

Abstract
The unfolded protein response (UPR) plays a central role in regulating endoplasmic reticulum (ER) and global cellular physiology in response to pathologic ER stress. The UPR is comprised of three signaling pathways activated downstream of the ER membrane proteins IRE1, ATF6, and PERK. Once activated, these proteins initiate transcriptional and translational signaling that functions to alleviate ER stress, adapt cellular physiology, and dictate cell fate. Imbalances in UPR signaling are implicated in the pathogenesis of numerous, etiologically-diverse diseases, including many neurodegenerative diseases, protein misfolding diseases, diabetes, ischemic disorders, and cancer. This has led to significant interest in establishing pharmacologic strategies to selectively modulate IRE1, ATF6, or PERK signaling to both ameliorate pathologic imbalances in UPR signaling implicated in these different diseases and define the importance of the UPR in diverse cellular and organismal contexts. Recently, there has been significant progress in the identification and characterization of UPR modulating compounds, providing new opportunities to probe the pathologic and potentially therapeutic implications of UPR signaling in human disease. Here, we describe currently available UPR modulating compounds, specifically highlighting the strategies used for their discovery and specific advantages and disadvantages in their application for probing UPR function. Furthermore, we discuss lessons learned from the application of these compounds in cellular and in vivo models to identify favorable compound properties that can help drive the further translational development of selective UPR modulators for human disease.
Keywords: unfolded protein response (UPR), endoplasmic reticulum (ER), proteostasis, small molecule, modulation, high-throughput screening (HTS), unfolded protein response (UPR), proteostasis, small molecule, endoplasmic reticulum stress (ER stress), stress response
The endoplasmic reticulum (ER) is associated with critical biological functions, including protein secretion, lipid synthesis, and calcium regulation (1–5). Therefore, proper regulation of ER function in response to a constantly changing physiologic environment is crucial for organismal survival. Consistent with this, defects in ER biology are linked to nearly all types of human disease, including cancer, diabetes, infectious disease, amyloid diseases, ischemic disorders, and neurodegenerative diseases (6–13). Considering the importance of the ER in disease pathogenesis, pharmacologic intervention to ameliorate pathologic imbalances in ER function has emerged as a promising therapeutic approach for a wide variety of disorders.
One of the most attractive strategies to alter ER function in the context of disease is through targeting the unfolded protein response (UPR)—the predominant stress-responsive signaling pathway responsible for regulating ER and cellular physiology following an ER insult (i.e. ER stress) (14–19). The UPR comprises three signaling pathways activated downstream of the ER stress–sensing transmembrane proteins inositol-requiring enzyme 1 (IRE1), protein kinase R–like endoplasmic reticulum kinase (PERK), and activating transcription factor 6 (ATF6) (Fig. 1) (16–20). These three signaling pathways are activated in response to diverse types of ER stress, including the accumulation of non-native proteins within the ER lumen and lipid disequilibrium within the ER membrane. Activation of these UPR pathways elicits transcriptional and translational remodeling of ER and global cellular physiology that functions to alleviate the ER stress and promote cellular adaption following an acute insult (Fig. 1). Through this activity, the UPR functions as a protective signaling pathway that is involved in regulating diverse aspects of cellular physiology, including maintenance of secretory proteostasis, proliferation, redox regulation, differentiation, and metabolism (14, 15). However, in response to chronic or severe ER insults that cannot be alleviated through protective remodeling, prolonged UPR activation leads to pro-apoptotic signaling (10, 17). Thus, the UPR serves a critical role in dictating both protective and apoptotic signaling in response to pathologic ER insults.
Figure 1.

The three ER stress–sensing proteins that activate UPR signaling. Activation of IRE1, PERK, and ATF6 promotes integrated signaling that translationally and transcriptionally remodels ER and cellular proteostasis.
Due to the importance of UPR signaling for regulating ER function, it is not surprising that alterations in UPR signaling contribute to human disease pathogenesis. For example, hypomorphic or “loss-of-function” mutations in the EIF2AK3 gene, which encodes the PERK protein, are associated with multiple diseases, including Wolcott–Rallison syndrome, progressive supranuclear palsy, and late-stage Alzheimer's disease (21–24). Similarly, environmental or aging-related deficiencies in UPR signaling contribute to diverse types of disease, including cardiovascular disorders and neurodegenerative diseases (10, 11). In contrast, overactivity of UPR signaling is also associated with disease pathogenesis. For example, overactive PERK signaling is implicated in many different neurodegenerative diseases (11, 25, 26). Similarly, chronic IRE1 activity is associated with atherosclerosis in mouse models (27). Thus, either too much or too little signaling through UPR signaling pathways can promote pathogenesis in the context of human disease. This effect may be best demonstrated in the hereditary vision disorder achromatopsia, where mutations in the ATF6 gene that either increase or decrease ATF6 activity are both causatively implicated in the impaired retinal development central to disease pathogenesis (28, 29).
The importance of altered UPR signaling in the pathogenesis of etiologically-diverse diseases makes these pathways attractive targets for therapeutic intervention (9, 30, 31). This has led to significant interest in establishing compounds that either activate or inhibit select UPR signaling pathways to provide new opportunities to define the therapeutic potential for targeting the UPR in human disease. Here, we discuss currently available compounds that target individual UPR pathways, specifically highlighting how they were discovered, their described mechanism of action, and their applicability for studying the importance of UPR signaling in cellular and in vivo models. In addition, we summarize lessons learned from these available UPR-modulating compounds to identify specific properties that confer increased translational potential for application in human disease to help guide the future development of next-generation compounds.
The IRE1 arm of the UPR
The IRE1 signaling pathway is the most highly conserved arm of the UPR, found in all organisms from yeast to humans (Fig. 1) (20, 32). Notably, it was the first UPR pathway to be identified and is likely the most well-studied. IRE1 is a type I ER membrane protein comprising three domains: an ER luminal domain, a cytosolic kinase domain, and a cytosolic RNase domain (Fig. 2, A and B) (33, 34). Mammals encode two different IRE1 isoforms: IRE1α and IRE1β. IRE1α is the predominant isoform associated with UPR signaling in most cell types, whereas IRE1β appears to primarily function through tissue-specific mechanisms and/or the regulation of IRE1α activity (35–37). For the purposes of this review, we primarily focus on the IRE1α isoform (herein referred to as IRE1 unless otherwise noted).
Figure 2.
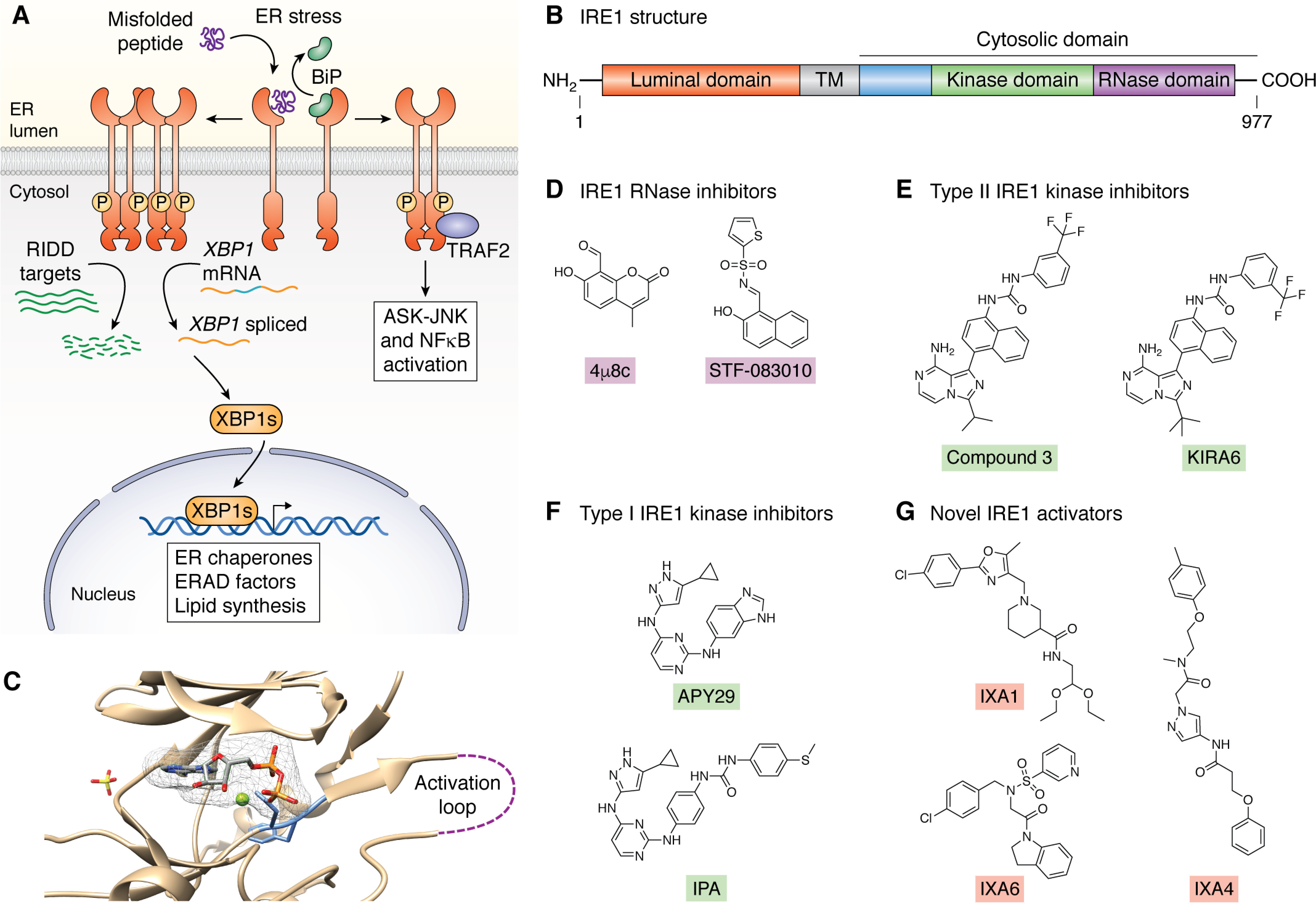
Pharmacologic targeting of the IRE1 UPR signaling pathway. A, simplified mechanism of ER stress–dependent activation and downstream signaling of the IRE1 UPR signaling pathway. B, domain architecture of IRE1, including the luminal domain, transmembrane domain (TM), and the cytosolic kinase and RNase domains. Enzymatic activities of key domains are as shown in A, where the cytosolic kinase domain of IRE1 participates in trans-autophosphorylation, and, upon activation, the RNase domain functions through both XBP1 mRNA splicing and RIDD. C, image showing the binding of ADP to the nucleotide-binding pocket of human IRE1 (48). D, structures of the IRE1 RNase active site inhibitors 4µ8c and STF-083010. E, structures of the Type II IRE1 kinase inhibitors Compound 3 and KIRA6 that inhibit both IRE1 kinase activity and RNase activity. F, structures of Type I IRE1 kinase inhibitors that inhibit IRE1 kinase activity while allosterically activating the IRE1 RNase. G, structures of the new IRE1/XBP1s-activating compounds IXA1, IXA4, and IXA6 identified through an HTS that prioritized transcriptional profiling.
IRE1 is activated in response to diverse cellular insults, including ER stress and lipid disequilibrium (38, 39). Despite being the most well-studied arm of the UPR, the molecular mechanism of IRE1 activation remains somewhat controversial, with multiple proposed models, all of which involve ER stress sensing by the IRE1 luminal domain. One prominent model suggests that IRE1 detects ER stress through dynamic interactions between the ER HSP70 chaperone binding immunoglobulin protein (BiP) and the IRE1 luminal domain through a process regulated by BiP co-chaperones, such as ER DNA J domain–containing protein 4 (ERdj4) (19, 40–42). In this model, BiP dissociates from the IRE1 luminal domain in response to the accumulation of misfolded proteins with the ER lumen, thus activating signaling through this pathway (Fig. 2A). However, another model proposes that IRE1 directly binds non-native protein conformations in a putative peptide-binding groove found in the IRE1 luminal domain, suggesting that IRE1 directly senses the accumulation of non-native proteins during ER stress (Fig. 2A) (43–45). Whereas the precise mechanism of IRE1 activation is still being scrutinized, the downstream events of IRE1 signaling are well-established to involve IRE1 oligomerization, autophosphorylation, and RNase activation (Fig. 2A) (38). IRE1 exists predominantly as a monomer in the ER membrane and in response to ER stress undergoes dimerization/oligomerization and subsequent trans-autophosphorylation (Fig. 2A) (18, 19, 38). Whereas both dimer and higher-order IRE1 oligomers have been described in vitro, it appears that clustering of IRE1 into higher-order assemblies parallels maximal activation as measured by downstream signaling outputs (46, 47). IRE1 autophosphorylation occurs at several sites on a conserved “activation loop” within its cytosolic kinase domain (Fig. 2C) (48). Extensive structural and biochemical studies on yeast and human IRE1 have characterized the displacement of this loop upon ATP binding, due to direct interactions of bound ADP with a conserved DFG motif in the kinase active site (Fig. 2C) (48). These events cause coordinated conformational changes through the IRE1 cytosolic domain, which allosterically activate the IRE1 RNase (48). This RNase domain cleaves the mRNA encoding X-box binding protein 1 (XBP1), which is then re-ligated by the RTCB RNA ligase, resulting in a frameshift in this transcript (Fig. 2A) (49–53). Spliced XBP1 mRNA (XBP1s) encodes the active transcription factor spliced XBP1 (XBP1s), which up-regulates transcriptional targets that promote ER proteostasis, including genes involved in ER-associated degradation (ERAD), ER chaperones and folding enzymes, and N-linked glycosylation (Fig. 2A) (50, 54). In addition, XBP1s has also been proposed to up-regulate transcripts involved in other biological pathways, including lipid biosynthesis (Fig. 2A) (54, 55). In general, XBP1s transcriptional signaling is a protective mechanism to promote ER proteostasis and cellular adaptation in response to acute stress.
Aside from inducing transcriptional changes via activation of XBP1s, IRE1 has other functionalities that play a role in stress-responsive signaling following ER stress. One of these additional functions is termed regulated IRE1-dependent decay (RIDD), in which the activated IRE1 RNase degrades a variety of ER-associated mRNAs (Fig. 2A) (56, 57). Whereas the specificity for these RIDD substrates is not yet understood, many putative RIDD targets have been identified, including scavenger receptor class A member 3 (SCARA3) and biogenesis of lysosomal organelles complex 1 (BLOS1) (57, 58). Unlike protective XBP1s transcriptional signaling, IRE1 RIDD activity has been implicated in both protective and pro-apoptotic signaling. For example, RIDD activity reduces incoming protein folding load within the ER and suppresses apoptotic signaling through degradation of mRNA encoding death receptor 5 (DR5) (59). Similarly, IRE1-dependent degradation of BLOS1 mRNA through RIDD promotes repositioning of late endosomes for degradation of protein aggregates (58). In contrast, RIDD has also been suggested to promote apoptotic signaling through the degradation of mRNA encoding protective UPR-regulated chaperones (e.g. BiP) and the degradation of anti-apoptotic pre-miRNAs (60, 61). Thus, RIDD activity appears to be involved in the transitioning of IRE1 signaling from adaptive to pro-apoptotic in response to severe or prolonged ER stress. Interestingly, IRE1-dependent degradation of BLOS1 and other RIDD targets has been suggested to involve signaling through the PERK arm of the UPR, although PERK activation on its own is not sufficient to promote RIDD, highlighting the importance of integration between UPR signaling pathways in regulating cellular responses to ER stress (62).
IRE1 also promotes signaling independent of its RNase activity. Active, phosphorylated IRE1 can bind tumor necrosis factor receptor–associated factor 2 (TRAF2) to promote apoptotic signaling downstream of the ASK-JNK signaling axis and inflammatory signaling downstream of nuclear factor κB (NFκB) (Fig. 2A) (63, 64). This IRE1-TRAF2 signaling promotes cell death and inflammation in response to severe or chronic ER insults associated with pathologic conditions, including fatty liver disease and neurodegeneration (65–67). This ability of IRE1 to promote cell death independent of its RNase activity is an important consideration when developing pharmacologic approaches to modulate IRE1 activity in the context of human disease.
The potential to influence diverse aspects of IRE1 signaling using pharmacologic approaches represents a unique opportunity to alter pathologic imbalances in UPR signaling implicated in diverse diseases. Below, we discuss the different types of compounds available to activate or inhibit IRE1 signaling and their application to probe IRE1 function in cellular and in vivo models of disease.
Preventing IRE1 signaling with RNase inhibitors
Whereas IRE1 activity is often protective during acute ER stress, chronic activity of this pathway has been associated with numerous disease etiologies and can support the persistence of certain cancers (68, 69). Thus, multiple strategies have been employed to develop potent inhibitors of IRE1 signaling. One such strategy focuses on developing compounds that block IRE1 RNase activity to prevent XBP1 splicing. To this end, high-throughput screening using in vitro FRET assays for XBP1 cleavage has been commonly utilized to identify these types of IRE1 RNase inhibitors. This approach has been used to identify classes of salicylaldehyde analogs (e.g. MK0186893) and umbelliferones (e.g. 4µ8c), as promising IRE1 inhibitors that selectively prevent IRE1 RNase activity but do not elicit cellular toxicity (Fig. 2D) (70, 71). Another IRE1 RNase inhibitor, STF-083010, was identified using a phenotypic cell-based high-throughput screen monitoring the activity of an XBP1-splicing luciferase reporter in human fibrosarcoma cells subjected to ER stress, demonstrating that cell-based strategies can also be applied to identify this class of inhibitor (Fig. 2D) (72). All of these IRE1 RNase inhibitors exhibit selectivity for IRE1 in cell-based models and have been shown to covalently modify residues in the IRE1 RNase domain via Schiff base formation (70). Importantly, 4µ8c and STF-083010, but not MK0186893, directly bind the IRE1 RNase domain without modification of the kinase domain, indicating that these compounds are able to block IRE1 signaling activated by the RNase domain (e.g. RIDD and XBP1 splicing), without interfering with IRE1 phosphorylation status (70). This specific activity provides a useful strategy to separate the enzymatic activities involved in IRE1 signaling and to probe the direct role of the RNase domain in the various IRE1 signaling functionalities.
