

Abstract
Encapsulated ferritins belong to the universally distributed ferritin superfamily, whose members function as iron detoxification and storage systems. Encapsulated ferritins have a distinct annular structure and must associate with an encapsulin nanocage to form a competent iron store that is capable of holding significantly more iron than classical ferritins. The catalytic mechanism of iron oxidation in the ferritin family is still an open question because of the differences in organization of the ferroxidase catalytic site and neighboring secondary metal-binding sites. We have previously identified a putative metal-binding site on the inner surface of the Rhodospirillum rubrum encapsulated ferritin at the interface between the two-helix subunits and proximal to the ferroxidase center. Here we present a comprehensive structural and functional study to investigate the functional relevance of this putative iron-entry site by means of enzymatic assays, MS, and X-ray crystallography. We show that catalysis occurs in the ferroxidase center and suggest a dual role for the secondary site, which both serves to attract metal ions to the ferroxidase center and acts as a flow-restricting valve to limit the activity of the ferroxidase center. Moreover, confinement of encapsulated ferritins within the encapsulin nanocage, although enhancing the ability of the encapsulated ferritin to undergo catalysis, does not influence the function of the secondary site. Our study demonstrates a novel molecular mechanism by which substrate flux to the ferroxidase center is controlled, potentially to ensure that iron oxidation is productively coupled to mineralization.
Keywords: ferritin, crystal structure, mass spectrometry (MS), X-ray crystallography, iron metabolism, iron, encapsulated ferritin, encapsulin, ferroxidase, native mass spectrometry
The ferritin superfamily consists of a number of structurally and functionally distinct members, each of which is built around a four-helix bundle scaffold that forms an iron-binding di-iron ferroxidase active site (the ferroxidase center, FOC) (1). The quaternary structure of these protein families shows a high degree of variation, from monomeric rubrerythrin proteins (2, 3), dimeric iron-mineralizing encapsulin (Enc)-associated firmicute (IMEF) proteins (4), dodecameric DNA Protection in Starved (DPS) cells proteins and DPS-like (DPSL) proteins (5–9), the 24-meric classical ferritins (Ftn) and bacterioferritins (10–13), and the decameric encapsulated ferritins (EncFtn) (14, 15). The rubrerythrin and DPS proteins play key roles in the protection of cells from oxidative stress, whereas DPS and the other members of the ferritin superfamily are vital iron stores (7, 16–22). These latter proteins oxidize and convert free Fe(II) to inert Fe(III) oxyhydroxide or Fe(III) phosphate mineral forms that provide a reservoir of bioavailable iron (23). The nanocages formed by the quaternary structure of DPS, DPSL, Ftn, and bacterioferritin allow iron oxidation and mineralization to occur within a biochemically privileged environment within the cell, which protects the host cell from iron-induced oxidative stress. Interestingly, the recently described IMEF and EncFtn proteins both have ferroxidase activity, but they do not form nanocage structures on their own; instead they must be localized to the lumen of encapsulin nanocages to form a competent iron store (4, 14, 15). EncFtn and IMEF are directed into encapsulins by a terminal localization sequence that binds to the interior wall of encapsulin. The EncFtn proteins form a distinct homodecameric annular assembly comprising a pentamer of dimers with D5 symmetry (Fig. 1A). Each EncFtn subunit is formed of two long anti-parallel helices (α1 and α2) followed by a shorter helix (α3) (Fig. 1B); this is in contrast to the classical ferritins, which have 4-helix subunits. The annular assembly has two classes of 2-fold symmetry forming two distinct dimer interfaces. One of the dimer assemblies contains the FOC across the dimer interface, with the two iron ions bound within the interface (Fig. 1C, FOC dimer). The other dimer forms a back-to-back conformation, exposing the metal ion binding residues, and is known as the nonFOC dimer (Fig. 1D) (24).
Figure 1.
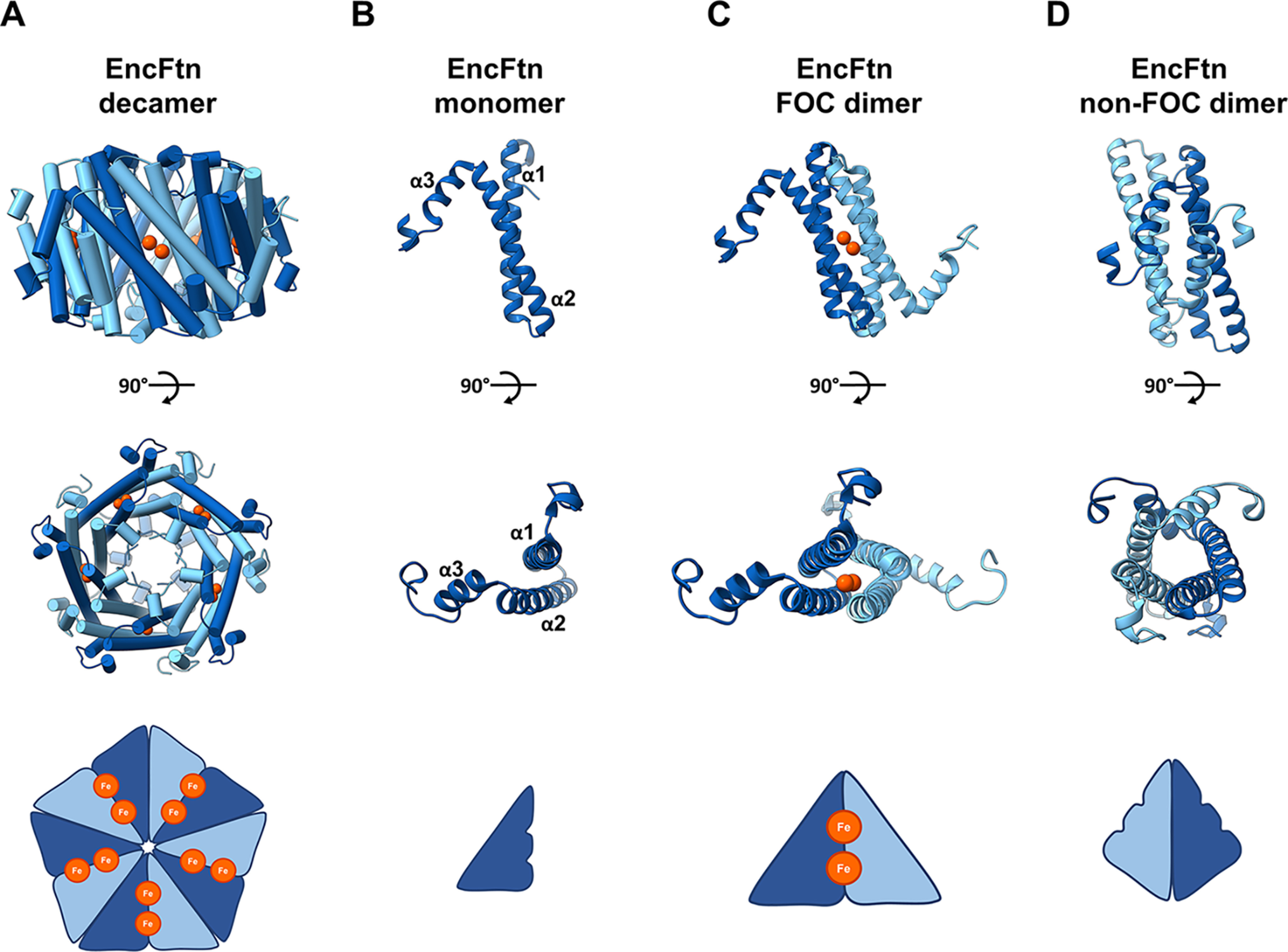
EncFtn subcomplex architecture. A, annular decamer structure of EncFtn shown in a side-on and top-down view, with a simplified representation of topology. The decamer structure has 10 iron ions bound (orange spheres). B, an EncFtn monomer with a simplified cartoon. C, the FOC dimer in cartoon with two coordinated iron ions (shown as orange spheres), and a simplified FOC cartoon is shown with two iron ions as orange circles. D, the nonFOC dimer with cartoon secondary structure and a simplified representation.
A model for the engagement of EncFtn decamers with the encapsulin nanocage has been proposed based on the crystallographic model of the Thermotoga maritima encapsulin complex (15, 25). In this model the C-terminal encapsulation sequence of the EncFtn interacts with a specific binding site on the encapsulin monomer, which is oriented to face the lumen of the nanocage (Fig. S1). This model places the central hole of the EncFtn decamer directly under the pores formed at the icosahedral 5-fold axis of the encapsulin nanocage, which would form a pathway for iron to transit across the encapsulin shell toward the putative metal entry site. The residues lining the 5-fold pore in the encapsulin nanocage are not well conserved between species, and based on crystallographic evidence, this pore is primarily made up of main-chain atoms, implying that this pore does not have a role as a selectivity filter.
The catalytic route to iron mineralization in the different ferritin iron stores is still a subject of vigorous debate (23, 26–30). Given the lack of absolute conservation in the structure of the ferroxidase center, it is likely that there is no single conserved mechanism for their activity (23, 26–30). All of the ferritin family proteins that have been structurally characterized have additional iron-binding sites proximal to the ferroxidase center. In the case of the sites proximal to the ferroxidase center identified in DPS and ferritins, these have been suggested to be iron-entry, -transit, and -exit points from the ferroxidase center (31–33), or the sites of electron transfer from Fe(II) substrates to the FOC (34). The subunit boundaries in the DPS and ferritin nanocages form a variety of symmetrical pores that have been suggested, or shown, to be entry and exit sites to the lumen for iron, water, and phosphate substrates (4, 10, 35–37). The proposed iron-entry site in DPSL is distinct from other ferritin family proteins, with an unusual arrangement of cysteine residues and a disulfide bond (8). Furthermore, an additional metal-binding site located on the inner surface of the dodecamer has been identified in members of the DPSL proteins; because of its location, this site is reminiscent of the third iron site in Escherichia coli ferritins and bacterioferritins and it is proposed to be involved in the iron translocation through the protein shell (9). Whatever their function, these secondary metal sites are essential to the correct functioning of the ferritin iron stores (38–40).
In our previous work we identified a number of conserved secondary metal-binding sites in EncFtn proteins (14, 15). A partially occupied metal ion site was present on the outer surface (proposed exit site) of the EncFtn decamer, and a highly occupied metal ion site was present on the inner face (proposed entry site), which is proximal to the ferroxidase center (Fig. 2, A and B). Both of these sites are found at dimer interfaces between the two-helix EncFtn subunits and are formed when the four-helical ferritin fold is reconstituted by the iron-mediated assembly of the EncFtn ferroxidase center (24). A comparison of these sites to secondary metal-binding sites in other ferritin family members highlights the fact that “C” site, first identified in the E. coli ferritin (41, 42), is topologically similar to the proposed iron-exit site identified in our previous work, whereas the entry site is unique to the EncFtn family (15). In our structure of the Rhodospirillum rubrum EncFtn protein, the entry site coordinates a calcium ion from the crystallization solution in a symmetrical arrangement of glutamic acid residues (Glu-31/Glu-34) and a tryptophan residue (Trp-38) contributed by each chain from the ferroxidase center dimer (24) (Fig. 2B). The metal coordination distances between the side-chain carboxylic acid oxygens and calcium ion are 2.1 Å for Ca–Glu-31–Ox and 2.7 Å for Glu34–Ox, which are similar to the Fe-Glu-Ox coordination distances seen in the FOC of 2.2–2.6 Å.
