

SUMMARY
Collective migration is essential for development, wound repair, and cancer metastasis. For most collective systems, “leader cells” determine both the direction and the power of the migration. It has remained unclear, however, how the highly polarized vertebrate epithelium migrates directionally during branching morphogenesis. We show here that, unlike in other systems, front-rear polarity of the mammary epithelium is set up by preferential cell proliferation in the front in response to the FGF10 gradient. This leads to frontal stratification, loss of apicobasal polarity, and leader cell formation. Leader cells are a dynamic population and move faster and more directionally toward the FGF10 signal than do follower cells, partly because of their intraepithelial protrusions toward the signal. Together, our data show that directional migration of the mammary epithelium is a unique multistep process and that, despite sharing remarkable cellular and molecular similarities, vertebrate and invertebrate epithelial branching are fundamentally distinct processes.
In Brief
Lu et al. demonstrate that directional migration of mammary epithelium is a unique multistep process that includes asymmetric stratification, loss of apicobasal polarity, and active migration stages. Leader cells are a dynamic population, which form intra-epithelial protrusions and move faster and more directionally than follower cells do toward the signal source.
Graphical Abstract
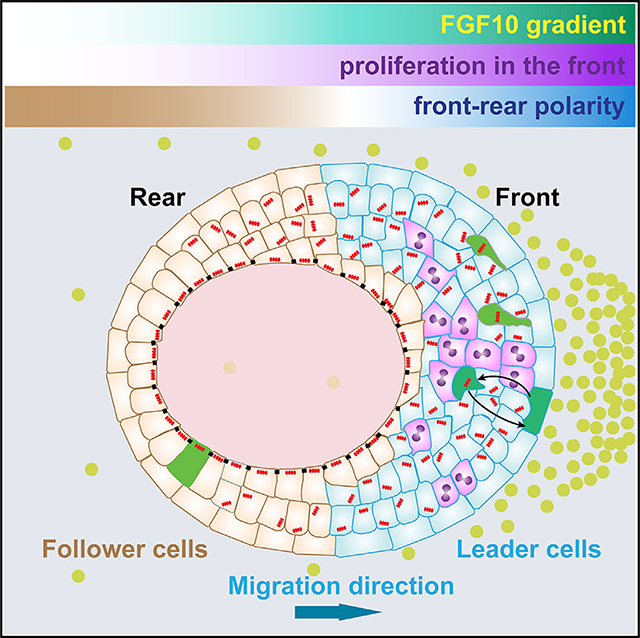
INTRODUCTION
Migration is a fundamental cell behavior in many biological processes (Friedl and Gilmour, 2009). Although migration of single cells is traditionally most studied and best understood, recent advances have highlighted the importance of collective migration, in which a group or a cluster of cells cooperate and coordinate their movements as a dominant process in development that includes epithelial branching, tissue repair, and cancer metastasis (Friedl and Gilmour, 2009; Scarpa and Mayor, 2016). Regardless whether cells migrate as individuals or a collective, migration is often directional and, most times, is triggered and guided by a gradient from an external cue, be it a soluble factor, an electrical field, or a mechanical force (Bear and Haugh, 2014; Petrie et al., 2009). Thus, a major theme in understanding directional migration is to determine how directionality, or “front-rear” polarity, is set up by the external cue and where the source of the force that powers the migration process is located (Mayor and Etienne-Manneville, 2016).
For both single cells and cell collectives, one of the earliest morphological signs of front-rear polarity is the polarized formation of actin-rich filopodia or lamellipodia at the leading edge of the migrating unit (Haeger et al., 2015). Once formed, these cellular extensions undergo sequential cycles of extension, adhesion, and contraction to generate traction force to power the migration process (Zegers and Friedl, 2014). Interestingly, although every cell that migrates individually forms these kinds of cellular extensions pointing toward the external cues, only one or a few cells at the migration front do so when they migrate as a collective (Friedl and Gilmour, 2009; Petrie et al., 2009). These cells are often referred to as “leader cells” because of their location at the leading position, whereas those in the rear are referred to as “follower cells” (Khalil and Friedl, 2010; Scarpa and Mayor, 2016; Theveneau and Linker, 2017). Importantly, for all known migrating collectives, leader cells are not only a manifestation of directionality but also the source of traction forces that power the migration process (Friedl and Gilmour, 2009; Haeger et al., 2015; Petrie et al., 2009). Indeed, in both Drosophila trachea during embryonic development and air sacs at larva stages, directional migration is driven by leader cells in response to fibroblast growth factor (FGF) cues and is essential for patterning the branch network (Affolter et al., 2003; Lu et al., 2006).
Compared with other collective systems, including fly trachea and air sacs, fibroblasts, and endothelial cells, directional migration of vertebrate epithelia has remained poorly understood. Unlike those other systems with loosely connected cells, vertebrate epithelia feature strong cell-cell junctions and adhesions (Friedl and Gilmour, 2009; Friedl and Mayor, 2017), which are required for maintaining apical-basal polarity but are also thought to be an important constraint against collective migration (Friedl and Gilmour, 2009; Friedl and Mayor, 2017). However, it has long been assumed that vertebrate epithelia, especially those from branched organs, including the lung, kidney, and mammary gland, undergo directional migration as a part of their ontogeny (Ewald et al., 2008; Lu et al., 2008; Lu and Werb, 2008). The assumption is in part based on the observation that invertebrate and vertebrate branching systems, which are non-homologous structures, share a surprising amount of cellular and molecular events or “deep homology” during organ formation (Affolter et al., 2003; Davies, 2002; Lu et al., 2006; Lu and Werb, 2008).
At present, almost all of what we know about vertebrate epithelial directional migration has been based on research using an in vitro model in which pieces of mammary gland epithelium, or organoids are cultured in a medium containing FGF2 (Ewald et al., 2008; Ewald et al., 2012; Fata et al., 2007; Huebner et al., 2014; Nguyen-Ngoc et al., 2012). Because FGF2 stimulates mammary organoids to form small branches, this model has been extensively used for understanding the basis of branching morphogenesis, a multi-step process that includes both directional migration and subsequent ductal elongation (Affolter et al., 2003; Lu et al., 2006; Lu and Werb, 2008). However, the use of FGF2-based model has yet to offer insight on how directionality is determined or the location of the source of force that powers mammary epithelial migration (Friedl and Gilmour, 2009). Part of this paradox might be explained by our recent surprising finding that mammary gland epithelium does not undergo directional migration toward FGF2. Here, we followed up on our observation that FGF10 induces directional migration and attempted to examine the underlying mechanism.
RESULTS
Mammary Gland Epithelium Undergoes Directional Migration toward FGF10 Signal
We first tested whether mammary stroma could induce epithelial directional migration. We harvested stromal cells, which were primarily fibroblasts, and aggregated them in a hanging-drop culture overnight. Aggregated stromal cells, referred to as “stromospheres,” were then juxtaposed with mammary organoid epithelium in a basal medium and were monitored via differential interference contrast (DIC) time-lapse microscopy (Figure 1A). We found that mammary epithelium moved toward the stromosphere during the following 48 h, suggesting the mammary stromal cells (Figures 1B–1B″′) can elicit directional collective migration of the corresponding epithelium. Interestingly, we also observed directional migration of the stromosphere toward the epithelium (Figures 1B–1B″′). Unlike the epithelium, the stromosphere sent out streams of fibroblasts that had pronounced cellular protrusions at the leading edge (Figures 1B–1B″′; Video S1) before movement of the stromosphere as a whole.
Figure 1. Mammary Gland Epithelium Undergoes Directional Migration toward the FGF10 Signal.

