cryo-FIB | ∼5 nm | Hours | Vitrification | None | None | ∼$1M | 21–23 |
| XRM | 80 nm | Hours | Chemical | Heavy metals | Ni-DAB | $1M–1.5M | 71 |
| SXT | 30 nm | Minutes | Vitrification | None | None | Synchrotron cost: $200M+; beamline cost: $2M–3M | 29, 48, 123 |
Soft X-ray tomography
Theory and instrumentation
On the electromagnetic spectrum, soft X-rays reside between the extreme UV and X-ray bands, from 250 eV to several thousand eV, corresponding to wavelengths of 5 nm to ∼2 Å (26). This range includes specifically well-defined L- and K-edges, for identifying biologically relevant elements, such as carbon, nitrogen, and oxygen. The so-called “water window” (Fig. 1C), from the carbon K-edge at 284 eV (4.38 nm) to the oxygen K-edge at 543 eV (2.34 nm), is of particular interest in biological studies as it offers intrinsic contrast between water and protein (27). Visualizing unstained cells and tissues at such energies would thus lead to strong X-ray absorption by macromolecules, such as lipid membranes or intracellular organelles, with only minimal attenuation by water molecules. Differences in the absorption relate to a parameter termed the linear absorption coefficient (LAC), whose constant values can be used to identify specific intracellular structures (27). This property presents the underlying principle of SXT, and it led to the development and refinement of the technique over the past 30 years (28). Over the past two decades, the use of SXT has expanded and, along with concurrent advances in detection technologies, has allowed for structural studies of samples, preserved in their native states, which were previously inaccessible.
Figure 1.
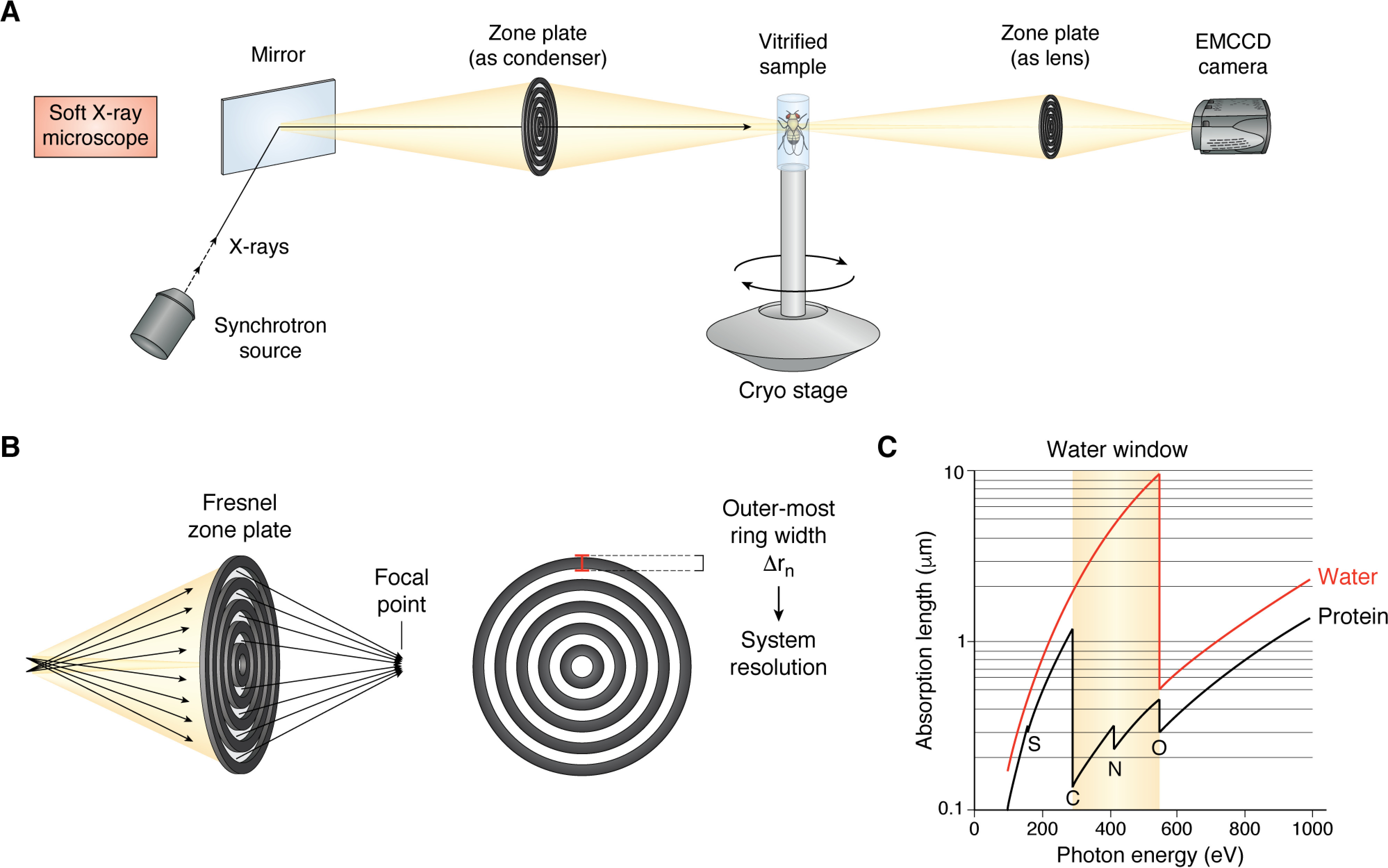
Soft X-ray microscope design and principle. A, diagrammatic representation of a soft X-ray microscope. X-rays from a synchrotron source are passed through a bend magnet and steered to the stage via a mirror. Fresnel zone plates are used as both the condenser and the lens. Vitrified samples are mounted on a cryo-stage, and images are collected directly on an EMCCD. B, a Fresnel zone plate consists of radially symmetric rings, with the outermost ring of the objective zone plate effectively determining the resolution of the system. C, K-absorption edges of carbon (284 eV) and oxygen (543 eV) form the “water window,” at which energies water (red trace) is transparent but protein (black trace) can generate sufficient contrast for imaging.
Like all microscopes, a soft X-ray microscope (SXM) is comprised of a source, a detector, and a means of driving the beam to the sample (condenser) and focusing it to the detector (objective). The X-ray source necessary for an SXM need not be bright, but it does require a large photon flux (29). These requirements have generally restricted SXM to facilities where synchrotron radiation can be passed to low-emittance storage rings, where a series of undulators or bending magnets allow soft X-rays to be filtered and utilized for high-resolution experiments (30, 31). The complexity and scale of synchrotron facilities have kept SXM systems for biological samples to less than a handful worldwide (32). At the moment, there are four such facilities, including the Advanced Light Source at Berkeley, the BESSY II in Berlin, the Diamond Light Source in the UK, and ALBA in Barcelona. Smaller soft X-ray sources have been developed over the past decade, using nitrogen or methanol plasma (33, 34), with the aim of producing a soft X-ray source suitable for a small laboratory. These sources, however, have not seen wide adoption, mainly due to their lower flux and the fact that unlike synchrotron radiation, plasma sources are not continuous. As such, data generated by them results in lower resolution and requires longer integration times.
SXM systems condense soft X-rays onto samples and collect emission through diffractive elements called zone plates (29, 35). These plates are comprised of alternating concentric rings of transparent and opaque “zones,” with the outermost zone width (Δrn) defining the maximum resolution of a system (Fig. 1B). Zone plate resolutions range from 60 to 25 nm (36, 37), allowing imaging depths of up to 15 μm. While offering enhanced resolution, zone plate use in soft X-ray microscopes does pose some constraints on imaging experiments. First, zone plates are very inefficient, with most of the radiation being absorbed or diffracted, allowing roughly 10% through to the sample (38). Second, acquisition geometry requires the objective plate to reside millimeters away from the sample, which limits the usable energies of the microscope. Finally, the use of zone plates has historically been an inflexible solution, as changing of optics to alter resolution or imaging depth necessitates the complete venting of the imaging chamber and careful realignment of all optical elements (29). Recent efforts have shown the utility of multiple microzone plates, thus allowing for a more flexible, multiresolution approach (39).
In contrast to the complex X-ray sources, soft X-ray microscopes have modest requirements for detection. As soft X-rays that pass through zone plates can be directly detected, there is no need for scintillation material. Thus, simple, back-thinned CCD cameras are sufficiently fast for data acquisition, and the ability to modulate on-chip gain levels and binning make such devices amenable for SXT imaging (Fig. 1A).
Among the chief advantages of SXT is that samples do not need to be chemically fixed or stained with exogenous contrasting agents. Such fixation protocols have been known to cause numerous artifacts, including uneven fixation, formation of aggregates, and morphological changes, such as dehydration (40–42). Yeast in particular, as described below, are notoriously unreceptive to chemical fixation, which results in dramatic morphological changes when compared with vitrified cells (43). As such, cells can be imaged in as close to their native states as possible. X-ray radiation readily damages cells (44), and most biological samples have to be vitrified to maintain their integrity during imaging. It has been reported that reducing sample temperatures to the cryogenic regime permits the acquisition of more than 1000 images without any evidence of radiation damage (45). Vitrification is typically achieved via plunge freezing into a cryogen such as liquid ethane, much like the method used in cryo-EM (46, 47), or by a cryogenic gas stream using N2-cooled helium (48). SXT of cryo-preserved cells thus requires the use of a cryo-stage to maintain the samples in a vitrified state. Depending on the vitrification approach and the stage design, this may require the use of vacuum chambers (29). Sample mounting is typically achieved by the use of either canonical-style TEM grids or custom glass capillaries, which provide the ability to visualize the samples from any rotational angle (19, 37). To facilitate reconstruction, most experiments employ gold bead fiducials, embedded on the exterior surface of imaging capillaries (49).
