

Abstract
Heavy metals released into the environment have a significant effect on respiratory health. Lung macrophages are important in mounting an inflammatory response to injury, but they are also involved in repair of injury. Macrophages develop mixed phenotypes in complex pathological conditions and polarize to a predominant phenotype depending on the duration and stage of injury and/or repair. Little is known about the reprogramming required for lung macrophages to switch between these divergent functions; therefore, understanding the mechanism(s) by which macrophages promote metabolic reprogramming to regulate lung injury is essential. Here, we show that lung macrophages polarize to a pro-inflammatory, classically activated phenotype after cadmium-mediated lung injury. Because metabolic adaptation provides energy for the diverse macrophage functions, these classically activated macrophages show metabolic reprogramming to glycolysis. RNA-Seq revealed up-regulation of glycolytic enzymes and transcription factors regulating glycolytic flux in lung macrophages from cadmium-exposed mice. Moreover, cadmium exposure promoted increased macrophage glycolytic function with enhanced extracellular acidification rate, glycolytic metabolites, and lactate excretion. These observations suggest that cadmium mediates the persistence of classically activated lung macrophages to exacerbate lung injury.
Keywords: macrophage, metal, lung injury, redox regulation, peroxisome proliferator-activated receptor (PPAR), cadmium
Environmental exposure to heavy metals is often overlooked in disease development (1, 2). Although smoking tobacco is an important source of heavy metal exposure in humans, cadmium exposure has been shown to double the risk of lung disease (2). The lung is the primary exposure route to environmental toxins and cadmium is absorbed more efficiently by the lungs with 50% of inhaled cadmium absorbed compared with 5% when orally ingested (3). Cadmium is widely distributed in the environment and natural air emission sources coming from volcanoes, airborne soil particles, and forest fires (3, 4). People residing near coal fired power plants, coke ovens, municipal waste, or strip mines are exposed to anthropogenic sources of cadmium due to movement of soil through wind, vehicles, and equipment (5).
Lung macrophages have a critical role in mounting an immune response to injury (6–8). Cadmium has been shown to suppress the innate immune response of macrophages. Lung macrophages are also essential in mediating remodeling of the alveolar wall that is necessary following lung injury. To accommodate these diverse functions, macrophages are highly plastic and are able to adapt to their local environment (9–12). Macrophages switch between phenotypically distinct subpopulations, the classically activated, pro-inflammatory and the alternatively activated, anti-inflammatory macrophages. Macrophage polarization is tightly regulated, and mixed populations exist in complex pathological conditions (9–12).
Peroxisome proliferator-activated receptor γ (PPARγ), a ligand-activated nuclear receptor, is essential for the differentiation of alveolar macrophages from fetal monocytes (13). PPARγ has been implicated as a negative regulator of the inflammatory response by inhibiting production of pro-inflammatory cytokines (14). PPARγ has also been reported to control the alternative activation of monocytes and macrophages (15).
With environmental levels of cadmium steadily rising (16) and the high morbidity and mortality associated with acute lung injury, understanding the pathogenesis and progression of cadmium-mediated lung injury is essential. Here, we aimed to identify the mechanism(s) regulating the metabolic reprogramming of lung macrophages that contributes to lung injury after cadmium exposure.
Results
Cadmium promoted the persistence of the pro-inflammatory macrophage phenotype
Lung macrophages have a critical role in mounting an immune response to injury (6–8). Cadmium has been shown to suppress the innate immune response of macrophages (6, 17). We have shown that cadmium inhibits lung macrophage host defense by inhibiting the Rho GTPase, Rac2 (6); however, it is not known if cadmium regulates the phenotypic switching of macrophages to influence lung repair. We found that cadmium promotes macrophage polarization to the pro-inflammatory, classically activated phenotype. TNFα and iNOS mRNA expression were increased 3-8–fold, respectively, in macrophages exposed to CdCl2 (Fig. 1, A and B). The alternatively activated markers, arginase 1, TGF-β1, IL-10, and PDGF-B were significantly reduced compared with vehicle exposed (Fig. 1, C–F).
Figure 1.
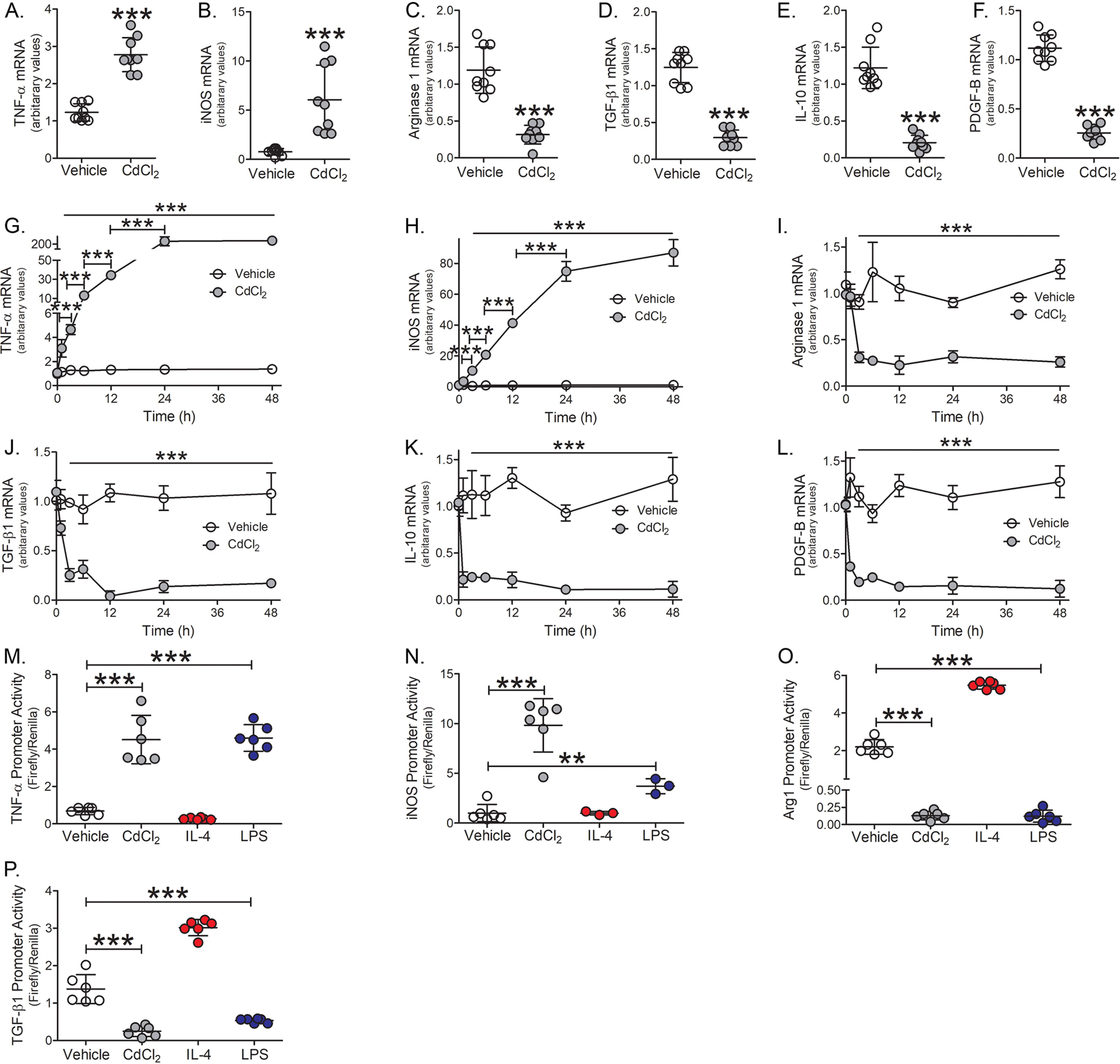
Cadmium promotes the persistence of the pro-inflammatory macrophage phenotype. THP-1 macrophages were exposed to vehicle or CdCl2 (50 μm, 3 h). A, TNFα; B, iNOS; C, arginase 1; D, TGF-β1; E, IL-10; and F, PDGF-B mRNA expression (n = 9). THP-1 cells were treated with vehicle or CdCl2 for the indicated times. G, TNFα; H, iNOS; I, arginase 1; J, TGF-β1; K, IL-10; and L, PDGF-B mRNA expression (n = 3). THP-1 cells were exposed to vehicle, CdCl2 (50 μm), IL-4 (20 ng/ml, negative control), or LPS (100 μg/ml, positive control) for 3 h. M, TNFα; N, iNOS; O, arginase 1; and P, TGF-β1 promoter activity were measured by luciferase assay (n = 6). **, p < 0.001; ***, p < 0.0001. Values are shown as mean ± S.D.
To determine whether cadmium mediated the persistence of a pro-inflammatory phenotype, macrophages were exposed to cadmium for 0, 1, 3, 6, 12, 24, or 48 h. Cadmium-exposed macrophages showed a time-dependent increase in TNFα and iNOS mRNA expression with maximal expression seen 24-48 h after exposure (Fig. 1, G and H). Arginase 1 and TGF-β1 expression were drastically reduced in macrophages exposed to cadmium for 3 h (Fig. 1, I and J), whereas IL-10 and PDGF-B were significantly reduced 1 h after cadmium exposure (Fig. 1, K and L). The reduction in anti-inflammatory gene expression from cadmium-exposed macrophages persisted over the duration of the time course.
