

Abstract
A large number of newly synthesized membrane proteins in the endoplasmic reticulum (ER) are assembled into multiprotein complexes, but little is known about the mechanisms required for assembly membrane proteins. It has been suggested that membrane chaperones might exist, akin to the molecular chaperones that stabilize and direct the assembly of soluble protein complexes, but the mechanisms by which these proteins would bring together membrane protein components is unclear. Here, we have identified that the tail length of the C-terminal transmembrane domains (C-TMDs) determines efficient insertion and assembly of membrane proteins in the ER. We found that membrane proteins with C-TMD tails shorter than ∼60 amino acids are poorly inserted into the ER membrane, which suggests that translation is terminated before they are recognized by the Sec61 translocon for insertion. These C-TMDs with insufficient hydrophobicity are post-translationally recognized and retained by the Sec61 translocon complex, providing a time window for efficient assembly with TMDs from partner proteins. Retained TMDs that fail to assemble with their cognate TMDs are slowly translocated into the ER lumen and are recognized by the ER-associated degradation (ERAD) pathway for removal. In contrast, C-TMDs with sufficient hydrophobicity or tails longer than ∼80 residues are quickly released from the Sec61 translocon into the membrane or the ER lumen, resulting in inefficient assembly with partner TMDs. Thus, our data suggest that C-terminal tails harbor crucial signals for both the insertion and assembly of membrane proteins.
Keywords: Sec61 translocon, membrane protein insertion, membrane protein assembly, translocation, membrane protein, protein assembly, protein degradation, endoplasmic reticulum (ER)
Membrane proteins represent one-third of the proteins encoded by the human genome and carry out essential cellular processes including molecular transport, signaling, and cell–cell communication (1, 2). Most membrane proteins utilize the co-translational pathway for insertion into the endoplasmic reticulum (ER). As the hydrophobic signal sequence or the first transmembrane domain (TMD) of a membrane protein emerges from the cytosolic ribosome, the signal recognition particle (SRP) captures it and delivers the ribosome nascent chain complex to the ER membrane via the SRP receptor (3–5). The SRP-bound TMD is then transferred to the Sec61 translocon channel in the ER membrane. The hydrophobic TMD of a nascent membrane protein engages the lateral gate of the channel, where it can sample the hydrophobic chains of phospholipids (6). Subsequently, the TMD partitions into the lipid phase as the nascent chain further elongates during translation. It is generally believed that TMDs of membrane proteins are sequentially inserted into the ER membrane (7).
It is estimated that over 40% of newly synthesized membrane proteins assemble into multiprotein complexes (8). Even though these multimembrane protein complexes serve fundamental functions in cells, the mechanisms that govern the assembly of individual membrane proteins into complexes are poorly understood. The assembly of newly synthesized membrane proteins faces several challenges in the two-dimensional ER membrane. For instance, unassembled membrane proteins can have nonproductive interactions, leading to misfolding, aggregation, and degradation (9, 10). Second, unassembled membrane proteins are a potential target for ER-associated degradation (ERAD) pathways for elimination before their assembly with partner proteins (11). Third, because the ER is the largest membrane network within eukaryotic cells, the newly synthesized subunits can move from the rough ER to the other part of the ER (12–14), thus diluting their concentration as well as reducing their chance to find each other. Last, proteins have often the propensity to form homooligomers because they are translated from the same polysome and their local subunit concentration is high, thus reducing the chance to assemble with the partner protein. In recent years, much attention has been focused on understanding the assembly of soluble protein complexes (15). These studies propose two prevailing models. First, the large protein complexes such as proteasome often have dedicated molecular chaperones that can shield the nascent proteins from inappropriate interactions and facilitate assembly with partner proteins (16). Second, the newly synthesized proteins can be co-translationally assembled into heterooligomeric complexes in eukaryotes (17).
In contrast to soluble protein complexes, less is known about the assembly of membrane protein complexes in the ER membrane, where the majority of the membrane proteins are synthesized in cells. Pioneering early studies have used the T-cell receptor (TCR) complex as a model membrane protein complex to investigate this problem (18). The opposite charge residues in TMDs of membrane proteins drive assembly by forming ionic bonds between subunits (18–20). The unassembled TCR subunits can be translocated completely into the ER lumen where they are recognized by ERAD components for degradation (21). However, it is unclear how these two different subunits find each other in the extensive ER membrane network after their synthesis and form a specific ionic pair. Moreover, this is even less understood for assembly of polytopic membrane protein complexes, but it is often proposed that membrane chaperones may mediate the assembly process. However, experimental evidence of membrane chaperones mediating the assembly of membrane protein complexes is lacking.
To reveal the biochemical features that are necessary for both assembly and degradation of membrane protein complexes, we investigated the assembly of the tail-anchored membrane protein insertion complex composed of WRB and CAML proteins (22–24). While this study was under preparation, two recent studies have reported that WRB plays a crucial role in fixing the topology of CAML (25, 26). In the absence of WRB, CAML exhibits two different topologies: one with both TMD2 and TMD3 translocated into the ER lumen and the other with only TMD2 translocated into the ER lumen (25, 26). The molecular mechanism by which newly synthesized WRB and CAML are brought together for efficient assembly is unclear. In this study, we identified that the Sec61 translocon can post-translationally retain membrane proteins. This retention mechanism depends on less hydrophobic C-TMDs with shorter tails. The Sec61 translocon-retained TMD provides a time window for efficient assembly with TMDs from partner membrane proteins. If they missed assembly with their partner TMDs, C-TMDs are slowly translocated into the ER lumen and are recognized for degradation by ERAD. Thus, our studies suggest that the C-terminal TMD and its flanking cytosolic tails are important biochemical features for the assembly of membrane protein complexes.
Results
The C-terminal TMD hydrophobicity plays a role in the degradation of WRB
To determine the biochemical features that are required for the assembly of multimembrane protein complexes, we sought for model membrane protein substrates that should fulfill three criteria. First, they must be relatively small and amenable to biochemical manipulations. Second, they should be quickly degraded when they failed to assemble. Third, degradation signals or degrons should be buried when they are assembled, but exposed when they fail to assemble. We reasoned that tail-anchored membrane protein insertase complex, WRB and CAML, may be ideal substrates to investigate this fundamental problem because both proteins are relatively small in their sizes (20 and 33 kDa, respectively) with each having three TMDs (Fig. 1A). We first tested whether these proteins are degraded when transiently expressed individually in HEK293 cells. The cycloheximide chase assay revealed that the expression of either WRB or CAML alone led to degradation in a proteasomal dependent manner because the turnover could be inhibited by the proteasomal inhibitor MG132 (Fig. 1, B and C). However, the co-expression of both WRB and CAML slowed their degradation compared with when they were expressed separately (Fig. 1D).
Figure 1.
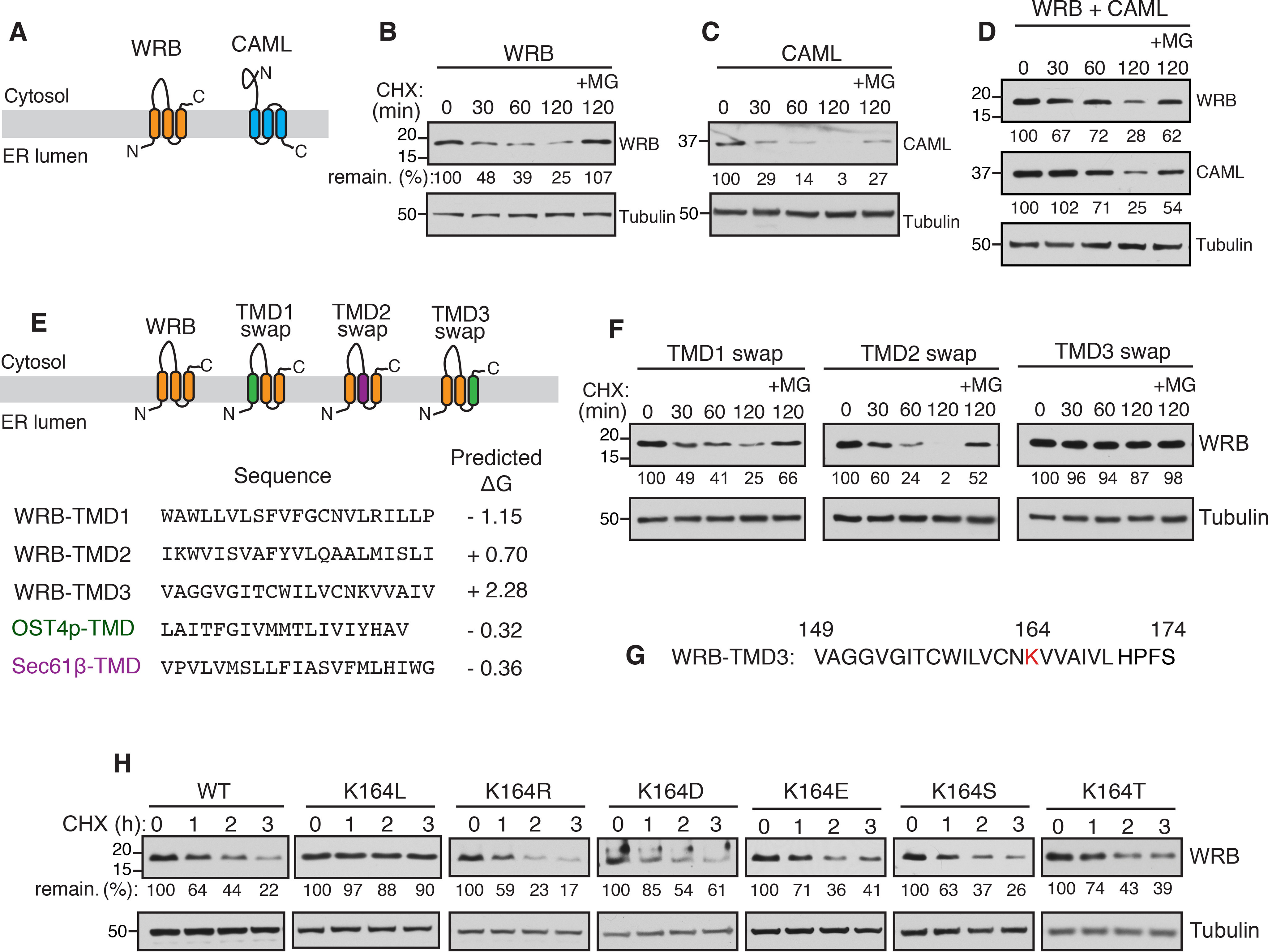
The C-TMD of WRB is recognized by ERAD due to its insufficient hydrophobicity. A, schematics showing the predicted topologies of WRB and CAML. B, WRB-FLAG was transfected into HEK293 cells. After 24 h of transfection, cells were treated with cycloheximide (CHX) for the indicated time points and analyzed by immunoblotting with an anti-FLAG antibody for WRB. Tubulin was immunoblotted as a loading control. The protein level at 0-h time point was taken as 100%, and the percentage of the remaining protein was calculated with respect to 0 h. C and D, the cycloheximide chase experiments were performed and analyzed for the indicated constructs as in B. E, schematics showing the swapping of TMDs of WRB with either the TMD from Ost4p for TMD1 and TMD3 swaps (shown in green) or the TMD from Sec61β for the TMD2 swap (shown in purple). F, the cycloheximide chase experiments were performed and analyzed for the indicated WRB swaps as in B. G, a single lysine (K164) is indicated in red in the amino acid sequence of WRB TMD3. H, the cycloheximide chase experiments were performed and analyzed for the indicated constructs as in B.