Figure 2.

Hypotheses for the functional role of the entry site in EncFtn. A, side-on view of the entry site and FOC within EncFtn. The metal-binding residues of the entry site (Glu-31 and Glu-34 of each subunit shown in purple and green, respectively) are shown coordinating a metal ion (gray sphere) within the dimer interface. Iron ions coordinated in the ferroxidase center (FOC) are depicted as red spheres with FOC residues in blue. B, top-down view of the entry site showing Glu-31, Glu-34, and Trp-38 of each subunit in purple, green, and pink, respectively. Glu-31 and Glu-34 are coordinating a divalent metal ion shown in gray. C, the proposed iron pathway in EncFtn. The black arrows indicate movement of Fe(II) ions (orange spheres) in EncFtn during their oxidation process to Fe(III) (brown sphere). Fe(II) enters the entry site, where the Glu-31 and Glu-34 side chains from both subunits are shown in purple and green. Fe(II) then proceeds to the FOC and then leaves EncFtn as Fe(III). Several hypotheses for the role played by the residues contributing to the entry site are shown in (D–F) (flow restriction valve (D), selectivity filter (E), and attraction (F). Ferrous and ferric ions are depicted as orange and brown circles, respectively, whereas noncognate species are in gray. The entry site glutamates are shown in orange and green stick representation, and the FOC residues are shown in shades of blue.
We hypothesize that this site acts as an entry site for iron ions approaching the ferroxidase center and acts in some way to control the access of metal ions to the active site of the protein (Fig. 2C). The glutamic acid residues in the entry site are arranged in such a way as to capture and coordinate an Fe(II) ion in an octahedral geometry before passing it to the FOC. The positioning of Trp-38 adjacent to this site suggests that the indole ring sterically constrains the position of the Glu-31 side chain to an optimal position for metal coordination. We have considered several hypotheses regarding the functional role of the entry site, including acting as a flow restrictor valve to limit the flow of metal ions to the FOC (“valve” function), an electrostatic funnel to direct metal ions to the FOC (“attraction” function), a discriminatory filter to avoid mis-metalation and consequent inhibition of the active site (“filter” function) (Fig. 2, D–F), or for protein stability and assembly (“stabilizing” function). It is also possible that these functions may overlap, and the entry site may play a number of key roles in the stability and activity of the encapsulated ferritins.
To understand the role of the entry site, a number of single-point amino acid variants of the R. rubrum EncFtn protein were produced to replace the residue of interest with nonmetal-binding residues. These variants were produced in the isolated R. rubrum EncFtn protein alone and in an R. rubrum Enc:EncFtn co-expression system. We produced and purified recombinant E31A, E34A, and W38G/A variants of EncFtn and the Enc:EncFtn complex and analyzed the solution and gas-phase behavior of these proteins to understand the influence of these variants on the stability of the EncFtn complex. We obtained X-ray crystal structures of the E31A and E34A variants of the EncFtn protein and found that their structures display altered entry site geometry and the abrogation of metal binding in this site. The functional role of the entry site was explored biochemically; we show that changes to the metal coordinating amino acids in this site enhance the ferroxidase activity of the EncFtn protein and increase its susceptibility to inhibition by competing zinc ions. Taken together, these results indicate that the entry site plays a minor role in stabilizing the oligomeric state of the EncFtn decamer and has a dual role as a site of metal ion attraction and as a flow restrictor valve to limit metal access to the ferroxidase center and thus slow the oxidation of ferrous ions to ferric ions.
Results
Changes to the metal ion entry site of encapsulated ferritins affect their oligomeric state in solution
To explore the functional roles of the residues in the entry site of the R. rubrum EncFtn, single-point variants were produced by site-directed mutagenesis, removing each metal-binding residue in turn and substituting each with alanine or glycine. The average molecular masses of the EncFtn proteins were confirmed by LC–MS (Table S1). The resulting variants were also generated in association with the encapsulin to produce Enc:EncFtn complexes.
Changes to the ferroxidase center affect both the assembly and activity of the R. rubrum EncFtn (EncFtn-WT) (15). To investigate whether changes to the entry site influence the quaternary structure of the EncFtn protein, each variant was subjected to size-exclusion chromatography using a HiLoad 16/60 Superdex 200 (S200) column calibrated with standards of known molecular mass (Fig. 3, A and B). Each of the variant EncFtn proteins eluted with a similar profile, with a major peak at 60 ml retention volume, a second peak at 75 ml, and the final peak at 82 ml (Fig. 3A, Fig. S2A, and Table S2). The E34A variant had significantly more protein in the 82 ml peak when compared with the EncFtn-WT and E31A variant, suggesting that this change affects the stability of the protein complex. Based on our prior work, these peaks correspond to decamer, tetramer, and dimer species, respectively (14, 24).
Figure 3.

Purification of recombinant EncFtn and Enc:EncFtn protein complexes. Recombinant EncFtn (A) and Enc:EncFtn (B) proteins were purified by anion exchange chromatography (Hi-Trap Q-Sepharose FF, GE Healthcare) and then subjected to size-exclusion chromatography using a Superdex 200 16/60 column (GE Healthcare) previously equilibrated with 50 mm Tris-HCl, pH 8.0, and 150 mm NaCl. The elution profiles in (A) reveal that all EncFtn proteins show a main peak at around 60 ml, diagnostic of oligomerization states close to 10-mer, and smaller peaks at ∼76 ml and 82 ml (indicative of smaller assembly states, Table S2). Enc:EncFtn protein complexes elute in a single peak around 46 ml, suggesting that EncFtn is compartmentalized within the encapsulin shell.
In contrast to the EncFtn variants, the Enc:EncFtn protein complexes all eluted as a single peak within the void volume of the S200 column, indicating the formation of protein complexes larger than 600,000 Da (separation range Mr for this column is 10,000–600,000 Da), hence they are not able to enter the matrix pores (Fig. 3B, Fig. S2B, and Table S2). SDS-PAGE analysis of peak fractions confirmed the presence of both the EncFtn and encapsulin proteins (Fig. S3A). We performed transmission EM to determine whether these complexes assembled into encapsulin nanocages (Fig. S2, D and E and Fig. S3B). All samples showed characteristic 25-nm encapsulin cages, with visible internal density for the EncFtn proteins. This is consistent with our previous observations of the R. rubrum Enc:EncFtn complex (15).
X-ray crystal structures of EncFtn variants show altered metal binding within the entry site
Given the observation that the E31A and E34A EncFtn variants display some differences in terms of oligomeric state and stability in solution, we determined the X-ray crystal structures of C-terminal hexahistidine tagged variants of these proteins (EncFtn-sH). Both the E31A and E34A variants crystallized in similar conditions to EncFtn-WT; however, we were unable to obtain crystals of either the W38G or W38A variants of the EncFtn protein, suggesting that removing this tryptophan destabilizes the quaternary structure of the protein sufficiently to present a barrier to crystallization.
The structures of the R. rubrum EncFtn-sH E31A and E34A variants were determined by X-ray crystallography to 2.66 and 2.19 Å resolution, respectively. They adopt the same overall quaternary fold as EncFtn-WT, with root-mean-square deviation Cα differences of 0.2 A2 over 91 Cα and 0.17 Å2 over 90 Cα for the E31A and E34A variants, respectively, and a root-mean-square deviation Cα of 0.23 Å2 over 90 Cα between the two variants. This indicates that these amino acid substitutions do not significantly alter the structure of the polypeptide backbone in the E31A and E34A variants. The position of side chains in the FOC and their metal coordination geometry are conserved for both variants, indicating that changes to the entry site do not fundamentally change the structure of the FOC.
Analysis of the entry site of the E31A variant shows that octahedral metal ion coordination seen for the WT protein is disrupted (Fig. 4), with a weak electron density feature at ∼4 Å distance from the carboxyl oxygens of Glu-34 (Fig. S4A). It was not possible to unambiguously model this feature, but it may be interpreted as a partially occupied divalent cation, such as calcium, which is present at 140 mm in the crystallization solution. Loss of the coordinating Glu-31 side-chain ligand causes Trp-38 to move when compared with the WT protein; this residue is found in two predominant conformations in this site, where it moves closer to the Glu-34 side chain (Supporting Movie 1). The change in metal ion binding when compared with the WT protein can be ascribed to the change in the coordination environment around this site. Similarly, the entry site of the E34A variant does not appear to coordinate any metal ions, the side chain of Glu-31 is flipped away by 120° from this site (Fig. 4 and Fig. S4B), and the Trp-38 indole ring is also moved away from this site when compared with EncFtn-WT (Supporting Movie 2). Together, the local structural changes seen for both entry site variants disrupt the metal coordination in this site when compared with the WT protein.
Figure 4.
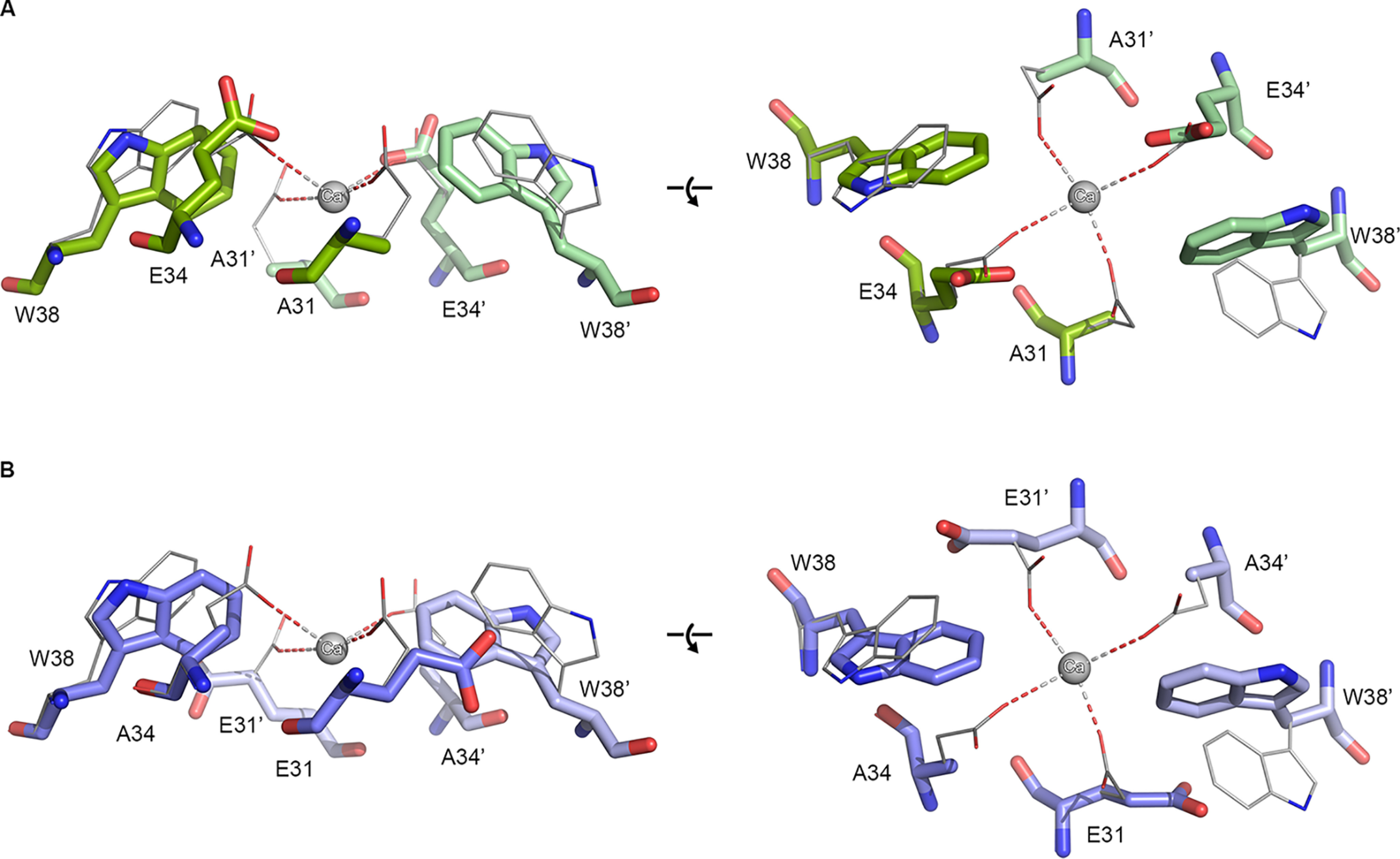
Comparison of side-chain positions in X-ray crystal structures of EncFtn entry site variants. Residues in the putative metal ion entry site for the X-ray crystal structures of the E31A (A, green) and E34A (B, blue) EncFtn variants are shown with the EncFtn-sH WT (gray) for comparison. Residues in the metal ion entry site are shown as stick representations with a sphere indicating the position of the calcium ion found coordinated in the WT protein. Residues in the entry site are labeled, with residues from the second monomer indicated with prime symbols. In the E31A variant, the Trp-38 side chain is moved toward the putative metal ion coordinating residues, whereas the Glu-34 side chain is in a similar conformation when compared with the WT. The Trp-38 side chain in E34A adopts a similar conformation to that seen in the E31A variant, whereas in the absence of the Glu-34 side chain, the Glu31A side chain rotates by 120° away from the putative metal-binding site.
