

Abstract
Manganese (Mn)-induced neurotoxicity resembles Parkinson's disease (PD), but the mechanisms underpinning its effects remain unknown. Mn dysregulates astrocytic glutamate transporters, GLT-1 and GLAST, and dopaminergic function, including tyrosine hydroxylase (TH). Our previous in vitro studies have shown that Mn repressed GLAST and GLT-1 via activation of transcription factor Yin Yang 1 (YY1). Here, we investigated if in vivo astrocytic YY1 deletion mitigates Mn-induced dopaminergic neurotoxicity, attenuating Mn-induced reduction in GLAST/GLT-1 expression in murine substantia nigra (SN). AAV5-GFAP-Cre-GFP particles were infused into the SN of 8-week–old YY1flox/flox mice to generate a region-specific astrocytic YY1 conditional knockout (cKO) mouse model. 3 weeks after adeno-associated viral (AAV) infusion, mice were exposed to 330 μg of Mn (MnCl2 30 mg/kg, intranasal instillation, daily) for 3 weeks. After Mn exposure, motor functions were determined in open-field and rotarod tests, followed by Western blotting, quantitative PCR, and immunohistochemistry to assess YY1, TH, GLAST, and GLT-1 levels. Infusion of AAV5-GFAP-Cre-GFP vectors into the SN resulted in region-specific astrocytic YY1 deletion and attenuation of Mn-induced impairment of motor functions, reduction of TH-expressing cells in SN, and TH mRNA/protein levels in midbrain/striatum. Astrocytic YY1 deletion also attenuated the Mn-induced decrease in GLAST/GLT-1 mRNA/protein levels in midbrain. Moreover, YY1 deletion abrogated its interaction with histone deacetylases in astrocytes. These results indicate that astrocytic YY1 plays a critical role in Mn-induced neurotoxicity in vivo, at least in part, by reducing astrocytic GLAST/GLT-1. Thus, YY1 might be a potential target for treatment of Mn toxicity and other neurological disorders associated with dysregulation of GLAST/GLT-1.
Keywords: adeno-associated viral (AAV), dopaminergic neurotoxicity, GLASTGLT-1, manganese Yin Yang 1, manganese, glutamate, astrocyte, Parkinson disease, animal model, adeno-associated viral vector, tyrosine hydroxylase
Manganese (Mn) is an essential trace element that is naturally found in the environment and is a necessary cofactor for several enzymes that support metabolism, development, and neuronal function (1). However, chronic exposure to Mn causes a neurological disorder referred to as manganism (2), sharing common clinical and pathological symptoms with Parkinson's disease (PD), such as dopaminergic dysfunction and motor deficits (2). Mn overexposure has been a public health concern due to its widespread industrial usage and risk for environmental contamination (3–5). High levels of Mn in South African mine workers have been associated with increased parkinsonian symptoms and poorer quality of life (4). Mn also lowers cognition and IQ in children (5) and adults (3, 6). Experimental animal studies have also shown that Mn impairs cognitive and memory function in mice (7–9), indicating it is a risk factor for neurodegenerative disorders associated with cognitive and motor functions.
Although numerous studies have reported pathological mechanisms of Mn-induced neurotoxicity, the molecular mechanisms are not completely understood. Mn induces oxidative stress (10), mitochondrial dysfunction (11), inflammation (12), and apoptotic cell death (13). In addition, Mn-induced excitotoxicity is critically involved in its neurotoxicity, as N-methyl-d-aspartate (NMDA) receptor antagonist MK801 attenuated its toxicity in the rat model (14).
Mn-induced excitotoxic neuronal injury may also be associated with astrocytic glutamate transporter impairment (15), but the mechanisms of Mn-induced dysregulation of glutamate neurotransmission has yet to be established. The glutamate aspartate transporter 1 (GLAST) and glutamate transporter 1 (GLT-1) are responsible for the majority of glutamate reuptake from the synaptic cleft, preventing the accumulation of excess glutamate (16–19). GLAST and GLT-1 may be used interchangeably with their human homologs excitatory amino acid transporters 1 (EAAT1) and 2 (EAAT2), respectively. Our previous studies have shown that Mn decreased astrocytic expression of both GLAST/EAAT1 and GLT-1/EAAT2 at the transcriptional level (20, 21).
As astrocytes predominantly express GLAST and GLT-1, impairment of these transporters can lead to excitotoxic neuronal injury (22, 23). In fact, dysregulation of astrocytic glutamate transporters GLAST and GLT-1 is associated with not only Mn toxicity, but also several other neurodegenerative diseases, such as Alzheimer's disease (AD) (24), PD (25), and ALS (26). Accordingly, various pharmacological agents have been explored to restore these transporters and treat neurological disorders associated with impairment of these transporters. For example, ceftriaxone, a β-lactam antibiotic, increases GLT-1 expression and affords protection against several neurotoxicants such as 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) (27) and 6-hydroxydopamine (6-OHDA) (28, 29), as well as pathological conditions such as ischemia (30), traumatic brain injury (31), and AD (32). We have previously reported that estrogenic compounds, such as 17β-estradiol and selective estrogen receptor modulators including tamoxifen and raloxifene, and arundic acid increased GLAST and GLT-1 mRNA/protein levels, and attenuated Mn-induced impairment of these transporters in astrocyte culture and in mice (15, 33–35). Histone deacetylase (HDAC) inhibitors, such as sodium butyrate and valproic acid, also attenuated Mn-induced dysregulation of GLAST/GLT-1 in astrocyte culture and dopaminergic neurotoxicity in mouse models, indicating that epigenetic modification could also play a significant role in Mn-induced neurotoxicity (34, 35).
Although pharmacological approaches may be effective in increasing expression and function of GLT-1/GLAST and neuroprotection, they may also exert other unwanted effects by binding to nonspecific sites. On the other hand, the identification of target genes that modulate GLT-1/GLAST can provide another avenue with more selective mechanisms, such as oligonucleotide therapeutics (i.e. those based on antisense RNAs, siRNA, and microRNA) (36). A transcription factor Yin Yang 1 (YY1) is known to activate or repress transcription of its target genes, depending on the cellular context and co-factor availability (37). Dysregulation of YY1 is implicated in various neurodegenerative disorders, such as PD, ALS, AD, Charcot-Marie-Tooth disease, and Rett syndrome (38–44). YY1 repressed GLAST promoter activity in chick cerebellar Bergmann glia cells (45), which was reversed by HDAC inhibitor valproic acid, indicating that YY1 interacts with HDACs to repress GLAST (46). YY1 also interacts with astrocyte elevated gene-1 (AEG-1) as a co-repressor to inhibit GLT-1 expression, leading to reduced glutamate uptake in astrocytes (47). Importantly, we have previously reported that Mn increased YY1 expression, which recruited the epigenetic modifiers HDACs to repress GLAST/GLT-1 by binding YY1 consensus sites in the GLT-1 and GLAST promoters in astrocytes (20, 21). This indicates that astrocytic YY1 might be a potential target for genetic manipulation to treat Mn toxicity, given that at least in vitro YY1 mediated Mn-induced decrease in GLT-1/GLAST expression and function (20, 21).
Adeno-associated viral (AAV) vectors are nonpathogenic and efficient for transgene expression in vivo (48). Proper selections of AAV serotype, gene promoter, and region of interest are critical for successful gene delivery. There are over 100 AAV serotypes that differ in the binding capacity of capsid proteins to specific cell surface receptors, which can transduce different cell types and brain regions in the CNS (48). Several AAV serotypes, such as AAV1, AAV2, AAV4, AAV5, AAV8, and AAV9, have been expressed in the central nervous system (CNS) (49). AAV vector serotypes 5 and 8 showed greater tropism and efficiency in expressing transgenes in astrocytes (48, 50). The glial fibrillary acidic protein (GFAP) promoter has been used to target astrocytes for AAV vector protein expression. Although GFAP is expressed in other cell types, such as ependymal cells during development, and radial glial progenitor cells in adult neurogenic niches (51), this gene is mainly expressed in astrocytes and its promoter is commonly used to express proteins in astrocytes (52). GFAP promoter-driven expression of Cre recombinase (Cre) is widely used and recognized for gene therapy as well as deletion of target genes in astrocytes (53). Accordingly, in the present study, the GFAP promoter was used to express Cre (AAV5-GFAP-Cre-GFP) and delete YY1 in the astrocytes of YY1 loxP mice. Because Mn preferentially accumulates in the basal ganglia, causes dopaminergic neuronal injury, and induces behavioral deficits and impaired motor coordination (35), we deleted astrocytic YY1 in the substantia nigra (SN), where dopaminergic neuronal cell bodies are localized.
Our results demonstrated that astrocytic YY1 deletion attenuated the Mn-induced impairment in locomotor activity and coordination. Moreover, this YY1 deletion attenuated Mn-induced reduction of TH-positive cells in SN, concomitant to the mitigation of Mn-induced down-regulation of GLAST and GLT-1 in SN, suggesting that Mn-induced activation of astrocytic YY1 may reduce expression of astrocytic GLT-1 and GLAST, leading to excitotoxic dopaminergic neuronal injury.
Results
Infusion of GFAP-Cre-GFP AAV5 particles into murine SN induces astrocyte-specific expression of GFAP-Cre recombinase
We used YY1-loxP homozygous mice (8 weeks old) to delete YY1 in astrocytes of the SN region of mouse brains to determine the effects of astrocytic YY1 in Mn-induced dopaminergic neurotoxicity. This model ensures that YY1 is deleted in the area where the AAV particles were infused (Fig. 1). We used equal numbers of male and female YY1 loxP mice, and they did not exhibit sex-specific effects in our experimental conditions (data not shown). After mice were randomly grouped, AAV5-GFAP-Cre-GFP vectors to express Cre recombinase or AAV5-GFAP-GFP vectors (as control vectors) were infused into both sides of the SN region of the mouse brain (Fig. 1A). This control vector was used to eliminate potential effects of the AAV vector injection itself (54). Because this YY1 loxP mouse model has an insertion of loxP-flanked sequences on exon 1 of YY1 (55), AAV5-GFAP-Cre vectors will delete exon 1 of the YY1 gene in the Cre recombinase (Cre)-expressing astrocytes, resulting in astrocyte-specific YY1 deletion (Fig. 1B).
Figure 1.

Schematic diagram of the experimental paradigm. A, coordinates of stereotaxic injection and bilateral infusion of AAV vectors into the SN of the mouse brain. B, genomic deletion of astrocytic YY1 by GFAP-Cre expression in astrocytes of the SN regions of YY1-loxP mice using a Cre-lox method. To induce astrocyte-specific YY1 deletion, AAV5-GFAP-Cre-GFP vectors were infused into the SN, using AAV5-GFAP-GFP vectors as control vectors. C, Mn exposure (30 mg/kg, intranasal instillation, daily for 3 weeks) after bilateral infusion of AAV5 particles into the SN regions of the mouse brain, as described under “Experimental procedures.” 3 weeks after AAV5 infusion, MnCl2 was administered intranasally into both nostrils alternately and distilled water was used as a vehicle.
Our previous in vitro studies have shown that astrocytic YY1 played a critical role in Mn-induced reduction of glutamate transporters GLT-1 and GLAST. Here, we tested if astrocytic YY1 deletion affords protective effects against Mn-induced dopaminergic neurotoxicity in vivo after 3 weeks of Mn exposure (Fig. 1C), concomitant with attenuation of Mn-induced repression of GLAST and GLT-1 in astrocytes.