(A) A schematic diagram depicts the experimental procedures during sample preparation, in vitro culture, and treatment methods. Mammary organoids and stromal fibroblasts were prepared from wild-type mice. Heparan sulfate beads were pre-soaked in fibroblast growth factors (FGFs) overnight and were briefly rinsed before use. Organoids were co-cultured with aggregated fibroblasts, the “stromospheres” in (B)–(B″′) and the beads pre-soaked in FGFs or BSA in (C)–(E) or in medium containing FGFs in (F)–(G′).
(B) Time course of in vitro co-cultures of epithelial organoids with stromospheres. Note the epithelium formed a cyst (hollow arrowhead) and underwent directional collective migration toward the stromospheres, whereas the stromospheres formed spiky extensions made of fibroblasts (filled arrowheads), often toward the direction of the epithelium (n = 5). White dotted outlines indicate the original positions of the organoid and stromosphere at time 0 h. Scale bars, 100 μm.
(C–E) Differential responses of epithelial organoids to beads pre-soaked in FGF10 (C), BSA (D), or FGF2 (E). (C)–(C″′) Time course of directional migration of epithelial organoid toward the FGF10 bead (n = 23). Organoid did not migrate toward the beads pre-soaked in BSA (D) and (D′) (n = 37) or FGF2 (E) and (E′), n = 7). Instead, when stimulated by FGF2, organoids formed huge cysts. Heparan acrylic beads of ~100 μm in diameter were juxtaposed with mammary organoids at a distance of ~100 μm. Scale bars, 100 μm.
(F and G) Epithelial organoid response to FGF10 (F) and (F′) (n = 5) or FGF2 (G) and (G′) (n = 33) when it was universally delivered in the medium. Note organoid epithelium formed branches after 5 days of culture in a medium containing FGF2 (G) and (G′). Scale bars, 100 μm.
One potential mechanism by which mammary stromal cells trigger epithelial directional migration is by secreting factors, for example, FGF10 and FGF2, both of which are known to be produced by mammary stromal fibroblasts, and turning on paracrine-signaling pathways. Indeed, we found that heparan-sulfate beads pre-soaked in FGF10 (Figure 1C–1C″′; Video S2), but not in bovine serum albumin (BSA; Figures 1D and 1D′), could attract organoid migration when organoids grew in size in both cases. Interestingly, mammary epithelium did not migrate toward beads pre-soaked in FGF2 but formed a cyst and grew greatly in size within the following 72 h (Figures 1E and 1E′). These results suggest that FGF10 acted as a chemo-attractant for mammary epithelium. We predicted that universal delivery of FGF10 would not attract epithelial movement. Indeed, we found that organoid epithelium did not migrate in the basal medium containing FGF10 (Figures 1F and 1F′). Curiously, however, organoids also did not grow under that condition, even though they did when FGF10 was delivered by beads (compare Figures 1C and 1F). As shown previously, mammary epithelium formed branches in the medium containing FGF2 after 3–5 days of incubation (Figures 1G and 1G′).
Together, these data show that stromal FGF10, but not FGF2, causes directional collective migration of mammary epithelium. Furthermore, because heparan-sulfate-beads deliver FGF ligands in a gradient (Makarenkova et al., 2009; Thotakura et al., 2019), the results show that it is the FGF10 gradient, rather the ligand per se, that promotes cell proliferation.
Front-Rear Polarity of the Epithelium Was Set Up by Differential Cell Proliferation
We characterized the kinetics and morphological changes during directional migration of organoid epithelium when stimulated by FGF10. We found that organoid epithelium increased in size over the 3-day time course (Figure 2A). Interestingly, the cells did not move, as shown by both their distances to the FGF10 bead (Figure 2B) and their speed (Figure 2C), until after ~30 h. Once they started moving, however, their speed appeared to increase with time. Using DIC time-lapse microscopy, we found a series of distinct changes in epithelial morphology during organoid migration. Specifically, we found that the epithelium first underwent a sealing process to form a cyst (Figures 2D and 2E). By the time the organoid was about to migrate at ~30 h, the side of the epithelium closer to the FGF10 bead, which we refer to as the “organoid front,” was noticeably thicker than the opposite, or rear, side (Figure 2F); this morphological feature was maintained throughout the migrating stages (Figure 2G).
Figure 2. Front-Rear Polarity of the Epithelium Is Set Up by Differential Cell Proliferation.

(A–C) Organoid size (A), distance to the bead (B), and speed (C), as measured every 5 h during the 3-day course of collective migration. Note that organoids started to move after ~30 h (n = 15).
(D–G′) Still DIC images of organoid during migration (D)–(G). The distinctive changes in organoid epithelial morphology imply the presence of substages during the migration process. Note that the increase in the thickness of the epithelial wall preceded migration. The white dotted line denotes lumen cavity. The red bar indicates the thickness of a normal epithelial duct at time 0. Scale bars, 100 μm. (D′)–(G′) DAPI nuclear staining indicates that changes in the thickness of the cyst epithelium are caused by an increased number of cells via stratification, rather than by cell enlargement. Scale bars, 20 μm.
(H–K) EdU incorporation based cell proliferation analysis between the cyst (E) and the stratification (F) stages. Quantification (K) indicates that more cells underwent proliferation in the front than in the rear of the organoid, suggesting stratification was not a result of cell rearrangement. Scale bars, 10 μm. Student’s t test, ***p < 0.001, 10 h, n = 10; 24 h, n = 8; 48 h, n = 9.
(L) Diagram indicates the distinct stages of epithelial migration, including the sealing, cyst, polarized cyst, and migrating cyst stages.
Using DAPI nuclear staining, we found that the thickening of the front side of the organoid resulted from an increase in the number of cells, rather than an enlargement of cell size in that area of the epithelium (Figures 2D′–2G′). To determine the cause of the increased cell numbers in the front of the organoid, we measured 5-ethynyl-2′-deoxyuridine (EdU) incorporation during organoid migration. We detected a spike of EdU incorporation before the thickening of the front epithelium, showing that cell proliferation, rather than cell rearrangement, of the epithelium causes cell numbers to increase, which results in the thickened epithelium in the organoid front (Figures 2H–2K).
Together, the results show that directional migration from FGF10 stimulation is a multi-stage process by which a simple epithelium forms a polarized cyst, which then undergoes epithelial sealing, differential cell proliferation and stratification, and subsequent migration (Figure 2L). Moreover, front-rear polarity, or directionality, is formed by preferential cell proliferation in the front epithelium.
Frontal Epithelial Stratification Is Required for Collective Directional Migration
Despite lacking direct proof from in vivo live imaging, it has long been speculated that the mammary gland shares similar processes, such as collective migration, with fly trachea and air sac systems during epithelial branching morphogenesis (Lu et al., 2006). The remarkable resemblance between the asymmetrically stratified migrating organoid and the epithelial terminal end buds (TEBs), whose characteristic stratification forms during the onset of postnatal mammary branching in mice at 3-weeks of age (Howard and Veltmaat, 2013), supports the speculation that TEBs may undergo collective migration in vivo. Therefore, we hypothesized that the transition from the terminal ends (TEs), which are simple epithelial duct ends, to TEBs also results from increased cell proliferation as we saw in vitro. With EdU-incorporation assays, we found that there was very little cell proliferation in the resting TE epithelium at the 2-week stage (Figures 3A and 3C). However, cell proliferation was greatly increased in the TEB epithelium (Figures 3B and 3C). These data suggest that the stratification of TEB epithelium is most likely, as in vitro migrating organoids, the result of increased cell proliferation and support the speculation that TEBs participate in collective migration in vivo.
Figure 3. Frontal Epithelial Stratification Is Required for Collective Directional Migration.

(A–C) Cell proliferation as examined by EdU (red) incorporation during the transformation of the mammary epithelium from a simple ductal epithelial end at the prepubertal (2 weeks, n = 7) stage (A) into a bulb-shaped stratified end bud at the pubertal (4 weeks, n = 4) stage (B). Quantification of cell proliferation (C). Scale bars, 30 μm.