Native MS reveals conformational heterogeneity in the EncFtn variants
To interrogate the stability and oligomeric states of the EncFtn variants, we performed native MS and ion-mobility MS experiments on samples purified by size-exclusion chromatography (Fig. 5) (14). In our initial studies we found that EncFtn-W38A was not amenable to native MS analysis because of poor solubility in native MS ammonium acetate buffer; consequently, native MS was performed on EncFtn-WT, EncFtn-E31A, EncFtn-E34A, and EncFtn-W38G.
Figure 5.

Native MS analysis of EncFtn variants. Native nanoelectrospray ionization spectra of EncFtn variants displaying decameric (pink triangles) and monomeric (blue circles) charge-state distributions. The elongated decameric charge-state distributions of WT and E34A are highlighted by an asterisk.
In native MS analysis, EncFtn-E31A behaves similarly to EncFtn-WT and presents solely as a decameric charge-state distribution centered around the 25+ charge state (Fig. 5, pink triangles). In contrast, under the same instrument conditions, EncFtn-E34A and EncFtn-W38G displayed both decameric and monomeric charge-state distributions (Fig. 5, highlighted with pink triangles and blue circles, respectively). The presence of monomeric high-charge states (8+ to 16+ for EncFtn-E34A and 8+ to 15+ for EncFtn-W38G) suggests a degree of gas-phase dissociation of the decameric complex during native MS analysis. Even with careful control of instrument conditions, this monomeric dissociation product could not be completely eliminated, suggesting that complexes of the EncFtn-E34A and EncFtn-W38G are less stable than EncFtn-WT. Thus, these initial observations suggest that loss of either Glu-34 or Trp-38 results in gas-phase destabilization of the decamer assembly, although we cannot rule out the observed difference in stability being due to different iron content within the FOC in each of these protein preparations. We also note that the decameric charge-state distributions of EncFtn-WT, EncFtn-E34A, and EncFtn-W38G are extended and include several additional higher charge, low-abundance charge states (Fig. 5, highlighted with an asterisk) when compared with our previous EncFtn native MS analysis of a truncated EncFtn-WT protein (15). We attribute their presence to the ability of the solvent-exposed encapsulation sequence to readily protonate in solution. The difference in gas-phase stability of the EncFtn variants was also evident when increasing the sampling cone voltage and/or activating the trap voltage of the mass spectrometer, which activates the complex and causes dissociation (Fig. S5).
To further investigate the gas-phase structure and the stability of the variants, ion-mobility collision-induced unfolding (CIU) experiments were performed (43, 44). Each gas-phase decamer protein complex was activated in a stepwise manner by incremental changes of the trap voltage prior to analysis of the conformation of the native protein ions using ion-mobility MS. The resulting ion-mobility profile (arrival time distributions) for an individual protein ion was then plotted as a function of activation voltage to produce a heat map to reveal discrete conformations and gas-phase unfolding transitions (Fig. S6, A–D). It is clear from these experiments that two major discrete conformations of each EncFtn variant are observed during CIU: a conformation with a compact structure and a second conformation with a more extended, unfolded structure (Fig. S6, A–D, labeled “C” and “E”, respectively, and Fig. S6E, i and ii). At minimal activation, EncFtn-WT, EncFtn-E34A, and EncFtn-E31A all exist in a single, compact decameric conformation with a drift time of between 10 and 11 ms, suggesting the same overall structure. As activation is increased, all three variants undergo a discrete transition to a more extended conformation (characterized by a drift time of 12.5 ms). A second, more complex transition to higher drift time is also observed at high activation (around 45 V) before dissociation of the complex to release monomeric subunits. The transition from the C to E states for each variant was quantified using CIU50 values (the collision voltage required to convert 50% of the compact protein form into the adjacent extended state) to give values of 35.2 V, 26.5 V, and 35.1 V for EncFtn-WT, EncFtn-E31A, and EncFtn-E34A, respectively. The close similarity of these CIU profiles strongly suggests similar gas-phase structures for the EncFtn-WT and the E31A and E34A variants (Fig. S6) with a slight destabilizing effect resulting from the mutation of the Glu-31 residue. In contrast, EncFtn-W38G occurs in the more extended conformation (12.5 ms) throughout the CIU experiment (Fig. S6D and Fig. S6Eiii), with the compact conformation only observed as a minor species at low activations. Because there is already less than 50% of compact conformation present in the lowest activating conditions, no CIU50 can be established for the transition from compact to extended state for W38G. EncFtn-W38G also undergoes a more complex transition from the extended conformation but with a lower CIU50 of 35.2 V compared with the other variants of ∼45 V. This is in accord with the native MS studies and suggests that although the W38G assembly shares similar overall structure to the WT and glutamate variants, the tryptophan substitution has a significant destabilizing effect on the decameric assembly. This increased structural/conformational heterogeneity in the tryptophan variant of EncFtn may account for our inability to obtain crystals in standard crystal screens.
The EncFtn metal entry site acts to attract metals to the FOC and restrict its catalytic activity
The changes in metal ion coordination observed in the entry sites in the crystal structures of the E31A and E34A variants suggest that these residues play a direct role in controlling the accessibility of the ferroxidase center to metal ions. To test our hypotheses about the functional role of this site (Fig. 2, D–F), we investigated the ferroxidase activity of all of the EncFtn variants produced, both in isolation and in complex with the encapsulin protein.
The ferroxidase activity of the WT EncFtn protein was confirmed to be comparable with that previously determined for the EncFtn-strepII variant (14). Surprisingly, all three variants displayed higher catalytic activity than the WT protein (Fig. 6). The EncFtn-E34A variant had a 2-fold higher activity level, whereas both the W38A and E31A mutations oxidize Fe(II) to Fe(III) around 5-fold faster, and their progress curves present a distinct shape in contrast to the WT protein (Fig. 6). To test the contribution of Trp-38 to the EncFtn catalytic activity, we tested the W38A variant, allowing for a direct comparison with the other variants. We also verified that the W38G and W38A variants had comparable activities in complex with the encapsulin protein (Fig. S2C).
Figure 6.

Ferroxidase activity of EncFtn and Enc:EncFtn complexes. A, EncFtn-WT (green line), EncFtn-E34A (pink line), EncFtn-E31A (blue line), and EncFtn-W38A (orange line) (10 µm, monomer) were incubated with 50 µm FeSO4.7H2O (10× molar equivalent Fe(II)/FOC), and progress curves of the oxidation of Fe(II) to Fe(III) was monitored at 315 nm at room temperature. The background oxidation of iron at 50 µm in enzyme-free control is shown for reference (dotted gray line). Solid lines represent the average (n = 3) of technical replicates, and shaded areas represent standard deviation from the mean. Protein and iron samples were prepared anaerobically in buffer H (10 mm HEPES, pH 8.0, and 150 mm NaCl) and 0.1% (v/v) HCl, respectively. B, Enc:EncFtn-WT (dark green line), Enc:EncFtn-E34A (dark pink line), Enc:EncFtn-E31A (dark blue line), and Enc:EncFtn-W38A (orange line) (25 µm Enc:EncFtn, corresponding to 10 µm EncFtn monomer) were incubated with 50 µm FeSO4.7H2O, and progress curves of the oxidation of Fe(II) to Fe(III) were monitored at 315 nm at room temperature. The background oxidation of iron at 50 µm in enzyme-free control is shown for reference (dotted gray line). Solid lines represent the average (n = 3) of technical replicates, and shaded areas represent standard deviation from the mean. Protein and iron samples were prepared anaerobically in buffer H (10 mm HEPES, pH 8.0, and 150 mm NaCl) and 0.1% (v/v) HCl, respectively. Comparison of data collected for W38A and W38G variants are shown in Fig. S2, including elution profiles from gel-filtration purification steps (A and B), ferroxidase activities of Enc:EncFtn-W38A and W38G (C), and Enc:EncFtn-W38A and W38G transmission electron micrographs (D and E).
The increase in activity observed by all of these variants suggests that the entry site plays a role in restricting the flow of substrate Fe(II) ions to the FOC. When the differences in enzymatic activity are considered in the context of the X-ray crystal structures, it is clear that the disruption of metal coordination in the entry site has a significant impact on the catalytic activity of the EncFtn protein. The EncFtn-E31A structure shows that, as a consequence of changes in metal ion coordination, the restriction in metal ion flow provided by this variant is partially removed when compared with EncFtn-WT. However, the residual glutamic acid, Glu-34, may still be able to coordinate metal ions, albeit with lower affinity than the WT enzyme, given the increased distance between these residues and lack of additional coordination from Glu-31 (Fig. 4).