To validate the sites of the viral vector infusion into the SN regions where dopaminergic neuronal cell bodies are located, we performed IHC 3 weeks after infusion of the vectors into the SN region of YY1-floxed mice. The results showed that the AAV5-GFAP-GFP vectors expressed the GFAP promoter-driven GFP protein as an indicator for the infused vectors in astrocytes of the SN regions where TH-expressing dopaminergic neurons are localized (Fig. 2A). YY1 expression was also co-localized with GFP-expressing astrocytes and TH-expressing dopaminergic neurons (Fig. 2B). Infusion of AAV5-GFAP-Cre-GFP vectors into the SN of YY1-floxed mice expressed Cre in GFAP-expressing astrocytes (Fig. 2C). We found that AAV5 was successfully injected, expressing GFP fluorescence protein in the SN region of the mouse brain (Fig. 2A). In addition, GFAP-expressing cells (astrocytes) were highly co-localized with Cre (Fig. 2C, red), indicating that Cre is expressed in astrocytes.
Figure 2.

Validation of AAV vector infusion into the murine SN and Cre recombinase expression in astrocytes. 3 weeks after AAV vector infusion, the mouse brain was perfused and coronal sections were stained with antibodies for GFAP and Cre as described under “Experimental procedures.” A, cells expressing GFP (green, 4 × magnification) depict the site of AAV vector infusion in SN regions where TH-expressing dopaminergic neurons are localized (red, TH; green, GFP; ×10 magnification). White arrows, GFP-expressing astrocytes in SN. B, YY1 is co-expressed with GFP-expressing astrocytes (top panel), and the bottom panel shows expression of YY1 (blue), TH-expressing dopaminergic neurons (red) and GFP-expressing astrocytes (green) in SN 3 weeks after AAV5-GFAP-GFP (GFAP-control) vector infusion (×20 magnification). White arrows show YY1 co-expressing with GFP. C, coronal sections immunostained with GFAP and Cre antibodies showed co-localized expression of GFAP (green) and Cre (red) in the SN region, indicating that Cre was expressed in astrocytes (×20 magnification). White arrows show Cre co-expressing with GFAP.
Intranasal instillation of Mn increased Mn levels in mouse brain regions
To determine whether intranasal delivery of Mn exposure reached the brain and increased Mn levels, particularly the midbrain, where the AAV vectors were infused, we measured Mn content in the midbrain, striatum, frontal cortex, hippocampus, and cerebellum by means of inductively coupled plasma (ICP)-MS/MS. The results showed that Mn levels were significantly increased in all brain regions except for the hippocampal regions (Fig. 3). Mn levels were increased about 2-fold, depending on the regions. The highest increase of Mn levels was in midbrain regions where dopaminergic cell bodies are localized, followed by the cerebellum, striatum, and cortex.
Figure 3.

Mn exposure via intranasal instillation increased Mn levels in various regions of the mouse brain. Mice were treated with MnCl2 (30 mg/kg, daily) for 3 weeks via intranasal instillation. Twenty-four hours after the last Mn treatment, mouse brain tissues were dissected, and Mn levels in each region were measured by ICP-MS/MS, as described under “Experimental procedures.” CX, frontal cortex; HP, hippocampus; ST, striatum; MB, midbrain; CB, cerebellum. **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 compared with each other by Student's t test; n = 7-10. Data are expressed as mean ± S.D.
Because alteration of Mn levels can modulate other metals that share the same transporters, Fe, Cu, and Zn contents were also measured in mouse brain regions after 3 weeks of Mn exposure via intranasal instillation. The results showed that elevated Mn levels in the brain significantly decreased levels of Fe, Cu, and Zn in the frontal cortex (Fig. 4). Mn also decreased Cu and Zn levels in the cerebellum (Fig. 4, B and C). In addition, Mn significantly decreased Cu levels in the hippocampus (Fig. 4B) and Zn levels in the midbrain (Fig. 4C).
Figure 4.

Mn elevation decreases accumulation of other divalent metals (Fe, Cu, and Zn) in the brain. A–C, after mice were treated daily with MnCl2 (30 mg/kg) for 3 weeks via intranasal instillation, levels of Fe (A), Cu (B), and Zn (C) were measured in various regions of the mouse brain by ICP-MS as described under “Experimental procedures.” CX, frontal cortex; HP, hippocampus; ST, striatum; MB, midbrain; CB, cerebellum. #, p < 0.05; ##, p < 0.01; ###, p < 0.001, compared with each other by Student's t test; n = 7–10. Data are expressed as mean ± S.D.
Mn-induced increase in YY1 expression and its interaction with HDACs were attenuated by astrocytic YY1 deletion in the SN region
Because infusion of AAV5-GFAP-Cre-GFP vectors into the SN region of the mouse brain leads to expression of Cre exclusively in astrocytes, we tested if astrocytic YY1 is deleted by Cre in astrocytes. We found that AAV5-GFAP-Cre vectors decreased cells co-expressing both GFAP and YY1 in the SN of the mouse brain, compared with the control AAV-GFAP-GFP vector infusion (Fig. 5A), indicating that YY1 levels in GFAP-expressing astrocytes were decreased by Cre expression. Other cell types adjacent to astrocytes (GFAP-expressing cells) were immunostained for YY1 expression (Fig. 5A, red), indicating that YY1 is deleted only in astrocytes. Moreover, astrocytic YY1 deletion by Cre in the SN region of YY1-loxP mice decreased YY1 mRNA and protein levels in the midbrain (Fig. 5, B and C). Mn increased YY1 mRNA and protein levels in the midbrain, whereas astrocytic YY1 deletion attenuated Mn-induced YY1 expression significantly (Fig. 5, B and C). Mn increased YY1 levels in the astrocytic YY1-deleted midbrain, indicating that these increased YY1 levels could be from other cell types, such as oligodendrocytes, microglia, and/or neurons (Fig. 5C).
Figure 5.
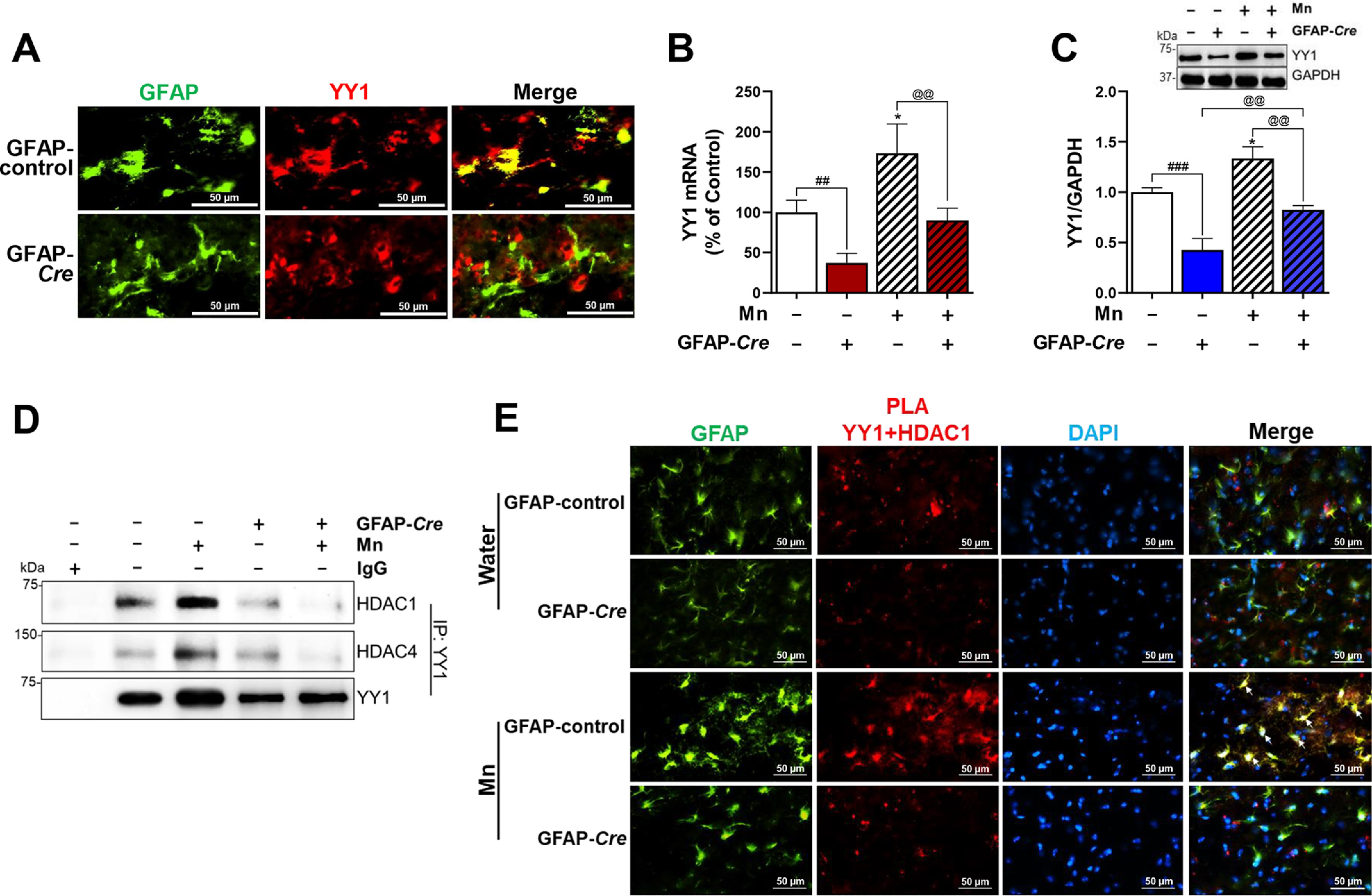
Astrocytic YY1 deletion by infusion of AAV-GFAP-Cre-GFP viral vectors into the SN attenuates Mn-increased YY1 expression and YY1 interaction with HDACs. A, after AAV infusion, coronal sections were stained with antibodies for GFAP and YY1, followed by IHC as described under “Experimental procedures.” Cells stained with GFAP (green) depict astrocytes and YY1 (red) indicates YY1 expression (×40 magnification). B and C, after Mn treatment, midbrain samples were extracted for total RNA and protein, followed by qPCR and Western blotting, as described under “Experimental procedures.” YY1 mRNA (B) and protein (C) levels in the midbrain were measured. GAPDH was used as loading controls of mRNA and protein. D, midbrain samples were tested for interaction of YY1 with HDAC1 and/or HDAC4 by co-IP as described under “Experimental procedures.” E, interaction of YY1 and HDAC1 in the SN of the mouse brain was determined by proximity ligation assay (×40 magnification). GFAP (green), YY1-HDAC1 proximity ligation signal (red), and DAPI (blue). *, p < 0.05 compared with the controls; ##, p < 0.01; ###, p < 0.001; @@, p < 0.01 compared with each other (one-way ANOVA followed by Tukey's post hoc test; n = 3). Data are expressed as mean ± S.D.
Because YY1 interaction with HDACs is involved in Mn-induced repression of GLT-1 and GLAST in in vitro settings (20, 21), we tested if astrocytic YY1 deletion in the SN also inhibits the YY1-HDAC complex. We used HDAC1 as representative class I HDACs and HDAC4 as class II. The results showed that Mn significantly increased interaction of YY1 with HDAC1 as well as HDAC4 in the SN/midbrain of mice infused with AAV5-GFAP-GFP mice (Fig. 5D). On the other hand, interactions of YY1 with HDAC1/4 were detected at low levels in both control and Mn-treated astrocytic YY1-deleted mice with AAV5-GFAP-Cre (Fig. 5D).