(D–G) Time course of epithelial organoid migration toward the FGF10 beads in the presence of cell proliferation inhibitor aphidicolin to block stratification. (D) Diagram illustrating the drug-treatment regimens. Aphidicolin was applied to the organoid either at the beginning, when it was a simple ductal epithelium (E) (n = 20), or after stratification was complete (F) (n = 20). Alternatively, the inhibitor was given to the organoid in the form of a TEB, which was a stratified epithelium (G) (n = 25). Scale bars, 50 μm. (E′)–(G′) Quantification of the percentages of organoids that migrated under the above treatment strategies. ****p < 0.0001. ns, not significant.
An important question is whether stratification, resulted from increased cell proliferation, is required for collective migration. We examined the effect of inhibiting cell proliferation and, thus, epithelial stratification, by adding aphidicolin to the medium either at the start or after stratification was completed during the FGF10-induced migration assay (Figure 3D). We found that, when added to the organoid as a simple ductal epithelium, aphidicolin treatment blocked organoid size increase and migration during the assay (Figures 3E and 3E’). However, if treatment was given after the epithelial stratification had been completed, then a majority (56.0%) of the organoids examined could undergo collective migration (Figures 3F and 3F’). Likewise, if the epithelium was already stratified from the start of the experiment, for example, when TEBs were used, a majority (56.4%) of samples could still effectively undergo collective migration, even if the treatment started at the beginning of the assay (Figures 3G and 3G’).
These data thus show that increased epithelial cell proliferation and, most likely, subsequent stratification during the polarizing stage, is required for collective migration of the mammary epithelium.
Apical-Basal Polarity Is Lost in Stratified Epithelium Both In Vitro and In Vivo
The above results seemed paradoxical when we considered that the migrating epithelial sheet in the 2D wound-healing assay is obviously not stratified. One explanation for this apparent paradox is that epithelial apical-basal polarity, maintained by tight junctions and characteristic cytoskeletal architecture, rather than stratification per se, is the actual barrier against directional migration. If so, epithelial polarity should be lost in, at least, the leader cells, which are thought to drive directional migration of the 2D epithelial sheet cells. To test that, we examined tight-junction-marker expression in the wound-healing assay. We found that tight junctions were present in the trailing cells of the migrating epithelial sheet, as shown by OCLN and ZO1 expression (Figures S1A, S1A1, S1B, and S1B1), but were absent from leading cells (Figures S1A, S1A2, S1B, and S1B2). These results are thus consistent with the notion that tight-junctions are inhibitory for leader cells.
Next, we used immunofluorescence microscopy to examine how tissue polarity may be modulated during the course of directional migration of mammary epithelium. As expected, the mammary epithelium is a simple columnar epithelium at the duct stage, as judged from its normal expression of proteins in the ZO1 complex and in Ezrin, both of which mark the apical domain of the mammary epithelium (Figures 4A and 4C), in E-cadherin (Figure 4E), which marks the basolateral domain, and in GM130 (Figure 4G), which marks the orientation of the Golgi complex. Interestingly, all of these tissue polarity markers lost their distinctive expression pattern upon epithelial stratification. Thus, in the migrating cyst epithelium, both the apical markers ZO1 and Ezrin were only present on the cells immediately facing the lumen and were no longer found on cells in the middle of the stratified epithelium (Figures 4B and 4D). E-cadherin was no longer basolateral but was instead found in all the cell boundaries in the stratified epithelium (Figure 4F). Likewise, rather than pointing toward the lumen, the Golgi complex, as marked by GM130, became randomly distributed in the migrating cyst epithelium (Figure 4H).
Figure 4. Apical-Basal Polarity Is Lost in Stratified Epithelium Both In Vitro and In Vivo.

(A–H) Immunofluorescence examination of tissue polarity markers in organoids at the duct (A, C, E, and G) and at the migrating cyst stages (B, D, F, and H). Insets are close-up views of the area in the white rectangles with dashed lines. Note ZO1 and Ezrin are normally expressed in the apical membrane (A) (n = 15) and (C) (n = 6) but are lost in cells of the stratified epithelium (B) (n = 5) and (D) (n = 9), except in those facing the lumen. E-cadherin is normally present in the basal-lateral membrane of simple ducts (E) (n = 9) but are found throughout the membrane of stratified epithelial cells (F) (n = 5). GM130 is often found toward the apical side in ductal epithelium (G) (n = 10), but that orientation becomes random in migrating organoids (H) (n = 5). Scale bars, 20 μm.
(I–P) Immunofluorescence examination of the above tissue polarity markers in vivo in the ductal epithelial TEs at prepubertal (2 weeks): (I) (n = 6); (K) (n = 3); (M) (n = 5); and (O) (n = 4) and in the TEBs at pubertal stages (4 weeks): (J) (n = 3); (L) (n = 3); (N) (n = 4); and (P) (n = 3). Insets are close-up views of the area in the white rectangles with dashed lines. Note the expression patterns of in vivo TEBs (J, L, N, and P) are similar to those of the migrating cysts in vitro (B, D, F, and H). Scale bars, 20 μm.
An important question is whether the epithelial stratification we observed in vitro reflects what occurs in vivo when directional migration is supposed to occur during pubertal mammary branching. To determine that, we compared the epithelial TEs at 2 weeks of age, when branching has yet to start, and in those at 4 weeks of age, when branching is well underway (Howard and Lu, 2014; Inman et al., 2015). We found that the 2-week TEBs had a simple epithelium with polarity markers correctly located (Figures 4I, 4K, 4M, and 4O). By contrast, the 4-week TEBs formed a stratified epithelium with cells did not show clear polarity (Figures 4J, 4L, 4N, and 4P). Thus, we conclude that in both the in vitro model and the in vivo branching, epithelial stratification is accompanied by polarity loss.
Taken together, our data suggest that epithelial polarity, as marked by tight junction expression, is inhibitory to directional migration. This migration barrier is removed by downregulation of tight junctions in leader cells of the epithelial sheet and by stratification of the mammary gland in the frontal epithelium in vitro and in the TEBs in vivo.
Frontal Cells Move Faster and More Directionally Than Do Rear Cells during Collective Migration
These results also suggest that the driver cells are most likely in the front-stratified epithelium that has lost tissue polarity. Therefore, we next sought to determine individual cell behavior of the mammary organoid epithelium during directional migration. We prepared organoid epithelium from female mice carrying the H2B-GFP transgene, which allowed us to monitor cell movements by tracking the nuclei for various durations during collective migration. We found that all the cells in the mammary organoid epithelium were actively moving, albeit at different speed, with different directional preferences and varying distances covered, regardless of whether the organoid was stimulated by FGF10 or BSA (Figures 5 and S2; Video S3). Thus, in organoids stimulated by BSA-soaked beads, which did not move, individual cells migrated at a speed of ~5 μm/h (Figures S2A–S2A″ and S2F). However, their directions of movement were random (Figure S2E), and we did not detect any differences in individual cell behavior between the front and rear cells of the organoids stimulated by BSA.
Figure 5. Front Cells Move Faster and More Directionally Than Rear Cells Do during Collective Migration.

(A–D″) Analysis of cell behavior during collective migration of organoid epithelium in response to the FGF10 bead. Note, beads were placed to the right of the organoid epithelium and outside the view. (A–D) Maximum intensity projections from 3D confocal videos of organoid epithelium derived from female mice carrying the H2B-GFP (green) transgene. Scale bars, 50 μm. (A′–D′) Time course of nuclei trajectories for the indicated durations. Purple and red colors of the time bar indicate the beginning and end of the video, respectively. Cells were tracked for 25, 49, 73, and 100 frames, respectively, in (A′), (B′), (C′), and (D′), respectively. (A″–D″) Cell displacement at the indicated times. Arrows represent cell displacement directions. Cell displacement was tracked at the indicated durations using 25, 49, 73, and 100 frames in (A″), (B″), (C″), and (D″), respectively.