The crystal structure of the EncFtn-E34A variant shows that the mutation of Glu-34 to Ala results in a reorganization of the entry site, with the side chain of Glu-31 reoriented in such a way that it is no longer correctly positioned for metal coordination, essentially removing any electrostatic attraction and flow restriction for this site (Fig. 4). A ferroxidase assay carried out with an EncFtn-E31A/E34A variant shows that its activity is comparable with that of EncFtn-E34A (Fig. S7). These data corroborate our hypothesis that in EncFtn-E34A, the flow-restricting function is completely abolished and no attraction is conveyed by the residual Glu-31. Because no flow restriction is provided at this level, we would expect to observe a higher activity; however the restructuring of this site results in a loss of attractive forces, and consequently only a modest increase in activity is observed when compared with EncFtn-WT. The difference in activity between EncFtn-E31A and EncFtn-E34A demonstrates that the electrostatic attraction provided by Glu-34 plays an important role in the attraction of metal ions to the FOC.
Because we were unable to produce crystals of the W38A variant, we could not determine the effect of removing this tryptophan residue on the structure of the entry site. Analysis of our crystal structures shows that the indole ring of the tryptophan may act to sterically constrain the Glu-31 side chain in the correct position for metal coordination. Consequently, it is possible that the removal of Trp-38 results in the Glu-31 side chain moving away from this site. Our ferroxidase assay data show that the Trp-38 variants have comparable activity profiles to the E31A variant; we therefore propose that the two variants possess similar entry-site structures. The broader destabilizing effects of the loss of the tryptophan side chain are harder to model, but the loss of a side chain of the size of the tryptophan indole may lead to further structural rearrangements around the internal dimer interfaces engaged by this residue.
The same ferroxidase assay was performed with the Enc:EncFtn complex, with WT and EncFtn variants (Fig. 6B). The deviations from WT behavior were compared with those observed in the absence of the encapsulin nanocompartment (Fig. 6A). Enzymatic reaction initial rates (v0) were calculated for each of the variants (in the first 200 s of the assay) and then divided by the initial rate of the corresponding WT protein (EncFtn-WT or Enc:EncFtn-WT) (Fig. S8, Table S3, Table S4, and Table S5). This allowed comparison of the assay data across the two contexts for the EncFtn enzyme (encapsulated versus nonencapsulated). Our data show that the E31A and W38A variants have a greater effect on activity within both systems (about 4-fold faster when compared with WT initial rate), whereas the E34A variant displays a 2-fold increase in the initial velocity v0 (Fig. S8 and Table S5). Therefore, the EncFtn variants and WT retain the same relative differences in their activities whether they are associated to the encapsulin shell or not. This observation suggests that the attraction and flow-restricting properties are exerted by the entry site and are not changed by the sequestration of the EncFtn within the encapsulin cage.
The ferroxidase activity of EncFtn is inhibited by zinc
To test the hypothesis that the entry site acts as a selectivity filter and is able to discriminate between metal ions approaching the FOC, ferroxidase assays were performed with the addition of zinc as a competitive inhibitor (45). The activity of EncFtn WT and variant proteins (10 μm, monomer) with 50 μm Fe(II) was assayed in the presence of 34 μm Zn(II) (Fig. 7A). The selected zinc concentration was previously determined to be the IC50 for WT EncFtn-strepII (14). This concentration was chosen to allow identification of the impact of inhibition across the set of variant proteins. In line with our previous observations, EncFtn-WT showed 54% inhibition (14) (Fig. 7, A and B). The activity of the three variant proteins (Glu-31, Glu-34, and Trp-38) showed a markedly increased relative inhibition when compared with their activity in the absence of zinc (88%, 66%, and 69%, respectively) (Fig. 7, A and B). The increased inhibition seen in the variants can be explained by the changes in metal coordination at the entry site. With all the variants, the abrogation of metal coordination at this site permits greater access to the FOC for competing metal ions because they are no longer effectively captured in the entry site. Although these data show that the entry site restricts the general accessibility of the FOC to competing metal ions, they do not directly address the question of whether the site is acting as a selectivity filter.
Figure 7.

Ferroxidase activity of EncFtn WT and variants is inhibited by zinc. A, comparisons between data shown in Fig. 6A, collected in the absence of competing metals, are here plotted with curves from ferroxidase assays of EncFtn WT and variants carried out using [protein] = 10 µm and [Fe(II)] = 50 µm in the presence of [Zn(II)] = 34 µm. The zinc concentration corresponds to a response inhibited by 50% for EncFtn-Strep under the same experimental conditions (14). EncFtn-WT is represented by a dark green line, EncFtn-E34A by a purple line, EncFtn-E31A by a pale blue line, and EncFtn-W38A by a red line. As a control, a mix of Fe(II) and Zn(II) salts (gray dotted line) was assayed in the absence of the enzyme. B, end-point data presented in (A) were plotted as columns to compare inhibition by zinc. Percentage of inhibition is shown above in corresponding columns. Color-coding is consistent with (A).
Steady-state fluorescence emission reveals zinc ion binding at the entry site
To determine whether zinc ions interact with the entry site and thus compete with iron for access to the FOC, intrinsic tryptophan fluorescence was exploited to study the Trp-38 microenvironment in the presence and absence of zinc sulfate. In addition to Trp-38, EncFtn possesses two additional tryptophan residues (Trp-72 and Trp-80), one phenylalanine (Phe-89), and two tyrosine residues (Tyr-39 and Tyr-87). Tyr-39 is located in close proximity to the FOC and Glu-31/Glu-34, whereas the other aromatic residues are >10 Å from both metal coordination sites. Therefore, any observed change in the protein fluorescence emission should be due to changes in the entry site, and hence to metal binding at this site. This hypothesis was tested by monitoring steady-state fluorescence emission of EncFtn proteins over time and upon addition of increasing sub-stoichiometric concentrations of Zn(II). The control experiment with EncFtn-W38A showed a hypsochromic shift of 8 nm in the emission maximum (λEM ∼ 322 nm compared with λEM ∼ 330 nm of EncFtn-WT) (Fig. S9A), suggesting a contribution to intrinsic fluorescence from more buried residues in the absence of Trp-38. Although this variant still possesses both glutamic acid residues in the entry site, no specific trend was observed upon zinc addition (Fig. 8).
Figure 8.

Intrinsic fluorescence of EncFtn WT and variants titrated with Zn(II). Fluorescence emission of EncFtn proteins (20 µm, corresponding to [entry site] = 10 µm) at various wavelength (330 nm for EncFtn-WT and -E31A, 355 nm for EncFtn-E34A and E31A/E34A, and 322 nm for EncFtn-W38A), with excitation at 280 nm, and following titration with ZnSO4.7H2O. A and B, fluorescence is quenched upon zinc addition, with inflection around 1 molar equivalents, suggesting perturbation of environment around Trp-38. C and D, in the absence of entry-site residues available for metal coordination, no quenching was observed. E, EncFtn-W38A was tested as the control. Symbols represent average and standard deviation (error bars) of experimental replicates, collected at equilibrium either by Scan (n >3) or Kinetic (n = 10) options on the Cary Eclipse software package. Data were normalized to allow comparison. Dotted gray lines were added at 1 molar equivalent of Zn(II) when inflection was observed.
Only WT EncFtn and the E31A variant showed quenching of their fluorescence emission on addition of zinc, with an inflection at ∼1 molar equivalent of Zn(II) per entry site (Fig. 8 and Fig. S9, B and C). Variants with abrogated metal coordination (E34A and E31A/E34A) did not show any specific trend or inflection upon zinc addition (Fig. 8 and Fig. S9F). Moreover, tryptophan fluorescence is highly influenced by the solvent polarity of the surrounding environment, and fluorescence emission spectra from both E34A and E31A/E34A variants exhibited a bathochromic shift of 25 nm (λEM ∼ 355 nm), implying an increasing polarity in Trp-38 surroundings (Fig. S9A). These observations suggest that upon metal coordination, the Trp-38 indole ring is restrained toward the gateway of the channel leading to FOC.
Thus, we propose that metal ions can indeed interact with the entry site when it is intact (WT) or partially present (E31A). In the absence of the entry site, Zn(II) will likely reach the FOC, an event that is not detectable with this assay but shown by ferroxidase analysis in the presence of Zn(II) where increased inhibition of all variants was observed.
Zn(II) displaces Fe(II) in the FOC and stabilizes the FOC dimer interface
Given the observation in the intrinsic tryptophan fluorescence experiments that zinc directly interacts with the entry site, we performed high-resolution native Fourier-transform ion cyclotron resonance (FT-ICR) MS to further probe whether this site was acting as a selectivity filter to restrict the passage of competing ions to the FOC. The high mass resolution of FT-ICR MS affords isotopic resolution of native protein complexes and, when applied to the study of metalloproteins, allows assignment of coordinated metals with high confidence. To further aid this analysis, we also employed an isotope depletion strategy (46). The depletion of 13C and 15N stable isotopes from the EncFtn protein sample (referred to as ID-EncFtn) results in a dramatically reduced isotope distribution in MS analysis. This simplified signal results in increased sensitivity and, most importantly for native MS analysis, reduces the occurrence of overlapping signals from proteoforms of similar mass, such as complete separation of sodiated and potassiated protein ions (a common observation in native protein MS). Thus, this strategy simplifies data interpretation and allows more confident and precise assignment of metal-bound species. Native FT-ICR analysis of apo-ID-EncFtn reveals that it exists exclusively as a monomer (Fig. 9A), as previously seen for EncFtn expressed in minimal media (15). Isotopic resolution confirms that there is no metal bound within the purified ID-EncFtn monomer; which is as predicted because of the limited bioavailability of metals in the minimal media required for the production of isotopically depleted proteins (Fig. S10A). Titration with Fe(II) prior to MS analysis results in the disappearance of the monomeric species and the appearance of a decameric charge-state distribution (Fig. 9B, pink triangles). This result is consistent with the Fe(II)-dependent assembly of EncFtn (15). Interestingly, titration with Zn(II) prior to native MS reveals a similar result (Fig. 9C, pink triangles), supporting our previous solution-phase observations that metal-mediated higher order assembly is not specific for iron (15). Loading ID-EncFtn with Fe(II) followed by the addition of Zn(II) results in a decameric charge distribution similar to that of the Zn(II)-mediated decamer (Fig. 9D). Isotopic resolution cannot be achieved for the decameric species because of heterogenous metal loading, but sufficient mass resolving power indicates that metal loading occurs to the decameric complex in each case (Fig. S10B). We used collision-induced dissociation to perform gas-phase disruption experiments to gain additional understanding of metal loading in ID-EncFtn. Iron-loaded ID-EncFtn dissociates predominantly into a high-charge monomer species, the so-called “typical” dissociation product of protein complexes (47). However, upon closer inspection, evidence of higher order collision-induced subcomplexes is present; a minor tetramer species is observed at m/z 3552 (charge 14+) (Fig. S11Ai, blue circles and purple diamond, respectively). This species has low signal-to-noise ratio; however, its depleted isotope distribution is similar to that of four ID-EncFtn monomers with two Fe(III) ions bound (Fig. S11Aii). We assign this species as two nonFOC dimers bound together by a di-iron containing FOC; this dissociation pathway is consistent with the proposed assembly of EncFtn (24) (Fig. S11Aiii).
Figure 9.

Native FT-ICR mass spectrum of ID-EncFtn. Native nanoelectrospray ionization mass spectra of apo-ID-EncFtn (A), ID-EncFtn titrated with Fe(II) (B), ID-EncFtn titrated with Zn(II) (C), and ID-EncFtn titrated with Fe(II) followed by Zn(II) (D). Oligomerization states are stressed with colored shapes, monomer in blue circles and decamer in pink triangles.