The proximity ligation assay (PLA) was also performed to determine whether interaction of YY1 and HDAC1 occurs in astrocytes of the SN. Results showed interaction of YY1 and HDAC1 in astrocytes as well as other cell types (Fig. 5E). Mn increased YY1-HDAC1 proximity ligation signals in astrocytes, but astrocytic YY1 deletion abrogated YY1-HDAC1 proximity ligation signals in astrocytes of the SN of Mn-treated mice (Fig. 5E).
Astrocytic YY1 deletion in the SN attenuated Mn-induced impairment of locomotor activity and motor coordination
Because nigrostriatal dopaminergic function regulates motor function, and Mn-induced dopaminergic neurotoxicity is associated with impairment of motor function (56, 57), we tested if astrocytic YY1 deletion in the SN attenuates Mn-induced motor deficits. Astrocytic YY1 deletion was bilaterally induced in murine SN by infusion of AAV5-GFAP-Cre vectors into both sides as to prevent potential motor impairments from a unilateral YY1 deletion (58). First, we determined if astrocytic YY1 deletion itself could modulate motor functions of mice at 3 weeks after AAV5-GFAP-Cre particle infusion, and we found no difference compared with the control AAV5-GFAP particle infusion (data not shown). To determine whether astrocytic YY1 deletion attenuates Mn-induced motor deficits, we assessed open-field movement on total distance traveled and motor coordination. As shown in Fig. 6, Mn decreased total movement and distance traveled (Fig. 6, A and B) as well as latency to fall from the rotarod (Fig. 6C), indicating that Mn impaired locomotor activity and motor coordination, respectively. On the other hand, astrocytic YY1 deletion in the SN regions attenuated Mn-induced impairment of behavior and motor coordination (Fig. 6).
Figure 6.

Deletion of astrocytic YY1 in SN attenuates Mn-induced impairment of locomotor activity and motor coordination. After Mn exposure (MnCl2, 30 mg/kg, intranasal instillation, daily for 3 weeks), locomotor activity and motor coordination were measured as described under “Experimental procedures.” A, the traces show one mouse's movement, and red dots depict vertical activity as a representative. B, total distance traveled, and C, time spent on the rotating rod. ###, p < 0.001 compared with the controls. @, p < 0.05; @@@, p < 0.001 compared with each other (one-way ANOVA followed by Tukey's post hoc test; n = 12). Data are expressed as mean ± S.D.
Astrocytic YY1 deletion attenuates Mn-induced decrease of dopaminergic TH expression in the SN of mouse brain
Given that Mn impairs locomotor activity in mice with concomitant reduction of TH expression in dopaminergic neurons in both in vitro and in vivo experimental models (10, 35), we determined if deletion of astrocytic YY1 modulates Mn-decreased TH-expressing dopaminergic neurons in SN. Expression of Cre with AAV5-GFAP-Cre-GFP vector infusion did not alter TH expression, showing similar TH expression to that of the control AAV5-GFAP-GFP vector infusion (Fig. 7A). Mn exposure decreased TH-positive cells in the SN of mice infused with AAV5-GFAP-GFP vectors (Fig. 7A), whereas astrocytic YY1 deletion by infusion of AAV5-GFAP-Cre-GFP vectors attenuated Mn-decreased TH-positive cells in the same region (Fig. 7A). Mn also decreased TH mRNA and protein levels, whereas astrocytic YY1 deletion attenuated Mn-decreased TH mRNA and protein levels in the midbrain (Fig. 7, B and C). Interestingly, astrocytic YY1 deletion in the SN region attenuated Mn-decreased TH mRNA and protein levels in the striatal regions of the mouse brain (Fig. 7, D and E), indicating that astrocytic YY1 deletion in the SN region affects the striatal region where SN's dopaminergic nerve terminals are localized.
Figure 7.

Deletion of astrocytic YY1 attenuates Mn-induced decrease of TH expression in SN/midbrain and striatum. A, after Mn exposure (MnCl2, 30 mg/kg, intranasal instillation, daily for 3 weeks), mice were perfused, and coronal sections of brain tissues were immunostained with TH antibody as described under “Experimental procedures.” The expression of TH protein was shown as red fluorescence signals (TRITC) in the SN of the mouse brain (×10 magnification). B–E, after Mn treatment, midbrain (B and C) and striatal (D and E) regions were processed for TH mRNA and protein levels by qPCR and Western blotting, respectively, as described under “Experimental procedures.” GAPDH was used as a loading control. *, p < 0.05; #, p < 0.05; ###, p < 0.001 compared with GFAP-GFP/vehicle; @, p < 0.05; @@@, p < 0.001 compared with each other (one-way ANOVA followed by Tukey's post hoc test; n = 3). Data are expressed as mean ± S.D.
Astrocytic YY1 deletion attenuates the Mn-induced decrease in GLT-1 and GLAST mRNA and protein levels in the SN of mouse brain
Next, we determined if the in vitro effects of YY1 deletion on GLT-1/GLAST are recapitulated in an in vivo mouse model. The results showed that Mn significantly decreased mRNA and protein levels of GLAST and GLT-1 in the SN/midbrain of mice treated with the control AAV5-GFAP-GFP vectors (Fig. 8, A–D). In addition, astrocytic YY1 deletion by infusion of the AAV5-GFAP-Cre-GFP vectors did not change GLAST and GLT-1 expression levels in the midbrain. However, Mn-decreased mRNA and protein levels of GLAST and GLT-1 in the midbrain were significantly attenuated by astrocytic YY1 deletion (Fig. 8, A–D).
Figure 8.
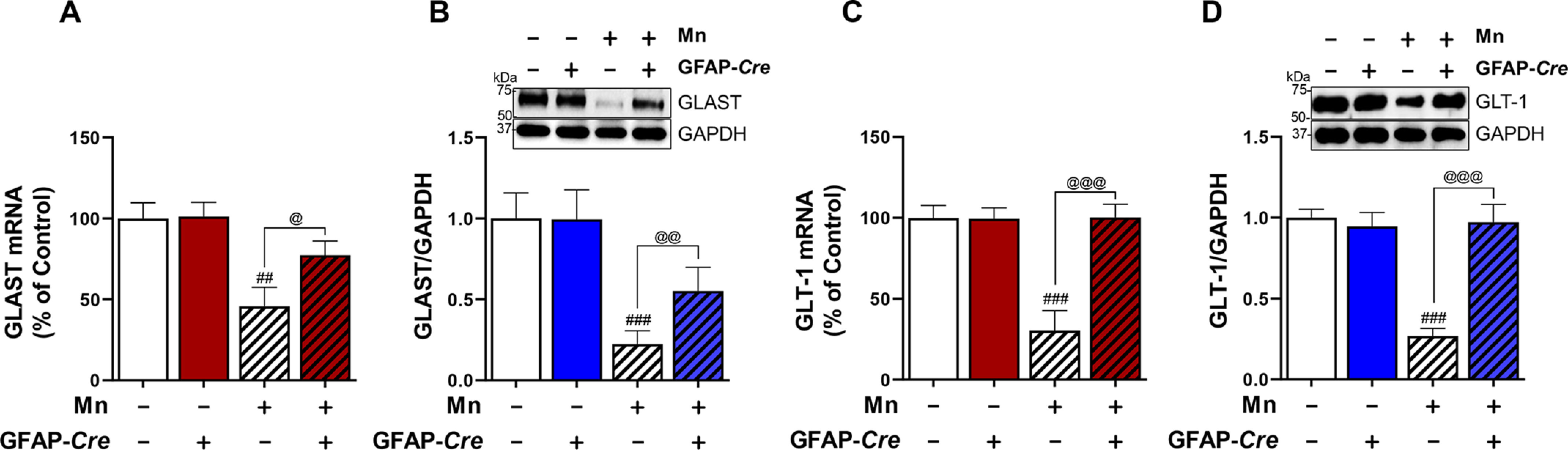
Astrocytic YY1 mediates Mn-induced down-regulation of astrocytic glutamate transporters in the midbrain of mice. After Mn exposure (MnCl2, 30 mg/kg, intranasal instillation, daily for 3 weeks), midbrain tissues of mice were processed to measure mRNA and protein levels by qPCR and Western blotting, respectively, as described under “Experimental procedures.” A and B, Mn decreased mRNA and protein levels of GLAST, whereas astrocytic YY1 deletion attenuated these Mn effects on GLAST in the midbrain region. C and D, Mn decreased mRNA and protein levels of GLT-1, whereas astrocytic YY1 deletion attenuated these Mn effects on GLT-1 in the midbrain region. GAPDH was used as a loading control. ##, p < 0.01; ###, p < 0.001 compared with the controls. @, p < 0.05; @@, p < 0.01; @@@, p < 0.01 compared with each other (one-way ANOVA followed by Tukey's post hoc test; n = 3). Bar graphs are expressed as mean ± S.D.
We also examined if astrocytic YY1 deletion modulated Mn-induced microglial activation. The results showed that Mn significantly increased Iba1-positive cells in the SN of control mice (Fig. 9). However, Mn-enhanced Iba1-positive cells were significantly attenuated in YY1-deleted astrocytes of the SN (Fig. 9).
Figure 9.

Astrocytic YY1 deletion attenuates Mn-induced microglial activation in the SN of the mouse brain. After Mn exposure (MnCl2, 30 mg/kg, intranasal instillation, daily for 3 weeks), mice were perfused, and coronal sections of brain tissues were immunostained with Iba1 antibody as described under “Experimental procedures.” The expression of Iba1 protein was shown as red fluorescent signals in the SN of the mouse brain (×40 magnification). Mn increased Iba1 fluorescence intensity in the SN of the mouse brain, whereas astrocytic YY1 deletion attenuated these Mn effects. **, p < 0.05 compared with the controls. @, p < 0.05 compared with each other (one-way ANOVA followed by Tukey's post hoc test; n = 3). Data are expressed as mean ± S.D.
Discussion
Our findings demonstrate, for the first time, that astrocytic YY1 deletion in the SN of YY1-floxed mice by infusing the AAV5-GFAP-Cre particles attenuated Mn-induced impairment of locomotor activity and TH reduction in dopaminergic neurons in the nigrostriatal pathway. Astrocytic YY1 deletion also attenuated the Mn-induced decrease in GLAST and GLT-1 mRNA/protein levels in the midbrain, indicating that astrocytic YY1 plays a critical role in Mn-induced dopaminergic neurotoxicity. The attenuating effects by astrocytic YY1 deletion might be associated with the attenuation of dysregulation of astrocytic glutamate transporters, and consequent glutamate overstimulation in the synaptic clefts, which leads to excitotoxic neuronal injury.
The infusion of AAV particles into the SN region of YY1 loxP mouse brain could be advantageous over the whole body deletion of astrocytic YY1, an astrocyte-specific YY1 conditional knockout (YY1 cKO) mouse model because manipulating gene expression in a specific region would only affect the region of interest compared with the YY1 cKO mouse model, which deletes astrocytic YY1 in the entire CNS.
AAV5 serotype among many serotypes works effectively to express Cre, which deletes loxP-flanked sequences in astrocytes under control of the astrocytic GFAP promoter (Fig. 2), corroborating a previous report (50) that this serotype efficiently expressed transgene in astrocytes. Although gene delivery to the CNS has largely been focused on neurons, targeting astrocytes might offer added advantages over neurons. It has been shown that AAV-mediated gene delivery of glial cell line-derived neurotrophic factor into astrocytes exerted protective effects against dopaminergic neurotoxicity induced by MPTP and 6-OHDA (52). Because gene delivery of neurotrophic factors to neuronal cells can be distributed through long-distance neuronal projections (52), targeting astrocytes could exert the same protection in dopaminergic neurons without spreading gene expression in the regions where neurons innervate (52). We used the GFAP promoter in AAV vector transduction into astrocytes, as it has been widely used to express transduced genes in astrocytes (53). Because GFAP is known to be expressed in other cell types such as radial glial progenitor cells and ependymal cells during developmental stages, the GFAP promoter-driven AAV infusion into the adult brain can eliminate this potential problem of expressing transgenes in other cell types. Taken together, our results indicate that AAV gene delivery strategies appear to develop cell-, region- and gene-specific targeted treatments.
