

Abstract
Structural analyses identified the central domain of ryanodine receptor (RyR) as a transducer converting conformational changes in the cytoplasmic platform to the RyR gate. The central domain is also a regulatory hub encompassing the Ca2+-, ATP-, and caffeine-binding sites. However, the role of the central domain in RyR activation and regulation has yet to be defined. Here, we mutated five residues that form the Ca2+ activation site and 10 residues with negatively charged or oxygen-containing side chains near the Ca2+ activation site. We also generated eight disease-associated mutations within the central domain of RyR2. We determined the effect of these mutations on Ca2+, ATP, and caffeine activation and Mg2+ inhibition of RyR2. Mutating the Ca2+ activation site markedly reduced the sensitivity of RyR2 to Ca2+ and caffeine activation. Unexpectedly, Ca2+ activation site mutation E3848A substantially enhanced the Ca2+-independent basal activity of RyR2, suggesting that E3848A may also affect the stability of the closed state of RyR2. Mutations in the Ca2+ activation site also abolished the effect of ATP/caffeine on the Ca2+-independent basal activity, suggesting that the Ca2+ activation site is also a critical determinant of ATP/caffeine action. Mutating residues with negatively charged or oxygen-containing side chains near the Ca2+ activation site significantly altered Ca2+ and caffeine activation and reduced Mg2+ inhibition. Furthermore, disease-associated RyR2 mutations within the central domain significantly enhanced Ca2+ and caffeine activation and reduced Mg2+ inhibition. Our data demonstrate that the central domain plays an important role in channel activation, channel regulation, and closed state stability.
Keywords: Ca2+ activation, Mg2+ inhibition, basal activity, ryanodine binding, calcium intracellular release, calcium imaging, ryanodine receptor, sarcoplasmic reticulum (SR), inositol trisphosphate receptor (InsP3R), calcium, calcium channel
Ryanodine receptor type 2 (RyR2) is an intracellular Ca2+ release channel that is expressed predominantly in the heart and brain and plays an essential role in many cellular processes, including muscle contraction, learning, and memory, by governing the release of Ca2+ from intracellular Ca2+ stores (1–9). RyR2 is normally activated through a mechanism known as Ca2+-induced Ca2+ release (CICR), in which an elevation of cytosolic Ca2+ opens the RyR2 channel, leading to a large Ca2+ release from the sarcoplasmic reticulum and the endoplasmic reticulum (10–12). Thus, activation of RyR2 by cytosolic Ca2+ is a critical step in the mechanism of CICR (13–15). Consistent with its important physiological roles, impaired Ca2+ activation of RyR2 has been associated with diseases in both the heart and brain, such as cardiac arrhythmias, cardiomyopathies, and intellectual disability (16–19). However, despite its physiological and pathological significance, the molecular mechanism underlying Ca2+ activation of RyR2 is not well defined.
It has long been recognized that RyR2 contains a high-affinity cytosolic Ca2+ activation site that mediates Ca2+ activation of RyR2 and CICR (10–12, 20–24). Major efforts have been focused on identifying residues that are responsible for Ca2+ activation of RyR2. Through site-directed mutagenesis and single-channel analyses, we showed that a point mutation E3987A in RyR2 dramatically reduced the sensitivity of RyR2 to activation by Ca2+ (25). We also reported that point mutation E3885A in RyR3 (corresponding to E3987A in RyR2) markedly decreased the Ca2+ sensitivity of RyR3 (26). Furthermore, point mutation E4032A in RyR1 (corresponding to E3987A in RyR2) abolished Ca2+-dependent activation of RyR1 (27, 28). These findings indicate that residue Glu-3987 in RyR2 and the corresponding residue Glu-4032 in RyR1 and Glu-3885 in RyR3 play an important role in Ca2+ activation of RyRs.
Recent success in solving near-atomic resolution 3D structures of RyR1 and RyR2 using cryo-EM has greatly advanced our understanding of the molecular basis of RyR activation (29–36). Comparisons of the open and closed states of RyR1 and RyR2 reveal that the central domains of RyR1 (amino acids 3668–4251) and RyR2 (amino acids 3613–4207) play an important role in channel activation (32–36). The putative Ca2+ binding site in RyR1 formed by residues Glu-3893, Glu-3967, Gln-3970, His-3895, and Thr-5001 in the core solenoid domain of RyR1, and the putative Ca2+ binding site in RyR2 formed by residues Glu-3848, Glu-3922, Gln-3925, His-3850, and Thr-4931 in the central domain of RyR2 have been identified (34–36). Interestingly, the RyR1–Glu-4032 or RyR2–Glu-3987 residue is located just beside the Ca2+ activation site. Although the RyR1–Glu-4032 or RyR2–Glu-3987 residue does not directly contribute to Ca2+ coordination, it is involved in H-bonding between the central domain and the C-terminal domain (CTD) that may stabilize the Ca2+ binding pocket. Furthermore, the putative ATP- and caffeine-binding sites have also been localized to regions very close to the Ca2+ activation site (34–36). Recent functional studies are consistent with the proposed locations of the Ca2+ and caffeine activation sites (37). For instance, mutations in the Ca2+ activation site (E3848A or E3922A in RyR2) diminished Ca2+ activation of RyR2, whereas mutation W4644A or W4644R in the caffeine-binding site abolished caffeine activation of RyR2 (37). Thus, the central domain is critically involved in RyR2 channel activation and regulation. It is also known that RyR is inactivated by Mg2+ (20, 22–24, 38), but the molecular basis of Mg2+-dependent inhibition of RyR is not well understood.
In addition to those Ca2+-coordinating residues, the central domain also contains a number of residues with negatively charged or oxygen-containing side chains clustered near the Ca2+ activation site (29–36). The significance of these residues in channel activation and regulation is unknown. The central domain also harbors a number of RyR2 mutations associated with catecholaminergic polymorphic ventricular tachycardia (CPVT) and sudden unexplained death (17, 39), but their functional effect has yet to be determined. It is also unclear how ATP and caffeine modulate Ca2+ activation of RyR2. To address these important questions, in the present study, we performed a systematic site-directed mutagenesis and structure-function relationship analysis of the central domain of RyR2. Our results revealed that the central domain plays a critical role not only in channel activation by Ca2+, ATP, and caffeine, but also in channel regulation by Mg2+ and in determining the stability of the closed state of the channel in the near absence of Ca2+. Our data also showed that disease-associated RyR2 mutations located within the central domain enhanced Ca2+ activation and reduced Mg2+ inhibition of RyR2. Thus, the central domain controls the activation and regulation of the RyR2 channel.
Results
Contribution of Ca2+-coordinating residues to Ca2+ and caffeine activation and basal activity of RyR2
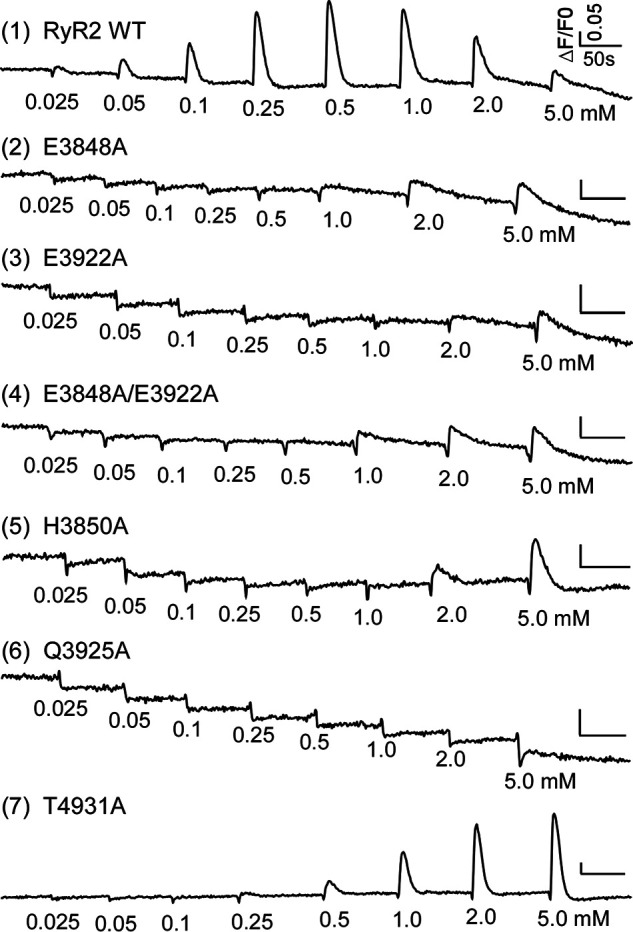