We hypothesized that the unassembled membrane proteins must expose a degradation signal or degron for recognition by ERAD. To investigate this, we focused on WRB as a model substrate. We reasoned that the degron must lie in one of three TMDs of WRB because WRB lacks a prominent luminal domain. Also, the cytosolic tryptophan-rich domain of WRB is functional without the rest of the protein (22). Therefore, we swapped one TMD at a time with a stable TMD from either Sec61β or Ost4p (Fig. 1E) based on the previously described protocol (27). Replacing the first TMD of WRB led to the degradation similar to the WT (Fig. 1F). The second TMD swap exhibited a slightly faster degradation. Remarkably, swapping the 3rd TMD resulted in the complete stabilization of WRB, suggesting that the 3rd TMD contains a degron (Fig. 1F). The close inspection of the amino acid sequence of the C-TMD revealed that it has a single positively charged lysine residue (Fig. 1G). To test whether the lysine residue in the C-TMD is required for the recognition by ERAD, we mutated lysine to the hydrophobic leucine residue and analyzed the residue by protein turnover assay. In agreement with our view, the degradation of WRB (K164L) was completely inhibited compared with the WT (Fig. 1H), confirming that the lysine residue in the C-TMD is crucial for degradation. To our surprise, replacing the lysine residue with either a charged residue or a hydrophilic residue also caused destabilization (Fig. 1H). These results suggested that the lysine residue in the C-TMD of WRB is not the direct signal for degradation, but rather indicated that the overall hydrophobicity of the C-TMD plays a role in recognition by ERAD factors for degradation.
C-TMDs of unassembled membrane proteins are translocated into the ER lumen
Because the C-TMD of WRB has a relatively low apparent free energy (ΔGapp = +2.28) (28, 29), we asked whether the C-TMD is properly inserted into the ER membrane. A negative value of free energy indicates that the sequence can be efficiently recognized as a TMD by the Sec61 translocon and integrated into the lipid bilayer. Conversely, a positive value of free energy indicates that the sequence is less efficiently inserted unless it is helped by interactions with neighboring TMDs (30). To test if the C-TMD of WRB is translocated into the ER lumen, we appended a glycosylation site (NFT) at the C terminus of WRB (Fig. S1A). Interestingly, about 37% of the C-TMD of WRB was translocated into the ER lumen (Fig. 2A). The glycosylated band of WRB was verified by the treatment with endoglycosidase H (Endo H). The pulse-chase experiment revealed that both the glycosylated (translocated) and nonglycosylated (inserted) forms of WRB were recognized by ERAD for degradation (Fig. 2B). We next asked whether the C-TMD translocation into the ER lumen is a common feature for unassembled membrane proteins. To address this, we looked for other membrane protein complexes. The Sec61 translocon is composed of three subunits, α, β, and γ. We reasoned that the C-TMD of the α subunit, which has a low apparent free energy of +2.95 (Fig. 2C), might translocate into the ER lumen if it failed to assemble with other two subunits. To test this, we added an N-glycosylation motif to the C terminus of Sec61α and monitored its translocation into the lumen. Consistent with our prediction, a small fraction of Sec61α was translocated into the ER lumen as shown by an Endo H-sensitive glycosylated band (Fig. 2C). We next tested the translocation of the C-TMD of TRAPγ, which forms a heterotrimeric complex with α and β subunits (31). The C-TMD of TRAPγ exhibits a low apparent free energy (ΔGapp = +2.06) due to the presence of several hydrophilic serine residues (Fig. 2D). About 50% of TRAPγ was translocated into the ER lumen as shown by the glycosylation assay (Fig. 2D). We further examined whether the translocation of the C-TMD of a polytopic membrane protein might be an indicator of defects in the assembly of TMDs within the same protein. To test this, we used peripheral myelin protein 22 (PMP22) as a model substrate because many patients affected by Charcot-Marie-Tooth disease possess mutations in TMDs of PMP22. We reasoned that introducing a patient mutation into the C-TMD of PMP22 (32) may lead to its translocation into the ER lumen due to defects in the assembly with neighboring TMDs. Although C-TMD of WT PMP22 was not translocated into the ER, the C-TMD of PMP22 carrying the positively charged arginine residue was translocated and thereby glycosylated in the ER lumen (Fig. 2E). Collectively, these findings suggest that translocation of the C-TMD is a general phenomenon for unassembled membrane proteins.
Figure 2.
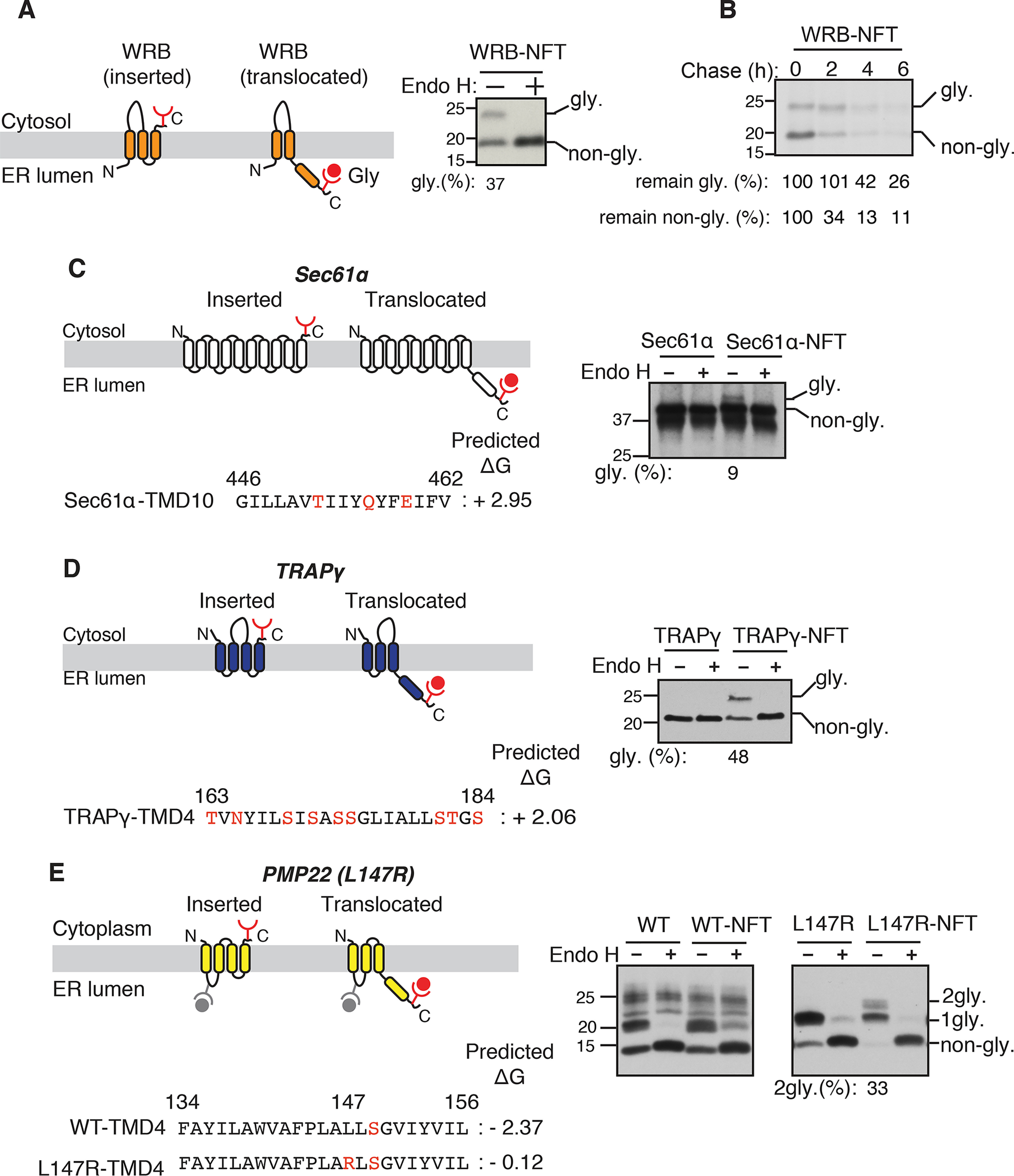
C-terminal TMDs of unassembled membrane proteins are translocated into the ER lumen. A, left panel: a schematic of two different topologies of WRB (inserted and translocated forms) is shown. The glycosylation acceptor site NFT is added after the C terminus HA tag of WRB. When the C-TMD is translocated into the ER lumen, the NFT site becomes glycosylated. Right panel, the lysate from WRBHA-NFT–transfected cells was treated without or with Endo H and analyzed by immunoblotting with an anti-HA antibody. The percentage of translocated form (glycosylated) from the total was quantified with ImageJ and shown under the blot. B, WRB-HA-NFT–transfected cells were metabolically labeled and chased for the indicated time points and analyzed by autoradiography after immunoprecipitation with anti-HA antibody beads. C, left panel: a diagram showing the topology of Sec61α with both inserted and translocated forms. The amino acid sequence of C-TMD (TMD10) of dog Sec61α and its apparent free energy value are shown. The hydrophilic amino acids in TMD10 are indicated in the red. Right panel, the lysates from either Sec61α-HA or Sec61α-HA-NFT–transfected cells were treated without or with Endo H and analyzed by immunoblotting with anti-HA antibody. The percentage of translocated form (glycosylated) from total was quantified with ImageJ and shown under the immunoblot. D, TRAPγ-HA-NFT and TRAPγ-HA were analyzed as in C. E, WT PMP22-HA and PMP22 (L147R)-HA with and without the C terminus glycosylation tag NFT were analyzed as in C. 2gly, indicates the C-TMD translocated and glycosylated form.