The Mn exposure paradigm used in the present study is highly relevant to studying Mn-induced neurotoxicity, as the motor deficits and neuropathology recapitulate many aspects of human manganism (59). The intranasal delivery of MnCl2 (800 μg) increased Mn levels in the rat brain as well as in the olfactory bulb and tubercle (60). In our study, intranasal instillation of Mn exposure (30 mg/kg of MnCl2 or 330 μg of Mn) for 3 weeks accumulated Mn in the mouse brain, particularly the basal ganglia including midbrain and striatum (Fig. 3), which are closely associated with Mn-induced motor deficits and neuropathology of manganism. This Mn exposure paradigm elicited dopaminergic neurotoxicity and movement impairment in mice (61). Moreover, this Mn exposure route increased Mn levels in olfactory bulbs, causing olfactory deficits (62). Mn exposure via this route can be transported directly into the brain through olfactory nerves without passing the blood-brain barrier, although some of it could reach the brain systemically (63, 64). Mn is known to accumulate in the SN of rats (65) and mice (66). Given that the SN is embedded in the midbrain, Mn can directly cause dopaminergic dysfunction. The highest increase of Mn levels in the midbrain supports the Mn's preferential neurotoxicity in the nigrostriatal pathway (57, 67). In addition to striatum and SN, Mn levels were significantly increased in the frontal cortex and cerebellum (Fig. 3), corroborating previous reports of Mn's distribution in the brain (67). The anatomical pathways connecting the olfactory bulb to various brain regions such as the cortex and basal ganglia (68) can explain increased Mn levels throughout the brain regions. Interestingly, hippocampal Mn levels were not significantly increased in our experimental paradigm of Mn exposure. Studies have shown that Mn accumulated more in the hippocampus of younger rats compared with older rats (69), indicating that distribution of Mn differs between mature (adult) and immature (neonatal) animals. Nevertheless, hippocampal Mn levels were significantly increased regardless of its route (70, 71). Thus, Mn accumulation in the hippocampus may be delayed compared with other regions depending on experimental conditions.
Our findings show that Mn accumulation in the mouse brain decreased levels of other divalent metals such as Fe, Cu, and Zn in several brain regions (Fig. 4). These divalent metals compete for the same transporters, including divalent metal transporter-1 (72), and therefore, Mn may inhibit the accumulation of these metals by inhibiting their transport. For example, Fe deficiency has been shown to increase brain Mn levels in humans and rodents (73, 74). Mn and Fe compete for transporters such as divalent metal transporter-1 and transferrin receptor for uptake (75). Mn accumulation in the brain has also been shown to decrease Cu levels, corroborating earlier studies (76, 77). On the other hand, studies showed that Mn increased or failed to change brain Cu levels (70, 78). Regarding Zn, because Mn is transported by the two ZIP proteins, ZIP8 and ZIP14 (79), Mn may compete with Zn for those transporters, resulting in inhibition of Zn uptake and accumulation in the mouse brain regions, such as midbrain, cortex, and cerebellum. Mn exposure also lowers Zn levels in the rat brain (67, 70, 80), which could lead to low activities of Zn-containing enzymes, such as acid phosphatase, alkaline phosphatase, and superoxide dismutase (80). These findings suggest that Mn can induce a deficiency of essential trace metals. Other studies reported that Mn increased brain Zn levels (75, 81) or had no effect on Zn levels in the rat brain (78). Interestingly, among Fe, Cu, and Zn, only Zn levels were decreased in the midbrain by our Mn exposure paradigm, suggesting further study is necessary to understand the differential effects of Mn exposure on the levels of other metals in the midbrain. This discrepancy could be due to different experimental conditions, models, and settings.
Impairment of the astrocytic GLAST and GLT-1 might be closely associated with Mn-induced dopaminergic neurotoxicity. Astrocytic glutamate transporters GLT-1 and GLAST play a critical role in glutamate uptake into astrocytes and clearance to prevent excess glutamate accumulation in the synapse and subsequent excitotoxic neuronal death (82). Dysregulation of GLT-1/GLAST and aberrant glutamate neurotransmission contributes to the pathogenesis of various neurological disorders including Mn-induced neurotoxicity and PD (10, 24, 83, 84). We have previously reported that Mn decreased expression of GLT-1 and GLAST mRNA and protein levels in astrocytic cultures (15). It appears that impairments of glutamate transporters are also associated with many other neurotoxins causing PD pathogenesis, such as 6-OHDA and MPTP (85). 6-OHDA decreased striatal GLT-1/GLAST levels (84) and increased glutamate activation on NMDA receptors in the rat model (86). The NMDA receptor antagonist Ro 25-6981 attenuated behavioral deficits of 6-OHDA and MPTP-treated animals (87), supporting the role of dysregulation of glutamate neurotransmission in PD pathogenesis. GLT-1 up-regulation was critical for protective effects of ginsenoside Rb1 on dopaminergic cells against MPTP-induced injury in mice (88). There is also mounting evidence supporting the important role of glutamate transporters in the glutamatergic excitotoxicity seen in PD patients (89, 90). Moreover, higher astrocytic GLT-1 expression was correlated with the improved cognitive function in human subjects with AD pathogenesis (91), underscoring the role of GLT-1 in neurodegenerative diseases.
Mn activated the transcription factor YY1, leading to decreased GLT-1/GLAST mRNA/protein levels in astrocyte culture (21), indicating a potential role for YY1 in Mn-induced neurotoxicity. Accordingly, the results from the present study confirm that deletion of astrocytic YY1 attenuated Mn-induced reduction of GLAST/GLT-1 and dopaminergic neuronal toxicity in an in vivo mouse model, suggesting that YY1 could be a highly relevant molecular target to treat Mn neurotoxicity. In addition to astrocytic GLT-1/GLAST, YY1 may directly repress TH expression in dopaminergic neurons, but astrocytic YY1, possibly via repression of astrocytic GLT-1/GLAST, plays a central role in Mn-induced dopaminergic neurotoxicity. It has been reported that increased YY1 levels were correlated with reduced GLT-1/EAAT2 levels in an ALS animal model (44). YY1 is essential in oligodendrocytic functions such as myelination (92), indicating that YY1 plays various roles in different cell types.
YY1 is known to interact with epigenetic regulators such as co-activators (i.e. histone acetyltransferases) and co-repressors (i.e. HDAC) (93). HDAC1 as representative of class I as well as HDAC4 of class II interacted with YY1 in midbrain/SN (Fig. 5D), indicating that HDACs serve as co-repressors of YY1 in repressing target genes including GLAST and GLT-1. YY1-HDAC complex regulated Mn-induced GLT-1/EAAT2 and GLAST/EAAT1 repression (20, 21). These findings suggest that interaction of YY1 and HDACs play a critical role in Mn-reduced astrocytic GLT-1/GLAST expression in the mouse brain, corroborating our previous studies in astrocyte cultures (20, 21).
In addition to the dysregulation of astrocytic glutamate transporters, astrocytic YY1 is involved in Mn-induced microglial activation (Fig. 9) as astrocytic YY1 deletion attenuated Mn effects. YY1 increased production of cytokines in astrocytes (94), indicating that YY1 can possibly produce and release some harmful factors such as cytokines, which may lead to activation of microglia. This is supported by the studies that astrocyte-microglia cross-talk plays an essential role in inflammatory responses in Mn toxicity (12).
Taken together, our findings that astrocytic YY1 deletion in the SN region attenuated Mn-induced reduction of GLT-1/GLAST mRNA/protein levels, with concomitant protection of dopaminergic neurons against Mn toxicity, clearly demonstrate the role of astrocytic YY1 in Mn-induced repression of GLAST and GLT-1 and dopaminergic neurotoxicity (Fig. 10). Because impairment of astrocytic glutamate transporters is also associated with many other neurodegenerative diseases, such as AD and PD, identifying cellular/molecular targets, potentially astrocytic YY1, to attenuate or restore astrocytic GLAST and GLT-1 expression and thereby, preventing excitotoxic neuronal injury is highly demanding. Our findings from the present study open unique avenues to explore therapeutic interventions targeting YY1 to treat Mn neurotoxicity, as well as other neurodegenerative diseases associated with the dysfunction of astrocytic glutamate transporters.
Figure 10.
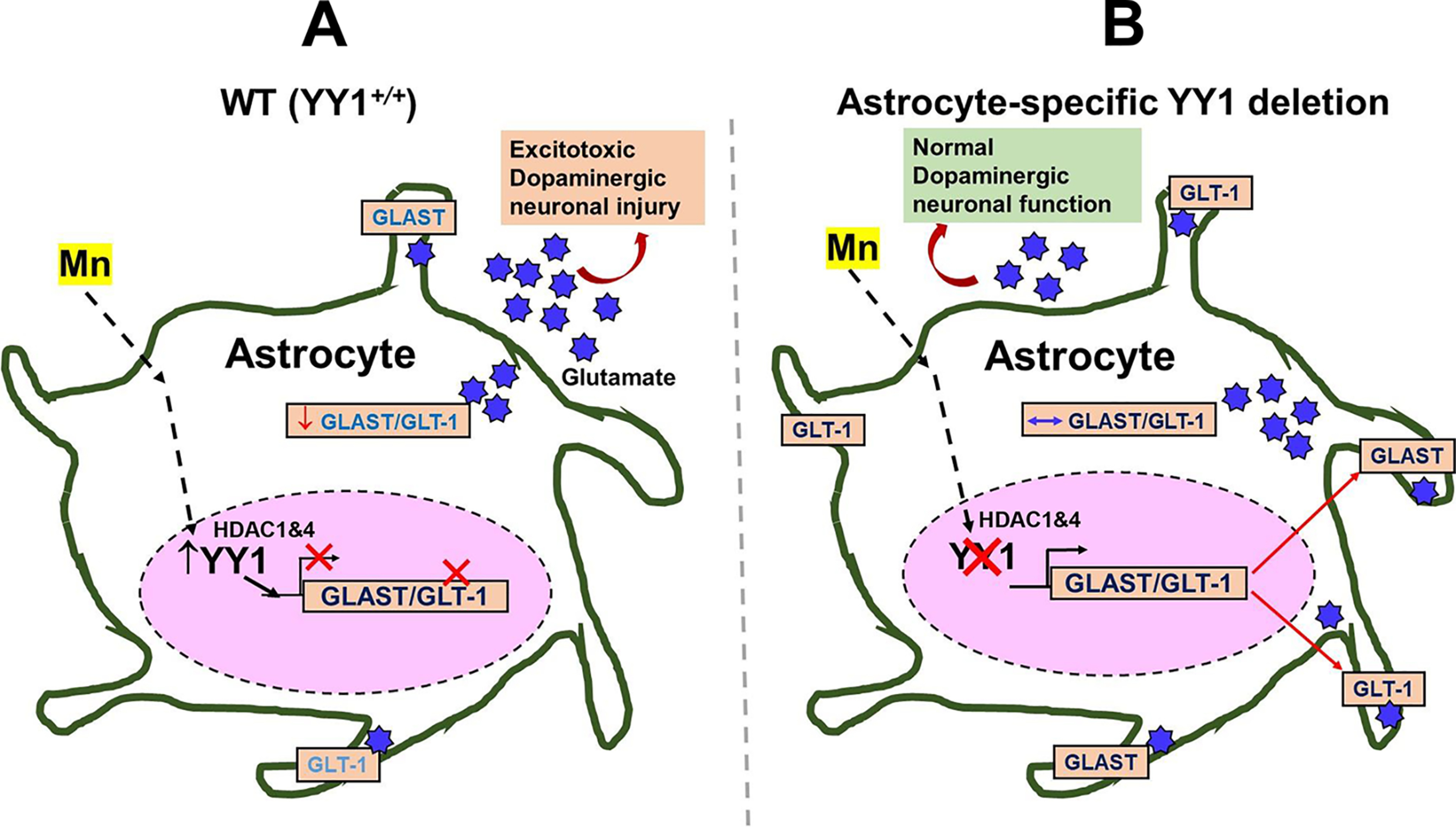
Schematic diagram of the proposed mechanism for YY1's role in Mn-induced GLAST and GLT-1 dysregulation in astrocytes. A, Mn-induced activation of YY1, which forms a repressor complex with HDACs, in astrocytes decreases levels of glutamate transporters GLAST/GLT-1 in the plasma membranes, resulting in extracellular glutamate accumulation leading to dopaminergic toxicity. B, the deletion of astrocytic YY1 attenuates the Mn-induced decrease in GLAST and GLT-1 by inhibiting YY1-HDAC activity. Because Mn-induced dysregulation of astrocytic glutamate transporters (GLAST and GLT-1) is closely associated with glutamate accumulation in the synaptic cleft, this mechanism could lead to Mn neurotoxicity in the SN region of the mouse brain, where dopaminergic cell bodies are located. The findings suggest that the deletion of astrocytic YY1 can be a critical target to attenuate Mn-induced dopaminergic toxicity.
