

Abstract
ADAMTSL2 mutations cause an autosomal recessive connective tissue disorder, geleophysic dysplasia 1 (GPHYSD1), which is characterized by short stature, small hands and feet, and cardiac defects. ADAMTSL2 is a matricellular protein previously shown to interact with latent transforming growth factor-β binding protein 1 and influence assembly of fibrillin 1 microfibrils. ADAMTSL2 contains seven thrombospondin type-1 repeats (TSRs), six of which contain the consensus sequence for O-fucosylation by protein O-fucosyltransferase 2 (POFUT2). O-fucose–modified TSRs are subsequently elongated to a glucose β1-3-fucose (GlcFuc) disaccharide by β1,3-glucosyltransferase (B3GLCT). B3GLCT mutations cause Peters Plus Syndrome (PTRPLS), which is characterized by skeletal defects similar to GPHYSD1. Several ADAMTSL2 TSRs also have consensus sequences for C-mannosylation. Six reported GPHYSD1 mutations occur within the TSRs and two lie near O-fucosylation sites. To investigate the effects of TSR glycosylation on ADAMTSL2 function, we used MS to identify glycan modifications at predicted consensus sequences on mouse ADAMTSL2. We found that most TSRs were modified with the GlcFuc disaccharide at high stoichiometry at O-fucosylation sites and variable mannose stoichiometry at C-mannosylation sites. Loss of ADAMTSL2 secretion in POFUT2−/− but not in B3GLCT−/− cells suggested that impaired ADAMTSL2 secretion is not responsible for skeletal defects in PTRPLS patients. In contrast, secretion was significantly reduced for ADAMTSL2 carrying GPHYSD1 mutations (S641L in TSR3 and G817R in TSR6), and S641L eliminated O-fucosylation of TSR3. These results provide evidence that abnormalities in GPHYSD1 patients with this mutation are caused by loss of O-fucosylation on TSR3 and impaired ADAMTSL2 secretion.
Keywords: ADAMTSL2, O-fucosylation, C-mannosylation, geleophysic dysplasia, TSR, glycosylation, glycoprotein secretion, extracellular matrix, cell biology, mass spectrometry, ADAMTSL2
ADAMTSL2 is a member of the A Disintegrin-like And Metalloprotease with Thrombospondin type I repeats (ADAMTS) superfamily. The mammalian ADAMTS family contains 19 secreted metalloproteases and seven matricellular ADAMTS-Like (ADAMTSL) members (1). ADAMTS proteins consistently have one or more thrombospondin type-1 repeats (TSRs), a cysteine-rich domain, a spacer region, and a variety of C-terminal modules in addition to the protease domain, whereas ADAMTSL proteins lack the protease domain. Although the functions of ADAMTSL proteins are less well understood, recent findings from genetic disorders suggested that some of them may operate within the context of fibrillin microfibrils or synaptic function (2–4). ADAMTSL2 is a secreted matricellular glycoprotein containing seven TSRs (Fig. 1) (5). ADAMTSL2 mutations were identified in patients with autosomal recessive geleophysic dysplasia (GPHYSD1, OMIM number 231050), a syndrome characterized by short stature, short tubular bones, thick skin, and early death caused by cardiopulmonary abnormalities (6). Some of the phenotypes, such as short stature and bones are also seen in Peters Plus Syndrome (PTRPLS, OMIM number 261540) patients (7), implying there might be similar molecular mechanisms contributing to GPHYSD1 and PTRPLS pathologies.
Figure 1.

ADAMTSL2 domain structure and predicted glycosylation sites on TSRs. Top, sequences surrounding the POFUT2 consensus sequence (C-X-X-S/T-C) from mouse ADAMTSL2 (mADAMTSL2) TSRs are shown. Red C indicates conserved cysteines, blue S or T indicates predicted O-fucosylated serine or threonine, underlined W indicates predicted C-mannosylated tryptophan, purple N indicates predicted N-glycosylated asparagine, and green W, S, and G identify residues corresponding to those mutated in GPHYSD. TSR6 is unusual in that the POFUT2 site overlaps with the N-glycan site. Middle, domain organization of mADAMTSL2 including signal peptide (sp), cysteine-rich domain (cys), spacer domain (spa), N-glycan rich domain (N-gly), PLAC domain, and seven TSRs (ovals). The TSRs containing consensus sequences for modification with O-fucose (red oval), C-mannose W-X-X-(W/C) (hatched), or both modifications (red and hatched) are indicated.
Six of 16 documented GPHYSD1 mutations have been identified within ADAMTSL2 TSRs (6, 8, 9). TSRs are small protein motifs of 50-60 amino acids and are characterized by six conserved cysteines forming three disulfide bonds (10). Three types of glycosylation are predicted on TSRs including C-mannosylation, N-linked glycosylation, and O-fucosylation, all of which occur in the endoplasmic reticulum (10–12). Each of the seven TSRs in mouse ADAMTSL2 contains at least one consensus sequence for glycosylation (Fig. 1). Six of the seven TSRs contain the W-X-X-(W/C) consensus sequence for potential C-mannosylation (Fig. 1) by C-mannosyltransferases (13). C-Mannosylation occurs co-translationally (14) and has been suggested to promote efficient folding and secretion of target proteins, such as ADAMTSL1 (13, 15, 16). Disrupting C-mannosylation by eliminating C-mannosyltransferases impairs secretion of target proteins (13, 17). N-Linked glycosylation is also largely co-translational and occurs on asparagine residues within the consensus sequence N-X-(S/T) (X is any amino acid other than proline) by oligosaccharyl transferase complex (18). N-Glycosylation is well-known to play a role in protein quality control and folding (19). ADAMTSL2 contains 10 N-glycosylation sites, one of which falls within TSR6 and overlaps with a predicted protein O-fucosyltransferase 2 (POFUT2) consensus sequence (Fig. 1).
TSRs are modified with an O-fucose on a serine (Ser) or threonine (Thr) within the consensus sequence C-X-X-(S/T)-C by POFUT2 (12). POFUT2 only modifies TSRs after they are folded and has been proposed to be a folding sensor for TSRs (20, 21). The O-fucose is typically elongated with a glucose (Glc) by β1,3-glucosyltransferase (B3GLCT), forming a glucose β1,3-fucose (GlcFuc) disaccharide on TSRs (10). Elimination of Pofut2 in mice leads to embryonic lethality with defects in gastrulation (22). In contrast, mutations in B3GLCT (previously named B3GALTL) lead to PTRPLS, a developmental disorder characterized by anterior eye chamber defects (specifically a malformation termed Peters Anomaly), short stature, craniofacial abnormalities, brachydactyly, as well as variable levels of cleft palate, and a range of cardiovascular, genitourinary, and cognitive defects (7, 23, 24). Similar craniofacial and skeletal abnormalities are observed in B3glct mutant mice, although without the PTRPLS eye defects (25). Knocking down or knocking out POFUT2 in HEK293T cells reduces the secre-tion of target proteins, including ADAMTSL1, ADAMTS9, ADAMTS13, and ADAMTS20 (20, 25–27). In fact, blocking secretion of ADAMTS9, which is required for early development in mice, appears to be the reason for the embryonic lethality in Pofut2-null mice (28). Interestingly, siRNA knockdown or CRISPR-Cas9 knockout of B3GLCT in HEK293T cells reduces secretion of some but not all POFUT2 targets, suggesting PTRPLS and phenotypes in B3glct-null mice may result from reduced secretion of a subset of POFUT2 targets. ADAMTSL2 secretion was reduced when either POFUT2 or B3GLCT were knocked down using siRNA (20), suggesting reduction of ADAMTSL2 secretion could help explain the shared skeletal phenotypes between GPHYSD1 and PTRPLS.