In stark contrast, ID-EncFtn with coordinated Zn(II) ions dissociates into monomer and dimer species (Fig. S11Bi, blue circles and green squares, respectively). Isotopic resolution was achieved for the dimer species and reveals that this species has a mass consistent with two ID-EncFtn monomers associated two Zn(II) ions (Fig. S11Bii). The presence of two metal ions bound within the dimer suggests that this species is the FOC dimer with the FOC occupied by two Zn(II) ions. There is no peak observed, consistent with an apo-dimer, nor a dimer with one, or three, Zn(II) ions bound. This “atypical” dissociation pathway was previously observed for EncFtn (15) (Fig. S11Biii). The differing dissociation pathways of zinc-coordinated ID-EncFtn and iron-coordinated ID-EncFtn suggests that zinc stabilizes the FOC interface and binds with a higher affinity than iron in the FOC catalytic center; this higher affinity is as predicted from the Irving-Williams series. This supports our observations from the ferroxidase assay that show zinc inhibiting the FOC. Analysis of ID-EncFtn first loaded with iron and then subsequently challenged with zinc reveals an identical dissociation pathway to that titrated solely with zinc (Fig. S11Ci). Analysis of the isotopic distribution of the dimer species shows the same dimer species seen when titrated with zinc alone (Fig. S11Cii). No iron-containing dissociated subcomplexes were observed in the mass spectrum, suggesting that in EncFtn-WT, zinc can efficiently displace iron in the FOC. Thus, taken together, our native MS and biochemical analysis suggest that zinc inhibition acts at the catalytic ferroxidase center and that zinc is not held within or discriminated against at the entry site (Fig. S11Ciii).
Discussion
We have shown that substitution of residues in the entry site triad (Glu-31, Glu-34, and Trp-38) with nonmetal-binding residues results in the enhancement of enzymatic activity (Fig. 6A). Our crystal structures support the conclusion that this is a consequence of the creation of a wider and more loosely coordinating entry site that allows the faster passage of metal ions to the FOC because of a reduced energy barrier at this site (Fig. 4). Furthermore, when these key residues are removed, the protein is more susceptible to inhibition by Zn(II).
Solution and gas-phase analyses of variant proteins showed that, in contrast to the FOC, removal of the metal coordinating ligands (E31A/E34A) did not have a large effect on protein oligomerization; however, removal of the Trp-38 side chain had a significant destabilizing effect on the quaternary structure of the protein. Investigation of the EncFtn intrinsic fluorescence emission properties upon zinc binding revealed coordination in the entry site when this is intact or partially formed (WT and E31A, respectively), confirming that this site acts as an additional metal-binding site prior to the FOC.
High-resolution MS analysis of ID-EncFtn metal binding reveals that ID-EncFtn decamer assembly can be induced by iron and zinc ions. Furthermore, zinc appears not to be trapped by the metal ion entry site but is bound within the FOC more tightly than iron and stabilizes the FOC dimer. When ID-EncFtn is loaded with iron and subsequently challenged with zinc, only zinc is observed bound in ID-EncFtn subcomplexes, indicating that zinc displaces iron from the FOC.
Taken together, these results imply that the entry site has a lower affinity for metal ions than the ferroxidase center and that, in accord with the Irving-Williams series, the FOC forms more stable complexes with zinc than iron. However, bacterial cells buffer metal concentrations via a polydisperse buffer in the order of the Irving-William series (48–51), resulting in highly competitive Zn(II) ions being buffered to lower available levels than Fe(II), thus presumably preventing the mis-metalation of ferritin sites in vivo.
These observations support the hypothesis that the entry site has a dual function, in attracting metal ions to the catalytic site and as a flow restrictor to limit the movement of metal ions to the FOC and thus slow the rate of iron oxidation (Fig. 2, D and F). Our results confirm that the ferroxidase center is indeed the active site of this enzyme, because the loss of the entry site enhances enzyme activity rather than abrogates it. Why the EncFtn protein has evolved a mechanism to effectively slow its activity is still an open question. There may be a tradeoff between substrate turnover and the availability of a sink for the electrons liberated from the iron in the ferroxidase center. The catalytic mechanism and electron sink for ferritins is still a subject of vigorous debate. The EncFtn family presents a unique structural arrangement of the active site, which will no doubt present a distinct mechanism to other members of the ferritin family when probed further (40). The entry site may well provide a brake to ensure productive coupling of iron oxidation and mineralization.
Encapsulation of the EncFtn protein within the encapsulin nanocage enhances the activity of all of the EncFtn variants (Fig. 6) but does not change the way in which modifications to the entry site affect the catalytic activity, indicating that the function of the entry site is not changed through association with the encapsulin shell. These results suggest that the encapsulin shell acts to guide productive iron ion interactions with the EncFtn protein; indeed, our previous work showed that the encapsulin shell interacts with a significant amount of iron in the absence of the EncFtn protein. This warrants future investigation into the ability of the encapsulin shell to conduct metal ions to the EncFtn active site and the role of the encapsulin shell pores in the access of substrates to the encapsulated enzyme.
Experimental procedures
Cloning
Expression constructs for the R. rubrum EncFtn protein (pACYCDuet-1-Rru_EncFtn) and Enc:EncFtn protein complex (pACYCDuet-1-Enc:EncFtn) were produced in the previous work (15).
Site-directed mutagenesis to generate the desired variants of EncFtn and Enc:EncFtn was conducted via the QuikChange® protocol (Stratagene) using primers listed in Table S6. pACYCDuet-1-EncFtn (15) or pACYCDuet-1-Enc:EncFtn (15) were used as templates to produce single mutants, whereas pACYCDuet-1-EncFtn-E31A was used to produce EncFtn-E31A/E34A double mutant.
Protein nomenclature used throughout the text is shown in Table S7. Primers used in this work for this purpose are listed in Table S6.
Protein production and purification
Expression plasmids for the R. rubrum EncFtn protein and Enc:EncFtn complex were transformed into competent E. coli BL21(DE3) cells, grown in LB medium supplemented with appropriate antibiotics, and expressed as described previously (14, 15). Protein sequences are listed in Table S8.
EncFtn and its variants were expressed and purified as described in (14). A single colony of E. coli BL21(DE3) cells transformed with protein expression plasmid was grown overnight at 37 °C in 10 ml of LB medium, supplemented with 35 μg/ml chloramphenicol or 50 μg/ml kanamycin, while shaking at 180 rpm.
The starting culture was then inoculated into 1 liter of LB medium and grown until OD600 = 0.5; then protein expression was induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside, the temperature was reduced to 30 °C, and the cells were incubated for a further 18 h. The cells were pelleted by centrifugation at 4000 × g for 25 min at 4 °C and washed with PBS before an additional centrifugation step.
EncFtn WT and variants cell pellets were resuspended in 10× (v/w) Q sepharose buffer A (QA) (50 mm Tris, pH 8.0) and lysed by sonication on ice (5 min of 30-s bursts of sonication at 35% amplitude and 30-s cool down). Cell lysate was clarified by ultracentrifugation at 16,000 × g for 25 min at 4 °C, followed by filtration using a 0.22-μm syringe filter (Millipore). EncFtn protein sample was applied to a 20-ml Q sepharose anion exchange column (Hi-Prep Q HP, GE Healthcare) pre-equilibrated with buffer QA. Unbound protein was then washed off with 10 volumes of buffer QA. Proteins were eluted with a linear gradient of 20 column volumes of buffers QA and Q sepharose buffer B (QB) (50 mm Tris, pH 8.0, and 1 m NaCl) to 100% QB. The protein of interest was identified by SDS-PAGE and concentrated using a Vivaspin centrifugal concentrator (MWCO 10 kDa, Sartorius) following the manufacturer's instructions. Concentrated sample was loaded onto a size-exclusion column (HiLoad 16/60 Superdex 200, GE Healthcare) pre-equilibrated with gel fitration buffer (GF) (50 mm Tris, pH 8.0, and 150 mm NaCl). Fractions were analyzed by 15% SDS-PAGE to assess purity and oligomerization state, and those containing the protein of interest were pooled and further concentrated as described before. Trace data for gel filtration experiments are available at doi. 10.6084/m9.figshare.9885557.
Enc:EncFtn WT and variant complex cell pellets were resuspended in 10× (v/w) lysis buffer (20 mm HEPES, pH 8.0, 2 mm MgCl2, 1 mg/ml lysozyme, and 20 units/ml benzonase). The cells were sonicated on ice (5 min at 30 s on/off cycles, 35% amplitude), followed by incubation for 30 min at 37 °C in a water bath. Cell lysate was clarified by centrifugation (35,000 × g, 30 min, 4 °C) and the supernatant was collected and incubated for 10 min at 80 °C in a water bath. After cooling, the sample was centrifuged again (30 min, 35,000 × g, 4 °C), and the supernatant was collected and filtered using a 0.22-μm syringe filter (Millipore). Enc:EncFtn protein complex sample was loaded onto a 20-ml anion exchange column (Hi-Prep Q HP, GE Healthcare) pre-equilibrated with buffer QA (50 mm Tris, pH 8.0). Unbound protein was washed with Buffer QA. A linear gradient elution of 20 column volumes was performed by mixing buffers QA and QB (50 mm Tris, pH 8.0, and 1 m NaCl) to 100% QB. Because Enc:EncFtn does not bind the anion exchange column, proteins of interest were found in flow-through sample, as confirmed by 15% SDS-PAGE. The protein was concentrated using a Vivaspin centrifugal concentrator and then applied to a size-exclusion column (HiLoad 16/60 Superdex 200, GE Healthcare) pre-equilibrated with buffer GF (50 mm Tris, pH 8.0, and 150 mm NaCl). Fractions of interest were run on a 15% SDS-PAGE and further concentrated using a Vivaspin centrifugal concentrator as before. After concentration, protein samples were analyzed by 10% Tricine-SDS-PAGE (52) to resolve bands around 30 kDa. Elution volumes of EncFtn and Enc:EncFtn complexes from the size-exclusion column are listed in Table S2.
His-tagged protein production and purification
Hexahistidine-tagged EncFtn-E31A and -E34A variants (EncFtn-sH) were produced in E. coli BL21(DE3) as described above and purified following the same protocols as previously described (15). A 5-ml HisTrap column (GE Healthcare) was equilibrated with buffer HisA (50 mm Tris, 500 ml NaCl, and 50 mm imidazole, pH 8.0) and loaded with the clarified cell lysate. Unbound proteins were washed with buffer HisA. A step gradient of 50 and 100% HisB (50 mm Tris, 500 ml NaCl, and 500 mm imidazole, pH 8.0) was performed to elute His-tagged proteins. Fractions containing the protein of interest were identified by 15% SDS-PAGE, pooled together, and concentrated by Vivaspin centrifugal concentrator (10 kDa MWCO). Protein sample was applied to a size-exclusion column (HiLoad 16/60 Superdex 200, GE Healthcare) pre-equilibrated with buffer GF (50 mm Tris, pH 8.0, and 150 mm NaCl). A 15% SDS-PAGE was carried out to assess purity and oligomerization state, and samples containing the protein were further concentrated as described before.