Experimental procedures
Materials
Reagents were purchased from Invitrogen unless stated otherwise. Manganese chloride (MnCl2) was obtained from Millipore Sigma. Antibodies for Cre-recombinase, YY1, Iba1, Alexa Fluor 405, 488, and 568 were obtained from Abcam (Cambridge, MA). Antibodies for GLT-1, GLAST, YY1, TH, and GAPDH were from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies for HDAC1 and HDAC4 were from Cell Signaling Technology (Danvers, MA). AAV particles of AAV5-GFAP-GFP and AAV5-GFAP-Cre-GFP were obtained from the University of North Carolina (UNC) Vector Core (Chapel Hill, NC). The PLA kit was from Millipore Sigma.
Animal husbandry
Animal protocols and experiments were approved by the Florida A&M University Institutional Animal Care and Use Committee. Briefly, heterozygous YY1-loxP (YY1flox/WT) male and female mice (stock no. 014649) from the Jackson Laboratory (Bar Harbor, ME) were used to generate a homozygous YY1-loxP (YY1flox/flox) at the Florida A&M University Animal Care Facility. After generating homozygous YY1-loxP mice, animals were housed (3-5 mice/cage) and maintained on a 12-h light/dark cycle at 22 ± 2 °C with food, water, and enrichment available ad libitum.
Stereotaxic infusion of AAV particles and Mn exposure
Forty-eight YY1-loxP homozygous mice (8-week–old, body weight: 23–24 g) were randomly assigned into 4 groups. Mice anesthetized with ketamine/xylazine (100 mg/kg) were placed in a stereotaxic apparatus (12 animals/group; 6 male/6 female). One μl of viral vectors were infused into the SN of mouse brain using a 10-μl Hamilton syringe (Stoeltling Inc., Wood Dale, IL) mounted to a microinjection pump at a flow rate of 0.2 μl/min. Briefly, stereotaxic injection coordinates for SN to bregma were: x = ±1.2 mm (medial to lateral), y = −3.0 mm (anterior to posterior), and z = −4.3 mm (dorsal to ventral). The needle remained in place for 5 additional minutes before its slow withdrawal to prevent reflux. Skin incisions were closed with a suture, and the animal was kept warm with a heating pad before being returned to the cage.
Mice were separated into four groups 1) AAV5-GFAP-Cre-GFP plus vehicle, 2) AAV5-GFAP-Cre-GFP plus Mn, 3) AAV5-GFAP-GFP plus vehicle, and 4) AAV5-GFAP-GFP plus Mn; 12 mice/group; n = 48). Astrocytic YY1 was deleted in YY1-loxP mice injected with AAV5-GFAP-Cre-GFP. We used a protocol for Mn treatment described in our previous study with slight modifications (34). For intranasal instillation, treatment with either Mn or distilled water (ddH2O) as a vehicle was administered once daily for 21 consecutive days. For the Mn-treated groups, 2 μl of MnCl2 (30 mg/kg, 330 μg of Mn) via intranasal instillation was administered into the left or right nostril alternatively. Mice in the control vehicle groups received 2 μl of ddH2O. Animals were placed under isoflurane-induced anesthesia for 3 min during pre- and post-instillation periods to sedate and prevent expulsion of Mn or distilled water from the nostril. The experimental paradigm is summarized in Fig. 1.
Locomotor activity and motor coordination
Open-field test for locomotor activity and rotarod test for motor coordination were performed as described in our previous study (95). Briefly, 24 h after last Mn exposure, locomotor activity was assessed in seamless open field activity arenas using Fusion (version 6.25) SuperFlex software (Omnitech Electronics, Columbus, OH). Each open-field arena was made of clear Plexiglas, measuring 27.3 × 27.3 × 20.3 cm, and was covered with a Plexiglas lid containing air holes. Each animal went through an acclimation period for 3 consecutive days (3 days before the last Mn exposure). To measure locomotor activity, each mouse was placed in the center of the open-field for acclimation for 30 min and then, the activity of each mouse was recorded for 30 min. The mean distance traveled was used to assess locomotor activity.
For motor coordination, mice were trained for 3 consecutive days, with 1 session consisting of 3 trials separated by 300 s resting periods in the AccuRotor rotarod system (Omnitech Electronics). 3 weeks after AAV infusion for the effect of astrocytic YY1 deletion as well as after 21 days of Mn treatment for the effects of Mn exposure, motor coordination was assessed by placing animals on the rod and trials were started when rod begins to rotate with speed, gradually increasing from 4 to 40 rpm up to 10 min by 0.1 revolutions/s. The fall latency, a time for subjects to fall from the rotarod at various speeds was assessed with Fusion Software (version 6.3) for AccuRotor (Omnitech). If the mouse persisted on the rod for the entire duration, the measurement value was recorded for 650 s. The mean duration for each group was used for comparison.
Immunohistochemistry (IHC)
The IHC procedures were performed as described in our previous study with a slight modification (34). Briefly, mice were perfused with 4% paraformaldehyde in PBS buffer, and brains were isolated and placed in 4% paraformaldehyde in PBS overnight, then soaked in 30% sucrose solution for 48 h, followed by immediately freezing tissue samples in dry ice until use. Brain tissue was sliced for frozen coronal sections in 20 μm thickness using a Cryostat (Thermo Fisher) to obtain the SN regions, spanning from the bregma −2.70 to −3.40 mm. Tissue sections (3 mice/group) were prepared for IHC to assess TH, YY1, GFP, and Cre expression in the SN region of the mouse brain. Tissue sections were washed twice with PBST (1 × PBS, 0.3% Triton X-100), followed by incubation with blocking buffer (1 × PBS, 5% normal goat serum, 0.3% Triton X-100) for 1 h at room temperature. Then, tissue sections on the slides were incubated with the primary antibody solution in a dark humidity chamber at 4 °C overnight. Antibodies for TH (1:250 dilution), YY1 (1:1000 dilution), and Cre (1:250 dilution) were diluted in blocking buffer. After overnight incubation, slides were washed with washing buffer 3 times for 5 min in each washing, followed by incubation with secondary antibody solution conjugated with Alexa Fluor 405 (blue, DAPI), 488 (green, FITC), and 568 (red, TRITC) fluorescent dyes (1:1000 dilution) for 2 h at room temperature in the dark chamber. Tissue sections were washed twice with washing buffer, followed by placing the mounting media between the coverslip and specimen. Fluorescence intensity of TH, GFP, YY1, and Cre expressions was assessed in the SN regions with a Ts2R fluorescence microscope (Nikon Instruments, Melville, NY), and relative fluorescence intensity was determined by ImageJ software (National Institutes of Health, Bethesda, MD). Microglial activation in the SN was also assessed using Iba1 (1:250 dilution).
Quantitative real-time PCR (qPCR)
Tissue samples (3 samples/group) were extracted from midbrain regions of mice. Briefly, total RNA was extracted from mouse brain tissue using the RNeasy Mini Kit (Qiagen, Valencia, CA). Purified RNA was transcribed to cDNA with the high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA). For quantitative real-time PCR (qPCR), the following primers were used: for GLT-1, 5-GCC AAT ACA ACC AAG GCA GTC-3' (forward) and 5'-TTC ATC CCG TCC TTG AAC TC-3' (reverse); for GLAST, 5-GAT CGG AAA CAT GAA GGA GC-3' (forward) and 5-CAA GAA GAG GAT GCC CAG AG-3' (reverse); for TH, 5'-CAC TAT GCC CAC CCC CAG-3' (forward) and 5'-CGC CGT CCA ATG AAC CTT-3' (reverse); for YY1, 5'-CTC CTG CAG CCC TGG GCG CAT C-3' (forward) and 5'-GGT AAG CCC TTT AGC GCC TC-3′ (reverse); and for GAPDH, 5'-TCA ACG GGA AGC CCA TCA-3' (forward) and 5'-CTC GTG GTT CAC ACC CAT CA-3' (reverse). A total volume of 25 μl for qPCR contained 1 μl of cDNA template of each sample 0.4 μm of primers and SsoAdvanced™ Universal SYBR® Green SuperMix (Bio-Rad) for each reaction. qPCR was performed in CFX96 real-time detection system (Bio-Rad) with one cycle at 95 °C for 10 min and 40 cycles at 95 °C for 15 s and 60 °C for 1 min. GAPDH was used to normalize all samples. The Bio-Rad CFX Manager Software was used for data analysis.
Western blotting analysis
For protein analysis, tissues (3 mice/group) were harvested from midbrain or striatal regions and used for protein extraction and further analysis. Brain tissue samples were homogenized in a radioimmunoprecipitation assay (RIPA) buffer containing protease inhibitors, followed by bicinchoninic acid assay. Equal amounts of proteins were run on 10% SDS-PAGE (YY1, TH, GLAST, and GLT-1), followed by Western blotting analysis. Antibodies for GLT-1 and GLAST (1:5000 dilution) and YY1 and TH (1:1000 dilution) were used, followed by horseradish peroxidase-conjugated secondary antibody (1:5000 dilution). Blots were developed using a chemiluminescence detection kit (Pierce, Rockford, IL), followed by blot imaging and quantification with the Bio-Rad ChemiDoc Imaging System and Image Laboratory Software version 5.2.1 (Bio-Rad), respectively.
Co-immunoprecipitation (Co-IP)
For co-IP experiments, 500 μg of protein extracts from midbrain were incubated with 2.5 μg of indicated antibodies and rocked at 4 °C for 2 h, followed by incubation with 20 μl of protein A + G-agarose beads (Santa Cruz) overnight at 4 °C on a rotating device. After washing the beads 3 times with RIPA buffer, 40 μl of sample loading buffer was added and boiled at 95 °C for 3 min. After centrifugation, the eluted proteins in the supernatant were used for SDS-PAGE and Western blotting.