Several GPHYSD1 mutations reduce secretion of ADAMTSL2, including a GPHYSD1 mutation adjacent to the O-fucosylation site in TSR6 (G811R) (6). Another GPHYSD1 mutation, S635L, falls within the POFUT2 consensus sequence in TSR3 from ADAMTSL2 (8). These results raised the possibility that altered O-fucosylation is responsible for reduced secretion of ADAMTSL2 in GHPYSD1 S635L and G811R patients. To address the relationship between GPHYSD1 mutations and glycosylation of ADAMTSL2, we used MS to analyze glycosylation on WT mouse ADAMTSL2, which shares 88% sequence identity with human ADAMTSL2 including complete conservation of consensus sequences for TSRs glycosylation. TSR3 and TSR6, in which these mutations occur, share sequence identity of 95 and 96%, respectively (Fig. S1). Mouse knockouts of Adamtsl2 phenocopy many of the defects observed in GPHYSD1 patients (4, 29), suggesting that mice provide an excellent model for studying this disease. In addition, we also examined the impact of GPHYSD1 mutations on glycosylation and secretion of ADAMTSL2 using cell-based secretion assays. We show that loss of O-fucosylation, such as in POFUT2−/− cells, eliminated secretion of mouse ADAMTLS2, whereas lack of extension to the GlcFuc disaccharide in B3GLCT−/− cells had no effect on secretion. We also demonstrated that GPHYSD1 analogous mutations in TSR3 (S641L) and TSR6 (G817R) reduced secretion of mouse ADAMTSL2. Significantly, we showed that the reduction in secretion of the S641L mutation results from reduced O-fucosylation of TSR3, providing a molecular explanation for GPHYSD1 in patients with this mutation.
Results
ADAMTSL2 TSRs were modified with O-fucose and C-mannose at variable stoichiometries
The seven ADAMTSL2 TSRs contain six consensus sites for O-fucosylation, eight consensus sites for C-mannosylation, and one consensus site for N-glycosylation (Fig. 1). To determine the glycoforms present on ADAMTSL2 TSRs and the stoichiometry of glycan modifications, we subjected purified mouse ADAMTSL2 (mADAMTSL2) to tryptic and chymotryptic digestions and analyzed digested fragments via nano-LC–MS/MS to identify glycopeptides containing the POFUT2 consensus sites as described under “Experimental procedures.” To determine the relative amounts of O-fucose (O-Fuc) monosaccharide, disaccharide, and unmodified peptide (naked), we generated extracted ion chromatograms (EICs) for each glycoform of the selected peptides and calculated peak areas for each (30). At predicted O-fucosylation sites, the elongated GlcFuc disaccharide was the major glycoform on TSR1, TSR3 and TSR7 (Fig. 2A, Fig. S2, A, B, D, H, and I). TSR5 was mainly unmodified with only a small percentage of GlcFuc disaccharide, suggesting that an innate characteristic of this TSR reduces recognition by POFUT2 (Fig. 2A, Fig. S2F). Surprisingly, TSR2 was predominantly modified with O-Fuc monosaccharide, suggesting this TSR is poorly modified by B3GLCT (Fig. 2A, Fig. S2C).
Figure 2.

ADAMTSL2 TSRs were modified with O-fucose-glucose disaccharide and C-mannose in varying stoichiometries. A, relative abundance of GlcFuc disaccharide, O-fuc monosaccharide, and unmodified glycoforms on peptides from TSRs containing the C-X-X-(S/T)-C O-fucose consensus site analyzed by mass spectral analyses. See Fig. S2 for spectra and EICs of each peptide. Data can be found in Tables S3, S4, S7, and S8. B, plasmids encoding TSP1-TSR3-MycHis6, ADAMTSL2-TSR6-MycHis6, or an empty vector (EV) were transfected into HEK293T cells grown in the presence of 6AF as described under “Experimental procedures.” The proteins were purified from the medium, digested with or without PNGase F, click reaction was performed with azide-biotin, separated by SDS-PAGE, and probed with anti-myc antibody (red) or streptavidin (green). C, relative abundance of C-mannosylated forms of peptides from TSRs containing W-X-X-(W/C) consensus sequence. See Fig. S2 for spectra and EICs of each peptide. Data can be found in Tables S3, S4, S7, and S8. D, domain map of mADAMTSL2 summarizing the distribution of O-fucosylation and C-mannosylation on TSRs. Red triangle represents fucose. Blue circle represents glucose. Green circle represent mannose. Hollow icons represent less than 50% modification of fucose (red hollow triangle), glucose (blue hollow circle), and mannose (green hollow circle).
Using MS, we were unable to detect peptides containing the TSR6 POFUT2 consensus site, even with multiple different protease and glycosidase digestions. TSR6 is unusual in that the POFUT2 consensus site overlaps with an N-glycosylation consensus site and therefore could be modified with either or both modifications (Fig. 1). As an alternative approach to determine whether TSR6 was O-fucosylated, we tested whether ADAMTSL2 TSR6 (TSL2-TSR6) expressed in HEK293T cells incorporated the bio-orthogonal probe 6-alkynylfucose (6AF). Previously we have shown that 6AF is efficiently transferred onto O-fucose sites in TSRs by POFUT2 and can be modified with azide-biotin using “click” chemistry, allowing for detection with streptavidin (31). 6AF was efficiently incorporated onto TSP1-TSR3, which is known to be O-fucosylated (31) but was not incorporated onto TSL2-TSR6 (Fig. 2B), demonstrating that TSR6 from ADAMTSL2 is not O-fucosylated. To determine whether TLS2-TSR6 was N-glycosylated, we tested for presence of N-glycans by treatment with PNGase F (Fig. 2B). TSL2-TSR6 size-shifted significantly following PNGase F treatment, suggesting that it is N-glycosylated. The complete shift of TSL2-TSR6 after PNGase F treatment suggested the N-glycosylation in TSR6 was stoichiometric. In contrast, TSR3 from human thrombospondin-1 (TSP1-TSR3), which lacks an N-glycosylation site, did not shift (Fig. 2B).
In contrast to POFUT2 consensus sites, C-mannosylation sites were modified at lower stoichiometries (Fig. 2C). For ADAMTSL2 TSR1, the most abundant C-mannosylation species were detected on the second and third tryptophans in the W-X-X-W-X-X-W-X-X-C motif, with very little modification of the first W (Fig. S2B). In contrast, TSRs 3-7 each have a single W-X-X-C C-mannosylation consensus motif (Fig. 1). Among these TSRs, significant levels of C-mannosylation were only detected on TSR3 and TSR7, with no modification detected on TSRs 4, 5, or 6 (Fig. S2, D–G and I). The glycan modifications are summarized in Fig. 2D.
ADAMTSL2 secretion requires TSR O-fucosylation but not extension to the GlcFuc disaccharide
Previous work demonstrated that siRNA knockdown of POFUT2 in HEK293T cells significantly reduced ADAMTSL2 secretion (20). To confirm the importance of TSR O-fucosylation for ADAMTSL2 secretion, we examined ADAMTSL2 secretion in Lec13 CHO cells grown in the presence or absence of fucose (Fig. 3A). Lec13 CHO cells have a mutation in GDP-mannose 4,6-dehydratase (32), one of the enzymes in the de novo GDP-fucose biosynthetic pathway, the predominant pathway for GDP-fucose synthesis and utilization in humans. The mutation can be rescued through the GDP-fucose salvage pathway by addition of fucose to the medium (20). In the absence of fucose, ADAMTSL2 was trapped in the cell lysate and was not secreted to the medium from Lec13 CHO cells, but secretion was restored when medium was supplemented with fucose (Fig. 3A), suggesting that loss of either TSR O-fucosylation or fucosylation of N-glycans impaired ADAMTSL2 secretion. To distinguish between these possibilities, we compared secretion of ADAMTSL2 in WT (Pro5) and mutant (Lec1) CHO cells (Fig. 3B). Lec1 cells have a mutation in Mgat1 that blocks processing of N-glycans to hybrid or complex types that could contain fucose (33). ADAMTSL2 was secreted from both Pro5 and Lec1 cells (Fig. 3B). Moreover, ADAMTSL2 secreted from the Lec1 cells migrated faster than that from the Pro5 cells, consistent with the lack of N-glycan processing. The efficient secretion of ADAMTSL2 lacking fucosylation on N-glycans suggested that loss of O-fucosylation on ADAMTSL2 TSR motifs was responsible for reduced secretion of ADAMTSL2 secretion from Lec13 CHO cells.