Isotopically depleted strepII-tagged protein production and purification
To produce isotopically depleted EncFtn proteins, competent E. coli BL21(DE3) cells were transformed with the required expression plasmid. A single colony was used to inoculate 10 ml of LB media supplemented with the appropriate antibiotic before overnight incubation at 37 °C. This was then used to inoculate 500 ml of 2× YT media incubated at 37 °C until an A600 0.6–0.8 was obtained, at which point the cell culture was centrifuged at 5000 × g at 4 °C for 20 min (200 ml/expression culture required). The pellet was washed with M9 salts solution (33.9 g/liter Na2HPO4, 15 g/liter KH2PO4, and 2.5 g/liter NaCl) and suspended in 100 ml of M9 minimal media. Isotopically depleted M9 minimal media was supplemented with 12C (99.9%)–glucose (Cambridge Isotope Laboratories) and 14N (99.99%)-ammonium sulfate. The M9 minimal media cultures were further incubated at 37 °C for 1 h before protein expression was induced and stored as previously described (46).
Isotopically depleted strepII-tagged EncFtn (ID-EncFtn) was purified as reported in He et al. (14). The cells were suspended in 10× (v/w) Strep-Trap wash buffer (W) (100 mm Tris, pH 8.0, and 150 mm NaCl), sonicated, and clarified by centrifugation. Cell lysate was loaded onto a Strep-Trap HP column (5 ml, GE Healthcare) and equilibrated according to the manufacturer's instructions, and unbound protein was washed out with buffer W (5 column volumes). ID-EncFtn was eluted by buffer E (100 mm Tris, pH 8.0, 150 mm NaCl, and 2.5 mm desthiobiotin).
Protein quantification
Both untagged and His-tagged EncFtn proteins were quantified using the Pierce BCA Assay kit (Thermo Fisher Scientific) following the manufacturer's instructions for the Test Tube procedure. Concentration of Enc:EncFtn proteins was determined using the Beer-Lambert equation, measuring absorbance at 280 nm with a Nanodrop Spectrophotometer (Thermo Fisher Scientific), and using an extinction coefficient calculated by the ProtParam tool on the Expasy platform entering a sequence composed of 3× Enc and 2× Enc-Ftn sequences. This ratio was determined by examination of SDS-PAGE gels of Enc:EncFtn proteins and densitometry of bands.
Transmission EM
Transmission EM imaging was performed as previously described (15) on purified Enc:EncFtn proteins. The proteins were diluted inbBuffer GF to a final concentration of 0.1 mg/ml before being spotted on glow-discharged 300-mesh carbon-coated copper grids. Excess liquid was removed by blotting with filter paper (Cytiva Life Sciences) before the sample was washed three times with distilled water prior to being stained with 0.2% uranyl acetate, blotted again, and air-dried. The grids were imaged using a Hitachi HT7800 transmission electron microscope and images were collected with an EMSIS Xarosa camera.
Protein crystallization and X-ray data collection
EncFtn-sH variants (E31A or E34A) were concentrated to 10 mg/ml (based on extinction coefficient calculation) and subjected to crystallization under similar conditions as the WT protein (Table S9). Crystallization drops were set up in 24-well Linbro plates using the hanging drop vapor diffusion method at 292 K. Glass coverslips were set up with 1–2 μl of protein mixed with 1 μl of well solution and sealed over 1 ml of well solution. Crystals appeared after 1 week to two months and were mounted using a LithoLoop (Molecular Dimensions Limited), transferred briefly to a cryoprotection solution containing well solution supplemented with 1 mm FeSO4 (in 0.1% (v/v) HCl) and 20% (v/v) PEG 200, and subsequently flash-cooled in liquid nitrogen.
All crystallographic datasets were collected at 10–100 eV above the experimentally determined Fe-Kα edge on the macromolecular crystallography beamlines at Diamond Light Source at 100 K using Pilatus 6M detectors. Diffraction data were integrated and scaled using XDS (53), and symmetry related reflections were merged with Aimless (54). Data collection statistics are shown in Table 1. The resolution cutoff used for structure determination and refinement was determined based on the CC1/2 criterion proposed by Karplus and Diederichs (55).
Table 1.
Data collection and refinement statistics
| Data collection | E31A* | E34A* |
|---|---|---|
| Wavelength | 1.74 | 1.74 |
| Resolution range | 49.40–2.66 (2.75–2.66) | 48.73–2.19 (2.27–2.19) |
| Space group | P 1 21 1 | P 1 21 1 |
| Unit cell (Å) a b c |
97.89 120.16 139.37 |
97.79 120.14 139.34 |
| β(°) | 95.33 | 95.20 |
| Total reflections | 310,119 (30,187) | 906,663 (86,277) |
| Unique reflections | 170,103 (16,471) | 31,4834 (30,221) |
| Multiplicity | 1.8 (1.8) | 2.9 (2.9) |
| Completeness (%) | 93.30 (90.35) | 96.37 (92.66) |
| Mean I/sigma(I) | 5.45 (1.40) | 8.04 (1.43) |
| Wilson B-factor (Å2) | 39.14 | 33.05 |
| Rmerge | 0.136 (0.657) | 0.094 (0.688) |
| Rmeas | 0.184 (0.878) | 0.116 (0.842) |
| Rpim | 0.123 (0.578) | 0.065 (0.478) |
| CC1/2 | 0.978 (0.517) | 0.995 (0.579) |
| CC* | 0.994 (0.826) | 0.999 (0.857) |
| Refinement | ||
| Reflections used in refinement | 170,031 (16,463) | 314,739 (30,212) |
| Reflections used for Rfree | 8,346 (794) | 15,590 (1548) |
| Rwork | 0.195 (0.297) | 0.184 (0.282) |
| Rfree | 0.236 (0.327) | 0.212 (0.315) |
| CCwork | 0.946 (0.748) | 0.959 (0.768) |
| CCfree | 0.914 (0.684) | 0.952 (0.689) |
| Model | ||
| Number of nonhydrogen atoms | 22,382 | 22,760 |
| macromolecules | 21,988 | 22,072 |
| ligands | 65 | 48 |
| solvents | 329 | 640 |
| Protein residues | 2680 | 2696 |
| RMS(bonds) (Å) | 0.007 | 0.005 |
| RMS(angles) (°) | 1.03 | 0.87 |
| Ramachandran | ||
| favored (%) | 99.69 | 99.77 |
| allowed (%) | 0.31 | 0.23 |
| outliers (%) | 0.00 | 0.00 |
| Rotamer outliers (%) | 0.26 | 1.07 |
| Clashscore | 4.79 | 3.01 |
| Average B-factor (Å2) | 41.64 | 40.53 |
| macromolecules | 41.66 | 40.52 |
| ligands | 55.57 | 46.17 |
| solvents | 37.08 | 40.62 |
| PDB ID | 6SUW | 6SV1 |
Statistics for the highest-resolution shell are shown in parentheses.
* Friedel pairs are treated as separate reflections for data analysis and refinement.
The structures of the EncFtn-sH variants were determined by molecular replacement using decameric WT protein (PDB ID: 5DA5) as the search model (15). A single solution comprising three decamers in the asymmetric unit was found by molecular replacement using Phaser (56). The initial model was rebuilt using Phenix.autobuild (57) followed by cycles of refinement with Phenix.refine (58), with manual rebuilding and model inspection in Coot (59). The final model was refined with isotropic B-factors, torsional NCS restraints, and anomalous group refinement to correctly model the iron ions in the ferroxidase center. The model was validated using MolProbity (60). Structural superimpositions were calculated using Coot (59). Crystallographic figures were generated with PyMOL (Schrödinger, LLC.). Model refinement statistics are shown in Table 1. The final models and experimental data are deposited in the PDB.
MS analysis
LC–MS, native MS, ion-mobility MS (IM-MS), and CIU experiments were performed on a Synapt G2 ion-mobility equipped Q-ToF instrument (Waters, Manchester, UK). For LC–MS analysis, an ACQUITY UPLC equipped with a reverse phase C4 Aeris Widepore 50 × 2.1-mm HPLC column (Phenomenex) equilibrated with 0.1 % formic acid in water. The sample was eluted over 10 minutes with a 5–95 % gradient of 0.1 % formic acid in acetonitrile. For LC–MS, samples were typically analyzed at 5 μm, and data analysis was performed using MassLynx v4.1 and MaxEnt deconvolution.
For native MS analysis, all protein samples were buffer-exchanged into ammonium acetate (100 mm, pH 8.0) using Micro Biospin Chromatography Columns (Bio-Rad) prior to analysis, and the resulting protein samples were analyzed at a typical final concentration of ∼5 μm (oligomer concentration). Nanoelectrospray ionization was employed using a Nanomate infusion robot (Advion Biosciences, Ithaca, NY). Instrument parameters were tuned to preserve noncovalent protein complexes and were consistent for the analysis of the EncFtn variants. After native MS optimization, parameters were as follows: nanoelectrospray voltage 1.55 kV, sample cone 100 V, extractor cone 0 V, trap collision voltage 4 V, source temperature 60 °C, and source backing pressure 4.5 mbar. IM-MS was performed using the traveling-wave mobility cell of the Synapt G2 with nitrogen drift gas. The typical conditions used for IM-MS were as follows: wave velocity 300 m/s, wave height 19 V, and sampling cone of 75 V. For CIU experiments, the samples were analyzed with incremental increasing trap voltages between 10–60 V using 2-V increments. Data were processed and CIU50 values were determined using CIUSuite (43) and CIUSuite 2 (44).
High-resolution native MS was performed on a 12T SolariX 2XR FT-ICR MS (Bruker Daltonics) equipped with a nanoelectrospray source. Because of the quadrupole range of the FT-ICR MS instrument used, a truncated and strep(II)-tagged version of EncFtn-WT was used for these experiments. The protein samples were buffer-exchanged into ammonium acetate (100 mm, pH 8.0) before direct infusion. Source conditions and ion optics were optimized to transmit native proteins ions and when required, continual accumulation of selected ions was employed to isolate charge states of interest. Typically, 2 Megaword data were collected in quadrupolar detection (2ω) mode to produce a 6-s free-induction decay, which resulted in a typical mass resolving power of ∼300,000. The resulting data were processed using Data Analysis (Bruker Daltonics). For metal titrations, fresh iron(II) chloride tetrahydrate or zinc acetate in 0.1% (v/v) HCl was added to ID-EncFtn (1:1 metal:protein concentration) before buffer exchange into ammonium acetate (100 mm, pH 8.0).
Ferroxidase assay of EncFtn proteins
Enzymatic activity of EncFtn proteins was tested by ferroxidase assay, as previously described (14). Fe(II) aliquots were prepared under anaerobic conditions by dissolving FeSO4·7H2O in 0.1% (v/v) HCl. Purified protein was diluted anaerobically in Buffer H (10 mm HEPES, pH 8.0, 150 mm NaCl), previously purged with gaseous nitrogen, to a final concentration of 10 μm (EncFtn).
Protein and ferrous iron samples were added to a quartz cuvette (Hellma) under aerobic conditions at a final concentration of 10 μm and 50 μm, respectively, corresponding to a FOC:Fe(II) ratio of 1:10.
Absorbance at 315 nm was recorded every second for 1500 s by a UV-visible spectrophotometer (PerkinElmer, Lambda 35), using the TimeDrive software. The same experiment was performed in the absence of the enzyme to determine the oxidation of ferrous salts by atmospheric oxygen.