The C-terminal cytosolic tail influences both translocation and proper insertion of C-TMDs
We investigated whether the translocation of C-TMD into the ER lumen is caused by its low hydrophobicity. To test this, we constructed variants of C-TMDs with either increasing hydrophobicity by introducing hydrophobic leucine residues or decreasing hydrophobicity by introducing hydrophilic residues (Fig. 3A). We analyzed the translocation of C-TMDs of WRB variants by the glycosylation assay combined with immunoblotting. C-TMDs of WRBs with insufficient hydrophobicity were translocated into the ER lumen as evidenced by glycosylated bands (Fig. 3B). The efficiency of the C-TMD translocation generally correlated with its hydrophobicity. For instance, about 90% of very hydrophilic C-TMD of WRB (DDRRK) translocated into the ER lumen. Strikingly, increasing the hydrophobicity of C-TMD did not prevent its translocation into the ER lumen, arguing that strong hydrophobicity of a C-TMD is not a determinant for insertion into the membrane (Fig. 3B). This result is further corroborated with the data derived from metabolically labeled cells expressing WRB variants as well as the data from an in vitro experiment where WRB variants were translated in the presence of rough microsomes (Fig. 3C, Fig. S2).
Figure 3.
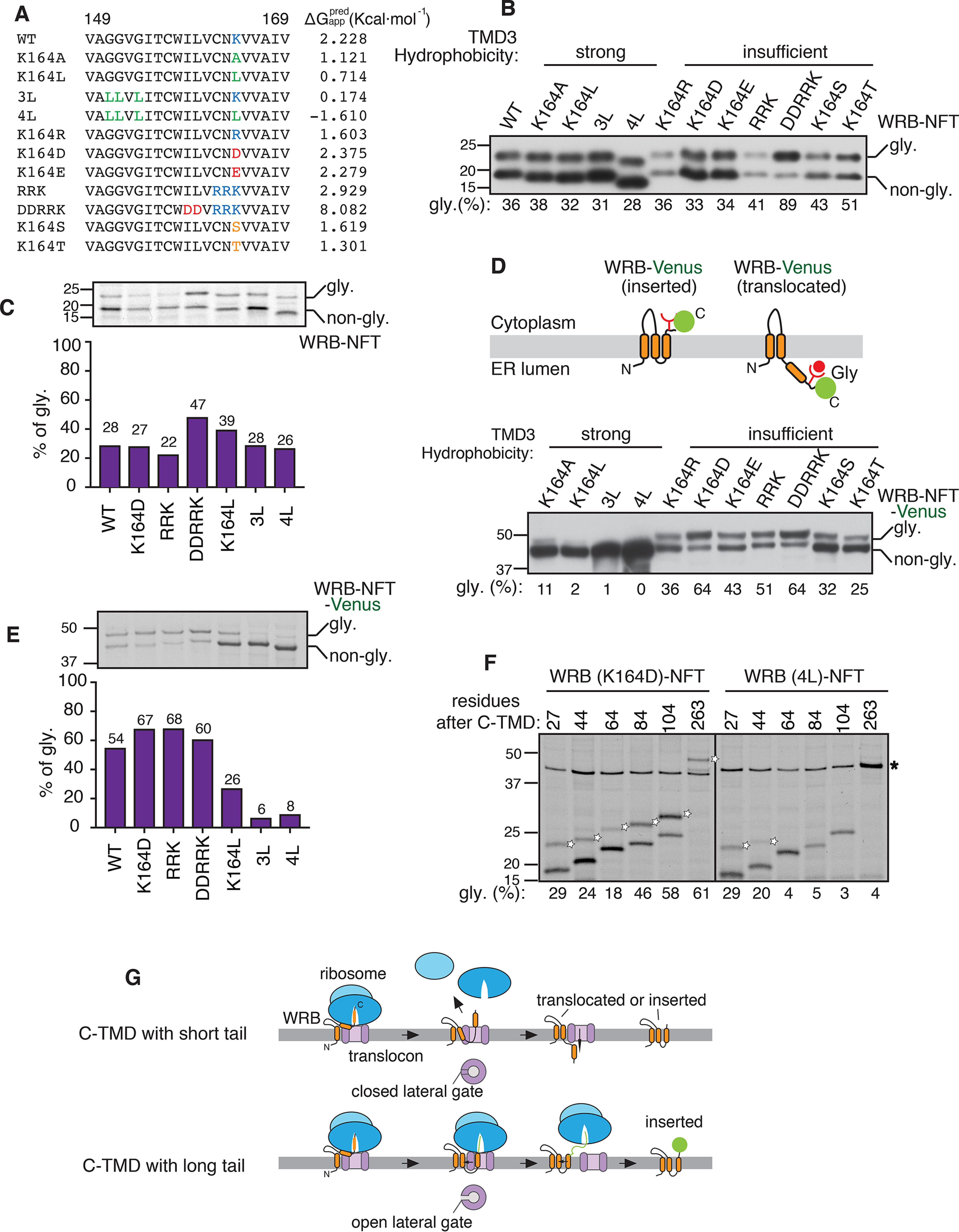
The cytosolic tail influences both insertion and translocation of the C-TMD. A, amino acid sequences of WRB TMD3 and its variants along with apparent free energy predictions. The mutations are highlighted using the following colors: hydrophobic residue, green; positively charged residues, blue; negatively charged residues, red; hydrophilic residues, orange. B, the indicated WRBHA-NFT variants were transfected into HEK293 cells and analyzed by immunoblotting with an anti-HA antibody. The percentage of translocated form (glycosylated) was quantified with ImageJ and shown under the blot. C, HEK293 cells expressing the indicated WRB-HA-NFT variants were metabolically labeled for 30 min and immunoprecipitated with anti-HA antibody beads. The immunoprecipitants were analyzed by autoradiography. The percentage of translocated form (glycosylated) was quantified and plotted in a bar graph (bottom). D, top panel: a schematic showing the two different topologies of WRB-NFT-Venus with inserted or translocated C-TMD. The glycosylation acceptor site NFT is added between WRB and Venus. Bottom panel, the indicated WRB-NFT-Venus variants were transfected into HEK293 cells and analyzed as in B. E, HEK293 cells expressing the indicated WRB-NFT-Venus variants were analyzed as in C. F, the indicated WRB-NFT-Venus constructs with C-terminal truncations were transfected and analyzed as in C. Empty stars denote translocated forms of WRB variants. Star indicates the nonspecific band. Please note that the nonspecific band (star) merges with the specific band of the full-length (263) construct of WRB (4L)-NFT. G, a schematic showing the C-terminal tail influencing the insertion of the C-TMD with strong hydrophobicity. Note that the open and closed states of the translocon lateral gate are influenced by the ribosome binding.
