

Abstract
The proton-coupled folate transporter (PCFT, SLC46A1) is required for folate intestinal absorption and transport across the choroid plexus. Recent work has identified a F392V mutation causing hereditary folate malabsorption. However, the residue properties responsible for this loss of function remains unknown. Using site-directed mutagenesis, we observed complete loss of function with charged (Lys, Asp, and Glu) and polar (Thr, Ser, and Gln) Phe-392 substitutions and minimal function with some neutral substitutions; however, F392M retained full function. Using the substituted-cysteine accessibility method (with N-biotinyl aminoethyl methanethiosulfonate labeling), Phe-392 mutations causing loss of function, although preserving membrane expression and trafficking, also resulted in loss of accessibility of the substituted cysteine in P314C-PCFT located within the aqueous translocation pathway. F392V function and accessibility of the P314C cysteine were restored by insertion of a G305L (suppressor) mutation. A S196L mutation localized in proximity to Gly-305 by homology modeling was inactive. However, when inserted into the inactive F392V scaffold, function was restored (mutually compensatory mutations), as was accessibility of the P314C cysteine residue. Reduced function, documented with F392H PCFT, was due to a 15-fold decrease in methotrexate influx Vmax, accompanied by a decreased influx Kt (4.5-fold) and Ki (3-fold). The data indicate that Phe-392 is required for rapid oscillation of the carrier among its conformational states and suggest that this is achieved by dampening affinity of the protein for its folate substrates. F392V and other inactivating Phe-392 PCFT mutations lock the protein in its inward-open conformation. Reach (length) and hydrophobicity of Phe-392 appear to be features required for full activity.
Keywords: methotrexate, proton-coupled folate transporter, PCFT, SLC46A1, hereditary folate malabsorption (HFM), reduced folate carrier, facilitative transporters, mutually compensatory mutations, suppressor mutations, homology modeling, folate, proton transport, transporter, mutant
The proton-coupled folate transporter (PCFT), a member (SLC46A1) of the superfamily of solute transporters, harnesses the proton gradient as the energy source to achieve uphill transport of folates into cells (1, 2). Both the affinity for folate substrates and the rate of oscillation of the carrier among its conformational states increase as the extracellular pH is decreased to ∼5.5, the optimal pH for this transporter (1, 3). The two major physiological roles for PCFT are (i) the intestinal absorption of folates at the brush-border membrane of the proximal jejunum, as is the case for the absorption of other substrates mediated by proton-coupled processes (4, 5), and (ii) transport of folates across the choroid plexus into the cerebrospinal fluid (2, 6). Loss of function mutations in the PCFT gene result in the rare autosomal recessive disorder hereditary folate malabsorption (HFM), which is characterized by systemic folate deficiency and a marked deficiency of folate in the cerebrospinal fluid (1, 2, 7, 8). PCFT is also highly expressed in a variety of carcinomas where it delivers antifolates to cancer cells within the acidic microenvironment of tumors (9, 10). Among the antifolates, pemetrexed and new-generation glycinamide ribonucleotide transformylase inhibitors have the highest affinity for PCFT (11–13).
Ongoing research is focused on characterizing the structure-function of this transporter. Insights have come from functional changes associated with PCFT mutations identified in subjects with HFM and from site-directed mutagenesis (2, 6). The substituted-cysteine accessibility method has been used to characterize the secondary structure, the accessibility of residues within transmembrane segments and defined elements of the aqueous translocation pathway. Residues that are involved in proton binding, proton coupling, extracellular gating, folate substrate binding, and carrier translocation rates have been identified (2, 14, 15). Homology models of PCFT in the inward-open (16, 17), and more recently in both the inward- and outward-open (14, 18, 19), conformations have been developed, the latter based upon the structures of the mammalian GLUT5 fructose transporter (20). These models have been useful in understanding the structural ramifications of functional studies in the absence of a crystal structure of the PCFT protein.
A recent study from this laboratory demonstrated the critical role that mutations at Asp-109, located within a GXXXDXXGRR/K motif (Motif A) within the cytoplasmic loop between the second and third transmembrane segments, play in achieving the outward-open conformation (15). Using the substituted-cysteine accessibility method, the loss of function associated with substitutions at Asp-109 locked the protein in an inward-open conformation, as manifested by the loss of accessibility of residues that are accessible in WT PCFT to a water-soluble sulfhydryl reagent. The current study, focused on the role of Phe-392 in PCFT function, complements a report of a F392V mutation in cohort of Japanese subjects with HFM (21). Although a loss of function was observed in this report, the basis for the functional loss was not pursued, nor was the role this residue plays in PCFT function clarified. The current study demonstrates that Phe-392 is critical to sustaining oscillation of the PCFT protein among its conformational states and suggests that this is achieved by dampening the affinity of the protein for its folate substrates. Like residues in Motif A in the first intracellular loop (15), mutations at Phe-392 that result in a marked loss of function lock the protein in an inward-open conformation that can be substantially reversed by the introduction of second mutations within the protein.
Results
Characterization of a Val substitution at Phe-392
A recent report described a F392V PCFT mutation in a subject with HFM (21). Phe-392, one of twelve fully conserved (human, monkey, horse, mouse, rat, dog, bovine, opossum, Xenopus, and zebrafish) phenylalanine residues, is located in the 11th transmembrane segment near the cytoplasmic interface (Fig. 1, green fill). Fig. 2A illustrates the lack of [3H] MTX MTX transport activity when F392V PCFT was transiently transfected into HeLa cells that lack endogenous PCFT or reduced folate carrier activity, at concentrations an order of magnitude below and above the influx Kt, the latter reflecting the influx Vmax. A Western blotting analysis (Fig. 2B) indicates that expression of the mutant PCFT in the crude membrane fraction and at the cell surface is comparable with that of PCFT-WT. Hence, the stability of the protein and its trafficking to the plasma membrane is intact; loss of function is attributed entirely to an intrinsic defect in the protein. To further explore the structural requirements at this residue, particularly in view of the loss of function with a relatively conservative mutation, a variety of other substitutions were assessed as indicated in the next section.
Figure 1.