Acquisition principles and parameters
A single SXM image is simply a projection image and has no z-depth. Thus, an SXT data set requires the sequential acquisition of images over a range of angular increments. The size of the specimen, along with resolution requirements and system limitations, dictates the necessary parameters for an optimal tomographic reconstruction. As images in a tomographic acquisition are attained in the axis perpendicular to sample rotation, and each reconstruction plane is independent of the rest, the resolution of the final tomogram is equal to the resolution perpendicular to the planes (50). The minimum number of equally spaced projection images required to generate a tomogram with isotropic voxels that is collected over an angular range is given by the Crowther criterion, n = πD/d, where D is the sample diameter, and d is the spatial resolution (9, 50, 51). In practice, this means that for a 180° rotation, images would be acquired at increments of 1–2°, equating to 90–180 individual projection images (48).
The manner in which the sample is mounted to the imaging stage plays a vital role in an instrument's ability to generate complete tomograms. SXM systems that use flat TEM-style grids are limited to tilts of ±70° (32) by the short working distance of the objective zone plate, which presents several disadvantages. First, the inability to rotate the sample to 180° results in the “missing wedge” problem, where there are insufficient data to generate a complete tomogram (49, 51). Flat grids, when rotated through an imaging beam, will have a variable cross-section of vitreous ice, with thicker sections at the limits of tilt (49). This variance results in an inability to consistently measure and corelate the LAC to specific intracellular structures. In contrast, using the glass capillary approach allows for a full 360° rotation, which in turn resolves the “missing wedge” problem (48). Unlike flat grids, however, the capillary mounting approach is generally incompatible with adherent cell types (52).
Among the main advantages of soft X-ray tomography is the sheer speed of data acquisition. Due to the high-intensity synchrotron sources used, and the relatively thin samples typically imaged, acquisition rates are on the order of 5–10 frames/s (19, 48). This allows an entire tomography data set to be acquired on the order of 20 min, significantly improving throughput and facilitating the use of high degrees of automation in some systems (32).
Applications of soft X-ray tomography
SXT has evolved into an important tool for studying subcellular structures that have otherwise been inaccessible or difficult to assay. One such structure is the eukaryotic nucleus, whose spatial organization has been recently linked to the modulation of gene expression (53, 54) as well as the development of various cancers (55–57). The resolution requirements and central location of the nucleus have, however, made topological analyses difficult. SXT has allowed the exploration of the relationship between the spatial organization and function of the nucleus in a minimally perturbed manner. In one particularly impressive study, Lomvardas and colleagues (58) illustrated that the preferential expression of a specific olfactory receptor gene over others was due to the protein's proximity to the nuclear envelope, and such spatial organization may underlie monogenic gene expression. A more recent study utilized the SXT approach to explore the organization of chromatin in primary hematopoietic cells, revealing that both the amount of euchromatin and the spatial organization of heterochromatin govern the transition between pluripotent and progenitor cell states (59).
The ultrastructure of yeast cells has been difficult to study, largely due to the thick cell wall, which prevents effective chemical fixation. Stripping of the cell wall leads to denaturation, altering the subcellular ultrastructure from the native state. High-pressure freezing and subsequent freeze substitution processing methods are effective, but time-consuming, and are ultimately limited by the data acquisition approaches. Furthermore, the divergent imaging approaches employed have contributed significant amounts of data regarding different yeast species yet have offered limited understanding of the three-dimensional architecture. Saccharomyces cerevisiae, specifically, has been studied using SEM (Fig. 2A (43)), TEM (Fig. 2B (43)), and FIB-SEM (Fig. 2C (60)). Whereas SEM is particularly well-suited to topographical analysis of the the outer cell well, it offers no insight into interior organelle structure. TEM, conversely, offers the best imaging resolution, at the expense of understanding three-dimensional organization. FIB-SEM provides excellent resolution and the ability to generate nanotomographic data, but long processing times and imaging have hindered these efforts. SXT has proven to be instrumental in overcoming these barriers and elucidating the internal mesoscale organization of several yeast species, including S. cerevisiae and Schizosaccharomyces pombe. Larabell and colleagues at the Advanced Light Source, who have been at the forefront of this area and pioneered the development of SXT, have, in a series of studies, beautifully shown the morphology, density, and organization of nuclei, mitochondria, and lipid droplets in yeast species at resolutions down to 60 nm, while imaging over the span of minutes, rather than hours (Fig. 2D) (19, 61–64).
Figure 2.
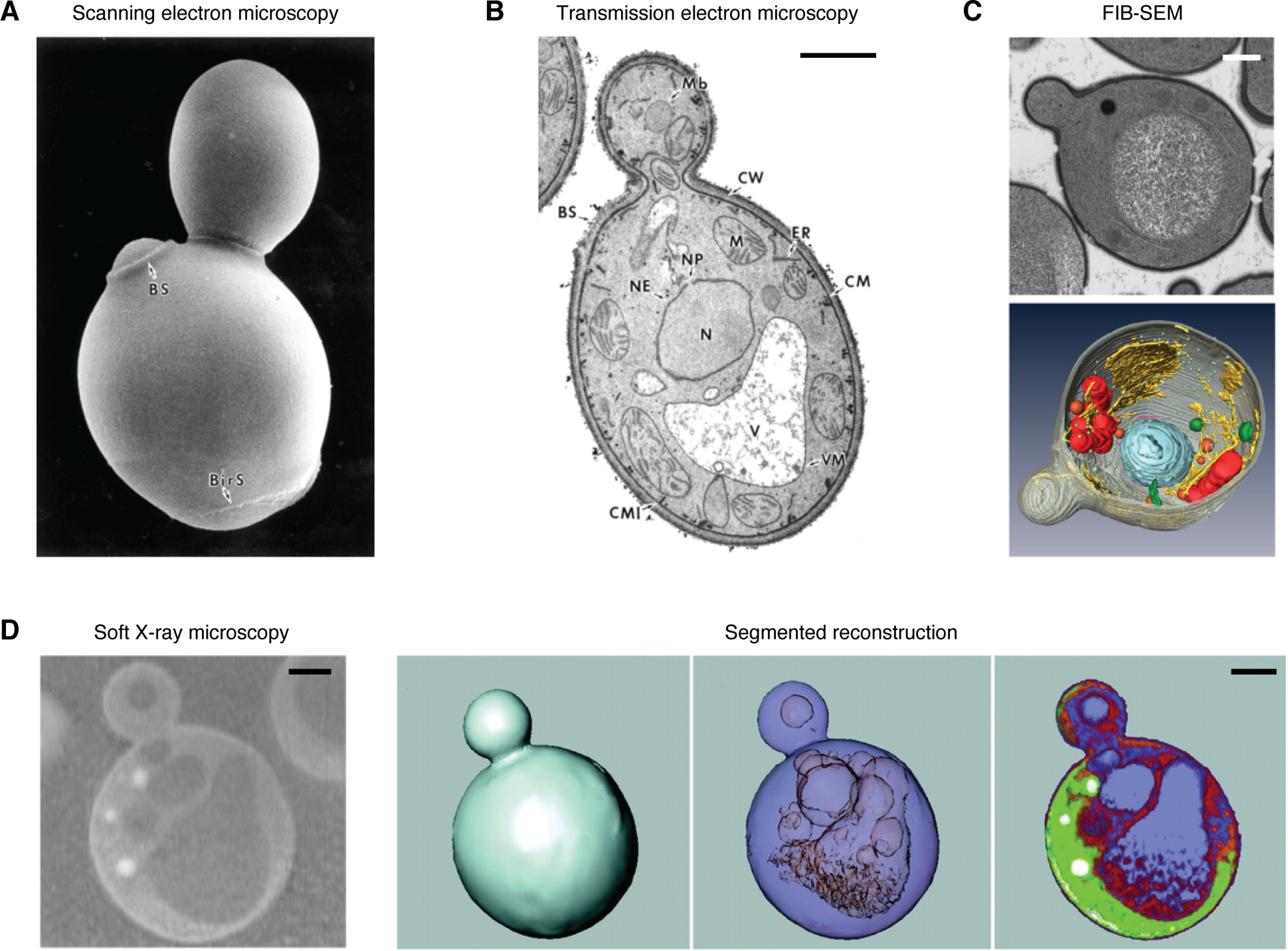
Comparison of imaging techniques used for analyzing S. cerevisiae. A, SEM image showing cell wall topography; B, TEM image demonstrating the high contrast and resolution attainable with TEM (both panels adapted from Fig. 1 of Osumi et al. (43)). C, FIB-SEM image shows the ability of this nanotomographic approach to generate high-resolution volumetric data. Top, raw, single-plane image; bottom, segmented reconstruction (adapted from Figs. 1 and 2 of Wei et al. (60)). D, SXM image (left) and segmented reconstruction of S. cerevisiae cell, showing the ability of SXT to resolve intracellular structures in minimally perturbed samples (adapted from Figs. 1 and 2 of Larabell and Le Gros (63). Scale bars, 1 μm.
The ability to study the three-dimensional organization of cells at nanoscale resolution using SXT has also been leveraged to elucidate the manner in which certain pathogenic microorganisms interact with eukaryotic cells. Using this approach, the structure of enveloped vaccinia virions and the localization of high-density regions, such as membrane-bound compartments, have been extrapolated (65). More recently, studies have identified endoplasmic reticulum membrane alterations that are associated with hepatitis C, showing colocalization between endoplasmic reticulum protrusions and viral RNA (66), as well as demonstrated that the initiation of herpes simplex type 1 infection evokes the generation of channels within the host nucleus that facilitate viral egress (67). These and other studies have demonstrated the ability of SXT to assay previously undetected or underappreciated cellular phenomena.