The same setup was adopted for recording the activity of EncFtn-E34A and EncFtn-E31A/E34A in a separate assay at a higher concentration (20 μm EncFtn and 100 μm Fe(II)) for comparison purposes. Data presented here are the mean of three technical replicates of time zero–subtracted progress curves with standard deviations calculated from the mean.
Calculation of the EncFtn enzymatic reaction initial rate (v0) was made by applying the Linear Regression tool in GraphPad (Prism8) on the absorbance at 315 nm measured for the first 200 s, when curves are still linear and following zero-order kinetics (Fig. S8 and Table S3). Slopes obtained from these curves correspond to the initial reaction rate. v0variant/v0wild type factors were calculated by dividing the initial rates of the variant enzymes by the initial rate of the WT protein (Table S5). Data for ferroxidase assays are available at doi. 10.6084/m9.figshare.9885575.
Ferroxidase assay of Enc:EncFtn protein complexes
Ferroxidase assay of Enc:EncFtn protein complexes was performed in the same conditions as those used for EncFtn but with different protein and iron salt concentration. Enc:EncFtn was diluted to a final concentration of 25 μm, which corresponds to 10 μm EncFtn based on a 3:2 Enc:EncFtn ratio, as previously observed by SDS-PAGE by our group (Fig. 1A in He et al. (15)). Fe(II) sample was diluted to 50 μm in the reaction system to maintain a final ratio of 1:10 FOC:Fe(II).
Calculation of the Enc-EncFtn enzymatic reaction initial rate (v0) and v0variant/v0wild type factors was carried out as described in the above section (Fig. S8, Table S4, and Table S5).
Zinc inhibition of ferroxidase activity of EncFtn proteins
Ferroxidase assay was performed as above with EncFtn (10 μm) and Fe(II) samples (50 μm) in the presence of 34 μm ZnSO4.7H2O. The chosen concentration corresponds to the IC50 value previously determined for EncFtn-StrepII under the same experimental conditions by our group (14). Oxygen-free metal samples were added to the quartz cuvette under aerobic conditions, followed by the protein sample. Data were replicated three times and means and standard deviation of time zero-subtracted progress curves were calculated. A negative control was performed by monitoring A315 of Zn(II) and Fe(II) salts mixed in the absence of enzyme.
Monitoring quenching of protein intrinsic fluorescence upon metal binding
Experiments were carried out aerobically in a quartz cuvette (Hellma) using a Cary Eclipse Fluorescence spectrophotometer. Protein was diluted in Buffer GF to a final concentration of 20 μm, corresponding to 10 μm for the entry site. ZnSO4×7H2O stock was prepared in deionized water. The instrument excitation wavelength was set to 280 nm, corresponding to maximum absorption for tryptophan residues.
Two distinct modes of detection were used; the Kinetic option on the Analysis software package was chosen to record protein fluorescence emission signal at specific wavelengths over time. Pre-scans were carried out to find the optimal emission wavelength (ranging from 322–355 nm) for each protein variant. Metal aliquots (0.15 or 0.3 molar equivalents) were added to the cuvette while pausing the data collection for fewer than 5 s. The final data represent an average of 10 data points at equilibrium upon each metal addition.
The Scan option of the Analysis software package was chosen to record emission in the 290–400 nm range, allowing detection of possible shifts in emission peak maximum upon metal addition. Spectra were recorded in triplicate at equilibrium. Intrinsic fluorescence data is available at: doi. 10.6084/m9.figshare.11920512.
Data availability
Data sets supporting this paper have been deposited in the appropriate public data repositories. Please see Experimental Procedures and tables for links to these.
Supplementary Material
Acknowledgments
We thank staff at the Newcastle University Electron Microscopy Research Services for assistance with transmission EM. We thank Diamond Light Source for beamtime (proposal mx9487) and the staff of beamlines I02, I03, and I24 for assistance with crystal testing and data collection. We thank Prof. Dominic Campopiano and Gregor Skeldon for constructive advice throughout this project.
This article contains supporting information.
Author contributions—C. P., J. R., D. H., D. J. C., and J. M.-W. conceptualization; C. P., J. R., D. J. C., and J. M.-W. data curation; C. P., J. R., D. H., A. B., K. J. W., D. J. C., and J. M.-W. formal analysis; C. P., J. R., D. J. C., and J. M.-W. validation; C. P., J. R., D. H., K. J. G., W. A. S., L. A., K. J. W., D. J. C., and J. M.-W. investigation; C. P., J. R., W. A. S., A. B., D. J. C., and J. M.-W. visualization; C. P., J. R., D. H., K. J. G., C. L. M., K. J. W., and D. J. C. methodology; C. P. and J. R. writing-original draft; C. P., J. R., D. H., K. J. W., D. J. C., and J. M.-W. writing-review and editing; K. J. G., C. L. M., and K. J. W. resources; D. J. C. and J. M.-W. supervision; D. J. C. and J. M.-W. funding acquisition; D. J. C. and J. M.-W. project administration.
Funding and additional information—This work was supported by Royal Society Research Grant RG130585 (to J. M.-W.), Biotechnology and Biological Sciences Research Council New Investigator Grant BB/N005570/1 (to J. M.-W., D. J. C., C. P., and W. A. S.), Newcastle University (J. M.-W.), the University of Edinburgh (D. J. C. and J. R.), Biotechnology and Biological Sciences Research Council EastBio DTP studentship BB/M010996/1 (to J. R.), the China Scholarship Council (D. H.), and the Wellcome Trust and Royal Society through Sir Henry Dale Fellowship 098375/Z/12/Z (to K. J. W.). Equipment for transmission electron microscopy was funded through the Biotechnology and Biological Sciences Research Council 17ALERT call BB/R013942/1 (to J. M.-W.). Fourier-transform ion cyclotron resonance instrumentation was funded by Biotechnology and Biological Sciences Research Council 17ALERT BB/R013993/1 (to D. J. C.).
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- Enc
- encapsulin
- EncFtn
- encapsulated ferritin
- FOC
- ferroxidase center
- CIU
- collision-induced unfolding
- FT-ICR
- Fourier-transform ion cyclotron resonance
- ID
- isotope depletion
- IMEF
- iron-mineralizing encapsulin-associated firmicute
- DPS
- dodecameric DNA protection in starved cells
- DPSL
- DPS-like
- sH
- hexahistidine
- IM-MS
- ion mobility MS.
References
- 1. Andrews S. C. (2010) The Ferritin-like superfamily: evolution of the biological iron storeman from a rubrerythrin-like ancestor. Biochim. Biophys. Acta 1800, 691–705 10.1016/j.bbagen.2010.05.010 [DOI] [PubMed] [Google Scholar]
- 2. Dillard B. D., Demick J. M., Adams M. W. W., and Lanzilotta W. N. (2011) A cryo-crystallographic time course for peroxide reduction by rubrerythrin from Pyrococcus furiosus. JBIC J. Biol. Inorg. Chem. 16, 949–959 10.1007/s00775-011-0795-6 [DOI] [PubMed] [Google Scholar]
- 3. Cardenas J. P., Quatrini R., and Holmes D. S. (2016) Aerobic lineage of the oxidative stress response protein rubrerythrin emerged in an ancient microaerobic, (hyper)thermophilic environment. Front. Microbiol. 7, 1822 10.3389/fmicb.2016.01822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Giessen T. W., Orlando B. J., Verdegaal A. A., Chambers M. G., Gardener J., Bell D. C., Birrane G., Liao M., and Silver P. A. (2019) Large protein organelles form a new iron sequestration system with high storage capacity. eLife. 8, e46070 10.7554/eLife.46070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Grant R. A., Filman D. J., Finkel S. E., Kolter R., and Hogle J. M. (1998) The crystal structure of Dps, a ferritin homolog that binds and protects DNA. Nat. Struct. Biol. 5, 294–303 10.1038/nsb0498-294 [DOI] [PubMed] [Google Scholar]
- 6. Pesek J., Büchler R., Albrecht R., Boland W., and Zeth K. (2011) Structure and mechanism of iron translocation by a Dps protein from Microbacterium arborescens. J. Biol. Chem. 286, 34872–34882 10.1074/jbc.M111.246108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Martinez A., and Kolter R. (1997) Protection of DNA during oxidative stress by the nonspecific DNA-binding protein Dps. J. Bacteriol. 179, 5188–5194 10.1128/jb.179.16.5188-5194.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gauss G. H., Benas P., Wiedenheft B., Young M., Douglas T., and Lawrence C. M. (2006) Structure of the DPS-like protein from Sulfolobus solfataricus reveals a bacterioferritin-like dimetal binding site within a DPS-like dodecameric assembly. Biochemistry 45, 10815–10827 10.1021/bi060782u [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gauss G. H., Reott M. A., Rocha E. R., Young M. J., Douglas T., Smith C. J., and Lawrence C. M. (2012) Characterization of the Bacteroides fragilis bfr gene product identifies a bacterial DPS-like protein and suggests evolutionary links in the ferritin superfamily. J. Bacteriol. 194, 15–27 10.1128/JB.05260-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Andrews S. C. (1998) Iron storage in bacteria. Adv. Microb. Physiol. 40, 281–351 10.1016/s0065-2911(08)60134-4 [DOI] [PubMed] [Google Scholar]
- 11. Pfaffen S., Abdulqadir R., Le Brun N. E., and Murphy M. E. P. (2013) Mechanism of ferrous iron binding and oxidation by ferritin from a pennate diatom. J. Biol. Chem. 288, 14917–14925 10.1074/jbc.M113.454496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Frolow F., Kalb A. J., and Yariv J. (1994) Structure of a unique twofold symmetric haem-binding site. Nat. Struct. Biol. 1, 453–460 10.1038/nsb0794-453 [DOI] [PubMed] [Google Scholar]
- 13. Harrison P. M., and Arosio P. (1996) The ferritins: molecular properties, iron storage function and cellular regulation. Biochim. Biophys. Acta 1275, 161–203 10.1016/0005-2728(96)00022-9 [DOI] [PubMed] [Google Scholar]
- 14. He D., Piergentili C., Ross J., Tarrant E., Tuck L. R., Mackay C. L., McIver Z., Waldron K. J., Clarke D. J., and Marles-Wright J. (2019) Conservation of the structural and functional architecture of encapsulated ferritins in bacteria and archaea. Biochem. J. 476, 975–989 10.1042/BCJ20180922 [DOI] [PubMed] [Google Scholar]
- 15. He D., Hughes S., Vanden-Hehir S., Georgiev A., Altenbach K., Tarrant E., Mackay C. L., Waldron K. J., Clarke D. J., and Marles-Wright J. (2016) Structural characterization of encapsulated ferritin provides insight into iron storage in bacterial nanocompartments. eLife. 5, e18972 10.7554/eLife.18972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ratnayake D. B., Wai S. N., Shi Y., Amako K., Nakayama H., and Nakayama K. (2000) Ferritin from the obligate anaerobe Porphyromonas gingivalis: purification, gene cloning and mutant studies The GenBank accession number for the sequence reported in this paper is AB016086. Microbiology 146, 1119–1127 10.1099/00221287-146-5-1119 [DOI] [PubMed] [Google Scholar]