Further examining the role of cadmium gene regulation, TNFα promoter activity showed nearly 4-fold increase in cadmium-exposed macrophages and was similar to LPS-exposed macrophages, which was used as a positive control (Fig. 1M). iNOS promoter activity increased nearly 10-fold in cadmium-exposed macrophages (Fig. 1N). In contrast, arginase 1 and TGF-β1 promoter activities were significantly reduced in cadmium-exposed macrophages (Fig. 1, O and P). Cadmium exposure did not alter macrophage apoptosis as no changes were detected in the mitochondrial or cytoplasmic localization of Bcl-2, cytochrome c, or Bax expression (Fig. S1, A and B). Moreover, caspase-3 activity remained unchanged over the duration in cadmium-exposed macrophages (Fig. S1C). These data suggest that cadmium induces a rapid and persistent polarization of macrophages to a pro-inflammatory phenotype.
Cadmium-induced lung injury and the pro-inflammatory phenotype of lung macrophages
To determine the biological relevance of our in vitro observations, we exposed WT mice to cadmium at the mean concentration found in bronchioalveolar lavage (BAL) fluid from cigarette smoke-exposed mice (6). BAL was performed 7 days later. Cadmium-exposed mice showed over 2.5-fold increase in the number of BAL cells (Fig. 2A) and greater than 95% of the cells were macrophages (Fig. 2B). Cadmium exposure induced the polarization of lung macrophages to a pro-inflammatory phenotype. Lung macrophages isolated from cadmium-exposed WT mice showed over 5-fold increase in TNFα and iNOS mRNA expression compared with vehicle-exposed (Fig. 2, C and D). In contrast, arginase 1, TGF-β1, IL-10, and PDGF-B mRNA expression were significantly reduced in lung macrophages from cadmium-exposed mice (Fig. 2, E–H). Cadmium exposure did not alter the expression or localization of Bcl-2, cytochrome c, or Bax in lung macrophages (Fig. S2, A and B) and caspase-3 activity remained unchanged between vehicle and cadmium-exposed mice (Fig. S2C).
Figure 2.
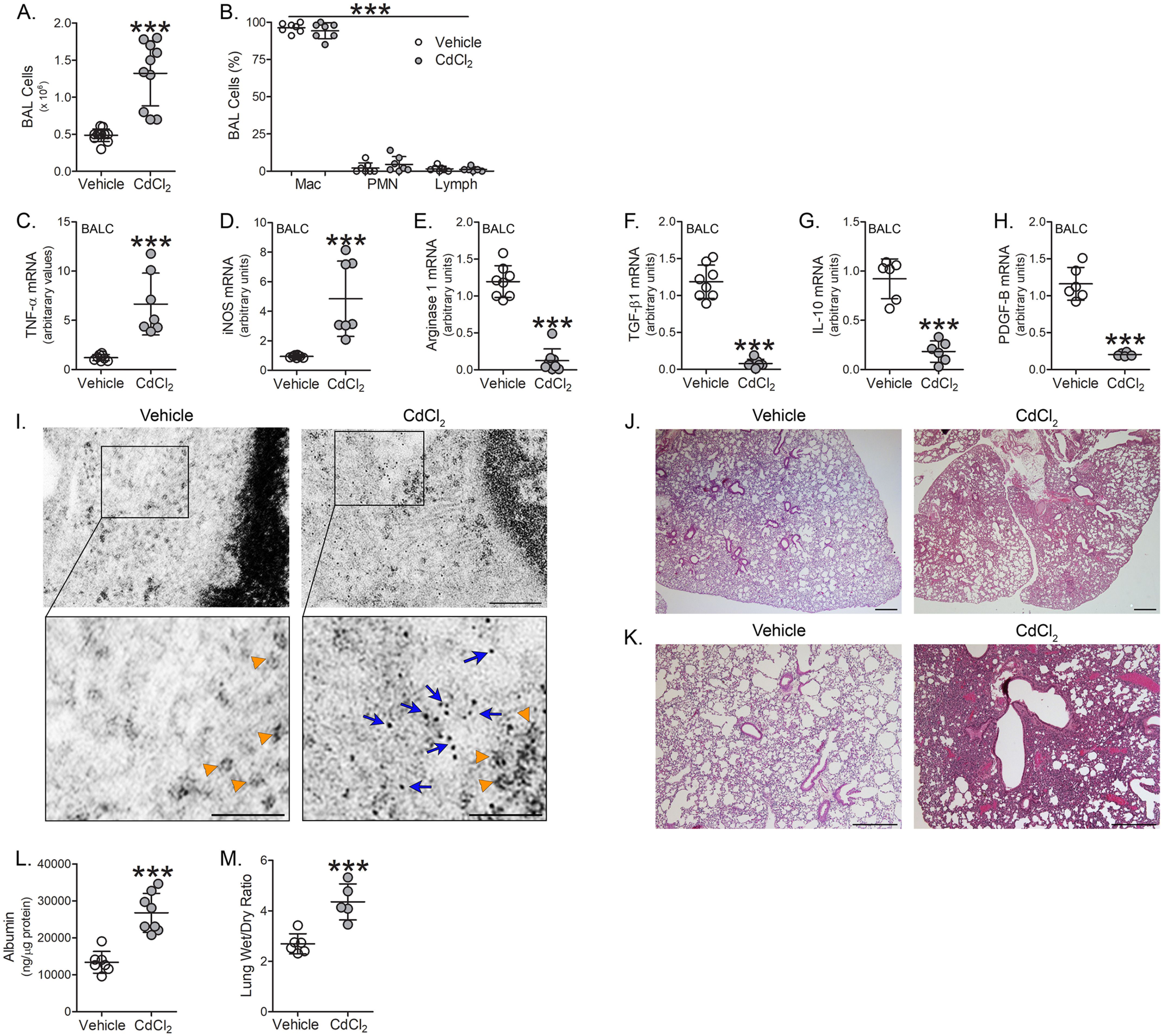
Cadmium induces lung injury and the pro-inflammatory phenotype of lung macrophages. WT mice were exposed to vehicle or CdCl2 (100 ng/kg, intratracheal). After 7 days, BAL was performed. A, total number of BAL cells (n = 10-11) and B, cell differential (n = 7) from exposed mice. C, TNFα; D, iNOS; E, arginase 1; F, TGF-β1; G, IL-10; and H, PDGF-B mRNA expression in isolated BAL cells from exposed mice (n = 6-8). I, representative TEM analysis of BAL cells from exposed mice. Orange arrowheads indicate ribosomes. Blue arrows indicate cadmium particles. Scale bars: 200 nm (main), 100 nm (insets) (n = 3). J and K, representative H & E staining of lung tissue from exposed mice (n = 3). Scale bars: 500 nm. L, albumin levels in BAL fluid from exposed mice (n = 7-8). M, wet to dry ratio of lung weight from exposed mice (n = 5-6). p < 0.0001. Mac, macrophage; PMN, polymorphonuclear leukocyte; Lymph, lymphocyte. Values are shown as mean ± S.D.
Because macrophages are phagocytic cells and to confirm that there is uptake and accumulation of cadmium in lung macrophages, transmission EM (TEM) analysis of isolated lung macrophages from cadmium-exposed mice revealed an accumulation of dark aggregates (∼1 nm in diameter, blue arrows, for reference ribosomes are indicated with orange arrowheads) found in the cytoplasm of macrophages, whereas this was absent in vehicle-exposed (Fig. 2I). Lung histology from cadmium-exposed mice revealed increased lobar consolidation associated with hemorrhage, whereas vehicle-exposed mice showed normal lung architecture (Fig. 2, J and K). These results were confirmed by increased albumin concentration in the BAL fluid indicating the presence of lung injury in cadmium-exposed mice (Fig. 2L). Moreover, the ratio of wet to dry lung weight was significantly increased in the cadmium-exposed mice (Fig. 2M). These results suggest a correlation between the cadmium-mediated lung macrophage phenotype and lung injury.
Cadmium-increased lung macrophage mitochondrial ROS
The redox-sensitive transcription factor, nuclear factor (NF)-κB, is a critical mediator of the macrophage inflammatory response and is required for pro-inflammatory gene expression (18–21). Using a promoter construct driven by NF-κB, we found that cadmium-exposed macrophages had significantly greater NF-κB–driven luciferase activity than vehicle exposed and the cadmium-induced increase in activity was significantly greater than LPS-exposed (Fig. 3A). Moreover, the transcription factors associated with classical activation, p-STAT1, and the p65 subunit of NF-κB, were localized to the nuclear fraction in macrophages exposed to cadmium, whereas p65 remained in the cytosol in vehicle-exposed (Fig. 3, B and C).
Figure 3.