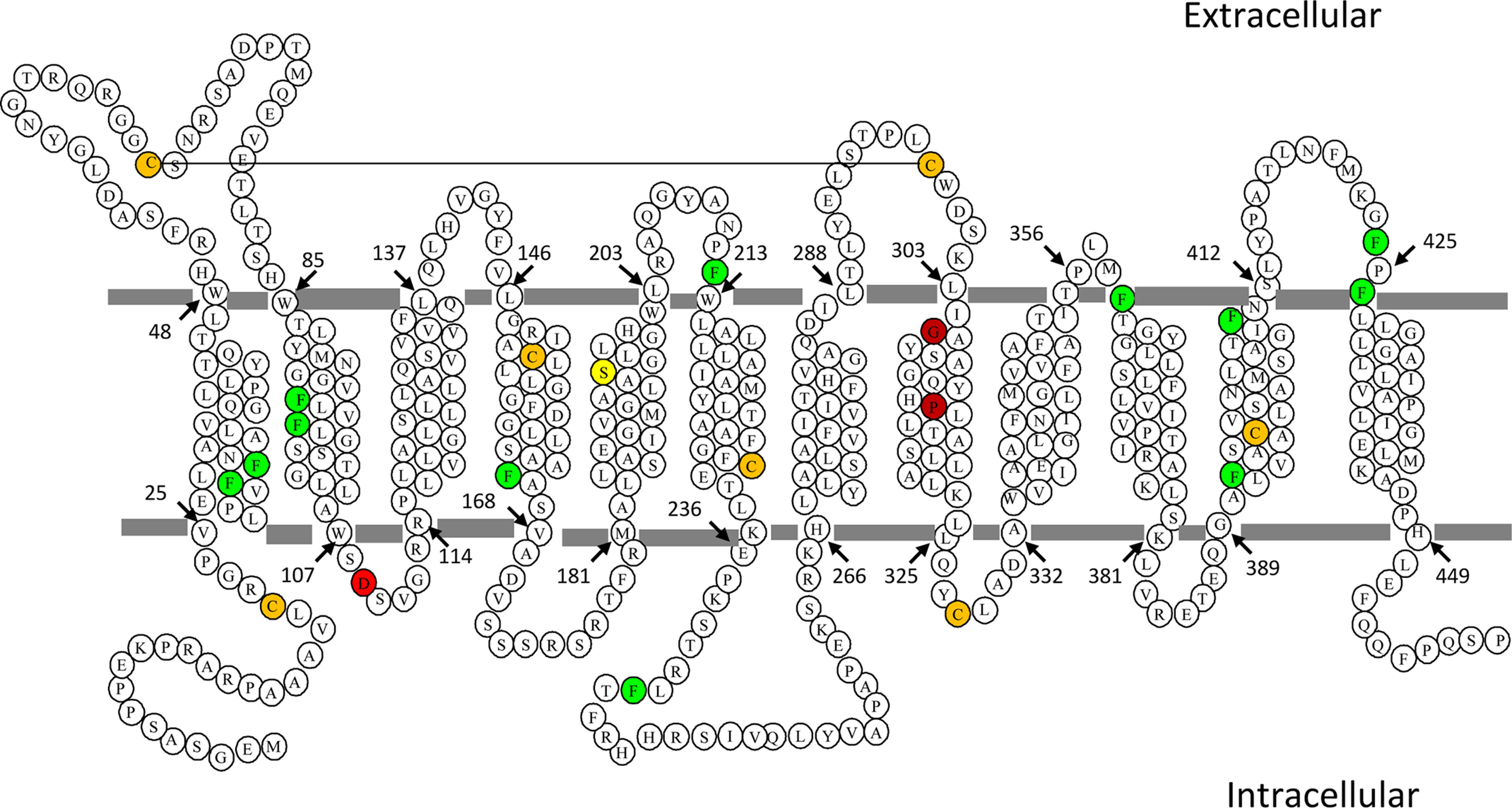
Location of phenylalanine and other residues relevant to the current study in human PCFT. Human PCFT has twelve transmembrane segments with its C terminus and N terminus oriented to the cytosol (22). The twelve fully conserved phenylalanine residues are noted in green whereas the seven native cysteine residues are noted in orange. The Asp-109, Gly-305, and Pro-314 residues are indicated in red; S196L is indicated in yellow. The solid line connecting Cys-66 and Cys-298 indicates the location of a disulfide bond in PCFT-WT (22).
Figure 2.
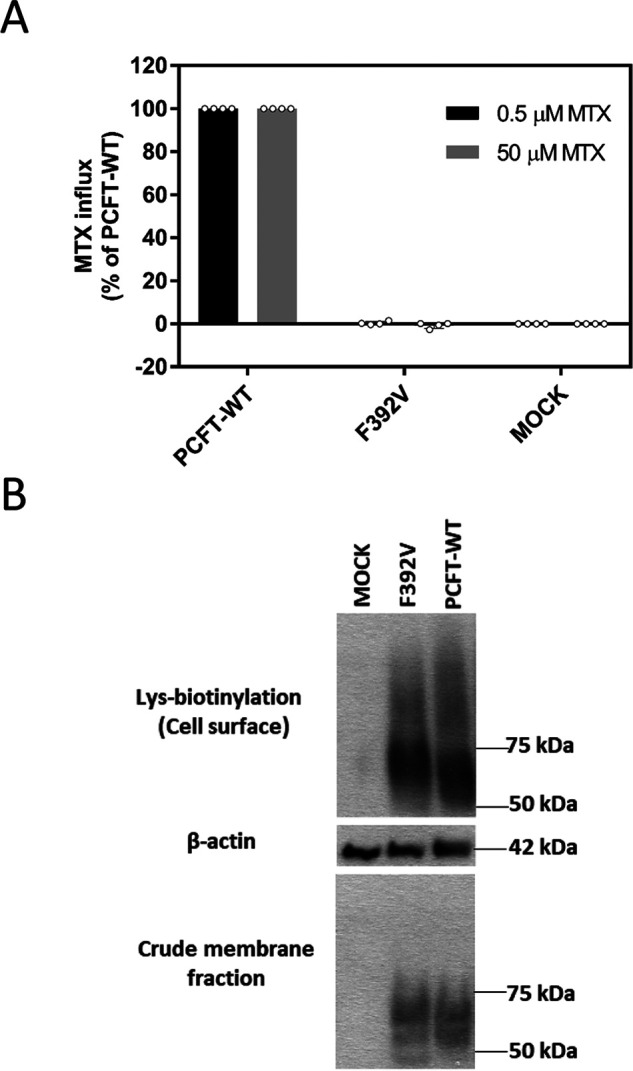
Function and expression of the F392V PCFT mutant. A, [3H] MTX influx was assessed in transient transfectants at pH 5.5 and 37 °C over 1 min at concentrations of 0.5 µm and 50 µm. Activity was normalized to PCFT-WT. Data are the mean ± S.D. from at least three independent experiments. B, the upper row indicates PCFT expression at the plasma membrane determined by biotinylation of lysine residues located at the exofacial region of the protein. The bottom row is PCFT expression in the crude membrane fraction. The middle row is actin expression, the loading control. The blots are representative of three independent experiments.
Impact of diverse substitutions at Phe-392 on PCFT expression and function
Structural requirements at the Phe-392 residue were evaluated by replacement with a spectrum of amino acids followed by assessment of expression and function. As indicated in Fig. 3A, all mutants were expressed at the cell surface at levels comparable with PCFT-WT. There was a complete loss of function (Fig. 3B) for all charged substitutions studied (Glu, Asp, and Lys) and Thr at the low and high concentrations; the His substitution retained 30% of PCFT-WT activity at the low concentration. Activity was also preserved primarily at the low concentration for Ala and residues with aromatic side chains (Ala, Trp, and Tyr). The specificity of the Phe-392 residue was further assessed with additional neutral substitutions. Again, none of the substitutions altered expression of mutants in the crude and membrane fractions (Fig. 3C). However, whereas there was marked loss of function with Leu, Ile, and Gln substitutions, there was complete retention of activity with the Met substitution (Fig. 3D).
Figure 3.

Function and expression of PCFT constructs with a spectrum of substitutions at Phe-392. A and C, the upper row is PCFT expression at the plasma membrane; the bottom row is PCFT expression in the crude membrane fraction. The middle row is the actin loading control. The blots are representative of three independent experiments. B and D, [3H] MTX influx was assessed in transient transfectants at pH 5.5 and 37 °C over 1 min at concentrations of 0.5 µm and 50 µm. Activity was normalized to that of PCFT-WT. Data are the mean ± S.D. from at least three independent experiments. *p < 0.05 based upon one-way analysis of variance comparing the mutants to PCFT-WT. E, a representative analysis of [3H] MTX influx kinetics mediated by PCFT-WT and the F392H mutant. Influx was determined at pH 5.5 over 1 min in transient transfectants. The data are representative of three independent experiments. F, MTSEA-biotin labeling of F392C in the absence or presence of membrane permeabilization by digitonin in a cysteine-less PCFT construct (-CL) with PCFT-CL and T240C-CL serving as negative and positive controls, respectively. PCFT expression in the crude membrane fraction is indicated in the bottom row. The images are representative of three independent experiments.