Importantly, use of IRE1 RNase inhibitors has shown promise as a potential therapeutic strategy to counteract disease pathogenesis associated with overactive IRE1 signaling. For example, both STF-083010 and 4µ8c prevent the systemic up-regulation of inflammatory factors IL-1β and IL-18 downstream of IRE1 in response to hyperlipidemia in mice, preventing the chronic “metainflammation” known to play a role in the development of metabolic disorders (27). Consistent with this, these compounds lower immune responses and atherosclerotic plaque development in mouse models of atherosclerosis (27). Additionally, 4µ8c and STF-083010 slow cancer cell proliferation in models of multiple myeloma, demonstrating the potential of inhibiting prosurvival UPR functionality in carcinogenic cells as a mode of chemotherapy (70, 72). Importantly, whereas both 4µ8c and STF-083010 are widely used and exhibit limited toxicity, 4µ8c has some reported off-target effects. For example, treatment of pancreatic β-cells with 4µ8c resulted in reduced insulin secretion independent of IRE1 RNase inhibition (73). In addition, 4µ8c appears to have antioxidant properties, demonstrated by decreases in angiotensin II–induced reactive oxygen species production (74). Therefore, whereas STF-083010, 4µ8c, and related analogs have distinguished themselves as useful tools to inhibit IRE1 RNase signaling, special consideration must be given when using 4µ8c for investigating the specific consequences of IRE1 inhibition in cellular and in vivo models.
Inhibiting IRE1 autophosphorylation with kinase inhibitors
Similar in vitro approaches to those described above have also been used to establish compounds that inhibit IRE1 kinase activity. Generally, kinase inhibitors are classified as Type I or Type II based on their ability to stabilize kinase active sites in opposing conformations (75). In the context of IRE1, Type II inhibitors have been shown to stabilize the IRE1 activation loop in a conformation that blocks both IRE1 autophosphorylation and allosteric activation of the IRE1 RNase (76). Thus, Type II kinase inhibitors are predicted to inhibit both IRE1 RNase-dependent activities (e.g. XBP1 splicing and RIDD) and RNase-independent functions dependent on IRE1 phosphorylation (e.g. TRAF2 binding). A class of pyrazolopyrimidine-based Type II kinase inhibitors was identified by FRET-based screening, leading to the development of Compound 3, which prevented XBP1 cleavage to a similar extent as the RNase inhibitor, STF-083010 (Fig. 2E) (76). Whereas Compound 3 was further validated to inhibit IRE1 in INS-1 cells, its selectivity for IRE1 remains to be defined in cell-based models (76). A second generation of IRE1 Type II kinase inhibitors, called kinase-inhibiting RNase attenuators (KIRAs), included KIRA6, which had the same mechanistic properties as Compound 3 (Fig. 2E) (77). However, KIRA6 afforded increased potency for inhibition of cytosolic IRE1 activity in vitro (77). Further characterization in INS-1 cells demonstrated that KIRA6 prevented IRE1 phosphorylation and XBP1 splicing induced by the ER stressor, tunicamycin, in a dose-dependent manner (77). Studies in other cellular and in vitro models, however, describe KIRA6-induced cell death at nanomolar concentrations and poor specificity against a panel of diverse kinases, thus demonstrating the need for validating the selectivity of these types of UPR modulators in multiple contexts (78, 79). Despite these challenges, KIRA6 and the related compound KIRA8 have been shown to reduce pancreatic damage associated with Type I diabetes, protect the retina against ER stress–induced apoptosis, and decrease Zika virus infection, demonstrating the potential for these types of kinase inhibitors to mitigate pathologic events associated with IRE1 signaling in disease models (77, 80, 81). However, further studies are still needed to validate the dependence of these observed benefits on the specific inhibition of IRE1 activity as opposed to off-target effects.
Allosteric activation of IRE1 RNase activity using kinase inhibitors
Type I kinase inhibitors have opposing effects to Type II inhibitors on the conformation of the activation loop within the IRE1 kinase active site (82, 83). Thus, Type I kinase inhibitors are predicted to allosterically activate the IRE1 RNase domain, while inhibiting IRE1 autophosphorylation. This class of kinase inhibitors unsurprisingly includes ATP mimics, such as the clinically approved kinase inhibitor sunitinib and the aminopyrazole APY29 (Fig. 2F) (46). Co-crystal structures of yeast Ire1 bound to APY29 demonstrated this mechanism of allosteric RNase activation by Type I kinase inhibitors (46). Subsequent studies with APY29 in INS-1 cells showed divergent effects of APY29 and the Type II IRE1 kinase inhibitor, Compound 3, on XBP1 splicing and multiple other downstream consequences of IRE1 signaling, further highlighting the distinct activities of these two classes of kinase inhibitors on IRE1 RNase activity (76). Importantly, cell-based studies indicated pleiotropic toxicity from APY29 treatment at low micromolar concentrations, making it difficult to apply this compound to diverse cellular contexts (77).
Targeted compound-engineering efforts have additionally resulted in the development of other IRE1 kinase inhibitors, such as IPA (Fig. 2F)(84). This compound binds the IRE1 kinase active site and stabilizes its active conformation to increase IRE1 oligomerization and subsequent RNase activation (84). However, like APY29, IPA demonstrated cellular toxicity at nanomolar concentrations in cells, limiting the potential for this compound to probe IRE1 activation in cellular and organismal models (84). This toxicity is likely associated with off-target binding of IPA to other kinases. Consistent with this, the IPA scaffold also bound to the PERK kinase active site to activate PERK signaling at low concentrations (<2 μm) and inhibit PERK signaling at higher concentrations (>2 μm) in HEK293T cells (84).
Type I and Type II IRE1 kinase inhibitors have proven very useful for deconvoluting the molecular mechanism of IRE1 activation and allow a unique opportunity to inhibit or activate IRE1 RNase signaling through kinase inhibition without directly targeting the RNase domain. However, due to the off-target activity of these compounds (likely associated with binding to other kinases), toxicity, and/or poor bioavailability, these compounds have proven less useful for determining the functional implications of IRE1 signaling in the context of complex cellular and in vivo systems, as compared with other IRE1-modulating compounds. Regardless, these compounds can be used in certain cellular contexts, as well as in tandem with compounds that directly modulate IRE1 RNase activity, to validate the role of IRE1 signaling in a variety of biological and pathological settings.
Phenotypic screening for IRE1-activating compounds with a novel mechanism
In addition to targeted screening for IRE1 activators, phenotypic screening provides a useful approach to identify compounds that activate specific aspects of IRE1 signaling. Recently, a cell-based phenotypic high-throughput screen (HTS) was applied to identify compounds that selectively activate the protective IRE1/XBP1s signaling arm of the UPR. This screening strategy prioritized transcriptional selectivity to identify compounds that show preferential activation of IRE1/XBP1s over other arms of the UPR or other stress-responsive signaling pathways. This HTS used an IRE1-dependent XBP1 splicing luciferase reporter to screen a >650,000-compound library to identify IRE1-activating compounds (85). Hits were then counterscreened against a luciferase reporter for the ATF6 UPR signaling pathway to identify compounds that preferentially activate IRE1/XBP1s signaling over other arms of the UPR. Through this screen, three compounds were identified, IXA1, IXA4, and IXA6, which were shown to selectively activate IRE1-dependent XBP1s signaling without significantly activating RIDD or TRAF2-dependent signaling (Fig. 2G). Importantly, these compounds were non-toxic in HEK293TREX cells (IC50 > 3 μm) (85). The selectivity of these compounds for the IRE1/XBP1s pathway was further defined by RNA-seq transcriptional profiling, confirming that these compounds do not activate other arms of the UPR or other stress-responsive signaling pathways (85). Importantly, these compounds increase IRE1/XBP1s signaling without inhibiting kinase activity, indicating that these compounds activate IRE1 through a mechanism distinct from the above-mentioned Type I kinase inhibitors (85). Although the mechanism of action for these compounds as well as in vivo efficacy remains to be established, these compounds have already proven useful for demonstrating the potential for pharmacologic IRE1 activation to reduce the production and toxicity of amyloid precursor protein proteolytic fragments in cell culture models (85).
The PERK arm of the UPR
The PERK UPR signaling pathway has proven to be an extremely attractive target for pharmacologic intervention. PERK is composed of an N-terminal luminal domain and a cytosolic effector kinase domain (Fig. 3, A and B) (86, 87). Like IRE1, PERK can be activated by both ER stress and lipid disequilibrium (39, 87). Interestingly, the mechanism of PERK activation is similar, although not identical, to that observed for IRE1. In response to ER stress, BiP dissociates from the PERK luminal domain, promoting oligomerization and autophosphorylation of the cytosolic PERK kinase domain (Fig. 3A) (40, 88, 89). However, unlike IRE1, BiP co-chaperones such as ERdj4 do not appear to be involved in the regulation of PERK signaling, highlighting subtle differences between the activation of these different ER stress sensors (41). Once activated, PERK primarily functions through phosphorylation of the α-subunit of eukaryotic initiation factor 2 (eIF2α) at residue Ser-51 (90), although other PERK kinase targets, such as nuclear factor erythroid 2–related factor 2 (NRF2), have also been reported (91). Phosphorylation of eIF2α disrupts protein translation by increasing the affinity of the eIF2 complex for the eIF2B GTP exchange factor. This prevents eIF2B activity, which is required for translation initiation (92, 93). As a result, ribosomal protein synthesis is globally reduced. This reduction protects the ER by reducing the load of newly synthesized, unfolded proteins entering into the ER lumen, allowing ER proteostasis factors (e.g. chaperones and folding enzymes) to engage existing misfolded ER proteins and facilitate their refolding or clearance through mechanisms including ERAD or autophagy (Fig. 3A) (86, 87, 94, 95). Apart from reducing folding load, PERK-dependent translational attenuation has also been shown to regulate other aspects of cellular physiology, including cell-cycle progression, mitochondrial protein import, and mitochondrial morphology, through mechanisms such as the increased degradation of short-lived proteins (96–99).
Figure 3.
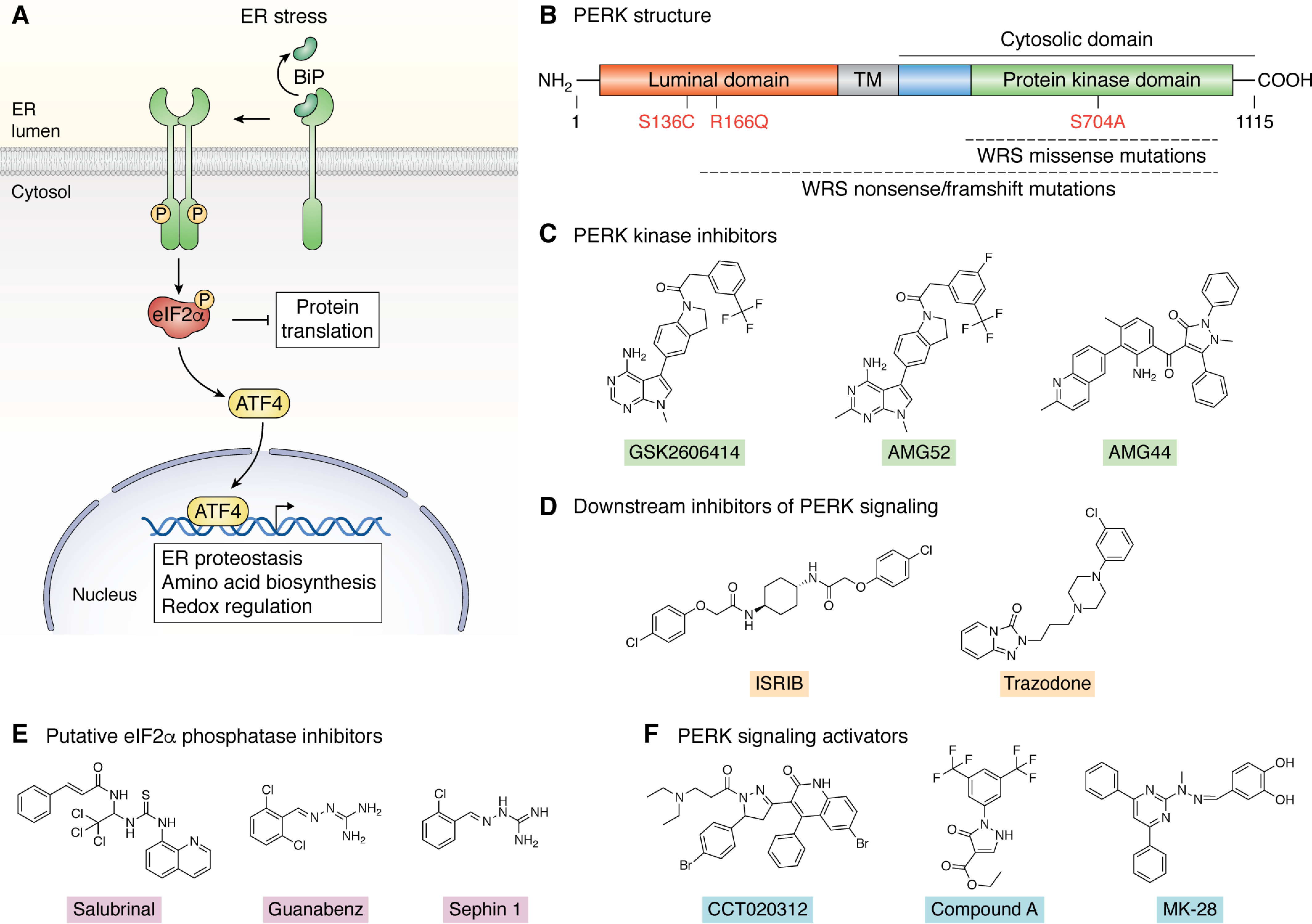
Pharmacologic PERK-modulating compounds. A, mechanism of activation and signaling for the PERK signaling arm of the UPR. B, domain architecture of PERK, including the luminal domain, transmembrane domain (TM), and cytosolic protein kinase domain. The protein kinase domain functions as shown in A through both PERK autophosphorylation and the phosphorylating eIF2α, the latter a key step of the PERK signaling cascade. C, structures of the PERK kinase inhibitors GSK2606414, AMG52, and AMG44. WRS, Wolcott–Rallison syndrome associated mutations are indicated. D, structures of the inhibitors of PERK signaling ISRIB and trazodone that block PERK signaling downstream of eIF2α phosphorylation. E, structures of the putative eIF2α phosphatase inhibitors salubrinal, guanabenz, and sephin 1. F, structures of PERK activators CCT020312, compound A, and MK-28 identified through phenotypic and computational screening approaches.
Whereas general protein synthesis is decreased by PERK-dependent eIF2α phosphorylation, a specific group of proteins is selectively translated under these conditions. These include the stress-responsive transcription factor activating transcription factor 4 (ATF4) (Fig. 3A) (100–102). The mRNA encoding this protein escapes translational inhibition afforded by eIF2α phosphorylation through upstream ORFs found in its 5′-UTR (103). ATF4 induces the expression of stress-responsive genes involved in diverse biological functions, including cellular redox, mitochondrial proteostasis, tRNA charging, and nutrient transport (Fig. 3A) (104, 105). Many of these genes are also transcriptionally regulated via upstream ORFs and thus are translated during PERK activation (103). ATF4 also induces the expression of the eIF2α phosphatase regulator subunit growth arrest and DNA damage–inducible protein 34 (GADD34), which is involved in dephosphorylating eIF2α and restoring translation in a negative feedback loop that suppresses PERK signaling (106, 107). Through this transcriptional activity, PERK promotes cellular adaptation and survival in response to acute insults.
However, severe or chronic ER stress promotes pro-apoptotic signaling downstream of PERK (10). One mechanism by which PERK promotes apoptosis is through the up-regulation of the transcription factor C/EBP-homologous protein 10 (CHOP) (108). CHOP induces the expression of multiple pro-apoptotic factors, including Bcl2-like protein 11 (BIM) and DR5 to activate intrinsic apoptotic signaling cascades and caspase activation (10, 59, 109–111). PERK can also promote cell death through other mechanisms, including increased oxidative stress associated with the recovery of protein synthesis following translational attenuation, increased expression of microRNAs that disrupt cellular metabolism, and the suppression of antiapoptotic factors, such as X-linked inhibitor of apoptosis (XIAP) (10, 104, 112, 113). Interestingly, this pro-apoptotic signaling downstream of PERK coordinates with other UPR signaling to dictate cell fate in response to severe ER insults. For example, the CHOP-regulated, pro-apoptotic DR5 mRNA is a target of the RIDD pathway, which functions to suppress apoptotic signaling (59). In response to prolonged ER stress, RIDD activity declines, releasing this “break” on apoptosis and allowing DR5-mediated apoptotic signaling. This type of integration provides a sophisticated mechanism to regulate cell fate in response to varying levels of ER stress.
