

Abstract
Ribonucleic acids (RNAs) play essential roles in living cells. Many of them fold into defined three-dimensional (3D) structures to perform functions. Recent advances in single-particle cryo-electron microscopy (cryo-EM) have enabled structure determinations of RNA to atomic resolutions. However, most RNA molecules are structurally flexible, limiting the resolution of their structures solved by cryo-EM. In modeling these molecules, several computational methods are limited by the requirement of massive computational resources and/or the low efficiency in exploring large-scale structural variations. Here we use hierarchical natural move Monte Carlo (HNMMC), which takes advantage of collective motions for groups of nucleic acid residues, to refine RNA structures into their cryo-EM maps, preserving atomic details in the models. After validating the method on a simulated density map of tRNA, we applied it to objectively obtain the model of the folding intermediate for the specificity domain of ribonuclease P from Bacillus subtilis and refine a flexible ribosomal RNA (rRNA) expansion segment from the Mycobacterium tuberculosis (Mtb) ribosome in different conformational states. Finally, we used HNMMC to model atomic details and flexibility for two distinct conformations of the complete genomic RNA (gRNA) inside MS2, a single-stranded RNA virus, revealing multiple pathways for its capsid assembly.
Keywords: RNA folding, molecular motions, conformational sampling, structural modeling, RNA virus
INTRODUCTION
Ribonucleic acids (RNAs) are important biological macromolecules that perform various functions inside the cell. In addition to acting as a carrier of genetic information (Crick 1970), RNAs play active roles in cell survival and development, such as regulation of gene expression (Garst et al. 2011; Ha and Kim 2014) and catalysis of protein synthesis (Cech et al. 1981; Guerrier-Takada et al. 1983; Schluenzen et al. 2000). Due to these essential roles, the malfunction of RNA is implicated in many diseases, including neurological diseases (Esteller 2011; Fenoglio et al. 2013), cardiovascular diseases (Uchida and Dimmeler 2015), and cancers (Huarte 2015).
To perform their diverse functions, many RNAs need to precisely fold into their three-dimensional (3D) structures. The folding process of RNA is hierarchical (Woodson 2010). The primary RNA sequence first forms secondary structures, such as helices, bulges, junctions, and loops, through local nucleotide interactions. These secondary structures then arrange into a tertiary structure, via long-range RNA interactions. While the secondary structures of RNA form within a short period of time (10−5 to 10−4 sec), the tertiary structures of RNA are formed much slower (10−2 to 10−1 sec), through a process that involves complex interactions between RNA secondary structures, as well as the interactions between RNAs and neighboring proteins (Crothers et al. 1974; Onoa and Tinoco 2004). In addition, the secondary structures of RNA are generally more stable compared to the tertiary structures (Mathews et al. 1999). Taking these facts into account, it is intuitive to model RNA structures in a hierarchical way. First, the secondary structures in RNA can be obtained by computational predictions, through methods such as comparative sequence analysis (Cannone et al. 2002; Mathews et al. 2010; Seetin and Mathews 2012) and/or free-energy minimization (Eddy 2004; Mathews and Turner 2006). The accuracy of these predictions can be improved by incorporating the experimental mapping data of secondary structures, using chemical/enzymatic probing and RNA sequencing (Loughrey et al. 2014; Smola et al. 2015; Lu and Chang 2016). Then the tertiary structures of RNA can be sampled by spatially arranging these RNA secondary structures.