(E) 3D rendering of cell tracks when plotted with a common origin. Cells were tracked for 86.75 h (244 frames). Note cell tracks were preferentially toward the right side where an FGF10 bead was located.
(F) Mean cell speeds, as calculated from nuclear trajectories in which track lengths were divided by time.
(G) Displacement lengths of analyzed cells. A total of eight organoids were analyzed, but only data from two representative organoids were combined to generate (F) and (G) (front cells, n = 32; rear cells, n = 47). Frame interval was 15 min. Unpaired Student’s t test was used for statistical analysis. ****p < 0.0001, ***p < 0.001.
By contrast, there was a clear difference in the cell behavior between the front and rear cells of organoids stimulated by FGF10. For example, we found front cells moved more preferentially toward the FGF10 signal, whereas rear cells moved randomly like cells in BSA-stimulated organoids (Figures 5A–5A″ and 5E). Moreover, front cells moved faster than the rear cells did (~10 μm/h versus ~7 μm/h). Interestingly, the rear cells of FGF10-stimulated organoid moved at a faster speed than did cells in the BSA-stimulated organoids. Not surprisingly, we found that front cells traveled a farther distance than rear cells did in organoids stimulated by FGF10 (Figures 5G, S2G, and S2I). It is noteworthy that the speed of organoid movement was one magnitude slower than that of single-cell movements, regardless of whether they were stimulated or not (compare Figures 2C and 5F; Figures S2F and S2H). Therefore, we did not subtract the organoid speed from individual cell speed in our calculations.
Together, our data show the main determinants of individual cell behavior during migration are the nature of the stimulant and the cell position in the organoid. They show that front cells move faster and more directionally than rear cells do during collective migration, suggesting that they are leader cells that power the migration process.
Leader Cells in the Migrating Epithelium Are a Dynamic Cell Population
A common feature of all existing models of collective migration is that leader cells locate at the migrating front. These leader cells are often a stable population of cells that send filopodium or lamellipodium extensions into the matrix to generate a pulling force that powers the entire migrating group. Therefore, we sought to characterize the cells at the migrating front and to determine whether they are the leader cells of the mammary organoid epithelium (Figure 6A). To that end, we harvested mammary organoids from mice carrying the R26RmTmG allele, which were then infected with adeno-Cre at a low infection rate, so only a few cells would be marked by eGFP protein expression before the organoids were subjected to the FGF10-based collective migration assay (Figure 6B).
Figure 6. Leader Cells in the Migrating Epithelium Are a Dynamic Cell Population.

(A) Schematic diagram depicting the hypothetical zone of leader cells at the epithelial invasion front.
(B) Schematic diagram showing the experimental procedure in sample preparation, adenoviral infection, and labeling of individual cells during time-lapse confocal imaging. Mammary organoids were harvested from R26RmTmG mice and were infected with adenoviral Cre. Cell movement was recorded during organoid migration.
(C–D′) 3D reconstructed views of individual cell movement tracked during the time frames indicated. Areas in rectangles with white dashed lines are shown in close-up views in (C′) and (D′). Arrows indicate the cells in question, with (C) and (C′) showing a cell initially at the leading position moved outside with time, whereas (D) and (D′) depict a cell initially outside of the leading position moved into it with time. Scale bars, 20 μm.
(E) Summary illustrating a lack of stable leader-cell population (green) during directional epithelial migration, in which, unlike in other collective migration systems, cells (blue and pink) can move in (pink cell) and out (blue cell) of the leading position of the epithelium.
We first evaluated this alternative method to mark individual cells for their usefulness in characterizing cell behavior during epithelial migration. We found that, on average, ~10 cells of an organoid of ~300 cells were labeled in green by ~20 h. The delay in eGFP expression presumably reflected the time that it took for the GFP reporter gene to be activated and expressed (Figure S3A). Consistent with the results showing a higher rate of cell proliferation in the front than in the organoid’s rear, we found that the number of green cells preferentially increased in the organoid front (Figures S3B–S3D). Green cells moved faster and more preferentially toward the front than did rear cells (Figures S3A–S3G). These results agree with the findings from the H2B-GFP system and thus validate their use for further characterizations of cell behavior during collective migration (Video S4).
Next, we determined whether cells at the migration front were a stable population. Interestingly, we found cells at the migrating front exited it and ended in the trailing zone (Figures 6C and 6C′; Video S5). Likewise, we also found cells initially in the trailing zone moved into the migrating front (Figures 6D and 6D′; Video S6). These data thus suggest that cells at the front could exit, whereas trailing cells could enter the leading zone. We did not observe filopodia, lamellipodia, or any other cellular extensions during organoid migration toward the FGF10 bead or mammary stromosphere (Figures 1B–1B″′). This is in contrast to the stromosphere, which, during migration, sent out streams of cells (Figures 1B–1B″′ and S4A) and actin-based lamellipodia (Figure S4B). To examine whether directional migration of mammary epithelium relies on actin cytoskeleton as other systems do, we treated organoids with either the actin-polymerization inhibitor latrunculin (Figures S4E and S4F) or the actin-depolymerization inhibitor jasplakinolide (Figures S4G and S4H). In both cases, the addition of the actin-polymerization or -depolymerization inhibitor completely abolished collective migration of the mammary epithelium, indicating that force generation in this model also depends on the actin-based cytoskeleton, similar to other systems.
Taken together, these data suggest that, unlike other collective migration systems, leader cells are a dynamic group in the frontal epithelium rather than a fixed population at the leading edge (Figure 6E).
Leader Cell Protrusions Are Oriented toward the FGF10 Source
It was possible that the organoid epithelium could be driven by “hidden” intra-epithelial protrusions that do not extend into the matrix but that could still generate a traction force by interacting with matrix. We, therefore, analyzed cell-shape changes using the above mosaic-mammary epithelium in which some cells were labeled by membranous eGFP expression (Figures 7A, 7B, and S5). To quantitatively assess the directions of intraepithelial protrusions, we assigned them to 45° bins, with the 0–180° axis aligned with the migration direction (Figures 7A′ and 7A″; Video S7). Interestingly, we found that the intra-epithelial protrusions of the front cells showed a strong preference toward the FGF10 beads (Figure 7B), whereas those of the rear cells did not (Figures S5A–S5A″ and S5B; Video S8). Neither the front nor the rear cell intra-epithelial protrusions showed any preferences toward a particular direction when beads were pre-soaked in BSA (Figures S5C–S5F). These data thus suggest that FGF10 has a role in orienting the intra-epithelial protrusions, which are likely to have a role in the generating traction force to power the collective migration of the mammary organoid.
Figure 7. Leader Cell Protrusions Are Oriented toward the FGF10 Source.

(A–A″) A representative 3D reconstruction of a front cell with protrusions, imaged during collective migration induced by FGF10. (A″) Overlaying method for assigning protrusions to 45° bins, with the 0–180° axis aligned with the migration direction. Scale bars, 20 μm.
(B) Protrusions per bin as quantified from front cells of the organoid. Two-sample Hotelling᾽s T-squared test showed a significant mean direction (p < 0.01). A total of 1,575 protrusions from 25 cells of 17 organoids were used for this analysis. See Method Details.
(C and D) Still DIC images of organoid during migration at the times indicated. Note that the FGF10 bead was pushed by the migrating organoid. White dotted line denotes the original position of the FGF10 bead. Green and red dots indicate the center of gravity of the bead at the beginning and end, respectively. Arrow indicates the distance and direction of bead displacement. Scale bars, 100 μm.