Considering the importance of PERK in dictating both adaptation and survival in response to ER stress, it is not surprising that both increases and decreases in PERK activity are implicated in diverse types of disease. For example, mutations in EIF2AK3 (the gene encoding PERK) that reduce or eliminate signaling through this UPR pathway are associated with Wolcott–Rallison syndrome and progressive supranuclear palsy (Fig. 3B) (22–24). In contrast, overactivity of PERK is observed in clinical samples and mouse models of many neurodegenerative diseases, including prion disease, Alzheimer's disease, and frontotemporal dementia (25). Because of this, there has been significant interest in establishing new strategies to pharmacologically inhibit or activate PERK signaling for different human diseases.
Inhibiting PERK autophosphorylation using kinase inhibitors
One of the earliest strategies to modulate PERK focused on targeting its kinase active site to inhibit the PERK autophosphorylation step required for activation. The initial compound that emerged from this approach was GSK2606414, which was shown to have >100-fold selectivity for the PERK kinase domain relative to other kinases tested (Fig. 3C) (114, 115). However, more recent studies have indicated that GSK2606414 and its close analog GSK2656157 can inhibit other kinases, such as receptor-interacting serine/threonine kinase (RIPK) and receptor protein tyrosine kinase (KIT), reflecting off-target activity often associated with targeting kinase active sites and potential complications when interpreting the direct cause of physiological changes from treatment by these compounds (78, 116). New analogs of these two compounds, such as AMG52, and novel quinoline-based PERK inhibitors, such as AMG44, have been recently engineered to decrease RIPK inhibition. However, these compounds show reduced activity for inhibiting PERK signaling in cellular models as compared with GSK2606414 (Fig. 3C) (117). Regardless, PERK kinase inhibitors (e.g. GSK2606414) have been widely used both in vitro and in vivo to define the pathologic and therapeutic implications of PERK signaling in diverse diseases. For example, administration of PERK kinase inhibitors reduced tumorigenesis in pancreatic and multiple myeloma tumor xenograft models and improved outcomes in mouse models of neurodegenerative diseases, including prion disease and frontotemporal dementia (114, 118–120). However, these compounds are still somewhat limited in their in vivo application as they have demonstrated dose-dependent defects, including weight loss and pancreatic toxicity (121, 122). Regardless, PERK kinase inhibitors have proven valuable for defining the therapeutic potential for inhibiting PERK in models of disease and continue to be explored for clinical use.
Pharmacologic activation of eIF2B to inhibit PERK signaling
As opposed to the direct targeting of the kinase active site, other strategies have employed unbiased cell-based phenotypic screens to identify compounds that inhibit PERK signaling. The most prominent compound to emerge from this approach is ISRIB (Fig. 3D). ISRIB was identified from an HTS, where >100,000 compounds were screened for inhibition of ER stress–dependent activation of a cell-based luciferase reporter for ATF4 translation (123). ISRIB showed high potency for selectively inhibiting PERK-regulated transcriptional and translational signaling without significantly affecting other arms of the UPR (123). Interestingly, ISRIB did not reduce PERK-dependent phosphorylation of eIF2α, indicating that this compound worked downstream of PERK kinase activity and likely functions by desensitizing cells to eIF2α phosphorylation. A consequence of this specific mechanism is that ISRIB can block eIF2α phosphorylation-dependent signaling induced by other stress-regulated eIF2α kinases, including general control nonderepressible 4 (GCN4), activated in response to nutrient deprivation, heme-regulated inhibitor (HRI), activated by oxidative or mitochondrial stress, and protein kinase R (PKR), activated by viral infection (92). This means care must be taken when assigning ISRIB-sensitive phenotypes specifically to PERK signaling under conditions where these other kinases may be active.
The biological target of ISRIB was identified using genetic screening approaches that showed disruption of specific subunits of eIF2B-desensitized cells to ISRIB (124, 125). Two subsequent cryo-EM structures of eIF2B bound to ISRIB showed that this molecule binds eIF2B at a regulatory site localized between its β and γ subunits, stabilizing the decameric enzyme complex in its active conformation (126, 127). This binding increases eIF2B dimerization and subsequent activation, allowing eIF2B to remain active even in the presence of phosphorylated eIF2α. This structural elucidation of ISRIB binding to eIF2B has enabled structure-based drug design to establish next-generation ISRIB analogs with improved efficacy and potency for biological applications.
Unlike PERK kinase inhibitors, ISRIB does not induce off-target activity, such as weight loss or pancreatic toxicity (121). This observation can be explained by the fact that ISRIB functions as a partial inhibitor of eIF2α phosphorylation-dependent signaling, with efficacy varying, depending on the amount and extent of eIF2α phosphorylation (121, 128). In response to mild to moderate acute insults, ISRIB is effective at inhibiting eIF2α-mediated signaling. However, chronic or severe insults show reduced sensitivity to ISRIB-dependent inhibition of eIF2α signaling. As a consequence, ISRIB can mitigate pathologic outcomes associated with moderate increases in eIF2α phosphorylation, while allowing for protective signaling through this pathway in tissues such as the pancreas that experience high levels of ER stress.
The relatively low toxicity associated with ISRIB has allowed this compound to be widely used to probe the importance of PERK activity and/or eIF2α phosphorylation in diverse in vivo and cellular models of disease. For example, ISRIB has been shown to improve cognitive function in mice, reduce stress granule formation, promote cytotoxicity in xenograft models of prostate cancer, and protect against neurodegeneration in rodent models of prion disease and vanishing white matter disease (121, 123, 129–131). Further, ISRIB has proven invaluable for probing the importance of eIF2α phosphorylation-mediated signaling in regulating diverse biological processes, including ER proteostasis and mitochondrial regulation (97, 132, 133). Taken together, the development of ISRIB has proven transformative for improving our understanding of both the pathologic and potential therapeutic implications of eIF2α-dependent signaling in the context of health and disease.
Despite the promise of ISRIB, a limiting factor in the further translational development of this compound is its poor solubility. Although new analogs of ISRIB, such as 2BAct, that show improved solubility and chemical properties continue to be developed (129), there is significant interest in identifying other compounds that target the PERK signaling pathway in ways analogous to ISRIB. One potential alternative is the antidepressant selective serotonin uptake inhibitor trazodone (Fig. 3D). Trazodone was identified as a UPR modulator in a phenotypic screen of a >1000-compound library enriched for Food and Drug Administration–approved drugs that monitored ER stress–induced developmental delays in Caenorhabditis elegans (134). Trazodone, like ISRIB, was shown to block eIF2α transcriptional and translational signaling downstream of eIF2α phosphorylation. However, unlike ISRIB, which activates eIF2B, trazodone is suggested to prevent eIF2α phosphorylation–dependent reductions in ternary complex formation required for translation initiation, although this mechanism remains to be formally defined (134). Despite this, initial studies show that treatment with trazodone mimics benefits observed with ISRIB, including reductions in both neurodegeneration in models of prion disease and tumor metastasis (134, 135). Whereas additional studies are required to fully appreciate the potential of trazodone as an inhibitor of eIF2α phosphorylation–dependent signaling (e.g. the possibility of separating selective serotonin uptake inhibitor activity from its effects on signaling downstream of eIF2α), the identification of trazodone further highlights the potential for targeting PERK signaling downstream of eIF2α phosphorylation as a promising strategy to intervene in disease.
Targeting protein phosphatases to enhance PERK-eIF2α signaling
Apart from inhibiting PERK-mediated eIF2α signaling, enhancing activity through this pathway also offers opportunities to improve pathologic outcomes in human disease. A significant challenge in developing compounds that activate PERK is the pro-apoptotic signaling induced by this pathway. To avoid pro-apoptotic signaling, strategies to increase PERK activity have primarily focused on targeting downstream aspects of PERK signaling, most notably the phosphatases responsible for dephosphorylating eIF2α. Dephosphorylation of eIF2α is mediated by complexes of protein phosphatase 1 (PP1), G-actin, and one of two regulatory subunits, CreP or GADD34 (136, 137). CreP (PPP1R15B) is constitutively expressed and functions to dephosphorylate eIF2α under basal conditions (138). In contrast, GADD34 (PPP1R15A) is a stress-activated eIF2α phosphatase regulatory subunit that is induced through transcriptional signaling activated by eIF2α phosphorylation and functions to dephosphorylate eIF2α as part of a negative feedback loop in the PERK signaling pathway (107, 139, 140). Thus, targeting the activity of CreP or GADD34 offers opportunities to increase eIF2α phosphorylation–dependent signaling by changing the dynamics of its dephosphorylation.
The first compound suggested to target these phosphatases was salubrinal, which was identified in a screen for compounds that block ER stress–induced apoptosis (Fig. 3E) (141). Treatment with salubrinal induced eIF2α phosphorylation and delayed dephosphorylation of eIF2α following acute stress. Although these results indicate that salubrinal impacts eIF2α dephosphorylation, its precise mechanism of action remains undefined. Despite this, salubrinal is widely used and has proven protective in cellular and in vivo models of diverse diseases, including viral infection (141), retinitis pigmentosa (142, 143), and familial ALS (144). However, treatment with salubrinal also has been shown to exacerbate neurotoxic eIF2α-dependent signaling in mouse models of prion disease, highlighting the challenges of activating this pathway in human disease (145).
Based on the protection afforded by genetic reductions of GADD34 in models of ALS and Charcot-Marie-Tooth disease (146, 147), there has been significant interest in identifying compounds that selectively inhibit GADD34 activity to delay translational recovery following acute insult. One of the first compounds proposed to selectively target GADD34 was the α2-adrenergic agonist guanabenz (Fig. 3E). Both guanabenz and its close analog sephin-1, which lacks α2-adrenergic activity, were previously shown to delay translation recovery following acute ER insult (Fig. 3E) (148, 149). Whereas initial biochemical studies suggested that this delay could be attributed to selective inhibition of GADD34 (148–150), other studies indicate that this compound does not inhibit GADD34-dependent phosphatase activity in vitro, casting doubt on this potential mechanism of action (151, 152). Further, the reductions in ER stress–associated signaling afforded by treatment with guanabenz and sephin-1 appear independent of eIF2α phosphorylation, suggesting that these compounds exert their effects through an alternative mechanism (152). Although the mechanism of action for these compounds remains to be established, guanabenz and sephin-1 are protective in mouse models of multiple diseases, including Charcot-Marie-Tooth, familial ALS, multiple sclerosis, and prion disease, reflecting their ability to improve pathologic outcomes in the context of different diseases (148, 149, 153, 154). That being said, it is important to use caution when interpreting the relationship between this protection and signaling through PERK and related pathways due to the lack of clarity regarding the mechanism of action for these compounds. Regardless, GADD34 remains an attractive therapeutic target for modulating eIF2α-dependent signaling in the context of health and disease.
PERK activators
Phenotypic screening has also been employed to identify compounds that activate the PERK pathway. Initial reports of a small molecule that selectively activates PERK signaling resulted from a high-throughput screen in human colon carcinoma cells focused on identifying compounds that halt cancer cell progression by activating the G1/S cell-cycle checkpoint (155). The identification of compound CCT020312 by this method was then linked to PERK signaling by microarray transcriptional profiling, revealing similarities between this compound and other positive effectors of eIF2α phosphorylation signaling (Fig. 3F) (155). CCT020312 did not appear to exhibit global UPR activation in cell culture models, and compound-dependent increases in eIF2α phosphorylation were shown to be PERK-dependent via RNAi depletion (155). However, the mechanism of PERK activation by CCT020312 remains undefined. Another phenotypic screen using a FRET-based reporter of eIF2α phosphorylation also identified promising pyrazole-carboxylate derivatives, including Compound A, which elicited eIF2α phosphorylation and NRF2 activation in a PERK activity–dependent manner (Fig. 3F) (156). Further, computational screening for compounds that activate the PERK kinase identified compound A4, which was subsequently shown to activate PERK in cell culture models and protect against HTT-associated toxicity in neuronal cells (157, 158). Subsequent biochemical studies indicated that this compound (and its more potent derivative MK-28) activates PERK signaling both in vitro and in vivo and protects against ER stress–associated insults (Fig. 3F) (159). Although the mechanism of action for MK-28 remains to be established, molecular modeling suggests that MK-28 could activate PERK kinase activity through allosteric regulation of the kinase active loop, suggesting a direct effect on the PERK kinase, although this remains to be formally tested (159).
The identification of these PERK-activating compounds provides new opportunities to activate PERK signaling in cellular and in vivo models. Initial results using these activators have shown protection in mouse models of diverse diseases, including mitochondrial diseases, tauopathy, and Huntington's disease, through mechanisms linked to both increased eIF2α phosphorylation and NRF2 activation (159–161). However, because the mechanism of action for these compounds and the selectivity of these compounds for PERK relative to other UPR or stress-responsive signaling pathways remain to be defined, care should be taken when relating biologic effects of these compounds specifically to PERK activation.
The ATF6 arm of the UPR
The last arm of the UPR to be identified was the ATF6 pathway. As with IRE1, humans encode two different ATF6 genes, ATF6α and ATF6β. ATF6β functions in a predominantly regulatory role, whereas ATF6α is the primary protein responsible for adapting cellular physiology in response to ER stress (162–169). Thus, we primarily discuss ATF6α in this review, which will be referred to as ATF6 herein. ATF6 is a type II transmembrane protein comprising an N-terminal bZIP transcription factor domain and a C-terminal ER luminal domain (Fig. 4, A and B). Unlike IRE1 and PERK, which rely on oligomerization and autophosphorylation for activation, ATF6 is activated through a different mechanism. In the absence of ER stress, ATF6 exists as monomers and disulfide-bound dimers/oligomers that are maintained by protein disulfide isomerases (PDIs) localized to the ER lumen (Fig. 4A) (170–172). Oxidized ATF6 is bound at its luminal domain by the ER HSP70 BiP and retained within the ER (Fig. 4A) (173, 174). In response to ER stress, ATF6 disulfides are reduced through a PDI-dependent mechanism, and BiP is released from the luminal domain, resulting in an increase in reduced monomeric ATF6. This reduced ATF6 monomer is then trafficked to the golgi and proteolytically processed by site 1 and site 2 proteases (S1P and S2P, respectively) (Fig. 4A) (170, 175–178). This releases the active, N-terminal ATF6 bZIP transcription factor domain, which dimerizes and localizes to the nucleus. This active ATF6 transcription factor elicits a transcriptional response that includes the up-regulation of multiple ER proteostasis factors (e.g. BiP) through binding ER stress–responsive elements (ERSEs) in target gene promoters (Fig. 4A) (54, 179, 180). Apart from ER proteostasis, ATF6 activation also transcriptionally regulates other aspects of cellular physiology, including cell growth and redox regulation, through the up-regulation of transcriptional targets, including RHEB and catalase, respectively (181–184). ATF6 transcriptional activity integrates with IRE1 signaling through multiple mechanisms, including the ATF6-dependent up-regulation of XBP1 and heterodimerization between the cleaved ATF6 transcription factor and XBP1s, which increases expression of genes involved in ER proteostasis, including ERAD factors (50, 54, 165). Through this transcriptional activity, ATF6 functions to promote adaptive remodeling of cellular physiology following ER stress.
Figure 4.
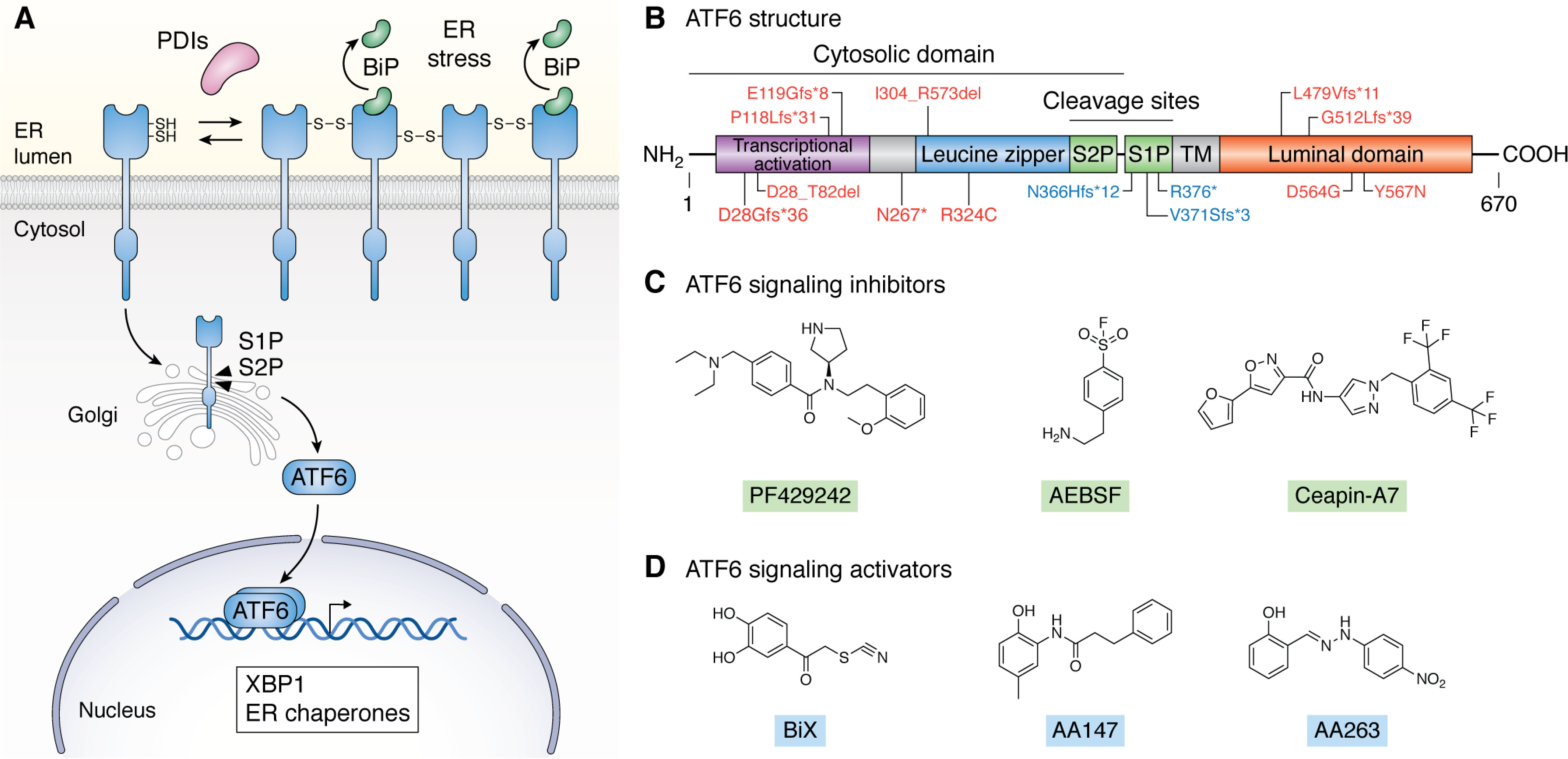
Identification and targeting of the ATF6 UPR signaling pathway. A, mechanism of ATF6 activation and downstream transcriptional signaling. B, domain architecture of ATF6, including the N-terminal bZIP transcription factor domain, the transmembrane domain (TM), and the luminal domain. Upon ATF6 activation, the N-terminal cytosolic domain containing the leucine zipper motif is liberated via processing in the golgi as shown in A, resulting in release of the active ATF6 transcription factor. Specific inhibiting (red) or activating (blue) mutations in ATF6 implicated in achromatopsia are indicated. C, structures of ATF6 inhibitors, including the S1P inhibitors PF429242 and AEBSF and the selective ATF6 inhibitor Ceapin-A7. D, structures of ATF6-activating compounds BiX, AA147, and AA263.