Recent 3D structural analyses revealed the RyR2 Ca2+ activation site that is formed by residues Glu-3848, Glu-3922, Gln-3925, His-3850, and Thr-4931 (35, 36) (Fig. 1A). To assess the role of these Ca2+-coordinating residues in RyR2 function, we mutated each of these residues to alanine (i.e. E3848A, E3922A, Q3925A, H3850A, and T4931A) in the mouse RyR2 and determined the Ca2+-dependent [3H]ryanodine binding to each of these RyR2 mutants with a wide range of Ca2+ concentrations (∼0.1–100 mm). Note that the mouse and human RyR2 proteins share >97% amino acid sequence identity. Because ryanodine only binds to the open state of RyR, [3H]ryanodine binding assay has widely been used to monitor RyR channel activity (27, 40–42). As shown in Fig. 1, there was little or no [3H]ryanodine binding to RyR2 WT in the near absence of Ca2+ (∼0.1 nm). This indicates that there is little or no Ca2+-independent basal activity of RyR2 WT. [3H]ryanodine binding to RyR2 WT was activated at ∼100 nm Ca2+ with an EC50 of 0.21 μm, maximized at ∼1 μm Ca2+, and slightly inactivated at >10 mm Ca2+ (Fig. 1, B, C, and E and Table 1). This is consistent with previous studies (25, 43). Unlike WT, the E3848A mutant displayed a complex Ca2+ dependence of [3H]ryanodine binding. In the near absence of Ca2+, the E3848A mutant exhibited a substantially higher level of [3H]ryanodine binding than WT (p < 0.0001) (Fig. 1, C and E). This indicates that the E3848A mutation markedly increases the Ca2+-independent basal activity of RyR2. Interestingly, different from WT, elevating Ca2+ concentration from ∼1 μm to ∼10 mm decreased (rather than increased) [3H]ryanodine binding to the E3848A mutant. This suggests the existence of a putative Ca2+ inactivation site(s) in RyR2 that is independent of residue Glu-3848. However, at Ca2+ concentrations >10 mm Ca2+, [3H]ryanodine binding to the E3848A mutant increased but did not saturate even at 100 mm Ca2+ (Fig. 1E and Table 1). Because we could not estimate the maximal (saturated) [3H]ryanodine binding to E3848A, we cannot accurately determine the EC50 of Ca2+-dependent activation of E3848A, but it is likely to be greater than 10 mm (Fig. 1E and Table 1). We propose that this increase in [3H]ryanodine binding at Ca2+ concentrations >10 mm reflects the Ca2+-dependent activation of the E3848A mutant, which would be dramatically reduced compared with that of the WT. The E3922A mutant also displayed a significantly higher level of basal [3H]ryanodine binding than the WT in the near absence of Ca2+ (∼0.1 nm). In addition, [3H]ryanodine binding to E3922A was activated at ∼10mM Ca2+ (Fig. 1, B, C, and E and Table 1). This indicates that, like the E3848A mutation, the E3922A mutation also significantly increases the Ca2+-independent basal activity and dramatically decreases the Ca2+-dependent activation of RyR2. We also assessed the effect of the E3838A/E3922A double mutation on the Ca2+ dependence of [3H]ryanodine binding. The E3848A/E3922A double mutation nearly completely abolished Ca2+-dependent activation of RyR2 (even by 100 mm Ca2+). The E3848A/E3922A mutant also displayed elevated Ca2+-independent basal [3H]ryanodine binding and Ca2+-dependent inhibition (by ∼1 μm to 10 mm Ca2+) of [3H]ryanodine binding (Fig. 1, B, C, and E and Table 1). The Q3925A and H3850A mutations also significantly increased the EC50 (158 μm for Q3925A and 0.78 μm for H3850A) of Ca2+-dependent activation of [3H]ryanodine binding to RyR2 but did not significantly affect Ca2+-independent basal [3H]ryanodine binding (Fig. 1, B, C, and F and Table 1). The T4931A mutation did not significantly alter the EC50 or the basal activity of [3H]ryanodine binding (Fig. 1, B, C, and F). Taken together, consistent with 3D structural analyses, our data indicate that residues Glu-3848, Glu-3922, and Gln-3925 are critical for Ca2+-dependent activation of RyR2. Our results also show, unexpectedly, that the E3848A and E3922A mutations could alter the stability of the closed state of RyR2 in the near absence of Ca2+.
Figure 1.

Effects of mutating the Ca2+-coordinating residues on Ca2+ and caffeine activation and basal activity of RyR2. A, 3D locations of the residues involved in Ca2+ coordination and the binding sites for ATP and caffeine in the RyR2 3D structure. The central domain, CTD, and U-motif are shown. All 3D structure images were generated from PDB 6JI0 using PyMOL. [3H]ryanodine binding to cell lysates prepared from HEK293 cells expressing the RyR2 WT or mutants was carried out at various Ca2+ concentrations (0.1 nm–100 mm). The amounts of [3H]ryanodine binding to WT or mutants at various Ca2+ concentrations were normalized to its own maximal binding (100%) for WT and each mutant. B, EC50 values of Ca2+ activation and (C) basal activity (in the near absence of Ca2+, ∼0.1 nm) of [3H]ryanodine binding to RyR2. D, the apparent EC50 values of caffeine-induced Ca2+ releases in HEK293 cells transfected with RyR2 WT or mutants. [3H]ryanodine binding to RyR2 WT, E3848A, E3922A, or E3848A/E3922A mutant (E) and to RyR2 WT, H3850A, Q3925A, or T4931A mutant (F). The relationships between caffeine-induced Ca2+ release and cumulative caffeine concentrations in HEK293 cells transfected with RyR2 WT, E3848A, E3922A, or E3848A/E3922A mutant (G) or with RyR2 WT, H3850A, Q3925A, or T4931A mutant (H). Data points shown are mean ± S.D. (error bars) from 3–5 separate experiments. *, p < 0.05; **, p < 0.01.
Table 1.
Effects of mutations on [3H]ryanodine binding to RyR2
| EC50 (μm) of Ca2+ activation | Adjusted p value | Basal activity (pmol/mg) | Adjusted p value | n number of separate experiments performed | |
|---|---|---|---|---|---|
| A. Mutations of Ca2+-coordinating residues | |||||
| RyR2 WT | 0.21 ± 0.02 | -- | 3.1 × 10−3 ± 1.7 × 10−3 | -- | 4 |
| E3848A | >1.0 × 104 | -- | 28.0 × 10−3 ± 2.8 × 10−3 | <0.0001 | 3 |
| E3848A/E3922A | -- | -- | 52.9 × 10−3 ± 4.1 × 10−3 | <0.0001 | 3 |
| H3850A | 0.78 ± 0.01 | <0.0001 | 0.4 × 10−3 ± 0.4 × 10−3 | 0.4700 | 3 |
| E3922A | 1.38 × 104 ± 0.25 × 104 | 0.0055 | 12.2 × 10−3 ± 3.1 × 10−3 | 0.0002 | 4 |
| Q3925A | 1.58 × 102 ± 0.24 × 102 | 0.0031 | 0.2 × 10−3 ± 0.2 × 10−3 | 0.3416 | 4 |
| T4931A | 0.28 ± 0.04 | 0.2359 | 1.5 × 10−3 ± 1.1 × 10−3 | 0.8839 | 3 |
| EC50 ANOVA summary: F = 426.3, p value < 0.0001; Basal activity ANOVA summary: F = 231.1, p value < 0.0001 | |||||
| B. Mutations of residues with negatively charged or oxygen-containing side chains | |||||
| RyR2 WT | 0.21 ± 0.02 | -- | 4.3 × 10−3 ± 2.1 × 10−3 | -- | 3 |
| T3929A | 0.29 ± 0.02 | 0.0071 | 3.5 × 10−3 ± 4.8 × 10−3 | 0.9998 | 3 |
| Q3932A | 0.28 ± 0.03 | 0.0231 | 7.2 × 10−3 ± 5.4 × 10−3 | 0.9964 | 3 |
| Q3933A | 0.18 ± 0.02 | 0.5725 | 17.3 × 10−3 ± 10.5 × 10−3 | 0.0869 | 3 |
| S3984A | 0.15 ± 0.03 | 0.0197 | 4.2 × 10−3 ± 1.7 × 10−3 | >0.9999 | 4 |
| N3989A | 0.24 ± 0.01 | 0.7655 | 4.4 × 10−3 ± 3.7 × 10−3 | >0.9999 | 3 |
| E4146A | 0.32 ± 0.03 | <0.0001 | 1.6 × 10−3 ± 2.2 × 10−3 | 0.9961 | 5 |
| Y4149S | 0.09 ± 0.02 | <0.0001 | 21.8 × 10−3 ± 5.7 × 10−3 | 0.0060 | 4 |
| T4934A | 0.29 ± 0.02 | 0.0035 | 9.7 × 10−3 ± 11.5 × 10−3 | 0.8642 | 3 |
| Q4936A | 0.12 ± 0.02 | 0.0004 | 70.1 × 10−3 ± 9.5 × 10−3 | <0.0001 | 3 |
| E4937A | 0.44 ± 0.01 | <0.0001 | 2.3 × 10−3 ± 2.5 × 10−3 | 0.9996 | 3 |
| EC50 ANOVA summary: F =59.8, p value < 0.0001; Basal activity ANOVA summary: F = 33.7, p value < 0.0001 | |||||
| C. Disease-associated mutations | |||||
| RyR2 WT | 0.21 ± 0.02 | -- | 5.8 × 10−3 ± 2.1 × 10−3 | -- | 3 |
| K3997E | 0.16 ± 0.01 | 0.0025 | 4.0 × 10−3 ± 0.3 × 10−3 | 0.9676 | 3 |
| M3999V | 0.06 ± 0.01 | <0.0001 | 27.0 × 10−3 ± 3.0 × 10−3 | <0.0001 | 3 |
| F4020L | 0.10 ± 0.01 | <0.0001 | 8.1 × 10−3 ± 1.1 × 10−3 | 0.8778 | 3 |
| N4097S | 0.15 ± 0.01 | 0.0002 | 4.6 × 10−3 ± 0.7 × 10−3 | 0.9971 | 3 |
| R4157Q | 0.14 ± 0.01 | <0.0001 | 8.4 × 10−3 ± 3.2 × 10−3 | 0.8026 | 3 |
| L4188P | 0.12 ± 0.02 | <0.0001 | 17.8 × 10−3 ± 6.4 × 10−3 | 0.0004 | 3 |
| T4196A | 0.11 ± 0.01 | <0.0001 | 20.3 × 10−3 ± 1.2 × 10−3 | <0.0001 | 3 |
| Q4201R | 0.11 ± 0.01 | <0.0001 | 22.4 × 10−3 ± 1.8 × 10−3 | <0.0001 | 3 |
| EC50 ANOVA summary: F =30.3, p value < 0.0001; Basal activity ANOVA summary: F = 28.4, p value < 0.0001 | |||||
Data are presented as mean ± S.D. The significance of differences in EC50 and basal activity between WT and mutants was evaluated by performing one-way ANOVA with Dunnett's multiple comparisons post hoc testing. A p value <0.05 was considered statistically significant.