X-ray microscopy
Despite the obvious advantages of SXT in terms of resolution and the visualization and localization of complex intracellular structures in near-native state, the technique requires high photon flux, which limits it to synchrotron X-ray sources and low X-ray energies, which are required to access the “water window,” which restricts the thickness of samples to 10–20 μm. Imaging much larger samples, such as whole organs or an intact embryo, is simply not feasible. Furthermore, whereas LAC measurements can be instrumental in identifying intracellular structures, labeling target proteins with specific stains allows for increased precision in identifying and localizing such targets. XRM, which utilizes a table-top hard X-ray source (>10 keV), along with different modes of magnification, offers the ability to perform such experiments.
Instrumentation
Micro-CT sources are relatively compact, comprised of an electron gun (cathode) focused on a metal target (anode) typically made of tungsten or copper, housed in an evacuated chamber. The X-ray intensity is related, in a linear fashion, to the current output to the cathode, whereas the potential difference between the cathode and anode dictates the energy of the emitted X-rays (68). This architecture permits the independent adjustment of both parameters, thus providing an extremely flexible imaging source. The geometrically divergent beam passes through the sample, which is typically mounted on a stage that has a large three-axis translational range and can rotate ±180°, and is then detected (69).
Standard micro-CT instruments (Fig. 3A) suffer from a fundamental limitation, in that due to the divergence of the beam, the magnification geometry is solely dependent on the ratio of the source-to-sample and sample-to-detector distances. This is significant, as high-resolution can only be achieved when the sample is located as close to the source as possible. This has the consequence of limiting the range of sample motion and restricts the size of a sample that can be easily imaged. Detection is performed either via a flat panel detector (which is typical in micro-CT) or via a set of high-numerical aperture objectives that incorporate a scintillation layer in their front elements to convert impinging radiation into visible photons, which are subsequently detected via an electron-multiplied CCD (EMCCD) camera (Fig. 3B) (70). This offers a secondary, optical magnification factor, allowing for what is termed “resolution at a distance,” or RaaD. This allows for substantially greater flexibility in the geometrical setup of an experiment, while still maintaining high resolution over a much larger range of sample sizes (71).
Figure 3.
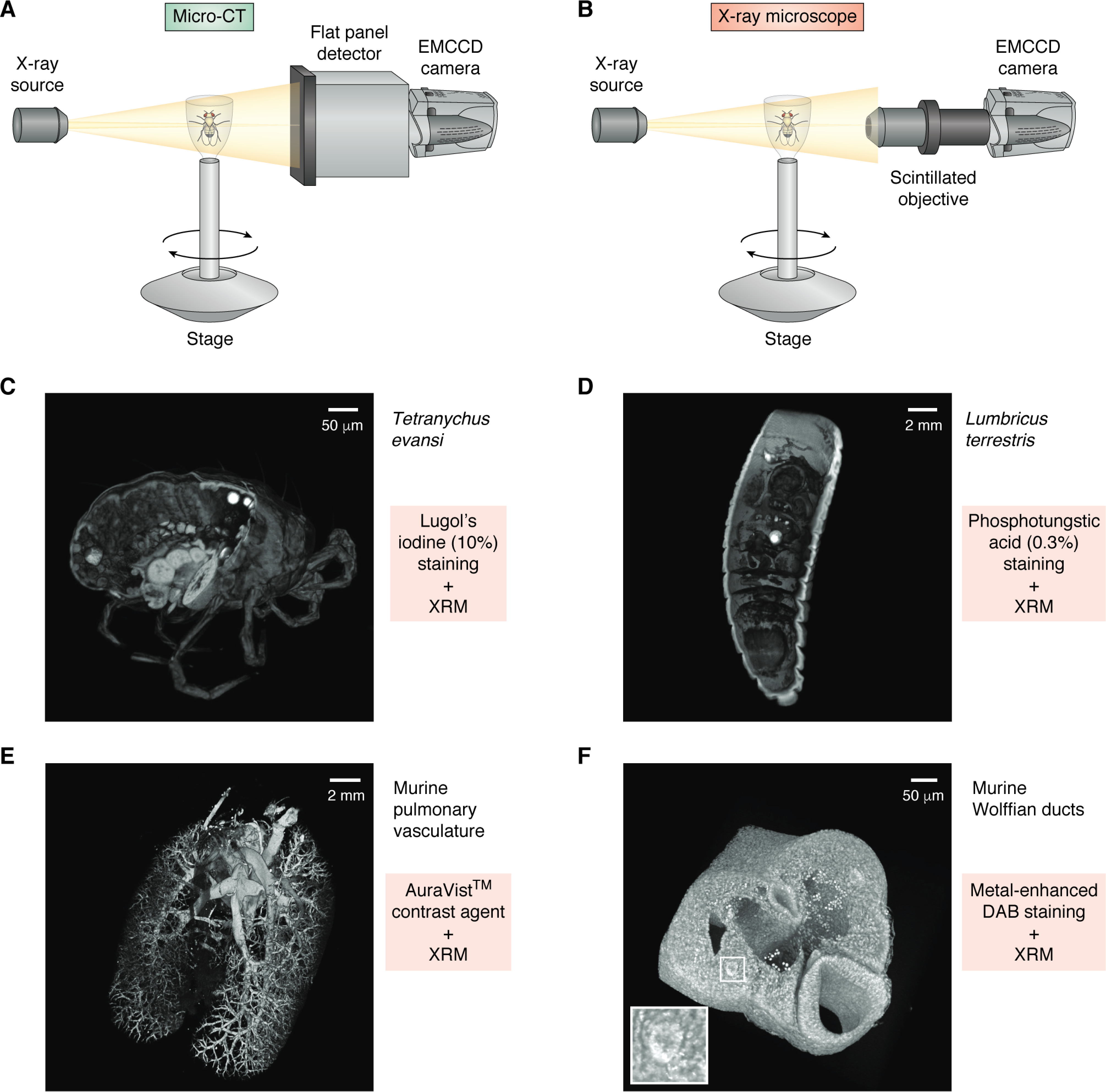
Micro-CT and X-ray microscope design and use of contrast agents. A, diagrammatic representation of a micro-CT system, comprised of an X-ray source and a flat panel detector coupled to an EMCCD camera. Samples are mounted to a rotating stage between the source and the detector. Magnification is solely dependent on geometric ratio of source to sample and sample to detector. B, an X-ray microscope is similar in design to a micro-CT but introduces a high-numerical aperture objective that utilizes a scintillator to detect X-rays and emit photons that are detected by the EMCCD. The objective adds a second magnification factor to an XRM system. C–F, examples of volume-rendered XRM tomograms of biological samples stained using different contrast agents (each tomogram is digitally cut to illustrate stain penetration). C, Tetranychus evansi, stained using 10% Lugol's iodine. D, Lumbricus terrestris, stained with 0.3% phosphotungstic acid. E, murine pulmonary vasculature filled with AuroVist™ contrast agent. F, metal-enhanced DAB staining of murine Wolffian ducts (white box and inset).
Contrast agents
In contrast with SXT, which utilizes the “water window” to image intrinsic contrast differences between subcellular compartments, attenuating the hard X-rays in XRM requires elements of higher atomic number. Thus, visualizing nonmineralized soft tissues and cells necessitates the use of exogenous contrast-enhancing staining agents. Such agents are usually comprised of heavy metal–based solutions, such as osmium tetroxide (72, 73), phosphotungstic acid (Fig. 3D) (72–74), phosphomolybdic acid (74), and various iodine-based solutions, including ethanol- (I2E), methanol- (I2M), or water-based (I2KI, Lugol's) (75). The latter has become particularly prominent due to its ease of use, low toxicity, cost efficacy, and reversibility and its systemic, yet differential, staining of most tissues. Lugol's iodine (Fig. 3C) has thus been used to study a variety of samples, including murine embryos (76) and internal organs (77), neural tissues (78–80), and muscle (81–84).
Whereas soluble, nonspecific stains offer ease of use and an ability to visualize all tissues within a given sample, directing labels to specific targets remains a fundamental goal in XRM tomography studies. A number of contrast agents, for example, have been utilized in an intravasal manner to study lymphatics and microvasculature. Among these are barium sulfate (85, 86) and lead oxide, the latter of which has been described as the “gold standard” of fine vasculature visualization (87), although some work has shown that these agents might not fill smaller vessels, thus leading to an underestimation of the total vasculature (88). More recent studies have employed different tracing agents, such as gold nanoparticles (Fig. 3E) (89), bismuth formulations (90, 91), and tantalum oxide (92), the results of which have shown great promise as tools for XRM vasculature analyses.
More specific targeting of proteins of interest has also been demonstrated with XRM, offering a way to directly quantify aggregation. One such approach uses immune precipitation to deposit metal ions in close proximity to the target of interest. Specifically, this method uses a horseradish peroxidase–conjugated secondary antibody that binds to a primary antibody and locally produces singlet oxygen. A silver metal-deposition enzyme metallography kit then interacts with the singlet oxygen, thus reducing silver ions from the solution and causing its deposition near the site of the primary antibody. Metscher and Müller (93) used this approach to visualize and study acetylated tubulin in a chick nervous system as well as localization of type-II collagen in developing limbs. A similar approach has used a nickel-enhanced 3,3-diaminobenzidine (Ni-DAB) deposition (94) in conjunction with OsO4 to explore changes in avian excretory system morphology in RET tyrosine kinase mutants (Fig. 3F) (95). A set of novel genetically encoded targeting approaches, including enhanced ascorbate peroxidase (APEX) (96) and mini singlet oxygen generator (MiniSOG) (97), were originally designed for use in EM, but they have also been utilized to study the organization and distribution of specific cell types and localize specific structures using XRM in correlative studies (25, 98).