(E) Organoid speed was measured every 4 h, starting with the bead-contacted organoid (n = 5). Note that organoid speed briefly slowed when it touched the bead before it accelerated again.
(F) A model of the FGF10 function during directional collective migration of the mammary gland epithelium. The FGF10 gradient promotes preferential cell proliferation (cell with purple nucleus) in the organoid front and thus sets up the “front-rear” polarity (1). As a result, the organoid front becomes stratified (dark beige cells) and loses tissue polarity (inner layer cells with GM130 irregular arrangement and loss of tight junction) (2). The FGF10 also orients intra-epithelial protrusions toward the signal source (F1 and F2) and coordinates movements (F3) of individual front leader cells (3). As a result, leader cells drive mammary organoids to undergo directional migration. Note that cell protrusions in the rear “passenger” cells are random (R). Moreover, cells lining the lumen are unlikely to be driver cells because they still show tissue polarity, as marked by the expression patterns of ZO1 and GM130 typically observed in the polarized epithelium.
These data suggest that leader cells in the front epithelium are drivers that power directional migration. If that is correct, then the force is unlikely to be a “pulling” force alone, which would have been the case had the leader cells been situating at the leading edge; rather, the driving force should be more likely a combination of both a “pulling” and a “pushing” force, depending on the relative position of each leader cell to their neighboring cells in the entire collective. Thus, a leader cell could both pull a trailing cell and push a cell at its front in the organoid. Indeed, we found that, when the migration assay was not stopped after the organoid had touched the FGF10-soaked bead, it would start “pushing” and migrate together with the bead (Figures 7C and 7D; Video S9). Interestingly, the speed of the organoid initially dropped upon touching the bead, but then, the organoid-bead combination regained speed in a kinetics similar to the migrating organoid alone (Figure 7E).
Taken together, these data show that the FGF10 gradient orients intra-epithelial protrusions of the front cells, and, as a result, leader cells drive epithelial directional migration by both a pushing and a pulling force.
DISCUSSION
Collective migration is a fundamental cell behavior essential for development, wound repair, and cancer metastasis. However, it has remained unclear as to whether vertebrate epithelium, characterized by abundant adherens and tight junctions, undergoes directional migration and, if it does, how its directionality is set up and where the driver cells are. Here, we show that directional migration of mammary epithelium is a multi-step process (Figure 7F). In the first step, front-rear polarity is set up when front epithelium undergoes increased cell proliferation and thickening. In the second step, front epithelium becomes stratified and partially loses apical-basal polarity, leading to the generation of leader cells. In the third step, leader cells, which are a dynamic, rather than a stable, population move faster and more directionally than do rear follower cells, extending their intra-epithelial protrusions along the direction of the FGF10 gradient, thus generating a coordinated force to power the epithelial migration toward the FGF10 signal. We show that a leader cell in the migrating mammary epithelium might pull or push the collective, depending on its relative position in the organoid and whether there are cells in front of it. Together, these results uncover a novel mechanism underlying directional collective migration of vertebrate epithelium (Figure 7F).
Directional Migration Is an Integral Part of Vertebrate Epithelial Branching but with a Distinct Cellular Basis from That of Invertebrate Systems
Branching morphogenesis is a fundamental process, essential for the formation of many invertebrate and vertebrate organs from Drosophila trachea and air sacs to mammalian lungs, kidneys, vasculature, pancreas, and many exocrine glands, including the mammary and salivary glands (Lu and Werb, 2008). Despite being non-homologous structures, the fly systems and the vertebrate branched organs share certain similar genetic requirements, including FGF signaling, during their ontogeny. As such, it has been speculated that these invertebrate and vertebrate organs share a similar cellular basis, such as directional migration, which is essential for branch initiation during fly trachea and air sac development (Lu et al., 2006).
Several lines of evidence from our work support that speculation. For example, we show that FGF10 functions as a chemoattractant for mammary epithelial organoids in the in vitro assay. Moreover, we show that the transition phases in which a simple ductal epithelium becomes a stratified, migrating organoid greatly resemble those occurring in vivo when TEB forms from the TE epithelium. These data are consistent with previous data showing that TEBs are a primary driver of mammary epithelial branching (Scheele et al., 2017). Finally, the results show that tissue polarity loss is accompanied by in vivo and in vitro stratification, which is required for directional migration. Our data thus support the notion that epithelial directional migration also occurs in vivo during mammary branching. However, definitive proof for that notion awaits future studies in which live imaging of vertebrate branching can be performed.
The data show that leader cells of the migrating mammary epithelium are a dynamic population that exert both a pushing and a pulling force, which is different from leader cells of the fly systems (Affolter and Weijer, 2005; Friedl and Gilmour, 2009; Scarpa and Mayor, 2016). Thus, despite sharing certain remarkable “deep homologies,” invertebrate and vertebrate epithelial branching is fundamentally different at the cellular and, most likely, the molecular level.
Modeling Distinct Aspects of Vertebrate Epithelial Branching by Different In Vitro Cultures
At present, almost all of what we know about epithelial directional migration has been based on the use of an in vitro model in which mammary organoid epithelium undergoes branching in a medium containing FGF2. Using that model, it has been shown that epithelial polarity is transiently lost in the stratified epithelium (Ewald et al., 2012), that cells at the end of each epithelial branch are a dynamic population (Ewald et al., 2008), that cells form intra-epithelial protrusions (Huebner et al., 2016), and that cell proliferation is not required for collective cell migration and elongation of the epithelial duct (Huebner et al., 2016). On the surface, those findings appear to overlap with some of the mechanisms we report here, by which the mammary epithelium undergoes directional migration. It is thus important to compare these findings from the literature with ours and to evaluate their similarities and differences.
Two criteria need to be borne in mind when we critically re-examine those findings. First, the biological processes on which those findings were made must directly concern the epithelial directional migration. Second, they must be able to answer, at least partially, one or both of the two main questions that concerned this study, i.e., how front-rear polarity is set up in the mammary epithelium and where the source of force is that powers the migration process. Our work shows that mammary epithelium does not undergo directional migration and that the FGF2-based branching model recapitulates more of the ductal elongation, a separate step of epithelial branching than that of directional migration (Zhang et al., 2014). Together, these data conclusively demonstrate that the aforementioned findings, unlike those made in the current analysis, do not relate to epithelial directional migration.
Thus, the dynamic cell movements observed in the FGF2-based branching model should not be considered epithelial directional migration or collective cell migration, which are interchangeable in most contexts (Friedl and Gilmour, 2009; Haeger et al., 2015; Scarpa and Mayor, 2016). Based on previous reports, the speed of individual cell movements are ~5 μm/h (Huebner et al., 2016), which is, as we report here, comparable to the speed of cell movements in the presence of BSA or the speed of rear cells in the presence of FGF10 beads (Figures 5F, S2F, and S2H). Therefore, the dynamic nature of individual cells being able to move at a baseline speed and, most likely, rearranging their relative positions are intrinsic properties of cells within the vertebrate epithelium and should not be interpreted as a part of directional migration.
Likewise, although loss of epithelial polarity has been observed in stratified epithelium in the FGF2-branching model (Ewald et al., 2012), that observation does not shed light on how polarity loss leads to the establishment of directionality and/or leader cells in the frontal epithelium, as presented in our current analysis. In fact, epithelial stratification has also been observed during oncogenic transformation of the mammary gland epithelium, even though it has been mechanistically unclear regarding what the subsequent consequences might be (Halaoui et al., 2017). It will be interesting to examine, in future studies, whether polarity loss during oncogenic stratification leads to increased epithelial collective migration, as our results here would predict.