We also assessed the effect of mutations of the Ca2+-coordinating residues on channel function by measuring caffeine-induced Ca2+ release in RyR2 WT or mutant expressing HEK293 cells (Fig. 1, D, G, and H, Fig. 2, and Table 2). The amplitude of caffeine-induced Ca2+ release in HEK293 cells transfected with RyR2-WT increased progressively with each cumulative addition of caffeine (from 0.05 to 1.0 mm) with an apparent EC50 of caffeine activation of ∼0.18 mm and then decreased with further cumulative additions of caffeine (from 2.5 and 5 mm), likely because of the depletion of the intracellular Ca2+ stores by the prior additions of caffeine (0.025–1.0 mm) (Fig. 1, G and H and Fig. 2). Consistent with their effect on Ca2+ activation of [3H]ryanodine binding, mutations E3848A, E3922A, E3848A/E3922A, Q3925A, and H3850A also markedly inhibited caffeine-induced Ca2+ release in HEK293 cells, shifting the caffeine response curve to the right (Fig. 1, D, G, and H, Fig. 2, and Table 2). Interestingly, the T4931A mutation also markedly inhibited caffeine activation (Fig. 1, D and H, Fig. 2, and Table 2), despite its small (insignificant) inhibitory effect on the Ca2+-dependent [3H]ryanodine binding. This suggests that the activation of RyR2 by Ca2+ and caffeine is not always the same.
Figure 2.
Effects of RyR2 Ca2+-coordinating residue mutations on caffeine-induced Ca2+ release in HEK293 cells. HEK293 cells were transfected with RyR2 WT or mutants of Ca2+-coordinating residues. The fluorescence intensity of the Fluo-3-loaded transfected cells was monitored continuously before and after each caffeine addition.
Table 2.
Effects of mutations on caffeine activation of RyR2
| Apparent EC50 (mm) | Adjusted p value | n number | |
|---|---|---|---|
| A. Mutations of Ca2+-coordinating residues | |||
| RyR2 WT | 0.19 ± 0.01 | -- | 4 |
| E3848A | 2.69 ± 0.22 | <0.0001 | 4 |
| E3848A/E3922A | 2.68 ± 0.17 | <0.0001 | 4 |
| H3850A | 3.98 ± 0.40 | <0.0001 | 4 |
| E3922A | 3.99 ± 0.03 | <0.0001 | 4 |
| Q3925A | 6.53 ±0.00 | <0.0001 | 4 |
| T4931A | 1.88 ± 0.13 | <0.0001 | 4 |
| ANOVA summary: F = 427.1, p value < 0.0001 | |||
| B. Mutations of residues with negatively charged or oxygen-containing side chains | |||
| RyR2 WT | 0.18 ± 0.02 | -- | 5 |
| T3929A | 0.36 ± 0.04 | 0.0003 | 5 |
| Q3932A | 0.41 ± 0.05 | 0.0008 | 5 |
| Q3933A | 0.19 ± 0.04 | 0.9956 | 5 |
| S3984A | 0.12 ± 0.01 | 0.0172 | 5 |
| N3989A | 0.19 ±0.07 | >0.9999 | 5 |
| E4146A | 1.01 ± 0.16 | 0.0019 | 5 |
| Y4149S | 0.09 ± 0.01 | 0.0021 | 5 |
| T4934A | 0.44 ± 0.05 | 0.0003 | 5 |
| Q4936A | 0.13 ± 0.02 | 0.0346 | 5 |
| E4937A | 1.05 ± 0.18 | 0.0024 | 5 |
| ANOVA summary: F = 93.0, p value < 0.0001 | |||
| C. Disease-associated mutations | |||
| RyR2 WT | 0.18 ± 0.01 | -- | 5 |
| K3997E | 0.17 ± 0.03 | 0.4697 | 5 |
| M3999V | 0.08 ± 0.01 | <0.0001 | 5 |
| F4020L | 0.10 ± 0.02 | <0.0001 | 5 |
| N4097S | 0.15 ± 0.01 | 0.0149 | 5 |
| R4157Q | 0.15 ± 0.02 | 0.0133 | 5 |
| L4188P | 0.07 ± 0.00 | <0.0001 | 5 |
| T4196A | 0.11 ± 0.01 | <0.0001 | 5 |
| Q4201R | 0.13 ± 0.01 | 0.0002 | 5 |
| ANOVA summary: F = 32.0, p value < 0.0001 | |||
Data are presented as mean ± S.D. The significance of differences in caffeine activation between WT and mutants was evaluated by performing one-way ANOVA with Dunnett's multiple comparisons post hoc testing. A p value <0.05 was considered statistically significant.
Effect of ATP and caffeine on [3H]ryanodine binding to RyR2 WT and Ca2+ activation site mutants
Structural analyses also revealed that the Ca2+ activation site is located near the ATP- and caffeine-binding sites and that the Ca2+-, ATP-, and caffeine-binding sites are interconnected through the CTD (Fig. 1A). Thus, it is possible that mutations in the Ca2+ activation site may alter the actions of ATP and caffeine in RyR2 channel gating. To test this idea, we assessed the effect of ATP/caffeine on [3H]ryanodine binding to Ca2+ activation site mutants. We found that ATP (3 mm) plus caffeine (3 mm) markedly increased the sensitivity of RyR2 WT to Ca2+ activation (Ca2+ sensitivity) and the basal activity (in the near absence of Ca2+, ∼0.1 nm) of RyR2 WT (Fig. 3, A, H, and I and Table 3). Similarly, ATP/caffeine enhanced both the Ca2+ sensitivity and basal activity of the T4931A mutant (Fig. 3, B, H, and I and Table 3). ATP/caffeine also dramatically increased the basal activity (at ∼0.1 nm Ca2+) of the E3848A, E3922A, or E3848A/E3922A mutants. The effect of ATP/caffeine on Ca2+ activation of these mutants could not be accurately determined because of their nonsaturated [3H]ryanodine binding. Nevertheless, the estimated thresholds of Ca2+ activation of [3H]ryanodine binding to these mutants in the absence and presence of ATP/caffeine appeared to be similar (∼10 mm) (Fig. 3, C–E, H, and I and Table 3). On the other hand, ATP/caffeine significantly enhanced the Ca2+ sensitivity of the H3850A and Q3925A mutants without significantly altering their basal activity (Fig. 3, F–I and Table 3). These observations indicate that ATP/caffeine significantly affects both the Ca2+ independent basal activity and the Ca2+-dependent activation (i.e. Ca2+ sensitivity) of RyR2. Our data also indicate that mutations of the Ca2+-coordinating residues can exert different effects on the actions of ATP and caffeine.
Figure 3.

Effects of ATP and caffeine on [3H]ryanodine binding to mutants of Ca2+-coordinating residues. [3H]ryanodine binding to cell lysates prepared from HEK293 cells expressing the RyR2 WT (A) or RyR2 mutants (B–G) was carried out at various Ca2+ concentrations (0.1 nm–100 mm) in the absence (control) or the presence of 3 mm ATP and 3 mm caffeine (caff). [3H]ryanodine binding to each construct was performed using the same amount of the same RyR2 WT or mutant cell lysate in the absence or presence of ATP/caffeine. The amount of [3H]ryanodine binding to RyR2 WT and each RyR2 mutant at various Ca2+ concentrations in the absence or presence of ATP/caffeine was normalized to the maximal binding observed among these two conditions (i.e. with or without ATP/caffeine). H, EC50 values of Ca2+ activation and (I) basal activity (in the near absence of Ca2+, ∼0.1 nm) of [3H]ryanodine binding to RyR2. Data points shown are mean ± S.D. (error bars) from three separate experiments. *, p < 0.05; **, p < 0.01.
Table 3.
Effect of ATP/caffeine on [3H]ryanodine binding to Ca2+-coordinating residue mutants of RyR2
| EC50 (μm) |
p value | Basal activity (pmol/mg) |
p value | n number | |||
|---|---|---|---|---|---|---|---|
| Control | +ATP/Caff | Control | +ATP/Caff | ||||
| RyR2 WT | 0.21 ± 0.02 | 0.03 ± 0.003 | 0.0043 | 3.3 × 10−3 ± 0.7 × 10−3 | 53.9 × 10−3 ± 2.4 × 10−3 | 0.0003 | 3 |
| E3848A | >1.0 × 104 | >1.0 × 104 | -- | 28.0 × 10−3 ± 2.8 × 10−3 | 122.0 × 10−3 ± 6.1 × 10−3 | 0.0002 | 3 |
| E3848A/E3922A | -- | -- | -- | 51.1 × 10−3 ± 4.3 × 10−3 | 150.4 × 10−3 ± 23.2 × 10−3 | 0.0153 | 3 |
| H3850A | 0.78 ± 0.09 | 0.33 ± 0.12 | 0.0234 | 0.2 × 10−3 ± 0.4 × 10−3 | 3.6 × 10−3 ± 2.8 × 10−3 | 0.1685 | 3 |
| E3922A | >1.0 × 104 | >1.0 × 104 | -- | 8.2 × 10−3 ± 1.6 × 10−3 | 154.6 × 10−3 ± 22.1 × 10−3 | 0.0073 | 3 |
| Q3925A | 1.54 × 102 ± 0.26 × 102 | 0.22 × 102 ± 0.11 × 102 | 0.0059 | 0.002 × 10−3 ± 0.003 × 10−3 | 4.5 × 10−3 ± 2.7 × 10−3 | 0.1000 | 3 |
| T4931A | 0.43 ± 0.09 | 0.13 ± 0.04 | 0.0153 | 1.6 × 10−3 ± 0.5 × 10−3 | 37.9 × 10−3 ± 9.3 × 10−3 | 0.0210 | 3 |
Data are presented as mean ± S.D. The significance of difference in EC50 and basal activity with and without 3 mm ATP/3 mm caffeine was assessed by two-tailed Student's t tests. A p value <0.05 was considered statistically significant.