Figure 3.

ADAMTSL2 secretion required O-fucosylation but not N-glycan fucosylation. Plasmids encoding ADAMTSL2-MycHis6 and hIgG were co-transfected into (A) Lec13-CHO cells grown in the presence (+) or absence (−) of 1 mm l-fucose or (B) into Lec1-CHO cells and Pro5-CHO cells. The medium and cell lysates were analyzed by Western blots probed with anti-myc (red) and anti-IgG (green) antibodies.
To further confirm the importance of TSR O-fucosylation for ADAMTSL2 secretion and determine the role of the B3GLCT-mediated extension to the disaccharide, we evaluated secretion of ADAMTSL2 using recently developed CRISPR-Cas9 knockouts of POFUT2 or B3GLCT in HEK293T cells (28, 34). ADAMTSL2 was efficiently secreted in WT HEK293T cells. In contrast, secretion was eliminated in POFUT2−/− cells (Fig. 4, A and B). Co-transfection with POFUT2 restored secretion of ADAMTSL2 in POFUT2−/− cells, demonstrating the specificity of the mutation. In contrast, knocking out B3GLCT did not affect ADAMTSL2 secretion (Fig. 4, A and B). Secretion of hIgG was not affected in either POFUT2−/− or B3GLCT−/− cell lines, suggesting that reduced secretion of ADAMTSL2 in POFUT2−/− cells did not result from a global defect in trafficking. Moreover, the loss of the upper band in the POFUT2 knockout cell lysate provided evidence that ADAMTSL2 lacking O-fucosylation was trapped in the endoplasmic reticulum and did not move to the Golgi where terminal processing of the N-glycans occurs (Fig. 4A, right-hand panel). Taken together (Figs. 3 and 4), O-fucosylation by POFUT2 on TSR motifs was essential for ADAMTSL2 secretion, but elongation of the O-fucose on ADAMTSL2 TSRs by B3GLCT was dispensable for proper secretion.
Figure 4.
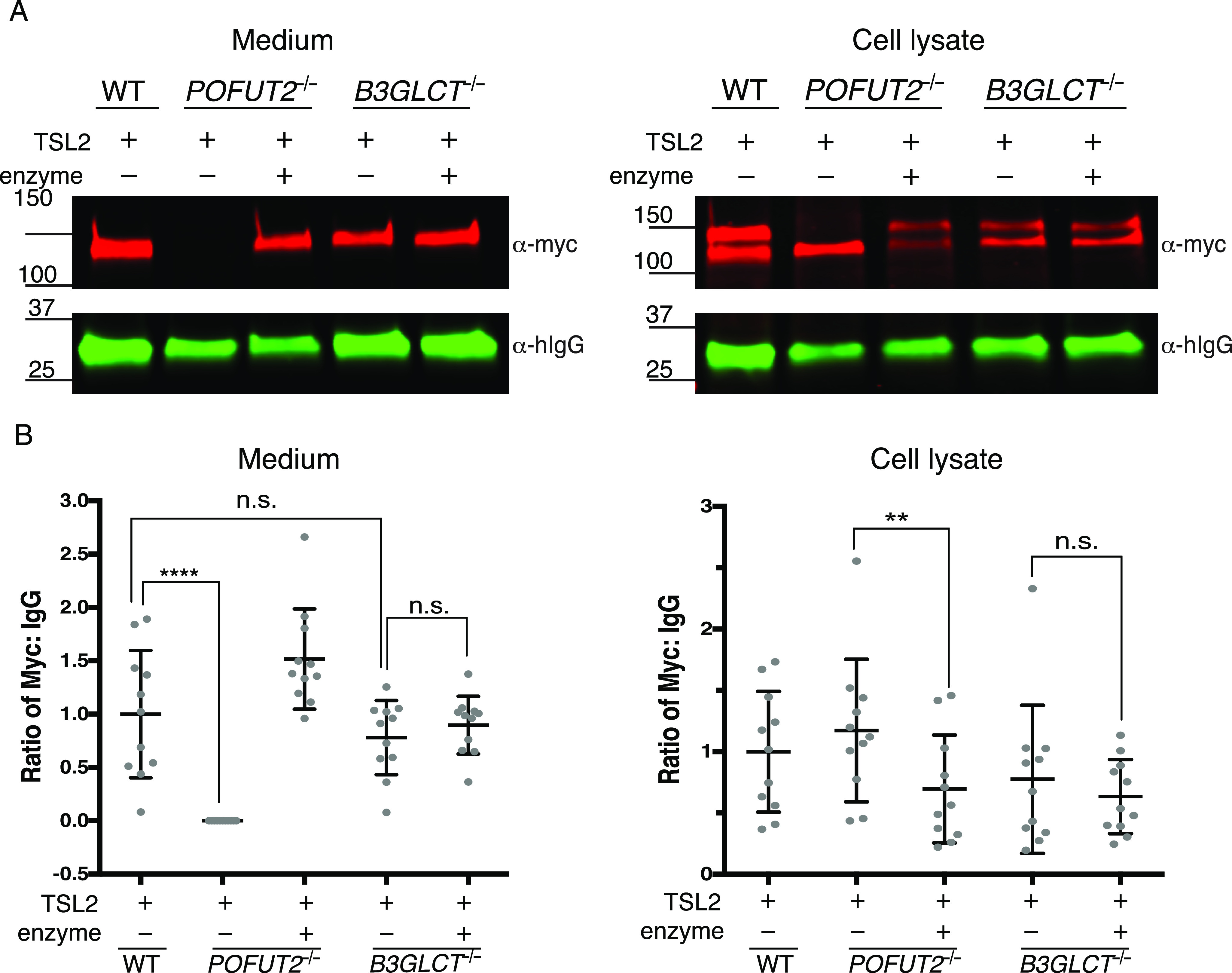
POFUT2 is essential for mADAMTSL2 secretion, but B3GLCT is not. A, plasmids encoding ADAMTSL2-MycHis6 (TSL2) and hIgG were co-transfected into WT, POFUT2-null (POFUT2−/−), or B3GLCT-null (B3GLCT−/−) HEK293T cells. Rescue experiments were performed by co-transfection with a plasmid encoding full-length POFUT2 or full-length B3GLCT (enzyme). Medium and cell lysate were analyzed by Western blots probed with anti-myc (red) to detect ADAMTSL2 (TSL2) and for transfection control anti-hIgG (green). B, quantification of Western blots from A. ADAMTSL2 signals were normalized to hIgG levels. Significance was analyzed with ordinary one-way ANOVA in Prism 7, n = 11 in biological replicates with independent experiments. ****, p < 0.0001; **, p < 0.01; n.s., not significant.
The S641L and G817R GPHYSD1 mutations impair ADAMTSL2 secretion, and the S641L mutation blocks O-fucosylation of ADAMTSL2 TSR3
Previous work by Le Goff et al. (6) showed that the human ADAMTSL2 GPHYSD1 G811R substitution reduced protein secretion. We examined whether introducing GPHYSD1 S635L or G811R equivalent mutations into mADAMTSL2 (S641L or G817R, respectively) similarly affected secretion. mADAMTSL2 S641L was secreted at less than 30% and G817R at less than 10% of WT mADAMTSL2 (Fig. 5). These data suggested that the GPHYSD1 phenotype caused by these mutations likely resulted from inappropriately low levels of ADAMTSL2 in the extracellular matrix.
Figure 5.

GPHYSD1 equivalent mutations S641L and G817R reduced secretion of mADAMTSL2. A, plasmids encoding WT, S641L-mutant, and G817R-mutant ADAMTSL2-MycHis6 were co-transfected with hIgG into WT HEK293T cells. The medium and cell lysate were analyzed by Western blots probed with anti-myc (red) and anti-IgG (green) antibodies. B, quantitation of Western blots from A as described in the legend to Fig. 4.
