

Abstract
Endocannabinoid signaling plays a regulatory role in various (neuro)biological functions. 2-arachidonoylglycerol (2-AG) is the most abundant endocannabinoid, and although its canonical biosynthetic pathway involving phosphoinositide-specific phospholipase C and diacylglycerol lipase α is known, alternative pathways remain unsettled. Here, we characterize a noncanonical pathway implicating glycerophosphodiesterase 3 (GDE3, from GDPD2 gene). Human GDE3 expressed in HEK293T cell membranes catalyzed the conversion of lysophosphatidylinositol (LPI) into monoacylglycerol and inositol-1-phosphate. The enzyme was equally active against 1-acyl and 2-acyl LPI. When using 2-acyl LPI, where arachidonic acid is the predominant fatty acid, LC-MS analysis identified 2-AG as the main product of LPI hydrolysis by GDE3. Furthermore, inositol-1-phosphate release into the medium occurred upon addition of LPI to intact cells, suggesting that GDE3 is actually an ecto-lysophospholipase C. In cells expressing G-protein–coupled receptor GPR55, GDE3 abolished 1-acyl LPI–induced signaling. In contrast, upon simultaneous ex-pression of GDE3 and cannabinoid receptor CB2, 2-acyl LPI evoked the same signal as that induced by 2-AG. These data strongly suggest that, in addition to degrading the GPR55 LPI ligand, GDE3 can act as a switch between GPR55 and CB2 signaling. Coincident with a major expression of both GDE3 and CB2 in the spleen, spleens from transgenic mice lacking GDE3 displayed doubling of LPI content compared with WT mice. Decreased production of 2-AG in whole spleen was also observed, supporting the in vivo relevance of our findings. These data thus open a new research avenue in the field of endocannabinoid generation and reinforce the view of GPR55 and LPI being genuine actors of the endocannabinoid system.
Keywords: endocannabinoid, lysophosphatidylinositol, 2-arachidonoylglycerol, monoacylglycerol, GPR55, CB1, CB2, lysophospholipid, phospholipase C, G-protein–coupled receptor (GPCR), CB1/CB2, glycerophosphodiesterase 3 (GDE3, GDPD2)
The endocannabinoid system includes two G-protein–coupled receptors (CB1 and CB2), their endogenous endocannabinoid ligands (mainly N-arachidonoylethanolamine (anandamide) and 2-arachidonoylglycerol (2-AG)), and various enzymes and transporters involved in the metabolism of endocannabinoids (1–10). Anandamide and other N-acylethanolamines are synthesized through a complex network of somewhat redundant pathways and enzymes (4, 5, 11, 12). 2-AG is a key lipid mediator in a number of physiological and pathophysiological situations, including control of synaptic transmission, neurodegeneration, inflammation, and immunity (6–10). The canonical pathway of 2-AG biosynthesis involves a phosphoinositide-specific phospholipase Cβ coupled to the α-isoform of diacylglycerol lipase (6–8). However, other routes for 2-AG production have been suggested but still remain hypothetical (6–8).
Another G-protein–coupled receptor, GPR55, is alternatively considered as a third cannabinoid receptor, displaying low sequence homology with CB1 and CB2 but good affinity for various cannabinoids (13–16). Lysophosphatidylinositol (LPI) was identified as the endogenous agonist of GPR55 (17), the 2-arachidonoyl molecular species displaying the highest biological activity (18). Notably, one LPI analog, lysophosphatidyl-β-d-glucoside, appears as a still more potent agonist of GPR55 (19, 20). Although other targets might also be involved, interaction of LPI with GPR55 is generally considered as the main mechanism of LPI involvement in cancer, metabolism, inflammation, and obesity (13–16).
Glycerophosphodiesterase 3 (GDE3) belongs to a group of recently identified mammalian glycerophosphodiesterases (21, 22). It contains six potential transmembrane α-helices and exposes to the cell surface an enzymatic domain able to hydrolyze glycerophosphoinositol (GPIns) into glycerol and inositol-1-phosphate (Ins1P) (Fig. 1A) (23). Rather than water-soluble glycerophosphodiesters, some of these enzymes (GDE1, GDE4, and GDE7) can hydrolyze monoacyl or diacyl derivatives, allowing the production of lipid mediators such as lysophosphatidic acid and anandamide (24–28). We postulated that LPI instead of GPIns could be the natural substrate of GDE3 (Fig. 1B). If so, this LPI-specific phospholipase C (LPI-PLC) would allow breakdown of the main endogenous ligand of GPR55. With LPI bearing an arachidonoyl group at the sn-2 position, GDE3 would also provide an alternative route for 2-AG production (Fig. 1B). Using recombinant human GDE3 expressed in HEK293T cells and exogenous LPI at physiological concentrations, we bring evidence that this is actually the case. During the preparation of this article, Tsutsumi et al. (29) published similar data reporting the same in vitro enzymatic properties of GDE3. In the present study, we further show that coexpression of GDE3 with GPR55 abrogates interaction of LPI with GPR55, whereas at the vicinity of CB2, GDE3 allows LPI to evoke signaling events identical to those induced by 2-AG. Finally, invalidation of mouse GDE3 gene (Gdpd2) resulted in accumulation of LPI together with a reduction of 2-AG content in the spleen, i.e. the organ displaying one of the highest expression levels of GDE3. This will stimulate further studies aimed at defining the biological function of GDE3 within the endocannabinoid system.
Figure 1.

Structure of PI, LPI, GPIns, and their cleavage products. A, the predominant molecular species of PI is 1-stearoyl-2-arachidonoyl-PI. The corresponding LPI (1-stearoyl and 2-arachidonoyl) are formed upon PI hydrolysis by phospholipase A2 (PLA2) and PLA1, respectively. Further deacylation of each LPI by lysophospholipases (LysoPlases) leads to water-soluble sn-glycero-3-phosphoinositol (GPIns), which is cleaved into glycerol and inositol-1-phosphate (Ins-1P) by GDE3. B, according to our working hypothesis, hydrolysis of 2-arachidonoyl LPI by lysoPLC generates 2-AG and Ins-1P or cyclic inositol-1,2-phosphate (cIns-1,2P). The cyclization occurring during hydrolysis is not a constant event and depends on the mechanism of each enzyme.
Results
GDE3 but not GDE2 is an LPI-specific PLC in vitro
To test our working hypothesis, HEK293T cells were transfected with plasmids allowing the expression of human GDE3 in fusion with GFP or with red fluorescent protein (RFP) at its C-terminal. Two other plasmids bore the R230A mutation previously shown to suppress hydrolytic activity of GDE3 against GPIns (23). All the fusion proteins displayed an exclusive localization in particulate fraction (Fig. 2A), as expected from the sequence of GDE3 (21–23). Moreover, confocal microscopy revealed the presence of WT and mutated GDE3 at the plasma membrane, whereas GFP alone remained cytosolic (Fig. 2, B–D).
Figure 2.
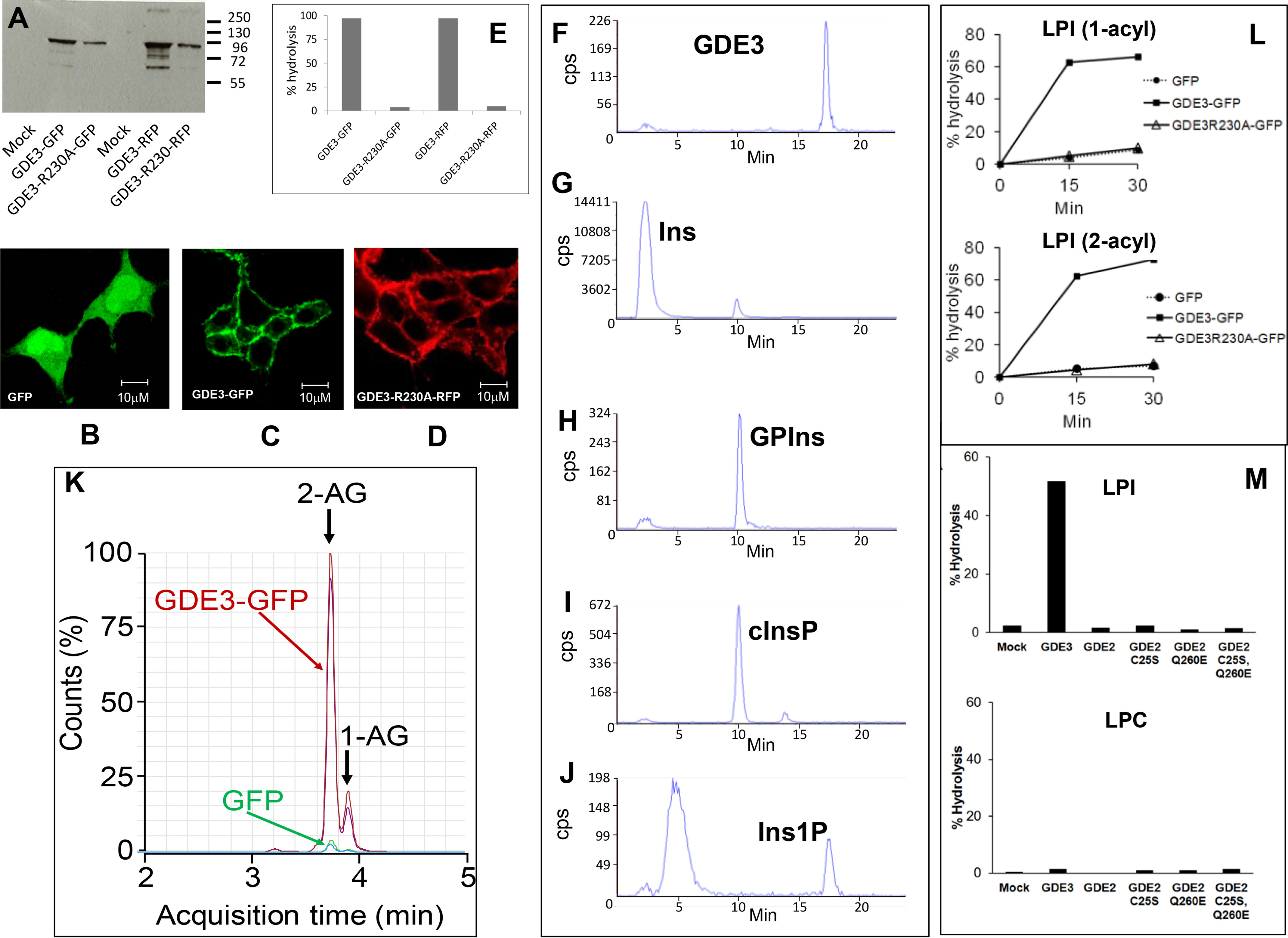
Identification of GDE3 as a lysoPLC specific for LPI. A, HEK293T cells were transfected with GFP or RFP (Mock), GDE3-GFP, GDE3-R230A-GFP, GDE3-RFP, or GDE3-R230A-RFP. Membranes were isolated and used for Western blotting analysis using anti-human GDPD2 mouse polyclonal antibody (Abnova). B–D, the cells were observed by confocal microscopy as described under “Experimental procedures”. E, membranes (10 µg of protein) were incubated with 50 µm of [3H]Ins-LPI (1-acyl) during 30 min in 0.1 ml of 100 mm Tris-HCl, pH 7.4, containing 2 mm CaCl2 for determination of lysoPLC activity. Data are individual values representative of five different experiments for GFP-tagged enzyme and one single experiment for RFP-tagged protein. F–J, membranes (10 µg of protein) prepared from HEK293T cells transfected with GDE3-GFP were incubated for 30 min with 50 µm [3H]Ins-LPI (1-acyl) in 0.1 ml of 100 mm Tris-HCl, pH 7.4, containing 2 mm CaCl2. After lipid extraction in the presence of 0.1 m HCl, the water-soluble extracts and standard solutions were loaded onto the HPLC column and analyzed as described. Standards loaded onto the HPLC column were pure [3H]Ins (G), GP[3H]Ins (H), cyclic [3H]Ins-1,2-phosphate (cInsP, I), or [3H]Ins-1-phosphate (Ins1P, J). The product generated by acid hydrolysis of cInsP and eluting as an early peak was not identified. Data are from one experiment representative of three experiments with very similar results. K, nonradioactive LPI (2-acyl, 50 µm) was incubated for 30 min with membranes (10 µg of protein) from HEK293T cells transfected either with GDE3-GFP or with GFP in 0.1 ml of 100 mm Tris-HCl, pH 7.4, containing 2 mm CaCl2. Lipids were extracted and analyzed by LC-MS (see “Experimental procedures”). Major and minor peaks at 3.7 and 3.8 min correspond to 2-AG and 1-AG, respectively, indicating that migration of arachidonic acid from sn-2 to sn-1 position was about 17%. The profiles of two independent experiments are shown. L, membranes (10 µg of protein) were isolated from HEK293T cells expressing GDE3-GFP, GDE3-R230A-GFP, or GFP alone. Substrate was [3H]Ins-LPI (1-acyl, 50 µm) or [3H]Ins-LPI (2-acyl, 50 µm), and lysoPLC activity was determined as described after incubation for indicated times in 0.1 ml of 100 mm Tris-HCl, pH 7.4, containing 2 mm CaCl2. Data (single determinations) are representative of two experiments with identical results. M, HEK293T cells were transfected with various vectors encoding GFP (Mock), GDE3-GFP (GDE3), GDE2-GFP (GDE2), or the various GDE2 mutants prepared as described under “Experimental procedures”. LysoPLC activity was determined using membrane fractions isolated from transfected cells (30 µg of protein) and incubated for 30 min with 50 µm [3H]Ins-LPI or 1 µm [3H]choline-LPC (1-acyl) in 0.1 ml of 100 mm Tris-HCl, pH 7.4, containing 2 mm CaCl2. Data (single values) are representative of two experiments with very similar results.