We asked why the C-TMD with strong hydrophobicity does not obey the biological hydrophobicity (29), as it is not efficiently inserted into the membrane. We reasoned that when the translation is terminated, most of the sequence of C-TMD and its tail might still be within the exit tunnel of the ribosome, which can accommodate ∼40 amino acids (33) (Fig. S1A). This might force the translocon to post-translationally recognize and insert the C-TMD into the lipid bilayer. We hypothesized this post-translational recognition of C-TMD by the Sec61 translocon might be slow and inefficient because the previous structural studies have shown that the lateral gate of the translocon is closed in the absence ribosome (34, 35). To test this idea, we increased the length of the cytosolic C-tail by appending a large Venus tag that comprises 238 amino acids (Fig. S1C). In support of our hypothesis, the C-TMD translocation of WRB-Venus variants with strong hydrophobicity was significantly prevented as shown by both immunoblotting and metabolic labeling results (Fig. 3, D and E). Consistent with the biological hydrophobicity scale (29), C-terminal TMDs with insufficient hydrophobicity were translocated into the ER lumen. Of note, WRB-Venus with insufficient hydrophobicity exhibited more efficient translocation into the ER lumen compared with WRB constructs with small tails (compare Fig. 3, C and E). These results suggest that the long cytosolic C-terminal tail is important for both efficient insertion of strong hydrophobic C-TMD and translocation of less hydrophobic C-TMD into the ER lumen.
We next wanted to determine the minimum C-terminal cytosolic tail length required for either insertion of strong hydrophobic C-TMDs or translocation of insufficient hydrophobic C-TMDs. To this end, we prepared WRB (K164D) constructs with varying C-terminal length and tested them for insertion or translocation by metabolic labeling and immunoprecipitation. We found ∼100 residues at the C terminus rendered efficient translocation of C-TMD with insufficient hydrophobicity (Fig. 4F). In contrast, about 60 residues at the C terminus were sufficient to prevent the translocation of C-TMDs with strong hydrophobicity (Fig. 3F). This result is consistent with the model that the presence of the ribosome at the translocon is crucial for efficient insertion of C-TMDs with strong hydrophobicity, presumably by trigging the opening of the lateral gate of the translocon (Fig. 3G). Collectively, these findings suggest that hydrophobicity alone is not sufficient for the insertion of a TMD and that the C-terminal tail length also influences the insertion of a TMD into the ER membrane.
Figure 4.

Insufficiently hydrophobic C-TMDs with short tails slowly translocate into the ER lumen. A, HEK293 cells were transfected with WRB-HA-NFT and metabolically labeled for 15 min and chased in the presence of a p97 ATPase inhibitor for the indicated time points. Cell lysates were immunoprecipitated with anti-HA antibody beads and analyzed by autoradiography. The percentage of translocated form, inserted form, and total signals were quantified from three independent experiments, and their mean ± S.D. are depicted in a graph. B-F, the indicated constructs were transfected and analyzed as in A, except that C, D, and F were quantified from two independent experiments.
Insufficient hydrophobic C-TMDs with short tails enable slow translocation into the ER lumen
We hypothesized that the translocation of C-TMD of WRB into the ER lumen may be slow to provide a time window for assembly with the partner protein CAML. To test this hypothesis, we monitored the dynamics of C-TMD translocation by performing pulse-chase experiments. The cells expressing WRB constructs with short C-terminal tails were briefly labeled and chased for 3 h. Also, we treated cells with a p97 ATPase inhibitor during the chase period to prevent retrotranslocation and degradation of WRB, thus allowing us to quantify the translocated C-TMD without losing the signal from degradation. A proportion of the C-TMD of WT WRB containing a positively charged residue translocated into the ER lumen even during labeling (0 h) as shown by glycosylation (Fig. 4A). Intriguingly, about 25% of the C-TMD of WRB was continuously post-translationally translocated into the ER lumen as reflected by increased glycosylation signals during the chase period (Fig. 4A). In contrast, the nonglycosylated inserted form was significantly reduced during the chase period. This reduction is likely caused by both continuous translocation into the ER lumen and degradation even in the presence of the p97 ATPase inhibitor. The inhibitor did not completely block the degradation as this was evidenced by the continuous loss of total signal during the chase period (Fig. 4A). The C-TMD containing a negative charge residue (K164D) also yielded a similar result (Fig. 4B). In sharp contrast, the translocation of C-TMD of WRB (K164L) with sufficient hydrophobicity occurred only during labeling, but did not translocate post-translationally into the ER lumen during the chase period (Fig. 4C). We then investigated whether the length of the C-tail influences the translocation dynamics of C-TMD into the ER lumen. In contrast to the short C-terminal tail, the C-TMD of WRB-Venus was not significantly translocated into the ER lumen during the chase period (Fig. 4D). Although the C-TMD of WRB (K164D)-Venus was not post-translationally translocated into the ER lumen during the chase period, its nonglycosylated inserted form was degraded even in the presence of the p97 ATPase inhibitor (Fig. 4E). Similar to the short C-tail (Fig. 4C), the C-TMD of WRB (K164L)-Venus with sufficient hydrophobicity translocated only during labeling but did not translocate post-translationally into the ER lumen during the chase period (Fig. 5F). Taken together our results suggest that both the hydrophobicity of C-TMD and the cytosolic tail length contribute to the slow translocation of C-TMD into the ER lumen.
Figure 5.
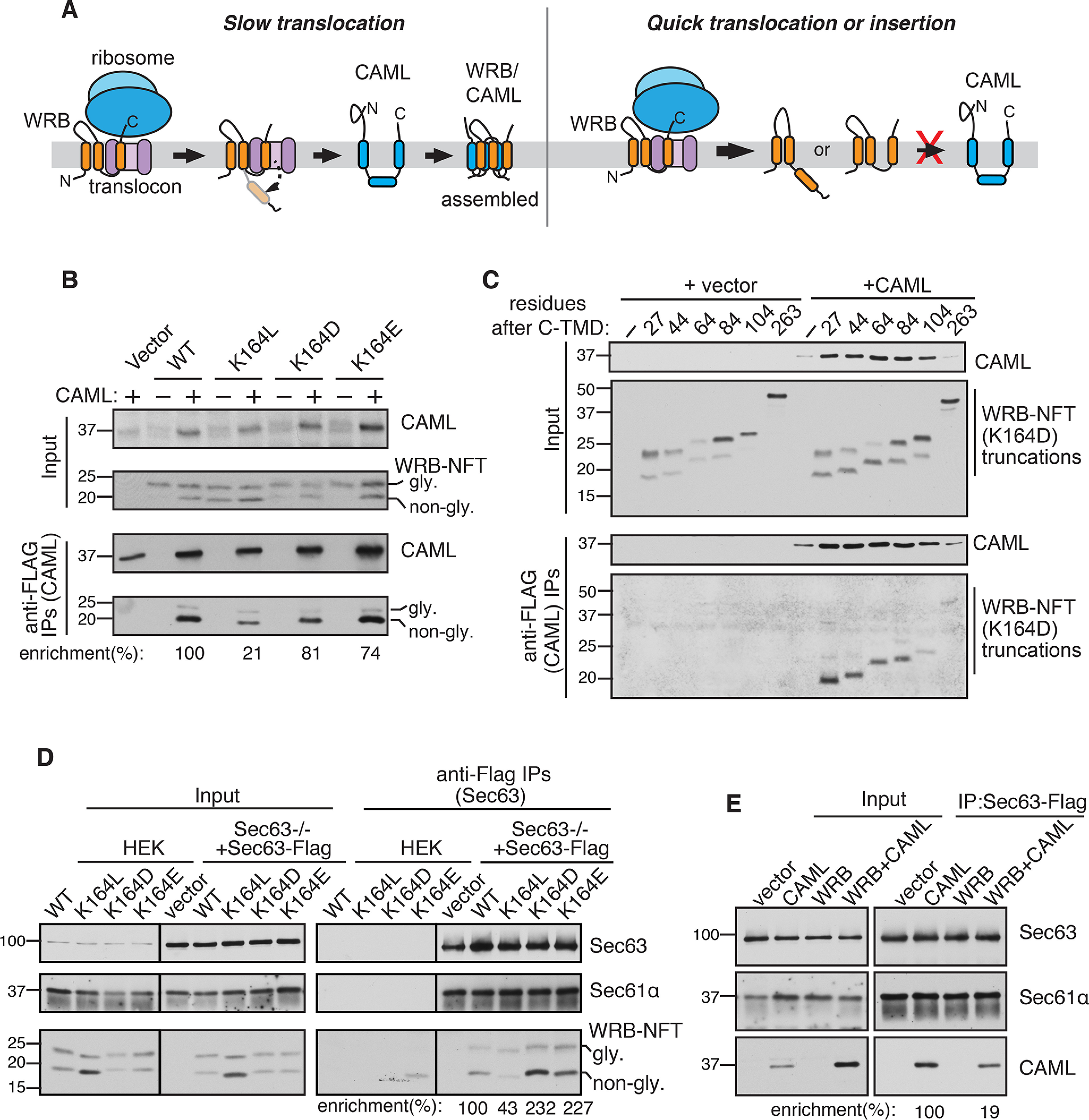
The C-TMD translocation rate determines the assembly efficiency with its partner TMD. A, left panel: a hypothetic model showing the slow translocation of insufficiently hydrophobic C-TMD of WRB provides a time window for efficient assembly with the partner TMD of CAML. The topology of unassembled CAML is adopted from recent studies (25, 26). Right panel, the C-TMD of WRB that either quickly inserts into the membrane or translocates into the ER lumen poorly assembles with the partner TMD of CAML. B, the indicated WRB-HA-NFT constructs were co-transfected with CAML-FLAG into HEK293 cells and immunoprecipitated with anti-FLAG beads. The resulting samples were analyzed by immunoblotting with anti-FLAG antibody (CAML) and anti-HA antibody (WRB). The band intensity was quantified by ImageJ, and the ratio of immunoprecipitates (IP) to inputs was calculated. The IP enrichment of WT WRB was taken as 100%. C, the indicated WRB-NFT-Venus truncations were co-transfected with CAMLFLAG into HEK293 cells and analyzed as in B. D, the indicated WRB-HA-NFT variants were transfected into Sec63−/− HEK293 cells stably expressing Sec63-FLAG and immunoprecipitated with anti-FLAG beads. The resulting samples were analyzed by immunoblotting with Sec63 antibodies, Sec61α antibodies, and HA antibodies (WRB-HA-NFT variants). The percentage of enrichment was quantified as in B. E, the indicated WRB-HA or/and CAML-HA plasmids were transfected into Sec63−/− HEK293 cells stably expressing Sec63-FLAG and immunoprecipitated using anti-FLAG beads. The resulting samples were analyzed by immunoblotting with Sec63 antibodies, Sec61α antibodies, and HA antibodies (CAML). The band intensity was quantified as in B.