Regardless of the staining modality employed, a key feature in data sets obtained using XRM is that they offer not only the spatial localization of structures and specific proteins, but also the density of aggregated contrast agent. These densities present a distinct advantage over other imaging modalities, as they directly correlate to the expressed protein they target (93). The use of protein-specific staining, in conjunction with silver deposition, can thus be utilized to quantify a target gene product within a given tomographic volume.
Correlative approaches
Using divergent imaging approaches to study a given sample is a tremendously powerful technique, allowing for analyses of a region of interest over different spatial and temporal scales, thus maximizing efficiency in resources and precious sample utilization and extending the precision and breadth of complementary techniques. Thus, correlative light and EM (CLEM) approaches have become indispensable in associating ultrastructural organization with protein localization and temporally finite events (e.g. gene expression) (99). The recent popularization of cryogenic EM (cryo-EM) (100, 101) has also led to the development of correlative workflows with cryo-fluorescence microscopy (cryo-CLEM) (102, 103). Both SXT and XRM have been shown to extend and enhance these correlative approaches, offering a bridge between light and EM, thus linking physiological processes, such as motility, metabolic transport, and ion exchange, to cellular ultrastructure.
As SXT necessitates the vitrification of cells to prevent radiation damage, correlative light workflows require the development and use of light systems capable of imaging fluorescence while maintaining cryogenic temperatures. Such cryo-fluorescence imaging has gained much interest, not only due to the emergence of tomographic cryo-EM studies, but also for the fact that cryogenic temperatures offer significant improvements in fluorophore stability (104), fluorescence yield (105), and resolution (106). Whereas cryo-CLEM with SXT has only been in use over the past decade, and developments in the workflow and technology are ongoing (49, 52, 107), the resulting data thus far have demonstrated the incredible potential for this approach. Applications have included identification of inactive X-chromosomes in female transformed thymic lymphoma cells (108), localization of vacuoles in yeast strains (109), and analysis of autophagosome formation in the endoplasmic reticulum of a mammalian cell (110). Correlation of the fluorescence and X-ray volumetric data are, however, dependent on the use of fiducial markers, which are typically fluorescent polystyrene beads that are capable of being visualized by both modalities.
Whereas correlative light and EM techniques have been developed and improved upon for over 4 decades (111), XRM offers a means to bridge the two modalities with increased speed, precision, and reproducibility. Fiducial-based image registration (112, 113) has been successfully used in CLEM studies, but due to the three-dimensional nature of the samples in question, this approach requires correlating six fiducials for localization, thus imposing limitations on the density of the markers. Most EM preparations involve secondary fixation with heavy metal solutions, such as osmium, lead, and uranium (114, 115), thus rendering the samples opaque. As X-rays are readily attenuated by these compounds, EM staining is especially advantageous in XRM, and the resultant tomogram can be used as a three-dimensional map to locate regions of interest for subsequent EM (116). This approach has been demonstrated with FIB-SEM nanotomography of materials (117), in bridging brightfield light microscopy to TEM in the assaying of adipose tissue (118), and through the use of genetically encoded tags in murine brain sections, which were subsequently imaged via serial block-face EM (25). More recent work has utilized XRM-dependent correlative approaches to an increasingly wide range of targets, from exploring ultrastructural properties of parasitic worms in the small bowel (119) to correlating histological and TEM imaging of Araneae papal organs (120) to linking intravital microscopy studies of mouse cerebral vasculature to FIB-SEM nanotomographic analysis (121). An added advantage of the high resolution, mesoscale imaging, and speed of XRM acquisition is the ability to employ the technique as a region of interest localization tool. As an example, in exploring osseous metastatic disease in whole bones via TEM, we employed XRM within our workflow to facilitate the location of metastases within long bones (Fig. 4). This allowed for targeted sectioning of the site of metastasis, which would be impractical to accomplish without the aid of the XRM data. It is apparent that since its recent incorporation into correlative studies, XRM has become an indispensable tool.
Figure 4.
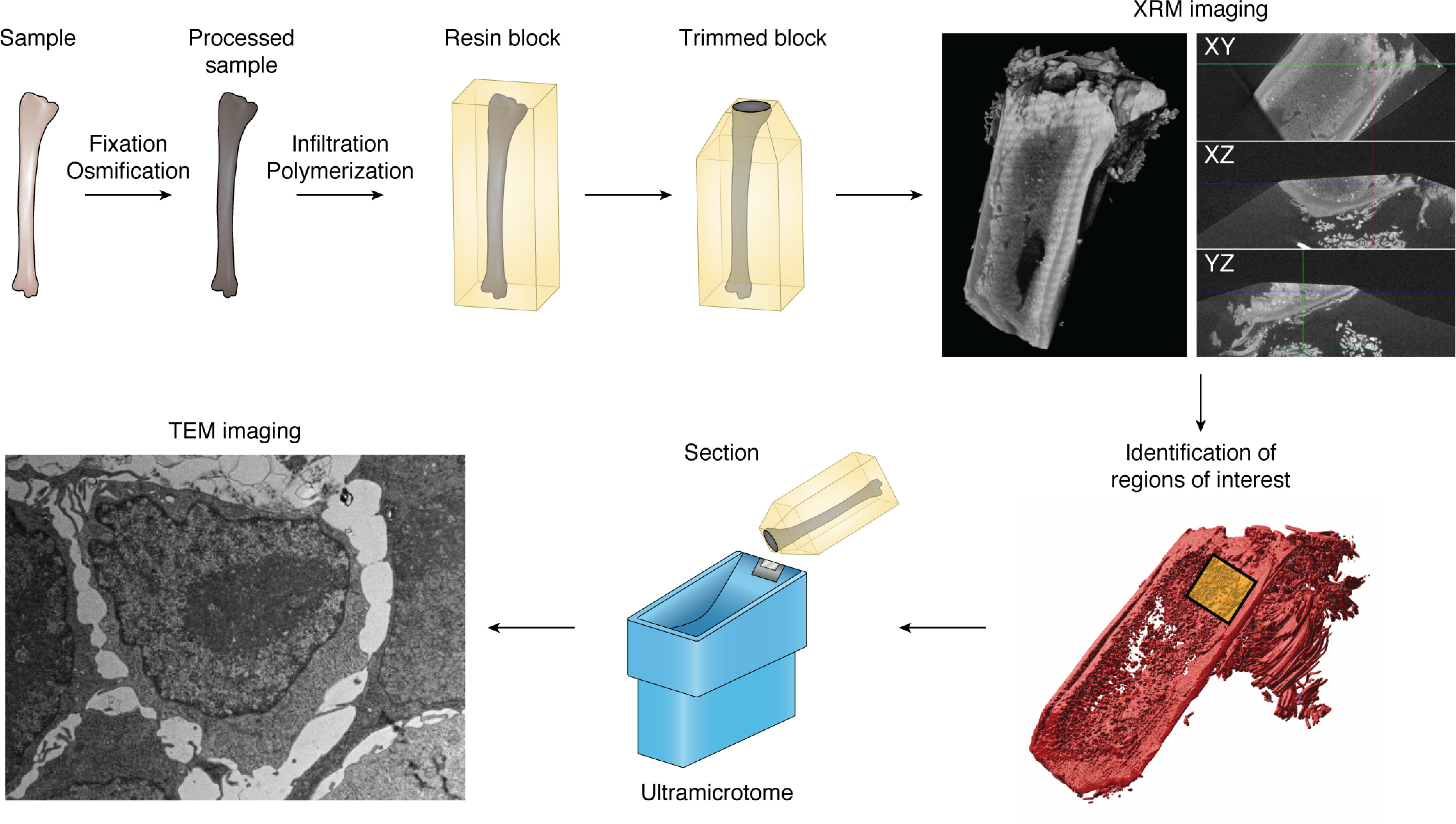
Correlative XRM–TEM workflow for spatial targeting of single cells in large sample volumes. An example workflow shows the generation of tomographic data using an XRM and using these tomograms to facilitate targeted sectioning and TEM imaging. A sample is first fixed in EM-grade fixatives and subsequently post-stained with heavy metals. The processed sample is embedded in a resin block and trimmed to facilitate imaging. Trimmed blocks are imaged on an XRM system, generating tomographic data, allowing for identification of a region of interest. Thin sections are thus generated from the targeted region and are imaged on a TEM.
Conclusions
The application of X-rays to study various biological samples is not a new technique. Indeed, some of the earliest X-ray imaging was performed over a century ago, and zone plate–based systems were being developed as early as the 1960s (122). The past 20 years, however, have seen a rapid expansion in the application, utilization, and integration of X-ray–based microscopy into workflows aimed to assay three-dimensional biological structures. Use of soft X-rays has been proven capable of studying minimally perturbed cells, with resolutions nearing that of EM, and it holds the exciting prospect of being able to thoroughly explore the cellular mesoscale architecture (123). The development and application of metallic stains for XRM and the ability to direct such staining to specific targets offer the promise of exploring the means by which the spatial organization of any protein of interest affects the spatiotemporal dynamic properties of cells. The physical properties of both SXT and XRM hold unique quantification opportunities, with linear absorption coefficients offering the ability to identify subcellular compartments (123) and stain densities in XRM presenting a unique means of measuring protein aggregation. Application of X-ray microscopy techniques in EM correlative studies have demonstrated the increased efficacy the approach offers and the resultant increases in sample throughput. Ongoing developments in targeted contrast agents, workflow integration, and system flexibility and expansion make SXT and XRM exciting tools for future studies of cellular organization and function.