Taken together, we describe a unique mechanism whereby the mammary epithelium undergoes directional migration. We await future studies aimed at the examination of whether that is a general principle, that other vertebrate epithelia, including those from the lung, kidney, and salivary gland, could also undergo directional migration.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Pengfei Lu (lvpf@shanghaitech.edu.cn).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
This study did not generate datasets. All raw data is available on request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse strains
Mice carrying the pTRE-H2BGFP allele (JAX Mice, #005104) and the R26RmT/mG reporter allele (JAX Mice, #007576) (Muzumdar et al., 2007) were purchased from the Jackson Laboratory. FVB mouse strains were purchased from Vital River Laboratory Animal Technology Co., Ltd. (Beijing). If it is not specifically mentioned, then 6 to 8 weeks old female mice were used to harvest the mammary gland organoid. Mice were housed and maintained in the Mouse Core Facility at the National Institute for Protein Science Center at Shanghai and Jiabo Biotechnology Co. Ltd. according to regulations from ShanghaiTech University’s Institutional Animal Care and Use Committee (IACUC# 2015SHT0006).
Cell culture
Female dog kidney epithelial cell line, Madin-Darby Canine Kidney (MDCK) cell, is a gift from Dr. James Edward Rothman. MDCK cells were cultured in DMEM medium (CORNING, Cat. No 10–103-CRV) supplemented with 10% (vol/vol) fetal bovine serum (FBS) (Hyclone, Cat. No SV30087) and penicillin (100 U/ml) /streptomycin (100μg/ml) (CORNING, Cat. No 30–002-CI) at 37°C and 5% CO2.
METHOD DETAILS
Preparation of primary mammary organoids and fibroblasts
Mouse mammary glands were finely chopped with two scalpels, ~300–400 times per mouse, and the mince was digested in 10 mL collagenase buffer [0.2% collagenase (Sigma, #C5138) and 0.2% trypsin (Thermo Fisher Scientific, #27250–018) in DMEM/F12 supplemented with 5% (vol/vol) FBS, 50 μg/mL gentamicin, 5 μg/mL insulin (all from Sigma)] for 22 min at 37°C. Adding 80 U DNase I, samples were pipetted up and down 10 times with a 2.5% BSA coated 10 mL pipette tip. Then samples were centrifuged at 560 × g, 10 minutes, and pellets were treated with DNase I (20 U/μL) for 5 min. After washing with DMEM/F12, pellets were resuspended in DMEM/F12 and subjected to short-pulse centrifugation at 450 × g (differential centrifugation). The supernatant was collected, and pellets were resuspended in DMEM/F12 for another round of differential centrifugation. After five rounds of differential centrifugation, pellets containing mammary organoids were resuspended in 1ml basal organoid culture medium [DMEM/F12, 100 μg/L insulin, 55 μg/L transferrin, 50ng/L selenium (ITS, Sigma, #I3146), 100 U/mL penicillin, and 100 μg/mL streptomycin] before they were subjected to subsequent procedures.
For adenoviral infection, mammary organoids harvested from R26RmTmG mice were infected with adenoviral Cre. 1μL of 4.6×10e8 pfu/mL adenovirus was mixed with organoids in 500uL basal organoid culture medium in ultra-low attachment surface plate (CORNING, #3473) for 40 minutes at 37°C and 5% CO2.
For fibroblast preparations, supernatant from each differential centrifugation was pooled and pelleted. The pellets were resuspended in fibroblast medium [DMEM, 10% (vol/vol) FBS, 100 μg/L insulin, 55 μg/L transferrin, 50ng/L selenium (ITS, Sigma, #I3146), 100 U/mL penicillin, and 100 μg/mL streptomycin], and seeded on a cell culture dish. After 30 min, when fibroblasts had already adhered to the dish whereas other cellular types remained in suspension, the medium was aspirated, and dish was washed twice with PBS before adding fresh medium. Fibroblast cultures were allowed to grow until they reached ~80% confluence before they were passaged. Only early passages (up to passage number 5) were used in experiments.
For stromosphere preparations, stromal fibroblasts were aggregated by hanging-drop overnight. They were then juxtaposed with organoids in Matrigel (Corning, #354230) at a distance of ~100 μm using a tungsten needle and cultured in basal medium for 48 h at 37°C (Figure 1A).
In vitro epithelial branching and migration assays
Heparan sulfate beads (Sigma, #H5263) of ~100–200 μm in sizes were picked and washed in 10μL 100μg/mL BSA (dissolved in PBS) or FGF10 solution overnight at 4°C. 15μL (for 24 well plate) or 4 μL (for 8 well chamber slide) of 80% Matrigel was used to coat the plates, which were then heated on a 37°C block for 1 minute. Mammary organoids were put next to beads pre-soaked in BSA or FGF10 at the distance of ~100 μm using a tungsten needle (Figure 1A). The plate was warmed up on a 37°C block for 8 minutes and added 1000μL (for 24 well plate) or 300 μL (for 8 well chamber slide) basal organoid culture medium. Samples were cultured in a 37°C incubator with 5% CO2 or transferred to a live imaging microscope for time-lapse imaging. Branching assay was done as described previously (Ewald et al., 2008). FGF2 (2.5nM, GenScript, #Z03116–50), or FGF10 (2.5nM, GenScript, #Z03155–50) were used in the 5-day culture.
Wound healing assays
60,000 MDCK epithelial cells were inoculated in each of an 8-well chamber slides coated with poly-L-Lysine (BBI Life Science, Cat. No E607015) according to the manufacturer’s protocol. Upon reaching confluency (after ~48 hours), a scratch wound to the monolayer of cells was made using a sterilized toothpick. Cells were incubated for another 21 hours, during which time the wound was healed before they were fixed in 4% paraformaldehyde for 15 min at 25°C. Cells were permeabilized in PBS containing 0.5% Triton X-100 for 30 minutes at 25°C, blocked for 2 hours before incubation with antibodies (ZO1; Invitrogen, #339111 and Occludin; Invitrogen, #331594) at 1:100 dilution overnight at 4°C. Microscopy was performed on a LSM800 Zeiss confocal microscope using Plan-Apo 20 × /0.8 or Plan-Apo 40 × /1.3 oil objective lenses. Image processing was done using ImageJ.
Cell Proliferation assays
Mice were injected with EdU (1 mg/100 g body weight) 4 hr. before sacrifice. Mammary epithelium was harvested and fixed in 4% paraformaldehyde for 45 min at 18°C and was stained with 100 μL staining solution (Guangzhou RiboBio Co., Ltd.C10371–1 Cell-Light TM Apollo 567 Stain Kit) for 45 minutes according to the manufacturer’s protocol. For in vitro proliferation analysis, organoids were incubated in medium containing with EdU (1:5000) (Guangzhou RiboBio Co., Ltd.C00054) for 4 hours. They were then fixed and stained as mentioned above. Microscopy was done using an LSM800 Zeiss confocal. We have ensured that the same settings were applied to the areas of experimental comparisons, for example, front versus rear, especially in the same batch of experiments. Briefly, confocal microscopy was performed on a LSM800 Zeiss confocal with a Plan-Apo 40 × /1.3 oil objective lens. For Z stacked image series, the images were collected every 0.45 μm. EdU staining solution and DAPI solution were freshly prepared right before staining. For each experiment we used lowest possible laser intensity to avoid over-exposure. Compared with samples collected at 10 hours and at 24 hours, those collected at 48 hours had the strongest signal so the laser intensity used for these samples had the lowest intensity. Specifically, for 10hr samples, laser wavelength for EdU was 561nm, intensity was 15.00%, GaAsP-PMT detector gain was 500V, image processing threshold range was 50–40000. Laser wavelength for DAPI was 405nm, intensity was 3.50%, GaAsP-PMT detector gain was 500V, image processing threshold range was 300–20000. For 24hr samples, laser wavelength for EdU was 561nm, intensity was 4.00%, GaAsP-PMT detector gain was 500V, image processing threshold range was 50–40000. Laser wavelength for DAPI was 405nm, intensity was 3.50%, GaAsP-PMT detector gain was 550V, image processing threshold range was 300–40000. For 48hr samples, laser wavelength for EdU 561nm, intensity was 0.50%, GaAsP-PMT detector gain was 500V, image processing threshold range was 50–40000. Laser wavelength for DAPI was 405nm, intensity was 3.50%, GaAsP-PMT detector gain was 550V, image processing threshold range was 300–30000. An average organoid was around 100 μm in diameter. While the X-Y plane covered the entire organoid, the Z stack covered around 60 μm of depth. Images from the middle planes were chosen for the figures. The optimal interval for Z stack was 0.45 μm.