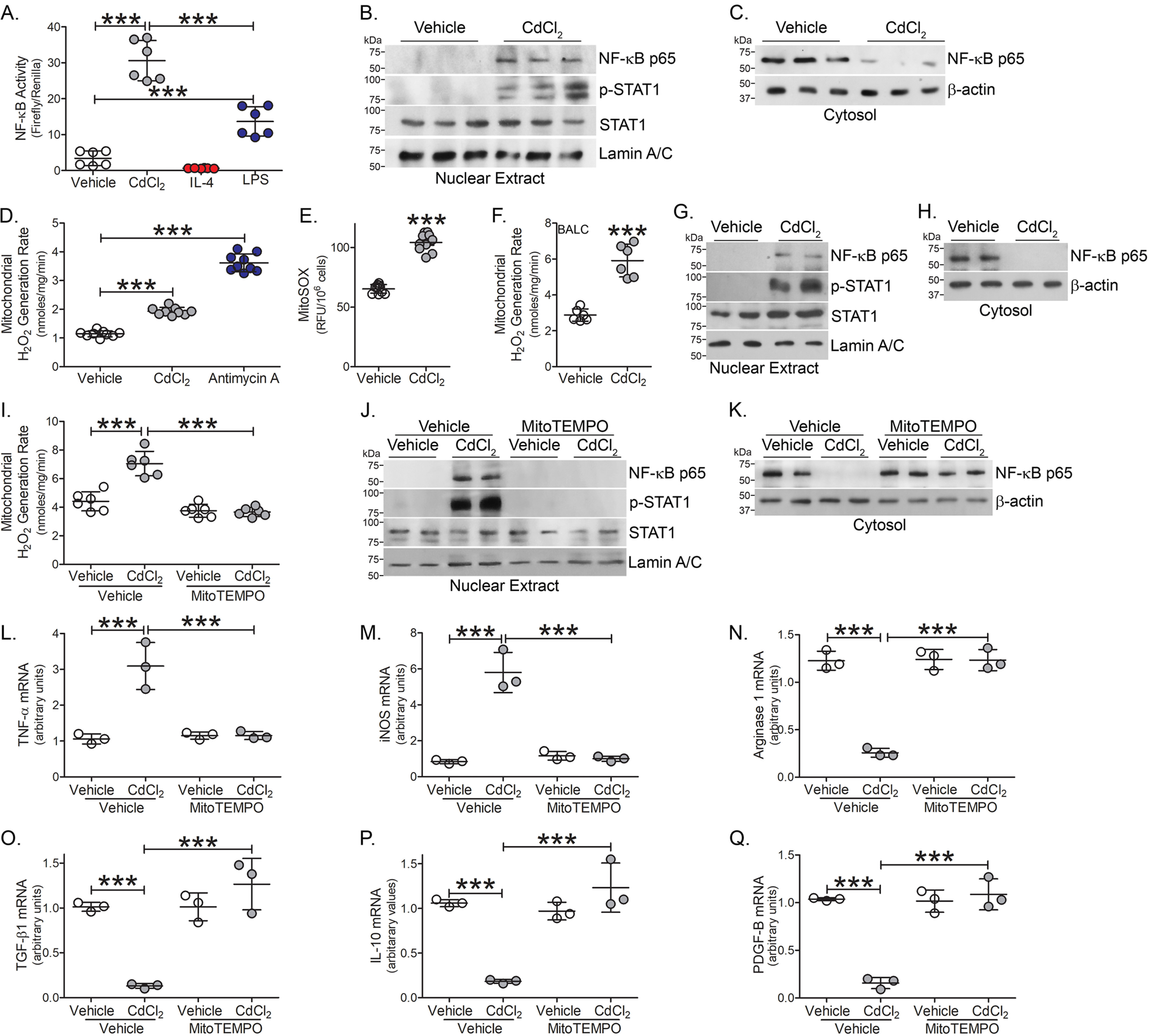
Cadmium increases lung macrophage mitochondrial ROS. A, NF- κB activity was measured by luciferase assay in THP-1 cells exposed to vehicle, CdCl2 (50 μm), IL-4 (20 ng/ml, negative control), or LPS (100 μg/ml, positive control) for 3 h (n = 6). Immunoblot analysis of exposed THP-1 cells in isolated (B) nuclear and (C) cytosolic extracts. mtROS generation in exposed THP-1 cells by (D) pHPA assay (n = 9) and (E) mitoSOX (antimycin A, 100 μm for 30 min, positive control) (n = 10). F, mtROS generation in BAL cells isolated from WT mice exposed to vehicle or CdCl2 for 7 days (n = 6). Immunoblot analysis of BAL cells isolated from exposed mice in (G) nuclear and (H) cytosolic extracts. THP-1 cells were treated with vehicle or mitoTEMPO (10 μm, 16 h) and exposed to vehicle or CdCl2 for 3 h. I, mtROS generation (n = 3). Immunoblot analysis in isolated (J) nuclear and (K) cytosolic fractions. L, TNFα; M, iNOS; N, arginase 1; O, TGF-β1; P, IL-10; and Q, PDGF-B mRNA expression (n = 3). ***, p < 0.0001. Values are shown as mean ± S.D.
Mitochondrial ROS (mtROS) are suggested to act as signal-transducing molecules that trigger inflammation by driving pro-inflammatory cytokine production (22, 23). We determined that cadmium-exposed macrophages showed a significant increase in mtROS generation (Fig. 3, D and E). These results were validated in vivo. The increased mtROS production in lung macrophages from cadmium-exposed mice (Fig. 3F) correlated with the activation of redox-regulated transcription factors. Lung macrophages from cadmium-exposed mice showed increased nuclear localization of the p65 subunit of NF-κB and p-STAT1 (Fig. 3G), whereas the p65 subunit of NF-κB remained in the cytosol in macrophages from vehicle-exposed mice (Fig. 3H).
Validating that cadmium-mediated regulation of transcription factor expression was induced by mtROS generation, cadmium-exposed macrophages were treated with mitoTEMPO, a specific scavenger of mtROS. MitoTEMPO treatment significantly reduced cadmium-mediated mtROS generation to the level seen in vehicle control (Fig. 3I). Nuclear localization of the p65 subunit of NF-κB and p-STAT1 were reduced with mitoTEMPO treatment, whereas the p65 subunit of NF-κB was increased in the cytosolic fraction with mitoTEMPO treatment in macrophages exposed to vehicle or cadmium. (Fig. 3, J and K).
Abrogating cadmium-mediated mtROS generation in macrophages influenced macrophage phenotypic switching. Treatment with mitoTEMPO reduced TNFα and iNOS gene expression to the level seen with vehicle alone (Fig. 3, L and M). In contrast, mitoTEMPO treatment rescued anti-inflammatory gene expression in cadmium-exposed macrophages similar to vehicle-exposed levels (Fig. 3, N–Q). These data suggest that cadmium-induced mtROS regulates redox-regulated transcription factors.
Cadmium promoted glycolytic flux in lung macrophages
Because hypoxia-inducible factor 1α (HIF-1α) is a critical regulator of macrophage inflammation (24), we determined that cadmium-exposed macrophages had increased HIF-1α expression in the nuclear fraction (Fig. 4A). Inhibiting cadmium-mediated mtROS generation with mitoTEMPO treatment abolished HIF-1α expression. Studies suggest classically activated macrophages utilize glycolytic metabolism, which can be rapidly activated to fuel responses for injury (25–27). Additionally, HIF-1α influences the metabolic reprogramming of macrophages to glycolysis (28). A critical activator of glucose metabolism, 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3), was similarly increased in macrophages exposed to cadmium and the expression of PFKFB3 was redox-regulated (Fig. 4B).
Figure 4.
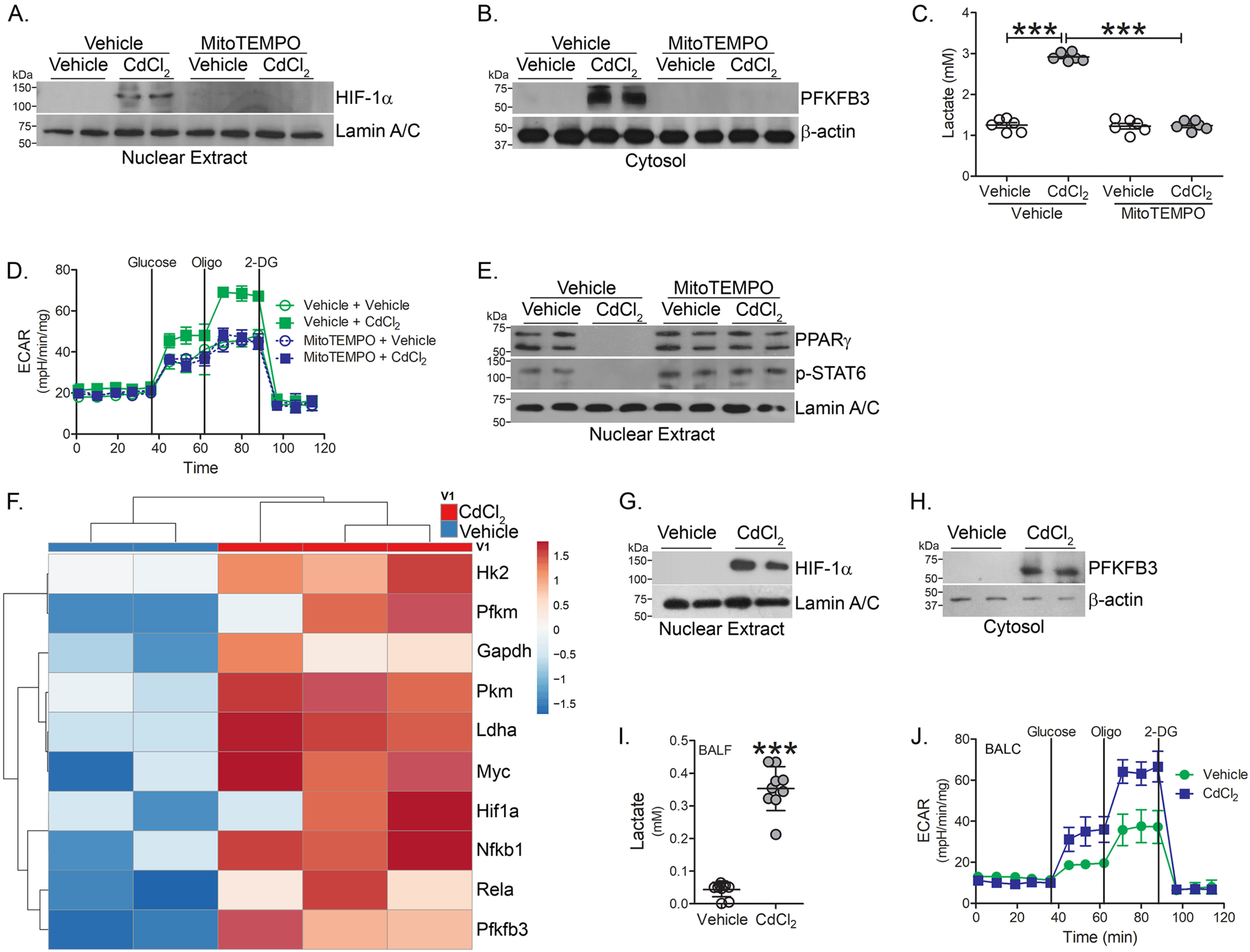
Cadmium promotes glycolytic flux in lung macrophages. THP-1 cells were treated with vehicle or mitoTEMPO (10 μm, 16 h) and exposed to vehicle or CdCl2 for 3 h. Immunoblot analysis in isolated (A) nuclear and (B) cytosolic extracts. C, lactate levels in condition media from exposed THP-1 cells (n = 6). D, ECAR in MH-S cells treated with vehicle or mitoTEMPO and exposed to vehicle or CdCl2 (n = 5). E, immunoblot analysis in isolated nuclear extracts from THP-1 cells. WT mice were exposed to vehicle or CdCl2 (100 ng/kg, intratracheal). After 7 days, BAL was performed. F, heatmap of RNA-Seq data of isolated BAL cells from exposed mice (n = 2-3). Immunoblot analysis of BAL cells from isolated (G) nuclear and (H) cytosolic fractions. I, lactate levels in BAL fluid from exposed mice (n = 9-10). J, ECAR in BAL cells from exposed mice (n = 5). ***, p < 0.0001. Values are shown as mean ± S.D.