Acknowledgments
This work utilized the resources of the Washington University Diabetes Research Center (supported by National Institutes of Health (NIH) Grant P30 DK020579, the Washington University Rheumatic Diseases Research Resource-based Center (NIH Grant P30 AR073752), the Washington University Musculoskeletal Research Center (NIH Grant P30 AR074992), and the Siteman Cancer Center of Barnes-Jewish Hospital and Washington University School of Medicine (NIH Grant P30 CA091842). The three-dimensional X-ray phase-contrast microscope for sub-micron quantitative biological imaging was acquired with the support of NIH Grant S10 OD021694.
Funding and additional information—This work was supported by Children's Discovery Institute of Washington University and St. Louis Children's Hospital Grants CDI-CORE-2015-505 and CDI-CORE-2019-813 (to J. A. J. F.), and Foundation for Barnes-Jewish Hospital Grants 3770 and 4642 (to J. A. J. F.).
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- SEM
- scanning EM
- SBF
- serial block-face
- FIB
- focused ion beam
- SXT
- soft X-ray tomography
- XRM
- X-ray microscopy
- LAC
- linear absorption coefficient
- SXM
- soft X-ray microscope
- TEM
- transmission EM
- Ni-DAB
- nickel-enhanced 3,3-diaminobenzidine
- CLEM
- correlative light and EM.
References
- 1. White J. G., Amos W. B., and Fordham M. (1987) An evaluation of confocal versus conventional imaging of biological structures by fluorescence light microscopy. J. Cell Biol. 105, 41–48 10.1083/jcb.105.1.41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brakenhoff G. J., van Spronsen E. A., van der Voort H. T., and Nanninga N. (1989) Three-dimensional confocal fluorescence microscopy. Methods Cell Biol. 30, 379–398 10.1016/s0091-679x(08)60987-5 [DOI] [PubMed] [Google Scholar]
- 3. Bayguinov P. O., Oakley D. M., Shih C. C., Geanon D. J., Joens M. S., and Fitzpatrick J. A. J. (2018) Modern laser scanning confocal microscopy. Curr. Protoc. Cytom. 85, e39 10.1002/cpcy.39 [DOI] [PubMed] [Google Scholar]
- 4. Power R. M., and Huisken J. (2017) A guide to light-sheet fluorescence microscopy for multiscale imaging. Nat. Methods 14, 360–373 10.1038/nmeth.4224 [DOI] [PubMed] [Google Scholar]
- 5. Denk W., Strickler J. H., and Webb W. W. (1990) Two-photon laser scanning fluorescence microscopy. Science 248, 73–76 10.1126/science.2321027 [DOI] [PubMed] [Google Scholar]
- 6. Padmanabhan K., Andrews S. E., and Fitzpatrick J. A. (2010) Multi-photon imaging. Curr. Protoc. Cytom. 2, 2.9 10.1002/0471142956.cy0209s54 [DOI] [PubMed] [Google Scholar]
- 7. Campagnola P. J., Wei M. D., Lewis A., and Loew L. M. (1999) High-resolution nonlinear optical imaging of live cells by second harmonic generation. Biophys. J. 77, 3341–3349 10.1016/S0006-3495(99)77165-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. De Rosier D. J., and Klug A. (1968) Reconstruction of three dimensional structures from electron micrographs. Nature 217, 130–134 10.1038/217130a0 [DOI] [PubMed] [Google Scholar]
- 9. Crowther R. A., De Rosier D. J., and Klug A. (1970) The reconstruction of a three-dimensional structure from projections and its application to electron microscopy. Proc. R. Soc. Lond. B Biol. Sci. 317, 319–340 [DOI] [PubMed] [Google Scholar]
- 10. Birch-Andersen A. (1955) Reconstruction of the nuclear sites of Salmonella typhimurium from electron micrographs of serial sections. J. Gen. Microbiol. 13, 327–329 10.1099/00221287-13-2-327 [DOI] [PubMed] [Google Scholar]
- 11. Stevens J. K., Davis T. L., Friedman N., and Sterling P. (1980) A systematic approach to reconstructing microcircuitry by electron microscopy of serial sections. Brain Res. 2, 265–293 10.1016/0165-0173(80)90010-7 [DOI] [PubMed] [Google Scholar]
- 12. Denk W., and Horstmann H. (2004) Serial block-face scanning electron microscopy to reconstruct three-dimensional tissue nanostructure. PLos Biol. 2, e329 10.1371/journal.pbio.0020329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Micheva K. D., and Smith S. J. (2007) Array tomography: a new tool for imaging the molecular architecture and ultrastructure of neural circuits. Neuron 55, 25–36 10.1016/j.neuron.2007.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Heymann J. A., Hayles M., Gestmann I., Giannuzzi L. A., Lich B., and Subramaniam S. (2006) Site-specific 3D imaging of cells and tissues with a dual beam microscope. J. Struct. Biol. 155, 63–73 10.1016/j.jsb.2006.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Knott G., Marchman H., Wall D., and Lich B. (2008) Serial section scanning electron microscopy of adult brain tissue using focused ion beam milling. J. Neurosci. 28, 2959–2964 10.1523/JNEUROSCI.3189-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hekking L. H., Lebbink M. N., De Winter D. A., Schneijdenberg C. T., Brand C. M., Humbel B. M., Verkleij A. J., and Post J. A. (2009) Focused ion beam-scanning electron microscope: exploring large volumes of atherosclerotic tissue. J. Microsc. 235, 336–347 10.1111/j.1365-2818.2009.03274.x [DOI] [PubMed] [Google Scholar]
- 17. He Q., Hsueh M., Zhang G., Joy D. C., and Leapman R. D. (2018) Biological serial block face scanning electron microscopy at improved z-resolution based on Monte Carlo model. Sci. Rep. 8, 12985 10.1038/s41598-018-31231-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kasthuri N., Hayworth K. J., Berger D. R., Schalek R. L., Conchello J. A., Knowles-Barley S., Lee D., Vázquez-Reina A., Kaynig V., Jones T. R., Roberts M., Morgan J. L., Tapia J. C., Seung H. S., Roncal W. G., et al. (2015) Saturated reconstruction of a volume of neocortex. Cell 162, 648–661 10.1016/j.cell.2015.06.054 [DOI] [PubMed] [Google Scholar]
- 19. Larabell C. A., and Nugent K. A. (2010) Imaging cellular architecture with X-rays. Curr. Opin. Struct. Biol. 20, 623–631 10.1016/j.sbi.2010.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shapiro D. A., Yu Y. S., Tyliszczak T., Cabana J., Celestre R., Chao W., Kaznatcheev K., Kilcoyne A. L. D., Maia F., Marchesini S., Meng Y. S., Warwick T., Yang L. L., and Padmore H. A. (2014) Chemical composition mapping with nanometre resolution by soft X-ray microscopy. Nat. Photonics 8, 765–776 10.1038/nphoton.2014.207 [DOI] [Google Scholar]
- 21. Schertel A., Snaidero N., Han H. M., Ruhwedel T., Laue M., Grabenbauer M., and Möbius W. (2013) Cryo FIB-SEM: volume imaging of cellular ultrastructure in native frozen specimens. J. Struct. Biol. 184, 355–360 10.1016/j.jsb.2013.09.024 [DOI] [PubMed] [Google Scholar]
- 22. Vidavsky N., Akiva A., Kaplan-Ashiri I., Rechav K., Addadi L., Weiner S., and Schertel A. (2016) Cryo-FIB-SEM serial milling and block face imaging: large volume structural analysis of biological tissues preserved close to their native state. J. Struct. Biol. 196, 487–495 10.1016/j.jsb.2016.09.016 [DOI] [PubMed] [Google Scholar]
- 23. Hoffman D. P., Shtengel G., Xu C. S., Campbell K. R., Freeman M., Wang L., Milkie D. E., Pasolli H. A., Iyer N., Bogovic J. A., Stabley D. R., Shirinifard A., Pang S., Peale D., Schaefer K., et al. (2020) Correlative three-dimensional super-resolution and block-face electron microscopy of whole vitreously frozen cells. Science 367, eaaz5357 10.1126/science.aaz5357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McDermott G., Le Gros M. A., and Larabell C. A. (2012) Visualizing cell architecture and molecular location using soft x-ray tomography and correlated cryo-light microscopy. Annu. Rev. Phys. Chem. 63, 225–239 10.1146/annurev-physchem-032511-143818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bushong E. A., Johnson D. D. Jr., Kim K. Y., Terada M., Hatori M., Peltier S. T., Panda S., Merkle A., and Ellisman M. H. (2015) X-ray microscopy as an approach to increasing accuracy and efficiency of serial block-face imaging for correlated light and electron microscopy of biological specimens. Microsc. Microanal. 21, 231–238 10.1017/S1431927614013579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Attwood D. T., Sakdinawat A., and Geniesse L. (2016) X-rays and Extreme Ultraviolet Radiation: Principles and Applications, 2nd Ed., pp. 27–59, Cambridge University Press, Cambridge, UK [Google Scholar]
- 27. Weiss D., Schneider G., Niemann B., Guttmann P., Rudolph D., and Schmahl G. (2000) Computed tomography of cryogenic biological specimens based on X-ray microscopic images. Ultramicroscopy 84, 185–197 10.1016/S0304-3991(00)00034-6 [DOI] [PubMed] [Google Scholar]
- 28. Jacobsen C. (1999) Soft x-ray microscopy. Trends Cell Biol. 9, 44–47 10.1016/S0962-8924(98)01424-X [DOI] [PubMed] [Google Scholar]
- 29. Carzaniga R., Domart M. C., Collinson L. M., and Duke E. (2014) Cryo-soft X-ray tomography: a journey into the world of the native-state cell. Protoplasma 251, 449–458 10.1007/s00709-013-0583-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Watanabe M., and Isoyama G. (1995) Synchrotron radiation and free electron lasers. in Dynamics during Spectroscopic Transitions: Basic Concepts, pp 506–568, Springer Verlag, Berlin [Google Scholar]