Influx kinetic changes associated with the Phe-to-His substitution
To further explore the basis for the loss of function with substitutions at Phe-392, influx kinetics were determined for F392H PCFT. This mutant was chosen because although function was markedly impaired, it was sufficient for accurate influx measurements over a broad concentration range. As illustrated in Fig. 3E and indicated in Table 1, the [3H] MTX influx Vmax for F392H PCFT was decreased 15-fold as compared with PCFT-WT, consistent with markedly impaired oscillation of the carrier, whereas the influx Kt was decreased 4.5-fold. The decrease in influx Kt for F392H can be attributed at least in part to an increase in the affinity of the transporter for this substrate as indicated by the much lower influx Ki relative to PCFT-WT for MTX (Table 1). The loss of function was much greater at the high versus low MTX concentrations for all the substitutions at Phe-392 where some function was retained. This is consistent with a decrease in influx Kt because a decrease in Vmax alone should result in a proportionate decrease in influx irrespective of substrate concentration. Hence, changes in influx kinetics observed with F392H likely reflect the pattern of change in influx for all mutant PCFTs that retain partial activity.
Table 1.
MTX influx kinetics for PCFT-WT and the F392H mutant
| Influx measurements | F392H | PCFT-WT | F392H/ PCFT-WT |
|---|---|---|---|
| MTX Kt (µm) | 0.65 ± 0.11 | 2.93 ± 0.59 | 4.5 ↓ |
| Vmax (pmol/mg protein/min) | 33 ± 5 | 497 ± 103 | 15 ↓ |
| MTX Ki (µm) | 0.55 ± 0.12 | 1.60 ± 0.27 | 2.9 ↓ |
Influx kinetic parameters (Vmax and Kt) were derived from the nonlinear regression best fit to the Michaelis-Menten equation. The MTX Ki was calculated for competitive inhibition based upon the inhibition of [3H] MTX influx by unlabeled MTX as described under “Experimental procedures”. MTX influx Vmax and Kt are the mean ± S.E. from four independent experiments. Influx Ki is the mean ± S.E. from at least four independent experiments.
Accessibility of Cys-substituted residues
Human PCFT has seven endogenous cysteine residues; of these, two (Cys-66 and Cys-298) are extracellular and form a spontaneous disulfide bond (Fig. 1, orange fill) (22). The remaining five cysteine residues are located either within the transmembrane segments or in the cytosol. Consistent with their location, the native cysteines lack accessibility to MTSEA-biotin (N-biotinyl aminoethyl methanethiosulfonate), a water-soluble sulfhydryl-reactive reagent that does not penetrate the cell membrane (22, 23). The cysteine residue in the F392C mutant was not accessible to MTSEA-biotin from the extracellular compartment, consistent with its location near the membrane-cytosol interface (not shown). However, this cysteine residue was labeled with MTSEA-biotin after permeabilization of the cell membrane. Hence, when F392C was introduced into a Cys-less PCFT scaffold (PCFT-CL), as indicated in Fig. 3F, the F392C-CL mutant was labeled only after the cells were permeabilized with digitonin, as was the case for the positive control, T240C-CL, located in the intracellular loop between the sixth and seventh transmembrane segments (22). Hence, the cysteine residue in the F392C-CL mutant is accessible only to the cytosol and appears to be located in the translocation pathway.
The impact of the Phe → Val mutation on the conformation of PCFT
The observation that F392V PCFT lost all function but the level of expression at the cell membrane was not altered as compared with PCFT-WT raised the possibility that this mutation may have locked the transporter in one of its conformational states, as observed for mutations in residues (Asp-109 and Gly-112) in the first intracellular loop (15). To evaluate this further, a second cysteine mutation, P314C, was introduced into the F392V mutant scaffold and function and accessibility assessed. Previous studies showed that (i) P314C is accessible to the membrane-impermeant MTSEA-biotin at room temperature, consistent with its accessibility to the extracellular milieu (18, 19); (ii) P314C accessibility is lost when PCFT is locked in the inward-open conformation (established when P314C was introduced into the D109A mutant); and (iii) P314C is highly active; indeed, its Vmax is ∼4- to 5-fold greater than that of PCFT-WT (18, 19, 24).
It can be seen in Fig. 4A that the P314C substitution alone markedly increased MTX influx at the saturating concentration, consistent with a marked increase in the Vmax. The much smaller change at the low MTX concentration is consistent with an increase in influx Kt as described above. However, when F392V was introduced into the P314C PCFT scaffold, influx was completely abolished. Fig. 4B indicates that introduction of P314C into the F392V scaffold does not alter PCFT expression in the crude membrane fraction or at the cell membrane. However, whereas the single Cys-substituted P314C was biotinylated by MTSEA-biotin, this was not the case when the Cys-substituted PCFT (P314C) was inserted into the F392V scaffold. Hence, the F392V mutation results in the loss of accessibility to MTSEA-biotin of the substituted cysteine located at P314C.
Figure 4.

The impact of the introduction of the Phe-392 mutations on the function and cysteine accessibility of a highly active P314C PCFT scaffold. A, [3H] MTX influx was assessed in transient transfectants at pH 5.5 and 37 °C over 1 min at concentrations of 0.5 µm and 50 µm. Activity was normalized to that of PCFT-WT. Data are the mean ± S.D. from at least three independent experiments. B, expression at the plasma membrane (upper row), cysteine biotinylation (middle row), and expression in the crude membrane fraction (bottom row). The black lines on the image represent repositioned lanes from the same Western blotting analysis. The image is representative of three independent analyses. C, [3H] MTX influx was assessed as described above. Activity was normalized to that of PCFT-WT. Data are the mean ± S.D. from at least three independent experiments. D, expression at the plasma membrane (upper row) and MTSEA-biotin biotinylation of the cysteine residue (middle row) of transient transfectants. Expression in the crude membrane fraction is indicated in the bottom row. The black line on the image represents a repositioned lane from the same Western blotting. The image is representative of three independent experiments.
To determine whether these changes are specific to the Val substitution, experiments were conducted with F392K and F392D as the inactivating residues (Fig. 3B). As indicated in Fig. 4C, when F392D or F392K were inserted into the P314C scaffold, function was lost. As indicated in Fig. 4D, expression of the double mutants at the cell surface and in the crude membrane extracts was comparable or slightly less than expression of the single mutants. However, whereas the single Cys-substituted P314C was biotinylated by MTSEA-biotin, accessibility of the Cys residue in the double mutants was lost. Taken together, these results imply that these loss-of-function mutations at Phe-392 also lock the protein in the inward-open conformation.
Impact of the introduction of the G305L substitution on F392V function and cysteine accessibility
In a prior study, introduction of G305L restored cysteine accessibility and partially restored function in the D109A/P314C and G112A/P314C double mutants that were locked in the inward-open conformation (15). Accordingly, studies were undertaken to determine whether introduction of G305L would reverse the F392V phenotype. G305L was introduced into (i) F392V to obtain G305L/F392V, (ii) P314C to obtain G305L/P314C, and (iii) P314C/P392V to obtain the G305L/P314C/F392V triple mutant. As indicated in Fig. 5A, introduction of G305L, which was itself partially active, restored function for F392V and more so for P314C/F392V PCFT. As indicated in Fig. 5B, expression at the cell surface of the P314C/F392V double mutant and the triple mutant were comparable, though less than P314C alone. However, whereas Cys-biotinylation of P314C/F392V was absent, accessibility of the Cys residue to MTSEA-biotin for both the P314C and G305L/P314C/F392V proteins was robust. P314C/G305L PCFT was less active than G305L alone or the other G305L mutants, even though expression was comparable with that of PCFT-WT (not shown).