To measure glycolytic flux, lactate excretion was measured in conditioned media from cadmium-exposed macrophages. Lactate was significantly increased in cell culture media from cadmium-exposed macrophages, whereas mitoTEMPO reduced lactate to control levels (Fig. 4C). Measuring glycolytic function via extracellular acidification rate (ECAR), cadmium-exposed macrophages showed increased ECAR compared with vehicle-exposed macrophages (Fig. 4D).
Studies indicate PPARγ plays a critical role in mediating the alternative activation of macrophages (15, 29). We determined that PPARγ expression was absent in CdCl2-exposed macrophages (Fig. 4E). Treating macrophages with mitoTEMPO induced alternative activation with STAT6 activation and PPARγ nuclear expression in cadmium-exposed macrophages. These data suggest that inhibiting mtROS generation in cadmium-exposed macrophages induces the phenotypic switching of macrophages and reduced glycolytic metabolism, a key feature of alternatively activated macrophages.
To further understand the bioenergetics in cadmium-exposed macrophages, RNA-Seq was performed in lung macrophages isolated from vehicle- and cadmium-exposed mice. Key glycolytic enzymes, hexokinase 2 (Hk2), phosphofructokinase (Pfkm), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), pyruvate kinase (PKM), and lactate dehydrogenase A (LDHA) were greater in cadmium-exposed lung macrophages (Fig. 4F). The transcription factors, c-myc and Hif1α, considered master regulators of glycolytic flux, were increased in lung macrophages from cadmium-exposed mice.
Because cellular metabolism plays an important role during macrophage polarization, we validated that HIF-1α was increased in lung macrophages from cadmium-exposed mice (Fig. 4G). Cadmium promoted a shift from mitochondrial oxidation toward glycolytic metabolism. Confirming the RNA-Seq data, lung macrophages isolated from cadmium-exposed mice showed increased PFKFB3 expression (Fig. 4H). Furthermore, BAL fluid from cadmium-exposed mice had elevated lactate levels, a measure of glycolytic flux (Fig. 4I). The increased flux was confirmed by an increase in ECAR in the cadmium-exposed mice (Fig. 4J). These data suggest that cadmium regulates the metabolic reprogramming to glycolysis, a critical bioenergetic characteristic in classically activated macrophages.
Cadmium altered macrophage glycolytic intermediates
Because cadmium treatment drastically reduced PPARγ expression and to understand the role of cadmium in promoting glycolysis, glycolytic intermediates and amino acid levels were measured by MS in macrophages expressing PPARγ and exposed to cadmium (Fig. 5A). Expression of PPARγ or treatment of cadmium did not alter glucose, glucose 6-phosphate, ribose 5-phosphate, or fructose 1,6-bisphosphate levels (Fig. 5, B–D). Although macrophages exposed to cadmium showed no change in glyceraldehyde 3-phosphate, PPARγ-expressing macrophages showed increased levels (Fig. 5E). Suggesting an increase in glycolysis, cadmium-exposed macrophages had a significant increase in 3-phosphoglycerate (Fig. 5F). Whereas serine levels remained unchanged, cysteine and glycine synthesis were reduced in cadmium-exposed and PPARγ-expressing macrophages. Further implicating enhanced glycolysis, phosphoenolpyruvate, and alanine levels were increased in cadmium-exposed macrophages and PPARγ expression reduced levels to that seen in controls (Fig. 5G). These studies suggest that cadmium promotes glycolysis in lung macrophages and PPARγ may provide a therapeutic mechanism to abrogate cadmium-mediated lung injury.
Figure 5.

Cadmium alters macrophage glycolytic intermediates. THP-1 cells expressing empty or PPARγ were treated with vehicle of CdCl2. A, schematic of the glycolytic pathway. B, glucose, immunoblot analysis; C, glucose 6-phosphate, ribose 5-phosphate; D, fructose 1,6-bisphosphate; E, glyceraldehyde 3-phosphate; F, 3-phosphoglycerate, serine, cysteine, glycine; and G, phosphoenolpyruvate, alanine levels by GC-TOF (n = 5). *, p < 0.01; **, p < 0.001; ***, p < 0.0001. Values are shown as mean ± S.D.
PPARγ induced the alternative phenotype in cadmium-exposed macrophages
Because PPARγ is a critical regulator of the anti-inflammatory phenotype in macrophages, we questioned if overexpression of PPARγ could alter the cadmium-mediated phenotypic switching of macrophages. Macrophages expressing PPARγ showed significantly reduced mtROS generation after cadmium exposure (Fig. 6A). The down-regulation of cadmium-mediated mtROS completely abrogated gene expression of TNFα and iNOS mRNA in cadmium-exposed macrophages expressing PPARγ (Fig. 6, B and C). The reverse was seen with the anti-inflammatory markers. PPARγ-expressing macrophages had significantly greater arginase 1, TGF-β1, IL-10, and PDGF-B mRNA expression (Fig. 6, D–G). More importantly, cadmium exposure did not alter expression of these genes in PPARγ-expressing macrophages.
Figure 6.