- 31. Fuchs M., Weingartner R., Popp A., Major Z., Becker S., Osterhoff J., Cortrie I., Zeitler B., Hörlein R., Tsakiris G. D., Schramm U., Rowlands-Rees T. P., Hooker S. M., Habs D., Krausz F., et al. (2009) Laser-driven soft-X-ray undulator source. Nat. Phys. 5, 826–829 10.1038/nphys1404 [DOI] [Google Scholar]
- 32. Duke E., Dent K., Razi M., and Collinson L. M. (2014) Biological applications of cryo-soft X-ray tomography. J. Microsc. 255, 65–70 10.1111/jmi.12139 [DOI] [PubMed] [Google Scholar]
- 33. Bertilson M., von Hofsten O., Vogt U., Holmberg A., and Hertz H. M. (2009) High-resolution computed tomography with a compact soft x-ray microscope. Opt. Express 17, 11057–11065 10.1364/oe.17.011057 [DOI] [PubMed] [Google Scholar]
- 34. Horne S. F., Silterra J., and Holber W. (2009) A compact soft X-ray microscope using an electrode-less Z-pinch source. J. Phys. Conf. Ser. 186, 12028 10.1088/1742-6596/186/1/012028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Baez A. V. (1961) Fresnel zone plate for optical image formation using extreme ultraviolet and soft X radiation. J. Opt. Soc. Am. 51, 405–412 10.1364/JOSA.51.000405 [DOI] [Google Scholar]
- 36. Schneider G., Guttmann P., Heim S., Rehbein S., Mueller F., Nagashima K., Heymann J. B., Müller W. G., and McNally J. G. (2010) Three-dimensional cellular ultrastructure resolved by X-ray microscopy. Nat. Methods 7, 985–987 10.1038/nmeth.1533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. McDermott G., Fox D. M., Epperly L., Wetzler M., Barron A. E., Le Gros M. A., and Larabell C. A. (2012) Visualizing and quantifying cell phenotype using soft X-ray tomography. Bioessays 34, 320–327 10.1002/bies.201100125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kirz J., and Attwood D. T. (2001) Zone plates. in X-ray Data Booklet (Thompson A. C., and Vaughan D., eds) 2nd Ed., Lawrence Berkeley National Laboratory, Berkeley, CA [Google Scholar]
- 39. Weinhardt V., Chen J. H., Ekman A. A., Guo J., Remesh S. G., Hammel M., McDermott G., Chao W., Oh S., Le Gros M. A., and Larabell C. A. (2020) Switchable resolution in soft X-ray tomography of single cells. PLoS ONE 15, e0227601 10.1371/journal.pone.0227601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hobot J. A., Villiger W., Escaig J., Maeder M., Ryter A., and Kellenberger E. (1985) Shape and fine structure of nucleoids observed on sections of ultrarapidly frozen and cryosubstituted bacteria. J. Bacteriol. 162, 960–971 10.1128/JB.162.3.960-971.1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Griffiths G., Brands R., Burke B., Louvard D., and Warren G. (1982) Viral membrane proteins acquire galactose in trans Golgi cisternae during intracellular transport. J. Cell Biol. 95, 781–792 10.1083/jcb.95.3.781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chandler D. E. (1984) Comparison of quick-frozen and chemically fixed sea-urchin eggs: structural evidence that cortical granule exocytosis is preceded by a local increase in membrane mobility. J. Cell Sci. 72, 23–36 [DOI] [PubMed] [Google Scholar]
- 43. Osumi M. (2012) Visualization of yeast cells by electron microscopy. J. Electron. Microsc. (Tokyo) 61, 343–365 10.1093/jmicro/dfs082 [DOI] [PubMed] [Google Scholar]
- 44. Sayre D., Kirz J., Feder R., Kim D. M., and Spiller E. (1976) Transmission microscopy of unmodified biological materials: comparative radiation dosages with electrons and ultrasoft X-ray photons. Ultramicroscopy 2, 337–349 10.1016/S0304-3991(76)91997-5 [DOI] [PubMed] [Google Scholar]
- 45. Le Gros M. A., McDermott G., and Larabell C. A. (2005) X-ray tomography of whole cells. Curr. Opin. Struct. Biol. 15, 593–600 10.1016/j.sbi.2005.08.008 [DOI] [PubMed] [Google Scholar]
- 46. Dubochet J., Adrian M., Chang J. J., Homo J. C., Lepault J., McDowall A. W., and Schultz P. (1988) Cryo-electron microscopy of vitrified specimens. Q. Rev. Biophys. 21, 129–228 10.1017/s0033583500004297 [DOI] [PubMed] [Google Scholar]
- 47. Baker L. A., and Rubinstein J. L. (2010) Radiation damage in electron cryomicroscopy. Methods Enzymol. 481, 371–388 10.1016/S0076-6879(10)81015-8 [DOI] [PubMed] [Google Scholar]
- 48. Parkinson D. Y., Epperly L. R., McDermott G., Le Gros M. A., Boudreau R. M., and Larabell C. A. (2013) Nanoimaging cells using soft X-ray tomography. Methods Mol. Biol. 950, 457–481 10.1007/978-1-62703-137-0_25 [DOI] [PubMed] [Google Scholar]
- 49. Cinquin B. P., Do M., McDermott G., Walters A. D., Myllys M., Smith E. A., Cohen-Fix O., Le Gros M. A., and Larabell C. A. (2014) Putting molecules in their place. J. Cell. Biochem. 115, 209–216 10.1002/jcb.24658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Weiß D., Schneider G., Vogt S., Guttmann P., Niemann B., Rudolph D., and Schmahl G. (2001) Tomographic imaging of biological specimens with the cryo transmission X-ray microscope. Nucl. Instr. Methods Phys. Res. A 467–468, 1308–1311 10.1016/S0168-9002(01)00648-9 [DOI] [Google Scholar]
- 51. Do M., Isaacson S. A., McDermott G., Le Gros M. A., and Larabell C. A. (2015) Imaging and characterizing cells using tomography. Arch. Biochem. Biophys. 581, 111–121 10.1016/j.abb.2015.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hagen C., Guttmann P., Klupp B., Werner S., Rehbein S., Mettenleiter T. C., Schneider G., and Grünewald K. (2012) Correlative VIS-fluorescence and soft X-ray cryo-microscopy/tomography of adherent cells. J. Struct. Biol. 177, 193–201 10.1016/j.jsb.2011.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Evans S. A., Horrell J., and Neretti N. (2019) The three-dimensional organization of the genome in cellular senescence and age-associated diseases. Semin. Cell Dev. Biol. 90, 154–160 10.1016/j.semcdb.2018.07.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Meaburn K. J. (2016) Spatial genome organization and its emerging role as a potential diagnosis tool. Front. Genet. 7, 134 10.3389/fgene.2016.00134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Easwaran H. P., and Baylin S. B. (2010) Role of nuclear architecture in epigenetic alterations in cancer. Cold Spring Harb. Symp. Quant. Biol. 75, 507–515 10.1101/sqb.2010.75.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lever E., and Sheer D. (2010) The role of nuclear organization in cancer. J. Pathol. 220, 114–125 10.1002/path.2651 [DOI] [PubMed] [Google Scholar]
- 57. Veltri R. W., and Christudass C. S. (2014) Nuclear morphometry, epigenetic changes, and clinical relevance in prostate cancer. Adv. Exp. Med. Biol. 773, 77–99 10.1007/978-1-4899-8032-8_4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Clowney E. J., LeGros M. A., Mosley C. P., Clowney F. G., Markenskoff-Papadimitriou E. C., Myllys M., Barnea G., Larabell C. A., and Lomvardas S. (2012) Nuclear aggregation of olfactory receptor genes governs their monogenic expression. Cell 151, 724–737 10.1016/j.cell.2012.09.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ugarte F., Sousae R., Cinquin B., Martin E. W., Krietsch J., Sanchez G., Inman M., Tsang H., Warr M., Passegué E., Larabell C. A., and Forsberg E. C. (2015) Progressive chromatin condensation and H3K9 methylation regulate the differentiation of embryonic and hematopoietic stem cells. Stem Cell Rep. 5, 728–740 10.1016/j.stemcr.2015.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wei D., Jacobs S., Modla S., Zhang S., Young C. L., Cirino R., Caplan J., and Czymmek K. (2012) High-resolution three-dimensional reconstruction of a whole yeast cell using focused-ion beam scanning electron microscopy. BioTechniques 53, 41–48 10.2144/000113850 [DOI] [PubMed] [Google Scholar]
- 61. Uchida M., McDermott G., Wetzler M., Le Gros M. A., Myllys M., Knoechel C., Barron A. E., and Larabell C. A. (2009) Soft X-ray tomography of phenotypic switching and the cellular response to antifungal peptoids in Candida albicans. Proc. Natl. Acad. Sci. U. S. A. 106, 19375–19380 10.1073/pnas.0906145106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Uchida M., Sun Y., McDermott G., Knoechel C., Le Gros M. A., Parkinson D., Drubin D. G., and Larabell C. A. (2011) Quantitative analysis of yeast internal architecture using soft X-ray tomography. Yeast 28, 227–236 10.1002/yea.1834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Larabell C. A., and Le Gros M. A. (2004) X-ray tomography generates 3-D reconstructions of the yeast, Saccharomyces cerevisiae, at 60-nm resolution. Mol. Biol. Cell 15, 957–962 10.1091/mbc.e03-07-0522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Walters A. D., Amoateng K., Wang R., Chen J. H., McDermott G., Larabell C. A., Gadal O., and Cohen-Fix O. (2019) Nuclear envelope expansion in budding yeast is independent of cell growth and does not determine nuclear volume. Mol. Biol. Cell 30, 131–145 10.1091/mbc.E18-04-0204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Carrascosa J. L., Chichón F. J., Pereiro E., Rodríguez M. J., Fernández J. J., Esteban M., Heim S., Guttmann P., and Schneider G. (2009) Cryo-X-ray tomography of vaccinia virus membranes and inner compartments. J. Struct. Biol. 168, 234–239 10.1016/j.jsb.2009.07.009 [DOI] [PubMed] [Google Scholar]