The Sec61 translocon complex serves as a holdase for insufficiently hydrophobic C-TMDs for efficient assembly with partner TMDs
We hypothesized that the slow translocation of C-TMD of WRB is important for efficient assembly with its partner TMD from CAML, which inserts incorrectly in the absence of WRB (25, 26) (Fig. 5A). Conversely, the C-TMD that either quickly inserts into the membrane or translocates into the ER lumen may not have sufficient time to efficiently assemble with its partner TMD from CAML. To test this, we performed CAML pulldown with WRB constructs that vary in their hydrophobicity of C-TMDs. We noticed that the nonglycosylated inserted form of WRB was selectively stabilized in cells co-expressing both CAML and WRB compared with cells expressing WRB alone (Fig. 5B, input). Indeed, CAML selectively co-immunoprecipitated with the nonglycosylated inserted form of WRB (Fig. 5B). Strikingly, the assembly of CAML with WRB (K164L), the C-TMD of which was quickly translocated (Fig. 5C), was significantly lower than WT WRB (Fig. 5B). Surprisingly, replacing lysine with a negatively charged residue in the C-TMD of WRB also assembled with CAML similar to the WT (Fig. 5B). This result suggests that the lysine residue in the C-TMD is not directly involved in the assembly with CAML, but rather it facilitates the slow translocation of the TMD into the ER lumen. We then investigated the effect of the C-terminal length of WRB on assembly with CAML. We hypothesized that WRB-Venus may not assemble efficiently with CAML because the larger C-terminal Venus tag facilitated a faster translocation of the C-TMD into the ER lumen (Fig. 4D). To test this, we compared CAML assembly with either WRB constructs with short tails or WRB constructs with large Venus tag. In support of our hypothesis, CAML efficiently assembled with WRB containing short C-tails, but inefficiently assembled with WRB containing large Venus tag (Fig. S3). We next asked whether the C-terminal length-dependent translocation observed in Fig. 3F would correlate with the assembly efficiency with CAML. To address this, we performed interaction studies between CAML and WRB constructs harboring varied length of C-terminal tails. Indeed, slow translocation WRB constructs that contain less than 84 residues at their C terminus assembled efficiently with CAML (Fig. 5C). In sharp contrast, faster translocation WRB constructs that contain more than 100 residues at their C terminus very poorly assembled with CAML. This conclusion is further corroborated by the observation that CAML levels were also stabilized upon co-expression of WRB constructs containing less than 84 residues at their C terminus (Fig. 5C, input).
These findings thus far suggest that insufficient hydrophobic C-TMDs with short tails are transiently retained by a membrane holdase and are translocated into the lumen when they failed to assemble with partner TMDs. We hypothesized that the Sec61 translocon may be a post-translational TMD holdase because it is responsible for recognizing the C-TMD right after its release from the ribosome. To selectively enrich the post-translationally retained C-TMD in the Sec61 translocon, we used the translocon-associated membrane protein Sec63 as a handle because it occludes the ribosome binding to the translocon (34, 36). Although Sec63 is known to facilitate post-translational translocation of proteins into the ER lumen (34, 37–39), it is dispensable for the C-TMD of WRB translocation into the ER lumen (Fig. S4). As expected, Sec63 formed a complex with the Sec61 translocon as it was efficiently co-immunoprecipitated with the translocon (Fig. 5D). In support of our hypothesis, C-TMDs of WRB constructs with insufficient hydrophobicity were enriched with the Sec63/Sec61 translocon complex. In contrast, the C-TMD (K164L) with sufficient hydrophobicity was weakly associated with the translocon even though it expressed higher than other constructs (Fig. 5D), thus explaining its poor assembly with CAML. We next asked whether CAML also transiently retained by the Sec61 translocon complex. Indeed, we found that CAML could interact with the Sec61 translocon complex in the absence of WRB, but this interaction was significantly reduced when CAML was co-expressed with WRB (Fig. 5E). Collectively, our results support a model in which insufficient hydrophobic C-TMDs with short tails are post-translationally retained and chaperoned by the Sec61 translocon, providing a sufficient time window for the assembly with partner TMDs.
Discussion
A large proportion of membrane proteins exist in multimembrane protein complexes and they serve fundamental roles in cell physiology (8). It is less understood how these newly synthesized membrane proteins find each other in the crowded and extensive network of the ER membrane, which includes many competing factors such as chaperone-linked quality control components (11, 14). In this study, we uncover new roles for the C-terminal tails of membrane proteins in the insertion and assembly of membrane proteins (Fig. 6).
Figure 6.

A model showing the roles of the C-terminal tail in membrane protein insertion and assembly. The C-TMD with a short tail is post-translationally recognized by the Sec61 translocon because the translation is terminated before the C-TMD contact the Sec61 translocon. The insufficiently hydrophobic C-TMD of WRB is transiently retained by the Sec61 translocon. This retention provides a time window to assemble with CAML, which is likely associated with the Sec61 translocon through its C-TMD or TMD2 (not shown). The topology of unassembled CAML is adopted from recent studies (25, 26). If the Sec61-retained TMD fails to be assembled with a partner TMD, it slowly translocates through the translocon into the ER lumen. The translocated TMD is recognized by the ERAD pathway for proteasomal degradation.
To reveal the biochemical features that are necessary for both membrane protein assembly and degradation, we chose to investigate the assembly of the tail-anchored membrane protein complex comprising WRB and CAML proteins because of their smaller sizes, which can be biochemically manipulated. Also, their assembly is mediated mainly through their transmembrane domains (23). Although WRB contains three TMDs with N terminus in the lumen and C terminus in the cytosol, CAML has three TMDs with N terminus in the cytosol and C terminus in the lumen (Fig. 6). Both proteins are robustly degraded when they failed to assemble in the ER membrane. However, coexpression of both membrane proteins increases their stability by shielding from recognition by ERAD. By performing a detailed domain mapping and mutagenesis studies, we identify that a single lysine residue in the C-TMD of WRB is responsible for degradation. Surprisingly, replacing lysine to a negatively charged or hydrophilic amino acid also causes the degradation of WRB, implicating that the overall hydrophobicity of the C-TMD is recognized by ERAD. Alternatively, similar to TCRα (40), the presence of a charged or hydrophilic residue in the C-TMD may prevent the formation of WRB oligomers that are not suitable for recognition by ERAD. Our data using an engineered N-linked glycosylation site at the C terminus of WRB revealed that the C-TMD of WRB is translocated into the ER lumen and thereby glycosylated. The translocated TMD of WRB is subsequently recognized for degradation by the ERAD pathway. Hence, it appears that the C-TMD translocation is one of the features displayed by unassembled polytopic membrane proteins. This notion is further supported by our data that either the C-TMD of TRAPγ or Sec61α translocate into the ER lumen in the absence of their partner proteins. Our data derived from introducing a patient mutation in PMP22 suggested that C-TMDs of polytopic membrane proteins that do not assemble with their neighboring TMDs can also translocate into the ER lumen. Because our studies were conducted using recombinantly expressed proteins, future studies are required to determine whether the C-TMD translocation into the ER lumen also occurs with the endogenously expressed membrane proteins.
Although the TMD translocation into the ER lumen has been reported by previous studies (21, 41–45), the biochemical features responsible for translocation remained unclear. We find that the translocation of C-TMD is not just due to less hydrophobic TMD, but also its C-terminal tail length influences the translocation of the TMD. Even a strong hydrophobic C-TMD does not efficiently insert into the membrane and thereby translocating into the ER lumen if the C terminus contains less than 60 amino acids. This is likely caused by the termination of translation before the C-TMD releases from the ribosome exit tunnel, which can accommodate ∼40 residues (33). Hence, the C-TMD is post-translationally recognized by the Sec61 translocon. However, the post-translational insertion of C-TMDs with short tails is inefficient because the lateral gate of the translocon is likely closed in the absence of the ribosome (34, 35, 46, 47). The C-terminal tail length plays a unique function in the case of C-TMDs with insufficient hydrophobicity. As expected, these hydrophilic C-TMDs translocate into the ER lumen, but their translocation was more efficient when their C-terminal was extended longer than 84 residues. At present, it is not clear why the presence of long C-terminal tails facilitates efficient translocation of C-TMDs of insufficient hydrophobicity into the ER lumen. One possible explanation is that the translating ribosome may push the C-TMDs of insufficient hydrophobicity with long tails into the ER lumen. Alternatively, the presence of long C-terminal tails on insufficient hydrophobic TMDs may recruit additional factors to the Sec61 translocon to facilitate the translocation of these TMDs into the ER lumen.