Figure 5.

Suppression of the locked phenotype with the introduction of G305L. A, [3H] MTX influx was assessed in transient transfectants at pH 5.5 and 37 °C over 1 min at concentrations of 0.5 µm and 50 µm. Activity was normalized to that of PCFT-WT. The y axis is plotted in two segments to better visualize the low activity of some double mutants. Data are the mean ± S.D. from at least three independent experiments. B, cysteine accessibility of the P314C/F392V double and P314C/F392V/G305L triple mutants. The upper row is lysine biotinylation at the cell membrane. The middle rows are MTSEA biotinylation of the cysteine residue and actin. The bottom row is expression in the crude membrane extract. The image is representative of three independent experiments. C, the impact of the introduction of G305L on transport activity of the F392D and F392K mutants. [3H] MTX influx was normalized to that of PCFT-WT. Data are the mean ± S.D. from at least three independent experiments. D, expression of the G305L/F392D and G305L/F392K double mutants. The upper row is lysine biotinylation at the cell membrane. The middle row is the actin loading control. The bottom row is expression in the crude membrane extract. The image is representative of three independent experiments. E, the impact of the insertion of G305L on the transport activity of the P314C/F392D double mutant. [3H] MTX influx was assessed as described above. Activity was normalized to that of PCFT-WT. Data are the mean ± S.D. from at least three independent experiments. F, the impact of the insertion of G305L on the accessibility of the cysteine moiety of the P314C/F392D double mutant. The upper row is lysine biotinylation at the cell membrane. The middle rows are cysteine biotinylation and actin. The bottom row is expression in the crude membrane extract. The image is representative of three independent experiments.
Likewise, as indicated in Fig. 5C, both G305L/F392D and G305L/F392K mutants exhibited some restoration of function. The extent of restoration of function would be greater when corrected for the decrease in PCFT expression at the cell surface. Expression of the F392-K or -D double mutants was similar to that of the single mutants in the crude membrane fraction, although the expression of G305L/F392D and G305L/F392K at the cell membrane was lower than the respective single mutants (Fig. 5D).
Focusing on F392D PCFT, for which there was a greater increase in function as compared with F392K when G305L was added, the G305L mutation was introduced into the P314C/F392D scaffold to obtain triple substitutions in the same protein. As indicated in Fig. 5E, function was also detected for the G305L/P314C/F392D triple mutant. The activity of G305L/F392D was included for the purpose of comparison. Expression of the triple mutant at the cell surface and in the crude membrane extract was similar to that of the double mutant. Whereas the Cys residue in the P314C/F392D mutant was not accessible to MTSEA-biotin, the Cys-substituted residue in the triple mutant was biotinylated by this reagent. Hence, the G305L mutation partially reversed the inward-open conformation of the protein resulting from the F392D substitution in human PCFT (Fig. 5F).
Impact of the introduction of the S196L substitution on F392V function and cysteine accessibility
In the previous report (15), homology modeling localized Ser-196 in the fifth transmembrane segment in proximity to Gly-305, and introduction of S196L was also shown to partially restore the function and cysteine accessibility of G109A/P314C PCFT. Fig. 6A illustrates the impact of the introduction of the S196L mutation into several PCFT scaffolds. It can be seen that, like F392V, a S196L substitution alone resulted in the loss of function, in contrast to G305L. However, the S196L/F392V double PCFT mutant was functional. Hence, the combination of two inactivating mutations resulted in restoration of function. Likewise, the S196L/P314C/F392V triple mutant was functional. As indicated in Fig. 6B, expression of S196L in the crude membrane fraction and at the cell membrane was comparable with that of PCFT-WT, F392V, and P314/F392V. Expression of S196L/F392V and the triple mutant were markedly decreased. When the decreased expression is taken into consideration, the restoration of function for both mutants would be even greater. When the accessibility to MTSEA-biotin of the various constructs containing P314C PCFT was assessed, only the triple mutant was detected despite the low level of expression at the cell membrane. Interestingly, the S196L mutant was also detected with MTSEA-biotin, although the signal was lost in the S196L/F392V double mutant that was functional. A possible basis for these observations is considered below.
Figure 6.
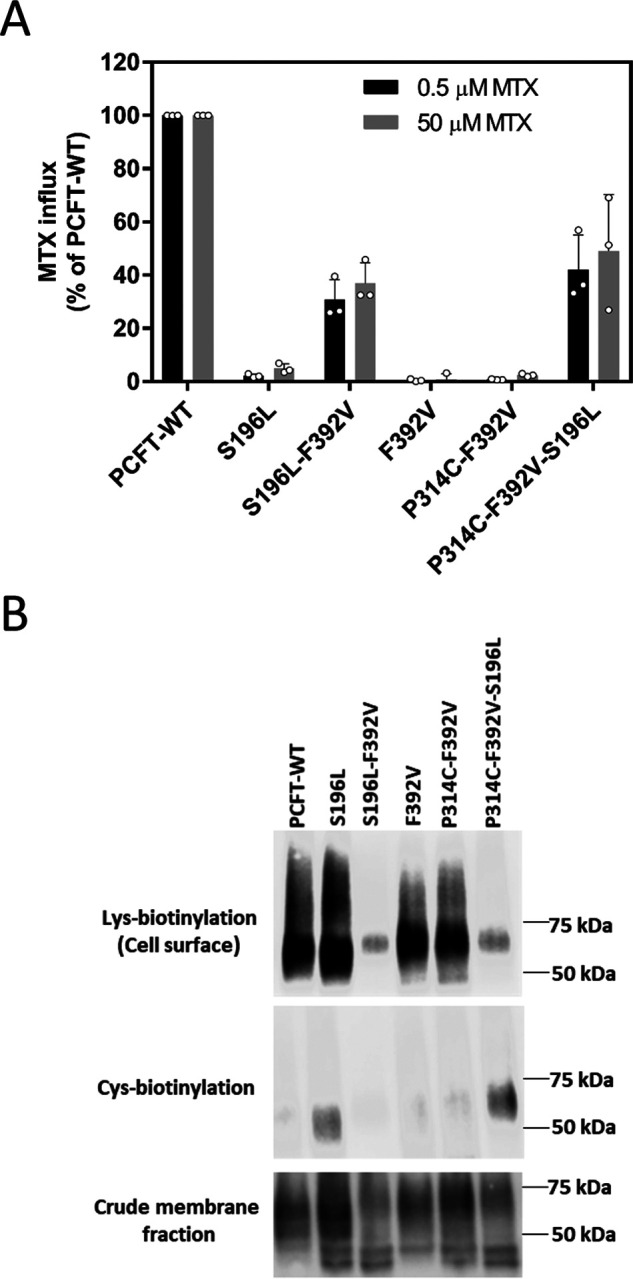
Suppression of the locked phenotype with the introduction of S196L. A, [3H] MTX influx was assessed in transient transfectants at pH 5.5 and 37 °C over 1 min at concentrations of 0.5 µm and 50 µm. Activity was normalized to that of PCFT-WT. Data are the mean ± S.D. from at least three independent experiments. B, expression of PCFT mutants. The upper row is lysine biotinylation at the cell membrane. The middle row is cysteine biotinylation. The bottom row is expression in the crude membrane extracts. The image is representative of three independent experiments.