Recent advances in cryo-electron microscopy (cryo-EM) have enabled the structural characterization of RNA molecules at unprecedented resolutions. One major advantage of using electrons to image RNA is that the phosphate backbone of RNA strongly scatters electrons, giving a much higher contrast compared to proteins. Moreover, better imaging hardware (Li et al. 2013; Ruskin et al. 2013; McMullan et al. 2014) and more robust image processing software (Grigorieff 2007; Tang et al. 2007; Scheres 2012; Moriya et al. 2017; Punjani et al. 2017) have enabled obtaining 3D density maps of ribosomal RNA (rRNA) to atomic (or near-atomic) resolutions (Fischer et al. 2015; Shalev-Benami et al. 2016; Zhang et al. 2016; Liu et al. 2017). Many protein–RNA complexes, for example, spliceosomes and telomerases, have also been determined to near-atomic or subnanometer resolutions (Jiang et al. 2015; Nguyen et al. 2015; Yan et al. 2015). New imaging strategies in cryo-EM, such as utilizing the direct-electron counting and phase contrast (Danev and Baumeister 2016; Danev et al. 2017; Khoshouei et al. 2017), may facilitate the imaging of smaller RNA molecules (Baird et al. 2010b; Zhang et al. 2018). Furthermore, viral genomic RNAs (gRNAs), generally long and flexible in solution, can also be imaged and characterized by cryo-EM (Gopal et al. 2012; Liu and Cheng 2015; Zhang et al. 2015), with recent breakthroughs to reveal defined gRNA 3D folds inside the capsids of single-stranded RNA bacteriophages (Gorzelnik et al. 2016; Koning et al. 2016; Zhong et al. 2016; Cui et al. 2017; Dai et al. 2017; Meng et al. 2019). Importantly, cryo-EM holds another advantage to study multiple states of a macromolecule in solution, allowing the mapping of an energy landscape in the conformational space (Dashti et al. 2014). Therefore, cryo-EM is gaining popularity for imaging and generating 3D density maps of RNA at increasingly higher resolutions under different functional states.
Several computational methods have been developed to build atomic models of RNA into the cryo-EM maps. This process generally involves two steps: (i) building a starting model; and (ii) refining it into a density map. If the target RNA has a homologous sequence to a known RNA template structure, comparative modeling can be used to generate a starting model (Rother et al. 2011). In cases where a homologous template is unavailable but the target RNA is short (100–300 nt), it is possible to predict the starting model ab initio (Das et al. 2010; Cheng et al. 2015). Otherwise, one can manually build fragments of the starting model then assemble them into the cryo-EM density before further refinements (Jossinet et al. 2010).
After building the starting model, refining the atomic coordinates of RNA into the cryo-EM map requires fitting as many atoms as possible into the density while keeping the proper stereochemistry of the model. Algorithms based on molecular dynamics calculations or simulated annealing optimizations are usually used in the model refinement (McGreevy et al. 2016; Liebschner et al. 2019). The accuracy of the refined model can be further improved by correcting errors in the RNA backbone (Chou et al. 2013). Most of these refinement tools need extensive computational resources and rely on high-quality initial models. If the starting model has a significant structural variation compared to the target structure, it leads to two major problems: (i) inability to dock the starting model into the density with the correct orientation for further refinement; and (ii) prolonged time for the model refinement due to the inefficiency in sampling large conformational changes between the starting and target structure. Therefore, there is a need for a tool that can objectively and efficiently sample RNA conformations from a starting model to facilitate the modeling process.
Recently, we have developed a method called hierarchical natural move Monte Carlo (HNMMC), which improves the conformational sampling efficiency in macromolecular modeling. Using natural move Monte Carlo with customizable hierarchical degrees of freedom (DOFs), we showed that the method can generate diverse canonical ensembles of macromolecular conformations (Sim et al. 2012). Implemented in the MOSAICS software package, HNMMC has been successfully used in several applications (Minary and Levitt 2014; Behrmann et al. 2015; Knapp et al. 2016; Krawczyk et al. 2016). The combination of MOSAICS and EMAN2, a cryo-EM data processing software package (Tang et al. 2007), resulted in a molecular refinement tool (named MOSAICS-EM), has been applied to the structure refinement of protein complexes from cryo-EM data (Zhang et al. 2012). Here we adapted MOSAICS-EM to refine RNA structures against cryo-EM maps. For example, a starting structural model of RNA can be obtained by comparative modeling, ab initio prediction, or manual building, and before the final refinement, HNMMC in MOSAICS-EM can sample the possible structural variations of this initial RNA model within the density map in a large scale, based on the customized secondary structural restraints, to maximize the coverage for a correct conformation.