Significant mouse and human genetic evidence highlights the unique potential for targeting ATF6 to intervene in human disease. Unlike IRE1 and PERK, genetic deletion of Atf6α or Atf6β in mice does not result in any prominent phenotype (165), although aged mice lacking Atf6α show rod and cone dysfunction in the eye (185) and increased sensitivity to ER stress (165, 186). However, combined deletion of both Atf6α and Atf6β is embryonic lethal, suggesting a potential overlap in developmental roles of these two ATF6 isoforms (165). Regardless, these results indicate that reducing ATF6 activity does not significantly impact organismal physiology in the absence of stress. Similarly, overexpression of the active ATF6 transcription factor domain in various mouse tissues is well-tolerated and is not associated with tissue-specific toxicity. Instead, increased ATF6 transcriptional activity has been shown to be protective in cellular and rodent models of multiple diseases, including diabetes, protein-misfolding diseases, myocardial infarction, and stroke (54, 182, 187–192). Further, mutations in ATF6 associated with the retinal disease achromatopsia have been shown to either inhibit or activate ATF6 transcriptional signaling (Fig. 4B) (28, 29, 185, 193). Although these mutations lead to impaired retinal development, to date, no other neurologic or systemic phenotype has been reported in these patients, despite having the mutant ATF6 ubiquitously expressed in all tissues. This suggests that increasing or decreasing ATF6 activity is likely to be well-tolerated in humans after retinal development. Collectively, these genetic results highlight the translational potential for pharmacologically targeting ATF6 in the context of diverse diseases. However, only recently has this potential been realized with the establishment of new compounds that selectively modulate ATF6 signaling.
Inhibiting ATF6 signaling by preventing trafficking and processing
The first pharmacologic approaches used to inhibit ATF6 signaling primarily focused on blocking S1P-dependent proteolysis of ATF6 in the golgi. Numerous S1P inhibitors, including PF429242 and AEBSF, have been and continue to be used to inhibit ATF6 signaling both in cells and in vivo (Fig. 4C) (175, 194). Whereas these compounds are effective at inhibiting ATF6 activation, pharmacologic inhibition of S1P also blocks the regulation of other stress-responsive transcription factors similarly dependent on this protease, such as SREBP (195). Thus, the potential impact on other signaling pathways must be considered when using S1P inhibitors for probing ATF6 activity in biological systems.
Recently, more selective ATF6 inhibitors were identified from a phenotypic HTS that sought to identify compounds that blocked activation of an ATF6-selective ERSE-luciferase reporter in cells treated with the global ER stress–activating compound thapsigargin. This screen identified a class of pyrazole amides called “Ceapins,” particularly Ceapin-A7, which inhibits ATF6 activation by stabilizing ATF6 oligomers within the ER lumen and preventing ER stress–dependent trafficking to the Golgi (Fig. 4C) (196, 197). The mechanistic basis of this inhibition was defined using a functional genomic strategy, which showed that Ceapin-A7 stabilizes ATF6 in the ER through the formation of neomorphic interactions between the N-terminal 90 residues of ATF6 and the peroxisome-localized membrane protein ABCD3 (198). Despite binding to ABCD3, Ceapin-A7 did not influence the activity of ABCD3, indicating that this Ceapin-A7 is likely to have few off-target activities associated with the stabilization of this protein-protein interaction. Whereas the in vivo efficacy and global selectivity of Ceapin-A7 remains to be demonstrated, this compound provides an important new pharmacologic tool to selectively block ATF6 activation in cell culture models.
Phenotypic screening for ATF6 activators
Overexpression of the active ATF6 transcription factor domain is protective in cellular and mouse models of numerous diseases, including myocardial infarction, stroke, protein-misfolding diseases, and obesity-associated metabolic disease (182, 188, 199, 200). However, few ATF6-activating compounds have been developed, likely owing to the difficulty in targeting ATF6 due to its lack of a known enzymatic function or small molecule–binding sites. To confront this challenge, ATF6 activator screens have primarily focused on phenotype-based approaches to identify compounds that activate this pathway.
The first such screen used a firefly luciferase (FLuc) reporter expressed downstream of the ERSE-containing BiP promoter—an ATF6-selective reporter identical to that used to identify the Ceapin compounds (201). Screening a 10,000-compound library, the compound BiX was identified to selectively promote the expression of the ATF6-regulated chaperone BiP through an ATF6-dependent mechanism in both cellular and in vivo models (Fig. 4D) (201–204). However, BiX did not show significant increases in other ATF6 target genes, likely reflecting a low level of ATF6 activation afforded by this compound (201). Regardless, BiX has proven valuable in probing the potential for ATF6-dependent increases in BiP to ameliorate pathologies associated with multiple pathologic conditions, including stroke and kidney ischemia in rodent models (202–204). Because the mechanism of action and selectivity of BiX for ATF6 remain largely undefined, the potential of this compound to specifically probe ATF6 activity in cellular and in vivo models is currently somewhat limited.
More recently, another screen for ATF6 activators utilized an analogous ERSE-Fluc reporter assay to identify compounds that selectively activate the ATF6 arm of the UPR (205). This screening strategy prioritized transcriptional selectivity at every step to enrich for compounds that showed preferential activation of the ATF6 transcriptional program over other arms of the UPR or other stress-responsive signaling pathways. Starting with >650,000 compounds, this screen identified highly selective ATF6-activating compounds, including AA147 and AA263, that preferentially activate the ATF6 transcriptional program to levels ∼50% that observed with global ER stressors (Fig. 4D) (205). Importantly, RNA-seq transcriptional profiling showed that these compounds do not increase expression of other stress-responsive signaling pathways, highlighting their selectivity for ATF6 (205). Follow-up mechanistic studies using medicinal chemistry and biochemistry demonstrated that these compounds activate ATF6 through a mechanism involving compound metabolic activation to an electrophile by ER oxidases followed by covalent modification of a subset of ER proteins, including PDIs involved in regulating ATF6 disulfide formation (Fig. 4A) (206). This selective PDI modification increases the ER concentration of reduced ATF6 monomers, allowing a subset of ATF6 to traffic to the golgi and undergo proteolytic activation.
These selective ATF6-activating compounds have proven valuable for probing ATF6 activity in multiple biological and pathologic contexts, including stem cell differentiation and mouse models of tissue-specific ischemia and reperfusion injury (207, 208). Importantly, in both cases, genetic disruption of ATF6 blocked compound-mediated effects, confirming that these compounds influence cellular physiology in these models through ATF6 activation. However, care must be taken when defining whether a compound's effects are mediated by ATF6, as two recent studies highlight how these ATF6-activating compounds can afford protection through an on-target, ATF6-independent mechanism involving compound metabolic activation and covalent modification of ER proteins (e.g. PDIs) (209, 210). Thus, whereas these compounds have provided new opportunities to probe ATF6 activity both in vitro and in vivo, it is important that this protection be paired with genetic or pharmacologic approaches that block ATF6 signaling to define the dependence of this protection on ATF6 activation, as opposed to upstream steps in the activation mechanism or potential off-target activities of these compounds.
Lessons learned from pharmacologic UPR modulators
As described above, there are many strategies for identifying and developing modulators of specific UPR signaling pathways. Each of these, however, comes with respective downstream challenges associated with defining mechanisms of action, compound specificity, and suitability for applying these compounds in diverse cellular and organismal models. For example, compounds that target IRE1 and PERK kinase sites often suffer from off-target activities that can confer toxicity in some cell types (77, 78). It is therefore paramount to characterize the extent of these off-target effects, via kinase activity assays and transcriptional profiling to define compound selectivity for specific UPR signaling pathways. Conversely, UPR modulators that have been identified via phenotypic screening, such as the ATF6 activator, AA147, and the PERK signaling inhibitor, ISRIB, have no initial indications as to protein target and mechanism of action. This can make targeted medicinal chemistry efforts to improve compound efficacy significantly less directed and therefore less efficient. This aspect of phenotypic screening additionally presents a challenge in evaluating cellular models for profiling compound activity, as cell types can significantly vary in genetic background and cellular physiology. Therefore, it is crucial that compounds identified via phenotypic screening be re-evaluated for arm-selective UPR activation in each new model system in which they are utilized. This consideration is important to ensure a model system includes all necessary protein targets at the appropriate abundance for selective UPR modulation by small molecules.
Despite these challenges, the wide variety of UPR-modulating compounds identified to date are beginning to reveal specific properties of the most useful compounds that can dictate further medicinal chemistry and drug discovery efforts for the continued development of these types of compounds for human disease. Below, we discuss some of the lessons learned from currently available UPR-modulating compounds to provide some guidance that can be applied when developing and prioritizing next generation compounds for use in probing the importance of UPR signaling in disease models and translating these compounds for the treatment of human disease.
Compound selectivity for specific UPR signaling pathways
One of the most important challenges in establishing arm-selective UPR activators or inhibitors is defining their selectivity for a specific UPR pathway. Off-target activity of UPR-modulating compounds significantly challenges the utility of these compounds to probe the physiologic, pathologic, and therapeutic implications of UPR signaling in diverse models. Similarly, toxicity associated with off-target activity limits the application of certain UPR-modulating compounds, as discussed above. Therefore, understanding compound selectivity and the limitations thereof is critical when identifying and developing new compounds that target individual UPR signaling pathways.
Over the past 5 years, multiple different strategies have been developed to probe the selectivity of compounds for specific stress-responsive transcriptional signaling pathways, including the three arms of the UPR. For example, the integration of reporters for multiple different UPR signaling pathways into the screening pipeline has proven useful in identifying compound selectivity for a specific pathway in a high-throughput format (85, 205). Similarly, incorporating cell-based toxicity screening early in an HTS pipeline provides a useful approach to remove compounds that are toxic through either on-target or off-target pathways (85, 205), although this toxicity could still emerge when compounds are applied to other cell models. Further, the development and implementation of transcriptional and kinase-profiling strategies has proven extremely useful for identifying compounds that are selective for a specific UPR pathway. Transcriptional profiling approaches, such as gene set enrichment analysis (GSEA), are powerful tools to demonstrate selective activation of UPR signaling pathways (211). However, whereas GSEA is highly effective at identifying modulation of UPR signaling pathways, this approach is challenged by the difficulty in separating the overlapping transcriptional targets and variable gene induction by different stress-responsive signaling pathways, such as those regulated by XBP1s and ATF6 (54) (Fig. 5A). A recent strategy to confront this challenge in the context of UPR activators includes normalizing induction of different UPR target genes to that observed upon treatment with global ER stressors (e.g. thapsigargin), providing a way to directly compare gene induction on a normalized scale (54, 212) (Fig. 5B). Using this approach, selectivity for IRE1/XBP1s and ATF6 can be partially deconvoluted, providing a transcriptional profiling strategy that can be applied to improve identification of selective activators for these two UPR pathways (212).
Figure 5.

Lessons learned from currently available UPR-modulating compounds. A, graph demonstrating the types of variable expression for ATF6, IRE1/XBP1s, and PERK/ATF4 transcriptional targets induced by specific UPR activators or global ER stressors (e.g. thapsigargin (Tg)). B, graph showing how normalization to thapsigargin-dependent expression can allow more accurate comparisons of target gene induction induced by different compounds. C, illustration showing how ISRIB functions to partially suppress pathologic PERK overactivity to improve outcomes in diverse diseases without inducing severe toxicity. D, graph showing the complexity in defining PK/PD for UPR-activating compounds, such as ATF6 activators. Whereas the parent compound is rapidly lost (PK), the initiated ATF6 transcriptional activity and consequences of this activation (PD) can maintain the protection even in the absence of the activator compound.
Importantly, transcriptional profiling also provides an opportunity to define compound selectivity beyond the UPR, to confirm that compounds do not globally activate other stress-responsive signaling pathways (e.g. the heat shock response or oxidative stress response) that could complicate phenotypic readouts. This is an important consideration with regard to the complex interplay between stress-responsive signaling pathways in pathologic and physiologic conditions. The development of high- to medium-throughput transcriptional profiling strategies (e.g. Drug-seq (213)) provides new opportunities to integrate this type of profiling either early in the screening pipeline or during subsequent medicinal chemistry development of “hit” compounds, allowing for improved capacity to identify compounds with a high level of specificity for a given UPR pathway.
Apart from transcriptional profiling, other proteome-wide profiling strategies can also be important for specific types of compounds. For example, compounds that bind to the IRE1 or PERK kinase domains should be profiled for activity against other kinases using different assays, such as the in vitro KinomeScan or a cell-based phosphokinase array, to confirm selectivity (214, 215). Further, proteomic strategies can be applied to identify potential post-transcriptional alterations in the proteome induced by on-target or off-target activity of UPR-modulating compounds, an important consideration when altering the activity of core proteostasis pathways involved in regulating proteome stability. Whereas no single cell-wide profiling approach can provide a complete picture for compound-dependent changes to cellular physiology, the increased application of these approaches when developing UPR-modulating compounds will increase our ability to define the most selective UPR-modulating compounds for use in probing the pathologic and therapeutic implication of UPR pathways in the context of health and disease.
Partial modulators that “reshape” the UPR response
Another notable challenge in developing UPR modulators is associated with the fact that both too much and too little signaling through a specific arm can lead to pathology (29). Thus, small molecules that completely inhibit or activate UPR pathways can both lead to toxicity. Whereas this on-target toxicity can be beneficial in the context of certain diseases, such as cancers, it can preclude the development of UPR-modulating compounds for other diseases due to severe side effects (e.g. pancreatic toxicity associated with PERK kinase inhibitors) (121). One solution to this problem lies in the development of UPR modulators that only partially alter signaling through a specific arm of the UPR. These types of compounds offer the unique opportunity to “reshape” the UPR response in a way that can often avoid toxicity associated with complete activation or inhibition of a given pathway. ISRIB is a good example of this type of partial modulation, as it is well-established as a partial inhibitor of eIF2α phosphorylation–dependent signaling (121). This partial inhibitory activity allows ISRIB to attenuate pathologic PERK signaling associated with moderate ER stress without affecting PERK-dependent regulation of insulin production required for pancreatic function (Fig. 5C) (121). Apart from ISRIB, new ATF6 and IRE1/XBP1s activators modulate these pathways to levels ∼40–60% that observed for global ER stressors, allowing for effective reshaping of these protective stress responses while minimizing potential on-target toxicity associated with high levels of signaling through these pathways (85, 205, 207). Ultimately, these results highlight the benefits of compounds that partially modulate signaling through a specific arm of the UPR, to reshape UPR signaling and mitigate pathologies associated with human disease.
Whereas it can be difficult to identify these types of partial modulators in HTS pipelines, multiple strategies can be applied downstream to identify compounds most suitable for this type of UPR modulation. Again, incorporating transcriptional profiling approaches into screening pipelines provides a useful tool to identify compounds that robustly and selectively activate or inhibit UPR pathways to moderate levels. Further, focusing on compounds that target proteins involved in regulating downstream aspects of UPR signaling pathways (e.g. eIF2B) and not core components of the signaling pathway (e.g. PERK or IRE1) also appears to be an effective strategy to develop these types of partial modulators. Last, tailoring compounds that modulate UPR signaling to levels optimized for a given phenotypic readout also can be used to identify compounds with the most effective level of signaling to improve pathogenic outcomes in a specific disease context. Thus, whereas it can be appealing to develop compounds that show the largest changes to UPR signaling, it is important to understand the extent of UPR modulation required and the extent tolerated when developing next generation compounds for specific disease indications.