The GPHYSD1 S641L and G817R mutations are located within and adjacent to the TSR3 and TSR6 POFUT2 consensus sites, respectively (Fig. 1). Given their location, reduced ADAMTSL2 secretion could result from defects in glycosylation. To test this hypothesis, we evaluated the glycosylation on mADAMTSL2 S641L using MS. Full-length WT and S641L-mutated mADAMTSL2 were subjected to chymotrypsin digestion and analysis by nano-LC–MS/MS. The most abundant form of the peptide from TSR3 of WT mADAMTSL2 was fully modified with one C-mannose and the GlcFuc disaccharide (Fig. 6A). In contrast, the most abundant form of the equivalent peptide containing the S641L mutation only had the C-mannose with no O-fucose glycan (Fig. 6A, Fig. S3). The mutation only affected O-fucosylation of TSR3 of ADAMTSL2; O-fucosylation and C-mannosylation of all other TSRs remained the same as WT ADAMTSL2 (Fig. S4). Hence, the S641L mutation only caused local loss of O-fucosylation in TSR3 without global effects on O-fucosylation or C-mannosylation on other TSRs in mADAMTSL2.
Figure 6.

GPHYSD1 S641L mutation reduced O-fucosylation resulting in reduced mADAMTSL2 secretion. A, relative abundance of glycoforms of the peptide containing the O-fucose consensus sequence from TSR3 generated from WT ADAMTSL2 or S641L-mutant ADAMTSL2 TSL2-TSR3 (S641L) chymotrypsin digestion. See Figs. S2D and S3 for spectra and EICs of each peptide, respectively. Data can be found in Tables S5, S6, S9, and S10. Statistics were performed with one-way ANOVA in Prism 7, n = 3 with biological replicates. B, plasmids encoding WT or T633A-mutant ADAMTSL2 were co-transfected with hIgG into WT HEK293T cells. The medium and cell lysate were analyzed by Western blots probed with anti-myc (red) and anti-IgG (green) antibodies. C, quantitation of Western blots from B using ordinary one-way ANOVA in Prism 7, n = 3 in biological replicates. **, p < 0.01; n.s., not significant.
Elimination of the O-fucose site on TSR3 reduces ADAMTSL2 secretion
To determine whether loss of O-fucosylation on TSR3 was responsible for the secretion defect, we mutated the O-fucosylated Thr to Ala (T643A) in full-length mADAMTSL2 and evaluated the effect of the mutation on secretion. The T643A mutated form was secreted ∼50% less than WT (Fig. 6, B and C), suggesting that much of the secretion defect of the S641L mutant was caused by loss of O-fucosylation on TSR3 rather than the S641L mutation itself.
Because the glycosylation sites in TSR6 could not be detected using MS, we analyzed whether the G817R mutation affected N-glycosylation and secretion by introducing G817R, as well as mutations to inhibit N-glycosylation (N813Q, T815V, N813Q/T815V) into mADAMTSL2-TSR6 (TSL2-TSR6) and labeled the proteins with 6AF as in Fig. 2B. TSL2-TSR6-G817R was secreted, although poorly compared with WT, and migrated at the same molecular weight with WT TSL2-TSR6 in both medium and cell lysate (Fig. 7, A and B). In contrast, disrupting the N-glycan site (N813Q, T815V, and N813Q/T815V) prevented secretion and resulted in a molecular weight shift downwards in cell lysates compared with WT TSL2-TSR6 and G817R (Fig. 7B). The reduction of secreted G817R protein (Fig. 7A) was consistent with our result that the G817R mutation also reduced full-length mADAMTSL2 secretion (Fig. 5). Interestingly, removal of N-glycosylation with N813Q did not restore O-fucosylation at threonine 815 (Fig. 7A). Taken together, these results show that the TSR6 G817R mutation did not disrupt N-glycosylation but reduced secretion of mADAMTSL2 without affecting the N-glycosylation status of TSR6.
Figure 7.

GPHYSD1 G817R mutation did not affect N-glycosylation in TSR6 and mutation of N-glycosylation site did not restore O-fucosylation. Plasmids encoding TSP1-TSR3-MycHis6, WT, N813Q, T815V, G817R, double mutant N813Q/T815V forms of ADAMTSL2-TSR6-MycHis6, or an empty vector (EV) were transfected into HEK293T cells grown in the presence of 6AF as described under “Experimental procedures.” The proteins were purified from the medium and cell lysate, click reaction performed with azide-biotin, separated by SDS-PAGE, and probed with anti-myc antibody (red) or streptavidin (green).
Discussion
Here we examined the effects of two GPHYSD1 mutations in ADAMTSL2 occurring within POFUT2 consensus sites, one in TSR3 (S641L) and the other in TSR6 (G817R). The corresponding human mutations (S635L and G811R, respectively) were both identified in GPHYSD1 patients who showed compound heterozygosity, i.e. the other ADAMTSL2 allele in each affected individual harbored a different mutation, W862X (6) and c.[1219C>T] (8), respectively. The W862X is predicted to produce a truncated peptide lacking TSR7 and C-terminal residues but is likely to lead to nonsense-mediated mRNA decay (6). The c.[1219C>T] mutation is a splice-site mutation that would block splicing of the exon encoding the N-glycan–rich domain, although this has not been verified experimentally (8). These two mutations, W862X and c.[1219C>T], are likely to be quite severe, resulting in loss of function from the respective alleles. Our data shows that the corresponding GPHYSD1 substitutions in mouse ADAMTSL2, S641L and G817R, significantly reduced secretion of ADAMTSL2, suggesting that low levels of ADAMTL2 in the extracellular matrix lead to GPHYSD1. The fact that the S641L mutation eliminated O-fucosylation of TSR3, and that elimination of O-fucosylation on TSR3 significantly reduced secretion of ADAMTSL2, suggests that loss of O-fucose on TSR3 is a major contributing factor to the defects observed in patients with the S635L mutation. In contrast, because the G817R mutation had no effect on the glycosylation status of TSR6, the reduction of mADAMTSL2 secretion of G817R appeared to be caused directly by the mutation.
PTRPLS patients display facies and skeletal defects similar to those seen in GPHYSD1. Similar skeletal defects were also observed in global knockouts of Adamtsl2 and B3glct in mice, such as shortened front and hind limbs and irregular shape of the skull (4, 25). Due to these similar resemblances, we hypothesized that ADAMTSL2 may be a prime B3GLCT target relevant to the skeletal defects observed in PTRPLS. Several GPHYSD1 missense mutations in addition to those described above have been reported to reduce secretion (6, 8, 9) suggesting this may be a major mechanism causing disease. Previous work from our laboratory showed that ADAMTSL2 secretion was lost when B3GLCT was knocked down using siRNA in HEK293T cells, supporting our hypothesis that ADAMTSL2 is a key factor for the overlapping phenotypes between PTRPLS and GPHYSD1 (20). In contrast, our data here indicated that the secretion of ADAMTSL2 was not affected by CRISPR-Cas9 deletion of B3GLCT in HEK293T cells. Comparing these two gene manipulation methods, siRNA has stronger off-target effects by triggering the translational repression and/or degradation of nontarget genes as well as nonsequence specific off-target effects (35). This can overwhelm the endogenous interaction between microRNA and the RNA-induced silencing complex (35). We attribute the loss of ADAMTSL2 secretion to nonspecific repression of global cellular networks in B3GLCT siRNA knockdown experiments. These data suggest that reduced secretion of ADAMTSL2 may not cause the overlapping phenotypes between GPHYSD1 and PTRPLS.