- 66. Romero-Brey I., Merz A., Chiramel A., Lee J. Y., Chlanda P., Haselman U., Santarella-Mellwig R., Habermann A., Hoppe S., Kallis S., Walther P., Antony C., Krijnse-Locker J., and Bartenschlager R. (2012) Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog. 8, e1003056 10.1371/journal.ppat.1003056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Myllys M., Ruokolainen V., Aho V., Smith E. A., Hakanen S., Peri P., Salvetti A., Timonen J., Hukkanen V., Larabell C. A., and Vihinen-Ranta M. (2016) Herpes simplex virus 1 induces egress channels through marginalized host chromatin. Sci. Rep. 6, 28844 10.1038/srep28844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zink F. E. (1997) X-ray tubes. Radiographics 17, 1259–1268 10.1148/radiographics.17.5.9308113 [DOI] [PubMed] [Google Scholar]
- 69. Bidola P., Morgan K., Willner M., Fehringer A., Allner S., Prade F., Pfeiffer F., and Achterhold K. (2017) Application of sensitive, high-resolution imaging at a commercial lab-based X-ray micro-CT system using propagation-based phase retrieval. J. Microsc. 266, 211–220 10.1111/jmi.12530 [DOI] [PubMed] [Google Scholar]
- 70. Martin T., and Koch A. (2006) Recent developments in X-ray imaging with micrometer spatial resolution. J. Synchrotron Radiat. 13, 180–194 10.1107/S0909049506000550 [DOI] [PubMed] [Google Scholar]
- 71. Merkle A. P., and Gelb J. (2013) The ascent of 3D X-ray microscopy in the laboratory. Micros. Today 21, 10–15 10.1017/S1551929513000060 [DOI] [Google Scholar]
- 72. Metscher B. D. (2009) MicroCT for developmental biology: a versatile tool for high-contrast 3D imaging at histological resolutions. Dev. Dyn. 238, 632–640 10.1002/dvdy.21857 [DOI] [PubMed] [Google Scholar]
- 73. Metscher B. D. (2009) MicroCT for comparative morphology: simple staining methods allow high-contrast 3D imaging of diverse non-mineralized animal tissues. BMC Physiol. 9, 11 10.1186/1472-6793-9-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Pauwels E., Van Loo D., Cornillie P., Brabant L., and Van Hoorebeke L. (2013) An exploratory study of contrast agents for soft tissue visualization by means of high resolution X-ray computed tomography imaging. J. Microsc. 250, 21–31 10.1111/jmi.12013 [DOI] [PubMed] [Google Scholar]
- 75. Gignac P. M., Kley N. J., Clarke J. A., Colbert M. W., Morhardt A. C., Cerio D., Cost I. N., Cox P. G., Daza J. D., Early C. M., Echols M. S., Henkelman R. M., Herdina A. N., Holliday C. M., Li Z., et al. (2016) Diffusible iodine-based contrast-enhanced computed tomography (diceCT): an emerging tool for rapid, high-resolution, 3-D imaging of metazoan soft tissues. J. Anat. 228, 889–909 10.1111/joa.12449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wong M. D., Spring S., and Henkelman R. M. (2013) Structural stabilization of tissue for embryo phenotyping using micro-CT with iodine staining. PLoS ONE 8, e84321 10.1371/journal.pone.0084321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. S J. M., Zanette I., Noël P. B., Cardoso M. B., Kimm M. A., and Pfeiffer F. (2015) Three-dimensional non-destructive soft-tissue visualization with X-ray staining micro-tomography. Sci. Rep. 5, 14088 10.1038/srep14088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Metscher B. D. (2013) Biological applications of X-ray microtomography: imaging micro-anatomy, molecular expression and organismal diversity. Microsc. Anal. (Am. Ed.) 27, 13–16 [Google Scholar]
- 79. Heimel P., Swiadek N. V., Slezak P., Kerbl M., Schneider C., Nürnberger S., Redl H., Teuschl A. H., and Hercher D. (2019) Iodine-enhanced micro-CT imaging of soft tissue on the example of peripheral nerve regeneration. Contrast Media Mol. Imaging 2019, 7483745 10.1155/2019/7483745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Gignac P. M., and Kley N. J. (2014) Iodine-enhanced micro-CT imaging: methodological refinements for the study of the soft-tissue anatomy of post-embryonic vertebrates. J. Exp. Zool. B Mol. Dev. Evol. 322, 166–176 10.1002/jez.b.22561 [DOI] [PubMed] [Google Scholar]
- 81. Holliday C. M., Tsai H. P., Skiljan R. J., George I. D., and Pathan S. (2013) A 3D interactive model and atlas of the jaw musculature of Alligator mississippiensis. PLoS ONE 8, e62806 10.1371/journal.pone.0062806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Düring D. N., Ziegler A., Thompson C. K., Ziegler A., Faber C., Müller J., Scharff C., and Elemans C. P. (2013) The songbird syrinx morphome: a three-dimensional, high-resolution, interactive morphological map of the zebra finch vocal organ. BMC Biol. 11, 1 10.1186/1741-7007-11-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kupczik K., Stark H., Mundry R., Neininger F. T., Heidlauf T., and Röhrle O. (2015) Reconstruction of muscle fascicle architecture from iodine-enhanced microCT images: a combined texture mapping and streamline approach. J. Theor. Biol. 382, 34–43 10.1016/j.jtbi.2015.06.034 [DOI] [PubMed] [Google Scholar]
- 84. Dickinson E., Stark H., and Kupczik K. (2018) Non-destructive determination of muscle architectural variables through the use of DiceCT. Anat. Rec. (Hoboken) 301, 363–377 10.1002/ar.23716 [DOI] [PubMed] [Google Scholar]
- 85. Lafage-Proust M. H., Roche B., Langer M., Cleret D., Vanden Bossche A., Olivier T., and Vico L. (2015) Assessment of bone vascularization and its role in bone remodeling. Bonekey Rep. 4, 662 10.1038/bonekey.2015.29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Blery P., Pilet P., Bossche A. V., Thery A., Guicheux J., Amouriq Y., Espitalier F., Mathieu N., and Weiss P. (2016) Vascular imaging with contrast agent in hard and soft tissues using microcomputed-tomography. J. Microsc. 262, 40–49 10.1111/jmi.12339 [DOI] [PubMed] [Google Scholar]
- 87. Bergeron L., Tang M., and Morris S. F. (2006) A review of vascular injection techniques for the study of perforator flaps. Plast. Reconstr. Surg. 117, 2050–2057 10.1097/01.prs.0000218321.36450.9b [DOI] [PubMed] [Google Scholar]
- 88. Kingston M. J., Perriman D. M., Neeman T., Smith P. N., and Webb A. L. (2016) Contrast agent comparison for three-dimensional micro-CT angiography: a cadaveric study. Contrast Media Mol. Imaging 11, 319–324 10.1002/cmmi.1695 [DOI] [PubMed] [Google Scholar]
- 89. Mahan M. M., and Doiron A. L. (2018) Gold nanoparticles as X-ray, CT, and multimodal imaging contrast agents: formulation, targeting, and methodology. J. Nanomater. 2018, 1–15 10.1155/2018/5837276 [DOI] [Google Scholar]
- 90. Swy E. R., Schwartz-Duval A. S., Shuboni D. D., Latourette M. T., Mallet C. L., Parys M., Cormode D. P., and Shapiro E. M. (2014) Dual-modality, fluorescent, PLGA encapsulated bismuth nanoparticles for molecular and cellular fluorescence imaging and computed tomography. Nanoscale 6, 13104–13112 10.1039/c4nr01405g [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Perera V. S., Hao J., Gao M., Gough M., Zavalij P. Y., Flask C., Basilion J. P., and Huang S. D. (2011) Nanoparticles of the novel coordination polymer KBi(H2O)2[Fe(CN)6]·H2O as a potential contrast agent for computed tomography. Inorg Chem. 50, 7910–7912 10.1021/ic200587s [DOI] [PubMed] [Google Scholar]
- 92. Chakravarty S., Hix J. M. L., Wiewiora K. A., Volk M. C., Kenyon E., Shuboni-Mulligan D. D., Blanco-Fernandez B., Kiupel M., Thomas J., Sempere L. F., and Shapiro E. M. (2020) Tantalum oxide nanoparticles as versatile contrast agents for X-ray computed tomography. Nanoscale 12, 7720–7734 10.1039/d0nr01234c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Metscher B. D., and Müller G. B. (2011) MicroCT for molecular imaging: quantitative visualization of complete three-dimensional distributions of gene products in embryonic limbs. Dev. Dyn. 240, 2301–2308 10.1002/dvdy.22733 [DOI] [PubMed] [Google Scholar]
- 94. Shu S. Y., Ju G., and Fan L. Z. (1988) The glucose oxidase-DAB-nickel method in peroxidase histochemistry of the nervous system. Neurosci. Lett. 85, 169–171 10.1016/0304-3940(88)90346-1 [DOI] [PubMed] [Google Scholar]