Incubation of membranes containing WT GDE3 with [3H]Ins-LPI (1-acyl) resulted in almost total hydrolysis of the substrate with appearance of water-soluble radioactivity (Fig. 2E). However, no LPI hydrolysis could be detected with GDE3 bearing the R230A mutation.
Using anion exchange HPLC, the radioactive water-soluble compound was identified as Ins1P (Fig. 2F). Exclusion of Ins (Fig. 2G) and GPIns (Fig. 2H) as water-soluble products of LPI cleavage eliminated the possibility of GDE3 being a lysophospholipase D or A, respectively. The GDE3 mechanism differed from that of bacterial PLC, which produced cyclic Ins-1,2-phosphate (Fig. 2I). This was partially converted into Ins1P upon acidic hydrolysis (Fig. 2J).
When nonradioactive LPI (2-acyl) was used as a substrate, 2-AG was identified by LC-MS as the main product of hydrolysis (Fig. 2K). GDE3 equally hydrolyzed 1-acyl and 2-acyl LPI species (Fig. 2L). Among five different phospholipids, LPI was the only lysophospholipid degraded by GDE3 (Table 1).
Table 1.
Specificity of GDE3 lysoPLC activity for LPI
The various lysophospholipids were obtained as described under “Experimental procedures”. They were added to membranes (20 µg of protein) isolated from HEK293 cells transfected with empty vector (Mock) or GDE3-GFP vector (GDE3) and incubated (final concentration 50 µm) for 60 min in 0.1 ml of 100 mm Tris-HCl, pH 7.4, containing 2 mm CaCl2. Lipids were then extracted and analyzed for the corresponding monoacylglycerol by LC-MS (see “Experimental procedures”). Data are individual values
| Substrates | Monoacylglycerol formed (pmol/assay) |
|
|---|---|---|
| Mock | GDE3 | |
| 2-acyl-sn-glycero-3-phosphoinositol | 35 | 1547 |
| 1-myristoyl-sn-glycero-3-phosphocholine | 0 | 0 |
| 1-myristoyl-sn-glycero-3-phosphoethanolamine | 0 | 0 |
| 1-myristoyl-sn-glycero-3-phosphoglycerol | 0 | 0 |
| 1-oleoyl-sn-glycero-3-phosphoserine | 1.4 | 3.3 |
GDE2 is another glycerophosphodiesterase displaying very common features with GDE3, i.e. six putative transmembrane segments and an active site oriented to the cell exterior (30). GDE2 and GDE3 were found to hydrolyze glycosyl-PI anchors, generating very specific signaling events important for neurodevelopment or cancer (30–33). However, GDE2 did not achieve hydrolysis of either LPI or lysophosphatidylcholine (LPC) (Fig. 2M). LPC was tested with the view that GDE2 was also reported to degrade sn-glycero-3-phosphocholine (34), thus suggesting that LPC might have been a natural substrate of GDE2. Intact GDE2 activity against glycosyl-PI anchors was previously shown to require reduction of an intracellular disulfide bridge (35). However, the C25S mutant of GDE2, which abrogates the inhibitory intracellular disulfide bridge of GDE2 (35), remained inactive toward the two substrates (Fig. 2M). Finally, the consensus sequence common to classical PLC and various GDEs (Fig. 3) indicated that a very constant glutamate residue at position 260 was selectively replaced by glutamine in GDE2. But again, the E260Q mutant of GDE2 did not reveal any hydrolytic activity toward the two lysophospholipid substrates (Fig. 2M). The same negative results were obtained with the GDE2 mutant bearing the double C25S/E260Q mutation (Fig. 2M)).
Figure 3.

Conserved sequences within catalytic domains of phospholipases C (PLC) and various glycerophosphodiesterases. Alignment and definition of the consensus sequence found in PLC (Con) is identical to that provided in Fig. S6 by Rao and Sockanathan. ecGLPQ is the periplasmic glycerophosphodiesterase from Escherichia coli; h, r, m, and c refer to human, rat, mouse and chicken, respectively. The most highly conserved residues are colored in red, exceptions being highlighted in yellow.
In conclusion, GDE2 enzymatic activity is restricted to glycosyl-PI anchors (30, 31), in contrast to GDE3, which actively hydrolyzes LPI.
GDE3 lysoPLC activity was maximal at pH 7.4 (9.0 ± 0.5 nmol × min−1 × mg−1, mean ± S.E., four determinations) and required [Ca2+] at mm range (Fig. 4). It increased up to 50 μm LPI and decreased afterward (Fig. 4E), indicating that it was restricted to monomers (critical micelle concentration of LPI is around 30–70 μm (36)), in a range of concentrations occurring in biological fluids (13).
Figure 4.

Enzymatic properties of lysoPLC activity of GDE3. Membranes were isolated from HEK293T cells transfected with GDE3-GFP or GFP and incubated in 0.1-ml final volume in the presence of [3H]Ins-LPI (1-acyl). LysoPLC activity was determined as described under “Experimental procedures”. A, proteins (10 µg) were incubated for 30 min with 50 µm LPI in 0.1 m Tris-HCl buffer at the indicated pH containing 2 mm CaCl2. Data are means ± S.E. of three determinations. B, proteins (10 µg) were incubated for indicated times with 50 µm LPI in 100 mm Tris-HCl buffer, pH 7.4, containing 2 mm CaCl2. Data are individual values from one experiment representative of three experiments with very similar results. C, increasing amounts of protein incubated for 30 min with 50 µm LPI in 100 mm Tris-HCl buffer, pH 7.4, containing 2 mm CaCl2. Data are individual values from one typical experiment. D, proteins (10 µg) were incubated for 60 min with 50 µm LPI in 100 mm Tris-HCl buffer, pH 7.4, containing or absent of 2 mm CaCl2. Data are expressed as % hydrolysis and are means ± S.E. of three determinations. E, proteins (10 µg) were incubated with increasing concentrations of LPI in 0.1 ml of 100 mm Tris-HCl, pH 7.4, containing 2 mm CaCl2. Data are from one experiment representative of three experiments with very similar results. CMC, critical micelle concentration.
Altogether, these data identify GDE3 as an LPI-specific PLC. This activity is supported by the same active site involved in GPIns hydrolysis (23). The main difference between the two substrates is that LPI can provide monoacylglycerol instead of glycerol. Because both 1-acyl and 2-acyl LPI species are equally cleaved, GDE3 might thus participate in an alternative pathway of 2-AG production.
GDE3 is an ecto-PLC
When [3H]Ins-LPI was added to confluent HEK293T cells expressing GDE3, a water-soluble radioactive compound accumulated in supernatant (Fig. 5A). With cells expressing the R230A mutant of GDE3, production of the radioactive water-soluble compound was abolished (Fig. 5A). Together with the fact that GDE3 is essentially localized at the plasma membrane (Fig. 2, C and D), and in agreement with structural predictions deduced from GDE3 sequence (21, 22), GDE3 thus appeared as an ecto-enzyme and is likely the ecto-PLC previously characterized in various cells (37–39).
Figure 5.

Determination of ecto-lysoPLC activity of GDE3, GDE2, and various GDE2 mutants. A, monolayers of HEK293T cells expressing GDE3 were seeded in 6-well dishes (9.5 cm2) and incubated with 11.25 µm [3H]Ins-LPI (1-acyl) added to the medium (2-ml final volume). At various times, 0.5 ml of supernatant was drawn from each well and lipids were extracted in the presence of 0.1 m HCl. The radioactivity (dpm) in upper water-methanol phase was then determined. Data are individual values from one experiment representative of three experiments with very similar results. B, HEK293T cells were transfected with GFP (Mock), GDE3-GFP (GDE3), GDE2-GFP (GDE2), or various mutants of GDE2 in fusion with GFP. Water-soluble radioactivity (cpm) was determined as in (A) on 0.5-ml aliquots submitted to the precipitation procedure using BSA and HClO4 (see “Experimental procedures”). Data are single values from one typical experiment.
The assay using intact cells was also applied to HEK293T cells transfected with GDE2 and the various mutants described above. Again, no activity could be detected against LPI (Fig. 5B), despite the fact that GDE2 also exposes its catalytic domain to the cell surface, where it hydrolyzes glycosyl-PI anchors (30, 31).
GDE3 acts as a switch between GPR55 and CB2 signaling
The various enzymatic properties described above suggested two possible nonexclusive functions for GDE3: inhibition of LPI-induced signaling by degradation of GPR55 ligand and activation of cannabinoid receptor via generation of 2-AG. We first verified that GDE3 and GPR55, on the one hand, and GDE3 and CB2, on the other hand, were colocalized in the plasma membrane upon expression in HEK293T cells (Fig. 6, A and E). In CHO cells coexpressing GDE3 and GPR55, the characteristic Ca2+ spikes evoked by LPI (40) were abolished by active GDE3 but not by inactive R230A GDE3 mutant (Fig. 6, B–D). In CHO cells coexpressing CB2 and WT GDE3, both 2-AG and LPI induced Gi signaling, i.e. dose-dependent inhibition of adenylate cyclase (41, 42) (Fig. 6F). In contrast, cells transfected with the R230A mutant of GDE3 responded only to 2-AG (Fig. 6G). The effects of LPI and 2-AG on adenylate cyclase were reversed by the CB2 inverse agonist AM630 (Fig. 6H). Altogether, these data indicate that GDE3 can function as a switch between GPR55 and CB2.