Discussion
Deciphering the functional role of PCFT residues mutated in HFM has contributed important insights into the properties of this transporter. A N411K mutation was localized within the external gate of the PCFT translocation pathway (14, 17). A P425R mutation revealed a difference in the binding pocket for MTX versus pemetrexed (25). R113-S and -C mutations pointed to an important role for the first intracellular loop in PCFT function, subsequently established with the observation that mutations in this region lock the protein in an inward-open conformation (7, 15, 26). An R376Q mutation suggested that this residue influences protein coupling and/or binding (27). The loss of function associated with a F392V mutation within the 11th transmembrane segment in subjects with HFM (21) has in this report identified this residue as critical to oscillation of the protein among its conformational states and, in particular, essential for achieving the outward-open formation.
Homology models of PCFT based upon the structures of the bovine and rodent GLUT5 fructose transporters (20) have provided insights into the structural basis for functional changes that occur with site-directed mutations of PCFT or mutations that occur in subjects with HFM (14). Exofacial residues associated with an external gate, which opens and closes as the carrier cycles, were previously defined and characterized; two are located in the 11th transmembrane segment (Ser-407 and Asn-411) (17). The latter, mutated (N411K) in a subject with HFM, resulted in markedly impaired function due largely to a decrease in influx Vmax (14). The homology model predicts that Phe-392 within the endofacial region of the helix undergoes substantial movement as the carrier shifts among its conformation states, as illustrated in Fig. 7. The Phe-392 side chain points into the aqueous translocation pathway in the inward-open configuration and moves further into the pathway as the protein shifts to the outward-open conformation. Hence, this residue is located within a region of the protein that can have a major impact on carrier function.
Figure 7.

Homology models of PCFT in the inward and outward conformations highlighting the Phe-392, Asp-109, Gly-305 and Ser-196 residues. Homology models of human PCFT in the outward-open (blue) and inward-open (green) conformations were generated based upon the crystal structures of the bovine and rat GLUT5 fructose transporters as described previously (18). Phe-392, located in the endofacial region of the 11th transmembrane segment (highlighted) points into the aqueous translocation pathway in the inward-open conformation and shifts further into the pathway in the outward-open conformation. Asp-109 in the second transmembrane segment is in proximity to Phe-392. Ser-196 in the fifth transmembrane segment is located in close proximity to Gly-305 in the exofacial region of the eighth transmembrane segment. A, planar view. B, a view into the protein from the extracellular compartment. C, a view into the protein from the intracellular compartment.
The data indicate that substitutions at Phe-392 (→ Val, → Lys, or → Asp) that result in a loss of function lock the protein in an inward-open conformation. This is based upon the loss of function and accessibility of the Cys residue when the highly active and fully accessible P314C in PCFT-WT is introduced into a variety of PCFT scaffolds: F392V/P314C, F392D/P314C, and F392K/P314C. This is similar to what was observed for residues (Asp-109 and Gly-112) in the first intracellular loop of PCFT (15). In the case of Asp-109 mutants, no substitution could be identified, including glutamate, in which there was any residual function (28). The Asp-109 and Gly-112 residues are located in a region of the protein characterized by Motif A (GXXXDXXGR(R/K)), which plays a key role in the function of solute transporters (29, 30). Whereas Phe-392 is not located in a region with a known predictive motif, recent structural analyses of the Escherichia coli YajR proton-coupled transporter indicate that there is an inter-domain helical bundle between the 11th and second transmembrane segments that is necessary to achieve the outward-facing conformation, and when residues in this region are disrupted, the protein stabilizes in its inward-open conformation (31). In the homology model of PCFT, Asp-109 in the loop between the second and third transmembrane segments is predicted to form a charge-pole interaction with the N-terminal end of the 11th transmembrane segment, further stabilizing the outward-facing conformation (Fig. 7). This may account for the similar phenotypes when substitutions are made in the Phe-392, Asp-109, or Gly-112 residues (15).
Some aspects of the structural requirements at Phe-392 are expected, as with the complete loss of function when this residue, located in the cell membrane, is replaced with charged (Asp, Arg, and Lys) or polar (Ser, Thr, and Gln) amino acids. Although it might be expected that aromatic residues would be preferred, there was only a low level of activity retained with these substitutions (Trp and Tyr). The F392M substitution was the only one that retained the full function of PCFT. The Met residue has the same hydrophobicity and exact same reach (length) as Phe. Other hydrophobic residues, with smaller lengths (Ala, Val, Ile, Leu, and Cys), were either inactive or retained only a small fraction of activity. Aromatic residues with longer lengths and less hydrophobic features than Phe (such as Tyr, which is larger and has a polar hydroxyl group, and Trp, which is hydrophobic but significantly larger than Phe), retained only a low level of activity. The modest preservation of activity for the F392H mutant is likely related to its hydrophobic aromatic imidazole side chain. These mutational studies suggest that the reach (length) and hydrophobicity of the Phe-392 residue are the two important features that must be maintained for full activity.
These studies also demonstrate that substitutions that lock Phe-392 in the inward-open conformation can be reversed, or “suppressed” in terms of function and accessibility to the extracellular compartment, by introduction of G305L (Fig. 7), which was shown to substantially reverse both parameters for the D109A and to a lesser extent the G112A locked forms (15). A similar “suppression” phenomenon has been observed for the loss of function of other transporters with mutations in Motif A (32–34). Based upon the homology model, G305L is located in the exofacial region of the eighth transmembrane segment remote from the 11th transmembrane segment in either conformation. As suggested for its impact on substitutions of residues between the second and third transmembrane segments, it is likely that substitutions with large residues at the Gly-305 position modify a latch-like conformational change because of mutations of Phe-392, Asp-109, or Gly-112 that lock the protein in an inward-open conformation. The previous study also demonstrated that introduction of a S196L substitution in the fifth transmembrane segment, predicted by homology modeling to be in close proximity to Gly-305 (Fig. 7), partially restored D109A function. The impact of the S196L substitution on F392V function was more complex. First, S196L PCFT alone had virtually no activity. However, S196L/F392V was active. Hence, combining two inactivating mutations overcame the structural change induced by either mutation and restored the function of the protein consistent with near-restoration of the WT protein structure. A “mutually compensatory mutation” as observed in this study, generated not by selection but based upon structural considerations, is highly fortuitous. These mutations are rare in general but occur during evolutionary selection in humans and microorganisms (35–37). Not only was S196L/F392V/P314C active, but the Cys moiety was now accessible to MTSEA-biotin and the extracellular compartment consistent with reversal of the locked conformation. Unexpected was the observation that the S196L substitution alone resulted in a signal when cells were exposed to MTSEA-biotin, suggesting that the structural change produced by this mutation made one or more of the native Cys residues accessible. Consistent with this was the loss of the signal when the protein contained both S196L and F392V substitutions and function was restored, presumably because of restoration to a more normal structure.