Our pulse-chase experiments show that insufficiently hydrophobic C-TMDs with short tails exhibit a slow translocation rate. However, our data do not exclude the possibility that the artificial glycosylation site introduced at the C terminus of WRB variants might be differentially recognized by the luminal glycosylation machinery. Alternative assays that are independent of the glycosylation machinery such as protease protection are needed to confirm if the C-TMD of WRB is translocated in hydrophobicity as well as a tail length-dependent manner. Our data are consistent with the previous study analyzing a single spanning membrane protein TCRα, which continuously post-translationally enters into the ER lumen (21). These data suggested that the slow translocation of less hydrophobic C-TMDs into the ER lumen may provide a time window for the assembly with the partner protein. Indeed, C-TMDs of WRB that slowly translocates into the ER lumen efficiently assemble with CAML. Previous studies have shown that a charged residue in a TMD of the membrane protein is often required for making an ionic bond with an opposite charge in the partner TMD of membrane protein (20, 21, 41). Our data suggest that charged residues in TMDs may also be evolved to interact with membrane protein holdases, but not necessarily to form ionic bonds between TMDs.
Our data suggest that the Sec61 translocon is responsible for transiently retaining insufficiently hydrophobic C-TMDs with shorter tails. The interaction between insufficiently hydrophobic C-TMDs of WRB and the Sec61 translocon correlates with the assembly efficiency with CAML. Interestingly, CAML also post-translationally associates with the Sec61 translocon, and the interaction is reduced when co-expressed with WRB. Based on recent studies showing the translocation of TMD2 of CAML into the ER lumen (Fig. 6) (25, 26), we speculate that the C-TMD (TMD3) of CAML is also retained in the Sec61 translocon to assemble with WRB. Alternatively, TMD2 of CAML may be retained in the Sec61 translocon with the C-TMD exposed to the cytosol. In this case, WRB may facilitate post-translational insertion of the C-TMD of CAML and the translocation of the C terminus into the ER lumen, as previously proposed by the Borgese group (26). In the absence of WRB, we predict that TMD2 and C-TMD of CAML are sequentially translocated through the Sec61 translocon into the ER lumen and recognized for degradation (Fig. 6), which is supported by the observation that both TMD2 and C-TMD of CAML are localized in the ER lumen (26). It remains to be understood how the Sec61 translocon-retained TMD is released to form a complex with a partner TMD in the ER membrane. We speculate that the C-TMD of WRB might dynamically sample the lipid environment via the lateral gate of the Sec61 translocon to find its potential TMD partner. Upon interaction, the C-TMD may be released from translocons. Interestingly, the endogenous level of CAML is estimated to be five times higher than WRB (48), suggesting that CAML has a higher chance to find and release WRB from the translocon. The mechanism that we discovered with WRB in this study is likely used by the assembly of membrane proteins that contain short C-terminal tails, including TRAPγ, and Sec61α. However, other mechanisms might exist to regulate the assembly of membrane proteins that contain longer C-terminal regions.
We propose that our findings of C-terminal length influencing insertion, translocation, and assembly of membrane proteins are important to understand the biogenesis of glycosylphosphatidylinositol (GPI)-anchored proteins (49). The GPI-anchor sequences are typically localized at the C terminus of GPI-anchored proteins. These GPI-anchored sequences are moderately hydrophobic with no or very short C-terminal tails. Similar to C-TMD of WRB, the GPI-anchored sequences are likely post-translationally recognized and slowly translocated into the ER lumen. This view is supported by the previous studies that GPI-anchored sequences cannot be inserted into the membrane even when extending their C terminus with additional cytosolic sequences (50). It is tempting to suggest that GPI-anchored sequences were also post-translationally retained by the Sec61 translocon, thus providing a kinetic time window for the GPI-anchor addition to the C-terminal end of a GPI-anchored protein by the GPI transamidase complex. However, this hypothesis needs to be tested in future studies.
Materials and methods
DNA constructs
All the constructs were created using the pcDNA/FRT/TO vector (Invitrogen). WRB-FLAG, WRB-HA, WRB-HA-NFT, CAML-FLAG, CAML-HA, PMP22-HA, PMP22-HA-NFT, TRAPγ-HA, TRAPγ-HA-NFT, Sec61α-HA, and Sec61α-HA-NFT were generated using standard molecular biology methods. pcDNA/FRT/TO containing WRB-TMD1-swap-FLAG was constructed by replacing WRB TMD1 (amino acids 9-29) with yeast Ost4pTMD (amino acids 10-28). WRB-TMD2-swap-FLAG was cloned by replacing WRB TMD2 (amino acids 100-120) with human Sec61β TMD (amino acids 71-91). WRB-TMD3-swap-FLAG was generated by replacing WRB TMD3 (amino acids 149-169) with yeast Ost4p TMD (amino acids 10-28). WRB-HA-NFT-Venus was created by overlap PCR with standard molecular biology methods. Although the Pfu polymerase (Agilent Technologies) was used for site-directed mutagenesis, the Phusion polymerase (New England Biolabs) was used for other PCR. The coding sequences of all constructs were verified by sequencing (Yale Keck DNA Sequencing) to preclude any sequence error.
Cell culture and the generation of CRISPR/Case9 knockout cells
293T and HEK293-Flp-In T-Rex cells (kindly provided by Dr. Ramanujan Hegde, MRC, United Kingdom) were cultured in high glucose Dulbecco's modified Eagle's medium with 10% fetal bovine serum and 100 units/ml of penicillin and 100 μg/ml of streptomycin at 5% CO2. Sec63−/− HEK293-Flp-In T-Rex cell lines were generated using the CRISPR/Cas9 system as previously described (51, 52). HEK293-Flp-In T-Rex cells were transfected with pSpCas9(BB)-2A-Puro and gRNA expression plasmid of Sec63 (GCCAGAGGTAGTATGTCGC). Cells were grown for 24 h and treated with 2.5 μg/ml of puromycin for 72 h to select the successfully transfected clones. Single-cell clones were isolated by plating at 0.5 cells/well in 96-well–plates. Sec63 knockouts were confirmed by immunoblotting. All the cell lines used in this study were not tested for mycoplasma, but many cell lines were used in immunofluorescence assays with Hoechst staining that should reveal the presence of mycoplasma. Cells were assumed to be authenticated by their respective suppliers and were not further confirmed in this study. However, knockout lines were routinely validated by immunoblotting.
To establish Sec63−/− HEK293 cells stably expressing Sec63-FLAG, 1.6 μg of pOG44 vector (Invitrogen) and 0.4 μg of pcDNA/FRT/TO-Sec63-FLAG were transfected into Sec63−/− cells in a well in the 6-well–plate using Lipofectamine 2000 (Invitrogen). After 24 h of transfection, cells were transferred to 10-cm dishes and selected with 150 μg/ml of hygromycin (Invitrogen) and 10 μg/ml of blasticidin (InvivoGen, San Diego, CA). The selection medium was replaced every 3 days until colonies appeared. The colonies were picked and the protein expression was evaluated by immunoblotting. Transfections in HEK293-Flp-In T-Rex cells were performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol but with either half or quarter amounts of the recommended DNA and Lipofectamine 2000.
Cycloheximide chase
WT HEK293-Flp-In T-Rex (0.15 × 106/well) were plated on polylysine pre-coated (0.15 mg/ml) 24-well–plates and transiently transfected with 400 or 800 ng (for co-expression experiments, 400 ng of each plasmid) of the indicated construct as described in the figure legends. Typically, expression of the indicated proteins were induced with doxycycline (200 ng/ml) for 24 h prior to treatment with cycloheximide (150 μg/ml). The treated cells were directly collected with 100 μl of 2× SDS sample buffer at the indicated time points. In some cases, cells were treated with both cycloheximide and MG132 (20 μm). Samples were then analyzed by immunoblotting with the indicated antibodies as described in the figure legends.
Glycosylation assay and Endo H treatment
HEK293-Flp-In T-Rex cells (0.15 × 106/well) were plated on polylysine (0.15 mg/ml)-coated 24-well–plates and transiently transfected with 400 ng of the indicated plasmids. Protein expression was induced with 200 ng/ml of doxycycline. After 24 h transfection and induction, cells were collected with 60 μl of SDS buffer (1% SDS, 0.1 m Tris, pH 8.0). The lysates were diluted with 60 μl of 2% β-mercaptoethanol, 0.1 m Tris (pH 6.8) and boiled at 100 °C for 5 min. The samples were then divided into two aliquots (60 μl for each), and were treated with or without 2 μl (1000 units) of Endo H (New England Biolabs) in a 100-μl reaction volume including 1× G5 buffer (New England Biolabs) at 37 °C for 4 h. The reaction was terminated by adding 50 μl of 5× SDS sample buffer and boiled for 5 min before analyzing by immunoblotting with anti-HA antibody.