Figure 6.
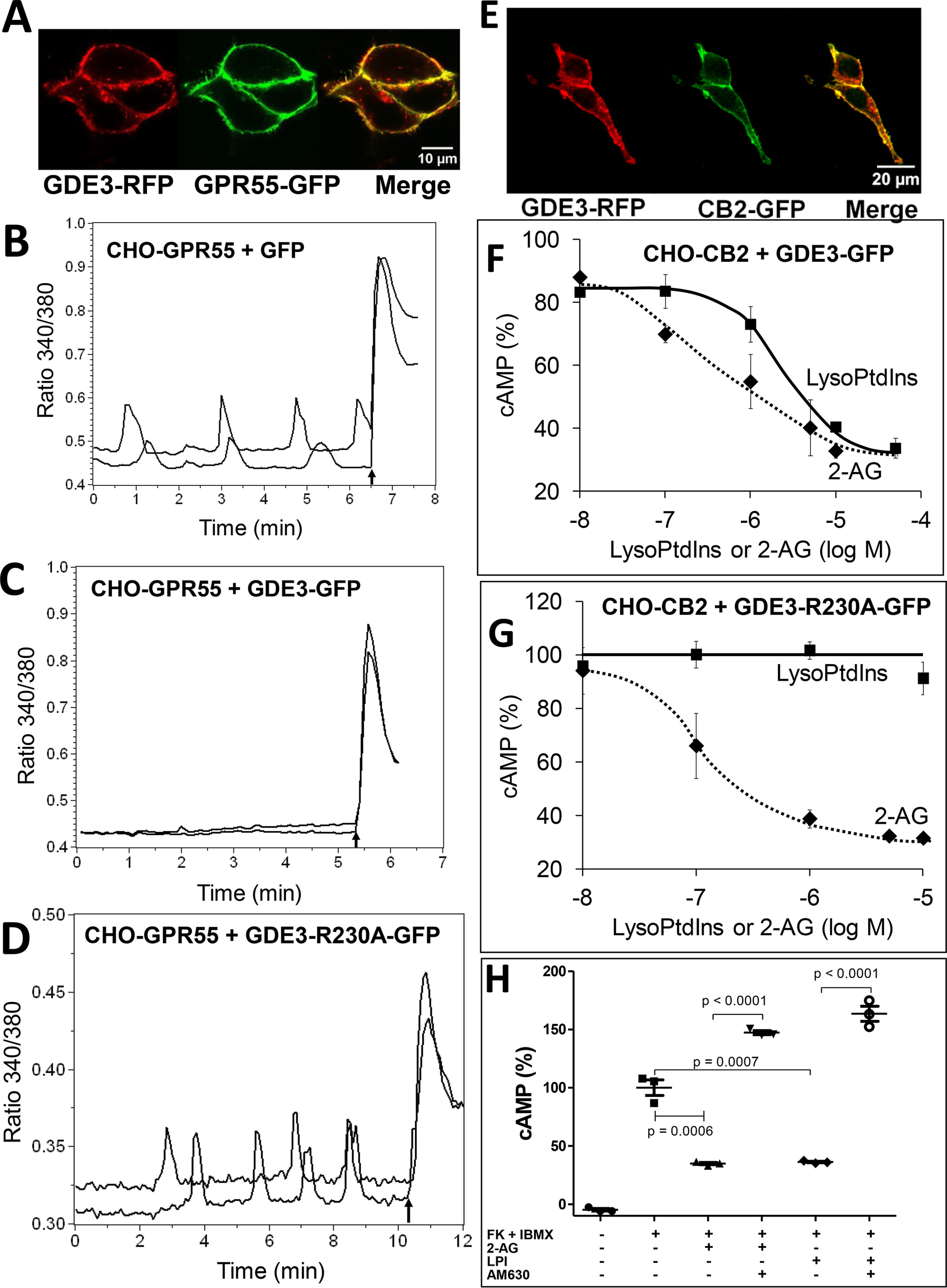
GDE3 acts as a switch between GPR55 and CB2 signaling. A, HEK293T cells were transfected with GPR55-GFP and GDE3-RFP and examined by confocal microscopy as described under “Experimental procedures”. B, CHO-GPR55 cells transfected with GFP as a control were loaded with Fura-2-AM, stimulated by 5 µm LPI (1-acyl), and examined for fluorescence ratio 340 nm/380 nm, reflecting cytoplasmic free calcium concentration ([Ca2+]i). Arrow indicates addition of 1 µm ionomycin. C, same as in (B) with CHO-GPR55 cells transfected with GDE3-GFP. D, same as in (B) with CHO-GPR55 cells transfected with GDE3-R230A-GFP. (B), (C), and (D) show two single cell traces. E, HEK293T cells were transfected with CB2-GFP and GDE3-RFP and examined by confocal microscopy as in (A). F, CHO-CB2 cells transfected with GDE3-GFP were incubated for 20 min with 7 µm forskolin plus 200 µm IBMX and various concentrations of 2-AG or LPI (2-acyl). Cellular cAMP content was determined as described under “Experimental procedures”. Data (means ± S.E., three determinations) are expressed as % of cAMP content determined in the presence of forskolin plus IBMX only. G, Same as in (F) with CHO-CB2 cells transfected with GDE3-R230-GFP. H, Same as in (F), except that CHO-CB2 cells were preincubated or not for 15 min with 10 µm AM630 before addition of forskolin plus IBMX, followed by 5 µm LPI (2-acyl) or 5 µm 2-AG. Data are means ± S.D. of three determinations. p, probability of significance according to unpaired t independent test.
Evidence that GDE3 LPI-PLC is functional in vivo
The next step was to investigate whether GDE3 could participate in vivo in a biological function regulated by the availability of 2-AG and/or LPI. Comparative mRNA expression of GDE3 (Gdpd2), CB1 (Cnr1), CB2 (Cnr2), and Gpr55 in various mouse tissues (Fig. 7A) revealed gross variations in the ratios between the expression levels of the four genes. GDE3 expression was the highest in smooth muscle (derived from the small intestine) and in the spleen. This is coherent with available data from various banks (GeneCards, Tabula muris) and with a previous publication also showing a high expression level of GDE3 in bone (43). Noticeably, a high level of GDE3 and CB2 was found in the spleen, which might represent an interesting organ for future studies aimed to explore a possible cooperation of the two genes.
Figure 7.

Expression of GDE3, CB1, CB2, and Gpr55 in various mouse tissues and lysoPLC activity in the brain and spleen. A, contents of mRNA encoding CB1 (Cnr1), CB2 (Cnr2), GDE3 (Gdpd2), and Gpr55 were determined in mouse tissues by quantitative real time PCR (see “Experimental procedures”). Data are expressed as normalized ratios relative to β-tubulin and are means ± S.E. of three determinations. B, brain and spleen membranes were prepared from WT or GDE3 KO mice as described under “Experimental procedures”. LysoPLC activity was determined in 30-min incubations using 10 µg (spleen) or 20 µg (brain) of protein and 50 µm [3H]Ins-LPI (1-acyl) as substrate in 0.1 m Tris-HCl buffer, pH 7.4, containing 2 mm CaCl2. Data are means ± S.D. (n = 5 for brain, n = 3 for spleen). C, after sacrifice, central nervous tissues were dissected to isolate cerebellum (Cer), cortex (Cor), hippocampus (Hip), mesencephalon (Mes), olfactory bulb (OB), spinal cord (SC), and striatum (Stri). LysoPLC activity was determined in crude membranes as in (B). Data (means ± S.D. (error bars)) are from four WT mice, except for hippocampus and striatum (n = 3), and from one KO mouse as a control.
In agreement with data on mRNA, LPI-PLC activity was 40-fold higher in the spleen compared with the brain (Fig. 7B). In both cases, about 85% of the activity determined in WT mice disappeared in Gdpd2-KO mice, indicating that GDE3 is the major if not the unique LPI-PLC in both organs. Indeed, basal activity could represent some cytosolic phosphatidylinositol (PI)–specific PLC or some lysophospholipase A remaining adsorbed on the membranes. We were surprised by the low activity detected in the brain given the fact that significant LPI-PLC activity was previously described in rat brain membranes (44–46). Because GDE2 is more abundant than GDE3 in neural tissues, we suspected GDE2 as the possible lysoPLC previously described in the brain. However, the negative data reported above together with the low activity determined in the brain eliminate this possibility. Finally, dissection did not reveal a striking increase of LPI-PLC activity in any region of the central nervous system (Fig. 7C).
To check whether LPI was a physiological substrate of GDE3 in vivo, LPI molecular species were determined by HPLC-MS in two organs (brain and spleen) from both WT and GDE3-KO mice. As shown in Fig. 8 (left side), all the main LPI molecular species were doubled in the spleens of mice invalidated for the Gdpd2 gene. In contrast, a subtle change was observed in brains from KO mice compared with WT animals, but this did not reach significance, in agreement with the 40-fold lower activity of LPI-PLC in the brain. Analysis of monoacylglycerol molecular species also revealed the lack of modification in brains from KO mice, the only difference observed in the spleen being a small but significant decrease of 2-AG (Fig. 8, right side) in mice lacking GDE3. As will be further commented in the discussion, these data bring strong evidence for in vivo lysoPLC activity on LPI from the spleen, its role in an alternative pathway of 2-AG production remaining compatible with our data.
Figure 8.

Quantification of molecular species of LPI and monoacylglycerol in brain and spleen from WT and GDE3 KO mice. Each organ was frozen immediately after harvesting. Frozen tissues were weighed and homogenized in 2 ml of water, followed by acidification with 0.24 ml of 2 m HCl and lipid extraction in 8 ml of dichloromethane (CH2Cl2) and 4 ml of methanol. Lipids from the organic phase were submitted to solid-phase extraction using silica and eluted with a hexane-isopropanol mixture for 2-AG and methanol for LPI. Lipids were then quantified by LC-MS as described under “Experimental procedures”. Data are expressed as nmol/g wet weight tissue and are means ± S.D. (error bars) of 17 WT and 18 KO mice. 16:0, palmitoyl-; 18:0, stearoyl-; 18:1, oleoyl-; 20:4, arachidonoyl-; p, probability of significance according to unpaired t independent test (see “Experimental procedures”).
Discussion
The present study unambiguously identifies LPI as a main substrate of GDE3 acting as an ecto-PLC, in close contact with extracellular medium providing neutral pH and mm Ca2+ concentration required for maximal activity. Using the same assay conditions, preferential hydrolysis of GPIns was previously reported (23) with an apparent Km of 97 μm, whereas it was ∼25 μm in the present study. A striking feature of GDE3 lysoPLC activity is its inhibition at substrate concentrations above the critical micelle concentration. This observation is surprising in view of historical studies showing that lipolytic enzymes differ from classical esterases by interfacial activation occurring just above the critical micelle concentration of substrate (47–49). This peculiar behavior of GDE3 is probably linked to its membrane localization, LPI concentrations above critical micelle concentration being rather deleterious to membrane structure (50).