- 17. Sztukowska M., Bugno M., Potempa J., Travis J., and Kurtz D. M. Jr (2002) Role of rubrerythrin in the oxidative stress response of Porphyromonas gingivalis. Mol. Microbiol. 44, 479–488 10.1046/j.1365-2958.2002.02892.x [DOI] [PubMed] [Google Scholar]
- 18. Almirón M., Link A. J., Furlong D., and Kolter R. (1992) A novel DNA-binding protein with regulatory and protective roles in starved Escherichia coli. Genes Dev. 6, 2646–2654 10.1101/gad.6.12b.2646 [DOI] [PubMed] [Google Scholar]
- 19. Choi S. H., Baumler D. J., and Kaspar C. W. (2000) Contribution of dps to acid stress tolerance and oxidative stress tolerance in Escherichia coli O157:H7. Appl. Environ. Microbiol. 66, 3911–3916 10.1128/aem.66.9.3911-3916.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nair S., and Finkel S. E. (2004) Dps protects cells against multiple stresses during stationary phase. J. Bacteriol. 186, 4192–4198 10.1128/JB.186.13.4192-4198.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chiancone E., and Ceci P. (2010) The multifaceted capacity of Dps proteins to combat bacterial stress conditions: detoxification of iron and hydrogen peroxide and DNA binding. Biochim. Biophys. Acta 1800, 798–805 10.1016/j.bbagen.2010.01.013 [DOI] [PubMed] [Google Scholar]
- 22. De Martino M., Ershov D., van den Berg P. J., Tans S. J., and Meyer A. S. (2016) Single-cell analysis of the dps response to oxidative stress. J. Bacteriol. 198, 1662–1674 10.1128/JB.00239-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chasteen N. D., and Harrison P. M. (1999) Mineralization in ferritin: an efficient means of iron storage. J. Struct. Biol. 126, 182–194 10.1006/jsbi.1999.4118 [DOI] [PubMed] [Google Scholar]
- 24. Ross J., Lambert T., Piergentili C., He D., Waldron K. J., Mackay C. L., Marles-Wright J., and Clarke D. J. (2020) Mass spectrometry reveals the assembly pathway of encapsulated ferritins and highlights a dynamic ferroxidase interface. Chem. Commun. 56, 3417–3420 10.1039/c9cc08130e [DOI] [PubMed] [Google Scholar]
- 25. Sutter M., Boehringer D., Gutmann S., Günther S., Prangishvili D., Loessner M. J., Stetter K. O., Weber-Ban E., and Ban N. (2008) Structural basis of enzyme encapsulation into a bacterial nanocompartment. Nat. Struct. Mol. Biol. 15, 939–947 10.1038/nsmb.1473 [DOI] [PubMed] [Google Scholar]
- 26. Xu B., and Chasteen N. (1991) Iron oxidation chemistry in ferritin. Increasing Fe/O2 stoichiometry during core formation. J. Biol. Chem. 266, 19965–19970 [PubMed] [Google Scholar]
- 27. Proulx-Curry P. M., and Chasteen N. D. (1995) Molecular aspects of iron uptake and storage in ferritin. Coord. Chem. Rev. 144, 347–368 10.1016/0010-8545(95)01148-I [DOI] [Google Scholar]
- 28. Yang X., Chen-Barrett Y., Arosio P., and Chasteen N. D. (1998) Reaction paths of iron oxidation and hydrolysis in horse spleen and recombinant human ferritins. Biochemistry 37, 9743–9750 10.1021/bi973128a [DOI] [PubMed] [Google Scholar]
- 29. Baaghil S., Lewin A., Moore G. R., and Le Brun N. E. (2003) Core formation in Escherichia coli bacterioferritin requires a functional ferroxidase center. Biochemistry 42, 14047–14056 10.1021/bi035253u [DOI] [PubMed] [Google Scholar]
- 30. Ebrahimi K. H., Hagedoorn P.-L., Jongejan J. A., and Hagen W. R. (2009) Catalysis of iron core formation in Pyrococcus furiosus ferritin. J. Biol. Inorg. Chem. 14, 1265–1274 10.1007/s00775-009-0571-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Theil E. C., Behera R. K., and Tosha T. (2013) Ferritins for chemistry and for life. Coord. Chem. Rev. 257, 579–586 10.1016/j.ccr.2012.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Masuda T., Goto F., Yoshihara T., and Mikami B. (2010) Crystal structure of plant ferritin reveals a novel metal binding site that functions as a transit site for metal transfer in ferritin. J. Biol. Chem. 285, 4049–4059 10.1074/jbc.M109.059790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Behera R. K., Torres R., Tosha T., Bradley J. M., Goulding C. W., and Theil E. C. (2015) Fe2+ substrate transport through ferritin protein cage ion channels influences enzyme activity and biomineralization. J. Biol. Inorg. Chem. 20, 957–969 10.1007/s00775-015-1279-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yao H., Wang Y., Lovell S., Kumar R., Ruvinsky A. M., Battaile K. P., Vakser I. A., and Rivera M. (2012) The structure of the BfrB–Bfd complex reveals protein–protein interactions enabling iron release from bacterioferritin. J. Am. Chem. Soc. 134, 13470–13481 10.1021/ja305180n [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bellapadrona G., Stefanini S., Zamparelli C., Theil E. C., and Chiancone E. (2009) iron translocation into and out of Listeria innocua dps and size distribution of the protein-enclosed nanomineral are modulated by the electrostatic gradient at the 3-fold “ferritin-like” pores. J. Biol. Chem. 284, 19101–19109 10.1074/jbc.M109.014670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu X. S., Patterson L. D., Miller M. J., and Theil E. C. (2007) Peptides selected for the protein nanocage pores change the rate of iron recovery from the ferritin mineral. J. Biol. Chem. 282, 31821–31825 10.1074/jbc.C700153200 [DOI] [PubMed] [Google Scholar]
- 37. Trefry A., and Harrison P. M. (1978) Incorporation and release of inorganic phosphate in horse spleen ferritin. Biochem. J. 171, 313–320 10.1042/bj1710313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Treffry A., Zhao Z., Quail M. A., Guest J. R., and Harrison P. M. (1998) How the presence of three iron binding sites affects the iron storage function of the ferritin (EcFtnA) of Escherichia coli. FEBS Lett. 432, 213–218 10.1016/S0014-5793(98)00867-9 [DOI] [PubMed] [Google Scholar]
- 39. Ebrahimi K. H., Bill E., Hagedoorn P.-L., and Hagen W. R. (2016) Spectroscopic evidence for the role of a site of the di-iron catalytic center of ferritins in tuning the kinetics of Fe(ii) oxidation. Mol. Biosyst. 12, 3576–3588 10.1039/c6mb00235h [DOI] [PubMed] [Google Scholar]
- 40. Hagen W. R., Hagedoorn P. L., and Honarmand Ebrahimi K. (2017) The workings of ferritin: a crossroad of opinions. Metallomics 9, 595–605 10.1039/c7mt00124j [DOI] [PubMed] [Google Scholar]
- 41. Hempstead P. D., Hudson A. J., Artymiuk P. J., Andrews S. C., Banfield M. J., Guest J. R., and Harrison P. M. (1994) Direct observation of the iron binding sites in a ferritin. FEBS Lett. 350, 258–262 10.1016/0014-5793(94)00781-0 [DOI] [PubMed] [Google Scholar]
- 42. Stillman T. J., Hempstead P. D., Artymiuk P. J., Andrews S. C., Hudson A. J., Treffry A., Guest J. R., and Harrison P. M. (2001) The high-resolution X-ray crystallographic structure of the ferritin (EcFtnA) of Escherichia coli; comparison with human H ferritin (HuHF) and the structures of the Fe(3+) and Zn(2+) derivatives. J. Mol. Biol. 307, 587–603 10.1006/jmbi.2001.4475 [DOI] [PubMed] [Google Scholar]
- 43. Eschweiler J. D., Rabuck-Gibbons J. N., Tian Y., and Ruotolo B. T. (2015) CIUSuite: a quantitative analysis package for collision induced unfolding measurements of gas-phase protein ions. Anal. Chem. 87, 11516–11522 10.1021/acs.analchem.5b03292 [DOI] [PubMed] [Google Scholar]
- 44. Polasky D. A., Dixit S. M., Fantin S. M., and Ruotolo B. T. (2019) CIUSuite 2: next-generation software for the analysis of gas-phase protein unfolding data. Anal. Chem. 91, 3147–3155 10.1021/acs.analchem.8b05762 [DOI] [PubMed] [Google Scholar]
- 45. Le Brun N. E., Andrews S. C., Guest J. R., Harrison P. M., Moore G. R., and Thomson A. J. (1995) Identification of the ferroxidase centre of Escherichia coli bacterioferritin. Biochem. J. 312, 385–392 10.1042/bj3120385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gallagher K. J., Palasser M., Hughes S., Mackay C. L., Kilgour D. P. A., and Clarke D. J. (2020) Isotope depletion mass spectrometry (ID-MS) for accurate mass determination and improved top-down sequence coverage of intact proteins. J. Am. Soc. Mass Spectrom. 31, 700–710 10.1021/jasms.9b00119 [DOI] [PubMed] [Google Scholar]
- 47. Hall Z., Hernández H., Marsh J. A., Teichmann S. A., and Robinson C. V. (2013) The role of salt bridges, charge density, and subunit flexibility in determining disassembly routes of protein complexes. Structure 21, 1325–1337 10.1016/j.str.2013.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Foster A. W., Osman D., and Robinson N. J. (2014) Metal preferences and metallation. J. Biol. Chem. 289, 28095–28103 10.1074/jbc.R114.588145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tottey S., Waldron K. J., Firbank S. J., Reale B., Bessant C., Sato K., Cheek T. R., Gray J., Banfield M. J., Dennison C., and Robinson N. J. (2008) Protein-folding location can regulate manganese-binding versus copper- or zinc-binding. Nature 455, 1138–1142 10.1038/nature07340 [DOI] [PubMed] [Google Scholar]
- 50. Fraústo da Silva J. J. R., and Williams R. J. P. (1991) The biological chemistry of the elements: the inorganic chemistry of life. Oxford Univ. Press [Google Scholar]
- 51. Reyes-Caballero H., Campanello G. C., and Giedroc D. P. (2011) Metalloregulatory proteins: metal selectivity and allosteric switching. Biophys. Chem. 156, 103–114 10.1016/j.bpc.2011.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schägger H. (2006) Tricine–SDS-PAGE. Nat. Protoc. 1, 16–22 10.1038/nprot.2006.4 [DOI] [PubMed] [Google Scholar]
- 53. Kabsch W. (2010) Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 133–144 10.1107/S0907444909047374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Evans P. R. (2011) An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr. D. Biol. Crystallogr. 67, 282–292 10.1107/S090744491003982X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Karplus P. A., and Diederichs K. (2012) Linking crystallographic model and data quality. Science 336, 1030–1033 10.1126/science.1218231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., and Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. Sect. D Biol. Crystallogr. 68, 352–367 10.1107/S0907444912001308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chen V., Arendall W., Headd J., Keedy D., Immormino R., Kapral G., Murray L., Richardson J., and Richardson D. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 12–21 10.1107/S0907444909042073 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data sets supporting this paper have been deposited in the appropriate public data repositories. Please see Experimental Procedures and tables for links to these.