Metabolic labeling and pulse-chase assay
HEK293-Flp-In T-Rex or Hrd1−/− cells (0.6 × 106/well) were plated on polylysine-coated (0.15 mg/ml) 6-well–plates and transiently transfected with 2 μg of the indicated plasmids. Expression of indicated proteins was induced with doxycycline (200 ng/ml) for 24 h prior to the chase. Cells were starved with nonMet/Cys medium including 10% dialyzed fetal bovine serum and doxycycline for 30 min. For the metabolic labeling assay in Fig. 3, cells were labeled with 80 μCi/ml of Express35S protein labeling mix for 15 min. For pulse-chase assays, cells were labeled with the 35S-labeling mix for 30 min and chased in Dulbecco's modified Eagle's medium supplemented with 2 mm methionine and 2 mm cysteine for the indicated time. For Fig. 4, 10 μm P97 inhibitor (NMS-873, Millipore Sigma) was added during starvation, labeling, and chase periods. The labeled cells were collected and lysed in RIPA buffer including a protease inhibitor mixture (Roche Applied Science). The lysate was cleared by centrifugation at 20,000 × g for 15 min. The supernatant was mixed with either anti-HA magnetic beads or anti-green fluorescent protein antibodies for 2 h with rotation in the cold room. After incubation, protein A-agarose was added to the samples containing anti-green fluorescent protein antibodies and incubated for 1 h. The immunoprecipitates were washed three times with 1 ml of RIPA buffer, and proteins were eluted with 2× SDS sample buffer and run on 7.5 or 10% Tris-Tricine SDS-PAGE gels. The gels were dried and analyzed by autoradiography.
In vitro translation and insertion
Transcripts encoding WRB-NFT-HA variants were obtained from in vitro transcription reactions as described previously (53). The transcripts were translated in the presence or absence of canine pancreas rough microsomes for 45 min at 32 °C. The translated products (10 μl) were denatured with 100 μl of Tris-SDS buffer (0.1 m Tris-HCl and 1% SDS) and diluted with the Triton buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, and 1% Triton X-100) before immunoprecipitating with anti-HA magnetic beads. The resulting samples were analyzed by SDS-PAGE and autoradiography.
Immunoprecipitation
To probe the interaction between CAML and WRB variants in Fig. 5, HEK293-Flp-In T-Rex cells (0.6 × 106/well) were plated on polylysine-coated 6-well–plates and transiently transfected with 1 μg of pcDNA/FRT/TO-CAML-FLAG and 1 μg of pcDNA/FRT/TO-WRB-HA variants and induced to the protein expression by treating cells with 200 ng/ml of doxycycline. After 24 h transfection, cells were washed and harvested in PBS. The cell pellets were obtained by centrifuging for 2 min at 10,000 × g. The cell pellets were lysed with 200 μl of Buffer A (2% digitonin, 150 mm NaCl, 50 mm Tris, pH 7.4, 2 mm MgAc, and 1× Protease inhibitor mixture) for 30 min on ice. The cell lysates were then diluted with 600 μl of Buffer B (0.1% digitonin, 150 mm NaCl, 50 mm Tris, pH 7.4) and cleared by centrifugation at 20,000 × g for 15 min. The supernatants were incubated with either anti-FLAG beads (Biolegend) or anti-HA magnetic beads (Thermo Scientific) for 1.5 h. The beads were then washed for 3 times with 1 ml of Buffer B. The bound material was eluted from the beads with 50 μl of 2× SDS sample buffer and boiled for 5 min. The samples were then analyzed by immunoblotting with the indicated antibodies.
To test the interaction between WRB variants and Sec63 in Fig. 5E, Sec63−/− cells stably expressing Sec63-FLAG (0.6 × 106/well) and HEK293-Flp-In T-Rex cells were plated on polylysine-coated 6-well–plates and transiently transfected with either 2 μg of WRB-HA-NFT variants or empty vector. After 24 h of transfection and induction with 200 ng/ml of doxycycline, the aforementioned digitonin-based immunoprecipitation protocol was used and analyzed by immunoblotting with anti-Sec63, anti-Sec61α, and anti-HA antibodies.
Quantification and statistical analysis
Quantification of Western blotting and autoradiographs were performed using ImageJ gel analysis/lane plotting. Error bars represent mean ± S.D. from two or three independent experiments. The apparent free energy (ΔG) values for TMDs were predicted using the online tool (http://dgpred.cbr.su.se/) (29).
Data availability
All raw data are available at reasonable request by contacting Dr. Malaiyalam Mariappan (malaiyalam.mariappan@yale.edu), Yale School of Medicine, all remaining data are contained within the article.
Supplementary Material
Acknowledgments
We are grateful to Dr. Ramanujan Hegde for Sec61α and Sec63 antibodies. We thank Dr. Zai-Rong Zhang for comments on the manuscript. We thank Jacob Culver and Dr. Xia Li for discussions and comments on the manuscript.
This article contains supporting information.
Author contributions—S. S. formal analysis; S. S. and M. M. validation; S. S. investigation; S. S. and M. M. methodology; S. S. and M. M. writing-original draft; S. S. and M. M. writing-review and editing; M. M. conceptualization; M. M. supervision; M. M. funding acquisition.
Funding and additional information—This work was supported by National Institutes of Health Grants 1R01GM117386 and 1R21AG056800 (to M. M.) and a start-up grant from the Yale School of Medicine. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that no competing interests exits.
- ER
- endoplasmic reticulum
- C-TMD
- C-terminal transmembrane domain
- ERAD
- ER-associated degradation
- SRP
- signal recognition particle
- TCR
- T-cell receptor
- Endo H
- endoglycosidase H
- PMP22
- peripheral myelin protein 22
- GPI
- glycosylphosphatidylinositol
- HA
- hemagglutinin
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine.
References
- 1. Krogh A., Larsson B., von Heijne G., and Sonnhammer E. L. (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580 10.1006/jmbi.2000.4315 [DOI] [PubMed] [Google Scholar]
- 2. Shao S., and Hegde R. S. (2011) Membrane protein insertion at the endoplasmic reticulum. Annu. Rev. Cell Dev. Biol. 27, 25–56 10.1146/annurev-cellbio-092910-154125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Grudnik P., Bange G., and Sinning I. (2009) Protein targeting by the signal recognition particle. Biol. Chem. 390, 775–782 10.1515/BC.2009.102 [DOI] [PubMed] [Google Scholar]
- 4. Park E., and Rapoport T. A. (2012) Mechanisms of Sec61/SecY-mediated protein translocation across membranes. Annu. Rev. Biophys. 41, 21–40 10.1146/annurev-biophys-050511-102312 [DOI] [PubMed] [Google Scholar]
- 5. Zhang X., and Shan S. O. (2014) Fidelity of cotranslational protein targeting by the signal recognition particle. Annu. Rev. Biophys. 43, 381–408 10.1146/annurev-biophys-051013-022653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li L., Park E., Ling J., Ingram J., Ploegh H., and Rapoport T. A. (2016) Crystal structure of a substrate-engaged SecY protein-translocation channel. Nature 531, 395–399 10.1038/nature17163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Skach W. R. (2007) The expanding role of the ER translocon in membrane protein folding. J. Cell Biol. 179, 1333–1335 10.1083/jcb.200711107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Juszkiewicz S., and Hegde R. S. (2018) Quality control of orphaned proteins. Mol. Cell 71, 443–457 10.1016/j.molcel.2018.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Balchin D., Hayer-Hartl M., and Hartl F. U. (2016) In vivo aspects of protein folding and quality control. Science 353, aac4354 10.1126/science.aac4354 [DOI] [PubMed] [Google Scholar]
- 10. Kramer G., Shiber A., and Bukau B. (2019) Mechanisms of cotranslational maturation of newly synthesized proteins. Annu. Rev. Biochem. 88, 337–364 10.1146/annurev-biochem-013118-111717 [DOI] [PubMed] [Google Scholar]
- 11. Brodsky J. L. (2012) Cleaning up: ER-associated degradation to the rescue. Cell 151, 1163–1167 10.1016/j.cell.2012.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Foresti O., Rodriguez-Vaello V., Funaya C., and Carvalho P. (2014) Quality control of inner nuclear membrane proteins by the Asi complex. Science 346, 751–755 10.1126/science.1255638 [DOI] [PubMed] [Google Scholar]
- 13. Khmelinskii A., Blaszczak E., Pantazopoulou M., Fischer B., Omnus D. J., Le Dez G., Brossard A., Gunnarsson A., Barry J. D., Meurer M., Kirrmaier D., Boone C., Huber W., Rabut G., Ljungdahl P. O., et al. (2014) Protein quality control at the inner nuclear membrane. Nature 516, 410–413 10.1038/nature14096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wu H., Carvalho P., and Voeltz G. K. (2018) Here, there, and everywhere: the importance of ER membrane contact sites. Science 361, eaan5835 10.1126/science.aan5835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schwarz A., and Beck M. (2019) The benefits of cotranslational assembly: a structural perspective. Trends Cell Biol. 29, 791–803 10.1016/j.tcb.2019.07.006 [DOI] [PubMed] [Google Scholar]
- 16. Le Tallec B., Barrault M. B., Courbeyrette R., Guérois R., Marsolier-Kergoat M. C., and Peyroche A. (2007) 20S proteasome assembly is orchestrated by two distinct pairs of chaperones in yeast and in mammals. Mol. Cell 27, 660–674 10.1016/j.molcel.2007.06.025 [DOI] [PubMed] [Google Scholar]