It remains to be shown whether GDE3 PLC interacts with LPI monomers directly from the extracellular space, in the same way as it is presumed to bind water-soluble GPIns, or whether LPI intercalates between lipids of the external membrane leaflet before reaching the GDE3 catalytic site. Both possibilities remain open, because a third specific substrate of GDE3 corresponds to glycosyl-PI anchors (32, 33), which are embedded in the lipid phase of the outer leaflet. This point deserves appropriate questioning because the two possibilities exist when comparing interaction of phospholipid ligands with their G-protein–coupled receptors: LPA1 receptor is the only example permitting direct extracellular interaction with lysophosphatidic acid, whereas S1P1, LPA6, and CB1 receptors bind their ligand after diffusion through a gap between transmembrane α-helices I and VII in the extracellular leaflet (51–53). The 3D structures of GPR55 and GDE3 have not yet been solved, thus leaving unknown how LPI interacts with both its receptor and its degrading enzyme.
Among possible functions of GDE3, its capacity to regulate the availability of LPI to its receptor might be of great relevance in a number of pathophysiological conditions such as cancer, obesity, neurodegeneration, or inflammation (13–16). This is supported by our signaling experiments showing an inhibition of GPR55 activation upon coexpression of GDE3 and by in vivo accumulation of LPI in spleens from Gdpd2 KO mice. Further studies should define how GDE3 is positioned relative to GPR55, i.e. either in the same membrane as in our experimental example or in neighboring cells. This will probably change with organs and tissues and should be defined in a number of experimental systems where GPR55 was shown to exert important (patho)physiological functions, including the brain and nervous system, endocrine pancreas, gut, and immune system, with a particular interest for those tissues, such as spleen, displaying the highest expression levels.
Our study also demonstrated that GDE3 LPI-PLC is equally active against 1-acyl and 2-acyl species of LPI, indicating that all forms of LPI are concerned, independent of the phospholipase (A1 or A2) involved in their production (16). If we focus on the 2-acyl forms of LPI, those will be particularly rich in arachidonic acid (13, 16), with two functional consequences: 2-arachidonoyl LPI is the most active LPI at GPR55 (18), and its hydrolysis by PLC will generate the endocannabinoid agonist 2-AG, thus providing an alternative pathway of 2-AG synthesis (Fig. 9A). As a first step of this pathway, the intracellular phospholipase A1 DDHD1 was suggested as a good candidate able to produce 2-arachidonoyl LPI (13, 54). This will require export of the lysophospholipid via the ABC transporter C1 (ABCC1) (55, 56). However, one cannot entirely exclude an extracellular PLA1, for instance, one member of the pancreatic lipase family, some of which are involved in the production of other phospholipid mediators such as 2-acyl LPA and lysophosphatidylserine (57). In this context, it is worth mentioning that pancreatic lipase related protein 2 (PLRP2), which was discovered as a guinea pig pancreatic phospholipase A1 but is also expressed in the hypothalamus and several immune cells (58, 59), was found to be very active against phosphatidylinositol (60).
Figure 9.

Role of GDE3 in 2-AG synthesis and as a switch between GPR55 and the cannabinoid receptor. A, the three major phosphoinositides, phosphatidylinositol (PI), PI 4-phosphate (PIP), and PI-bisphosphate are in permanent interconversion through four steps of phosphorylation-dephosphorylation. The canonical pathway of 2-AG synthesis involves PIP2 cleavage by PLCβ, followed by removal of stearic acid from the sn-1 position of diacylglycerol (DAG) by DAG lipase α (DAGLα). The alternative pathway requires PI hydrolysis by a phospholipase A1 (PLA1), either DDHD1 for an intracellular reaction or an extracellular lipase. GDE3 then produces 2-AG from the 2-arachidonoyl LPI species. B, as shown by data from Fig. 6, GDE3 is able to convert GPR55 signaling evoked by LPI into CB2 activation by 2-AG. The three proteins are not represented in the same membrane because the depicted exchanges might involve membranes from the same or from neighboring cells. Although not shown in this study, CB1 could also be emphasized in the switching event.
As also depicted in Fig. 9A, 2-AG is currently produced by a canonical pathway involving the sequential action of PLCβ on phosphatidylinositol (4, 5)-bisphosphate and diacylglycerol (DAG) lipase α on the resulting diacylglycerol (6–8). This pathway has been well established as supporting retrograde inhibition of synaptic transmission by CB1, as shown by specific knockout of DAG lipase α gene (61, 62) or by using specific inhibitors (63). Based on the low activity of GDE3 lysoPLC in the brain and the lack of 2-AG increase in Gdpd2 KO mice, the alternative pathway suggested by the present study is not predicted to play any role in the central nervous system, at least at the level of synaptic transmission. In contrast, the low but significant decrease of 2-AG occurring in spleens from mice lacking GDE3 suggests that the alternative pathway might be functional in this organ or in the immune system in general. However, data based on the specific accumulation of a mediator or its lack of variation in KO mice are to be considered with caution for two main reasons: 2-AG is rapidly degraded by at least three enzymes (64) and might escape accurate determination, and whole-organ analysis deals with different pools and might thus be unable to detect significant variation of a minor, very localized production of 2-AG. As discussed by van Esbroeck et al. (65), this could be the case of Abhd6 DAG lipase activity. As another example, conditional deletion of colony stimulating factor 1 (Csf1) selectively in spleen red pulp fibroblasts dramatically altered red pulp macrophage content without any change in the level of circulating Csf1 (66). The same reason might also explain the small but nonsignificant increase of brain LPI observed in KO mice. Fine dissection of the brain is a very delicate procedure because of rapid metabolic changes occurring postmortem, as shown for instance for 2-AG (67), and was thus not further emphasized.
From our preliminary observations, GDE3 KO mice do not display any obvious phenotype. Hence, definitive answers concerning the possible role of GDE3 in various tissues might come from the observation in KO mice of subtle functional changes known to involve endocannabinoids and/or the GPR55 axis. This should be the object of future studies. Another crucial development of our present finding will be to identify which specific cells express GDE3 in the various concerned tissues. Because of the lack of a specific antibody, this has not been feasible in the present study. However, one can speculate that GDE3 might deserve some very specific functions within the immune system when comparing, for instance, the high expression level observed in the spleen compared with the very low signal detected in the lymph node (Fig. 7A). From available literature data (68), Gdpd2 is preferentially expressed in astrocytes compared with neurons, opening the view that GDE3 could be an interesting player in neuroinflammation (69). Finally, the high level of GDE3 in smooth muscle depicted in Fig. 7A will deserve further investigation in the future, because the cDNA extract used in that experiment (provided by Takara Bio USA) was actually obtained from small intestine mucosa and might just reflect the high expression level of Gdpd2 in enterocytes (70).
The interpretation of our signaling studies suggesting GDE3 could act as a switch between GPR55 and CB1 is schematized in Fig. 9B. Although definitive proof that this regulation occurs in vivo is still lacking, this main conclusion is proposed as a working hypothesis. Two specific remarks can be made: 1) a similar switch is also suggested for 2-arachidonoyl LPA through the action of ecto-lipid phosphate phosphatases, which display a membrane topological organization reminiscent of GDE3 (13, 71–73), and 2) the observed switch reinforces the link between the endocannabinoid system and GPR55, which was shown to form heteromers with both CB1 and CB2 (74, 75).
Finally, three additional points are worth mentioning. First, in their recent study also describing GDE3 as an ecto-LPI-PLC, Tsutsumi et al. (29) provided evidence that the same switch between GPR55 and cannabinoid signaling could be involved in the maturation of osteoblasts. They also suggested that GDE3 might provide 2-oleoylglycerol from the corresponding LPI to stimulate the fat sensor receptor GPR119. We did not find any accumulation of 2-oleoylglycerol in the two organs from GDE3 KO mice explored for monoacylglycerol content. However, the gut and pancreas seem more appropriate for future studies dealing with that question. The second point concerns lysophosphatidyl-β-d-glucoside, which displays a high degree of structural similarity with LPI (Fig. S1) and was shown to modulate astrocyte-dependent guidance of spinal cord sensory axons via GPR55 (19, 20). Unfortunately, this compound was not available for our present study, but it will be interesting to elucidate whether this rather close analog of LPI is also a substrate of GDE3. Last, as mentioned before, GDE2 and GDE3 are also known to hydrolyze glycosyl-PI anchors in various systems, with functional consequences related to the release of cell surface proteins such as RECK, glypican, urokinase receptor, or CNTFRα (30–33). If the interpretation of the data is straightforward for GDE2, which did not reveal any activity against lysophospholipids, the situation is more complex for GDE3. In particular, the two latter studies dealing with GDE3 observed a decreased proliferation promoted by GDE3 in two different cell models (32, 33). It will be interesting to revisit the contribution of LPI-PLC activity to these events, keeping in mind the role of the LPI-GPR55 axis in the control of cell proliferation (13–16, 55, 76).
In conclusion, although an alternative pathway of 2-AG synthesis was suggested in a number of reviews, current evidence in this paper that GDE3 acts as an ecto-PLC specific of LPI both in vitro and in vivo should stimulate a number of studies aimed at identifying functional relevance of this novel pathway. Molecular tools and transgenic mice models will certainly reveal interesting consequences of this signaling switch occurring within the endocannabinoid system.
Experimental procedures
Plasmids
ORF DNA sequences encoding human GDE3 (NM_017711.3, hGDPD2), murine GDE2 (NM_201352, mGDPD5), and human CB2 (NM_001841, hCNR2) were obtained from Thermo Fisher Scientific ABgene; human GPR55 (NM_005683.2) was from OriGene Technologies.
GDE3 ORF was used either in pCMV-SPORT6 expression vector or subcloned into pEGFP-N1 or pmRFP1-N1 vector (Clontech Laboratories). Corresponding plasmids will be referred to as GDE3, GDE3-GFP, and GDE3-RFP, respectively. An R230A mutant of GDE3 was prepared using the QuikChangeTM site-directed mutagenesis kit (Stratagene) and subcloned into pEGFP-N1 or pmRFP1-N1 vector as described above. These will be referred to as GDE3-R230A, GDE3-R230A-GFP, and GDE3-R230A-RFP, respectively.
Similarly, GPR55 in pCMV6-XL5 vector, GDE2 in pCMV-SPORT6 vector, and CB2 in pPCR-Script Amp SK+ vector were subcloned into pEGFP-N1 vector and used as GPR55-GFP, GDE2-GFP, and CB2-GFP, respectively.
Primers used for subcloning and for mutagenesis are listed in Table S1.
Two GDE2 mutants (C25S and Q260E) in pEGFP-N1 vector were prepared by GeneCust (Ellange, Luxembourg).
Cell culture
HEK293T cells were maintained in complete DMEM, 10% (v/v) FCS, and 1% (w/v) penicillin-streptomycin. The cells were transfected with lipofectamine (Invitrogen, Thermo Fisher Scientific) according to manufacturer's instructions.