Phenotypic selectivity of UPR-modulating compounds
Another important consideration when applying UPR-modulating compounds to cellular and in vivo models is the challenges associated with attributing a phenotype to the activation or inhibition of a particular UPR pathway. One approach to confront this challenge is to use genetic strategies to disrupt a specific UPR signaling pathway to confirm compound-dependent activity through that pathway. For example, cardiac-specific knockout of Atf6 blocked the protection against myocardial infarction afforded by the ATF6-activating compound AA147, indicating that this protection is mediated through a mechanism involving ATF6 activation (207). However, whereas this type of genetic approach is useful, it is important to consider challenges that can result from compensation of UPR (and other) signaling pathways associated with chronic loss or depletion of an ER stress–sensing protein or downstream effector. With the development of new compounds that selectively activate or inhibit all three arms of the UPR, it is now becoming possible to use different combinations of UPR-modulating compounds to probe the dependence of compound-mediated phenotypes on a given pathway, although to date this approach is best suited for cell-based studies. For example, combinations of IRE1 activators and inhibitors were used to confirm that the reduction in Aβ production afforded by the activator IXA4 depended on IRE1 activity (85). When applying pharmacologic strategies of this type, it is important, when possible, to use multiple compounds that activate or inhibit UPR signaling at different regulatory steps to minimize potential complications that can result from off-target activity. For example, the use of both ISRIB and GSK2606414 is a better strategy to define the dependence of a given phenotype on PERK signaling relative to use of either compound alone (133). Importantly, with this type of phenotypic profiling, it is possible that specific compounds could be found to promote protection through a mechanism independent of UPR activity. For example, the ATF6-activating compound AA147 reduces secretion of amyloidogenic light chain and protects against viral infection through a mechanism involving covalent modification of proteins such as ER-resident PDIs (209, 210). However, whereas this “on-target” protein modification is crucial for the activation of ATF6 by AA147, both of these downstream effects have been described to be independent of ATF6 activity (209, 210). Thus, whereas we can define the dependence of a given compound-dependent effect on a specific UPR pathway, it remains critical to establish compound mechanism of action to fully understand the physiologic impact of these compounds in different systems.
Defining compound PK and PD for UPR-modulating compounds
Finally, it is important when developing next generation UPR-modulating compounds to establish new tools that can function in disease relevant in vivo models, such as mice. New compounds should be characterized for their activity in vivo to maximize benefit across different models and allow for global understanding of how targeting UPR pathways impacts organismal physiology in health and disease. Whereas the in vivo activity of UPR inhibitor compounds can generally be determined using traditional pharmacokinetic (PK)/pharmacodynamic (PD) strategies, defining the in vivo PK/PD for UPR-activating compounds is more challenging. These types of compounds only have to initiate the adaptive transcriptional signaling pathway to promote a phenotypic effect, so optimizing PK may not be the most important aspect to follow for compound development, although it is always important to understand the metabolic breakdown of compounds. Instead, understanding the PD of these compounds (e.g. the tissue activity and duration of pathway activation) is likely the more important aspect of compound development. Understanding PD will allow for the establishment of doses and dosing regimens optimized to promote protective remodeling in relevant tissues without leading to potential toxicity associated with chronic activation of a given pathway. One example of this effect is observed with the ATF6 activator AA147, where it was shown that this compound is rapidly cleared from the blood but results in ATF6 transcriptional activity that persists for ∼3 days following IV injection (Fig. 5D) (207). Thus, when considering the development of UPR-activating compounds, it is important to understand how these compounds impact different tissues and the duration of this effect to allow for the identification of compounds with the best properties for in vivo applications.
Moving forward
Whereas many new UPR-modulating compounds still require further characterization to define selectivity and/or mechanism of action, many of these compounds can already be applied in a variety of experimental settings to probe the effects of UPR signaling in diverse physiologic and pathologic contexts. For example, potent and selective inhibitors for each UPR signaling pathway can be applied in combination with global ER stressors to probe the individual roles of IRE1, ATF6, and PERK signaling in regulating ER and cellular physiology. Further, combinatorial strategies pairing activators and inhibitors for a given UPR signaling arm can be applied to elucidate the biological importance of individual UPR pathways in cellular and organismal models. As we continue to implement new screening pipelines and medicinal chemistry efforts to establish next-generation compounds with improved activity and/or translational potential, it is important to integrate new strategies to define selectivity and/or potency of these compounds, allowing the identification of compounds with the most potential to influence a given phenotype or disease indication. In addition, it is critical to develop optimized dosing paradigms that provide sufficient activity to mitigate specific pathologic outcomes while minimizing potentially deleterious on- or off-target activity associated with signaling through a given pathway. It is now abundantly clear that pharmacologic targeting of UPR signaling pathways represents a promising strategy to intervene in etiologically-diverse diseases, with some compounds (e.g. ISRIB) demonstrating significant potential for multiple disorders. Thus, as we continue to develop and employ UPR-modulating compounds using the strategies described above, we will improve our ability to probe the importance of UPR signaling in diverse physiologic and disease contexts using pharmacologic approaches. Further, through these efforts, we will be able to optimize compounds for translational development to treat numerous diseases.
Acknowledgments
We thank Evan Powers, Jessica Rosarda, Dorian Rosen, Aparijita Madhavan, Justine Lebeau, Valerie Perea, Bibiana Rius, and Belle Noxon for critical reading of the manuscript. Further, we acknowledge that there is an additional body of important work done by colleagues pertaining to pharmacologic targeting of the UPR, which we omitted from this review in the interests of clarity and brevity.
Funding and additional information—This work was supported by National Institutes of Health grants AG046495 (to R. L. W.) and AG063489 (to J. M. D. G.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—R. L. W. is an inventor on patents filed for the ATF6 activators AA147 and AA263 described in this review. Both R. L. W. and J. M. D. G. are inventors on a patent filed for the IRE1/XBP1s activator IXA4 described in this review.
- ER
- endoplasmic reticulum
- UPR
- unfolded protein response
- BiP
- binding immunoglobulin protein
- ERAD
- ER-associated degradation
- RIDD
- regulated IRE1-dependent decay
- PDI
- protein disulfide isomerase
- S1P
- site 1 protease
- S2P
- site 2 protease
- ERSE
- ER stress–responsive element
- AEBSF
- 4-benzenesulfonyl fluoride hydrochloride
- GSEA
- gene set enrichment analysis
- PK
- pharmacokinetic
- PD
- pharmacodynamic.
References
- 1. Fagone P., and Jackowski S. (2009) Membrane phospholipid synthesis and endoplasmic reticulum function. J. Lipid Res. 50, S311–S316 10.1194/jlr.R800049-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Raffaello A., Mammucari C., Gherardi G., and Rizzuto R. (2016) Calcium at the center of cell signaling: interplay between endoplasmic reticulum, mitochondria, and lysosomes. Trends Biochem. Sci. 41, 1035–1049 10.1016/j.tibs.2016.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Malhotra J. D., and Kaufman R. J. (2007) The endoplasmic reticulum and the unfolded protein response. Semin. Cell Dev. Biol. 18, 716–731 10.1016/j.semcdb.2007.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Owusu B. Y., Zimmerman K. A., and Murphy-Ullrich J. E. (2018) The role of the endoplasmic reticulum protein calreticulin in mediating TGF-β-stimulated extracellular matrix production in fibrotic disease. J. Cell Commun. Signal. 12, 289–299 10.1007/s12079-017-0426-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pinton P., Giorgi C., Siviero R., Zecchini E., and Rizzuto R. (2008) Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 27, 6407–6418 10.1038/onc.2008.308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang S., and Kaufman R. J. (2012) The impact of the unfolded protein response on human disease. J. Cell Biol. 197, 857–867 10.1083/jcb.201110131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cnop M., Foufelle F., and Velloso L. A. (2012) Endoplasmic reticulum stress, obesity and diabetes. Trends Mol. Med. 18, 59–68 10.1016/j.molmed.2011.07.010 [DOI] [PubMed] [Google Scholar]
- 8. Chen J. J., Genereux J. C., and Wiseman R. L. (2015) Endoplasmic reticulum quality control and systemic amyloid disease: impacting protein stability from the inside out. IUBMB Life 67, 404–413 10.1002/iub.1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Plate L., and Wiseman R. L. (2017) Regulating secretory proteostasis through the unfolded protein response: from function to therapy. Trends Cell Biol. 27, 722–737 10.1016/j.tcb.2017.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hetz C., and Papa F. R. (2018) The unfolded protein response and cell fate control. Mol. Cell 69, 169–181 10.1016/j.molcel.2017.06.017 [DOI] [PubMed] [Google Scholar]
- 11. Hetz C., and Saxena S. (2017) ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 13, 477–491 10.1038/nrneurol.2017.99 [DOI] [PubMed] [Google Scholar]
- 12. Back S. H., and Kaufman R. J. (2012) Endoplasmic reticulum stress and type 2 diabetes. Annu. Rev. Biochem. 81, 767–793 10.1146/annurev-biochem-072909-095555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Thiebaut A. M., Hedou E., Marciniak S. J., Vivien D., and Roussel B. D. (2019) Proteostasis during cerebral ischemia. Front Neurosci 13, 637 10.3389/fnins.2019.00637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang K., and Kaufman R. J. (2004) Signaling the unfolded protein response from the endoplasmic reticulum. J. Biol. Chem. 279, 25935–25938 10.1074/jbc.R400008200 [DOI] [PubMed] [Google Scholar]
- 15. Bernales S., Papa F. R., and Walter P. (2006) Intracellular signaling by the unfolded protein response. Annu. Rev. Cell Dev. Biol. 22, 487–508 10.1146/annurev.cellbio.21.122303.120200 [DOI] [PubMed] [Google Scholar]
- 16. Schroder M., and Kaufman R. J. (2005) ER stress and the unfolded protein response. Mutat. Res. 569, 29–63 10.1016/j.mrfmmm.2004.06.056 [DOI] [PubMed] [Google Scholar]
- 17. Hetz C., Zhang K., and Kaufman R. J. (2020) Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 21, 421–438 10.1038/s41580-020-0250-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Karagöz G. E., Acosta-Alvear D., and Walter P. (2019) The unfolded protein response: detecting and responding to fluctuations in the protein-folding capacity of the endoplasmic reticulum. Cold Spring Harb. Perspect. Biol. 11, a033886 10.1101/cshperspect.a033886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Preissler S., and Ron D. (2019) Early events in the endoplasmic reticulum unfolded protein response. Cold Spring Harb. Perspect. Biol. 11, a033894 10.1101/cshperspect.a033894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hollien J. (2013) Evolution of the unfolded protein response. Biochim. Biophys. Acta 1833, 2458–2463 10.1016/j.bbamcr.2013.01.016 [DOI] [PubMed] [Google Scholar]
- 21. Bell M. C., Meier S. E., Ingram A. L., and Abisambra J. F. (2016) PERK-opathies: an endoplasmic reticulum stress mechanism underlying neurodegeneration. Curr. Alzheimer Res. 13, 150–163 10.2174/1567205013666151218145431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Delepine M., Nicolino M., Barrett T., Golamaully M., Lathrop G. M., and Julier C. (2000) EIF2AK3, encoding translation initiation factor 2-α kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nat. Genet. 25, 406–409 10.1038/78085 [DOI] [PubMed] [Google Scholar]
- 23. Yuan S. H., Hiramatsu N., Liu Q., Sun X. V., Lenh D., Chan P., Chiang K., Koo E. H., Kao A. W., Litvan I., and Lin J. H. (2018) Tauopathy-associated PERK alleles are functional hypomorphs that increase neuronal vulnerability to ER stress. Hum. Mol. Genet. 27, 3951–3963 10.1093/hmg/ddy297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Höglinger G. U., Melhem N. M., Dickson D. W., Sleiman P. M., Wang L. S., Klei L., Rademakers R., de Silva R., Litvan I., Riley D. E., van Swieten J. C., Heutink P., Wszolek Z. K., Uitti R. J., Vandrovcova J., et al. (2011) Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat. Genet. 43, 699–705 10.1038/ng.859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hughes D., and Mallucci G. R. (2019) The unfolded protein response in neurodegenerative disorders—therapeutic modulation of the PERK pathway. FEBS J. 286, 342–355 10.1111/febs.14422 [DOI] [PubMed] [Google Scholar]
- 26. Remondelli P., and Renna M. (2017) The endoplasmic reticulum unfolded protein response in neurodegenerative disorders and its potential therapeutic significance. Front. Mol. Neurosci. 10, 187 10.3389/fnmol.2017.00187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tufanli O., Telkoparan Akillilar P., Acosta-Alvear D., Kocaturk B., Onat U. I., Hamid S. M., Çimen I., Walter P., Weber C., and Erbay E. (2017) Targeting IRE1 with small molecules counteracts progression of atherosclerosis. Proc. Natl. Acad. Sci. U. S. A. 114, E1395–E404 10.1073/pnas.1621188114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ansar M., Santos-Cortez R. L., Saqib M. A., Zulfiqar F., Lee K., Ashraf N. M., Ullah E., Wang X., Sajid S., Khan F. S., Amin-Ud-Din M., and University of Washington Center for Mendelian Genomics (2015) Mutation of ATF6 causes autosomal recessive achromatopsia. Hum. Genet. 134, 941–950 10.1007/s00439-015-1571-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chiang W.-C., Chan P., Wissinger B., Vincent A., Skorczyk-Werner A., Krawczyński M. R., Kaufman R. J., Tsang S. H., Héon E., Kohl S., and Lin J. H. (2017) Achromatopsia mutations target sequential steps of ATF6 activation. Proc. Natl. Acad. Sci. U. S. A. 114, 400–405 10.1073/pnas.1606387114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Glembotski C. C., Rosarda J. D., and Wiseman R. L. (2019) Proteostasis and beyond: ATF6 in ischemic disease. Trends Mol. Med. 25, 538–550 10.1016/j.molmed.2019.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hetz C., Axten J. M., and Patterson J. B. (2019) Pharmacological targeting of the unfolded protein response for disease intervention. Nat. Chem. Biol. 15, 764–775 10.1038/s41589-019-0326-2 [DOI] [PubMed] [Google Scholar]
- 32. Nikawa J., and Yamashita S. (1992) IRE1 encodes a putative protein kinase containing a membrane-spanning domain and is required for inositol phototrophy in Saccharomyces cerevisiae. Mol. Microbiol. 6, 1441–1446 10.1111/j.1365-2958.1992.tb00864.x [DOI] [PubMed] [Google Scholar]
- 33. Tirasophon W., Welihinda A. A., and Kaufman R. J. (1998) A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 12, 1812–1824 10.1101/gad.12.12.1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cox J. S., Shamu C. E., and Walter P. (1993) Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 73, 1197–1206 10.1016/0092-8674(93)90648-A [DOI] [PubMed] [Google Scholar]
- 35. Bertolotti A., Wang X., Novoa I., Jungreis R., Schlessinger K., Cho J. H., West A. B., and Ron D. (2001) Increased sensitivity to dextran sodium sulfate colitis in IRE1β-deficient mice. J. Clin. Invest. 107, 585–593 10.1172/JCI11476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Grey M. J., Cloots E., Simpson M. S., LeDuc N., Serebrenik Y. V., De Luca H., De Sutter D., Luong P., Thiagarajah J. R., Paton A. W., Paton J. C., Seeliger M. A., Eyckerman S., Janssens S., and Lencer W. I. (2020) IRE1β negatively regulates IRE1α signaling in response to endoplasmic reticulum stress. J. Cell Biol. 219, e201904048 10.1083/jcb.201904048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tsuru A., Fujimoto N., Takahashi S., Saito M., Nakamura D., Iwano M., Iwawaki T., Kadokura H., Ron D., and Kohno K. (2013) Negative feedback by IRE1β optimizes mucin production in goblet cells. Proc. Natl. Acad. Sci. U. S. A. 110, 2864–2869 10.1073/pnas.1212484110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shamu C. E., and Walter P. (1996) Oligomerization and phosphorylation of the Ire1p kinase during intracellular signaling from the endoplasmic reticulum to the nucleus. EMBO J. 15, 3028–3039 10.1002/j.1460-2075.1996.tb00666.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Volmer R., van der Ploeg K., and Ron D. (2013) Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc. Natl. Acad. Sci. U. S. A. 110, 4628–4633 10.1073/pnas.1217611110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bertolotti A., Zhang Y., Hendershot L. M., Harding H. P., and Ron D. (2000) Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2, 326–332 10.1038/35014014 [DOI] [PubMed] [Google Scholar]