- 95. Hoshi M., Reginensi A., Joens M. S., Fitzpatrick J. A. J., McNeill H., and Jain S. (2018) Reciprocal spatiotemporally controlled apoptosis regulates Wolffian duct cloaca fusion. J. Am. Soc. Nephrol. 29, 775–783 10.1681/ASN.2017040380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Martell J. D., Deerinck T. J., Sancak Y., Poulos T. L., Mootha V. K., Sosinsky G. E., Ellisman M. H., and Ting A. Y. (2012) Engineered ascorbate peroxidase as a genetically encoded reporter for electron microscopy. Nat. Biotechnol. 30, 1143–1148 10.1038/nbt.2375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Shu X., Lev-Ram V., Deerinck T. J., Qi Y., Ramko E. B., Davidson M. W., Jin Y., Ellisman M. H., and Tsien R. Y. (2011) A genetically encoded tag for correlated light and electron microscopy of intact cells, tissues, and organisms. PLoS Biol. 9, e1001041 10.1371/journal.pbio.1001041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Ng J., Browning A., Lechner L., Terada M., Howard G., and Jefferis G. (2016) Genetically targeted 3D visualisation of Drosophila neurons under electron microscopy and X-ray microscopy using miniSOG. Sci. Rep. 6, 38863 10.1038/srep38863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. de Boer P., Hoogenboom J. P., and Giepmans B. N. (2015) Correlated light and electron microscopy: ultrastructure lights up! Nat. Methods 12, 503–513 10.1038/nmeth.3400 [DOI] [PubMed] [Google Scholar]
- 100. Nogales E. (2016) The development of cryo-EM into a mainstream structural biology technique. Nat. Methods 13, 24–27 10.1038/nmeth.3694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Lyumkis D. (2019) Challenges and opportunities in cryo-EM single-particle analysis. J. Biol. Chem. 294, 5181–5197 10.1074/jbc.REV118.005602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Sartori A., Gatz R., Beck F., Rigort A., Baumeister W., and Plitzko J. M. (2007) Correlative microscopy: bridging the gap between fluorescence light microscopy and cryo-electron tomography. J. Struct. Biol. 160, 135–145 10.1016/j.jsb.2007.07.011 [DOI] [PubMed] [Google Scholar]
- 103. van Driel L. F., Valentijn J. A., Valentijn K. M., Koning R. I., and Koster A. J. (2009) Tools for correlative cryo-fluorescence microscopy and cryo-electron tomography applied to whole mitochondria in human endothelial cells. Eur. J. Cell Biol. 88, 669–684 10.1016/j.ejcb.2009.07.002 [DOI] [PubMed] [Google Scholar]
- 104. Moerner W. E., and Orrit M. (1999) Illuminating single molecules in condensed matter. Science 283, 1670–1676 10.1126/science.283.5408.1670 [DOI] [PubMed] [Google Scholar]
- 105. Kaufmann R., Schellenberger P., Seiradake E., Dobbie I. M., Jones E. Y., Davis I., Hagen C., and Grünewald K. (2014) Super-resolution microscopy using standard fluorescent proteins in intact cells under cryo-conditions. Nano Lett. 14, 4171–4175 10.1021/nl501870p [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Metzger M., Konrad A., Skandary S., Ashraf I., Meixner A. J., and Brecht M. (2016) Resolution enhancement for low-temperature scanning microscopy by cryo-immersion. Opt. Express 24, 13023–13032 10.1364/OE.24.013023 [DOI] [PubMed] [Google Scholar]
- 107. Smith E. A., Cinquin B. P., McDermott G., Le Gros M. A., Parkinson D. Y., Kim H. T., and Larabell C. A. (2013) Correlative microscopy methods that maximize specimen fidelity and data completeness, and improve molecular localization capabilities. J. Struct. Biol. 184, 12–20 10.1016/j.jsb.2013.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Smith E. A., McDermott G., Do M., Leung K., Panning B., Le Gros M. A., and Larabell C. A. (2014) Quantitatively imaging chromosomes by correlated cryo-fluorescence and soft x-ray tomographies. Biophys. J. 107, 1988–1996 10.1016/j.bpj.2014.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Le Gros M. A., McDermott G., Uchida M., Knoechel C. G., and Larabell C. A. (2009) High-aperture cryogenic light microscopy. J. Microsc. 235, 1–8 10.1111/j.1365-2818.2009.03184.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Duke E. M. H., Razi M., Weston A., Guttmann P., Werner S., Henzler K., Schneider G., Tooze S. A., and Collinson L. M. (2014) Imaging endosomes and autophagosomes in whole mammalian cells using correlative cryo-fluorescence and cryo-soft X-ray microscopy (cryo-CLXM). Ultramicroscopy 143, 77–87 10.1016/j.ultramic.2013.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Ellisman M. H., Deerinck T. J., Shu X., and Sosinsky G. E. (2012) Picking faces out of a crowd: genetic labels for identification of proteins in correlated light and electron microscopy imaging. Methods Cell Biol. 111, 139–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Kukulski W., Schorb M., Welsch S., Picco A., Kaksonen M., and Briggs J. A. (2011) Correlated fluorescence and 3D electron microscopy with high sensitivity and spatial precision. J. Cell Biol. 192, 111–119 10.1083/jcb.201009037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Schellenberger P., Kaufmann R., Siebert C. A., Hagen C., Wodrich H., and Grünewald K. (2014) High-precision correlative fluorescence and electron cryo microscopy using two independent alignment markers. Ultramicroscopy 143, 41–51 10.1016/j.ultramic.2013.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Bozzola J. J., and Russell L. D. (1999) Electron Microscopy: Principles and Techniques for Biologists, 2nd Ed., Jones and Bartlett, Sudbury, MA [Google Scholar]
- 115. Deerinck T. J., Bushong E. A., Lev-Ram V., Shu X., Tsien R. Y., and Ellisman M. H. (2010) Enhancing serial block-face scanning electron microscopy to enable high resolution 3-D nanohistology of cells and tissues. Microsc. Microanal. 16, 1138–1139 10.1017/S1431927610055170 [DOI] [Google Scholar]
- 116. Joens M. S., Geanon D. J., Bayguinov P. O., Lavine K., and Fitzpatrick J. A. J. (2018) Navigating the fog: a correlated XRM and FIB-SEM imaging pipeline for the rapid and precise spatial targeting of rare structures in biological samples. Microsc. Microanal. 24, 2312–2313 10.1017/S1431927618012047 [DOI] [Google Scholar]
- 117. Burnett T. L., McDonald S. A., Gholinia A., Geurts R., Janus M., Slater T., Haigh S. J., Ornek C., Almuaili F., Engelberg D. L., Thompson G. E., and Withers P. J. (2014) Correlative tomography. Sci. Rep. 4, 4711 10.1038/srep04711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Sengle G., Tufa S. F., Sakai L. Y., Zulliger M. A., and Keene D. R. (2013) A correlative method for imaging identical regions of samples by micro-CT, light microscopy, and electron microscopy: imaging adipose tissue in a model system. J. Histochem. Cytochem. 61, 263–271 10.1369/0022155412473757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. O'Sullivan J. D. B., Cruickshank S. M., Starborg T., Withers P. J., and Else K. J. (2020) Characterisation of cuticular inflation development and ultrastructure in Trichuris muris using correlative X-ray computed tomography and electron microscopy. Sci. Rep. 10, 5846–5846 10.1038/s41598-020-61916-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Dederichs T. M., Müller C. H. G., Sentenská L., Lipke E., Uhl G., and Michalik P. (2019) The innervation of the male copulatory organ of spiders (Araneae)—a comparative analysis. Front. Zool. 16, 39–39 10.1186/s12983-019-0337-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Karreman M. A., Mercier L., Schieber N. L., Solecki G., Allio G., Winkler F., Ruthensteiner B., Goetz J. G., and Schwab Y. (2016) Fast and precise targeting of single tumor cells in vivo by multimodal correlative microscopy. J. Cell Sci. 129, 444–456 10.1242/jcs.181842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Kirz J., and Jacobsen C. (2009) The history and future of X-ray microscopy. J. Phys. Conf. Ser. 186, 012001 10.1088/1742-6596/186/1/012001 [DOI] [Google Scholar]
- 123. Ekman A. A., Chen J.-H., Guo J., McDermott G., Le Gros M. A., and Larabell C. A. (2017) Mesoscale imaging with cryo-light and X-rays: larger than molecular machines, smaller than a cell. Biol. Cell 109, 24–38 10.1111/boc.201600044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Rust M. J., Bates M., and Zhuang X. (2006) Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 3, 793–795 10.1038/nmeth929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Betzig E., Patterson G. H., Sougrat R., Lindwasser O. W., Olenych S., Bonifacino J. S., Davidson M. W., Lippincott-Schwartz J., and Hess H. F. (2006) Imaging intracellular fluorescent proteins at nanometer resolution. Science 313, 1642–1645 10.1126/science.1127344 [DOI] [PubMed] [Google Scholar]
- 126. Meyer J. C., Girit C. O., Crommie M. F., and Zettl A. (2008) Imaging and dynamics of light atoms and molecules on graphene. Nature 454, 319–322 10.1038/nature07094 [DOI] [PubMed] [Google Scholar]
- 127. Lifshin E., Kandel Y. P., and Moore R. L. (2014) Improving scanning electron microscope resolution for near planar samples through the use of image restoration. Microsc. Microanal. 20, 78–89 10.1017/S1431927613013688 [DOI] [PubMed] [Google Scholar]
- 128. Xu C. S., Hayworth K. J., Lu Z., Grob P., Hassan A. M., García-Cerdán J. G., Niyogi K. K., Nogales E., Weinberg R. J., and Hess H. F. (2017) Enhanced FIB-SEM systems for large-volume 3D imaging. Elife 6, e25916 10.7554/eLife.25916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Shi Y., Wang L., Zhang J., Zhai Y., and Sun F. (2017) Determining the target protein localization in 3D using the combination of FIB-SEM and APEX2. Biophys. Rep. 3, 92–99 10.1007/s41048-017-0043-x [DOI] [PMC free article] [PubMed] [Google Scholar]