- 17. Shiber A., Döring K., Friedrich U., Klann K., Merker D., Zedan M., Tippmann F., Kramer G., and Bukau B. (2018) Cotranslational assembly of protein complexes in eukaryotes revealed by ribosome profiling. Nature 561, 268–272 10.1038/s41586-018-0462-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Manolios N., Bonifacino J. S., and Klausner R. D. (1990) Transmembrane helical interactions and the assembly of the T cell receptor complex. Science 249, 274–277 10.1126/science.2142801 [DOI] [PubMed] [Google Scholar]
- 19. Bonifacino J. S., Cosson P., and Klausner R. D. (1990) Colocalized transmembrane determinants for ER degradation and subunit assembly explain the intracellular fate of TCR chains. Cell 63, 503–513 10.1016/0092-8674(90)90447-M [DOI] [PubMed] [Google Scholar]
- 20. Cosson P., Lankford S. P., Bonifacino J. S., and Klausner R. D. (1991) Membrane protein association by potential intramembrane charge pairs. Nature 351, 414–416 10.1038/351414a0 [DOI] [PubMed] [Google Scholar]
- 21. Feige M. J., and Hendershot L. M. (2013) Quality control of integral membrane proteins by assembly-dependent membrane integration. Mol. Cell 51, 297–309 10.1016/j.molcel.2013.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vilardi F., Lorenz H., and Dobberstein B. (2011) WRB is the receptor for TRC40/Asna1-mediated insertion of tail-anchored proteins into the ER membrane. J. Cell Sci. 124, 1301–1307 10.1242/jcs.084277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vilardi F., Stephan M., Clancy A., Janshoff A., and Schwappach B. (2014) WRB and CAML are necessary and sufficient to mediate tail-anchored protein targeting to the ER membrane. PLoS ONE 9, e85033 10.1371/journal.pone.0085033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yamamoto Y., and Sakisaka T. (2012) Molecular machinery for insertion of tail-anchored membrane proteins into the endoplasmic reticulum membrane in mammalian cells. Mol. Cell 48, 387–397 10.1016/j.molcel.2012.08.028 [DOI] [PubMed] [Google Scholar]
- 25. Inglis A. J., Page K. R., Guna A., and Voorhees R. M. (2020) Differential modes of orphan subunit recognition for the WRB/CAML complex. Cell Rep. 30, 3691–3698.e5 10.1016/j.celrep.2020.02.084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Carvalho H. J. F., Del Bondio A., Maltecca F., Colombo S. F., and Borgese N. (2019) The WRB subunit of the Get3 receptor is required for the correct integration of its partner CAML into the ER. Sci. Rep. 9, 11887 10.1038/s41598-019-48363-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang F., Chan C., Weir N. R., and Denic V. (2014) The Get1/2 transmembrane complex is an endoplasmic-reticulum membrane protein insertase. Nature 512, 441–444 10.1038/nature13471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hessa T., Kim H., Bihlmaier K., Lundin C., Boekel J., Andersson H., Nilsson I., White S. H., and von Heijne G. (2005) Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature 433, 377–381 10.1038/nature03216 [DOI] [PubMed] [Google Scholar]
- 29. Hessa T., Meindl-Beinker N. M., Bernsel A., Kim H., Sato Y., Lerch-Bader M., Nilsson I., White S. H., and von Heijne G. (2007) Molecular code for transmembrane-helix recognition by the Sec61 translocon. Nature 450, 1026–1030 10.1038/nature06387 [DOI] [PubMed] [Google Scholar]
- 30. Ojemalm K., Halling K. K., Nilsson I., and von Heijne G. (2012) Orientational preferences of neighboring helices can drive ER insertion of a marginally hydrophobic transmembrane helix. Mol. Cell 45, 529–540 10.1016/j.molcel.2011.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lang S., Pfeffer S., Lee P. H., Cavalie A., Helms V., Forster F., and Zimmermann R. (2017) An update on Sec61 channel functions, mechanisms, and related diseases. Front. Physiol. 8, 887 10.3389/fphys.2017.00887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Navon R., Seifried B., Gal-On N. S., and Sadeh M. (1996) A new point mutation affecting the fourth transmembrane domain of PMP22 results in severe de novo Charcot-Marie-Tooth disease. Hum. Genet. 97, 685–687 10.1007/BF02281883 [DOI] [PubMed] [Google Scholar]
- 33. Voss N. R., Gerstein M., Steitz T. A., and Moore P. B. (2006) The geometry of the ribosomal polypeptide exit tunnel. J. Mol. Biol. 360, 893–906 10.1016/j.jmb.2006.05.023 [DOI] [PubMed] [Google Scholar]
- 34. Wu X., Cabanos C., and Rapoport T. A. (2019) Structure of the post-translational protein translocation machinery of the ER membrane. Nature 566, 136–139 10.1038/s41586-018-0856-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Voorhees R. M., and Hegde R. S. (2016) Structure of the Sec61 channel opened by a signal sequence. Science 351, 88–91 10.1126/science.aad4992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Itskanov S., and Park E. (2019) Structure of the posttranslational Sec protein-translocation channel complex from yeast. Science 363, 84–87 10.1126/science.aav6740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Deshaies R. J., Sanders S. L., Feldheim D. A., and Schekman R. (1991) Assembly of yeast Sec proteins involved in translocation into the endoplasmic reticulum into a membrane-bound multisubunit complex. Nature 349, 806–808 10.1038/349806a0 [DOI] [PubMed] [Google Scholar]
- 38. Matlack K. E., Misselwitz B., Plath K., and Rapoport T. A. (1999) BiP acts as a molecular ratchet during posttranslational transport of prepro-α factor across the ER membrane. Cell 97, 553–564 10.1016/S0092-8674(00)80767-9 [DOI] [PubMed] [Google Scholar]
- 39. Panzner S., Dreier L., Hartmann E., Kostka S., and Rapoport T. A. (1995) Posttranslational protein transport in yeast reconstituted with a purified complex of Sec proteins and Kar2p. Cell 81, 561–570 10.1016/0092-8674(95)90077-2 [DOI] [PubMed] [Google Scholar]
- 40. Soetandyo N., Wang Q., Ye Y., and Li L. (2010) Role of intramembrane charged residues in the quality control of unassembled T-cell receptor α-chains at the endoplasmic reticulum. J. Cell Sci. 123, 1031–1038 10.1242/jcs.059758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Roushar F. J., Gruenhagen T. C., Penn W. D., Li B., Meiler J., Jastrzebska B., and Schlebach J. P. (2019) Contribution of cotranslational folding defects to membrane protein homeostasis. J. Am. Chem. Soc. 141, 204–215 10.1021/jacs.8b08243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Buck T. M., and Skach W. R. (2005) Differential stability of biogenesis intermediates reveals a common pathway for aquaporin-1 topological maturation. J. Biol. Chem. 280, 261–269 10.1074/jbc.M409920200 [DOI] [PubMed] [Google Scholar]
- 43. Kim S. J., and Skach W. R. (2012) Mechanisms of CFTR folding at the endoplasmic reticulum. Front. Pharmacol. 3, 201 10.3389/fphar.2012.00201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rabeh W. M., Bossard F., Xu H., Okiyoneda T., Bagdany M., Mulvihill C. M., Du K., di Bernardo S., Liu Y., Konermann L., Roldan A., and Lukacs G. L. (2012) Correction of both NBD1 energetics and domain interface is required to restore ΔF508 CFTR folding and function. Cell 148, 150–163 10.1016/j.cell.2011.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Coelho J. P. L., Stahl M., Bloemeke N., Meighen-Berger K., Alvira C. P., Zhang Z. R., Sieber S. A., and Feige M. J. (2019) A network of chaperones prevents and detects failures in membrane protein lipid bilayer integration. Nat. Commun. 10, 672 10.1038/s41467-019-09912-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Voorhees R. M., Fernández I. S., Scheres S. H., and Hegde R. S. (2014) Structure of the mammalian ribosome-Sec61 complex to 3.4 Å resolution. Cell 157, 1632–1643 10.1016/j.cell.2014.05.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Voorhees R. M., and Hegde R. S. (2016) Toward a structural understanding of co-translational protein translocation. Curr. Opin. Cell Biol. 41, 91–99 10.1016/j.ceb.2016.04.009 [DOI] [PubMed] [Google Scholar]
- 48. Colombo S. F., Cardani S., Maroli A., Vitiello A., Soffientini P., Crespi A., Bram R. J., Benfante R., and Borgese N. (2016) Tail-anchored protein insertion in mammals: function and reciprocal interactions of the two subunits of the TRC40 receptor. J. Biol. Chem. 291, 18855 10.1074/jbc.A115.707752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kinoshita T., and Fujita M. (2016) Biosynthesis of GPI-anchored proteins: special emphasis on GPI lipid remodeling. J. Lipid Res. 57, 6–24 10.1194/jlr.R063313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dalley J. A., and Bulleid N. J. (2003) The endoplasmic reticulum (ER) translocon can differentiate between hydrophobic sequences allowing signals for glycosylphosphatidylinositol anchor addition to be fully translocated into the ER lumen. J. Biol. Chem. 278, 51749–51757 10.1074/jbc.M303978200 [DOI] [PubMed] [Google Scholar]
- 51. Ran F. A., Hsu P. D., Wright J., Agarwala V., Scott D. A., and Zhang F. (2013) Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 10.1038/nprot.2013.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mali P., Yang L., Esvelt K. M., Aach J., Guell M., DiCarlo J. E., Norville J. E., and Church G. M. (2013) RNA-guided human genome engineering via Cas9. Science 339, 823–826 10.1126/science.1232033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mariappan M., Li X., Stefanovic S., Sharma A., Mateja A., Keenan R. J., and Hegde R. S. (2010) A ribosome-associating factor chaperones tail-anchored membrane proteins. Nature 466, 1120–1124 10.1038/nature09296 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw data are available at reasonable request by contacting Dr. Malaiyalam Mariappan (malaiyalam.mariappan@yale.edu), Yale School of Medicine, all remaining data are contained within the article.