- 41. Amin-Wetzel N., Saunders R. A., Kamphuis M. J., Rato C., Preissler S., Harding H. P., and Ron D. (2017) A J-protein co-chaperone recruits BiP to monomerize IRE1 and repress the unfolded protein response. Cell 171, 1625–1637.e13 10.1016/j.cell.2017.10.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Amin-Wetzel N., Neidhardt L., Yan Y., Mayer M. P., and Ron D. (2019) Unstructured regions in IRE1α specify BiP-mediated destabilisation of the luminal domain dimer and repression of the UPR. Elife 8, e50793 10.7554/eLife.50793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Karagöz G. E., Acosta-Alvear D., Nguyen H. T., Lee C. P., Chu F., and Walter P. (2017) An unfolded protein-induced conformational switch activates mammalian IRE1. Elife 6, e30700 10.7554/eLife.30700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Credle J. J., Finer-Moore J. S., Papa F. R., Stroud R. M., and Walter P. (2005) On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proc. Natl. Acad. Sci. U. S. A. 102, 18773–18784 10.1073/pnas.0509487102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gardner B. M., and Walter P. (2011) Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science 333, 1891–1894 10.1126/science.1209126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Korennykh A. V., Egea P. F., Korostelev A. A., Finer-Moore J., Zhang C., Shokat K. M., Stroud R. M., and Walter P. (2009) The unfolded protein response signals through high-order assembly of Ire1. Nature 457, 687–693 10.1038/nature07661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li H., Korennykh A. V., Behrman S. L., and Walter P. (2010) Mammalian endoplasmic reticulum stress sensor IRE1 signals by dynamic clustering. Proc. Natl. Acad. Sci. U. S. A. 107, 16113–16118 10.1073/pnas.1010580107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ali M. M., Bagratuni T., Davenport E. L., Nowak P. R., Silva-Santisteban M. C., Hardcastle A., McAndrews C., Rowlands M. G., Morgan G. J., Aherne W., Collins I., Davies F. E., and Pearl L. H. (2011) Structure of the Ire1 autophosphorylation complex and implications for the unfolded protein response. EMBO J. 30, 894–905 10.1038/emboj.2011.18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sidrauski C., and Walter P. (1997) The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell 90, 1031–1039 10.1016/S0092-8674(00)80369-4 [DOI] [PubMed] [Google Scholar]
- 50. Yoshida H., Matsui T., Yamamoto A., Okada T., and Mori K. (2001) XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891 10.1016/S0092-8674(01)00611-0 [DOI] [PubMed] [Google Scholar]
- 51. Jurkin J., Henkel T., Nielsen A. F., Minnich M., Popow J., Kaufmann T., Heindl K., Hoffmann T., Busslinger M., and Martinez J. (2014) The mammalian tRNA ligase complex mediates splicing of XBP1 mRNA and controls antibody secretion in plasma cells. EMBO J. 33, 2922–2936 10.15252/embj.201490332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lu Y., Liang F. X., and Wang X. (2014) A synthetic biology approach identifies the mammalian UPR RNA ligase RtcB. Mol. Cell 55, 758–770 10.1016/j.molcel.2014.06.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kosmaczewski S. G., Edwards T. J., Han S. M., Eckwahl M. J., Meyer B. I., Peach S., Hesselberth J. R., Wolin S. L., and Hammarlund M. (2014) The RtcB RNA ligase is an essential component of the metazoan unfolded protein response. EMBO Rep. 15, 1278–1285 10.15252/embr.201439531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shoulders M. D., Ryno L. M., Genereux J. C., Moresco J. J., Tu P. G., Wu C., Yates J. R. 3rd, Su A. I., Kelly J. W., and Wiseman R. L. (2013) Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Rep. 3, 1279–1292 10.1016/j.celrep.2013.03.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sriburi R., Jackowski S., Mori K., and Brewer J. W. (2004) XBP1: a link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J. Cell Biol. 167, 35–41 10.1083/jcb.200406136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hollien J., and Weissman J. S. (2006) Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 313, 104–107 10.1126/science.1129631 [DOI] [PubMed] [Google Scholar]
- 57. Hollien J., Lin J. H., Li H., Stevens N., Walter P., and Weissman J. S. (2009) Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 186, 323–331 10.1083/jcb.200903014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bae D., Moore K. A., Mella J. M., Hayashi S. Y., and Hollien J. (2019) Degradation of Blos1 mRNA by IRE1 repositions lysosomes and protects cells from stress. J. Cell Biol. 218, 1118–1127 10.1083/jcb.201809027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lu M., Lawrence D. A., Marsters S., Acosta-Alvear D., Kimmig P., Mendez A. S., Paton A. W., Paton J. C., Walter P., and Ashkenazi A. (2014) Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science 345, 98–101 10.1126/science.1254312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Han D., Lerner A. G., Vande Walle L., Upton J. P., Xu W., Hagen A., Backes B. J., Oakes S. A., and Papa F. R. (2009) IRE1α kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 138, 562–575 10.1016/j.cell.2009.07.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Upton J. P., Wang L., Han D., Wang E. S., Huskey N. E., Lim L., Truitt M., McManus M. T., Ruggero D., Goga A., Papa F. R., and Oakes S. A. (2012) IRE1α cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science 338, 818–822 10.1126/science.1226191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Moore K., and Hollien J. (2015) Ire1-mediated decay in mammalian cells relies on mRNA sequence, structure, and translational status. Mol. Biol. Cell 26, 2873–2884 10.1091/mbc.E15-02-0074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Urano F., Wang X., Bertolotti A., Zhang Y., Chung P., Harding H. P., and Ron D. (2000) Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287, 664–666 10.1126/science.287.5453.664 [DOI] [PubMed] [Google Scholar]
- 64. Tam A. B., Mercado E. L., Hoffmann A., and Niwa M. (2012) ER stress activates NF-κB by integrating functions of basal IKK activity, IRE1 and PERK. PLoS ONE 7, e45078 10.1371/journal.pone.0045078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lee H., Noh J. Y., Oh Y., Kim Y., Chang J. W., Chung C. W., Lee S. T., Kim M., Ryu H., and Jung Y. K. (2012) IRE1 plays an essential role in ER stress-mediated aggregation of mutant huntingtin via the inhibition of autophagy flux. Hum. Mol. Genet. 21, 101–114 10.1093/hmg/ddr445 [DOI] [PubMed] [Google Scholar]
- 66. Hu P., Han Z., Couvillon A. D., Kaufman R. J., and Exton J. H. (2006) Autocrine tumor necrosis factor α links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1α-mediated NF-κB activation and down-regulation of TRAF2 expression. Mol. Cell Biol. 26, 3071–3084 10.1128/MCB.26.8.3071-3084.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wang J., He W., Tsai P. J., Chen P. H., Ye M., Guo J., and Su Z. (2020) Mutual interaction between endoplasmic reticulum and mitochondria in nonalcoholic fatty liver disease. Lipids Health Dis. 19, 72 10.1186/s12944-020-01210-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wang M., and Kaufman R. J. (2014) The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 14, 581–597 10.1038/nrc3800 [DOI] [PubMed] [Google Scholar]
- 69. Chevet E., Hetz C., and Samali A. (2015) Endoplasmic reticulum stress-activated cell reprogramming in oncogenesis. Cancer Discov. 5, 586–597 10.1158/2159-8290.CD-14-1490 [DOI] [PubMed] [Google Scholar]
- 70. Cross B. C., Bond P. J., Sadowski P. G., Jha B. K., Zak J., Goodman J. M., Silverman R. H., Neubert T. A., Baxendale I. R., Ron D., and Harding H. P. (2012) The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc. Natl. Acad. Sci. U. S. A. 109, E869–E878 10.1073/pnas.1115623109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Volkmann K., Lucas J. L., Vuga D., Wang X., Brumm D., Stiles C., Kriebel D., Der-Sarkissian A., Krishnan K., Schweitzer C., Liu Z., Malyankar U. M., Chiovitti D., Canny M., Durocher D., et al. (2011) Potent and selective inhibitors of the inositol-requiring enzyme 1 endoribonuclease. J. Biol. Chem. 286, 12743–12755 10.1074/jbc.M110.199737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Papandreou I., Denko N. C., Olson M., Van Melckebeke H., Lust S., Tam A., Solow-Cordero D. E., Bouley D. M., Offner F., Niwa M., and Koong A. C. (2011) Identification of an Ire1α endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood 117, 1311–1314 10.1182/blood-2010-08-303099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sato H., Shiba Y., Tsuchiya Y., Saito M., and Kohno K. (2017) 4mu8C inhibits insulin secretion independent of IRE1α RNase activity. Cell Struct. Funct. 42, 61–70 10.1247/csf.17002 [DOI] [PubMed] [Google Scholar]
- 74. Chan S. M. H., Lowe M. P., Bernard A., Miller A. A., and Herbert T. P. (2018) The inositol-requiring enzyme 1 (IRE1α) RNAse inhibitor, 4micro8C, is also a potent cellular antioxidant. Biochem. J. 475, 923–929 10.1042/BCJ20170678 [DOI] [PubMed] [Google Scholar]
- 75. Liu Y., and Gray N. S. (2006) Rational design of inhibitors that bind to inactive kinase conformations. Nat. Chem. Biol. 2, 358–364 10.1038/nchembio799 [DOI] [PubMed] [Google Scholar]
- 76. Wang L., Perera B. G., Hari S. B., Bhhatarai B., Backes B. J., Seeliger M. A., Schurer S. C., Oakes S. A., Papa F. R., and Maly D. J. (2012) Divergent allosteric control of the IRE1α endoribonuclease using kinase inhibitors. Nat. Chem. Biol. 8, 982–989 10.1038/nchembio.1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ghosh R., Wang L., Wang E. S., Perera B. G., Igbaria A., Morita S., Prado K., Thamsen M., Caswell D., Macias H., Weiberth K. F., Gliedt M. J., Alavi M. V., Hari S. B., Mitra A. K., et al. (2014) Allosteric inhibition of the IRE1α RNase preserves cell viability and function during endoplasmic reticulum stress. Cell 158, 534–548 10.1016/j.cell.2014.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Mahameed M., Wilhelm T., Darawshi O., Obiedat A., Tommy W. S., Chintha C., Schubert T., Samali A., Chevet E., Eriksson L. A., Huber M., and Tirosh B. (2019) The unfolded protein response modulators GSK2606414 and KIRA6 are potent KIT inhibitors. Cell Death Dis. 10, 300 10.1038/s41419-019-1523-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Harnoss J. M., Le Thomas A., Shemorry A., Marsters S. A., Lawrence D. A., Lu M., Chen Y. A., Qing J., Totpal K., Kan D., Segal E., Merchant M., Reichelt M., Ackerly Wallweber H., Wang W., et al. (2019) Disruption of IRE1α through its kinase domain attenuates multiple myeloma. Proc. Natl. Acad. Sci. U. S. A. 116, 16420–16429 10.1073/pnas.1906999116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Chu H., Yuen T. T. T., Chik K. K. H., Yuan S., Shuai H., Zou Z., Wang Y., Zhu Z., Yang D., Poon V. K. M., Chan C. C. S., Zhou J., Yin F., Kok K. H., Yuen K. Y., et al. (2020) Targeting the inositol-requiring enzyme-1 pathway efficiently reverts Zika virus-induced neurogenesis and spermatogenesis marker perturbations. ACS Infect. Dis. 6, 1745–1758 10.1021/acsinfecdis.9b00526 [DOI] [PubMed] [Google Scholar]
- 81. Morita S., Villalta S. A., Feldman H. C., Register A. C., Rosenthal W., Hoffmann-Petersen I. T., Mehdizadeh M., Ghosh R., Wang L., Colon-Negron K., Meza-Acevedo R., Backes B. J., Maly D. J., Bluestone J. A., and Papa F. R. (2017) Targeting ABL-IRE1α signaling spares ER-stressed pancreatic β cells to reverse autoimmune diabetes. Cell Metab. 25, 883–897.e8 10.1016/j.cmet.2017.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Papa F. R., Zhang C., Shokat K., and Walter P. (2003) Bypassing a kinase activity with an ATP-competitive drug. Science 302, 1533–1537 10.1126/science.1090031 [DOI] [PubMed] [Google Scholar]
- 83. Lin J. H., Li H., Yasumura D., Cohen H. R., Zhang C., Panning B., Shokat K. M., Lavail M. M., and Walter P. (2007) IRE1 signaling affects cell fate during the unfolded protein response. Science 318, 944–949 10.1126/science.1146361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Mendez A. S., Alfaro J., Morales-Soto M. A., Dar A. C., McCullagh E., Gotthardt K., Li H., Acosta-Alvear D., Sidrauski C., Korennykh A. V., Bernales S., Shokat K. M., and Walter P. (2015) Endoplasmic reticulum stress-independent activation of unfolded protein response kinases by a small molecule ATP-mimic. Elife 4, e05434 10.7554/eLife.05434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Grandjean J. M. D., Madhavan A., Cech L., Seguinot B. O., Paxman R. J., Smith E., Scampavia L., Powers E. T., Cooley C. B., Plate L., Spicer T. P., Kelly J. W., and Wiseman R. L. (2020) Pharmacologic IRE1/XBP1s activation confers targeted ER proteostasis reprogramming. Nat. Chem. Biol. 16, 1052–1061 10.1038/s41589-020-0584-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Shi Y., Vattem K. M., Sood R., An J., Liang J., Stramm L., and Wek R. C. (1998) Identification and characterization of pancreatic eukaryotic initiation factor 2 α-subunit kinase, PEK, involved in translational control. Mol. Cell Biol. 18, 7499–7509 10.1128/mcb.18.12.7499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Harding H. P., Zhang Y., and Ron D. (1999) Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397, 271–274 10.1038/16729 [DOI] [PubMed] [Google Scholar]
- 88. Carrara M., Prischi F., Nowak P. R., Kopp M. C., and Ali M. M. (2015) Noncanonical binding of BiP ATPase domain to Ire1 and Perk is dissociated by unfolded protein CH1 to initiate ER stress signaling. Elife 4, e03522 10.7554/eLife.03522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Ma K., Vattem K. M., and Wek R. C. (2002) Dimerization and release of molecular chaperone inhibition facilitate activation of eukaryotic initiation factor-2 kinase in response to endoplasmic reticulum stress. J. Biol. Chem. 277, 18728–18735 10.1074/jbc.M200903200 [DOI] [PubMed] [Google Scholar]
- 90. Scheuner D., Song B., McEwen E., Liu C., Laybutt R., Gillespie P., Saunders T., Bonner-Weir S., and Kaufman R. J., (2001) Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol. Cell 7, 1165–1176 10.1016/S1097-2765(01)00265-9 [DOI] [PubMed] [Google Scholar]
- 91. Cullinan S. B., Zhang D., Hannink M., Arvisais E., Kaufman R. J., and Diehl J. A. (2003) Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell Biol. 23, 7198–7209 10.1128/mcb.23.20.7198-7209.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Costa-Mattioli M., and Walter P. (2020) The integrated stress response: From mechanism to disease. Science 368, eaat5314 10.1126/science.aat5314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Pakos-Zebrucka K., Koryga I., Mnich K., Ljujic M., Samali A., and Gorman A. M. (2016) The integrated stress response. EMBO Rep. 17, 1374–1395 10.15252/embr.201642195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Gupta S., McGrath B., and Cavener D. R. (2010) PERK (EIF2AK3) regulates proinsulin trafficking and quality control in the secretory pathway. Diabetes 59, 1937–1947 10.2337/db09-1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Kondratyev M., Avezov E., Shenkman M., Groisman B., and Lederkremer G. Z. (2007) PERK-dependent compartmentalization of ERAD and unfolded protein response machineries during ER stress. Exp. Cell Res. 313, 3395–3407 10.1016/j.yexcr.2007.07.006 [DOI] [PubMed] [Google Scholar]
- 96. Brewer J. W., and Diehl J. A. (2000) PERK mediates cell-cycle exit during the mammalian unfolded protein response. Proc. Natl. Acad. Sci. U. S. A. 97, 12625–12630 10.1073/pnas.220247197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Lebeau J., Saunders J. M., Moraes V. W. R., Madhavan A., Madrazo N., Anthony M. C., and Wiseman R. L. (2018) The PERK arm of the unfolded protein response regulates mitochondrial morphology during acute endoplasmic reticulum stress. Cell Rep. 22, 2827–2836 10.1016/j.celrep.2018.02.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Rainbolt T. K., Atanassova N., Genereux J. C., and Wiseman R. L. (2013) Stress-regulated translational attenuation adapts mitochondrial protein import through Tim17A degradation. Cell Metab. 18, 908–919 10.1016/j.cmet.2013.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Rainbolt T. K., Saunders J. M., and Wiseman R. L. (2014) Stress-responsive regulation of mitochondria through the ER unfolded protein response. Trends Endocrinol. Metab. 25, 528–537 10.1016/j.tem.2014.06.007 [DOI] [PubMed] [Google Scholar]