Proliferation inhibition assays
Aphidicolin was used at 10 μM and added at the start of culture or after the asymmetric stratification stage (Figure 3E). Time-lapse movies were collected for 2–3 days from the time of inhibitor addition and analyzed to determine the migration ratio. Migration was scored as successful when the center of gravity of the organoid moved out the initial leading front after a three-day culture. Migration ratio was calculated based on every five cultured organoids.
Confocal image Acquisition and Time-lapse imaging
Still confocal images were acquired using a Leica SP8 STED 3X Leica microscope using a HC PL APO CS2 40x/1.30 OIL objective. The magnification is 40x. Image resolution was 1,024 × 1,024 square pixels. Red-green-blue (RGB) images were assembled using Fiji software. ZO1 uses laser line 631nm, intensity 15%, HyD detector gain 100%, image processing threshold range is 400–2000. Ezrin uses laser line 631nm, intensity 30%, HyD detector gain 120V, image processing threshold range is 200–2000. E-cadherin uses laser line 631nm, intensity 15%, HyD detector gain 100%, image processing threshold range is 300–2500. GM130 uses laser line 488nm, intensity 15%, HyD detector gain 100%, image processing threshold range is 200–1500.
For time-lapse imaging, DIC (differential interference contrast) microscopy was done on a Zeiss Cell Observer SD spinning disk confocal microscope by using an EC Plan-Neofluar 10x/0.3 Ph1 M27 objective lens (Item no.: 420341–9911-000), an Analy DIC Transmission light reflector, a sCMOS camera at 37°C with 5% CO2. Images were taken every 15 minutes using ZEN (blue edition) software.
Confocal live imaging was done on a Zeiss Cell Observer SD spinning disk confocal microscope and a Leica TCS SP5 confocal microscope. To minimize phototoxicity and avoid photobleaching, we used the lowest possible incident power for each laser without noticeable sacrifice on the signal. Specifically, the Zeiss microscope was used for individual cell behavior and H2B-GFP samples were used, the excitation and emission wavelengths for the laser were 488nm and 520–550nm, respectively. incident power was 1%. The Leica microscope was used for live imaging on individual cell behavior and mTmG samples were used. The excitation and emission wavelengths for one laser were 488nm and 494–543nm, respectively. incident power was 11%; the excitation and emission wavelengths for another laser were 561nm and 603–671nm, respectively. incident power was 15%; The Zeiss microscope was used for live imaging on cell protrusions and mTmG samples were used. The excitation and emission wavelengths for one laser were 488nm and 500–554nm, respectively, and incident power was 5.5%; the excitation and emission wavelengths for another laser were 561nm and 615–675nm, respectively, and incident power was 3%.
Tracking of organoids, cells, and nuclei
ImageJ was used to measure organoid size, distance traveled, and speed. Center of gravity of organoids was used for these measurements. Nuclei tracking was done on cells expressing the H2B-GFP transgene. They were tracked automatically using the Spots function of Imaris software (Bitplane). Nuclear center was marked with a spot in each frame and spots were then connected over time. Cells carrying the R26RmT/mG reporter allele also using the Spots function of Imaris. Mean cell speed was calculated by the total track length divided by tracking time, and displacement length was the length of cell displacement. Unpaired t test was used for assessing the statistical significance of cell speed and displacement lengths of the front or the rear cells of organoids. A single-factor ANOVA was used for assessing the statistical significance of cell speed and displacement length between different organoids.
Cellular protrusion analysis
Protrusions were analyzed based on 3D reconstructions of GFP expression of organoids carrying the R26RmT/mG reporter allele. Images were captured every 15 or 30 mins for 48–96h and analyzed using Imaris (Bitplane). Protrusion data were collected using an eight-section pie with deviations every 45° on a transparent circular sheet. The 0 to 180° axis was aligned with the direction of organoid migration and pie center was placed over the center of each analyzed cell. Two criteria were used to choose the cells for analysis. First, the area is not crowded with labeled cells, and the selected cell does not touch other cells (if a cell of interest touches other cells, that makes it difficult to define the protrusion). Second, the selected cell’s protrusion touches the surface of the organoid, which can be determined with the Imaris software (Bitplane). To avoid any bias, we analyzed all cells that meet these two criteria and found only 25 cells from 17 organoids that met these two criteria. The number of protrusions per bin per cell was counted every 15 or 30 min for a minimum of 6 hours. Protrusion data were plotted in sector charts using RStudio. Two-sample Hotelling᾽s T-squared test was used to determine the significance of a weighted mean direction. The null hypothesis is that there is no mean direction.
QUANTIFICATION AND STATISTICAL ANALYSIS
Sample size for each figure is denoted in the figure legends. Statistical significance between conditions was assessed by two-tailed Student’s t tests. For multiple group comparison, one-way ANOVA analysis was performed. For sector charts, two-sample Hotelling᾽s T-squared test was performed. All error bars represent SEM, and significance is denoted as *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001. n.s. denotes not significant.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| ZO-1 Monoclonal Antibody (ZO1-1A12), FITC | Invitrogen | Cat#33-9111; RRID: AB_2533148 |
| Occludin Monoclonal Antibody (OC-3F10), Alexa Fluor 594 | Invitrogen | Cat#331594; RRID: AB_2532186 |
| Ezrin Antibody | Cell Signaling Technology | Cat#3145S; RRID: AB_2100309 |
| Anti-E-CADHERIN Monoclonal Antibody, Unconjugated, Clone ECCD-2 | Innovative Research | Cat#13-1900; RRID: AB_86571 |
| GM130 antibody | BD Biosciences | Cat#560257; RRID: AB_1645351 |
| Bacterial and Virus Strains | ||
| Adeno-Cre-GFP | This paper Obio Technology (Shanghai) Corp., Ltd. | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| EdU | Guangzhou RiboBio Co., Ltd. | Cat#C00054 |
| Cell-Light EdU Apollo567 In Vitro Flow Cytometry Kit | Guangzhou RiboBio Co., Ltd. | Cat#C10371-1 |
| FGF2 | GenScript | Cat#Z03116-50 |
| FGF10 | GenScript | Cat#Z03155-50 |
| Critical Commercial Assays | ||
| Heparan sulfate beads | Sigma | Cat#H5263 |
| Matrigel | Corning | Cat#354230 |
| Experimental Models: Cell Lines | ||
| Female dog kidney epithelial cell line, Madin-Darby Canine Kidney (MDCK) cells | kind gift from Dr. James Edward Rothman | N/A |
| Experimental Models: Organisms/Strains | ||
| Mice carrying the pTRE-H2BGFP allele | The Jackson Laboratory | JAX: 005104 |
| Mice carrying the R26RmT/mG reporter allele | Muzumdar et al., 2007; The Jackson Laboratory | JAX: 007576 |
| FVB mouse strains | Vital River Laboratory Animal Technology Co., Ltd.(Beijing) | Vital River: 215 |
| Software and Algorithms | ||
| ImageJ | https://imagej.net/Welcome | RRID: SCR_003070 |
| Imaris | https://imaris.oxinst.com/packages | RRID: SCR_007370 |
| GraphPad Prism | https://www.graphpad.com/ | RRID: SCR_002798 |
Highlights.