Our mass spectral analyses showed that ADAMTSL2 is O-fucosylated at most of the predicted sites in each of its TSR domains, further validating the reliability of the POFUT2 consensus sequence: C-X-X-(S/T)-C. TSRs 1, 3, and 7 were all modified and elongated with the O-fucose disaccharide at high stoichiometries, whereas TSR2 was predominantly monosaccharide, and TSRs 5 and 6 were unfucosylated (Fig. 2A). The S641L mutation completely eliminated O-fucosylation of TSR3, suggesting the replacement of Ser by Leu in the POFUT2 consensus sequence (C-L-R-T-C) severely reduced recognition of this site. Prior work in our laboratory analyzed the effects of mutations in the consensus sequence (C-Xa-Xb-(S/T)-C) using a model TSR (TSR3 from human thrombospondin 1, TSP1-TSR3) (36). Consistent with our S641L results, an Leu in the Xa position significantly reduced O-fucosylation of TSP1-TSR3 (36). TSR6 has overlapping N-glycosylation and O-fucosylation consensus sequences: C-N-T-T-C. Our results showed that TSR6 is N-glycosylated but not O-fucosylated, but that eliminating the N-glycosylation site by mutating the Asn to a Gln failed to restore O-fucosylation. This was surprising, as we assumed that the addition of the N-glycan, which occurs prior to protein folding, was blocking O-fucosylation by POFUT2, which occurs after TSR folding (20). This result suggested that mutating the Asn to Gln in the TSR6 POFUT2 consensus sequence (C-Q-T-T-C) reduced modification by POFUT2. A Gln in the Xa position also significantly reduced O-fucosylation in TSP1–TSR3, which may explain why the N813Q mutant was not O-fucosylated in TSR6. Our results here confirm the importance of the amino acid in the Xa position in the consensus in determining the efficiency of O-fucosylation by POFUT2. Alternatively, the lack of O-fucosylation on TSR5 or -6 could be the result of overexpression of ADAMTSL2 in a cell-culture system or that TSR6 was analyzed as a single TSR instead of in the context of the full-length protein.
Of the TSRs where we have analyzed stoichiometry of modification with the GlcFuc disaccharide (ADAMTS9 (37), ADAMTS20 (25), and ADAMTS17 (34)), TSR2 of ADAMTSL2 is the first TSR we have seen that is only modified with O-fucose monosaccharide. The only other TSR that is partially modified with the glucose elongation is TSR6 from ADAMTS20 (25). The consensus sequence for TSR2 of ADAMTSL2 is C-S-A-T-C, whereas that for TSR6 of ADAMTS20 is C-T-A-T-C. It will be interesting to see if this similarity contributes to the reduced modification of O-fucose by B3GLCT at these sites.
All of the TSRs in ADAMTSL2 besides TSR2 have the consensus sequence W-X-X-C for C-mannosylation, and TSR1 contains the extended W-X-X-W-X-X-W-X-X-C consensus sequence (Fig. 1). TSR1, -3, and -7 were modified at 1 or multiple W-X-X-(W/C) tryptophans, whereas TSRs 4-6 lacked any C-mannose modifications. The W-X-X-W sites have been reported to be modified by the C-mannosyltransferase DPY19L1, and the W-X-X-C sites by DPY19L3 (13). Because TSRs 4-6 were not modified, additional factors must be controlling recognition of these W-X-X-C sites by DPY19L3. Recent research has shown that C-mannosylated tryptophans with the W-X-X-W-X-X-W have a stabilizing effect on a TSR from UNC-5, with the mannose on the first Trp having a stronger effect than the mannose on the second Trp (13, 16). Because the first Trp of the W-X-X-W-X-X-W-X-X-C site on TSR1 of ADAMTSL2 is poorly modified, we anticipate the stabilizing effects from C-mannosylation would be low for ADAMTSL2 TSRs. In contrast, four out of six TSRs in mADAMTSL2 had high stoichiometry of O-fucosylation, adding weight to the importance of O-fucosylation for stabilization of ADAMTSL2 TSRs.
Our analysis of ADAMTSL2 GPHYSD1-related mouse mutations provided evidence that disruption of TSR-specific O-fucosylation may contribute to protein secretion defects that lead to potential disease mechanisms and pathology. Substoichiometric O-fucosylation of highly conserved TSRs suggested that modulation of O-fucosylation was important for efficient trafficking of TSR-containing proteins. In light of the current interest in development of therapeutics with TSR motifs as targets (38), it is essential to advance our knowledge of TSR motif function by further investigating the functional role of O-fucosylation and the mechanistic role of glucose-fucose elongation.
Experimental procedures
Plasmids and mutagenesis
pcDNA3.1-mADAMTSL2-Myc-His6 was described previously (5) and used for expression of mouse ADAMTSL2 and its analysis. The plasmid expressing TSR6 was created by PCR amplification using primers: 5'-CGTACGAAGCTTCCCACTGGCTGGCTCAAG-3' and 5'-GGGCCCTCCTCGAGAGCAGTGCTCTC-3' incorporating HindIII and XhoI restriction sites (underlined). Amplicons were digested with HindIII and XhoI and ligated into pSecTag2C/hygroC (Invitrogen). The plasmid encoding TSR3 of human thrombospondin 1 (hTSP1-TSR3) with a C-terminal Myc-His6 tag (pSecTag2-TSP1-TSR3-Myc-His6) was described previously (39). Mouse ADAMTSL2 substitutions S641L (for Cell-based secretion assay), and N813Q, T815V, G817R, and N813Q/T815V in TSR6 were made by PCR-mediated site-directed mutagenesis using primers listed in Table S1 and Herculase II (Agilent), following the manufacturer's instructions. PCR products were digested with DpnI at 37 °C overnight then transfermed into DH5α-competent cells (Invitgrogen). cDNA sequences were verified by sequencing the entire insert using primers listed in Table S2. Mouse ADAMTSL2 S641L (for protein purification and mass spectral analysis) was generated by PCR-mediated site-directed mutagenesis using primers: 5'-GTGAGTGCCTGCGTACCTGTGGTGAGGGCCATCAGTTCCG-3' and 5'-CAGGTACGCAGGCACTCACTCCAGCTGCTGGTCTCCCAC-3'. This PCR was performed with CloneAmp HiFi Premix (Takara Bio, USA) following the manufacturer's instructions. PCR product digestion and transformation were the same as described above. cDNA sequence was verified by sequenceing the entire insert using primers in Table S2.
Protein expression and purification
HEK293T cells (ATCC) were cultured in Dulbecco's modified Eagle's medium (DMEM, GE Healthcare Life Sciences) supplemented with 10% bovine calf serum (BCS, Hyclone) and 1% penicillin plus streptomycin antibiotic (pen+strep, Sigma-Aldrich). Cells were seeded in DMEM with 10% BCS into 10-cm dishes and grown to 80% confluence before transfection. Before transfection, medium was changed to 6 ml of Opti-MEM (Gibco) for each 10-cm dish. The cells were transiently transfected with mouse ADAMTSL2 WT or S641L plasmid using 5 μg of plasmid, 30 μl of 1 mg/ml of polyethylenimine (PEI) (40), and 500 μl of Opti-MEM. The plasmid, PEI, and Opti-MEM mixture was incubated at room temperature for 15 min before addition to the cells. The culture medium was collected by centrifuging at 3900 rpm, 4 °C for 10 min after 3 days incubation at 37 °C. The supernatant was filtered through a 0.45- μm filter and purified using nickel-nitrilotriacetic acid (Qiagen) affinity chromatography at 4 °C. The proteins were eluted in TBS containing 250 mm imidazole and stored in −20 °C until use.
Cell-based secretion assays
WT and mutated ADAMTSL2 plasmids were transiently transfected into Lec1 or Pro5 cells using 0.8 μg of plasmid with 0.2 μg of hIgG control plasmid DNA with 6 μl of PEI transfection reagent in 100 μl of Opti-MEM and incubated at room temperature for 20 min. Cells at 80% confluence in 35-mm wells were washed with 2 ml of PBS, then incubated in 1 ml of Opti-MEM with transfection mixture added dropwise and cultured for 4 h before a medium change with 1 ml of fresh Opti-MEM. CRISPR-Cas9 HEK293T knockouts of POFUT2 and B3GLCT were generated as described previously by Benz et al. (28) and Hubmacher et al. (34), respectively. Cells were seeded in 2 ml (4.25 × 106 cells/ml) of DMEM supplemented with 10% BCS in 6-well–plates, incubated overnight and transfected when 80% confluent. Transient co-transfection of POFUT2−/−, B3GLCT−/−, and WT HEK293T cells used 1.2 μg of mouse ADAMTSL2 plasmid or pSecTag2/hygroC empty vector as a control, 0.1 μg of hIgG as a control for normalization of secreted protein, 0.24 μg of POFUT2 or B3GLCT plasmid for rescue of secretion, or empty vector as a control with 9.8 μl of PEI in 154 μl of Opti-MEM incubated at room temperature for 15 min. Before transfection, the old media was changed to 700 μl of Opti-MEM and the transfection mixture was added dropwise and cultured for 48 h at 37 °C before collection for Western blotting analysis.