Proximity ligation assays
Proximity ligation was performed in 4% paraformaldehyde-fixed tissues according to the manufacturer's protocols. Briefly, SN coronal tissue sections were incubated with primary antibodies for YY1 and HDAC1 at 4 °C overnight. Secondary antibodies conjugated with oligonucleotides were applied and incubated at 37 °C for 1 h. Ligation solution, consisting of two oligonucleotides and ligase, was added and incubated at 37 °C for 30 min to hybridize oligonucleotides with two proximity ligation probes, resulting in closed loops if the two proteins are in close proximity. Amplification solution, consisting of nucleotides and fluorescently labeled oligonucleotides, was added together with polymerase and incubated at 37 °C for 100 min. Fluorescent-labeled oligonucleotides were hybridized to the amplified products. The protein–protein interactions were visible as distinct red fluorescent signals, which were analyzed by fluorescence microscopy.
ICP-MS analysis
YY1-loxP homozygous mice (8-week–old mice, body weight: 23–24 g, n = 20) were randomly separated into 2 groups: 1) control and 2) Mn (10 mice/group). After Mn treatment (30 mg/kg MnCl2, intranasal instillation, both sides alternatively, daily for 3 weeks), mice were euthanized, and brain tissues were immediately removed and dissected. Tissue samples from different brain regions including the frontal cortex, hippocampus, striatum, midbrain, and cerebellum were placed in microcentrifuge tubes and stored in −80 °C until analysis. Tissue samples (n = 10/group) from control and Mn-treated groups were prepared using the microwave-assisted acid digestion method. Briefly, samples were digested in a solution containing 100 μg/liter of germanium (Ge) in 1% nitric acid and 0.5 ml of H2O2 for 30 min at 220 °C in a microwave system, followed by cooling down of the mixture to room temperature. After complete digestion, samples were diluted with water. Metal concentrations of samples including Mn, iron (Fe), copper (Cu), and zinc (Zn) were determined by the Agilent 8800 triple quadrupole ICP-MS (Agilent Technologies, Waldbronn, Germany) and calculated as mg/kg of tissue.
Statistical analysis
Data were expressed as a mean ± S.D. Data analyses were carried out with GraphPad software (GraphPad, San Diego, CA). Statistical differences between two groups were determined by Student's t test, and comparisons between multiple groups were carried out by one-way analysis of variance (ANOVA) followed by Tukey's post hoc test. Statistical significance was set at p < 0.05.
Data availability
All data are available within the manuscript.
Author contributions—E. P. data curation; E. P. software; E. P., J. J., and M. S. formal analysis; E. P., D-S. S., and M. A. validation; E. P. and J. J. visualization; E. P., J. J., A. R., I. N-D., G. A., J. B., M. S., T. S., D-S. S., and M. A. methodology; E. P. writing-original draft; E. L. conceptualization; E. L. resources; E. L. supervision; E. L. funding acquisition; E. L. investigation; E. L. project administration.
Funding and additional information—This work was supported by NIEHS National Institutes of Health Grants R01 ES024756 (to E. L.), U54 MD007582 (to E. L.), SC1 CA200519 (to D. S.), R01 ES10563 (to M. A.), R01 ES07331 (to M. A.), and R01 ES020852 (to M. A.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article..
- PD
- Parkinson's disease
- NMDA
- N-methyl-d-aspartate
- GLAST
- glutamate aspartate transporter 1
- GLT-1
- glutamate transporter 1
- EAAT
- excitatory amino acid transporters
- AD
- Alzheimer's disease
- MPTP
- 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- 6-OHDA
- 6-hydroxydopamine
- HDAC
- histone deacetylase
- YY1
- Yin Yang 1
- AAV
- adeno-associated viral
- CNS
- central nervous system
- GFAP
- glial fibrillary acidic protein
- SN
- substantia nigra
- Cre
- Cre recombinase
- ICP
- inductively coupled plasma
- PLA
- proximity ligation assay
- TH
- tyrosine hydroxylase
- cKO
- conditional knockout
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- IHC
- immunohistochemistry
- DAPI
- 4′,6-diamidino-2-phenylindole
- TRITC
- tetramethylrhodamine isothiocyanate
- qPCR
- quantitative PCR
- co-IP
- co-immunoprecipitation
- ANOVA
- analysis of variance.
References
- 1. Keen C. L., Ensunsa J. L., Watson M. H., Baly D. L., Donovan S. M., Monaco M. H., and Clegg M. S. (1999) Nutritional aspects of manganese from experimental studies. Neurotoxicology 20, 213–223 [PubMed] [Google Scholar]
- 2. Kwakye G. F., Paoliello M. M., Mukhopadhyay S., Bowman A. B., and Aschner M. (2015) Manganese-induced Parkinsonism and Parkinson's disease: shared and distinguishable features. Int. J. Environ. Res. Public Health 12, 7519–7540 10.3390/ijerph120707519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bowler R. M., Kornblith E. S., Gocheva V. V., Colledge M. A., Bollweg G., Kim Y., Beseler C. L., Wright C. W., Adams S. W., and Lobdell D. T. (2015) Environmental exposure to manganese in air: Associations with cognitive functions. Neurotoxicology 49, 139–148 10.1016/j.neuro.2015.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dlamini W. W., Nelson G., Nielsen S. S., and Racette B. A. (2020) Manganese exposure, parkinsonian signs, and quality of life in South African mine workers. Am. J. Ind. Med. 63, 36–43 10.1002/ajim.23060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kullar S. S., Shao K., Surette C., Foucher D., Mergler D., Cormier P., Bellinger D. C., Barbeau B., Sauvé S., and Bouchard M. F. (2019) A benchmark concentration analysis for manganese in drinking water and IQ deficits in children. Environ. Int. 130, 104889 10.1016/j.envint.2019.05.083 [DOI] [PubMed] [Google Scholar]
- 6. Klos K. J., Chandler M., Kumar N., Ahlskog J. E., and Josephs K. A. (2006) Neuropsychological profiles of manganese neurotoxicity. Eur. J. Neurol. 13, 1139–1141 10.1111/j.1468-1331.2006.01407.x [DOI] [PubMed] [Google Scholar]
- 7. Song Q., Deng Y., Yang X., Bai Y., Xu B., Liu W., Zheng W., Wang C., Zhang M., and Xu Z. (2016) Manganese-disrupted interaction of dopamine D1 and NMDAR in the striatum to injury learning and memory ability of mice. Mol. Neurobiol. 53, 6745–6758 10.1007/s12035-015-9602-7 [DOI] [PubMed] [Google Scholar]
- 8. Peres T. V., Eyng H., Lopes S. C., Colle D., Gonçalves F. M., Venske D. K., Lopes M. W., Ben J., Bornhorst J., Schwerdtle T., Aschner M., Farina M., Prediger R. D., and Leal R. B. (2015) Developmental exposure to manganese induces lasting motor and cognitive impairment in rats. Neurotoxicology 50, 28–37 10.1016/j.neuro.2015.07.005 [DOI] [PubMed] [Google Scholar]
- 9. Guilarte T. R., and Chen M. K. (2007) Manganese inhibits NMDA receptor channel function: implications to psychiatric and cognitive effects. Neurotoxicology 28, 1147–1152 10.1016/j.neuro.2007.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kim J., Pajarillo E., Rizor A., Son D. S., Lee J., Aschner M., and Lee E. (2019) LRRK2 kinase plays a critical role in manganese-induced inflammation and apoptosis in microglia. PLoS ONE 14, e0210248 10.1371/journal.pone.0210248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sarkar S., Malovic E., Harischandra D. S., Ngwa H. A., Ghosh A., Hogan C., Rokad D., Zenitsky G., Jin H., Anantharam V., Kanthasamy A. G., and Kanthasamy A. (2018) Manganese exposure induces neuroinflammation by impairing mitochondrial dynamics in astrocytes. Neurotoxicology 64, 204–218 10.1016/j.neuro.2017.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tjalkens R. B., Popichak K. A., and Kirkley K. A. (2017) Inflammatory activation of microglia and astrocytes in manganese neurotoxicity. Adv. Neurobiol. 18, 159–181 10.1007/978-3-319-60189-2_8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Alaimo A., Gorojod R. M., and Kotler M. L. (2011) The extrinsic and intrinsic apoptotic pathways are involved in manganese toxicity in rat astrocytoma C6 cells. Neurochem. Int. 59, 297–308 10.1016/j.neuint.2011.06.001 [DOI] [PubMed] [Google Scholar]
- 14. Brouillet E. P., Shinobu L., McGarvey U., Hochberg F., and Beal M. F. (1993) Manganese injection into the rat striatum produces excitotoxic lesions by impairing energy metabolism. Exp. Neurol. 120, 89–94 10.1006/exnr.1993.1042 [DOI] [PubMed] [Google Scholar]
- 15. Lee E. S., Sidoryk M., Jiang H., Yin Z., and Aschner M. (2009) Estrogen and tamoxifen reverse manganese-induced glutamate transporter impairment in astrocytes. J. Neurochem. 110, 530–544 10.1111/j.1471-4159.2009.06105.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ke T., Sidoryk-Wegrzynowicz M., Pajarillo E., Rizor A., Soares F. A. A., Lee E., and Aschner M. (2019) Role of astrocytes in manganese neurotoxicity revisited. Neurochem. Res. 44, 2449–2459 10.1007/s11064-019-02881-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pines G., Danbolt N. C., Bjørås M., Zhang Y., Bendahan A., Eide L., Koepsell H., Storm-Mathisen J., Seeberg E., and Kanner B. I. (1992) Cloning and expression of a rat brain L-glutamate transporter. Nature 360, 464–467 10.1038/360464a0 [DOI] [PubMed] [Google Scholar]
- 18. Storck T., Schulte S., Hofmann K., and Stoffel W. (1992) Structure, expression, and functional analysis of a Na+-dependent glutamate/aspartate transporter from rat brain. Proc. Natl. Acad. Sci. U.S.A. 89, 10955–10959 10.1073/pnas.89.22.10955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tanaka K. (1993) Expression cloning of a rat glutamate transporter. Neurosci. Res. 16, 149–153 10.1016/0168-0102(93)90082-2 [DOI] [PubMed] [Google Scholar]
- 20. Karki P., Kim C., Smith K., Son D. S., Aschner M., and Lee E. (2015) Transcriptional regulation of the astrocytic excitatory amino acid transporter 1 (EAAT1) via NF-κB and yin yang 1 (YY1). J. Biol. Chem. 290, 23725–23737 10.1074/jbc.M115.649327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Karki P., Webb A., Smith K., Johnson J. Jr., Lee K., Son D. S., Aschner M., and Lee E. (2014) Yin Yang 1 is a repressor of glutamate transporter EAAT2, and it mediates manganese-induced decrease of EAAT2 expression in astrocytes. Mol. Cell Biol. 34, 1280–1289 10.1128/MCB.01176-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Danbolt N. C., Storm-Mathisen J., and Kanner B. I. (1992) An [Na+ + K+]coupled l-glutamate transporter purified from rat brain is located in glial cell processes. Neuroscience 51, 295–310 10.1016/0306-4522(92)90316-T [DOI] [PubMed] [Google Scholar]