Chinese hamster ovary (CHO) cells stably transfected with CB2 ORF (CHO-CB2) or GPR55 ORF (CHO-GPR55) were kindly provided by Dr. Christine Labit-Lebouteiller (Sanofi-Aventis, Toulouse, France). CHO-CB2 cell line was cultured in MEM without ribo-deoxyribonucleoside supplemented by 10% (v/v) dialyzed FCS and 1% (w/v) penicillin-streptomycin. CHO-GPR55 cells were maintained in F12 medium complemented with 10% (v/v) FCS, blasticidin S (30 μg/ml), and hygromycin B (500 μg/ml).
Animals
Male mice with Gdpd2 disruption on X-chromosome (Gdpd2-/0, referred to as GDE3 KO) and female Gdpd2+/− mice (Lexicon, TF0250) were purchased from Taconic. They were backcrossed onto C57BL/6J (Charles River, France) for 10 generations. They were housed in our animal facility and males of 8–12 weeks of age were used for all experiments, which were performed in accordance with national and European regulations and institutional guidelines. Mouse experimental protocols were approved by the local ethics committee (approval CEEA-122 2014-62).
Crude membrane preparation
Cells were scrapped at 4 °C in lysis buffer containing 100 mm Tris-HCl (pH 7.4), 5 mm EDTA, 0.5 mm PMSF, 50 μg/ml leupeptin, and 10 μg/ml aprotinin and disrupted by sonication at 4 °C. The supernatant was centrifuged at 100,000 × g for 1 h. The pellet was suspended by short sonication in lysis buffer.
Mice were sacrificed by cervical dislocation and tissues were homogenized at 4 °C in lysis buffer using Ultraturax. Homogenates were centrifuged at 1,500 × g for 20 min, the supernatant was then centrifuged at 100,000 × g for 1 h, and the pellet was suspended in lysis buffer as described above for cell membranes. Protein concentration was evaluated according to Bradford (77).
Western blotting
Membrane samples obtained from transfected HEK293T cells (10 μg of protein) were separated by SDS-PAGE (10%, w/v), transferred to nitrocellulose, and incubated overnight at 4 °C in a 1:1,000 dilution of affinity-purified anti-human GDPD2 mouse polyclonal antibody (Abnova). Rabbit anti-mouse IgG coupled to horseradish peroxidase (Sigma-Aldrich) was used as a secondary antibody at 1:10,000 dilution.
Synthesis of [3H]Ins-LPI
[Inositol-2-3H]-PI ([3H]Ins-PI) was purchased from PerkinElmer France and mixed with soybean PI (Avanti Polar Lipids). [3H]Ins-PI (0.25 GBq × mmol−1) was incubated with Crotalus adamanteus phospholipase A2 and 1-acyl-[3H]LPI was obtained as previously published (37). To prepare 2-acyl-[3H]LPI, bovine liver PI (1.25 mmol) was mixed with [3H]Ins-PI and incubated for 20 min at 37 °C with lipase from Rhizopus arrhizus (Sigma-Aldrich, 10 mg) in 100 mm borate buffer (pH 6.5) containing 5 mm CaCl2 and 0.2% (w/v) sodium deoxycholate. After lipid extraction according to Bligh and Dyer (78), the upper water/methanol phase was loaded onto a C18 Sep-PakTM cartridge and 2-acyl-[3H]Ins-LPI was eluted with pure methanol. Cold 2-acyl LPI used in experiments dealing with cAMP determination in CHO-CB2 cells was prepared by the same method with omission of [3H]Ins-PI. Purity of the two LPI was assessed by TLC on silica gel (Merck) using chloroform/methanol/water (65/25/4, v/v/v) as a solvent after coloration with Zinzadze reagent (79) and their concentration was measured by total phosphorus determination using a modification (80) of the procedure of Fiske and Subbarow (81).
Synthesis of [3H]LPC
Phosphatidylcholine, l-α-Dipalmitoyl, and [Choline-Methyl-3H] ([3H]phosphatidylcholine, 0.25 GBq × mmol−1) from PerkinElmer France were converted into [3H]LPC by Crotalus adamanteus phospholipase A2 as described above for [3H]LPI.
LysoPLC activity
For routine in vitro determination of LPI-PLC activity, [3H]LPI was incubated at 37 °C with membrane protein in 0.1 ml of 100 mm Tris-HCl (pH 7.4) containing 2 mm CaCl2. Incubation time, membrane protein, and substrate concentrations are indicated in the figure legends. The reaction was stopped by successive addition of 0.2 ml of BSA (Sigma-Aldrich, 2%, w/v) and 0.3 ml of 10% (v/v) HClO4. The mixture was incubated for 15 min at 4 °C and centrifuged for 5 min at 10,000 × g at 4 °C. Supernatant radioactivity was determined by scintillation counting.
Alternatively, [3H]LPC (1 μm final concentration) was used as substrate, and the reaction proceeded in the same way as with [3H]LPI.
Ecto-LPI-PLC activity and [3H]LPI uptake were assayed upon addition of [3H]LPI to confluent cell cultures as described by Volwerk et al. (37).
HPLC analysis of water-soluble compounds generated by GDE3
At the end of the incubation, the lipids were extracted (78) in the presence of 0.1 m HCl, the upper water-methanol layer was dried under nitrogen and dissolved in water, and the extracts were then loaded onto an HPLC anion exchange column (Partisphere SAX, Whatman) essentially as described (82). Briefly, the elution was performed at a flow rate of 1 ml/min using the following gradient: 0% 0.1 m (NH4)2HPO4 for 5 min, 0–10% 0.1 m (NH4)2HPO4 for 10 min, 10–30% 0.1 m (NH4)2HPO4 for 40 min, 30–100% 0.1 m (NH4)2HPO4 for 50 min, and 50–55% 0.1 m (NH4)2HPO4 for 55 min. The radioactivity was quantified using a continuous flow in-line scintillation detector (Packard 500TR).
The following standards were prepared to compare with the chromatographic behavior of the water-soluble product of [3H]LPI: pure [3H]Ins (PerkinElmer France), GP[3H]Ins obtained by methylamine hydrolysis (83) of [3H]Ins-LPI, cyclic [3H]Ins-1,2-phosphate (cInsP) produced by incubation of [3H]Ins-PI with Bacillus cereus PLC (84), or [3H]Ins-1-phosphate (Ins1P) generated upon incubation of [3H]cInsP with 1M HCl for 3 min at 80 °C (84). In that case, an unknown degradation product was also rapidly eluted from the column (see Fig. 2J).
Mass spectrometric quantification of 2-AG generated in vitro
Nonlabeled 2-acyl LPI (50 μm) was incubated with membranes from HEK293T cells transfected with GFP or GDE3-GFP (20 μg) for 60 min at 37 °C. At the end of the reaction, 1 μg of 1-heptadecanoyl-sn-glycerol (Avanti Polar) was added and lipids were extracted (78) using CH2Cl2 instead of CHCl3. The lower organic phase was used for LC-MS/MS analysis.
The same procedure was applied with four other lysophospholipids purchased from Avanti Polar: 1-myristoyl-sn-glycero-3-phosphocholine, 1-myristoyl-sn-glycero-3-phosphoethanolamine, 1-myristoyl-sn-glycero-3-phosphoglycerol, and 1-oleoyl-sn-glycero-3-phosphoserine.
LC-MS/MS analysis of 2-AG and 1-AG was performed as previously described (85). Briefly, 2-AG and 1-AG were separated on a ZorBAX SB-C18 column (2.1 mm, 50 mm; 1.8 μm) using Agilent 1290 Infinity HPLC system coupled to an ESI-triple quadruple G6460 mass spectrometer (Agilent Technologies). Data were acquired in Single Reaction Monitoring mode with optimized conditions (ion optics and collision energy). Peak detection, integration, and quantitative analysis were done using Mass Hunter Quantitative analysis software (Agilent Technologies) based on calibration lines built with commercially available standards (Cayman Chemical).
Quantification of 2-AG and LPI levels in the spleen and brain
Tissues (spleen and brain) were homogenized in water (2 ml) and lipids were extracted, following acidification, in the presence of internal standards, by adding 8 ml of dichloromethane (CH2Cl2) and 4 ml of methanol (MeOH). Following vigorous mixing and sonication, the samples were centrifuged and the organic layer was recovered and dried under a stream of N2. The resulting lipid extracts were purified by solid-phase extraction using silica and eluted with a hexane-isopropanol mixture for 2-AG and methanol for LPI. The resulting lipid fractions were analyzed by HPLC-MS using an LTQ-Orbitrap mass spectrometer (Thermo Fisher Scientific) coupled to an Accela HPLC system (Thermo Fisher Scientific). Analyte separation was achieved using a C-18 Phenomenex pre-column and a Kinetex LC-18 column (5 μm, 4.6 × 150 mm) (Phenomenex). The mass spectrometer was calibrated for mass accuracy before each series of injections. For data acquisition and processing, the Xcalibur® software (Thermo Fisher Scientific) was used. The signals of the lipids were normalized using the signal obtained for the corresponding internal standard (2-AG-d5 and 17:1 LPI).
For 2-AG, as previously described (86), mobile phases A and B consisted of methanol-H2O-acetic acid 75:24.9:0.1 (v/v/v) and methanol-acetic acid 99.9:0.1 (v/v), respectively. The gradient (0.5 ml/min) was designed as follows: transition from 100% A to 100% B linearly over 15 min, followed by 45 min at 100% B and subsequent re-equilibration at 100% A. An atmospheric pressure chemical ionization source was used, with the vaporizing temperature set at 300 °C; the corona discharge current was set at 5 μA. The capillary temperature and voltage were set at 250 °C and 20 V, respectively.
For LPI, as previously described (87), mobile phases A and B consisted of methanol-H2O-ammonium hydroxide 50:49.9:0.1 (v/v/v) and methanol-ammonium hydroxide 99.9:0.1 (v/v), respectively. The gradient (0.4 ml/min) was designed as follows: starting at 100% A and reaching linearly 100% B in 30 min, this was followed by 15 min at 100% B before re-equilibrating at 100% A. LPI species were analyzed in the negative mode. The ESI spray voltage was set at 5.0 kV, the capillary temperature was set at 270°C, and the sheath gas flow was set at 40 arbitrary units.
RT-qPCR
RNA and cDNA panels were from Takara Bio USA. Reverse transcription was performed with 1 μg of total RNA using M-MLV (Promega) and 25 μm Hexamers (Fermentas) in a 20-μl final reaction volume, according to the manufacturer's protocol. Quantitative PCR was performed using LightCycler 480 DNA SYBR Green I Master reaction mix (Roche LifeScience) with primers provided from Genecopoeia, USA.
Reactions were done on 96-well plates. The amplification program involved initial denaturation at 95 °C for 5 min, followed by 40 cycles at 95 °C for 10 s and at 60 °C for 40 s, using the LightCycler® 480 System. The reference gene tubulin was used for normalization and the method of 2−ΔΔCt was applied for comparison.
cAMP determination
CHO-CB2 cells were seeded in 48-well plates at 50,000 cells/well. The day after, they were transfected with GFP plasmid (mock conditions), GDE3-GFP, or GDE3-R230A-GFP using lipofectamine LTX (Invitrogen) and incubated for 48 h at 37 °C at 5% CO2 in humidified air. The cells were stimulated by forskolin (7 μm)/3-isobutyl-1-methylxanthine (IBMX, 200 μm) for 20 min at 37 °C in the absence or presence of 2-AG, 2-acyl LPI, and/or AM630 at concentrations indicated in figure legends. The incubations were stopped by addition of 0.2 ml of 0.1 m HCl and cAMP was determined using direct cAMP ELISA kit (Enzo Life Sciences).