Labeling with 6-alkynylfucose
To label proteins with 6AF, transfected cells were cultured in 200 μm alkynyl fucose (Invitrogen). Cycloaddition click reactions were performed to label fucosylated proteins as previously described (31). Medium and lysates were incubated with 1 mm copper sulfate and 2 mm sodium ascorbate in PBS with 0.1 mm azide-biotin and 0.1 mm tris(benzyltriazolymethyl)amine for 1 h at room temperature. Precipitant in the reaction mixture was removed by centrifugation, and samples were analyzed by immunoblot. For the PNGase F digest in Fig. 2B, plasmids encoding hTSP1-TSR3 or mADAMTSL2-TSR6 were transfected into HEK293T cells using Lipofectamine 2000 as described by the manufacturer (ThermoFisher), and the cells were cultured in 200 μm alkynyl fucose for 48 h. The proteins were purified from the medium using nickel-nitrilotriacetic acid-agarose and digested with PNGase F in 100 mm Tris-HCl, pH 8.0, 2% Nonidet P-40, for 4 h at 37 °C. The samples were then subjected to click chemistry with final concentrations of 20 μm azide-biotin, 500 μm THPTA, 100 μm CuSO4, and 5 mm sodium ascorbate for 20 min at room temperature. The reaction was stopped by heating to 100 °C in 1× Laemmli sample buffer.
Western blotting
Samples from all secretion and fucosylation assays were analyzed by immunoblot. Full-length ADAMTSL2 was resolved using either 10 or 4–20% gradient (the latter when full-length ADAMTSL2 was co-transfected with hIgG) SDS-PAGE, single TSRs by 15% SDS-PAGE, and proteins were transferred onto 0.45-μm nitrocellulose membrane. Membranes were blocked for 1 h at room temperature in 5% nonfat milk in TBS with 0.1% Tween 20, and then probed with anti-myc antibody (Santa-Cruz, sc-40, 9E10) at 4 °C overnight. Membranes were incubated in the dark at room temperature for 1 h with Alexa Fluor IRDye®680RD goat anti-mouse (LI-COR, catalog number 926-68070) and IRDye®800CW goat (polyclonal) anti-human IgG (H + L) (LI-COR catalog number 925-32232). Fucosylation assay blots were incubated with Alexa Fluor 680 goat anti-mouse IgG with streptavidin IRDye800 (Rockland Immunochemicals). Immunoblots were visualized and band intensities were quantified on an Odyssey Imager (LI-COR) software.
Mass spectral analysis
Recombinant full-length mouse ADAMTSL2-Myc-His6 WT or S641L mutant were expressed and purified as described above. Approximately 1 μg of purified protein was denatured in 8 m urea and 0.4 m ammonium bicarbonate. Samples were reduced with 10 mm tris(2-carboxyethyl)phosphine (Thermo Fisher) for 5 min at 50 °C, alkylated with 30 mm iodoacetamide in the dark for 30 min at room temperature, and diluted with mass spectrometer grade H2O to a final urea concentration of 2 m. Samples were then digested with 0.5 μg of trypsin (cleaves C-terminal to lysines or arginines, Sigma-Aldrich) or chymotrypsin (cleaves C-terminal to tryptophan, phenylalanine, tyrosine, leucine, or isoleucine, Sigma-Aldrich) for 4 h at 37 °C, acidified with 5% formic acid, sonicated for 20 min, then desalted using C18 ZipTips (EMD Millipore), eluted in 50% acetonitrile, 0.1% acetic acid mixture. Samples were diluted to 2 ng/μl in 20% acetonitrile with 0.1% formic acid prior to injection into the mass spectrometer. 10 ng of each sample was injected and detected on a Q-Exactive Orbitrap mass spectrometer (Thermo Fisher) equipped with an Easy nano-LC HPLC system with a C18 EasySpray PepMap RSLC C18 column (50 μm × 15 cm, Thermo Fisher Scientific). Separation of glycopeptides was carried out using a 30-min binary gradient consisting of solvent A (0.1% formic acid in water) and solvent B (90% acetonitrile and 0.1% formic acid in water) with a constant flow rate of 300 nl/min. The resulting spectra were acquired in the positive polarity/ion mode over a range of 350-2000 m/z at a resolution of 35,000 with an automatic gain control target value of 1 × 106. The top 10 most abundant precursor ions in each full MS scan were isolated and subjected to higher energy collision-induced dissociation-tandem MS (HCD-MS/MS) and fragmented with a normalized collision energy of 27%, an automatic gain control target value of 2 × 105 with an isolation window of 3 m/z at a fragment resolution of 17,500 and dynamic exclusion enabled. Peak lists and .raw data files were generated using Xcalibur software set to its default settings. Raw data files were analyzed using Proteome Discoverer 2.1.0.81 (Thermo Fisher) and were searched against a mouse ADAMTSL2 database (accession number Q7TSK7 version 1 (October 1, 2003)). Byonic software version 2.10.5 (Protein Metrics) was used as a module inside Proteome Discoverer for identifying peptides with glycan modifications. Fixed modification was carbamidomethyl on cysteines; variable modifications were Hex on tryptophan, oxidation on methionine, dHex on serine and threonine, dHexHex on serine and threonine. Five missed cleavages were permitted. Mass tolerance for precursor ions was set to 10 ppm and mass tolerance for fragment ions was set to 20 ppm. Due to the lability of the fucose-peptide bond in high energy collision dissociation experiments, Byonic is frequently unable to correctly assign the O-fucosylated Ser/Thr residue in a peptide. All assignments are based on the well-documented consensus sequence for O-fucosylation of Group 1 TSRs: C1-X-X-(S/T)-C2 (21). Note that all of the peptides identified by Byonic with an O-fucose modification contained this consensus sequence. Protein and peptide false discovery rates were set to a threshold of 1% and calculated in Byonic software version 2.10.5 (Protein Metrics) using the 2-dimensional target decoy strategy as described (41). EIC for all peptides were generated using Xcalibur Qual Browser 4.0.27.19 (Thermo Fisher). The glycoform distribution on each TSR was quantified based on area under the curve of each EIC for all biological and technical replicates.
Experimental design and statistical rationale
See Table 1.
Table 1.
Experimental design and statistical rationale
| Fig. 2A | Fig. 2C | Fig. 4 | Fig. 5 | Fig. 6A | Fig. 6C | |
|---|---|---|---|---|---|---|
| Sample sizea | 6 | 6 | 11 | 3 | 3 | 6 |
| Control sizeb | 3 | 3 | 11 | 3 | 3 | 6 |
| Biological replicatesc | 3 | 3 | 11 | 3 | 3 | 6 |
| Statistical methodsd | NAe | NA | Ordinary one-way ANOVA | Ordinary one-way ANOVA | NA | t test |
a Sample size: total number of samples performed and presented for each figure.
b Control size: number of controls for each experiment.
c Biological replicate: number of biological replicates for each figure.
d Statistical methods: types of statistical methods that were performed for each figure. All statistical analyses were performed in Prism 7. All error bars in each figure represents mean ± S.D.
e NA, not applicable.
Data availability
The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (42) partner repository with the data set identifier PXD018902.
Supplementary Material
Acknowledgments
We thank members of the Holdener and Haltiwanger labs for their insights and helpful discussions.
This article contains supporting information.
Author contributions—A. Z., S. J. B., D. V., T.-W. L., A. T., S. G., B. C. H., and R. S. H. formal analysis; A. Z., S. J. B., C.-L. M., D. V., T.-W. L., A. T., and S. G. investigation; A. Z., S. J. B., C.-L. M., D. V., T.-W. L., A. T., S. G., S. S. A., B. C. H., and R. S. H. methodology; A. Z., S. J. B., B. C. H., and R. S. H. writing-original draft; A. Z., S. J. B., S. S. A., B. C. H., and R. S. H. project administration; A. Z., S. J. B., C.-L. M., D. V., T.-W. L., A. T., S. G., S. S. A., B. C. H., and R. S. H. writing-review and editing; A. Z., S. J. B., C.-L. M., D. V., T.-W. L., A. T., S. S. A., B. C. H., and R. S. H. data curation; S. G. and S. S. A. resources; S. S. A., B. C. H., and R. S. H. conceptualization; S. S. A., B. C. H., and R. S. H. supervision; S. S. A., B. C. H., and R. S. H. funding acquisition.