Effect of mutating residues with negatively charged or oxygen-containing side chains near the Ca2+ activation site on Ca2+ and caffeine activation and basal activity of RyR2
In addition to the Ca2+-coordinating residues, there are a number of residues with negatively charged and oxygen-containing side chains that are clustered near the Ca2+ activation site (Fig. 4A). These include Thr-3929, Gln-3932, Gln-3933, Ser-3984, Asn-3989, Glu-4146, Tyr-4149, Thr-4934, Gln-4936, and Glu-4937. The functional significance of these residues is unclear. To this end, we mutated each of these residues and determined their effect on Ca2+ activation and Mg2+ inhibition of RyR2 using the [3H]ryanodine binding assay. As shown in Fig. 4, the T3929A, Q3932A, E4146A, T4934A, and E4937A mutations significantly suppressed the Ca2+ activation but without significantly affecting the basal activity of RyR2 (Fig. 4, B, C, and E–H and Table 1), whereas mutations Y4149S and Q4936A significantly increased the Ca2+ sensitivity and basal activity of RyR2 (Fig. 4, B, C, G, and H and Table 1). The S3984A mutation increased the Ca2+ sensitivity but not the basal activity of RyR2 (Fig. 4, B, C, and F and Table 1). On the other hand, mutations Q3933A and N3989A had no significant effect on the Ca2+ activation or basal activity of RyR2 (Fig. 4, B, C, E, and F and Table 1). Thus, these Ca2+ noncoordinating residues Thr-3929, Gln-3932, Glu-4146, Thr-4934, Glu-4937, Tyr-4149, Gln-4936, and Ser-3984 are also important for Ca2+ activation of RyR2, whereas residues Tyr-4149 and Gln-4936 are important for stabilizing the closed state of RyR2 in the near absence of Ca2+ (∼0.1 nm) (Fig. 4, B and C and Table 1). We also performed caffeine-induced Ca2+ release assays in HEK293 cells transfected with these mutations to determine their effect on the activation of RyR2 by caffeine. Consistent with their effects on Ca2+-dependent [3H]ryanodine binding to RyR2, mutations T3929A, Q3932A, E4146A, T4934A, and E4937A suppressed, whereas mutations S3984A, Y4149S, and Q4936A increased caffeine activation of RyR2. Mutations Q3933A and N3989A had no significant effect on caffeine activation (Fig. 4, D and I–L, Table 2, and Fig. 5).
Figure 4.

Effects of mutations of residues with negatively charged and oxygen-containing side chains near the Ca2+ activation site on [3H]ryanodine binding. A, 3D locations of residues with negatively charged and oxygen-containing side chains near the Ca2+ activation site within the central domain. [3H]ryanodine binding to cell lysate prepared from HEK293 cells expressing the RyR2 WT and mutants was carried out at various Ca2+ concentrations (0.1 nm–0.1 mm). The amounts of [3H]ryanodine binding at various Ca2+ concentrations were normalized to the maximal binding (100%). B, EC50 values of Ca2+ activation and (C) basal activity (in the near absence of Ca2+, ∼0.1 nm) of [3H]ryanodine binding to RyR2. D, the apparent EC50 values of caffeine-induced Ca2+ releases in HEK293 cells transfected with RyR2 WT or mutants. [3H]ryanodine binding to RyR2 WT, T3929A, Q3932A, and Q3933A (E), to RyR2 WT, S3984A, N3989A, and E4146A (F), to RyR2 WT, Y4149S, and T4934A (G), and to RyR2 WT, Q4936A, and E4937A (H). The relationships between caffeine-induced Ca2+ release and cumulative caffeine concentrations in HEK293 cells transfected with RyR2 WT, T3929A, Q3932A, and Q3933A (I), with RyR2 WT, S3984A, N3989A, and E4146A (J), with RyR2 WT, Y4149S, and T4934A (K), and with RyR2 WT, Q4936A, and E4937A (L). Data points shown are mean ± S.D. (error bars) from 3–5 separate experiments. *, p < 0.05; **, p < 0.01.
Figure 5.

Effects of RyR2 mutations of residues with negatively charged or oxygen-containing side chains on caffeine-induced Ca2+ release in HEK293 cells. HEK293 cells were transfected with RyR2 WT or mutants of residues with negatively charged or oxygen-containing side chains. The fluorescence intensity of the Fluo-3-loaded transfected cells was monitored continuously before and after each caffeine addition.
Effect of mutating residues with negatively charged or oxygen-containing side chains in the central domain on Mg2+ inhibition of RyR2
We next determined whether mutating residues with negatively charged and oxygen-containing side chains in the central domain affects the inhibition of RyR2 by Mg2+. To minimize the influence of Ca2+ on Mg2+-dependent inhibition of RyR2, we activated RyR2 by ATP (3 mm) plus caffeine (3 mm) in the near absence of Ca2+ (∼0.1 nm) and determined the effect of Mg2+ (2 mm) on [3H]ryanodine binding to each of the ATP/caffeine activated mutants. Under these conditions, Mg2+ exerted >80% inhibition on the RyR2 WT channel (Fig. 6A). However, Mg2+-dependent inhibition of [3H]ryanodine binding to mutants Y4149S, E4937A, E3848A, Q4936A, and S3984A was markedly reduced compared with that of [3H]ryanodine binding to RyR2 WT (Fig. 6A). On the other hand, mutations E3922A, Q3933A, T4934A, N3989A, Q3932A, and T3929A had no significant effect on the inhibition of [3H]ryanodine binding to RyR2 by Mg2+. Interestingly, mutation T4931A slightly increased Mg2+-dependent inhibition of [3H]ryanodine binding (Fig. 6A). The effect of mutations H3850A and Q3925A on Mg2+ inhibition of RyR2 cannot be determined, because they displayed little or no [3H]ryanodine binding under these conditions. Taken together, these data indicate that residue Glu-3848, located in the Ca2+ activation site, and residues Ser-3984, Tyr-4149, Gln-4936, and Glu-4937, located near the Ca2+ activation site, are important for Mg2+-dependent inhibition of RyR2 (Fig. 6B).
Figure 6.

Effects of Mg2+ on [3H]ryanodine binding to RyR2 WT and mutants. A, percentage of Mg2+ inhibition of [3H]ryanodine binding. [3H]ryanodine binding to cell lysates prepared from HEK293 cells expressing the RyR2 WT and mutants was carried out at 0.1 nm [Ca2+] and 2 mm Mg2+. ATP (3 mm) and caffeine (3 mm) were included to stimulate [3H]ryanodine binding. B, 3D locations of mutations that reduce Mg2+ inhibition. Data points shown are mean ± S.D. (error bars) from three separate experiments. Dashed red line indicates the percentage of Mg2+ inhibition in WT. *, p < 0.05; **, p < 0.01.
Disease-associated mutations in the central domain affect Ca2+ and caffeine activation and Mg2+ inhibition of RyR2
The central domain harbors a large number of disease-causing RyR2 mutations (Fig. 7A). RyR2 mutations K3997E (39, 44), F4020L (45), R4157Q (39, 46), T4196A (47), and Q4201R (48) were identified in individuals presenting with syncope, CPVT, and/or sudden unexplained death. The L4188P mutation was found in a 15-year-old girl with a history of seizure-like episodes often triggered by anxiety (49). The N4097S mutation was found in a sudden unexplained death individual through molecular autopsy (50). The M3999V mutation was found in the CPVT post-mortem panel in ClinVar (National Center for Biotechnology Information. ClinVar; VCV000201320.2). To understand the effect of these disease-associated RyR2 mutations on channel function, we generated each of these mutations. Ca2+-dependent [3H]ryanodine binding was carried out to determine their effect on Ca2+ activation and Mg2+ inhibition of RyR2. All of these mutations significantly enhanced the sensitivity of RyR2 to Ca2+ activation (Fig. 7, B and E–G and Table 1). Furthermore, M3999V, L4188P, T4196A, and Q4201R mutations, but not K3997E, F4020L, N4097S, and R4157Q mutations, increased the basal activity of RyR2 (Fig. 7, C and E–G and Table 1). This is consistent with the effect of other disease-associated RyR2 mutations located in the central domain (51). We also determined their effect on the activation of RyR2 by caffeine. Consistent with their effects on Ca2+-dependent [3H]ryanodine binding to RyR2, mutations M3999V, F4020L, N4097S, R4157Q, L4188P, T4196A, and Q4201R significantly enhanced caffeine activation of RyR2. Interestingly, the K3997E mutation had no significant effect on caffeine activation of RyR2, although it slightly reduced the EC50 of Ca2+ activation of [3H]ryanodine binding to RyR2, (Fig. 7, D and H–J, Table 2, and Fig. 8). The effect of these disease-associated mutations on Mg2+-dependent inhibition of RyR2 is shown in Fig. 9. Mutations M3999V, F4020L, T4196A, L4188P, Q4201R, N4097S, and R4157Q significantly reduced the Mg2+-dependent inhibition of RyR2, whereas mutation K3997E had no effect on the inhibition of RyR2 by Mg2+. These results suggest that enhanced Ca2+ and caffeine activation and reduced Mg2+ inhibition are common defects of central domain disease–associated RyR2 mutations.