Calcium flux
CHO-GPR55 (60,000 cells) were seeded in 35-mm glass base dishes (Iwaki America) in complete medium. The day after, they were transfected with GFP, GDE3-GFP, or GDE3-R230A-GFP using lipofectamine LTX (Invitrogen) according to the manufacturer's instructions. After 48 h of incubation, cells in Hanks' balanced salt solution were loaded with 10 μm Fura-2 acetoxymethyl ester (Molecular Probes) for 45 min at 37 °C. The cells were stimulated by LPI diluted in Hanks' balanced salt solution (5 μm final concentration), followed by ionomycin (1 μm final concentration). Fluorescence was measured on a Zeiss Axiovert 200M inverted microscope equipped with a CCD camera (CoolSNAP HQ, Photometrics, Tucson, AZ), an arc xenon lamp, and a computer-controlled monochromator (CAIRN Optoscan, Kent, UK) at 37 °C, 5% CO2. The cells were consecutively excited at 340 and 380 nm at 10-s intervals by means of the monochromator, and emission at 510 nm was collected with the CCD camera. The camera output was analyzed using the custom calcium-imaging software MetaFluor (Universal Imaging, West Chester, PA), allowing to calculate the 340:380 ratio for each individual cell at every time point.
Confocal experiments
Cells plated on glass cover slips were transfected as described above and fixed for 10 min at room temperature with 3% paraformaldehyde. The samples were mounted and examined using a Carl Zeiss LSM 710 confocal microscope (Carl Zeiss, Jena, Germany) with a 63× Plan-Apochromat objective (Numerical Aperture 1.4, oil). An argon laser at 488 nm was used to detect Alexa 488 fluorochrome. To detect red fluorescence, a diode laser at 641 nm was used.
Statistical analysis
Values are given as means ± S.E. or S.D. The statistical significance of differences was estimated by unpaired t indepen-dent test after verifying a Gaussian distribution of data according to D'Agostino-Pearson omnibus normality test and a nonsignificant difference of variances between samples. When normality could not be demonstrated, Mann Whitney nonparametric test was used. All tests were available from GraphPad Prism software. Differences were considered as significant at p < 0.05.
Sequence analysis of PLC and GDE in Fig. 3 was achieved according to Rao and Sockanathan (88).
Data availability
All data are contained within the article and in the supporting information.
Supplementary Material
Acknowledgments
We are indebted to Dr. C. Labit-Lebouteiller (Sanofi-Aventis, Toulouse, France) for the gift of CHO-CB2 and CHO-GPR55 cells. We thank the cellular imaging platform (S. A., A. Canivet, and D. Daviaud), the animal house staff members (INSERM UMS06, Regional Center for Functional Exploration and Experimental Resources, Toulouse, France), and the MetaToul lipidomic facility (I2MC, P. Le Faouder, J. Bertrand-Michel). Special thanks are also due to Daniel Dunia for great help in dissecting mouse brains.
This article contains supporting information.
Author contributions—F. B.-M., J.-P. S., and H. C. conceptualization; F. B.-M., V. P., J. M., G. C., N. B., B. P., G. G. M., and H. C. data curation; F. B.-M., J. M., G. C., B. P., and J.-L. D. formal analysis; F. B.-M., V. P., S. A., J. M., G. C., N. B., B. P., and G. G. M. investigation; F. B.-M., V. P., S. A., J. M., G. C., N. B., B. P., and G. G. M. methodology; F. B.-M., B. P., G. G. M., J.-L. D., and H. C. writing-original draft; J. A., J.-L. D., and J.-P. S. funding acquisition; J. A., J.-L. D., and J.-P. S. validation; J.-P. S. and H. C. supervision; J.-P. S. project administration.
Funding and additional information—This study was funded by institutional grants from Inserm, CNRS, and Paul Sabatier University.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- 2-AG
- 2-arachidonoyglycerol
- GDE
- glycerophosphodiesterase
- LPI
- lysophosphatidylinositol
- CB
- cannabinoid receptor
- GPIns
- glycerophosphoinositol
- Ins1P
- inositol-1-phosphate
- PLC
- phospholipase C
- RFP
- red fluorescent protein
- PLRP
- pancreatic lipase related protein
- DAG
- diacylglycerol
- PI
- phosphatidylinositol
- IBMX
- 3-isobutyl-1-methylxanthine
- PLA
- phospholipase A
- LPC
- lysophosphatidylcholine.
References
- 1. Mechoulam R., Hanuš L. O., Pertwee R., and Howlett A. C. (2014) Early phytocannabinoid chemistry to endocannabinoids and beyond. Nat. Rev. Neurosci. 15, 757–764 10.1038/nrn3811 [DOI] [PubMed] [Google Scholar]
- 2. Maccarrone M., Bab I., Bíró T., Cabral G. A., Dey S. K., Di Marzo V., Konje J. C., Kunos G., Mechoulam R., Pacher P., Sharkey K. A., and Zimmer A. (2015) Endocannabinoid signaling at the periphery: 50 years after THC. Trends Pharmacol. Sci. 36, 277–296 10.1016/j.tips.2015.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Piomelli D. (2014) More surprises lying ahead. The endocannabinoids keep us guessing. Neuropharmacology 76, 228–234 10.1016/j.neuropharm.2013.07.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hussain Z., Uyama T., Tsuboi K., and Ueda N. (2017) Mammalian enzymes responsible for the biosynthesis of N-acylethanolamines. Biochim. Biophys. Acta 1862, 1546–1561 10.1016/j.bbalip.2017.08.006 [DOI] [PubMed] [Google Scholar]
- 5. Tsuboi K., Uyama T., Okamoto Y., and Ueda N. (2018) Endocannabinoids related N-acylethanolamines: biological activities and metabolism. Inflamm. Regen. 38, 28 10.1186/s41232-018-0086-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sugiura T., Kishimoto S., Oka S., and Gokoh M. (2006) Biochemistry, pharmacology and physiology of 2-arachidonoylglycerol, an endogenous cannabinoid receptor ligand. Prog. Lipid Res. 45, 405–446 10.1016/j.plipres.2006.03.003 [DOI] [PubMed] [Google Scholar]
- 7. Baggelaar M. P., Maccarrone M., and van der Stelt M. (2018) 2-Arachidonoylglycerol: a signaling lipid with manifold actions in the brain. Prog. Lipid Res. 71, 1–17 10.1016/j.plipres.2018.05.002 [DOI] [PubMed] [Google Scholar]
- 8. Cristino L., Bisogno T., and Di Marzo V. (2020) Cannabinoids and the expanded endocannabinoid system in neurological disorders. Nat. Rev. Neurol. 16, 9–29 10.1038/s41582-019-0284-z [DOI] [PubMed] [Google Scholar]
- 9. Ohno-Shosaku T., and Kano M. (2014) Endocannabinoid-mediated retrograde modulation of synaptic transmission. Curr. Opin. Neurobiol. 29, 1–8 10.1016/j.conb.2014.03.017 [DOI] [PubMed] [Google Scholar]
- 10. Cabral G. A., Ferreira G. A., and Jamerson M. J. (2015) Endocannabinoids and the immune system in health and disease. Handb. Exp. Pharmacol. 231, 185–211 10.1007/978-3-319-20825-1_6 [DOI] [PubMed] [Google Scholar]
- 11. Simon G. M., and Cravatt B. F. (2010) Characterization of mice lacking candidate N-acyl ethanolamine biosynthetic enzymes provides evidence for multiple pathways that contribute to endocannabinoid production in vivo. Mol. Biosyst. 6, 1411–1418 10.1039/c000237b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Inoue M., Tsuboi K., Okamoto Y., Hidaka M., Uyama T., Tsutsumi T., Tanaka T., Ueda N., and Tokumura A. (2017) Peripheral tissue levels and molecular species compositions of N-acyl-phosphatidylethanolamine and its metabolites in mice lacking N-acyl-phosphatidylethanolamine-specific phospholipase D. J. Biochem. 162, 449–458 10.1093/jb/mvx054 [DOI] [PubMed] [Google Scholar]
- 13. Yamashita A., Oka S., Tanikawa T., Hayashi Y., Nemoto-Sasaki Y., and Sugiura T. (2013) The actions and metabolism of lysophosphatidylinositol, an endogenous agonist for GPR55. Prostaglandins Other Lipid Mediat. 107, 103–116 10.1016/j.prostaglandins.2013.05.004 [DOI] [PubMed] [Google Scholar]
- 14. Yang H., Zhou J., and Lehmann C. (2016) GPR55 - A putative “type 3” cannabinoid receptor in inflammation. J. Basic Clin. Physiol. Pharmacol. 27, 297–302 10.1515/jbcpp-2015-0080 [DOI] [PubMed] [Google Scholar]
- 15. Arifin S. A., and Falasca M. (2016) Lysophosphatidylinositol signalling and metabolic diseases. Metabolites 6, 6 10.3390/metabo6010006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Alhouayek M., Masquelier J., and Muccioli G. G. (2018) Lysophosphatidylinositols, from cell membrane constituents to GPR55 ligands. Trends Pharmacol. Sci. 39, 586–604 10.1016/j.tips.2018.02.011 [DOI] [PubMed] [Google Scholar]
- 17. Oka S., Nakajima K., Yamashita A., Kishimoto S., and Sugiura T. (2007) Identification of GPR55 as a lysophosphatidylinositol receptor. Biochem. Biophys. Res. Commun. 362, 928–934 10.1016/j.bbrc.2007.08.078 [DOI] [PubMed] [Google Scholar]
- 18. Oka S., Toshida T., Maruyama K., Nakajima K., Yamashita A., and Sugiura T. (2009) 2-Arachidonoyl-sn-glycero-3-phosphoinositol: a possible natural ligand for GPR55. J. Biochem. 145, 13–20 10.1093/jb/mvn136 [DOI] [PubMed] [Google Scholar]
- 19. Guy A. T., Nagatsuka Y., Ooashi N., Inoue M., Nakata A., Greimel P., Inoue A., Nabetani T., Murayama A., Ohta K., Ito Y., Aoki J., Hirabayashi Y., and Kamiguchi H. (2015) Glycerophospholipid regulation of modality-specific sensory axon guidance in the spinal cord. Science 349, 974–977 10.1126/science.aab3516 [DOI] [PubMed] [Google Scholar]
- 20. Guy A. T., Kano K., Ohyama J., Kamiguchi H., Hirabayashi Y., Ito Y., Matsuo I., and Greimel P. (2019) Preference for glucose over inositol headgroup during lysolipid activation of G protein-coupled receptor 55. ACS Chem. Neurosci. 10, 716–727 10.1021/acschemneuro.8b00505 [DOI] [PubMed] [Google Scholar]
- 21. Yanaka N. (2007) Mammalian glycerophosphodiester phosphodiesterases. Biosci. Biotechnol. Biochem. 71, 1811–1818 10.1271/bbb.70062 [DOI] [PubMed] [Google Scholar]
- 22. Corda D., Mosca M. G., Ohshima N., Grauso L., Yanaka N., and Mariggiò S. (2014) The emerging physiological roles of the glycerophosphodiesterase family. FEBS J. 281, 998–1016 10.1111/febs.12699 [DOI] [PubMed] [Google Scholar]