Funding and additional information—This work was supported by National Institutes of Health Grants CA123071, HD096030, and HD090156 (to R. S. H. and B. C. H.) and the Georgia Research Alliance. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- ADAMTSL2
- A disintegrin-like and metalloprotease with thrombospondin type I repeats-like 2
- 6AF
- 6-alkynylfucose
- B3GLCT
- β1,3-glucosyltransferase
- Cas9
- CRISPR-associated protein 9
- CHO
- Chinese hamster ovary
- CRISPR
- clustered regularly interspaced short palindromic repeats
- dHex
- deoxy-hexose
- EIC
- extracted ion chromatogram
- GPHYSD1
- geleophysic dysplasia 1
- HEK293T
- human embryonic kidney 293T
- Hex
- hexose
- O-Fuc
- O-linked fucose
- PEI
- polyethylenimine
- POFUT2
- protein O-fucosyltransferase 2
- PTRPLS
- Peters Plus Syndrome
- TBS
- Tris-buffered saline
- TSR
- thrombospondin type-1 repeats
- PEI
- polyethylenimine
- BCS
- bovine calf serum
- DMEM
- Dulbecco's modified Eagle's medium
- PNGase F
- peptide N-glycosidase F
- ANOVA
- analysis of variance
References
- 1. Apte S. S. (2009) A disintegrin-like and metalloprotease (reprolysin-type) with thrombospondin type 1 motif (ADAMTS) superfamily: functions and mechanisms. J. Biol. Chem. 284, 31493–31497 10.1074/jbc.R109.052340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hubmacher D., and Apte S. S. (2015) ADAMTS proteins as modulators of microfibril formation and function. Matrix Biol. 47, 34–43 10.1016/j.matbio.2015.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pinan-Lucarré B., Tu H., Pierron M., Cruceyra P. I., Zhan H., Stigloher C., Richmond J. E., and Bessereau J. L. (2014) C. elegans punctin specifies cholinergic versus GABAergic identity of postsynaptic domains. Nature 511, 466–470 10.1038/nature13313 [DOI] [PubMed] [Google Scholar]
- 4. Delhon L., Mahaut C., Goudin N., Gaudas E., Piquand K., Le Goff W., Cormier-Daire V., and Le Goff C. (2019) Impairment of chondrogenesis and microfibrillar network in Adamtsl2 deficiency. FASEB J. 33, 2707–2718 10.1096/fj.201800753RR [DOI] [PubMed] [Google Scholar]
- 5. Koo B. H., Le Goff C., Jungers K. A., Vasanji A., O'Flaherty J., Weyman C. M., and Apte S. S. (2007) ADAMTS-like 2 (ADAMTSL2) is a secreted glycoprotein that is widely expressed during mouse embryogenesis and is regulated during skeletal myogenesis. Matrix Biol. 26, 431–441 10.1016/j.matbio.2007.03.003 [DOI] [PubMed] [Google Scholar]
- 6. Le Goff C., Morice-Picard F., Dagoneau N., Wang L. W., Perrot C., Crow Y. J., Bauer F., Flori E., Prost-Squarcioni C., Krakow D., Ge G., Greenspan D. S., Bonnet D., Le Merrer M., Munnich A., et al. (2008) ADAMTSL2 mutations in geleophysic dysplasia demonstrate a role for ADAMTS-like proteins in TGF-beta bioavailability regulation. Nat. Genet. 40, 1119–1123 10.1038/ng.199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Weh E., Reis L. M., Tyler R. C., Bick D., Rhead W. J., Wallace S., McGregor T. L., Dills S. K., Chao M. C., Murray J. C., and Semina E. V. (2014) Novel B3GALTL mutations in classic Peters plus syndrome and lack of mutations in a large cohort of patients with similar phenotypes. Clin. Genet. 86, 142–148 10.1111/cge.12241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Allali S., Le Goff C., Pressac-Diebold I., Pfennig G., Mahaut C., Dagoneau N., Alanay Y., Brady A. F., Crow Y. J., Devriendt K., Drouin-Garraud V., Flori E., Genèvieve D., Hennekam R. C., Hurst J., et al. (2011) Molecular screening of ADAMTSL2 gene in 33 patients reveals the genetic heterogeneity of geleophysic dysplasia. J. Med. Genet. 48, 417–421 10.1136/jmg.2010.087544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Piccolo P., Sabatino V., Mithbaokar P., Polishchuck E., Law S. K., Magraner-Pardo L., Pons T., Polishchuck R., and Brunetti-Pierri N. (2019) Geleophysic dysplasia: novel missense variants and insights into ADAMTSL2 intracellular trafficking. Mol. Genet. Metab. Rep. 21, 100504 10.1016/j.ymgmr.2019.100504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Haltiwanger R. S., Wells L., Freeze H. H., and Stanley P. (2015) Other classes of eukaryotic glycans in Essentials of Glycobiology (Varki A., Cummings R. D., Esko J. D., Stanley P., Hart G. W., Aebi M., Darvill A. G., Kinoshita T., Packer N. H., Prestegard J. H., Schnaar R. L., and Seeberger P. H., eds) pp. 151–160, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 11. Hofsteenge J., Huwiler K. G., Macek B., Hess D., Lawler J., Mosher D. F., and Peter-Katalinic J. (2001) C-Mannosylation and O-fucosylation of the thrombospondin type 1 module. J. Biol. Chem. 276, 6485–6498 10.1074/jbc.M008073200 [DOI] [PubMed] [Google Scholar]
- 12. Luo Y., Koles K., Vorndam W., Haltiwanger R. S., and Panin V. M. (2006) Protein O-fucosyltransferase 2 adds O-fucose to thrombospondin type 1 repeats. J. Biol. Chem. 281, 9393–9399 10.1074/jbc.M511975200 [DOI] [PubMed] [Google Scholar]
- 13. Shcherbakova A., Tiemann B., Buettner F. F., and Bakker H. (2017) Distinct C-mannosylation of netrin receptor thrombospondin type 1 repeats by mammalian DPY19L1 and DPY19L3. Proc. Natl. Acad. Sci. U.S.A. 114, 2574–2579 10.1073/pnas.1613165114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Krieg J., Hartmann S., Vicentini A., Gläsner W., Hess D., and Hofsteenge J. (1998) Recognition signal for C-mannosylation of Trp-7 in RNase 2 consists of sequence Trp-X-X-Trp. Mol. Biol. Cell 9, 301–309 10.1091/mbc.9.2.301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang L. W., Leonhard-Melief C., Haltiwanger R. S., and Apte S. S. (2009) Post-translational modification of thrombospondin type-1 repeats in ADAMTS-like 1/punctin-1 by C-mannosylation of tryptophan. J. Biol. Chem. 284, 30004–30015 10.1074/jbc.M109.038059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shcherbakova A., Preller M., Taft M. H., Pujols J., Ventura S., Tiemann B., Buettner F. F., and Bakker H. (2019) C-Mannosylation supports folding and enhances stability of thrombospondin repeats. Elife 8, e52978 10.7554/eLife.52978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Buettner F. F., Ashikov A., Tiemann B., Lehle L., and Bakker H. (2013) C. elegans DPY-19 is a C-mannosyltransferase glycosylating thrombospondin repeats. Mol. Cell 50, 295–302 10.1016/j.molcel.2013.03.003 [DOI] [PubMed] [Google Scholar]