- 23. Haugeto O., Ullensvang K., Levy L. M., Chaudhry F. A., Honoré T., Nielsen M., Lehre K. P., and Danbolt N. C. (1996) Brain glutamate transporter proteins form homomultimers. J. Biol. Chem. 271, 27715–27722 10.1074/jbc.271.44.27715 [DOI] [PubMed] [Google Scholar]
- 24. Jacob C. P., Koutsilieri E., Bartl J., Neuen-Jacob E., Arzberger T., Zander N., Ravid R., Roggendorf W., Riederer P., and Grünblatt E. (2007) Alterations in expression of glutamatergic transporters and receptors in sporadic Alzheimer's disease. J. Alzheimers Dis. 11, 97–116 10.3233/jad-2007-11113 [DOI] [PubMed] [Google Scholar]
- 25. Ferrarese C., Zoia C., Pecora N., Piolti R., Frigo M., Bianchi G., Sala G., Begni B., Riva R., and Frattola L. (1999) Reduced platelet glutamate uptake in Parkinson's disease. J. Neural Transm. 106, 685–692 10.1007/s007020050189 [DOI] [PubMed] [Google Scholar]
- 26. Rothstein J. D., Van Kammen M., Levey A. I., Martin L. J., and Kuncl R. W. (1995) Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann. Neurol. 38, 73–84 10.1002/ana.410380114 [DOI] [PubMed] [Google Scholar]
- 27. Hsu C. Y., Hung C. S., Chang H. M., Liao W. C., Ho S. C., and Ho Y. J. (2015) Ceftriaxone prevents and reverses behavioral and neuronal deficits in an MPTP-induced animal model of Parkinson's disease dementia. Neuropharmacology 91, 43–56 10.1016/j.neuropharm.2014.11.023 [DOI] [PubMed] [Google Scholar]
- 28. Chotibut T., Davis R. W., Arnold J. C., Frenchek Z., Gurwara S., Bondada V., Geddes J. W., and Salvatore M. F. (2014) Ceftriaxone increases glutamate uptake and reduces striatal tyrosine hydroxylase loss in 6-OHDA Parkinson's model. Mol. Neurobiol. 49, 1282–1292 10.1007/s12035-013-8598-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Leung T. C., Lui C. N., Chen L. W., Yung W. H., Chan Y. S., and Yung K. K. (2012) Ceftriaxone ameliorates motor deficits and protects dopaminergic neurons in 6-hydroxydopamine-lesioned rats. ACS Chem. Neurosci. 3, 22–30 10.1021/cn200072h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Verma R., Mishra V., Sasmal D., and Raghubir R. (2010) Pharmacological evaluation of glutamate transporter 1 (GLT-1) mediated neuroprotection following cerebral ischemia/reperfusion injury. Eur. J. Pharmacol. 638, 65–71 10.1016/j.ejphar.2010.04.021 [DOI] [PubMed] [Google Scholar]
- 31. Cui C., Cui Y., Gao J., Sun L., Wang Y., Wang K., Li R., Tian Y., Song S., and Cui J. (2014) Neuroprotective effect of ceftriaxone in a rat model of traumatic brain injury. Neurol. Sci. 35, 695–700 10.1007/s10072-013-1585-4 [DOI] [PubMed] [Google Scholar]
- 32. Fan S., Xian X., Li L., Yao X., Hu Y., Zhang M., and Li W. (2018) Ceftriaxone improves cognitive function and upregulates GLT-1-related glutamate-glutamine cycle in APP/PS1 mice. J. Alzheimers Dis. 66, 1731–1743 10.3233/JAD-180708 [DOI] [PubMed] [Google Scholar]
- 33. Karki P., Webb A., Smith K., Lee K., Son D. S., Aschner M., and Lee E. (2013) cAMP response element-binding protein (CREB) and nuclear factor κB mediate the tamoxifen-induced up-regulation of glutamate transporter 1 (GLT-1) in rat astrocytes. J. Biol. Chem. 288, 28975–28986 10.1074/jbc.M113.483826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pajarillo E., Johnson J. Jr., Kim J., Karki P., Son D. S., Aschner M., and Lee E. (2018) 17β-Estradiol and tamoxifen protect mice from manganese-induced dopaminergic neurotoxicity. Neurotoxicology 65, 280–288 10.1016/j.neuro.2017.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Karki P., Hong P., Johnson J. Jr., Pajarillo E., Son D. S., Aschner M., and Lee E. Y. (2018) Arundic acid increases expression and function of astrocytic glutamate transporter EAAT1 via the ERK, Akt, and NF-κB pathways. Mol. Neurobiol. 55, 5031–5046 10.1007/s12035-017-0709-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Takakura K., Kawamura A., Torisu Y., Koido S., Yahagi N., and Saruta M. (2019) The clinical potential of oligonucleotide therapeutics against pancreatic cancer. Int. J. Mol. Sci. 20, e3331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Galvin K. M., and Shi Y. (1997) Multiple mechanisms of transcriptional repression by YY1. Mol. Cell Biol. 17, 3723–3732 10.1128/mcb.17.7.3723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Aubry S., Shin W., Crary J. F., Lefort R., Qureshi Y. H., Lefebvre C., Califano A., and Shelanski M. L. (2015) Assembly and interrogation of Alzheimer's disease genetic networks reveal novel regulators of progression. PLoS ONE 10, e0120352 10.1371/journal.pone.0120352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bedrosian T. A., Quayle C., Novaresi N., and Gage F. H. (2018) Early life experience drives structural variation of neural genomes in mice. Science 359, 1395–1399 10.1126/science.aah3378 [DOI] [PubMed] [Google Scholar]
- 40. Forlani G., Giarda E., Ala U., Di Cunto F., Salani M., Tupler R., Kilstrup-Nielsen C., and Landsberger N. (2010) The MeCP2/YY1 interaction regulates ANT1 expression at 4q35: novel hints for Rett syndrome pathogenesis. Hum. Mol. Genet. 19, 3114–3123 10.1093/hmg/ddq214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nowak K., Lange-Dohna C., Zeitschel U., Gunther A., Luscher B., Robitzki A., Perez-Polo R., and Rossner S. (2006) The transcription factor Yin Yang 1 is an activator of BACE1 expression. J. Neurochem. 96, 1696–1707 10.1111/j.1471-4159.2006.03692.x [DOI] [PubMed] [Google Scholar]
- 42. Ratajewski M., and Pulaski L. (2009) YY1-dependent transcriptional regulation of the human GDAP1 gene. Genomics 94, 407–413 10.1016/j.ygeno.2009.08.014 [DOI] [PubMed] [Google Scholar]
- 43. Tiwari P. C., and Pal R. (2017) The potential role of neuroinflammation and transcription factors in Parkinson disease. Dialogues Clin. Neurosci. 19, 71–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yin X., Wang S., Qi Y., Wang X., Jiang H., Wang T., Yang Y., Wang Y., Zhang C., and Feng H. (2018) Astrocyte elevated gene-1 is a novel regulator of astrogliosis and excitatory amino acid transporter-2 via interplaying with nuclear factor-κB signaling in astrocytes from amyotrophic lateral sclerosis mouse model with hSOD1(G93A) mutation. Mol. Cell Neurosci. 90, 1–11 10.1016/j.mcn.2018.05.004 [DOI] [PubMed] [Google Scholar]
- 45. Rosas S., Vargas M. A., López-Bayghen E., and Ortega A. (2007) Glutamate-dependent transcriptional regulation of GLAST/EAAT1: a role for YY1. J. Neurochem. 101, 1134–1144 10.1111/j.1471-4159.2007.04517.x [DOI] [PubMed] [Google Scholar]
- 46. Aguirre G., Rosas S., López-Bayghen E., and Ortega A. (2008) Valproate-dependent transcriptional regulation of GLAST/EAAT1 expression: involvement of Ying-Yang 1. Neurochem. Int. 52, 1322–1331 10.1016/j.neuint.2008.01.015 [DOI] [PubMed] [Google Scholar]
- 47. Lee S. G., Kim K., Kegelman T. P., Dash R., Das S. K., Choi J. K., Emdad L., Howlett E. L., Jeon H. Y., Su Z. Z., Yoo B. K., Sarkar D., Kim S. H., Kang D. C., and Fisher P. B. (2011) Oncogene AEG-1 promotes glioma-induced neurodegeneration by increasing glutamate excitotoxicity. Cancer Res. 71, 6514–6523 10.1158/0008-5472.CAN-11-0782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hammond S. L., Leek A. N., Richman E. H., and Tjalkens R. B. (2017) Cellular selectivity of AAV serotypes for gene delivery in neurons and astrocytes by neonatal intracerebroventricular injection. PLoS ONE 12, e0188830 10.1371/journal.pone.0188830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Powell S. K., Khan N., Parker C. L., Samulski R. J., Matsushima G., Gray S. J., and McCown T. J. (2016) Characterization of a novel adeno-associated viral vector with preferential oligodendrocyte tropism. Gene Ther. 23, 807–814 10.1038/gt.2016.62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Griffin J. M., Fackelmeier B., Fong D. M., Mouravlev A., Young D., and O'Carroll S. J. (2019) Astrocyte-selective AAV gene therapy through the endogenous GFAP promoter results in robust transduction in the rat spinal cord following injury. Gene Ther. 26, 198–210 10.1038/s41434-019-0075-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kempermann G. (2011) Seven principles in the regulation of adult neurogenesis. Eur. J. Neurosci. 33, 1018–1024 10.1111/j.1460-9568.2011.07599.x [DOI] [PubMed] [Google Scholar]
- 52. Drinkut A., Tereshchenko Y., Schulz J. B., Bähr M., and Kügler S. (2012) Efficient gene therapy for Parkinson's disease using astrocytes as hosts for localized neurotrophic factor delivery. Mol. Ther. 20, 534–543 10.1038/mt.2011.249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chow L. M., Zhang J., and Baker S. J. (2008) Inducible Cre recombinase activity in mouse mature astrocytes and adult neural precursor cells. Transgenic Res. 17, 919–928 10.1007/s11248-008-9185-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Albert K., Voutilainen M. H., Domanskyi A., Piepponen T. P., Ahola S., Tuominen R. K., Richie C., Harvey B. K., and Airavaara M. (2019) Downregulation of tyrosine hydroxylase phenotype after AAV injection above substantia nigra: caution in experimental models of Parkinson's disease. J Neurosci. Res. 97, 346–361 10.1002/jnr.24363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Affar el B., Gay F., Shi Y., Liu H., Huarte M., Wu S., Collins T., Li E., and Shi Y. (2006) Essential dosage-dependent functions of the transcription factor yin yang 1 in late embryonic development and cell cycle progression. Mol. Cell Biol. 26, 3565–3581 10.1128/MCB.26.9.3565-3581.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Guilarte T. R., Chen M. K., McGlothan J. L., Verina T., Wong D. F., Zhou Y., Alexander M., Rohde C. A., Syversen T., Decamp E., Koser A. J., Fritz S., Gonczi H., Anderson D. W., and Schneider J. S. (2006) Nigrostriatal dopamine system dysfunction and subtle motor deficits in manganese-exposed non-human primates. Exp. Neurol. 202, 381–390 10.1016/j.expneurol.2006.06.015 [DOI] [PubMed] [Google Scholar]
- 57. Ordoñez-Librado J. L., Anaya-Martínez V., Gutierrez-Valdez A. L., Colín-Barenque L., Montiel-Flores E., and Avila-Costa M. R. (2010) Manganese inhalation as a Parkinson disease model. Parkinsons Dis. 2011, 612989 10.4061/2011/612989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Iancu R., Mohapel P., Brundin P., and Paul G. (2005) Behavioral characterization of a unilateral 6-OHDA-lesion model of Parkinson's disease in mice. Behav. Brain Res. 162, 1–10 10.1016/j.bbr.2005.02.023 [DOI] [PubMed] [Google Scholar]
- 59. Lee J. W. (2000) Manganese intoxication. Arch. Neurol. 57, 597–599 10.1001/archneur.57.4.597 [DOI] [PubMed] [Google Scholar]