- 23. Corda D., Kudo T., Zizza P., Iurisci C., Kawai E., Kato N., Yanaka N., and Mariggiò S. (2009) The developmentally regulated osteoblast phosphodiesterase GDE3 is glycerophosphoinositol-specific and modulates cell growth. J. Biol. Chem. 284, 24848–24856 10.1074/jbc.M109.035444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Simon G. M., and Cravatt B. F. (2008) Anandamide biosynthesis catalyzed by the phosphodiesterase GDE1 and detection of glycerophospho-N-acyl ethanolamine precursors in mouse brain. J. Biol. Chem. 283, 9341–9349 10.1074/jbc.M707807200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tsuboi K., Okamoto Y., Rahman I. A., Uyama T., Inoue T., Tokumura A., and Ueda N. (2015) Glycerophosphodiesterase GDE4 as a novel lysophospholipase D: a possible involvement in bioactive N-acylethanolamine biosynthesis. Biochim. Biophys. Acta 1851, 537–548 10.1016/j.bbalip.2015.01.002 [DOI] [PubMed] [Google Scholar]
- 26. Ohshima N., Kudo T., Yamashita Y., Mariggiò S., Araki M., Honda A., Nagano T., Isaji C., Kato N., Corda D., Izumi T., and Yanaka N. (2015) New members of the mammalian glycerophosphodiester phosphodiesterase family: GDE4 and GDE7 produce lysophosphatidic acid by lysophospholipase D activity. J. Biol. Chem. 290, 4260–4271 10.1074/jbc.M114.614537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Aoyama C., Horibata Y., Ando H., Mitsuhashi S., Arai M., and Sugimoto H. (2019) Characterization of glycerophosphodiesterase 4-interacting molecules Gαq/11 and Gβ, which mediate cellular lysophospholipase D activity. Biochem. J. 476, 3721–3736 10.1042/BCJ20190666 [DOI] [PubMed] [Google Scholar]
- 28. Rahman I. A., Tsuboi K., Hussain Z., Yamashita R., Okamoto Y., Uyama T., Yamazaki N., Tanaka T., Tokumura A., and Ueda N. (2016) Calcium-dependent generation of N-acylethanolamines and lysophosphatidic acids by glycerophosphodiesterase GDE7. Biochim. Biophys. Acta 1861, 1881–1892 10.1016/j.bbalip.2016.09.008 [DOI] [PubMed] [Google Scholar]
- 29. Tsutsumi T., Matsuda R., Morito K., Kawabata K., Yokota M., Nikawadori M., Inoue-Fujiwara M., Kawashima S., Hidaka M., Yamamoto T., Yamazaki N., Tanaka T., Shinohara Y., Nishi H., and Tokumura A. (2020) Identification of human glycerophosphodiesterase 3 as an ecto phospholipase C that converts the G protein-coupled receptor 55 agonist lysophosphatidylinositol to bioactive monoacylglycerols in cultured mammalian cells. Biochim. Biophys. Acta 1865, 158761 10.1016/j.bbalip.2020.158761 [DOI] [PubMed] [Google Scholar]
- 30. Park S., Lee C., Sabharwal P., Zhang M., Meyers C. L., and Sockanathan S. (2013) GDE2 promotes neurogenesis by glycosylphosphatidylinositol-anchor cleavage of RECK. Science 339, 324–328 10.1126/science.1231921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Matas-Rico E., van Veen M., Leyton-Puig D., van den Berg J., Koster J., Kedziora K. M., Molenaar B., Weerts M. J., de Rink I., Medema R. H., Giepmans B. N., Perrakis A., Jalink K., Versteeg R., and Moolenaar W. H. (2016) Glycerophosphodiesterase GDE2 promotes neuroblastoma differentiation through glypican release and is a marker of clinical outcome. Cancer Cell 30, 548–562 10.1016/j.ccell.2016.08.016 [DOI] [PubMed] [Google Scholar]
- 32. van Veen M., Matas-Rico E., van de Wetering K., Leyton-Puig D., Kedziora K. M., De Lorenzi V., Stijf-Bultsma Y., van den Broek B., Jalink K., Sidenius N., Perrakis A., and Moolenaar W. H. (2017) Negative regulation of urokinase receptor activity by a GPI-specific phospholipase C in breast cancer cells. eLife 6, 23649 10.7554/eLife.23649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dobrowolski M., Cave C., Levy-Myers R., Lee C., Park S., Choi B. R., Xiao B., Yang W., and Sockanathan S. (2020) GDE3 regulates oligodendrocyte precursor proliferation via release of soluble CNTFRα. Development 147, dev180695 10.1242/dev.180695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gallazzini M., Ferraris J. D., and Burg M. B. (2008) GDPD5 is a glycerophosphocholine phosphodiesterase that osmotically regulates the osmoprotective organic osmolyte GPC. Proc. Natl. Acad. Sci. U. S. A. 105, 11026–11031 10.1073/pnas.0805496105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yan Y., Sabharwal P., Rao M., and Sockanathan S. (2009) The antioxidant enzyme Prdx1 controls neuronal differentiation by thiol-redox-dependent activation of GDE2. Cell 138, 1209–1221 10.1016/j.cell.2009.06.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bondarenko A. I., Malli R., and Graier W. F. (2011) The GPR55 agonist lysophosphatidylinositol directly activates intermediate-conductance Ca2+-activated K+ channels. Pflügers Arch. 462, 245–255 10.1007/s00424-011-0977-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Volwerk J. J., Birrell G. B., Hedberg K. K., and Griffith O. H. (1992) A high level of cell surface phosphatidylinositol-specific phospholipase C activity is characteristic of growth-arrested 3T3 fibroblasts but not of transformed variants. J. Cell. Physiol. 151, 613–622 10.1002/jcp.1041510322 [DOI] [PubMed] [Google Scholar]
- 38. Birrel G. B., Hedberg K. K., Volwerk J. J., and Griffith O. H. (1993) Differential expression of phospholipase C specific for inositol phospholipids at the cell surface of rat glial cells and REF52 rat embryo fibroblasts. J. Neurochem. 60, 620–625 10.1111/j.1471-4159.1993.tb03193.x [DOI] [PubMed] [Google Scholar]
- 39. Birrell G. B., Hedberg K. K., Barklis E., and Griffith O. H. (1997) Partial isolation from intact cells of a cell surface-exposed lysophosphatidylinositol-phospholipase C. J. Cell. Biochem. 65, 550–564 [DOI] [PubMed] [Google Scholar]
- 40. Henstridge C. M., Balenga N. A., Schröder R., Kargl J. K., Platzer W., Martini L., Arthur S., Penman J., Whistler J. L., Kostenis E., Waldhoer M., and Irving A. J. (2010) GPR55 ligands promote receptor coupling to multiple signalling pathways. Br. J. Pharmacol. 160, 604–614 10.1111/j.1476-5381.2009.00625.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sugiura T., Kondo S., Kishimoto S., Miyashita T., Nakane S., Kodaka T., Suhara Y., Takayama H., and Waku K. (2000) Evidence that 2-arachidonoylglycerol but not N-palmitoylethanolamine or anandamide is the physiological ligand for the cannabinoid CB2 receptor. Comparison of the agonistic activities of various cannabinoid receptor ligands in HL-60 cells. J. Biol. Chem. 275, 605–612 10.1074/jbc.275.1.605 [DOI] [PubMed] [Google Scholar]
- 42. Gonsiorek W., Lunn C., Fan X., Narula S., Lundell D., and Hipkin R. W. (2000) Endocannabinoid 2-arachidonyl glycerol is a full agonist through human type 2 cannabinoid receptor: antagonism by anandamide. Mol. Pharmacol. 57, 1045–1050 [PubMed] [Google Scholar]
- 43. Yanaka N., Imai Y., Kawai E., Akatsuka H., Wakimoto K., Nogusa Y., Kato N., Chiba H., Kotani E., Omori K., and Sakurai N. (2003) Novel membrane protein containing glycerophosphodiester phosphodiesterase motif is transiently expressed during osteoblast differentiation. J. Biol. Chem. 278, 43595–43602 10.1074/jbc.M302867200 [DOI] [PubMed] [Google Scholar]
- 44. Ueda H., Kobayashi T., Kishimoto M., Tsutsumi T., and Okuyama H. (1993) A possible pathway of phosphoinositide metabolism through EDTA-insensitive phospholipase A1 followed by lysophosphoinositide-specific phospholipase C in rat brain. J. Neurochem. 61, 1874–1881 10.1111/j.1471-4159.1993.tb09829.x [DOI] [PubMed] [Google Scholar]
- 45. Tsutsumi T., Kobayashi T., Ueda H., Yamauchi E., Watanabe S., and Okuyama H. (1994) Lysophosphoinositide-specific phospholipase C in rat brain synaptic plasma membranes. Neurochem. Res. 19, 399–406 10.1007/BF00967316 [DOI] [PubMed] [Google Scholar]
- 46. Tsutsumi T., Kobayashi T., Miyashita M., Watanabe S., Homma Y., and Okuyama H. (1995) A lysophosphoinositide-specific phospholipase C distinct from other phospholipase C families in rat brain. Arch. Biochem. Biophys. 317, 331–336 10.1006/abbi.1995.1171 [DOI] [PubMed] [Google Scholar]
- 47. Sarda L., and Desnuelle P. (1958) Actions of pancreatic lipase on esters in emulsions. Biochim. Biophys. Acta 30, 513–521 10.1016/0006-3002(58)90097-0 [DOI] [PubMed] [Google Scholar]
- 48. Pieterson W. A., Vidal J. C., Volwerk J. J., and de Haas G. H. (1974) Zymogen-catalyzed hydrolysis of monomeric substrates and the presence of recognition site for lipid-water interfaces in phospholipase A2. Biochemistry 13, 1455–1460 10.1021/bi00704a021 [DOI] [PubMed] [Google Scholar]
- 49. Mouchlis V. D., and Dennis E. A. (2019) Phospholipase A2 catalysis and lipid mediator lipidomics. Biochim. Biophys. Acta 1864, 766–771 10.1016/j.bbalip.2018.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gaits F., Salles J. P., and Chap H. (1997) Dual effect of lysophosphatidic acid on proliferation of glomerular mesangial cells. Kidney Int. 51, 1022–1027 10.1038/ki.1997.143 [DOI] [PubMed] [Google Scholar]
- 51. Blaho V. A., and Chun J. (2018) 'Crystal' clear? receptor structure insights and controversies. Trends Pharmacol. Sci. 39, 953–966 10.1016/j.tips.2018.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shao Z., Yin J., Chapman K., Grzemska M., Clark L., Wang J., and Rosenbaum D. M. (2016) High-resolution crystal structure of the human CB1 cannabinoid receptor. Nature 540, 602–606 10.1038/nature20613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kumar K. K., Shalev-Benami M., Robertson M. J., Hu H., Banister S. D., Hollingsworth S. A., Latorraca N. R., Kato H. E., Hilger D., Maeda S., Weis W. I., Farrens D. L., Dror R. O., Malhotra S. V., Kobilka B. K., et al. (2019) Structure of a signaling cannabinoid receptor 1-G protein complex. Cell 176, P448–P458.E12 10.1016/j.cell.2018.11.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yamashita A., Kumazawa T., Koga H., Suzuki N., Oka S., and Sugiura T. (2010) Generation of lysophosphatidylinositol by DDHD domain containing 1 (DDHD1): possible involvement of phospholipase D/phosphatidic acid in the activation of DDHD1. Biochim. Biophys. Acta 1801, 711–720 10.1016/j.bbalip.2010.03.012 [DOI] [PubMed] [Google Scholar]