- 18. Gavel Y., and von Heijne G. (1990) Sequence differences between glycosylated and non-glycosylated Asn-X-Thr/Ser acceptor sites: implications for protein engineering. Protein Eng. 3, 433–442 10.1093/protein/3.5.433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Parodi A., Cummings R. D., and Aebi M. (2015) Glycans in glycoprotein quality control. in Essentials of Glycobiology (Varki A., Cummings R. D., Esko J. D., Stanley P., Hart G. W., Aebi M., Darvill A. G., Kinoshita T., Packer N. H., Prestegard J. H., Schnaar R. L., and Seeberger P. H., eds) pp. 503–511, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 20. Vasudevan D., Takeuchi H., Johar S. S., Majerus E., and Haltiwanger R. S. (2015) Peters Plus Syndrome mutations disrupt a noncanonical ER quality-control mechanism. Curr. Biol. 25, 286–295 10.1016/j.cub.2014.11.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Holdener B. C., and Haltiwanger R. S. (2019) Protein O-fucosylation: structure and function. Curr. Opin. Struct. Biol. 56, 78–86 10.1016/j.sbi.2018.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Du J., Takeuchi H., Leonhard-Melief C., Shroyer K. R., Dlugosz M., Haltiwanger R. S., and Holdener B. C. (2010) O-fucosylation of thrombospondin type 1 repeats restricts epithelial to mesenchymal transition (EMT) and maintains epiblast pluripotency during mouse gastrulation. Dev. Biol. 346, 25–38 10.1016/j.ydbio.2010.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Oberstein S. A. J. L., Kriek M., White S. J., Kalf M. E., Szuhai K., den Dunnen J. T., Breuning M. H., and Hennekam R. C. M. (2006) Peters Plus Syndrome is caused by mutations in B3GALTL, a putative glycosyltransferase. Am. J. Hum. Genet. 79, 562–566 10.1086/507567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hess D., Keusch J. J., Oberstein S. A. L., Hennekam R. C. M., and Hofsteenge J. (2008) Peters Plus Syndrome is a new congenital disorder of glycosylation and involves defective O-glycosylation of thrombospondin type 1 repeats. J. Biol. Chem. 283, 7354–7360 10.1074/jbc.M710251200 [DOI] [PubMed] [Google Scholar]
- 25. Holdener B. C., Percival C. J., Grady R. C., Cameron D. C., Berardinelli S. J., Zhang A., Neupane S., Takeuchi M., Jimenez-Vega J. C., Uddin S. M. Z., Komatsu D. E., Honkanen R., Dubail J., Apte S. S., Sato T., et al. (2019) ADAMTS9 and ADAMTS20 are differentially affected by loss of B3GLCT in a mouse model of Peters Plus Syndrome. Hum. Mol. Genet. 28, 4053–4066 10.1093/hmg/ddz225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang L. W., Dlugosz M., Somerville R. P., Raed M., Haltiwanger R. S., and Apte S. S. (2007) O-fucosylation of thrombospondin type 1 repeats in ADAMTS-like-1/punctin-1 regulates secretion: implications for the ADAMTS superfamily. J. Biol. Chem. 282, 17024–17031 10.1074/jbc.M701065200 [DOI] [PubMed] [Google Scholar]
- 27. Ricketts L. M., Dlugosz M., Luther K. B., Haltiwanger R. S., and Majerus E. M. (2007) O-Fucosylation is required for ADAMTS13 secretion. J. Biol. Chem. 282, 17014–17023 10.1074/jbc.M700317200 [DOI] [PubMed] [Google Scholar]
- 28. Benz B. A., Nandadasa S., Takeuchi M., Grady R. C., Takeuchi H., LoPilato R. K., Kakuda S., Somerville R. P. T., Apte S. S., Haltiwanger R. S., and Holdener B. C. (2016) Genetic and biochemical evidence that gastrulation defects in Pofut2 mutants result from defects in ADAMTS9 secretion. Dev. Biol. 416, 111–122 10.1016/j.ydbio.2016.05.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hubmacher D., Wang L. W., Mecham R. P., Reinhardt D. P., and Apte S. S. (2015) Adamtsl2 deletion results in bronchial fibrillin microfibril accumulation and bronchial epithelial dysplasia: a novel mouse model providing insights into geleophysic dysplasia. Dis. Models Mech. 8, 487–499 10.1242/dmm.017046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Leonhard-Melief C., and Haltiwanger R. S. (2010) O-Fucosylation of thrombospondin type 1 repeats. Methods Enzymol. 480, 401–416 10.1016/S0076-6879(10)80018-7 [DOI] [PubMed] [Google Scholar]
- 31. Al-Shareffi E., Chaubard J. L., Leonhard-Melief C., Wang S. K., Wong C. H., and Haltiwanger R. S. (2013) 6-alkynyl fucose is a bioorthogonal analog for O-fucosylation of epidermal growth factor-like repeats and thrombospondin type-1 repeats by protein O-fucosyltransferases 1 and 2. Glycobiology 23, 188–198 10.1093/glycob/cws140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ripka J., Adamany A., and Stanley P. (1986) Two Chinese hamster ovary glycosylation mutants affected in the conversion of GDP-mannose to GDP-fucose. Arch. Biochem. Biophys. 249, 533–545 10.1016/0003-9861(86)90031-7 [DOI] [PubMed] [Google Scholar]
- 33. Stanley P., Caillibot V., and Siminovitch L. (1975) Selection and characterization of eight phenotypically distinct lines of lectin-resistant Chinese hamster ovary cell. Cell 6, 121–128 10.1016/0092-8674(75)90002-1 [DOI] [PubMed] [Google Scholar]
- 34. Hubmacher D., Schneider M., Berardinelli S. J., Takeuchi H., Willard B., Reinhardt D. P., Haltiwanger R. S., and Apte S. S. (2017) Unusual life cycle and impact on microfibril assembly of ADAMTS17, a secreted metalloprotease mutated in genetic eye disease. Sci. Rep. 7, 41871 10.1038/srep41871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Boettcher M., and McManus M. T. (2015) Choosing the right tool for the job: RNAi, TALEN, or CRISPR. Mol. Cell 58, 575–585 10.1016/j.molcel.2015.04.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Valero-González J., Leonhard-Melief C., Lira-Navarrete E., Jiménez-Osés G., Hernández-Ruiz C., Pallarés M. C., Yruela I., Vasudevan D., Lostao A., Corzana F., Takeuchi H., Haltiwanger R. S., and Hurtado-Guerrero R. (2016) A proactive role of water molecules in acceptor recognition by protein O-fucosyltransferase 2. Nat. Chem. Biol. 12, 240–246 10.1038/nchembio.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dubail J., Vasudevan D., Wang L. W., Earp S. E., Jenkins M. W., Haltiwanger R. S., and Apte S. S. (2016) Impaired ADAMTS9 secretion: a potential mechanism for eye defects in Peters Plus Syndrome. Sci. Rep. 6, 33974 10.1038/srep33974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lawler P. R., and Lawler J. (2012) Molecular basis for the regulation of angiogenesis by thrombospondin-1 and -2. Cold Spring Harb. Perspect. Med. 2, a006627 10.1101/cshperspect.a006627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Luo Y., Nita-Lazar A., and Haltiwanger R. S. (2006) Two distinct pathways for O-fucosylation of epidermal growth factor-like or thrombospondin type 1 repeats. J. Biol. Chem. 281, 9385–9392 10.1074/jbc.M511974200 [DOI] [PubMed] [Google Scholar]
- 40. Thomas M., Lu J. J., Ge Q., Zhang C., Chen J., and Klibanov A. M. (2005) Full deacylation of polyethylenimine dramatically boosts its gene delivery efficiency and specificity to mouse lung. Proc. Natl. Acad. Sci. U.S.A. 102, 5679–5684 10.1073/pnas.0502067102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bern M. W., and Kil Y. J. (2011) Two-dimensional target decoy strategy for shotgun proteomics. J. Proteome Res. 10, 5296–5301 10.1021/pr200780j [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D. J., Inuganti A., Griss J., Mayer G., Eisenacher M., Perez E., Uszkoreit J., Pfeuffer J., Sachsenberg T., Yilmaz S., et al. (2019) The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47, D442–D450 10.1093/nar/gky1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (42) partner repository with the data set identifier PXD018902.