- 100. Vattem K. M., and Wek R. C. (2004) Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. U. S. A. 101, 11269–11274 10.1073/pnas.0400541101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Lu P. D., Harding H. P., and Ron D. (2004) Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J. Cell Biol. 167, 27–33 10.1083/jcb.200408003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Jousse C., Bruhat A., Carraro V., Urano F., Ferrara M., Ron D., and Fafournoux P. (2001) Inhibition of CHOP translation by a peptide encoded by an open reading frame localized in the chop 5'UTR. Nucleic Acids Res. 29, 4341–4351 10.1093/nar/29.21.4341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Young S. K., and Wek R. C. (2016) Upstream open reading frames differentially regulate gene-specific translation in the integrated stress response. J. Biol. Chem. 291, 16927–16935 10.1074/jbc.R116.733899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Han J., Back S. H., Hur J., Lin Y. H., Gildersleeve R., Shan J., Yuan C. L., Krokowski D., Wang S., Hatzoglou M., Kilberg M. S., Sartor M. A., and Kaufman R. J. (2013) ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 15, 481–490 10.1038/ncb2738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Harding H. P., Zhang Y., Zeng H., Novoa I., Lu P. D., Calfon M., Sadri N., Yun C., Popko B., Paules R., Stojdl D. F., Bell J. C., Hettmann T., Leiden J. M., and Ron D. (2003) An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 11, 619–633 10.1016/S1097-2765(03)00105-9 [DOI] [PubMed] [Google Scholar]
- 106. Novoa I., Zeng H., Harding H. P., and Ron D. (2001) Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2α. J. Cell Biol. 153, 1011–1022 10.1083/jcb.153.5.1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Ma Y., and Hendershot L. M. (2003) Delineation of a negative feedback regulatory loop that controls protein translation during endoplasmic reticulum stress. J. Biol. Chem. 278, 34864–34873 10.1074/jbc.M301107200 [DOI] [PubMed] [Google Scholar]
- 108. Marciniak S. J., Yun C. Y., Oyadomari S., Novoa I., Zhang Y., Jungreis R., Nagata K., Harding H. P., and Ron D. (2004) CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 18, 3066–3077 10.1101/gad.1250704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Puthalakath H., O'Reilly L. A., Gunn P., Lee L., Kelly P. N., Huntington N. D., Hughes P. D., Michalak E. M., McKimm-Breschkin J., Motoyama N., Gotoh T., Akira S., Bouillet P., and Strasser A. (2007) ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 129, 1337–1349 10.1016/j.cell.2007.04.027 [DOI] [PubMed] [Google Scholar]
- 110. Zhang L., Lopez H., George N. M., Liu X., Pang X., and Luo X. (2011) Selective involvement of BH3-only proteins and differential targets of Noxa in diverse apoptotic pathways. Cell Death Differ. 18, 864–873 10.1038/cdd.2010.152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Yamaguchi H., and Wang H. G. (2004) CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J. Biol. Chem. 279, 45495–45502 10.1074/jbc.M406933200 [DOI] [PubMed] [Google Scholar]
- 112. Hiramatsu N., Messah C., Han J., LaVail M. M., Kaufman R. J., and Lin J. H. (2014) Translational and posttranslational regulation of XIAP by eIF2α and ATF4 promotes ER stress-induced cell death during the unfolded protein response. Mol. Biol. Cell 25, 1411–1420 10.1091/mbc.E13-11-0664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Hiramatsu N., Chiang K., Aivati C., Rodvold J. J., Lee J. M., Han J., Chea L., Zanetti M., Koo E. H., and Lin J. H. (2020) PERK-mediated induction of microRNA-483 disrupts cellular ATP homeostasis during the unfolded protein response. J. Biol. Chem. 295, 237–249 10.1074/jbc.RA119.008336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Axten J. M., Medina J. R., Feng Y., Shu A., Romeril S. P., Grant S. W., Li W. H., Heerding D. A., Minthorn E., Mencken T., Atkins C., Liu Q., Rabindran S., Kumar R., Hong X., Goetz A., et al. (2012) Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-p yrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J Med Chem 55, 7193–7207 10.1021/jm300713s [DOI] [PubMed] [Google Scholar]
- 115. Axten J. M., Romeril S. P., Shu A., Ralph J., Medina J. R., Feng Y., Li W. H., Grant S. W., Heerding D. A., Minthorn E., Mencken T., Gaul N., Goetz A., Stanley T., Hassell A. M., Gampe R. T., Atkins C., and Kumar R. (2013) Discovery of GSK2656157: an optimized PERK inhibitor selected for preclinical development. ACS Med. Chem. Lett. 4, 964–968 10.1021/ml400228e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Rojas-Rivera D., Delvaeye T., Roelandt R., Nerinckx W., Augustyns K., Vandenabeele P., and Bertrand M. J. M. (2017) When PERK inhibitors turn out to be new potent RIPK1 inhibitors: critical issues on the specificity and use of GSK2606414 and GSK2656157. Cell Death Differ. 24, 1100–1110 10.1038/cdd.2017.58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Smith A. L., Andrews K. L., Beckmann H., Bellon S. F., Beltran P. J., Booker S., Chen H., Chung Y. A., D'Angelo N. D., Dao J., Dellamaggiore K. R., Jaeckel P., Kendall R., Labitzke K., Long A. M., Materna-Reichelt S., Mitchell P., et al. (2015) Discovery of 1H-pyrazol-3(2H)-ones as potent and selective inhibitors of protein kinase R-like endoplasmic reticulum kinase (PERK). J. Med. Chem. 58, 1426–1441 10.1021/jm5017494 [DOI] [PubMed] [Google Scholar]
- 118. Atkins C., Liu Q., Minthorn E., Zhang S. Y., Figueroa D. J., Moss K., Stanley T. B., Sanders B., Goetz A., Gaul N., Choudhry A. E., Alsaid H., Jucker B. M., Axten J. M., and Kumar R. (2013) Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 73, 1993–2002 10.1158/0008-5472.CAN-12-3109 [DOI] [PubMed] [Google Scholar]
- 119. Radford H., Moreno J. A., Verity N., Halliday M., and Mallucci G. R. (2015) PERK inhibition prevents tau-mediated neurodegeneration in a mouse model of frontotemporal dementia. Acta Neuropathol. 130, 633–642 10.1007/s00401-015-1487-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Moreno J. A., Halliday M., Molloy C., Radford H., Verity N., Axten J. M., Ortori C. A., Willis A. E., Fischer P. M., Barrett D. A., and Mallucci G. R. (2013) Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci. Transl. Med. 5, 206ra138 10.1126/scitranslmed.3006767 [DOI] [PubMed] [Google Scholar]
- 121. Halliday M., Radford H., Sekine Y., Moreno J., Verity N., Le Quesne J., Ortori C. A., Barrett D. A., Fromont C., Fischer P. M., Harding H. P., Ron D., and Mallucci G. R. (2015) Partial restoration of protein synthesis rates by the small molecule ISRIB prevents neurodegeneration without pancreatic toxicity. Cell Death Dis. 6, e1672 10.1038/cddis.2015.49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Yu Q., Zhao B., Gui J., Katlinski K. V., Brice A., Gao Y., Li C., Kushner J. A., Koumenis C., Diehl J. A., and Fuchs S. Y. (2015) Type I interferons mediate pancreatic toxicities of PERK inhibition. Proc. Natl. Acad. Sci. U. S. A. 112, 15420–15425 10.1073/pnas.1516362112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Sidrauski C., Acosta-Alvear D., Khoutorsky A., Vedantham P., Hearn B. R., Li H., Gamache K., Gallagher C. M., Ang K. K., Wilson C., Okreglak V., Ashkenazi A., Hann B., Nader K., Arkin M. R., et al. (2013) Pharmacological brake-release of mRNA translation enhances cognitive memory. Elife 2, e00498 10.7554/eLife.00498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Sidrauski C., Tsai J. C., Kampmann M., Hearn B. R., Vedantham P., Jaishankar P., Sokabe M., Mendez A. S., Newton B. W., Tang E. L., Verschueren E., Johnson J. R., Krogan N. J., Fraser C. S., Weissman J. S., et al. (2015) Pharmacological dimerization and activation of the exchange factor eIF2B antagonizes the integrated stress response. Elife 4, e07314 10.7554/eLife.07314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Sekine Y., Zyryanova A., Crespillo-Casado A., Fischer P. M., Harding H. P., and Ron D. (2015) Stress responses: mutations in a translation initiation factor identify the target of a memory-enhancing compound. Science 348, 1027–1030 10.1126/science.aaa6986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Zyryanova A. F., Weis F., Faille A., Alard A. A., Crespillo-Casado A., Sekine Y., Harding H. P., Allen F., Parts L., Fromont C., Fischer P. M., Warren A. J., and Ron D. (2018) Binding of ISRIB reveals a regulatory site in the nucleotide exchange factor eIF2B. Science 359, 1533–1536 10.1126/science.aar5129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Tsai J. C., Miller-Vedam L. E., Anand A. A., Jaishankar P., Nguyen H. C., Renslo A. R., Frost A., and Walter P. (2018) Structure of the nucleotide exchange factor eIF2B reveals mechanism of memory-enhancing molecule. Science 359, eaaq0939 10.1126/science.aaq0939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Rabouw H. H., Langereis M. A., Anand A. A., Visser L. J., de Groot R. J., Walter P., and van Kuppeveld F. J. M. (2019) Small molecule ISRIB suppresses the integrated stress response within a defined window of activation. Proc. Natl. Acad. Sci. U. S. A. 116, 2097–2102 10.1073/pnas.1815767116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Wong Y. L., LeBon L., Basso A. M., Kohlhaas K. L., Nikkel A. L., Robb H. M., Donnelly-Roberts D. L., Prakash J., Swensen A. M., Rubinstein N. D., Krishnan S., McAllister F. E., Haste N. V., O'Brien J. J., Roy M., et al. (2019) eIF2B activator prevents neurological defects caused by a chronic integrated stress response. Elife 8, e42940 10.7554/eLife.42940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Sidrauski C., McGeachy A. M., Ingolia N. T., and Walter P. (2015) The small molecule ISRIB reverses the effects of eIFα phosphorylation on translation and stress granule assembly. Elife 4, e05033 10.7554/eLife.05033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Wong Y. L., LeBon L., Edalji R., Lim H. B., Sun C., and Sidrauski C. (2018) The small molecule ISRIB rescues the stability and activity of vanishing white matter disease eIF2B mutant complexes. Elife 7, e32733 10.7554/eLife.32733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Quiros P. M., Prado M. A., Zamboni N., D'Amico D., Williams R. W., Finley D., Gygi S. P., and Auwerx J. (2017) Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol. 216, 2027–2045 10.1083/jcb.201702058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Romine I. C., and Wiseman R. L. (2019) PERK signaling regulates extracellular proteostasis of an amyloidogenic protein during endoplasmic reticulum stress. Sci. Rep. 9, 410 10.1038/s41598-018-37207-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Halliday M., Radford H., Zents K. A. M., Molloy C., Moreno J. A., Verity N. C., Smith E., Ortori C. A., Barrett D. A., Bushell M., and Mallucci G. R. (2017) Repurposed drugs targeting eIF2α-P-mediated translational repression prevent neurodegeneration in mice. Brain 140, 1768–1783 10.1093/brain/awx074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Harvey R. F., Poyry T. A. A., Stoneley M., and Willis A. E. (2019) Signaling from mTOR to eIF2α mediates cell migration in response to the chemotherapeutic doxorubicin. Sci. Signal. 12, eaaw6763 10.1126/scisignal.aaw6763 [DOI] [PubMed] [Google Scholar]
- 136. Chen R., Rato C., Yan Y., Crespillo-Casado A., Clarke H. J., Harding H. P., Marciniak S. J., Read R. J., and Ron D. (2015) G-actin provides substrate-specificity to eukaryotic initiation factor 2α holophosphatases. Elife 4, e04871 10.7554/eLife.04871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Chambers J. E., Dalton L. E., Clarke H. J., Malzer E., Dominicus C. S., Patel V., Moorhead G., Ron D., and Marciniak S. J. (2015) Actin dynamics tune the integrated stress response by regulating eukaryotic initiation factor 2α dephosphorylation. Elife 4, e04872 10.7554/eLife.04872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Jousse C., Oyadomari S., Novoa I., Lu P., Zhang Y., Harding H. P., and Ron D. (2003) Inhibition of a constitutive translation initiation factor 2α phosphatase, CReP, promotes survival of stressed cells. J. Cell Biol. 163, 767–775 10.1083/jcb.200308075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Novoa I., Zhang Y., Zeng H., Jungreis R., Harding H. P., and Ron D. (2003) Stress-induced gene expression requires programmed recovery from translational repression. EMBO J. 22, 1180–1187 10.1093/emboj/cdg112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Brush M. H., Weiser D. C., and Shenolikar S. (2003) Growth arrest and DNA damage-inducible protein GADD34 targets protein phosphatase 1 α to the endoplasmic reticulum and promotes dephosphorylation of the α subunit of eukaryotic translation initiation factor 2. Mol. Cell Biol. 23, 1292–1303 10.1128/mcb.23.4.1292-1303.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Boyce M., Bryant K. F., Jousse C., Long K., Harding H. P., Scheuner D., Kaufman R. J., Ma D., Coen D. M., Ron D., and Yuan J. (2005) A selective inhibitor of eIF2α dephosphorylation protects cells from ER stress. Science 307, 935–939 10.1126/science.1101902 [DOI] [PubMed] [Google Scholar]
- 142. Yoshida T., Ozawa Y., Suzuki K., Yuki K., Ohyama M., Akamatsu W., Matsuzaki Y., Shimmura S., Mitani K., Tsubota K., and Okano H. (2014) The use of induced pluripotent stem cells to reveal pathogenic gene mutations and explore treatments for retinitis pigmentosa. Mol. Brain 7, 45 10.1186/1756-6606-7-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Athanasiou D., Aguila M., Bellingham J., Kanuga N., Adamson P., and Cheetham M. E. (2017) The role of the ER stress-response protein PERK in rhodopsin retinitis pigmentosa. Hum. Mol. Genet. 26, 4896–4905 10.1093/hmg/ddx370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Saxena S., Cabuy E., and Caroni P. (2009) A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat. Neurosci. 12, 627–636 10.1038/nn.2297 [DOI] [PubMed] [Google Scholar]
- 145. Moreno J. A., Radford H., Peretti D., Steinert J. R., Verity N., Martin M. G., Halliday M., Morgan J., Dinsdale D., Ortori C. A., Barrett D. A., Tsaytler P., Bertolotti A., Willis A. E., Bushell M., et al. (2012) Sustained translational repression by eIF2α-P mediates prion neurodegeneration. Nature 485, 507–511 10.1038/nature11058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. D'Antonio M., Musner N., Scapin C., Ungaro D., Del Carro U., Ron D., Feltri M. L., and Wrabetz L. (2013) Resetting translational homeostasis restores myelination in Charcot-Marie-Tooth disease type 1B mice. J. Exp. Med. 210, 821–838 10.1084/jem.20122005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Wang L., Popko B., and Roos R. P. (2014) An enhanced integrated stress response ameliorates mutant SOD1-induced ALS. Hum. Mol. Genet. 23, 2629–2638 10.1093/hmg/ddt658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Das I., Krzyzosiak A., Schneider K., Wrabetz L., D'Antonio M., Barry N., Sigurdardottir A., and Bertolotti A. (2015) Preventing proteostasis diseases by selective inhibition of a phosphatase regulatory subunit. Science 348, 239–242 10.1126/science.aaa4484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Tsaytler P., Harding H. P., Ron D., and Bertolotti A. (2011) Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science 332, 91–94 10.1126/science.1201396 [DOI] [PubMed] [Google Scholar]
- 150. Carrara M., Sigurdardottir A., and Bertolotti A. (2017) Decoding the selectivity of eIF2α holophosphatases and PPP1R15A inhibitors. Nat. Struct. Mol. Biol. 24, 708–716 10.1038/nsmb.3443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Crespillo-Casado A., Claes Z., Choy M. S., Peti W., Bollen M., and Ron D. (2018) A Sephin1-insensitive tripartite holophosphatase dephosphorylates translation initiation factor 2α. J. Biol. Chem. 293, 7766–7776 10.1074/jbc.RA118.002325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Crespillo-Casado A., Chambers J. E., Fischer P. M., Marciniak S. J., and Ron D. (2017) PPP1R15A-mediated dephosphorylation of eIF2α is unaffected by Sephin1 or Guanabenz. Elife 6, e26109 10.7554/eLife.26109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Thapa S., Abdelaziz D. H., Abdulrahman B. A., and Schatzl H. M. (2020) Sephin1 reduces prion infection in prion-infected cells and animal model. Mol. Neurobiol. 57, 2206–2219 10.1007/s12035-020-01880-y [DOI] [PubMed] [Google Scholar]