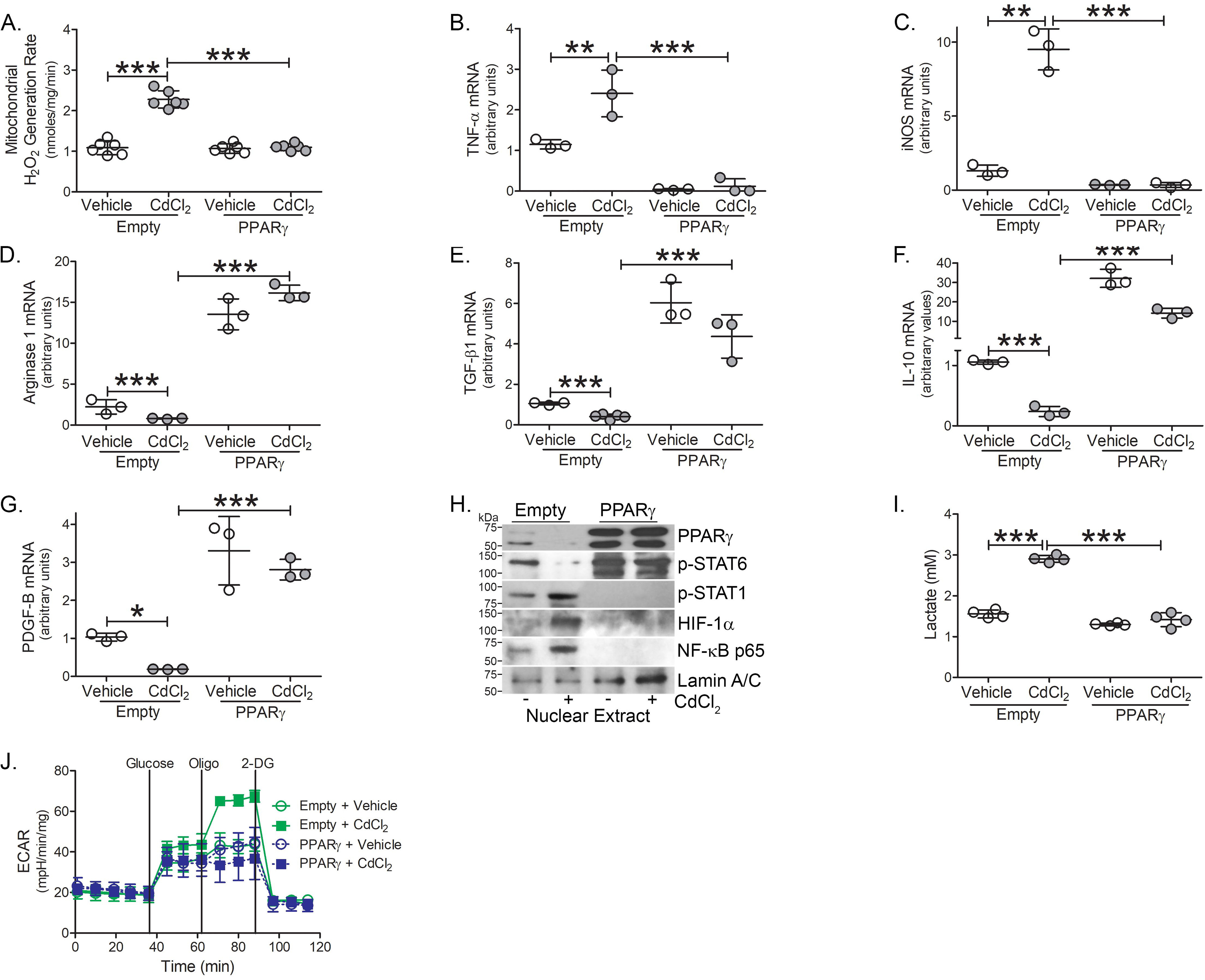
PPARγ induces the alternative phenotype in cadmium-exposed macrophages. THP-1 cells expressing empty or PPARγ were treated with vehicle of CdCl2. A, mtROS generation (n = 6). B, TNFα; C, iNOS; D, arginase 1; E, TGF-β1; F, IL-10; and G, PDGF-B mRNA expression (n = 3). H, Immunoblot analysis in the nuclear extract. I, lactate levels in conditioned media from treated THP-1 cells (n = 4). J, ECAR analysis (n = 5). *, p < 0.01; **, p < 0.001; ***, p < 0.0001. Values are shown as mean ± S.D.
Because PPARγ regulated macrophage phenotypic switching in cadmium-exposed macrophages, we determined if PPARγ altered the activation of redox-regulated transcription factors. Macrophages expressing PPARγ showed an absence of the transcription factors associated with classical activation, p-STAT1, and the p65 subunit of NF-κB in the nuclear fraction in macrophages exposed to cadmium, whereas p-STAT6 was highly up-regulated (Fig. 6H). Furthermore, PPARγ-expressing macrophages decreased HIF-1α expression, showed reduced lactate levels, and failed to undergo metabolic reprogramming toward glycolysis (Fig. 6, I and J). Cadmium exposure showed no effect in the presence of PPARγ. Taken together, these studies suggest that PPARγ-mediated phenotypic switching is associated with metabolic reprogramming in macrophages exposed to cadmium.
Discussion
Altered amino acid metabolism is a hallmark in defining macrophage phenotype. Pro-inflammatory macrophages convert l-arginine to nitric oxide by increasing iNOS activity, whereas anti-inflammatory macrophages have increased arginase 1, which converts l-arginine to urea (25). Classically activated, pro-inflammatory macrophages utilize glycolytic metabolism, which can be rapidly activated to fuel responses for injury (24, 25, 28). Although relatively inefficient in ATP production, the conversion of pyruvate into lactate is essential to restore NAD+ and maintain flux through the glycolytic pathway. Glycolytic flux is controlled by several enzymes, including hexokinase, phosphofructokinase, and pyruvate kinase. Although the nonoxidative part of glycolysis did not show accumulation of intermediates, oxidative glycolytic intermediates as well as exit products (alanine) were increased, suggesting that enzyme flux capacity was reached for pyruvate kinase and potentially GAPDH. This view is strengthened by the lack of significant differences of exit pathways, such as serine metabolism or pentose phosphate pathway intermediates. Moreover, changes in the concentration of metabolites can regulate flux via allosteric regulation of enzymes (26). Here, we show that cadmium-mediated lung injury results in the persistence of classically activated lung macrophages and induces the metabolic reprogramming of these cells to glycolysis to exacerbate lung injury.
The exposure of cadmium is closely associated with the development of lung diseases. Cadmium is one of the metal compounds present in cigarette smoke and each pack of cigarettes contains 30 μg of cadmium, but nearly 3 μg of cadmium is present in a single cigarette (27). In total, cigarette smokers have at least twice as much cadmium in body content than nonsmokers, and cadmium has no physiologic role in humans. Cadmium has an extremely long t1/2, up to 15-20 years (27). Thus, the physiologic effect of cadmium may be acute as well as being prolonged. Our data demonstrates the accumulation of cadmium in lung macrophages in an acute exposure; however, the accumulation in macrophages is not known in the prolonged setting.
Cadmium is not redox-active, but the generation of ROS is a critical mediator for cadmium-triggered tissue injury (30). Studies implicate cadmium-induced oxidative stress is mediated through cellular redox disruption by depletion of antioxidant enzymes (31). We show that cadmium-mediated mtROS generation regulates the polarization of macrophages, and studies indicate that mtROS are known to sustain inflammation by mediating pro-inflammatory cytokine secretion (32). Our data indicate that quenching mtROS induced the macrophage phenotype to become alternatively activated. Inhibiting cadmium-mediated mtROS reduced ECAR and may induce metabolic reprogramming to oxidative phosphorylation.
HIF-1α is a key transcription factor that regulates cell metabolism (24). Oxidative stress has been implicated in HIF-1α signaling, and mtROS has been shown to stabilize and activate HIF-1α (33, 34). There is some controversy on the role of cadmium and ROS in the regulation of HIF-1α. Cadmium-mediated ROS increased HIF-1α levels to induce malignant transformation of bronchial epithelial cells (35), whereas another study suggested that the inhibition of HIF-1α by cadmium was not secondary to oxidative stress (36). Thus, the regulation of HIF-1α by cadmium may be cell- and/or stimulus-specific.
The link between HIF-1α and NF-κB is also controversial. HIF-1α has been suggested to regulate NF-κB (37), and activation of HIF-1α may result from the inhibition of NF-κB (31). Other studies suggest NF-κB is a direct modulator of HIF-1α transcription during inflammatory conditions, such as hypoxia (38). Our data extended these findings by showing that cadmium-mediated mtROS generation regulates NF-κB activation and increases HIF-1α expression to potentially regulate metabolic reprogramming to glycolysis.
Evidence suggests that lactate competes with glucose as a mitochondrial substrate in the lung, and type II alveolar epithelial cells have been shown to readily utilize lactate (39). The effect of enhanced glycolysis is cell type-specific as increased glycolysis has been shown to protect alveolar epithelial cells from lung injury in an LPS model (40). Further studies are warranted to better understand the cross-talk between macrophages and alveolar epithelial cells, as we suggest the increased lactate is macrophage-derived and may play a critical role in lung injury.
PPARγ is a key regulator of lipid metabolism and inflammation and is expressed in various cell types throughout the human body. Our data indicate PPARγ may be a novel target for regulating macrophage metabolic reprogramming and influencing lung repair after cadmium exposure. PPARγ can be activated by synthetic ligands. Rosiglitizone and pioglitazone belong to the thiazolidinedione class of PPARγ agonists and are potent insulin-sensitizing drugs. Rosiglitazone has been shown to reduce bleomycin-induced lung fibrosis via inhibition of TGF-β1–mediated differentiation of lung fibroblasts (41, 42). Rosiglitazone decreased pulmonary artery remodeling in a rat model of pulmonary hypertension (43). Moreover, pioglitazone prevented LPS-induced acute lung injury in mice (44). Although the use of thiazolidinediones is associated with many undesirable side effects, such as weight gain, edema, and heart disease in diabetic patients (45), new classes of PPARγ agonists may be able to effectively target specific cells and tissues preserving the efficacy of these drugs whereas reducing their side effects.
Taken together, we show that cadmium-mediated mtROS in macrophages regulates redox-regulated transcription factors to maintain the persistence of a pro-inflammatory phenotype. These observations also suggest that the cadmium-induced pro-inflammatory phenotype in macrophages may hinder the resolution of lung injury.
Experimental procedures
Mice
All animal protocols were approved by the Institutional Animal Care and Use Committee of the University of Alabama at Birmingham and were performed in accordance with NIH guidelines. WT C57BL6 mice were purchased from JAX Laboratory (Bar Harbor, ME). 8- to 12-week–old male and female mice were intratracheally administered 100 ng/kg of CdCl2 or saline, as a vehicle control, after being anesthetized with 3% isoflurane using a precision Fortec vaporizer. Mice were fed ad libitum standard chow and kept at 12-h light/12-h dark cycles. Mice were euthanized 7 days after exposure and BAL was performed.