Figure 7.

Effects of disease-associated RyR2 mutations on [3H]ryanodine binding. A, 3D locations of disease-associated mutations in the central domain of RyR2. [3H]ryanodine binding to cell lysate prepared from HEK293 cells expressing the RyR2 WT or RyR2 disease-causing mutations was carried out at various Ca2+ concentrations (0.1 nm–0.1 mm). The amounts of [3H]ryanodine binding at various Ca2+ concentrations were normalized to the maximal binding (100%). B, EC50 values of Ca2+ activation and (C) basal activity (in the near absence of Ca2+, ∼0.1 nm) of [3H]ryanodine binding to RyR2. D, the apparent EC50 values of caffeine-induced Ca2+ releases in HEK293 cells transfected with RyR2 WT or mutants. [3H]ryanodine binding to RyR2 WT, K3997E, M3999V, and F4020L (E), to RyR2 WT, N4097S, L4188P, and R4157Q (F), and to RyR2 WT, T4196A, and Q4201R (G). The relationships between caffeine-induced Ca2+ release and cumulative caffeine concentrations in HEK293 cells transfected with RyR2 WT, K3997E, M3999V, and F4020L (H), with RyR2 WT, N4097S, L4188P, and R4157Q (I), and with RyR2 WT, T4196A, and Q4201R (J). Data points shown are mean ± S.D. (error bars) from 3–5 separate experiments. *, p < 0.05; **, p < 0.01.
Figure 8.

Effects of CPVT/sudden death–associated RyR2 mutations on caffeine-induced Ca2+ release in HEK293 cells. HEK293 cells were transfected with RyR2 WT or CPVT/sudden death–associated RyR2 mutants. The fluorescence intensity of the Fluo-3-loaded transfected cells was monitored continuously before and after each caffeine addition.
Figure 9.

Effects of Mg2+ on [3H]ryanodine binding to RyR2 WT and disease-associated mutants. Percentage of Mg2+ inhibition on [3H]ryanodine binding. [3H]ryanodine binding to cell lysates prepared from HEK293 cells expressing the RyR2 WT and disease-associated mutants was carried out at 0.1 nm Ca2+ and 2 mm Mg2+. ATP (3 mm) and caffeine (3 mm) were added to stimulate [3H]ryanodine binding. Data points shown are mean ± S.D. (error bars) from three separate experiments. Dashed red line indicates the percentage of Mg2+ inhibition in WT. *, p < 0.05; **, p < 0.01.
Discussion
The overall 3D structure of RyR consists of a large cytoplasmic assembly and a channel domain, which are connected by the central domain. Thus, the central domain is believed to be the primary transducer that integrates structural changes in the cytoplasmic assembly to the gating of the channel pore (29–36). Furthermore, recent structural studies mapped the Ca2+-, ATP-, and caffeine-binding sites within or near the central domain (34–36). This suggests that the central domain also serves as a signaling hub that controls the activity of RyR. Indeed, mutations of the putative Ca2+ activation site within the central domain abolished or markedly diminished Ca2+ activation of RyR2 (37). Consistent with these studies, we also found that mutating the Ca2+-coordinating residues (Glu-3848, Glu-3922, and Gln-3925) of RyR2 to alanine dramatically reduced Ca2+ activation of RyR2. The double mutation E3848A/E3922A nearly completely abolished Ca2+ activation of RyR2. Mutating the other two residues involved in Ca2+ coordination (His-3850 and Thr-4931) has a relatively lower effect on Ca2+ activation of RyR2. Surprisingly, mutation E3848A also markedly enhanced the basal activity of RyR2 in the near absence of Ca2+. This suggests that the E3848A mutation may destabilize the closed state of RyR2, resulting in spontaneous channel opening in the absence of activating Ca2+, but the underlying mechanism is unknown. It is possible that in the absence of Ca2+, the negatively charged side chains of the Ca2+-coordinating residues would tend to move away from each other because of electrostatic repulsion, which may contribute to the stabilization of the closed state of the channel. Removal of the negative charge from Glu-3848 (in the E3848A mutant) would permit the E3848A mutant reside to move closer to other Ca2+-coordinating residues, which may mimic the Ca2+-bound Glu-3848 state and thus destabilize the closed state. Further studies are needed to test this hypothesis. Interestingly, the enhanced basal activity in the E3848A mutant channel was inhibited by increasing concentrations of Ca2+ (∼1 μm–1 mm). This suggests the presence of a putative Ca2+-dependent inactivation site(s) that is different from the Glu-3848/Glu-3922–based high-affinity Ca2+ activation site. However, the identity, molecular mechanism, and physiological relevance of this putative Ca2+-dependent inactivation site(s) have yet to be investigated. Taken together, these observations suggest that residue Glu-3848 not only controls Ca2+ activation but also determines the basal activity of the channel in the near absence of Ca2+.
The close proximity of the Ca2+-, ATP-, and caffeine-binding sites within the central domain suggests that these ligands may interact with one another and interdependently modulate the activity of RyR2. Indeed, we found that ATP/caffeine increased both the basal activity and the Ca2+ sensitivity of the RyR2 WT and T4931A mutant channels. On the other hand, ATP/caffeine increased mainly the Ca2+-independent basal activity but had a relatively lower effect on the threshold or sensitivity of Ca2+ activation (by high concentrations of Ca2+) of [3H]ryanodine binding to the E3848A and E3922A mutants (Fig. 3, C, D, and H). This suggests that the action of ATP/caffeine in the Ca2+-dependent activation of RyR2 depends on Glu-3848 and Glu-3922 but that the action of ATP/caffeine in the Ca2+-independent basal activity of RyR2 does not. In contrast, ATP/caffeine increased predominantly the Ca2+ sensitivity, but not the basal activity, of the H3850A and Q3925A mutants. This suggests that the H3850A and Q3925A mutations strongly stabilized the closed state of the channel in the near absence of Ca2+, opposite to the effect of the E3848A mutation. Collectively, these data indicate that ATP and caffeine affect both the Ca2+-dependent activation and Ca2+-independent basal activity of RyR2 and that, vice versa, the Ca2+ activation site also has a major role in determining the actions of ATP/caffeine. These results also suggest that the Ca2+ activation site not only plays a critical role in Ca2+ activation but is also involved in controlling the stability of the closed state of RyR2.
It is of note that adjacent to the Ca2+ activation site, there is a cluster of residues with negatively charged or oxygen-containing side chains. The exact roles of these residues in RyR2 function are unknown. We previously reported that mutating one of these residues (E3987A) in RyR2 and the corresponding residue in RyR3 (E3885A) markedly reduced the Ca2+ activation of the channel (25, 26). Structural analysis suggested that although the Glu-3987 residue is not directly involved in Ca2+ coordination, it may play a role in the formation of the Ca2+ binding pocket by stabilizing the interface between the central domain and the CTD (34–36). Our current work showed that mutations T3929A, Q3932A, E4146A, T4934A, and E4937A near residue Glu-3987 also significantly decreased, whereas S3984A, Y4149S, and Q4936A significantly increased Ca2+ activation of RyR2, but their effect on Ca2+ activation is much lower compared with that of the Ca2+ coordination site mutations. Thus, these residues are unlikely to be directly involved in Ca2+ binding.
Although the residues with negatively charged or oxygen-containing side chains adjacent to the Ca2+ activation site may not play a major role in Ca2+ activation of RyR2, they may be involved in regulation of the channel by other cations, such as Mg2+. It is well established that Mg2+ inhibits the RyR channel (20, 23, 24, 38, 52), but the molecular basis of Mg2+-dependent inhibition of RyR is unknown. It has been proposed that Mg2+ inactivates RyR by binding to the Ca2+ activation site (20, 23, 24, 38, 52). Interestingly, the E3848A mutant that markedly reduced the Ca2+ activation site remained sensitive to both Ca2+ and Mg2+ inhibition. This suggests that Mg2+ may bind to another site, in addition to the Ca2+ activation site, to inactivate the RyR2 channel. To test this idea, we systematically assessed the effect of mutating the residues with negatively charged or oxygen-containing side chains located in the central domain on Mg2+ inhibition of RyR2. We found that mutations Y4149S, E4937A, E3848A, Q4936A, and S3984A significantly reduced Mg2+-dependent inhibition of RyR2. Interestingly, residues Tyr-4149, Glu-4937, Gln-4936, and Ser-3984 are located in close proximity and could potentially form a binding pocket (Fig. 6B). These results suggest that the central domain may also be involved in Mg2+ inhibition and Ca2+ activation of RyR2. However, it is possible that these mutations could alter Mg2+-dependent inhibition of RyR2 via an allosteric mechanism. High-resolution structural analysis will be needed to determine whether these residues are involved in Mg2+ binding.