- 154. Chen Y., Podojil J. R., Kunjamma R. B., Jones J., Weiner M., Lin W., Miller S. D., and Popko B. (2019) Sephin1, which prolongs the integrated stress response, is a promising therapeutic for multiple sclerosis. Brain 142, 344–361 10.1093/brain/awy322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Stockwell S. R., Platt G., Barrie S. E., Zoumpoulidou G., Te Poele R. H., Aherne G. W., Wilson S. C., Sheldrake P., McDonald E., Venet M., Soudy C., Elustondo F., Rigoreau L., Blagg J., Workman P., et al. (2012) Mechanism-based screen for G1/S checkpoint activators identifies a selective activator of EIF2AK3/PERK signalling. PLoS ONE 7, e28568 10.1371/journal.pone.0028568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Xie W., Pariollaud M., Wixted W. E., Chitnis N., Fornwald J., Truong M., Pao C., Liu Y., Ames R. S., Callahan J., Solari R., Sanchez Y., Diehl A., and Li H. (2015) Identification and characterization of PERK activators by phenotypic screening and their effects on NRF2 activation. PLoS ONE 10, e0119738 10.1371/journal.pone.0119738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Wang H., Blais J., Ron D., and Cardozo T. (2010) Structural determinants of PERK inhibitor potency and selectivity. Chem. Biol. Drug Des. 76, 480–495 10.1111/j.1747-0285.2010.01048.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Leitman J., Barak B., Benyair R., Shenkman M., Ashery U., Hartl F. U., and Lederkremer G. Z. (2014) ER stress-induced eIF2-α phosphorylation underlies sensitivity of striatal neurons to pathogenic huntingtin. PLoS ONE 9, e90803 10.1371/journal.pone.0090803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Ganz J., Shacham T., Kramer M., Shenkman M., Eiger H., Weinberg N., Iancovici O., Roy S., Simhaev L., Da'adoosh B., Engel H., Perets N., Barhum Y., Portnoy M., Offen D., et al. (2020) A novel specific PERK activator reduces toxicity and extends survival in Huntington's disease models. Sci. Rep. 10, 6875 10.1038/s41598-020-63899-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Bruch J., Xu H., Rosler T. W., De Andrade A., Kuhn P. H., Lichtenthaler S. F., Arzberger T., Winklhofer K. F., Muller U., and Hoglinger G. U. (2017) PERK activation mitigates tau pathology in vitro and in vivo. EMBO Mol. Med. 9, 371–384 10.15252/emmm.201606664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Balsa E., Soustek M. S., Thomas A., Cogliati S., Garcia-Poyatos C., Martin-Garcia E., Jedrychowski M., Gygi S. P., Enriquez J. A., and Puigserver P. (2019) ER and nutrient stress promote assembly of respiratory chain supercomplexes through the PERK-eIF2α axis. Mol. Cell. 74, 877–890.e6 10.1016/j.molcel.2019.03.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Yoshida H., Okada T., Haze K., Yanagi H., Yura T., Negishi M., and Mori K. (2000) ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol. Cell Biol. 20, 6755–6767 10.1128/mcb.20.18.6755-6767.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Haze K., Okada T., Yoshida H., Yanagi H., Yura T., Negishi M., and Mori K. (2001) Identification of the G13 (cAMP-response-element-binding protein-related protein) gene product related to activating transcription factor 6 as a transcriptional activator of the mammalian unfolded protein response. Biochem. J. 355, 19–28 10.1042/0264-6021:3550019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Yoshida H., Okada T., Haze K., Yanagi H., Yura T., Negishi M., and Mori K. (2001) Endoplasmic reticulum stress-induced formation of transcription factor complex ERSF including NF-Y (CBF) and activating transcription factors 6α and 6β that activates the mammalian unfolded protein response. Mol. Cell Biol. 21, 1239–1248 10.1128/MCB.21.4.1239-1248.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Yamamoto K., Sato T., Matsui T., Sato M., Okada T., Yoshida H., Harada A., and Mori K. (2007) Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6α and XBP1. Dev. Cell 13, 365–376 10.1016/j.devcel.2007.07.018 [DOI] [PubMed] [Google Scholar]
- 166. Thuerauf D. J., Marcinko M., Belmont P. J., and Glembotski C. C. (2007) Effects of the isoform-specific characteristics of ATF6 α and ATF6 β on endoplasmic reticulum stress response gene expression and cell viability. J. Biol. Chem. 282, 22865–22878 10.1074/jbc.M701213200 [DOI] [PubMed] [Google Scholar]
- 167. Thuerauf D. J., Morrison L., and Glembotski C. C. (2004) Opposing roles for ATF6α and ATF6β in endoplasmic reticulum stress response gene induction. J. Biol. Chem. 279, 21078–21084 10.1074/jbc.M400713200 [DOI] [PubMed] [Google Scholar]
- 168. Forouhan M., Mori K., and Boot-Handford R. P. (2018) Paradoxical roles of ATF6α and ATF6β in modulating disease severity caused by mutations in collagen X. Matrix Biol. 70, 50–71 10.1016/j.matbio.2018.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Pieper L. A., Strotbek M., Wenger T., Olayioye M. A., and Hausser A. (2017) ATF6β-based fine-tuning of the unfolded protein response enhances therapeutic antibody productivity of Chinese hamster ovary cells. Biotechnol. Bioeng. 114, 1310–1318 10.1002/bit.26263 [DOI] [PubMed] [Google Scholar]
- 170. Nadanaka S., Okada T., Yoshida H., and Mori K. (2007) Role of disulfide bridges formed in the luminal domain of ATF6 in sensing endoplasmic reticulum stress. Mol. Cell Biol. 27, 1027–1043 10.1128/MCB.00408-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Nadanaka S., Yoshida H., and Mori K. (2006) Reduction of disulfide bridges in the lumenal domain of ATF6 in response to glucose starvation. Cell Struct. Funct. 31, 127–134 10.1247/csf.06024 [DOI] [PubMed] [Google Scholar]
- 172. Koba H., Jin S., Imada N., Ishikawa T., Ninagawa S., Okada T., Sakuma T., Yamamoto T., and Mori K. (2020) Reinvestigation of disulfide-bonded oligomeric forms of the unfolded protein response transducer ATF6. Cell Struct. Funct. 45, 9–21 10.1247/csf.19030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Shen J., Chen X., Hendershot L., and Prywes R. (2002) ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev Cell 3, 99–111 10.1016/S1534-5807(02)00203-4 [DOI] [PubMed] [Google Scholar]
- 174. Shen J., Snapp E. L., Lippincott-Schwartz J., and Prywes R. (2005) Stable binding of ATF6 to BiP in the endoplasmic reticulum stress response. Mol. Cell Biol. 25, 921–932 10.1128/MCB.25.3.921-932.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Ye J., Rawson R. B., Komuro R., Chen X., Dave U. P., Prywes R., Brown M. S., and Goldstein J. L. (2000) ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 6, 1355–1364 10.1016/S1097-2765(00)00133-7 [DOI] [PubMed] [Google Scholar]
- 176. Haze K., Yoshida H., Yanagi H., Yura T., and Mori K. (1999) Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 10, 3787–3799 10.1091/mbc.10.11.3787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Oka O. B., van Lith M., Rudolf J., Tungkum W., Pringle M. A., and Bulleid N. J. (2019) ERp18 regulates activation of ATF6α during unfolded protein response. EMBO J. 38, e100990 10.15252/embj.2018100990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Higa A., Taouji S., Lhomond S., Jensen D., Fernandez-Zapico M. E., Simpson J. C., Pasquet J. M., Schekman R., and Chevet E. (2014) Endoplasmic reticulum stress-activated transcription factor ATF6α requires the disulfide isomerase PDIA5 to modulate chemoresistance. Mol. Cell Biol. 34, 1839–1849 10.1128/MCB.01484-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Yamamoto K., Yoshida H., Kokame K., Kaufman R. J., and Mori K. (2004) Differential contributions of ATF6 and XBP1 to the activation of endoplasmic reticulum stress-responsive cis-acting elements ERSE, UPRE and ERSE-II. J. Biochem. 136, 343–350 10.1093/jb/mvh122 [DOI] [PubMed] [Google Scholar]
- 180. Adachi Y., Yamamoto K., Okada T., Yoshida H., Harada A., and Mori K. (2008) ATF6 is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum. Cell Struct. Funct. 33, 75–89 10.1247/csf.07044 [DOI] [PubMed] [Google Scholar]
- 181. Blackwood E. A., Hofmann C., Santo Domingo M., Bilal A. S., Sarakki A., Stauffer W., Arrieta A., Thuerauf D. J., Kolkhorst F. W., Muller O. J., Jakobi T., Dieterich C., Katus H. A., Doroudgar S., and Glembotski C. C. (2019) ATF6 regulates cardiac hypertrophy by transcriptional induction of the mTORC1 activator, Rheb. Circ. Res. 124, 79–93 10.1161/CIRCRESAHA.118.313854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Jin J. K., Blackwood E. A., Azizi K., Thuerauf D. J., Fahem A. G., Hofmann C., Kaufman R. J., Doroudgar S., and Glembotski C. C. (2017) ATF6 decreases myocardial ischemia/reperfusion damage and links ER stress and oxidative stress signaling pathways in the heart. Circ. Res. 120, 862–875 10.1161/CIRCRESAHA.116.310266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Allen D., and Seo J. (2018) ER stress activates the TOR pathway through Atf6. J. Mol. Signal. 13, 1 10.5334/1750-2187-13-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Schewe D. M., and Aguirre-Ghiso J. A. (2008) ATF6α-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proc. Natl. Acad. Sci. U. S. A. 105, 10519–10524 10.1073/pnas.0800939105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Kohl S., Zobor D., Chiang W. C., Weisschuh N., Staller J., Gonzalez Menendez I., Chang S., Beck S. C., Garcia Garrido M., Sothilingam V., Seeliger M. W., Stanzial F., Benedicenti F., Inzana F., Heon E., et al. (2015) Mutations in the unfolded protein response regulator ATF6 cause the cone dysfunction disorder achromatopsia. Nat. Genet. 47, 757–765 10.1038/ng.3319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Wu J., Rutkowski D. T., Dubois M., Swathirajan J., Saunders T., Wang J., Song B., Yau G. D., and Kaufman R. J. (2007) ATF6α optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev. Cell 13, 351–364 10.1016/j.devcel.2007.07.005 [DOI] [PubMed] [Google Scholar]
- 187. Martindale J. J., Fernandez R., Thuerauf D., Whittaker R., Gude N., Sussman M. A., and Glembotski C. C. (2006) Endoplasmic reticulum stress gene induction and protection from ischemia/reperfusion injury in the hearts of transgenic mice with a tamoxifen-regulated form of ATF6. Circ. Res. 98, 1186–1193 10.1161/01.RES.0000220643.65941.8d [DOI] [PubMed] [Google Scholar]
- 188. Yu Z., Sheng H., Liu S., Zhao S., Glembotski C. C., Warner D. S., Paschen W., and Yang W. (2017) Activation of the ATF6 branch of the unfolded protein response in neurons improves stroke outcome. J. Cereb. Blood Flow Metab. 37, 1069–1079 10.1177/0271678X16650218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Chen X., Zhang F., Gong Q., Cui A., Zhuo S., Hu Z., Han Y., Gao J., Sun Y., Liu Z., Yang Z., Le Y., Gao X., Dong L. Q., Gao X., et al. (2016) Hepatic ATF6 increases fatty acid oxidation to attenuate hepatic steatosis in mice through peroxisome proliferator-activated receptor α. Diabetes 65, 1904–1915 10.2337/db15-1637 [DOI] [PubMed] [Google Scholar]
- 190. Ozcan L., Ghorpade D. S., Zheng Z., de Souza J. C., Chen K., Bessler M., Bagloo M., Schrope B., Pestell R., and Tabas I. (2016) Hepatocyte DACH1 is increased in obesity via nuclear exclusion of HDAC4 and promotes hepatic insulin resistance. Cell Rep. 15, 2214–2225 10.1016/j.celrep.2016.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Chen J. J., Genereux J. C., Qu S., Hulleman J. D., Shoulders M. D., and Wiseman R. L. (2014) ATF6 activation reduces the secretion and extracellular aggregation of destabilized variants of an amyloidogenic protein. Chem. Biol. 21, 1564–1574 10.1016/j.chembiol.2014.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Cooley C. B., Ryno L. M., Plate L., Morgan G. J., Hulleman J. D., Kelly J. W., and Wiseman R. L. (2014) Unfolded protein response activation reduces secretion and extracellular aggregation of amyloidogenic immunoglobulin light chain. Proc. Natl. Acad. Sci. U. S. A. 111, 13046–13051 10.1073/pnas.1406050111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Skorczyk-Werner A., Chiang W.-C., Wawrocka A., Wicher K., Jarmuż-Szymczak M., Kostrzewska-Poczekaj M., Jamsheer A., Płoski R., Rydzanicz M., Pojda-Wilczek D., Weisschuh N., Wissinger B., Kohl S., Lin J. H., and Krawczyński M. R. (2017) Autosomal recessive cone-rod dystrophy can be caused by mutations in the ATF6 gene. Eur. J. Hum. Genet. 25, 1210–1216 10.1038/ejhg.2017.131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Okada T., Haze K., Nadanaka S., Yoshida H., Seidah N. G., Hirano Y., Sato R., Negishi M., and Mori K. (2003) A serine protease inhibitor prevents endoplasmic reticulum stress-induced cleavage but not transport of the membrane-bound transcription factor ATF6. J. Biol. Chem. 278, 31024–31032 10.1074/jbc.M300923200 [DOI] [PubMed] [Google Scholar]
- 195. Ye J. (2020) Transcription factors activated through RIP (regulated intramembrane proteolysis) and RAT (regulated alternative translocation. J. Biol. Chem. 295, 10271–10280 10.1074/jbc.REV120.012669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Gallagher C. M., Garri C., Cain E. L., Ang K. K., Wilson C. G., Chen S., Hearn B. R., Jaishankar P., Aranda-Diaz A., Arkin M. R., Renslo A. R., and Walter P. (2016) Ceapins are a new class of unfolded protein response inhibitors, selectively targeting the ATF6α branch. Elife 5, e11878 10.7554/eLife.11878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Gallagher C. M., and Walter P. (2016) Ceapins inhibit ATF6α signaling by selectively preventing transport of ATF6α to the Golgi apparatus during ER stress. Elife 5, e11880 10.7554/eLife.11880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Torres S. E., Gallagher C. M., Plate L., Gupta M., Liem C. R., Guo X., Tian R., Stroud R. M., Kampmann M., Weissman J. S., and Walter P. (2019) Ceapins block the unfolded protein response sensor ATF6α by inducing a neomorphic inter-organelle tether. Elife 8, e46595 10.7554/eLife.46595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Kaneko M., Koike H., Saito R., Kitamura Y., Okuma Y., and Nomura Y. (2010) Loss of HRD1-mediated protein degradation causes amyloid precursor protein accumulation and amyloid-β generation. J. Neurosci. 30, 3924–3932 10.1523/JNEUROSCI.2422-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Wang Y., Vera L., Fischer W. H., and Montminy M. (2009) The CREB coactivator CRTC2 links hepatic ER stress and fasting gluconeogenesis. Nature 460, 534–537 10.1038/nature08111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Kudo T., Kanemoto S., Hara H., Morimoto N., Morihara T., Kimura R., Tabira T., Imaizumi K., and Takeda M. (2008) A molecular chaperone inducer protects neurons from ER stress. Cell Death Differ. 15, 364–375 10.1038/sj.cdd.4402276 [DOI] [PubMed] [Google Scholar]
- 202. Oida Y., Izuta H., Oyagi A., Shimazawa M., Kudo T., Imaizumi K., and Hara H. (2008) Induction of BiP, an ER-resident protein, prevents the neuronal death induced by transient forebrain ischemia in gerbil. Brain Res. 1208, 217–224 10.1016/j.brainres.2008.02.068 [DOI] [PubMed] [Google Scholar]
- 203. Prachasilchai W., Sonoda H., Yokota-Ikeda N., Ito K., Kudo T., Imaizumi K., and Ikeda M. (2009) The protective effect of a newly developed molecular chaperone-inducer against mouse ischemic acute kidney injury. J. Pharmacol. Sci. 109, 311–314 10.1254/jphs.08272sc [DOI] [PubMed] [Google Scholar]
- 204. Oida Y., Hamanaka J., Hyakkoku K., Shimazawa M., Kudo T., Imaizumi K., Yasuda T., and Hara H. (2010) Post-treatment of a BiP inducer prevents cell death after middle cerebral artery occlusion in mice. Neurosci. Lett. 484, 43–46 10.1016/j.neulet.2010.08.015 [DOI] [PubMed] [Google Scholar]
- 205. Plate L., Cooley C. B., Chen J. J., Paxman R. J., Gallagher C. M., Madoux F., Genereux J. C., Dobbs W., Garza D., Spicer T. P., Scampavia L., Brown S. J., Rosen H., Powers E. T., Walter P., et al. (2016) Small molecule proteostasis regulators that reprogram the ER to reduce extracellular protein aggregation. Elife 5, e15550 10.7554/eLife.15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Paxman R., Plate L., Blackwood E. A., Glembotski C., Powers E. T., Wiseman R. L., and Kelly J. W. (2018) Pharmacologic ATF6 activating compounds are metabolically activated to selectively modify endoplasmic reticulum proteins. Elife 7, e37168 10.7554/eLife.37168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Blackwood E. A., Azizi K., Thuerauf D. J., Paxman R. J., Plate L., Kelly J. W., Wiseman R. L., and Glembotski C. C. (2019) Pharmacologic ATF6 activation confers global protection in widespread disease models by reprograming cellular proteostasis. Nat. Commun. 10, 187 10.1038/s41467-018-08129-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Kroeger H., Grimsey N., Paxman R., Chiang W. C., Plate L., Jones Y., Shaw P. X., Trejo J., Tsang S. H., Powers E., Kelly J. W., Wiseman R. L., and Lin J. H. (2018) The unfolded protein response regulator ATF6 promotes mesodermal differentiation. Sci. Signal. 11, eaan5785 10.1126/scisignal.aan5785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Rius B., Mesgarzadeh J. S., Romine I. C., Paxman R. J., Kelly J. W., and Wiseman R. L. (2020) ER proteostasis regulators reduce amyloidogenic light chain secretion through an on-target, ATF6-independent mechanism. bioRxiv 10.1101/2020.05.15.098145 [DOI] [PMC free article] [PubMed]
- 210. Almasy K. M., Davies J. P., Lisy S. M., Tirgar R., Tran S. C., and Plate L. (2020) Dengue and Zika virus infection are impaired by small molecule ER proteostasis regulator 147 in an ATF6-independent manner. bioRxiv 10.1101/2020.05.15.098624 [DOI] [PMC free article] [PubMed]
- 211. Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., and Mesirov J. P. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 102, 15545–15550 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Grandjean J. M. D., Plate L., Morimoto R. I., Bollong M. J., Powers E. T., and Wiseman R. L. (2019) Deconvoluting stress-responsive proteostasis signaling pathways for pharmacologic activation using targeted RNA sequencing. ACS Chem. Biol. 14, 784–795 10.1021/acschembio.9b00134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Ye C., Ho D. J., Neri M., Yang C., Kulkarni T., Randhawa R., Henault M., Mostacci N., Farmer P., Renner S., Ihry R., Mansur L., Keller C. G., McAllister G., Hild M., et al. (2018) DRUG-seq for miniaturized high-throughput transcriptome profiling in drug discovery. Nat. Commun. 9, 4307 10.1038/s41467-018-06500-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Miduturu C. V., Deng X., Kwiatkowski N., Yang W., Brault L., Filippakopoulos P., Chung E., Yang Q., Schwaller J., Knapp S., King R. W., Lee J. D., Herrgard S., Zarrinkar P., and Gray N. S. (2011) High-throughput kinase profiling: a more efficient approach toward the discovery of new kinase inhibitors. Chem. Biol. 18, 868–879 10.1016/j.chembiol.2011.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Neradil J., Kyr M., Polaskova K., Kren L., Macigova P., Skoda J., Sterba J., and Veselska R. (2019) Phospho-protein arrays as effective tools for screening possible targets for kinase inhibitors and their use in precision pediatric oncology. Front. Oncol. 9, 930 10.3389/fonc.2019.00930 [DOI] [PMC free article] [PubMed] [Google Scholar]