- 60. Gianutsos G., Morrow G. R., and Morris J. B. (1997) Accumulation of manganese in rat brain following intranasal administration. Fundam. Appl. Toxicol. 37, 102–105 10.1093/toxsci/37.2.102 [DOI] [PubMed] [Google Scholar]
- 61. Johnson J. Jr., Pajarillo E., Karki P., Kim J., Son D. S., Aschner M., and Lee E. (2018) Valproic acid attenuates manganese-induced reduction in expression of GLT-1 and GLAST with concomitant changes in murine dopaminergic neurotoxicity. Neurotoxicology 67, 112–120 10.1016/j.neuro.2018.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Foster M. L., Rao D. B., Francher T., Traver S., and Dorman D. C. (2018) Olfactory toxicity in rats following manganese chloride nasal instillation: a pilot study. Neurotoxicology 64, 284–290 10.1016/j.neuro.2017.09.004 [DOI] [PubMed] [Google Scholar]
- 63. Brenneman K. A., Wong B. A., Buccellato M. A., Costa E. R., Gross E. A., and Dorman D. C. (2000) Direct olfactory transport of inhaled manganese (54MnCl2) to the rat brain: toxicokinetic investigations in a unilateral nasal occlusion model. Toxicol. Appl. Pharmacol. 169, 238–248 10.1006/taap.2000.9073 [DOI] [PubMed] [Google Scholar]
- 64. Dorman D. C., Struve M. F., Wong B. A., Dye J. A., and Robertson I. D. (2006) Correlation of brain magnetic resonance imaging changes with pallidal manganese concentrations in rhesus monkeys following subchronic manganese inhalation. Toxicol. Sci. 92, 219–227 10.1093/toxsci/kfj209 [DOI] [PubMed] [Google Scholar]
- 65. Robison G., Sullivan B., Cannon J. R., and Pushkar Y. (2015) Identification of dopaminergic neurons of the substantia nigra pars compacta as a target of manganese accumulation. Metallomics 7, 748–755 10.1039/c5mt00023h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lin M., Colon-Perez L. M., Sambo D. O., Miller D. R., Lebowitz J. J., Jimenez-Rondan F., Cousins R. J., Horenstein N., Aydemir T. B., Febo M., and Khoshbouei H. (2020) Mechanism of manganese dysregulation of dopamine neuronal activity. J. Neurosci. 40, 5871–5891 10.1523/JNEUROSCI.2830-19.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Scheuhammer A. M., and Cherian M. G. (1981) The influence of manganese on the distribution of essential trace elements: I. regional distribution of Mn, Na, K, Mg, Zn, Fe, and Cu in rat brain after chronic Mn exposure. Toxicol. Appl. Pharmacol. 61, 227–233 10.1016/0041-008X(81)90412-9 [DOI] [PubMed] [Google Scholar]
- 68. Palouzier-Paulignan B., Lacroix M. C., Aimé P., Baly C., Caillol M., Congar P., Julliard A. K., Tucker K., and Fadool D. A. (2012) Olfaction under metabolic influences. Chem. Senses 37, 769–797 10.1093/chemse/bjs059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Dorman D. C., Struve M. F., Vitarella D., Byerly F. L., Goetz J., and Miller R. (2000) Neurotoxicity of manganese chloride in neonatal and adult CD rats following subchronic (21-day) high-dose oral exposure. J. Appl. Toxicol. 20, 179–187 [DOI] [PubMed] [Google Scholar]
- 70. Lai J. C., Chan A. W., Leung T. K., Minski M. J., and Lim L. (1992) Neurochemical changes in rats chronically treated with a high concentration of manganese chloride. Neurochem. Res. 17, 841–847 10.1007/BF00993259 [DOI] [PubMed] [Google Scholar]
- 71. Lai J. C., Leung T. K., Lim L., Chan A. W., and Minski M. J. (1991) Effects of chronic manganese treatment on rat brain regional sodium-potassium-activated and magnesium-activated adenosine triphosphatase activities during development. Metab. Brain Dis. 6, 165–174 10.1007/BF00996908 [DOI] [PubMed] [Google Scholar]
- 72. Garrick M. D., Dolan K. G., Horbinski C., Ghio A. J., Higgins D., Porubcin M., Moore E. G., Hainsworth L. N., Umbreit J. N., Conrad M. E., Feng L., Lis A., Roth J. A., Singleton S., and Garrick L. M. (2003) DMT1: a mammalian transporter for multiple metals. Biometals 16, 41–54 10.1023/A:1020702213099 [DOI] [PubMed] [Google Scholar]
- 73. Erikson K. M., Syversen T., Steinnes E., and Aschner M. (2004) Globus pallidus: a target brain region for divalent metal accumulation associated with dietary iron deficiency. J. Nutr. Biochem. 15, 335–341 10.1016/j.jnutbio.2003.12.006 [DOI] [PubMed] [Google Scholar]
- 74. Garcia S. J., Gellein K., Syversen T., and Aschner M. (2007) Iron deficient and manganese supplemented diets alter metals and transporters in the developing rat brain. Toxicol. Sci. 95, 205–214 10.1093/toxsci/kfl139 [DOI] [PubMed] [Google Scholar]
- 75. Garcia S. J., Gellein K., Syversen T., and Aschner M. (2006) A manganese-enhanced diet alters brain metals and transporters in the developing rat. Toxicol. Sci. 92, 516–525 10.1093/toxsci/kfl017 [DOI] [PubMed] [Google Scholar]
- 76. Fu S., O'Neal S., Hong L., Jiang W., and Zheng W. (2015) Elevated adult neurogenesis in brain subventricular zone following in vivo manganese exposure: roles of copper and DMT1. Toxicol. Sci. 143, 482–498 10.1093/toxsci/kfu249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Abdel-Magied N., Abdel-Aziz N., Shedid S. M., and Ahmed A. G. (2019) Modulating effect of tiron on the capability of mitochondrial oxidative phosphorylation in the brain of rats exposed to radiation or manganese toxicity. Environ. Sci. Pollut. Res. Int. 26, 12550–12562 10.1007/s11356-019-04594-4 [DOI] [PubMed] [Google Scholar]
- 78. Reaney S. H., Bench G., and Smith D. R. (2006) Brain accumulation and toxicity of Mn(II) and Mn(III) exposures. Toxicol. Sci. 93, 114–124 10.1093/toxsci/kfl028 [DOI] [PubMed] [Google Scholar]
- 79. Chen P., Chakraborty S., Mukhopadhyay S., Lee E., Paoliello M. M., Bowman A. B., and Aschner M. (2015) Manganese homeostasis in the nervous system. J. Neurochem. 134, 601–610 10.1111/jnc.13170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Singh J., Husain R., Tandon S. K., Seth P. K., and Chandra S. V. (1974) Biochemical and histopathological alterations in early manganese toxicity in rats. Environ. Physiol. Biochem. 4, 16–23 [PubMed] [Google Scholar]
- 81. Chen M. T., Yiin S. J., Sheu J. Y., and Huang Y. L. (2002) Brain lipid peroxidation and changes of trace metals in rats following chronic manganese chloride exposure. J. Toxicol. Environ. Health A 65, 305–316 10.1080/15287390252800882 [DOI] [PubMed] [Google Scholar]
- 82. Lin C. L., Kong Q., Cuny G. D., and Glicksman M. A. (2012) Glutamate transporter EAAT2: a new target for the treatment of neurodegenerative diseases. Future Med. Chem. 4, 1689–1700 10.4155/fmc.12.122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Li S., Mallory M., Alford M., Tanaka S., and Masliah E. (1997) Glutamate transporter alterations in Alzheimer disease are possibly associated with abnormal APP expression. J. Neuropathol. Exp. Neurol. 56, 901–911 10.1097/00005072-199708000-00008 [DOI] [PubMed] [Google Scholar]
- 84. Chung E. K., Chen L. W., Chan Y. S., and Yung K. K. (2008) Downregulation of glial glutamate transporters after dopamine denervation in the striatum of 6-hydroxydopamine-lesioned rats. J. Comp. Neurol. 511, 421–437 10.1002/cne.21852 [DOI] [PubMed] [Google Scholar]
- 85. Holmer H. K., Keyghobadi M., Moore C., and Meshul C. K. (2005) l-DOPA-induced reversal in striatal glutamate following partial depletion of nigrostriatal dopamine with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Neuroscience 136, 333–341 10.1016/j.neuroscience.2005.08.003 [DOI] [PubMed] [Google Scholar]
- 86. Nandhu M. S., Paul J., Kuruvila K. P., Abraham P. M., Antony S., and Paulose C. S. (2011) Glutamate and NMDA receptors activation leads to cerebellar dysfunction and impaired motor coordination in unilateral 6-hydroxydopamine lesioned Parkinson's rat: functional recovery with bone marrow cells, serotonin and GABA. Mol. Cell Biochem. 353, 47–57 10.1007/s11010-011-0773-x [DOI] [PubMed] [Google Scholar]
- 87. Löschmann P. A., De Groote C., Smith L., Wullner U., Fischer G., Kemp J. A., Jenner P., and Klockgether T. (2004) Antiparkinsonian activity of Ro 25-6981, a NR2B subunit specific NMDA receptor antagonist, in animal models of Parkinson's disease. Exp. Neurol. 187, 86–93 10.1016/j.expneurol.2004.01.018 [DOI] [PubMed] [Google Scholar]
- 88. Qu S., Meng X., Liu Y., Zhang X., and Zhang Y. (2019) Ginsenoside Rb1 prevents MPTP-induced changes in hippocampal memory via regulation of the alpha-synuclein/PSD-95 pathway. Aging (Albany NY) 11, 1934–1964 10.18632/aging.101884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Ambrosi G., Cerri S., and Blandini F. (2014) A further update on the role of excitotoxicity in the pathogenesis of Parkinson's disease. J. Neural Transm. 121, 849–859 10.1007/s00702-013-1149-z [DOI] [PubMed] [Google Scholar]
- 90. Mironova Y. S., Zhukova I. A., Zhukova N. G., Alifirova V. M., Izhboldina O. P., and Latypova A. V. (2018) Parkinson's disease and glutamate excitotoxicity. Zh. Nevrol. Psikhiatr. Im. S S Korsakova 118, 50–54 10.17116/jnevro201811806250 [DOI] [PubMed] [Google Scholar]
- 91. Kobayashi E., Nakano M., Kubota K., Himuro N., Mizoguchi S., Chikenji T., Otani M., Mizue Y., Nagaishi K., and Fujimiya M. (2018) Activated forms of astrocytes with higher GLT-1 expression are associated with cognitive normal subjects with Alzheimer pathology in human brain. Sci. Rep. 8, 1712 10.1038/s41598-018-19442-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. He Y., Dupree J., Wang J., Sandoval J., Li J., Liu H., Shi Y., Nave K. A., and Casaccia-Bonnefil P. (2007) The transcription factor Yin Yang 1 is essential for oligodendrocyte progenitor differentiation. Neuron 55, 217–230 10.1016/j.neuron.2007.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Yao Y. L., Yang W. M., and Seto E. (2001) Regulation of transcription factor YY1 by acetylation and deacetylation. Mol. Cell Biol. 21, 5979–5991 10.1128/mcb.21.17.5979-5991.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Waters M. R., Gupta A. S., Mockenhaupt K., Brown L. N., Biswas D. D., and Kordula T. (2019) RelB acts as a molecular switch driving chronic inflammation in glioblastoma multiforme. Oncogenesis 8, 37 10.1038/s41389-019-0146-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Johnson J. Jr., Pajarillo E. A. B., Taka E., Reams R., Son D. S., Aschner M., and Lee E. (2018) Valproate and sodium butyrate attenuate manganese-decreased locomotor activity and astrocytic glutamate transporters expression in mice. Neurotoxicology 64, 230–239 10.1016/j.neuro.2017.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data are available within the manuscript.