We have previously shown that disease-associated RyR2 mutations located in the central domain increase the Ca2+ activation of RyR2 (51). We have now characterized additional RyR2 central domain mutations that are associated with CPVT and sudden death. Consistent with our previous work, we found that CPVT-associated mutations M3999V, F4020L, N4097S, R4157Q, L4188P, T4196A, and Q4201R (except for K3997E) significantly enhanced Ca2+ and caffeine activation and reduced Mg2+-dependent inhibition of RyR2. However, the exact molecular mechanisms by which these disease-associated RyR2 mutations alter Ca2+ and caffeine activation and Mg2+ inhibition of RyR2 have yet to be defined. A close examination of the locations of these mutations in the 3D structure of RyR2 revealed some clues. Among the central domain mutations characterized, the M3999V mutation exerted the strongest effect on RyR2 function by markedly increasing Ca2+ and caffeine activation and the basal activity of the channel. We speculate that this Met-3999 residue may potentially form hydrophobic interactions with Leu-3986, Leu-3982, Trp-3941, and Leu-4105, which may stabilize the adjacent Ca2+ binding pocket (Fig. 10A). Mutating Met-3999 to valine (M3999V) may strength these hydrophobic interactions, which may favor Ca2+ activation. The K3997E mutation that is located in the same helix as M3999V may alter the interaction among Lys-3997, Asn-3992, and Met-4109, and the Ca2+ activation of RyR2 in a similar manner but to a different extent (Fig. 10B). We further speculate that the Phe-4020 residue may potentially interact with Arg-4086 via cation-π interactions (53) and with Phe-4016 and Leu-4023 via hydrophobic interactions (Fig. 10C). Mutation F4020L could disrupt the cation-π interactions, which may allosterically alter the confirmation of the central domain and the confirmation of the Ca2+ activation site. We also hypothesize that the N4097S mutation may alter potential interactions among Asn-4097, Lys-3976, and Val-3979 and thus the confirmation of the adjacent CTD and Ca2+ activation site (Fig. 10D). Mutations R4157Q, L4188P, T4196A, and Q4201R are all located in the U-motif (Fig. 10E). This U-motif is the part of the central domain that grasps the CTD, forming a direct pathway transducing conformational changes from the central domain to the channel gate through the CTD and the S6 helix. We speculate that mutation R4157Q may alter potential interactions among residues Arg-4157, Phe-4920, and Ile-4152 (Fig. 10F), whereas mutation L4188P may affect potential interactions among residues Leu-4188, Val-4176, and Phe-4172 (Fig. 10G). Furthermore, we speculate that mutation T4196A may disturb potential interactions among residues Thr-4196, Phe-4192, and Leu-4919, whereas potential interactions with residue Q4201R are unclear (Fig. 10H). Thus, each of these mutations may alter the confirmation of the U-motif or the interactions between the U-motif and CTD, which may in turn affect the transduction of Ca2+- and caffeine-induced conformational changes to the channel gate or the stability of the channel gate itself, thereby altering Ca2+ and caffeine activation or the basal activity of RyR2. Clearly, further structural and functional analyses are required to validate these speculations and to define the molecular mechanisms of actions of each of these disease-associated RyR2 mutations.
Figure 10.

3D locations of disease-associated mutations and neighboring residues in the central domain of RyR2. A, 3D location of mutation M3999V and potentially interacting residues. B, 3D location of mutation K3997E and potentially interacting residues. C, 3D location of mutation F4020L and potentially interacting residues. D, 3D location of mutation N4097S and potentially interacting residues. E, 3D locations of mutations R4157Q, L4188P, T4196A, and Q4201R within the U-motif. F, 3D location of mutation R4157Q and potentially interacting residues. G, 3D location of mutation L4188P and potentially interacting residues. H, 3D locations of mutations T4196A and Q4201R and potentially interacting residues. Disease-associated mutations are colored in magenta and their potentially interacting residues in yellow.
Our current and previous studies (51) consistently demonstrate that enhanced Ca2+ activation and/or reduced Mg2+ inhibition of RyR2 represent a common defect of RyR2 mutations located in the central domain. However, the exact mechanisms by which RyR2 mutations in the central domain increase the propensity for CPVT and sudden death are unknown. Because activation of RyR2 by cytosolic Ca2+ is a critical component of the CICR mechanism that controls cardiac muscle contraction, enhanced RyR2 sensitivity to cytosolic Ca2+ activation or reduced sensitivity to Mg2+ inhibition would result in increased CICR sensitivity. An increased CICR sensitivity would enhance the propensity for the generation and propagation of arrhythmogenic spontaneous Ca2+ waves. Thus, it is possible that by increasing Ca2+ activation or decreasing Mg2+ inhibition of RyR2, the RyR2 central domain mutations may enhance CICR sensitivity and thus the propensity for spontaneous Ca2+ wave-evoked delayed afterdepolarizations, and triggered activity, and thus CPVT and sudden death.
The effect of the central domain mutations on caffeine activation was assessed and compared with their effect on the Ca2+ activation of RyR2. Notably, mutations that suppressed Ca2+ activation also suppressed caffeine activation, whereas mutations that enhanced Ca2+ activation also enhanced caffeine activation of RyR2. On the other hand, the effect of the central domain mutations on the Ca2+-independent basal activity of RyR2 has no correlation with that on caffeine activation. Because the caffeine-binding site differs from the Ca2+ activation site, some mutations may affect caffeine activation but not Ca2+ activation of RyR2. Indeed, we found that the T4931A mutation markedly suppressed caffeine activation of RyR2 but had little or no effect on Ca2+ activation of [3H]ryanodine binding. This indicates that although there is a close relationship between Ca2+ and caffeine activation, caffeine activation does not always reflect Ca2+ activation of RyR2.
In summary, our present study demonstrates that the central domain is a pivotal signaling hub that not only controls Ca2+ activation and basal activity of RyR2 but also determines Mg2+ inhibition of RyR2. The central domain also controls the modulation of the channel by ATP and caffeine and the stability of the closed state of RyR2 in the near absence of Ca2+. We also reveal that ATP and caffeine can enhance RyR2 channel activity in a Ca2+-dependent and Ca2+-independent manner. Our work also provides novel insights into the mechanism of RyR2 central domain mutations linked to cardiac arrhythmias and sudden death.
Experimental procedures
Materials
[3H]ryanodine was purchased from PerkinElmer. Ryanodine was purchased from Abcam. Caffeine was obtained from Sigma-Aldrich. ATP was obtained from EMD Millipore.
Construction of RyR2 mutations
The central domain RyR2 mutations, E3848A, E3922A, H3850A, Q3925A, T4931A, T3929A, Q3932A, Q3933A, S3984A, N3989A, E4146A, Y4149S, T4934A, Q4936A, E4937A, M3999V, F4020L, R4157Q, L4188P, T4196A, K3997E, N4097S, and Q4201R, were generated by the overlap extension method using PCR (25, 54). Briefly, the NruI-NotI (in the vector) fragment containing the mutation was obtained by overlapping PCR and used to replace the corresponding WT fragment in the full-length mouse RyR2 cDNA in pcDNA5. All mutations were confirmed by DNA sequencing.
Preparation of HEK293 cell lysates
HEK293 cells were transfected with RyR2 WT or central domain mutant cDNAs using the calcium phosphate precipitation method as described previously (25, 55). Twenty-four hours after transfection, the cells were harvested and resuspended in the lysis buffer containing 25 mm Tris, 50 mm HEPES, pH 7.4, 137 mm NaCl, 1% CHAPS, 0.5% egg phosphatidylcholine, 2.5 mm DTT, and a protease inhibitor mix (1 mm benzamidine, 2 μg/ml leupeptin, 2 μg/ml pepstatin A, 2 μg/ml aprotinin, and 0.5 mm PMSF) on ice for 60 min. Cell lysates were obtained after removing insolubilized materials by centrifugation.
[3H]ryanodine binding assay
Equilibrium [3H]ryanodine binding to cell lysates was performed as described previously (25, 55) with some modifications. Cell lysates were incubated with 5 nm [3H]ryanodine at 37 °C for 2 h in 300 μl of a binding solution containing 500 mm KCl, 25 mm Tris, and 50 mm HEPES, pH 7.4. For testing the effects of RyR2 modulators on channel function, Ca2+ (0.1 nm–100 mm), Mg2+ (2 mm), ATP (3 mm), and caffeine (3 mm) were added to the incubation solution. Free [Ca2+] was adjusted by EGTA and CaCl2 solutions using the computer program of Fabiato and Fabiato (56). Free [Mg2+] was adjusted by EGTA and MgCl2 solutions according to the Maxchelator program. At the completion of incubation, the samples were diluted with 5 ml of ice-cold washing buffer containing 25 mm Tris, pH 8.0, and 250 mm KCl and filtered through Whatman GF/B filters presoaked with 1% polyethylenimine. The filters were washed immediately with 2 × 5 ml of the same buffer. The amount of [3H]ryanodine retained in the filters was determined by liquid scintillation counting. Specifically bound [3H]ryanodine was calculated by subtracting nonspecific binding that was determined in the presence of 50 μm unlabeled ryanodine. All binding assays were performed in duplicate. [3H]ryanodine binding data were fitted with the Hill equation using the Prism 8 (GraphPad Software), from which EC50 values for Ca2+ activation were calculated.
Caffeine-induced Ca2+ release in HEK293 cells
The free cytosolic Ca2+ concentration in transfected HEK293 cells was measured using the fluorescence Ca2+ indicator dye Fluo-3 AM (Molecular Probes). HEK293 cells grown on 100-mm tissue culture dishes for 18–20 h after subculture were transfected with 12–16 μg of RyR2 WT or mutant cDNAs. Cells grown for 18–20 h after transfection were washed four times with PBS and incubated in KRH (Krebs–Ringer–Hepes, 125 mm NaCl, 5 mm KCl, 1.2 mm KH2PO4, 6 mm glucose, 1.2 mm MgCl2, 2 mm CaCl2, and 25 mm HEPES, pH 7.4) buffer without MgCl2 and CaCl2 at room temperature for 40 min and at 37 °C for 40 min. After being detached from culture dishes by pipetting, the cells were collected by centrifugation at 1,000 rpm for 2 min in a Beckman TH-4 rotor. Cell pellets were loaded with 5 μm Fluo-3 AM in high glucose DMEM at room temperature for 30 min, followed by washing with KRH buffer plus 2 mm CaCl2 and 1.2 mm MgCl2 (KRH+ buffer) three times and resuspended in 150 μl of KRH+ buffer plus 0.1 mg/ml BSA and 250 μm sulfinpyrazone. The Fluo-3 AM loaded cells were added to 2 ml (final volume) of KRH+ buffer in a cuvette. The fluorescence intensity of Fluo-3 AM at 530 nm was measured before and after repeated cumulative additions of various concentrations of caffeine (0.025–5mm) in an SLM Aminco series 2 luminescence spectrometer with 480 nm excitation at 25 °C (SLM Instruments). The peak levels of each caffeine-induced Ca2+ release were determined and normalized to the highest level (100%) of caffeine-induced Ca2+ release for each experiment. The normalized data of the ascending part of the caffeine dose response curve were fitted with the Hill equation to calculate the apparent EC50 value of caffeine activation for each construct using the curve fitting module of Prism 8 (GraphPad Software).