The data revealed that substitutions at Phe-392 resulted in a marked increase in the affinity of the protein for its folate substrates along with a decrease in the influx Vmax. Although the Kt obtained for a solute transporter is a complex term that may be influenced by more than the binding constant, the concurrent Ki determination confirmed that the Kt change largely reflected alterations in the affinity of PCFT for its substrates. The data indicate that the increase in substrate binding at this residue is associated with a decrease in the mobility of the protein. The data suggest a paradigm in which the integrity of the Phe-392 residue in PCFT-WT is critical to maintaining a high level of flexibility of the transporter, and this requires that the protein is in a reduced state for its substrates. With substitutions at this site, the affinity for folate substrates is increased and the protein becomes more constrained; this results in a marked fall in the rate at which the carrier oscillates among its conformational states, and when the loss of function is complete as occurs with some substitutions, the protein becomes stabilized in its inward-open conformation. This relationship between binding affinity and flexibility has been observed with an E. coli lactose permease mutant (38, 39) and the evolution of high-affinity antibodies (40). The marked inverse changes in influx kinetic parameters for a spectrum of substrates of the organic cation transporter 2 were attributed to differences in the rate of dissociation of bound substrates from the protein limiting the maximum rate of carrier cycling (41). The kinetic and binding changes observed for the Phe-392 mutants contrasts with marked increases in influx Kt, Ki, and Vmax observed for the P314C and Y315C PCFT mutations located at a breakpoint in the eighth transmembrane segment (18, 24). Following the same paradigm, these residues maintain the protein in a high-affinity state for its folate/antifolate substrates and thereby constrain the mobility of the protein. When mutated, binding is diminished and the carrier relaxes, allowing a greater degree of flexibility of the protein and a higher rate of conformational change as the carrier transports its substrates into and out of the cell.
Experimental procedures
Chemicals
[3′,5′,7-3H] MTX and [3H] pemetrexed were obtained from Moravek Biochemicals, Inc. EZ-Link Sulfo-NHS-LC-biotin[sulfosuccinimidyl-6-(biotinamido) hexanoate], streptavidin agarose beads, and dithiothreitol were obtained from Thermo Fisher Scientific. The sulfhydryl-reactive MTSEA-biotin was obtained from Biotium. Protease inhibitor mixture was purchased from Roche Applied Science.
Cell lines, culture conditions, and transient transfection
The HeLa-R1-11 cell line, which lacks PCFT and reduced folate carrier expression, was the recipient for all transient transfections (9, 42). These cells were maintained in RPMI 1640 medium under 5% CO2 with 10% FBS (Gemini Bio-Products), 100 units/ml penicillin, and 100 μg streptomycin. For transport studies, 3 × 105 cells were seeded in 17-mm glass vials (Research Products International); for Western blotting analyses, 5 × 105 cells/well were seeded in six-well plates. 2 days later, the cells were transfected with PCFT constructs (0.8 μg/vial or 1.6 μg/well, respectively) using lipofectamine 2000 (Invitrogen) in serum- and antibiotic-free RPMI 1640 medium.
Mutants and site-directed mutagenesis
Most of the mutations were introduced into a PCFT-WT template unless specified with suffix “CL”, which indicates a cysteine-less template lacking all seven native cysteine residues (17, 22). The P314C PCFT mutant was described previously (18, 19, 24). Single substitutions were introduced individually on the appropriate PCFT template, and additional mutations were introduced into the single or double mutants using the Quickchange II XL site-directed mutagenesis kit (Stratagene California). All PCFT mutants were tagged at the C terminus with HA. The coding sequence of all PCFT mutants was verified at the Albert Einstein Cancer Center Genomics and Computational Analysis Shared Resource.
Membrane transport
The cells were washed with HBS (HEPES-buffered saline: 20 mm HEPES, 140 mm NaCl, 5 mm KCl, 2 mm MgCl2, and 5 mm dextrose, pH 7.4) and pre-incubated in this buffer for 20 min at 37 °C before initiating uptake of [3H] MTX (a folate surrogate, specifically labeled, stable, and relatively inexpensive) over 1 min at 37 °C (an initial rate). Transport was stopped by the addition of ice-cold HBS; the cells were then washed three times with ice-cold HBS, then dissolved in 0.2 m NaOH (0.5 ml) by heating at 65 °C. One portion (0.4 ml) was assayed on a liquid scintillation spectrometer; 20 μl was used for the BCA protein determination (Thermo Fisher Scientific). MTX influx Kt and Vmax were determined by nonlinear regression analysis of influx as a function of the extracellular substrate concentration based on the Michaelis-Menten equation using prism software (Version 7 for Windows; GraphPad Software). MTX Ki was calculated from the Michaelis-Menten equation for competitive inhibition, . This was based upon the inhibition of [3H] MTX influx by unlabeled MTX, where I is the concentration of inhibitor, S is concentration of [3H] MTX, Kt is the concentration at which influx is half of the maximum rate (Vmax), and f is the fraction of influx in the presence/absence of the inhibitor.
Analysis of PCFT at the cell surface and accessibility of Cys-substituted residues by biotinylation
PCFT expression at the plasma membrane was assessed with EZ-Link Sulfo-NHS-LC-biotin, which is membrane impermeant and targets lysine residues accessible to the extracellular compartment. Accessibility of Cys-substituted residues was probed with the membrane-impermeant sulfhydryl-reactive MTSEA-biotin (17, 22). For both types of biotinylation, transient transfectants were incubated with sulfo-NHS-LC-biotin (0.5 mg/ml) or MTSEA-biotin (0.2 mg/ml) in HBS at room temperature for 30 min, following which the cells were washed twice in HBS at room temperature before the addition of 0.7 ml of ice-cold hypotonic buffer (0.5 mm Na2HPO4 and 0.1 mm EDTA, pH 7.0) containing protease inhibitor mixture. The cells were then detached from the plates with a disposable cell lifter and centrifuged at 16,000 × g for 10 min at 4 °C. The pellets were dissolved in 0.4 ml of lysis buffer (50 mm Tris-base, 150 mm NaCl, 1% Nonidet P-40, and 0.5% sodium deoxycholate, pH 7.4), and 50 μl was collected as a crude membrane sample for assay of total PCFT protein. The remaining suspension was mixed on a rotisserie for 0.5–1 h at 4 °C before centrifugation at 16,000 × g for 15 min at 4 °C. The supernatant was mixed on a rotisserie overnight at 4 °C with 50 μl of pre-washed streptavidin agarose beads. The beads were then washed twice with lysis buffer and an additional two times with lysis buffer containing 2% SDS, each with a 20-min mix on a rotisserie at room temperature. Protein bound to the beads was released by heating at 100 °C for 5 min in 2× SDS-PAGE sample loading buffer with dithiothreitol (70 μl).
The crude membrane fractions, after mixing (1:1) with the 2× SDS-PAGE sample loading buffer at room temperature and release of surface proteins from the beads, were resolved on 4–20% SDS-PAGE (Bio-Rad). Proteins were transferred to an Immobilon-P Transfer Membrane (Millipore) and were blocked with 10% dry milk in Tris-buffered saline with Tween (20 mm Tris, 135 mm NaCl, and 1% Tween 20, pH 7.6) overnight at 4 °C. The blots were probed with polyclonal anti-HA antibody (Sigma-Aldrich, H6908) or monoclonal anti-actin antibody (Sigma-Aldrich, A5441), followed by a second antibody-HRP conjugate (Cell Signaling Technology). The blots were developed with Western Lightning Plus-ECL (PerkinElmer). For assessment of biotinylation following membrane permeabilization, transient transfectants were incubated with digitonin (50 μg/ml) in HBS for 5 min before MTSEA-biotin labeling.
Homology models
Homology models for human PCFT in the inside-open and outside-open conformations were developed based upon the recently solved bovine and rat GLUT5 templates (20) in the outward-open and inward-open conformations, respectively, as recently reported in detail (14, 15, 18, 19).
Statistical Analysis
Statistical analyses were performed with GraphPad Prism version 7 applying one-way analysis of variance or the multiple t test.
Data availability
All data are contained within the article.