Cell culture
Human monocyte (THP-1) and mouse alveolar macrophage (MH-S) cell lines were obtained from American Type Culture Collection (Manassas, VA). Macrophages were maintained in RPMI 1640 media (Thermo Fisher Scientific) with 10% fetal bovine serum and penicillin/streptomycin supplements. All experiments were conducted in RPMI containing 0.5% serum. Cells were treated with vehicle or 50 μm CdCl2 for 3 h or the indicated time. Macrophages were treated with IL-4 (20 ng/ml, negative control) or LPS (100 μg/ml) as a positive control.
Quantitative real-time PCR
Total RNA was isolated, reverse transcribed, and quantitative real-time PCR was performed as described previously using previously published primer sets (46, 47). Data were calculated by the cycle threshold (ΔΔCT) method, normalized to β-actin or HPRT, and expressed in arbitrary units.
Plasmids, transfections, and luciferase assays
The PPARγ plasmid (8895) and iNOS promoter (19296) were purchased from Addgene (48, 49). TNFα promoter, arginase 1 promoter/enhancer, TGF-β1 promoter, and NF-κB gene expression were evaluated using luciferase reporter plasmids as previously described (19, 46, 47). The correct reading frame and sequence was verified by the Heflin Center Genomics Core at the University of Alabama. Cells were transfected using X-treme GENE 9 Transfection Reagent (Sigma) according to the manufacturer's protocol. Renilla and firefly luciferase activity was determined in cell lysates using the Dual Luciferase reporter assay kit (Promega) and normalized to control (firefly).
TEM
BAL cells were fixed in 2.5% paraformaldehyde and 2.5% glutaraldehyde in Sorenson's phosphate buffer as previously described (46). Cells were processed and sectioned with a diamond knife (Diatome, Electron Microscopy Sciences) at 70-80 nm and sections were placed on copper mesh grids. Sections were stained with uranyl acetate and lead citrate for contrast and viewed on a Tecnai Twin 120kv TEM (FEI).
ECAR
ECAR was determined using a Seahorse XF24 bioanalyzer (Seahorse Bioscience). In brief, 7.5 × 104 macrophages per well were subjected to ECAR measurement in the XF24 extracellular flux analyzer with sequential additions of the following conditions: glucose (10 mm), oligomycin (0.5 μm), and 2-deoxyglucose (50 μm).
Albumin
Albumin levels were determined in BAL fluid using the Mouse Albumin ELISA Kit (Immunology Consultants Laboratory) according to the manufacturer's protocol. Samples were diluted 1/500,000.
Lactate
Lactate was measured in conditioned media and BAL fluid using the Lactate Colorimetric/Fluorometric Assay Kit (BioVision) according to the manufacturer's protocol.
Next-generation RNA-Seq
mRNA-Seq was performed on the Illumina NextSeq500 as described by the manufacturer (Illumina Inc., San Diego, CA). Briefly, RNA quality was assessed using the Agilent 2100 Bioanalyzer. RNA with an RNA Integrity Number of ≥7.0 was used for sequencing library preparation. RNA passing quality control was converted to a sequencing ready library using the NEBNext Ultra II Directional RNA library kit as per the manufacturer's instructions (New England Biolaba, Ipswich, MA). The cDNA libraries were quantitated using quantitative PCR in a Roche LightCycler 480 with the Kapa Biosystems kit for Illumina library quantitation (Kapa Biosystems, Woburn, MA) prior to cluster generation. Cluster generation was performed according to the manufacturer's recommendations for onboard clustering (Illumina, San Diego, CA). 30-35 million paired end 75-bp sequencing reads were generated per sample for transcript level abundance.
Data assessment for RNA-Seq
STAR (version 2.5.3a) was used to align the raw RNA-Seq fastq reads to the reference genome from Gencode. Following alignment, HTSeq-count was used to count the number of reads mapping to each gene. Normalization and differential expression were applied to the count files using DESeq2. Data have been submitted to the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus database (GEO) with GEO accession number GSE155166.
Metabolite analysis
Glycolytic and amino acid intermediates were analyzed by Gas Chromatography Time-of-Flight mass spectrometry (GC-TOF) as previously described (50). Briefly, the Gerstel CIS4–with dual multipurpose sample injector and Agilent 6890 GC-Pegasus III TOF MS was used for analysis. Injector conditions include: 50 to 275 °C final temperature at a rate of 12 °C/s and holding for 3 min. Injection volume was 0.5 μl with 10 μl/s injection speed on a splitless injector with purge time of 25 s. GC conditions included: 30 m long, 0.25-mm inner diameter Rtx-5Sil MS column (0.25 μm 95% dimethyl, 5% diphenyl polysiloxane film) with an additional 10-m integrated guard column (Restek, Bellefonte PA). 99.99% pure helium with built-in purifier (Airgas, Radnor PA) was set at a constant flow of 1 ml/min and the oven temperature was held constant at 50 °C for 1 min, ramped at 20 °C/min to 330 °C, and held constant for 5 min. The Leco Pegasus IV TOF-MS was controlled by the Leco Chroma TOF software versus 2.32 (St. Joseph, MI). The transfer line temperature was set to 280 °C. Electron impact ionization at 70 V was employed with an ion source temperature of 250 °C. The acquisition rate was 17 spectra/s, with a scan mass range of 85-500 Da.
Data assessment for metabolomics
Raw data files are preprocessed directly after data acquisition and stored as Chroma TOF-specific *.peg files, as generic *.txt result files and additionally as generic ANDI MS *.cdf files as previously described (50). ChromaTOF versus 2.32 is used for data preprocessing without smoothing, 3 s peak width, baseline subtraction just above the noise level, and automatic mass spectral deconvolution and peak detection at signal/noise levels of 5:1 throughout the chromatogram. Apex masses are reported for use in the BinBase algorithm. Result *.txt files are exported to a data server with absolute spectra intensities and further processed by a filtering algorithm implemented in the metabolomics BinBase database.
Isolation of mitochondria and cytoplasm fractions
Mitochondria were isolated by lysing the cells in mitochondria buffer containing 10 mm Tris, pH 7.8, 0.2 mm EDTA, 320 mm sucrose, and protease inhibitors. Lysates were homogenized using a Kontes Pellet Pestle Motor and centrifuged at 2000 × g for 8 min at 4 °C. The supernatant was removed and incubated at 4 °C, and the pellet was lysed, homogenized, and centrifuged again. The two supernatants were pooled and centrifuged at 12,000 × g for 15 min at 4 °C. The pellet was washed in the mitochondrial buffer twice and then resuspended in mitochondria buffer without sucrose (10).
Isolation of nuclear fraction
Nuclear isolation was performed by resuspending cells in a lysis buffer (10 mm HEPES, 10 mm KCl, 2 mm MgCl2, 2 mm EDTA) for 15 min on ice. Nonidet P-40 (10%) was added to lyse the cells, and the cells were centrifuged at 4 °C at 14,000 rpm. The nuclear pellet was resuspended in an extraction buffer (50 mm HEPES, 50 mm KCl, 300 mm NaCl, 0.1 mm EDTA, 10% glycerol) for 20 min on ice. After centrifuging at 4 °C at 14,000 rpm, the supernatant was collected as nuclear extract (10).
Determination of mitochondrial ROS generation
Mitochondrial H2O2 production was determined fluorometrically by p-hydroxyphenylacetic acid (pHPA) assay. Freshly isolated mitochondria were incubated in phenol red-free Hanks' balanced salt solution supplemented with 6.5 mm glucose, 1 mm HEPES, 6 mm sodium bicarbonate, 1.6 mm pHPA, and 0.95 μg/ml of HRP Fluorescence of pHPA-dimer was measured using a spectrofluorometer at excitation of 320 nm and emission of 400 nm (46). Treatment of cells with antimycin A (100 μm for 30 min) was used as a positive control. MitoSOX, a mitochondrial superoxide indicator (Thermo Fisher Scientific), was used to detect mitochondrial superoxide anion according to the manufacturer's protocol. Equal numbers of cells were subjected to fluorescent reading (excitation, 510 nm; emission, 580 nm) as previously described (51).
Immunoblot analysis
Primary antibodies used were: Bax (2772S), Bcl-2 (3498), cytochrome c (4272), HIF-1α (79233), Lamin A/C (2032), PFKPB3 (13123), phospho-STAT1 (7649), phospho-STAT6 (9361), STAT1 (9172), VDAC (4866) (Cell Signaling); β-actin (A5441) (Sigma); NF-κB p65 (sc-372) (Santa Cruz Biotechnology); and PPARγ (A0270) (ABclonal).
Caspase-3 activity analysis
Caspase-3 activity was measured using EnzChek Caspase-3 Assay Kit Number 2 (Molecular Probes) according to the manufacturer's protocol. Cells were lysed in 1× lysis buffer, subjected to a freeze-thaw cycle, centrifuged to remove cellular debris, and loaded into individual microplate wells. The 2× reaction buffer with substrate was immediately added to the samples, and fluorescence was measured (excitation/emission 496/520 nm). A supplied inhibitor was used as a negative control in all experiments (11).
Statistics
Statistical comparisons were performed using a Student's t test when only two groups of data are presented, or one-way analysis of variance with a Tukey's post hoc test or two-way analysis of variance followed by Bonferroni post-test when multiple data groups are presents. All statistical analysis was expressed as mean ± S.D. and p < 0.05 was considered to be significant. GraphPad Prism statistical software was used for all analysis.
Data availability
All data are contained within the manuscript. Data have been submitted to the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus database (GEO) with GEO accession number GSE155166.
Supplementary Material
Acknowledgments
We thank Dr. David Crossman, University of Alabama at Birmingham, for assistance with RNA-seq.