Statistical analysis
All data shown are means ± S.D. One-way analysis of variance (ANOVA) followed by Dunnett's multiple comparisons test or Student's t test (two-tailed) was performed using GraphPad Prism version 8 to assess the difference between mean values. A p value <0.05 was considered statistically significant.
Data availability
All data are contained within the article.
Author contributions—W. G., B. S., R. W., and S. W. C. conceptualization; W. G., J. P. E., and R. W. data curation; W. G. and B. S. formal analysis; W. G., B. S., and S. W. C. investigation; W. G., B. S., and R. W. methodology; W. G., R. W., and S. W. C. writing-original draft; B. S., J. P. E., and S. W. C. writing-review and editing; J. P. E. and S. W. C. project administration; S. W. C. resources; S. W. C. supervision; S. W. C. funding acquisition.
Funding and additional information—This work was supported by the Canadian Institutes of Health Research Grant PJT-155940 (to S. R. W. C.), the Heart and Stroke Foundation of Canada Grant G-19-0026444 (to S. R. W. C.), and the Heart and Stroke Foundation Chair in Cardiovascular Research (to S. R. W. C.). W. G. is a recipient of the Alberta Innovates-Health Solutions Graduate Studentship Award, and B. S. is a recipient of the Heart and Stroke Foundation of Canada Junior Fellowship Award and the AIHS Fellowship Award.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- RyR2
- cardiac ryanodine receptor
- CICR
- Ca2+-induced Ca2+ release
- CTD
- C-terminal domain
- CPVT
- catecholaminergic polymorphic ventricular tachycardia
- KRH
- Krebs-Ringer-Hepes
- ANOVA
- analysis of variance.
References
- 1. Coronado R., Morrissette J., Sukhareva M., and Vaughan D. M. (1994) Structure and function of ryanodine receptors. Am. J. Physiol. 266, C1485–C1504 10.1152/ajpcell.1994.266.6.C1485 [DOI] [PubMed] [Google Scholar]
- 2. Meissner G. (1994) Ryanodine receptor/Ca2+ release channels and their regulation by endogenous effectors. Annu. Rev. Physiol. 56, 485–508 10.1146/annurev.ph.56.030194.002413 [DOI] [PubMed] [Google Scholar]
- 3. Ogawa Y. (1994) Role of ryanodine receptors. Crit. Rev. Biochem. Mol. Biol. 29, 229–274 10.3109/10409239409083482 [DOI] [PubMed] [Google Scholar]
- 4. Sorrentino V. (1995) The ryanodine receptor family of intracellular calcium release channels. Adv. Pharmacol. 33, 67–90 10.1016/S1054-3589(08)60666-3 [DOI] [PubMed] [Google Scholar]
- 5. Sutko J. L., and Airey J. A. (1996) Ryanodine receptor Ca2+ release channels: does diversity in form equal diversity in function? Physiol. Rev. 76, 1027–1071 10.1152/physrev.1996.76.4.1027 [DOI] [PubMed] [Google Scholar]
- 6. Zucchi R., and Ronca-Testoni S. (1997) The sarcoplasmic reticulum Ca2+ channel/ryanodine receptor: modulation by endogenous effectors, drugs and disease states. Pharmacol. Rev. 49, 1–51 [PubMed] [Google Scholar]
- 7. Fill M., and Copello J. A. (2002) Ryanodine receptor calcium release channels. Physiol. Rev. 82, 893–922 10.1152/physrev.00013.2002 [DOI] [PubMed] [Google Scholar]
- 8. Bers D. M. (2002) Cardiac excitation-contraction coupling. Nature 415, 198–205 10.1038/415198a [DOI] [PubMed] [Google Scholar]
- 9. Van Petegem F. (2012) Ryanodine receptors: structure and function. J. Biol. Chem. 287, 31624–31632 10.1074/jbc.R112.349068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ebashi S. (1991) Excitation-contraction coupling and the mechanism of muscle contraction. Annu. Rev. Physiol. 53, 1–16 10.1146/annurev.ph.53.030191.000245 [DOI] [PubMed] [Google Scholar]
- 11. Endo M. (1977) Calcium release from the sarcoplasmic reticulum. Physiol. Rev. 57, 71–108 10.1152/physrev.1977.57.1.71 [DOI] [PubMed] [Google Scholar]
- 12. Fabiato A. (1985) Time and calcium dependence of activation and inactivation of calcium-induced release of calcium from the sarcoplasmic reticulum of a skinned canine cardiac Purkinje cell. J. Gen. Physiol. 85, 247–289 10.1085/jgp.85.2.247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cannell M. B., and Soeller C. (1997) Numerical analysis of ryanodine receptor activation by L-type channel activity in the cardiac muscle diad. Biophys. J. 73, 112–122 10.1016/S0006-3495(97)78052-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cheng H., Lederer M. R., Lederer W. J., and Cannell M. B. (1996) Calcium sparks and [Ca2+]i waves in cardiac myocytes. Am. J. Physiol. 270, C148–C159 10.1152/ajpcell.1996.270.1.C148 [DOI] [PubMed] [Google Scholar]
- 15. Stern M. D., Song L. S., Cheng H., Sham J. S., Yang H. T., Boheler K. R., and Ríos E. (1999) Local control models of cardiac excitation-contraction coupling. A possible role for allosteric interactions between ryanodine receptors. J. Gen. Physiol. 113, 469–489 10.1085/jgp.113.3.469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. MacLennan D. H., and Chen S. R. (2009) Store overload-induced Ca2+ release as a triggering mechanism for CPVT and MH episodes caused by mutations in RYR and CASQ genes. J. Physiol. 587, 3113–3115 10.1113/jphysiol.2009.172155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Priori S. G., and Chen S. R. (2011) Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis. Circ. Res. 108, 871–883 10.1161/CIRCRESAHA.110.226845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Denniss A., Dulhunty A. F., and Beard N. A. (2018) Ryanodine receptor Ca2+ release channel post-translational modification: Central player in cardiac and skeletal muscle disease. Int. J. Biochem. Cell Biol. 101, 49–53 10.1016/j.biocel.2018.05.004 [DOI] [PubMed] [Google Scholar]
- 19. Lieve K. V. V., Verhagen J. M. A., Wei J., Bos J. M., van der Werf C., Rosés i Noguer F., Mancini G. M. S., Guo W., Wang R., van den Heuvel F., Frohn-Mulder I. M. E., Shimizu W., Nogami A., Horigome H., Roberts J. D., et al. (2019) Linking the heart and the brain: Neurodevelopmental disorders in patients with catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 16, 220–228 10.1016/j.hrthm.2018.08.025 [DOI] [PubMed] [Google Scholar]
- 20. Meissner G., Darling E., and Eveleth J. (1986) Kinetics of rapid Ca2+ release by sarcoplasmic reticulum. Effects of Ca2+, Mg2+, and adenine nucleotides. Biochemistry 25, 236–244 10.1021/bi00349a033 [DOI] [PubMed] [Google Scholar]
- 21. Laver D. R., Roden L. D., Ahern G. P., Eager K. R., Junankar P. R., and Dulhunty A. F. (1995) Cytoplasmic Ca2+ inhibits the ryanodine receptor from cardiac muscle. J. Membr. Biol. 147, 7–22 10.1007/BF00235394 [DOI] [PubMed] [Google Scholar]
- 22. Meissner G., Rios E., Tripathy A., and Pasek D. A. (1997) Regulation of skeletal muscle Ca2+ release channel (ryanodine receptor) by Ca2+ and monovalent cations and anions. J. Biol. Chem. 272, 1628–1638 10.1074/jbc.272.3.1628 [DOI] [PubMed] [Google Scholar]
- 23. Meissner G. (2004) Molecular regulation of cardiac ryanodine receptor ion channel. Cell Calcium 35, 621–628 10.1016/j.ceca.2004.01.015 [DOI] [PubMed] [Google Scholar]
- 24. Laver D. R. (2018) Regulation of the RyR channel gating by Ca2+ and Mg2+. Biophys. Rev. 10, 1087–1095 10.1007/s12551-018-0433-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li P., and Chen S. R. (2001) Molecular basis of Ca2+ activation of the mouse cardiac Ca2+ release channel (ryanodine receptor). J. Gen. Physiol. 118, 33–44 10.1085/jgp.118.1.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen S. R., Ebisawa K., Li X., and Zhang L. (1998) Molecular identification of the ryanodine receptor Ca2+ sensor. J. Biol. Chem. 273, 14675–14678 10.1074/jbc.273.24.14675 [DOI] [PubMed] [Google Scholar]
- 27. Du G. G., and MacLennan D. H. (1998) Functional consequences of mutations of conserved, polar amino acids in transmembrane sequences of the Ca2+ release channel (ryanodine receptor) of rabbit skeletal muscle sarcoplasmic reticulum. J. Biol. Chem. 273, 31867–31872 10.1074/jbc.273.48.31867 [DOI] [PubMed] [Google Scholar]
- 28. O'Brien J. J., Feng W., Allen P. D., Chen S. R. W., Pessah I. N., and Beam K. G. (2002) Ca2+ activation of RyR1 is not necessary for the initiation of skeletal-type excitation-contraction coupling. Biophys. J. 82, 2428–2235 10.1016/S0006-3495(02)75586-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Efremov R. G., Leitner A., Aebersold R., and Raunser S. (2015) Architecture and conformational switch mechanism of the ryanodine receptor. Nature 517, 39–43 10.1038/nature13916 [DOI] [PubMed] [Google Scholar]