FGF10 signal promotes proliferation in the front and sets up “front-rear” polarity
Frontal epithelium stratifies, loses apicobasal polarity, and forms leader cells
Leader cells are dynamic and form intra-epithelial protrusions toward the FGF10 signal
Organoid epithelium migrates directionally up the FGF10-concentration gradient
ACKNOWLEDGMENTS
We thank the Mouse Core Facility at the National Institute for Protein Science Center at Shanghai and Jiabo Biotechnology. We also thank the Molecular Imaging Core Facility (MICF) at ShanghaiTech University. This work was supported by grants from the Thousand Young Talents Program of China (to J.S.), the National Natural Science Foundation of China (NSF) of China grants 81970102 (to J.S.) and 31671494 (to P.L.); the National Institutes of Health (NIH) grant R35-DE026602 (to O.K.); the Ministry of Science and Technology of China grant 2017YFA0103502 (to P.L.); and start-up grants from ShanghaiTech University (to C.A., J.S., and P.L.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108246.
REFERENCES
- Affolter M, and Weijer CJ (2005). Signaling to cytoskeletal dynamics during chemotaxis. Dev. Cell 9, 19–34. [DOI] [PubMed] [Google Scholar]
- Affolter M, Bellusci S, Itoh N, Shilo B, Thiery JP, and Werb Z (2003). Tube or not tube: remodeling epithelial tissues by branching morphogenesis. Dev. Cell 4, 11–18. [DOI] [PubMed] [Google Scholar]
- Bear JE, and Haugh JM (2014). Directed migration of mesenchymal cells: where signaling and the cytoskeleton meet. Curr. Opin. Cell Biol. 30, 74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies JA (2002). Do different branching epithelia use a conserved developmental mechanism? BioEssays 24, 937–948. [DOI] [PubMed] [Google Scholar]
- Ewald AJ, Brenot A, Duong M, Chan BS, and Werb Z (2008). Collective epithelial migration and cell rearrangements drive mammary branching morphogenesis. Dev. Cell 14, 570–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewald AJ, Huebner RJ, Palsdottir H, Lee JK, Perez MJ, Jorgens DM, Tauscher AN, Cheung KJ, Werb Z, and Auer M (2012). Mammary collective cell migration involves transient loss of epithelial features and individual cell migration within the epithelium. J. Cell Sci. 125, 2638–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fata JE, Mori H, Ewald AJ, Zhang H, Yao E, Werb Z, and Bissell MJ (2007). The MAPK(ERK-1,2) pathway integrates distinct and antagonistic signals from TGFalpha and FGF7 in morphogenesis of mouse mammary epithelium. Dev. Biol. 306, 193–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl P, and Gilmour D (2009). Collective cell migration in morphogenesis, regeneration and cancer. Nat. Rev. Mol. Cell Biol. 10, 445–457. [DOI] [PubMed] [Google Scholar]
- Friedl P, and Mayor R (2017). Tuning collective cell migration by cell-cell junction regulation. Cold Spring Harb. Perspect. Biol. 9, a029199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeger A, Wolf K, Zegers MM, and Friedl P (2015). Collective cell migration: guidance principles and hierarchies. Trends Cell Biol. 25, 556–566. [DOI] [PubMed] [Google Scholar]
- Halaoui R, Rejon C, Chatterjee SJ, Szymborski J, Meterissian S, Muller WJ, Omeroglu A, and McCaffrey L (2017). Progressive polarity loss and luminal collapse disrupt tissue organization in carcinoma. Genes Dev. 31, 1573–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard BA, and Lu P (2014). Stromal regulation of embryonic and postnatal mammary epithelial development and differentiation. Semin. Cell Dev. Biol. 25–26, 43–51. [DOI] [PubMed] [Google Scholar]
- Howard BA, and Veltmaat JM (2013). Embryonic mammary gland development; a domain of fundamental research with high relevance for breast cancer research: preface. J. Mammary Gland Biol. Neoplasia 18, 89–91. [DOI] [PubMed] [Google Scholar]
- Huebner RJ, Lechler T, and Ewald AJ (2014). Developmental stratification of the mammary epithelium occurs through symmetry-breaking vertical divisions of apically positioned luminal cells. Development 141, 1085–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huebner RJ, Neumann NM, and Ewald AJ (2016). Mammary epithelial tubes elongate through MAPK-dependent coordination of cell migration. Development 143, 983–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inman JL, Robertson C, Mott JD, and Bissell MJ (2015). Mammary gland development: cell fate specification, stem cells and the microenvironment. Development 142, 1028–1042. [DOI] [PubMed] [Google Scholar]
- Khalil AA, and Friedl P (2010). Determinants of leader cells in collective cell migration. Integr. Biol. 2, 568–574. [DOI] [PubMed] [Google Scholar]
- Lu P, and Werb Z (2008). Patterning mechanisms of branched organs. Science 322, 1506–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P, Sternlicht MD, and Werb Z (2006). Comparative mechanisms of branching morphogenesis in diverse systems. J. Mammary Gland Biol. Neoplasia 11, 213–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P, Ewald AJ, Martin GR, and Werb Z (2008). Genetic mosaic analysis reveals FGF receptor 2 function in terminal end buds during mammary gland branching morphogenesis. Dev. Biol. 321, 77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarenkova HP, Hoffman MP, Beenken A, Eliseenkova AV, Meech R, Tsau C, Patel VN, Lang RA, and Mohammadi M (2009). Differential interactions of FGFs with heparan sulfate control gradient formation and branching morphogenesis. Sci. Signal. 2, ra55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayor R, and Etienne-Manneville S (2016). The front and rear of collective cell migration. Nat. Rev. Mol. Cell Biol. 17, 97–109. [DOI] [PubMed] [Google Scholar]
- Muzumdar MD, Tasic B, Miyamichi K, Li L, and Luo L (2007). A global double-fluorescent Cre reporter mouse. Genesis 45, 593–605. [DOI] [PubMed] [Google Scholar]
- Nguyen-Ngoc KV, Cheung KJ, Brenot A, Shamir ER, Gray RS, Hines WC, Yaswen P, Werb Z, and Ewald AJ (2012). ECM microenvironment regulates collective migration and local dissemination in normal and malignant mammary epithelium. Proc. Natl. Acad. Sci. USA 109, E2595–E2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrie RJ, Doyle AD, and Yamada KM (2009). Random versus directionally persistent cell migration. Nat. Rev. Mol. Cell Biol. 10, 538–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpa E, and Mayor R (2016). Collective cell migration in development. J. Cell Biol. 212, 143–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheele CL, Hannezo E, Muraro MJ, Zomer A, Langedijk NS, van Oudenaarden A, Simons BD, and van Rheenen J (2017). Identity and dynamics of mammary stem cells during branching morphogenesis. Nature 542, 313–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theveneau E, and Linker C (2017). Leaders in collective migration: are front cells really endowed with a particular set of skills? F1000Res. 6, 1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thotakura S, Basova L, and Makarenkova HP (2019). FGF Gradient Controls Boundary Position Between Proliferating and Differentiating Cells and Regulates Lacrimal Gland Growth Dynamics. Front. Genet. 10, 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zegers MM, and Friedl P (2014). Rho GTPases in collective cell migration. Small GTPases 5, e28997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Martinez D, Koledova Z, Qiao G, Streuli CH, and Lu P (2014). FGF ligands of the postnatal mammary stroma regulate distinct aspects of epithelial morphogenesis. Development 141, 3352–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate datasets. All raw data is available on request.