This article contains supporting information.
Author contributions—J. L. L-C. and A. B. C. conceptualization; J. L. L-C. and A. B. C. resources; J. L. L-C., L. G., and O. F. data curation; J. L. L-C., L. G., O. F., and A. B. C. formal analysis; J. L. L-C. and A. B. C. funding acquisition; J. L. L-C. validation; J. L. L-C. and L. G. investigation; J. L. L-C. and A. B. C. visualization; J. L. L-C., L. G., and O. F. methodology; J. L. L-C. writing-original draft; J. L. L-C., O. F., and A. B. C. writing-review and editing; A. B. C. project administration.
Funding and additional information—This work was supported by a Parker B. Francis fellowship and American Lung Association Grant RG-507440 (to J. L. C.), National Institutes of Health Grants 2R01ES015981-12 and P42ES027723, and Department of Veteran Affairs Grant 1 I01 CX001715-01 (to A. B. C.), and Comprehensive Cancer Center Core Grant P30CA031148. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors have declared that no conflict of interest with the contents of this article.
- PPARγ
- peroxisome proliferator-activated receptor γ
- BAL
- bronchioalveolar lavage
- TNF
- tumor necrosis factor
- PDGF
- platelet-derived growth factor
- TEM
- transmission EM
- IL
- interleukin
- mtROS
- mitochondrial ROS
- HIF-1α
- hypoxia-inducible factor 1α
- PFKFB3
- 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3
- ECAR
- extracellular acidification rate
- HPRT
- hypoxanthine-guanine phosphoribosyltransferase
- LPS
- lipopolysaccharide
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- ROS
- reactive oxygen species
- pHPA
- p-hydroxyphenylacetic acid.
References
- 1. Lampe B. J., Park S. K., Robins T., Mukherjee B., Litonjua A. A., Amarasiriwardena C., Weisskopf M., Sparrow D., and Hu H. (2008) Association between 24-hour urinary cadmium and pulmonary function among community-exposed men: the VA Normative Aging Study. Environ. Health Perspect. 116, 1226–1230 10.1289/ehp.11265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mannino D. M., Holguin F., Greves H. M., Savage-Brown A., Stock A. L., and Jones R. L. (2004) Urinary cadmium levels predict lower lung function in current and former smokers: data from the Third National Health and Nutrition Examination Survey. Thorax 59, 194–198 10.1136/thorax.2003.012054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Faroon O., Ashizawa A., Wright S., Tucker P., Jenkins K., Ingerman L., and Rudisill C. (2012) Agency for Toxic Substances and Disease Registry (ATSDR) toxicological profiles. in Toxicological profile for cadmium, Agency for Toxic Substances and Disease Registry (US), Atlanta, GA: [PubMed] [Google Scholar]
- 4. UNEP (2008) Interim review of scientific information on cadmium. United Nations Environment Program, Geneva [Google Scholar]
- 5. Raja R., Nayak A. K., Shukla A. K., Rao K. S., Gautam P., Lal B., Tripathi R., Shahid M., Panda B. B., Kumar A., Bhattacharyya P., Bardhan G., Gupta S., and Patra D. K. (2015) Impairment of soil health due to fly ash-fugitive dust deposition from coal-fired thermal power plants. Environ. Monit. Assess. 187, 679 10.1007/s10661-015-4902-y [DOI] [PubMed] [Google Scholar]
- 6. Larson-Casey J. L., Gu L., Jackson P. L., Briles D. E., Hale J. Y., Blalock J. E., Wells J. M., Deshane J. S., Wang Y., Davis D., Antony V. B., Massicano A. V. F., Lapi S. E., and Carter A. B. (2018) Macrophage Rac2 is required to reduce the severity of cigarette smoke-induced pneumonia. Am. J. Respir. Crit. Care Med. 198, 1288–1301 10.1164/rccm.201712-2388OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Broug-Holub E., Toews G. B., van Iwaarden J. F., Strieter R. M., Kunkel S. L., Paine R. 3rd., and Standiford T. J. (1997) Alveolar macrophages are required for protective pulmonary defenses in murine Klebsiella pneumonia: elimination of alveolar macrophages increases neutrophil recruitment but decreases bacterial clearance and survival. Infect. Immun. 65, 1139–1146 10.1128/IAI.65.4.1139-1146.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Knapp S., Leemans J. C., Florquin S., Branger J., Maris N. A., Pater J., van Rooijen N., and van der Poll T. (2003) Alveolar macrophages have a protective antiinflammatory role during murine pneumococcal pneumonia. Am. J. Respir. Crit. Care Med. 167, 171–179 10.1164/rccm.200207-698OC [DOI] [PubMed] [Google Scholar]
- 9. Martinez F. O., and Gordon S. (2014) The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 6, 13 10.12703/P6-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. He C., Ryan A. J., Murthy S., and Carter A. B. (2013) Accelerated development of pulmonary fibrosis via Cu,Zn-superoxide dismutase-induced alternative activation of macrophages. J. Biol. Chem. 288, 20745–20757 10.1074/jbc.M112.410720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Larson-Casey J. L., Murthy S., Ryan A. J., and Carter A. B. (2014) Modulation of the mevalonate pathway by Akt regulates macrophage survival and development of pulmonary fibrosis. J. Biol. Chem. 289, 36204–36219 10.1074/jbc.M114.593285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Murthy S., Adamcakova-Dodd A., Perry S. S., Tephly L. A., Keller R. M., Metwali N., Meyerholz D. K., Wang Y., Glogauer M., Thorne P. S., and Carter A. B. (2009) Modulation of reactive oxygen species by Rac1 or catalase prevents asbestos-induced pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 297, L846–L855 10.1152/ajplung.90590.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schneider C., Nobs S. P., Kurrer M., Rehrauer H., Thiele C., and Kopf M. (2014) Induction of the nuclear receptor PPAR-γ by the cytokine GM-CSF is critical for the differentiation of fetal monocytes into alveolar macrophages. Nat. Immunol. 15, 1026–1037 10.1038/ni.3005 [DOI] [PubMed] [Google Scholar]
- 14. Jiang C., Ting A. T., and Seed B. (1998) PPAR-γ agonists inhibit production of monocyte inflammatory cytokines. Nature 391, 82–86 10.1038/34184 [DOI] [PubMed] [Google Scholar]
- 15. Odegaard J. I., Ricardo-Gonzalez R. R., Goforth M. H., Morel C. R., Subramanian V., Mukundan L., Red Eagle A., Vats D., Brombacher F., Ferrante A. W., and Chawla A. (2007) Macrophage-specific PPARγ controls alternative activation and improves insulin resistance. Nature 447, 1116–1120 10.1038/nature05894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Satarug S., Garrett S. H., Sens M. A., and Sens D. A. (2010) Cadmium, environmental exposure, and health outcomes. Environ. Health Perspect. 118, 182–190 10.1289/ehp.0901234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cox J. N., Rahman M. A., Bao S., Liu M., Wheeler S. E., and Knoell D. L. (2016) Cadmium attenuates the macrophage response to LPS through inhibition of the NF-κB pathway. Am. J. Physiol. Lung Cell Mol. Physiol. 311, L754–L765 10.1152/ajplung.00022.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Carter A. B., Knudtson K. L., Monick M. M., and Hunninghake G. W. (1999) The p38 mitogen-activated protein kinase is required for NF-κB-dependent gene expression: the role of TATA-binding protein (TBP). J. Biol. Chem. 274, 30858–30863 10.1074/jbc.274.43.30858 [DOI] [PubMed] [Google Scholar]
- 19. Carter A. B., and Hunninghake G. W. (2000) A constitutive active MEK → ERK pathway negatively regulates NF-κB-dependent gene expression by modulating TATA-binding protein phosphorylation. J. Biol. Chem. 275, 27858–27864 10.1074/jbc.M003599200 [DOI] [PubMed] [Google Scholar]
- 20. Carter A. B., Monick M. M., and Hunninghake G. W. (1998) Lipopolysaccharide-induced NF-κB activation and cytokine release in human alveolar macrophages is PKC-independent and TK- and PC-PLC- dependent. Am. J. Respir. Cell Mol. Biol. 18, 384–391 10.1165/ajrcmb.18.3.2972 [DOI] [PubMed] [Google Scholar]
- 21. Barnes P. J., and Karin M. (1997) Nuclear factor-κB: a pivotal transcription factor in chronic inflammatory diseases. N. Engl. J. Med. 336, 1066–1071 10.1056/NEJM199704103361506 [DOI] [PubMed] [Google Scholar]
- 22. Naik E., and Dixit V. M. (2011) Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J. Exp. Med. 208, 417–420 10.1084/jem.20110367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tephly L. A., and Carter A. B. (2007) Constitutive NADPH oxidase and increased mitochondrial respiratory chain activity regulate chemokine gene expression. Am. J. Physiol. Lung Cell Mol. Physiol. 293, L1143–L1155 10.1152/ajplung.00114.2007 [DOI] [PubMed] [Google Scholar]