- 55. Piñeiro R., Maffucci T., and Falasca M. (2011) The putative cannabinoid receptor GPR55 defines a novel autocrine loop in cancer cell proliferation. Oncogene 30, 142–152 10.1038/onc.2010.417 [DOI] [PubMed] [Google Scholar]
- 56. Ruban E. L., Ferro R., Arifin S. A., and Falasca M. (2014) Lysophosphatidylinositol: a novel link between ABC transporters and G-protein-coupled receptors. Biochem. Soc. Trans. 42, 1372–1377 10.1042/BST20140151 [DOI] [PubMed] [Google Scholar]
- 57. Aoki J., Inoue A., Makide K., Saiki N., and Arai H. (2007) Structure and function of extracellular phospholipase A1 belonging to the pancreatic lipase gene family. Biochimie 89, 197–204 10.1016/j.biochi.2006.09.021 [DOI] [PubMed] [Google Scholar]
- 58. Chap H. (2016) Forty five years with membrane phospholipids, phospholipases and lipid mediators: a historical perspective. Biochimie 125, 234–249 10.1016/j.biochi.2016.04.002 [DOI] [PubMed] [Google Scholar]
- 59. Gilleron M., Lepore M., Layre E., Cala-De Paepe D., Mebarek N., Shayman J. A., Canaan S., Mori L., Carrière F., Puzo G., and De Libero G. (2016) Lysosomal lipases PLRP2 and LPLA2 process mycobacterial multi-acylated lipids and generate T cell stimulatory antigens. Cell Chem. Biol. 23, 1147–1156 10.1016/j.chembiol.2016.07.021 [DOI] [PubMed] [Google Scholar]
- 60. Fauvel J., Chap H., Roques V., and Douste-Blazy L. (1984) Substrate specificity of two cationic lipases with high phospholipase A1 activity purified from guinea pig pancreas. II. Studies on glycerophospholipids. Biochim. Biophys. Acta 792, 72–78 10.1016/0005-2760(84)90284-4 [DOI] [PubMed] [Google Scholar]
- 61. Gao Y., Vasilyev D. V., Goncalves M. B., Howell F. V., Hobbs C., Reisenberg M., Shen R., Zhang M. Y., Strassle B. W., Lu P., Mark L., Piesla M. J., Deng K., Kouranova E. V., Ring R. H., et al. (2010) Loss of retrograde endocannabinoid signaling and reduced adult neurogenesis in diacylglycerol lipase knock-out mice. J. Neurosci. 30, 2017–2024 10.1523/JNEUROSCI.5693-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tanimura A., Yamazaki M., Hashimotodani Y., Uchigashima M., Kawata S., Abe M., Kita Y., Hashimoto K., Shimizu T., Watanabe M., Sakimura K., and Kano M. (2010) The endocannabinoid 2-arachidonoylglycerol produced by diacylglycerol lipase α mediates retrograde suppression of synaptic transmission. Neuron 65, 320–327 10.1016/j.neuron.2010.01.021 [DOI] [PubMed] [Google Scholar]
- 63. Ogasawara D., Deng H., Viader A., Baggelaar M. P., Breman A., den Dulk H., van den Nieuwendijk A. M., Soethoudt M., van der Wel T., Zhou J., Overkleeft H. S., Sanchez-Alavez M., Mori S., Nguyen W., Conti B., et al. (2016) Rapid and profound rewiring of brain lipid signaling networks by acute diacylglycerol lipase inhibition. Proc. Natl. Acad. Sci. U. S. A. 113, 26–33 10.1073/pnas.1522364112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Blankman J. L., Simon G. M., and Cravatt B. F. (2007) A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem. Biol. 14, 1347–1356 10.1016/j.chembiol.2007.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. van Esbroeck A. C. M., Kantae V., Di X., van der Wel T., den Dulk H., Stevens A. F., Singh S., Bakker A. T., Florea B. I., Stella N., Overkleeft H. S., Hankemeier T., and van der Stelt M. (2019) Identification of α,β-hydrolase domain containing protein 6 as a diacylglycerol lipase in Neuro-2a cells. Front. Mol. Neurosci. 12, 286 10.3389/fnmol.2019.00286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bellomo A., Mondor I., Spinelli L., Lagueyrie M., Stewart B. J., Brouilly N., Malissen B., Clatworthy M. R., and Bajénoff M. (2020) Reticular fibroblasts expressing the transcription factor WT1 define a stromal niche that maintains and replenishes splenic red pulp macrophages. Immunity 53, 127–142 10.1016/j.immuni.2020.06.008 [DOI] [PubMed] [Google Scholar]
- 67. Brose S. A., Golovko S. A., and Golovko M. Y. (2016) Brain 2-arachidonoylglycerol levels are dramatically and rapidly increased under acute ischemia-injury which is prevented by microwave irradiation. Lipids 51, 487–495 10.1007/s11745-016-4144-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhang Y., Chen K., Sloan S. A., Bennett M. L., Scholze A. R., O'Keeffe S., Phatnani H. P., Guarnieri P., Caneda C., Ruderisch N., Deng S., Liddelow S. A., Zhang C., Daneman R., Maniatis T., et al. (2014) An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 34, 11929–11947 10.1523/JNEUROSCI.1860-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Colombo E., and Farina C. (2016) Astrocytes: key regulators of neuroinflammation. Trends Immunol. 37, 608–620 10.1016/j.it.2016.06.006 [DOI] [PubMed] [Google Scholar]
- 70. Dávalos-Salas M., Montgomery M. K., Reehorst C. M., Nightingale R., Ng I., Anderton H., Al-Obaidi S., Lesmana A., Scott C. M., Ioannidis P., Kalra H., Keerthikumar S., Tögel L., Rigopoulos A., Gong S. J., et al. (2019) Deletion of intestinal Hdac3 remodels the lipidome of enterocytes and protects mice from diet-induced obesity. Nat. Commun. 10, 5291 10.1038/s41467-019-13180-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Nakane S., Oka S., Arai S., Waku K., Ishima Y., Tokumura A., and Sugiura T. (2002) 2-Arachidonoyl-sn-glycero-3-phosphate, an arachidonic acid-containing lysophosphatidic acid: occurrence and rapid enzymatic conversion to 2-arachidonoyl-sn-glycerol, a cannabinoid receptor ligand, in rat brain. Arch. Biochem. Biophys. 402, 51–58 10.1016/S0003-9861(02)00038-3 [DOI] [PubMed] [Google Scholar]
- 72. Brindley D. N., and Pilquil C. (2009) Lipid phosphate phosphatases and signaling. J. Lipid Res. 50, S225–S230 10.1194/jlr.R800055-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Tang X., Benesch M. G. K., and Brindley D. N. (2015) Lipid phosphate phosphatases and their roles in mammalian physiology and pathology. J. Lipid Res. 56, 2048–2060 10.1194/jlr.R058362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kargl J., Balenga N., Parzmair G. P., Brown A. J., Heinemann A., and Waldhoer M. (2012) The cannabinoid receptor CB1 modulates the signaling properties of the lysophosphatidylinositol receptor GPR55. J. Biol. Chem. 287, 44234–44248 10.1074/jbc.M112.364109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Moreno E., Andradas C., Medrano M., Caffarel M. M., Pérez-Gómez E., Blasco-Benito S., Gómez-Cañas M., Pazos M. R., Irving A. J., Lluís C., Canela E. I., Fernández-Ruiz J., Guzmán M., McCormick P. J., and Sánchez C. (2014) Targeting CB2-GPR55 receptor heteromers modulates cancer cell signaling. J. Biol. Chem. 289, 21960–21972 10.1074/jbc.M114.561761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Andradas C., Caffarel M. M., Pérez-Gómez E., Salazar M., Lorente M., Velasco G., Guzmán M., and Sánchez C. (2011) The orphan G protein-coupled receptor GPR55 promotes cancer cell proliferation via ERK. Oncogene 30, 245–252 10.1038/onc.2010.402 [DOI] [PubMed] [Google Scholar]
- 77. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 10.1016/0003-2697(76)90527-3 [DOI] [PubMed] [Google Scholar]
- 78. Bligh E. G., and Dyer W. J. (1959) A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 10.1139/y59-099 [DOI] [PubMed] [Google Scholar]
- 79. Dittmer J. C., and Lester R. L. (1964) A simple, specific spray for the detection of phospholipids on thin-layer chromatograms. J. Lipid Res. 5, 126–127 [PubMed] [Google Scholar]
- 80. Böttcher C. J. F., van Gent C. M., and Pries C. (1961) A rapid and sensitive sub-micro phosphorus determination. Anal. Chim. Acta 24, 203–204 10.1016/0003-2670(61)80041-X [DOI] [Google Scholar]
- 81. Fiske C.H., and Subbarow Y. (1925) The coloric determination of phosphorus. J. Biol. Chem. 66, 375–400 [Google Scholar]
- 82. Payrastre B. (2004) Phosphoinositides: lipid kinases and phosphatases. Methods Mol. Biol. 273, 201–212 10.1385/1-59259-783-1:201 [DOI] [PubMed] [Google Scholar]
- 83. Clarke N. G., and Dawson R. M. C. (1981) Alkaline O leads to N-transacylation. A new method for the quantitative deacylation of phospholipids. Biochem. J. 195, 301–306 10.1042/bj1950301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Michell R. H., and Allan D. (1975) Inositol cyclic phosphate as a product of phosphatidylinositol breakdown by phospholipase C (Bacillus cereus). FEBS Lett. 53, 302–304 10.1016/0014-5793(75)80041-X [DOI] [PubMed] [Google Scholar]
- 85. Zoerner A. A., Batkai S., Suchy M. T., Gutzki F. M., Engeli S., Jordan J., and Tsikas D. (2012) Simultaneous UPLC–MS/MS quantification of the endocannabinoids 2-arachidonoyl glycerol (2AG), 1-arachidonoyl glycerol (1AG), and anandamide in human plasma: minimization of matrix-effects, 2AG/1AG isomerization and degradation by toluene solvent extraction. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 883–884, 161–171 10.1016/j.jchromb.2011.06.025 [DOI] [PubMed] [Google Scholar]
- 86. Mutemberezi V., Masquelier J., Guillemot-Legris O., and Muccioli G. G. (2016) Development and validation of an HPLC-MS method for the simultaneous quantification of key oxysterols, endocannabinoids, and ceramides: variations in metabolic syndrome. Anal. Bioanal. Chem. 408, 733–745 10.1007/s00216-015-9150-z [DOI] [PubMed] [Google Scholar]
- 87. Masquelier J., and Muccioli G. G. (2016) Development and validation of a specific and sensitive HPLC-ESI-MS method for quantification of lysophosphatidylinositols and evaluation of their levels in mice tissues. J. Pharm. Biomed. Anal. 126, 132–140 10.1016/j.jpba.2016.04.011 [DOI] [PubMed] [Google Scholar]
- 88. Rao M., and Sockanathan S. (2005) Transmembrane protein GDE2 induces motor neuron differentiation in vivo. Science 309, 2212–2215 10.1126/science.1117156 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are contained within the article and in the supporting information.