Author contributions—H.-Q. Z., M. N., I. D. G., and R. Z. conceptualization; H.-Q. Z., M. N., K. L., A. F., and R. Z. data curation; H.-Q. Z., M. N., K. L., S. A., A. F., I. D. G., and R. Z. formal analysis; H.-Q. Z. and I. D. G. validation; H.-Q. Z., M. N., K. L., S. A., A. F., I. D. G., and R. Z. methodology; H.-Q. Z., I. D. G., and R. Z. writing-original draft; H.-Q. Z., I. D. G., and R. Z. writing-review and editing; A. F. and I. D. G. funding acquisition; I. D. G. supervision.
Funding and additional information—This work was supported by National Institutes of Health Grants CA082621 (to I. D. G.) and GMI18709 (to A. F.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- PCFT
- proton-coupled folate transporter
- HFM
- hereditary folate malabsorption
- CL
- Cys-less
- MTX
- methotrexate
- MTSEA-biotin
- N-biotinyl aminoethyl methanethiosulfonate
- HBS
- HEPES-buffered saline.
References
- 1. Qiu A., Jansen M., Sakaris A., Min S. H., Chattopadhyay S., Tsai E., Sandoval C., Zhao R., Akabas M. H., and Goldman I. D. (2006) Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption. Cell 127, 917–928 10.1016/j.cell.2006.09.041 [DOI] [PubMed] [Google Scholar]
- 2. Zhao R., Aluri S., and Goldman I. D. (2017) The proton-coupled folate transporter (PCFT-SLC46A1) and the syndrome of systemic and cerebral folate deficiency of infancy: hereditary folate malabsorption. Mol. Aspects Med. 53, 57–72 10.1016/j.mam.2016.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Qiu A., Min S. H., Jansen M., Malhotra U., Tsai E., Cabelof D. C., Matherly L. H., Zhao R., Akabas M. H., and Goldman I. D. (2007) Rodent intestinal folate transporters (SLC46A1): secondary structure, functional properties, and response to dietary folate restriction. Am. J. Physiol. Cell Physiol. 293, C1669–C1678 10.1152/ajpcell.00202.2007 [DOI] [PubMed] [Google Scholar]
- 4. Anderson C. M., and Thwaites D. T. (2010) Hijacking solute carriers for proton-coupled drug transport. Physiology (Bethesda) 25, 364–377 10.1152/physiol.00027.2010 [DOI] [PubMed] [Google Scholar]
- 5. Thwaites D. T. (2013) Go with the flow - membrane transport in the gut. Curr. Opin. Pharmacol. 13, 843–846 10.1016/j.coph.2013.09.019 [DOI] [PubMed] [Google Scholar]
- 6. Visentin M., Diop-Bove N., Zhao R., and Goldman I. D. (2014) The intestinal absorption of folates. Annu. Rev. Physiol. 76, 251–274 10.1146/annurev-physiol-020911-153251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhao R., Min S. H., Qiu A., Sakaris A., Goldberg G. L., Sandoval C., Malatack J. J., Rosenblatt D. S., and Goldman I. D. (2007) The spectrum of mutations in the PCFT gene, coding for an intestinal folate transporter, that are the basis for hereditary folate malabsorption. Blood 110, 1147–1152 10.1182/blood-2007-02-077099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kronn D., and Goldman I. D. (2017) Hereditary Folate Malabsorption. in GeneReviews [Internet] (Pagon R. A., Adam M. P., Ardinger H. H., Wallace S. E., Amemiya A., Bean L. J. H., Bird T. D., Dolan C. R., Fong C. T., Smith R. J. H., and Stephens K., eds.) pp. 1993–2020, University of Washington, Seattle, WA: [PubMed] [Google Scholar]
- 9. Zhao R., Gao F., Hanscom M., and Goldman I. D. (2004) A prominent low-pH methotrexate transport activity in human solid tumor cells: Contribution to the preservation of methotrexate pharmacologic activity in HeLa cells lacking the reduced folate carrier. Clin. Cancer Res. 10, 718–727 10.1158/1078-0432.ccr-1066-03 [DOI] [PubMed] [Google Scholar]
- 10. Desmoulin S. K., Wang L., Hales E., Polin L., White K., Kushner J., Stout M., Hou Z., Cherian C., Gangjee A., and Matherly L. H. (2011) Therapeutic targeting of a novel 6-substituted pyrrolo[2,3-d]pyrimidine thienoyl antifolate to human solid tumors based on selective uptake by the proton-coupled folate transporter. Mol Pharmacol. 80, 1096–1107 10.1124/mol.111.073833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhao R., Qiu A., Tsai E., Jansen M., Akabas M. H., and Goldman I. D. (2008) The proton-coupled folate transporter: impact on pemetrexed transport and on antifolate activities as compared with the reduced folate carrier. Mol. Pharmacol. 74, 854–862 10.1124/mol.108.045443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Desmoulin S. K., Hou Z., Gangjee A., and Matherly L. H. (2012) The human proton-coupled folate transporter: biology and therapeutic applications to cancer. Cancer Biol. Ther. 13, 1355–1373 10.4161/cbt.22020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Matherly L. H., Hou Z., and Gangjee A. (2018) The promise and challenges of exploiting the proton-coupled folate transporter for selective therapeutic targeting of cancer. Cancer Chemother. Pharmacol. 81, 1–15 10.1007/s00280-017-3473-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Aluri S., Zhao R., Lubout C., Goorden S. M. I., Fiser A., and Goldman I. D. (2018) Hereditary folate malabsorption due to a mutation in the external gate of the proton-coupled folate transporter-SLC46A1. Blood Advances 2, 61–69 10.1182/bloodadvances.2017012690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Aluri S., Zhao R., Lin K., Shin D. S., Fiser A., and Goldman I. D. (2019) Substitutions that lock and unlock the proton-coupled folate transporter (PCFT-SLC46A1) in an inward-open conformation. J. Biol. Chem. 294, 7245–7258 10.1074/jbc.RA118.005533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lasry I., Berman B., Straussberg R., Sofer Y., Bessler H., Sharkia M., Glaser F., Jansen G., Drori S., and Assaraf Y. G. (2008) A novel loss of function mutation in the proton-coupled folate transporter from a patient with hereditary folate malabsorption reveals that Arg 113 is crucial for function. Blood 112, 2055–2061 10.1182/blood-2008-04-150276 [DOI] [PubMed] [Google Scholar]
- 17. Zhao R., Najmi M., Fiser A., and Goldman I. D. (2016) Identification of an extracellular gate for the proton-coupled folate transporter (SLC46A1) by cysteine cross-linking. J. Biol. Chem. 291, 8162–8172 10.1074/jbc.M115.693929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Aluri S., Zhao R., Fiser A., and Goldman I. D. (2017) Residues in the eighth transmembrane domain of the proton-coupled folate transporter (SLC46A1) play an important role in defining the aqueous translocation pathway and in folate substrate binding. Biochim. Biophys. Acta 1859, 2193–2202 10.1016/j.bbamem.2017.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Aluri S., Zhao R., Fiser A., and Goldman I. D. (2018) Substituted-cysteine accessibility and cross-linking identify an exofacial cleft in the 7th and 8th helices of the proton-coupled folate transporter (SLC46A1). Am. J. Physiol. Cell Physiol. 314, C289–C296 10.1152/ajpcell.00215.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nomura N., Verdon G., Kang H. J., Shimamura T., Nomura Y., Sonoda Y., Hussien S. A., Qureshi A. A., Coincon M., Sato Y., Abe H., Nakada-Nakura Y., Hino T., Arakawa T., Kusano-Arai O., et al. (2015) Structure and mechanism of the mammalian fructose transporter GLUT5. Nature 526, 397–401 10.1038/nature14909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tozawa Y., Abdrabou S. S. M. A., Nogawa-Chida N., Nishiuchi R., Ishida T., Suzuki Y., Sano H., Kobayashi R., Kishimoto K., Ohara O., Imai K., Naruto T., Kobayashi K., Ariga T., and Yamada M. (2019) A deep intronic mutation of c.1166-285 T > G in SLC46A1 is shared by four unrelated Japanese patients with hereditary folate malabsorption (HFM). Clin. Immunol. 208, 108256 10.1016/j.clim.2019.108256 [DOI] [PubMed] [Google Scholar]