- 30. Zalk R., Clarke O. B., Des Georges A., Grassucci R. A., Reiken S., Mancia F., Hendrickson W. A., Frank J., and Marks A. R. (2015) Structure of a mammalian ryanodine receptor. Nature 517, 44–49 10.1038/nature13950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yan Z., Bai X. C., Yan C., Wu J., Li Z., Xie T., Peng W., Yin C. C., Li X., Scheres S. H., Shi Y., and Yan N. (2015) Structure of the rabbit ryanodine receptor RyR1 at near-atomic resolution. Nature 517, 50–55 10.1038/nature14063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bai X. C., Yan Z., Wu J., Li Z., and Yan N. (2016) The central domain of RyR1 is the transducer for long-range allosteric gating of channel opening. Cell Res. 26, 995–1006 10.1038/cr.2016.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Peng W., Shen H., Wu J., Guo W., Pan X., Wang R., Chen S. R., and Yan N. (2016) Structural basis for the gating mechanism of the type 2 ryanodine receptor RyR2. Science 354, aah5324 10.1126/science.aah5324 [DOI] [PubMed] [Google Scholar]
- 34. Des Georges A., Clarke O. B., Zalk R., Yuan Q., Condon K. J., Grassucci R. A., Hendrickson W. A., Marks A. R., and Frank J. (2016) Structural basis for gating and activation of RyR1. Cell 167, 145–157e117 10.1016/j.cell.2016.08.075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gong D., Chi X., Wei J., Zhou G., Huang G., Zhang L., Wang R., Lei J., Chen S. R. W., and Yan N. (2019) Modulation of cardiac ryanodine receptor 2 by calmodulin. Nature 572, 347–351 10.1038/s41586-019-1377-y [DOI] [PubMed] [Google Scholar]
- 36. Chi X., Gong D., Ren K., Zhou G., Huang G., Lei J., Zhou Q., and Yan N. (2019) Molecular basis for allosteric regulation of the type 2 ryanodine receptor channel gating by key modulators. Proc. Natl. Acad. Sci. U. S. A. 116, 25575–25582 10.1073/pnas.1914451116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Murayama T., Ogawa H., Kurebayashi N., Ohno S., Horie M., and Sakurai T. (2018) A tryptophan residue in the caffeine-binding site of the ryanodine receptor regulates Ca2+ sensitivity. Commun. Biol. 1, 98 10.1038/s42003-018-0103-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Laver D. R., Baynes T. M., and Dulhunty A. F. (1997) Magnesium inhibition of ryanodine-receptor calcium channels: evidence for two independent mechanisms. J. Membr. Biol. 156, 213–229 10.1007/s002329900202 [DOI] [PubMed] [Google Scholar]
- 39. Medeiros-Domingo A., Bhuiyan Z. A., Tester D. J., Hofman N., Bikker H., van Tintelen J. P., Mannens M. M., Wilde A. A., and Ackerman M. J. (2009) The RYR2-encoded ryanodine receptor/calcium release channel in patients diagnosed previously with either catecholaminergic polymorphic ventricular tachycardia or genotype negative, exercise-induced long QT syndrome: a comprehensive open reading frame mutational analysis. J. Am. Coll. Cardiol. 54, 2065–2074 10.1016/j.jacc.2009.08.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Inui M., Saito A., and Fleischer S. (1987) Isolation of the ryanodine receptor from cardiac sarcoplasmic reticulum and identity with the feet structures. J. Biol. Chem. 262, 15637–15642 [PubMed] [Google Scholar]
- 41. Chen S. R., Li P., Zhao M., Li X., and Zhang L. (2002) Role of the proposed pore-forming segment of the Ca2+ release channel (ryanodine receptor) in ryanodine interaction. Biophys. J. 82, 2436–2247 10.1016/S0006-3495(02)75587-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang R., Zhang L., Bolstad J., Diao N., Brown C., Ruest L., Welch W., Williams A. J., and Chen S. R. W. (2003) Residue Gln4863 within a predicted transmembrane sequence of the Ca2+ release channel (ryanodine receptor) is critical for ryanodine interaction. J. Biol. Chem. 278, 51557–51565 10.1074/jbc.M306788200 [DOI] [PubMed] [Google Scholar]
- 43. Du G. G., and MacLennan D. H. (1999) Ca2+ inactivation sites are located in the COOH-terminal quarter of recombinant rabbit skeletal muscle Ca2+ release channels (ryanodine receptors). J. Biol. Chem. 274, 26120–26126 10.1074/jbc.274.37.26120 [DOI] [PubMed] [Google Scholar]
- 44. Walsh R., Peters N. S., Cook S. A., and Ware J. S. (2014) Paralogue annotation identifies novel pathogenic variants in patients with Brugada syndrome and catecholaminergic polymorphic ventricular tachycardia. J. Med. Genet. 51, 35–44 10.1136/jmedgenet-2013-101917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Postma A. V., Denjoy I., Kamblock J., Alders M., Lupoglazoff J. M., Vaksmann G., Dubosq-Bidot L., Sebillon P., Mannens M. M., Guicheney P., and Wilde A. A. (2005) Catecholaminergic polymorphic ventricular tachycardia: RYR2 mutations, bradycardia, and follow up of the patients. J. Med. Genet. 42, 863–870 10.1136/jmg.2004.028993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Broendberg A. K., Nielsen J. C., Bjerre J., Pedersen L. N., Kristensen J., Henriksen F. L., Bundgaard H., and Jensen H. K. (2017) Nationwide experience of catecholaminergic polymorphic ventricular tachycardia caused by RyR2 mutations. Heart 103, 901–909 10.1136/heartjnl-2016-310509 [DOI] [PubMed] [Google Scholar]
- 47. Tester D. J., Arya P., Will M., Haglund C. M., Farley A. L., Makielski J. C., and Ackerman M. J. (2006) Genotypic heterogeneity and phenotypic mimicry among unrelated patients referred for catecholaminergic polymorphic ventricular tachycardia genetic testing. Heart Rhythm 3, 800–805 10.1016/j.hrthm.2006.03.025 [DOI] [PubMed] [Google Scholar]
- 48. Laitinen P. J., Brown K. M., Piippo K., Swan H., Devaney J. M., Brahmbhatt B., Donarum E. A., Marino M., Tiso N., Viitasalo M., Toivonen L., Stephan D. A., and Kontula K. (2001) Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation 103, 485–490 10.1161/01.cir.103.4.485 [DOI] [PubMed] [Google Scholar]
- 49. LaPage M. J., Russell M. W., Bradley D. J., and Dick M. II (2012) Novel ryanodine receptor 2 mutation associated with a severe phenotype of catecholaminergic polymorphic ventricular tachycardia. J. Pediatr. 161, 362–364 10.1016/j.jpeds.2012.04.013 [DOI] [PubMed] [Google Scholar]
- 50. Tester D. J., Spoon D. B., Valdivia H. H., Makielski J. C., and Ackerman M. J. (2004) Targeted mutational analysis of the RyR2-encoded cardiac ryanodine receptor in sudden unexplained death: a molecular autopsy of 49 medical examiner/coroner's cases. Mayo Clin. Proc. 79, 1380–1384 10.4065/79.11.1380 [DOI] [PubMed] [Google Scholar]
- 51. Xiao Z., Guo W., Sun B., Hunt D. J., Wei J., Liu Y., Wang Y., Wang R., Jones P. P., Back T. G., and Chen S. R. (2016) Enhanced cytosolic Ca2+ activation underlies a common defect of central domain cardiac ryanodine receptor mutations linked to arrhythmias. J. Biol. Chem. 291, 24528–24537 10.1074/jbc.M116.756528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Laver D. R., and Honen B. N. (2008) Luminal Mg2+, a key factor controlling RYR2-mediated Ca2+ release: cytoplasmic and luminal regulation modeled in a tetrameric channel. J. Gen. Physiol. 132, 429–446 10.1085/jgp.200810001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gallivan J. P., and Dougherty D. A. (1999) Cation-π interactions in structural biology. Proc. Natl. Acad. Sci. U. S. A. 96, 9459–9464 10.1073/pnas.96.17.9459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ho S. N., Hunt H. D., Horton R. M., Pullen J. K., and Pease L. R. (1989) Site-directed mutagenesis by overlap extension using the polymerase chain reaction Gene 77, 51–59 10.1016/0378-1119(89)90358-2 [DOI] [PubMed] [Google Scholar]
- 55. Guo W., Sun B., Xiao Z., Liu Y., Wang Y., Zhang L., Wang R., and Chen S. R. (2016) The EF-hand Ca2+ binding domain is not required for cytosolic Ca2+ activation of the cardiac ryanodine receptor. J. Biol. Chem. 291, 2150–2160 10.1074/jbc.M115.693325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fabiato A., and Fabiato F. (1979) Calculator programs for computing the composition of the solutions containing multiple metals and ligands used for experiments in skinned muscle cells. J. Physiol. 75, 463–505 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data are contained within the article.