- 24. Cramer T., Yamanishi Y., Clausen B. E., Förster I., Pawlinski R., Mackman N., Haase V. H., Jaenisch R., Corr M., Nizet V., Firestein G. S., Gerber H. P., Ferrara N., and Johnson R. S. (2003) HIF-1α is essential for myeloid cell-mediated inflammation. Cell 112, 645–657 10.1016/S0092-8674(03)00154-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Duffield J. S., Forbes S. J., Constandinou C. M., Clay S., Partolina M., Vuthoori S., Wu S., Lang R., and Iredale J. P. (2005) Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J. Clin. Invest. 115, 56–65 10.1172/JCI200522675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Link H., Fuhrer T., Gerosa L., Zamboni N., and Sauer U. (2015) Real-time metabolome profiling of the metabolic switch between starvation and growth. Nat. Methods 12, 1091–1097 10.1038/nmeth.3584 [DOI] [PubMed] [Google Scholar]
- 27. Hendrick D. J. (2004) Smoking, cadmium, and emphysema. Thorax 59, 184–185 10.1136/thx.2003.018432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tannahill G. M., Curtis A. M., Adamik J., Palsson-McDermott E. M., McGettrick A. F., Goel G., Frezza C., Bernard N. J., Kelly B., Foley N. H., Zheng L., Gardet A., Tong Z., Jany S. S., Corr S. C., et al. (2013) Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 496, 238–242 10.1038/nature11986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bouhlel M. A., Derudas B., Rigamonti E., Dièvart R., Brozek J., Haulon S., Zawadzki C., Jude B., Torpier G., Marx N., Staels B., and Chinetti-Gbaguidi G. (2007) PPARγ activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 6, 137–143 10.1016/j.cmet.2007.06.010 [DOI] [PubMed] [Google Scholar]
- 30. Wang Y., Fang J., Leonard S. S., and Rao K. M. (2004) Cadmium inhibits the electron transfer chain and induces reactive oxygen species. Free Radic. Biol. Med. 36, 1434–1443 10.1016/j.freeradbiomed.2004.03.010 [DOI] [PubMed] [Google Scholar]
- 31. Carbia-Nagashima A., Gerez J., Perez-Castro C., Paez-Pereda M., Silberstein S., Stalla G. K., Holsboer F., and Arzt E. (2007) RSUME, a small RWD-containing protein, enhances SUMO conjugation and stabilizes HIF-1α during hypoxia. Cell 131, 309–323 10.1016/j.cell.2007.07.044 [DOI] [PubMed] [Google Scholar]
- 32. Bulua A. C., Simon A., Maddipati R., Pelletier M., Park H., Kim K. Y., Sack M. N., Kastner D. L., and Siegel R. M. (2011) Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J. Exp. Med. 208, 519–533 10.1084/jem.20102049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chandel N. S., Maltepe E., Goldwasser E., Mathieu C. E., Simon M. C., and Schumacker P. T. (1998) Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. U.S.A. 95, 11715–11720 10.1073/pnas.95.20.11715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chandel N. S., McClintock D. S., Feliciano C. E., Wood T. M., Melendez J. A., Rodriguez A. M., and Schumacker P. T. (2000) Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1α during hypoxia: a mechanism of O2 sensing. J. Biol. Chem. 275, 25130–25138 10.1074/jbc.M001914200 [DOI] [PubMed] [Google Scholar]
- 35. Jing Y., Liu L. Z., Jiang Y., Zhu Y., Guo N. L., Barnett J., Rojanasakul Y., Agani F., and Jiang B. H. (2012) Cadmium increases HIF-1 and VEGF expression through ROS, ERK, and AKT signaling pathways and induces malignant transformation of human bronchial epithelial cells. Toxicol. Sci. 125, 10–19 10.1093/toxsci/kfr256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chun Y. S., Choi E., Kim G. T., Choi H., Kim C. H., Lee M. J., Kim M. S., and Park J. W. (2000) Cadmium blocks hypoxia-inducible factor (HIF)-1-mediated response to hypoxia by stimulating the proteasome-dependent degradation of HIF-1α. Eur. J. Biochem. 267, 4198–4204 10.1046/j.1432-1327.2000.01453.x [DOI] [PubMed] [Google Scholar]
- 37. Walmsley S. R., Print C., Farahi N., Peyssonnaux C., Johnson R. S., Cramer T., Sobolewski A., Condliffe A. M., Cowburn A. S., Johnson N., and Chilvers E. R. (2005) Hypoxia-induced neutrophil survival is mediated by HIF-1α-dependent NF-kappaB activity. J. Exp. Med. 201, 105–115 10.1084/jem.20040624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rius J., Guma M., Schachtrup C., Akassoglou K., Zinkernagel A. S., Nizet V., Johnson R. S., Haddad G. G., and Karin M. (2008) NF-κB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1α. Nature 453, 807–811 10.1038/nature06905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lottes R. G., Newton D. A., Spyropoulos D. D., and Baatz J. E. (2015) Lactate as substrate for mitochondrial respiration in alveolar epithelial type II cells. Am. J. Physiol. Lung Cell Mol. Physiol. 308, L953–961 10.1152/ajplung.00335.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tojo K., Tamada N., Nagamine Y., Yazawa T., Ota S., and Goto T. (2018) Enhancement of glycolysis by inhibition of oxygen-sensing prolyl hydroxylases protects alveolar epithelial cells from acute lung injury. FASEB J. 32, 2258–2268 10.1096/fj.201700888R [DOI] [PubMed] [Google Scholar]
- 41. Genovese T., Cuzzocrea S., Di Paola R., Mazzon E., Mastruzzo C., Catalano P., Sortino M., Crimi N., Caputi A. P., Thiemermann C., and Vancheri C. (2005) Effect of rosiglitazone and 15-deoxy-Δ12,14-prostaglandin J2 on bleomycin-induced lung injury. Eur. Respir. J. 25, 225–234 10.1183/09031936.05.00049704 [DOI] [PubMed] [Google Scholar]
- 42. Kheirollahi V., Wasnick R. M., Biasin V., Vazquez-Armendariz A. I., Chu X., Moiseenko A., Weiss A., Wilhelm J., Zhang J.-S., Kwapiszewska G., Herold S., Schermuly R. T., Mari B., Li X., Seeger W., et al. (2019) Metformin induces lipogenic differentiation in myofibroblasts to reverse lung fibrosis. Nat. Commun. 10, 2987 10.1038/s41467-019-10839-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Joseph T., Crossno J., Garat C. V., Reusch J. E. B., Morris K. G., Dempsey E. C., McMurtry I. F., Stenmark K. R., and Klemm D. J. (2007) Rosiglitazone attenuates hypoxia-induced pulmonary arterial remodeling. Am. J. Physiol. Lung Cell. Mol. Physiol. 292, L885–L897 10.1152/ajplung.00258.2006 [DOI] [PubMed] [Google Scholar]
- 44. Grommes J., Mörgelin M., and Soehnlein O. (2012) Pioglitazone attenuates endotoxin-induced acute lung injury by reducing neutrophil recruitment. Eur. Respir. J. 40, 416–423 10.1183/09031936.00091011 [DOI] [PubMed] [Google Scholar]
- 45. Nesto R. W., Bell D., Bonow R. O., Fonseca V., Grundy S. M., Horton E. S., Le Winter M., Porte D., Semenkovich C. F., Smith S., Young L. H., and Kahn R. (2004) Thiazolidinedione use, fluid retention, and congestive heart failure: a consensus statement from the American Heart Association and American Diabetes Association. Diabetes Care 27, 256–263 10.2337/diacare.27.1.256 [DOI] [PubMed] [Google Scholar]
- 46. Larson-Casey J. L., Deshane J. S., Ryan A. J., Thannickal V. J., and Carter A. B. (2016) Macrophage Akt1 kinase-mediated mitophagy modulates apoptosis resistance and pulmonary fibrosis. Immunity 44, 582–596 10.1016/j.immuni.2016.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Larson-Casey J. L., Vaid M., Gu L., He C., Cai G. Q., Ding Q., Davis D., Berryhill T. F., Wilson L. S., Barnes S., Neighbors J. D., Hohl R. J., Zimmerman K. A., Yoder B. K., Longhini A. L. F., et al. (2019) Increased flux through the mevalonate pathway mediates fibrotic repair without injury. J. Clin. Invest. 129, 4962–4978 10.1172/JCI127959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hauser S., Adelmant G., Sarraf P., Wright H. M., Mueller E., and Spiegelman B. M. (2000) Degradation of the peroxisome proliferator-activated receptor γ is linked to ligand-dependent activation. J. Biol. Chem. 275, 18527–18533 10.1074/jbc.M001297200 [DOI] [PubMed] [Google Scholar]
- 49. Lowenstein C. J., Alley E. W., Raval P., Snowman A. M., Snyder S. H., Russell S. W., and Murphy W. J. (1993) Macrophage nitric oxide synthase gene: two upstream regions mediate induction by interferon γ and lipopolysaccharide. Proc. Natl. Acad. Sci. U.S.A. 90, 9730–9734 10.1073/pnas.90.20.9730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fiehn O., Wohlgemuth G., Scholz M., Kind T., Lee D. Y., Lu Y., Moon S., and Nikolau B. (2008) Quality control for plant metabolomics: reporting MSI-compliant studies. Plant J. 53, 691–704 10.1111/j.1365-313X.2007.03387.x [DOI] [PubMed] [Google Scholar]
- 51. Gu L., Larson-Casey J. L., and Carter A. B. (2017) Macrophages utilize the mitochondrial calcium uniporter for profibrotic polarization. FASEB J. 31, 3072–3083 10.1096/fj.201601371R [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are contained within the manuscript. Data have been submitted to the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus database (GEO) with GEO accession number GSE155166.