- 22. Zhao R., Unal E. S., Shin D. S., and Goldman I. D. (2010) Membrane topological analysis of the proton-coupled folate transporter (PCFT-SLC46A1) by the substituted cysteine accessibility method. Biochemistry 49, 2925–2931 10.1021/bi9021439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shin D. S., Zhao R., Fiser A., and Goldman I. D. (2013) Role of the fourth transmembrane domain in proton-coupled folate transporter function as assessed by the substituted cysteine accessibility method. Am. J. Physiol. Cell Physiol. 304, C1159–C1167 10.1152/ajpcell.00353.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Visentin M., Unal E. S., Najmi M., Fiser A., Zhao R., and Goldman I. D. (2015) Identification of Tyr residues that enhance folate substrate binding and constrain oscillation of the proton-coupled folate transporter (PCFT-SLC46A1). Am. J. Physiol. Cell Physiol. 308, C631–C641 10.1152/ajpcell.00238.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shin D. S., Zhao R., Yap E. H., Fiser A., and Goldman I. D. (2012) A P425R mutation of the proton-coupled folate transporter causing hereditary folate malabsorption produces a highly selective alteration in folate binding. Am. J. Physiol. Cell Physiol. 302, C1405–C1412 10.1152/ajpcell.00435.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lasry I., Berman B., Glaser F., Jansen G., and Assaraf Y. G. (2009) Hereditary folate malabsorption: a positively charged amino acid at position 113 of the proton-coupled folate transporter (PCFT/SLC46A1) is required for folic acid binding. Biochem. Biophys. Res. Commun. 386, 426–431 10.1016/j.bbrc.2009.06.007 [DOI] [PubMed] [Google Scholar]
- 27. Mahadeo K., Diop-Bove N., Shin D., Unal E., Teo J., Zhao R., Chang M. H., Fulterer A., Romero M. F., and Goldman I. D. (2010) Properties of the Arg376 residue of the proton-coupled folate transporter (PCFT-SLC46A1) and a glutamine mutant causing hereditary folate malabsorption. Am. J. Physiol. Cell Physiol. 299, C1153–C1161 10.1152/ajpcell.00113.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shin D. S., Min S. H., Russell L., Zhao R., Fiser A., and Goldman I. D. (2010) Functional roles of aspartate residues of the proton-coupled folate transporter (PCFT; SLC46A1); a D156Y mutation causing hereditary folate malabsorption. Blood 116, 5162–5169 10.1182/blood-2010-06-291237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Griffith J. K., Baker M. E., Rouch D. A., Page M. G., Skurray R. A., Paulsen I. T., Chater K. F., Baldwin S. A., and Henderson P. J. (1992) Membrane transport proteins: implications of sequence comparisons. Curr. Opin. Cell Biol. 4, 684–695 10.1016/0955-0674(92)90090-Y [DOI] [PubMed] [Google Scholar]
- 30. Jessen-Marshall A. E., Paul N. J., and Brooker R. J. (1995) The conserved motif, GXXX(D/E)(R/K)XG[X](R/K)(R/K), in hydrophilic loop 2/3 of the lactose permease. J. Biol. Chem. 270, 16251–16257 10.1074/jbc.270.27.16251 [DOI] [PubMed] [Google Scholar]
- 31. Jiang D., Zhao Y., Wang X., Fan J., Heng J., Liu X., Feng W., Kang X., Huang B., Liu J., and Zhang X. C. (2013) Structure of the YajR transporter suggests a transport mechanism based on the conserved motif A. Proc. Natl. Acad. Sci. U. S. A. 110, 14664–14669 10.1073/pnas.1308127110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yamaguchi A., Inagaki Y., and Sawai T. (1995) Second-site suppressor mutations for the Asp-66–>Cys mutant of the transposon Tn10-encoded metal-tetracycline/H+ antiporter of Escherichia coli. Biochemistry 34, 11800–11806 10.1021/bi00037a018 [DOI] [PubMed] [Google Scholar]
- 33. Jessen-Marshall A. E., Parker N. J., and Brooker R. J. (1997) Suppressor analysis of mutations in the loop 2–3 motif of lactose permease: evidence that glycine-64 is an important residue for conformational changes. J. Bacteriol. 179, 2616–2622 10.1128/jb.179.8.2616-2622.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cain S. M., Matzke E. A., and Brooker R. J. (2000) The conserved motif in hydrophilic loop 2/3 and loop 8/9 of the lactose permease of Escherichia coli. Analysis of suppressor mutations. J. Membr. Biol. 176, 159–168 10.1007/s00232001085 [DOI] [PubMed] [Google Scholar]
- 35. Hartl D. L., and Taubes C. H. (1996) Compensatory nearly neutral mutations: selection without adaptation. J. Theor. Biol. 182, 303–309 10.1006/jtbi.1996.0168 [DOI] [PubMed] [Google Scholar]
- 36. Chua P. K., Wen Y. M., and Shih C. (2003) Coexistence of two distinct secretion mutations (P5T and I97L) in hepatitis B virus core produces a wild-type pattern of secretion. J. Virol. 77, 7673–7676 10.1128/jvi.77.13.7673-7676.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mateu M. G., and Fersht A. R. (1999) Mutually compensatory mutations during evolution of the tetramerization domain of tumor suppressor p53 lead to impaired hetero-oligomerization. Proc. Natl. Acad. Sci. U. S. A. 96, 3595–3599 10.1073/pnas.96.7.3595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Smirnova I. N., and Kaback H. R. (2003) A mutation in the lactose permease of Escherichia coli that decreases conformational flexibility and increases protein stability. Biochemistry 42, 3025–3031 10.1021/bi027329c [DOI] [PubMed] [Google Scholar]
- 39. Nie Y., Smirnova I., Kasho V., and Kaback H. R. (2006) Energetics of ligand-induced conformational flexibility in the lactose permease of Escherichia coli. J. Biol. Chem. 281, 35779–35784 10.1074/jbc.M607232200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Thorpe I. F., and Brooks C. L. III (2007) Molecular evolution of affinity and flexibility in the immune system. Proc. Natl. Acad. Sci. U. S. A. 104, 8821–8826 10.1073/pnas.0610064104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Severance A. C., Sandoval P. J., and Wright S. H. (2017) Correlation between apparent substrate affinity and OCT2 transport turnover. J. Pharmacol. Exp. Ther. 362, 405–412 10.1124/jpet.117.242552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Diop-Bove N. K., Wu J., Zhao R., Locker J., and Goldman I. D. (2009) Hypermethylation of the human proton-coupled folate transporter (SLC46A1) minimal transcriptional regulatory region in an antifolate-resistant HeLa cell line. Mol. Cancer Ther. 8, 2424–2431 10.1158/1535-7163.MCT-08-0938 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data are contained within the article.