In this paper, we first established and validated HNMMC refinement with a known structure of tRNA. We then used it to automatically model the structure of the folding intermediate of the specific domain of ribonuclease P from Bacillus subtilis. In addition, we refined a structural model of an rRNA expansion segment, also known as the handle, in the ribosome of Mycobacterium tuberculosis (Mtb), into its cryo-EM maps of 40 different states. We then applied this tool to build the atomic model of the gRNA fragment (2394–2604) from a single-stranded RNA virus, MS2, and finally expanded the same strategy to obtain two models of the complete gRNA with distinct conformations, which reveal multiple pathways for the viral assembly.
RESULTS
Optimize HNMMC refinement using the simulated data of tRNA
The challenges in refining macromolecular structure using Markov Chain Monte Carlo (MC) simulations are usually the large number of DOFs. To overcome this problem, we group residues from a macromolecule into different levels of segments, representing different DOFs in the molecule. The grouped residues in each segment, termed natural move, translate and rotate collectively and can fluctuate individually within the segment. Therefore, these natural moves are usually constructed from the rigid or semi-rigid natural organizations of a molecule, such as helices of an RNA. For example, the cloverleaf-like tRNA structure is modeled as four natural moves with each helix as one segment. The centers, allowing each helix to rotate and translate, were defined within the connecting residues (Fig. 1A). An energy function, which combines the molecular energy and the weighted EM energy, was used for the MC simulation with a modulated temperature profile (see Materials and Methods; Supplemental Fig. 1).
FIGURE 1.
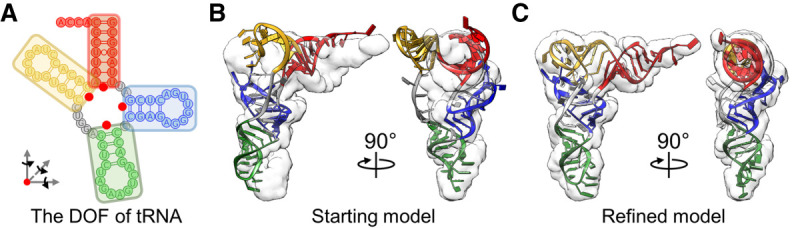
HNMMC refinement of a deformed tRNA. (A) The DOF of a tRNA. Each helical stem (red, blue, green, and yellow) of a tRNA is defined as a natural move (indicated in each box), which can translate and rotate independently, with each red dot as a center of rotation and translation. Two flexible unpaired loops are colored gray. (B) The starting tRNA model with 8.72-Å C4′ RMSD to the target structure fitted in the target map. (C) The refined model with the lowest total energy (Etotal) has a C4′ RMSD of 1.37 Å to the target structure. Each helical stem of the tRNA model is colored red, blue, green, and yellow, respectively, based on the DOF defined in A. The transparent density is Gaussian-blurred from the target structure to 5-Å resolution.
To evaluate and optimize the protocol of HNMMC refinement, different weights of the EM energy and different resolutions of the density map were used. Five target maps of tRNA, Gaussian-blurred to resolutions from 5 Å to 30 Å (Supplemental Table 1), were generated from the crystal structure (PDB 1EHZ) (Shi and Moore 2000) by EMAN2 (Tang et al. 2007). An initial model of tRNA was deformed from the crystal structure by MC simulation at a high temperature (300,000K), resulting in a C4′ root-mean-square deviation (RMSD) with 8.27 Å (Fig. 1B), which was subsequently refined into the density maps.
The best-refined result was from the EM weight of 200 and the map at a 5-Å resolution, which confers the smallest C4′ RMSD (1.37 Å) to the target structure (Fig. 1C; Supplemental Table 1; Supplemental Movie S1). The synchronous fluctuation of the total energy and C4′ RMSD with the annealing temperature indicates that the refinement is capable of escaping from local energy minima efficiently (Supplemental Fig. 1).
The refinement results of using different EM weights and map resolutions show that the higher resolution of the map yields a better-refined model. Increasing the EM weight can lead to a better-refined result when the map resolution is low. The refinement performs best when the EM weight is 200. Using this weight, all the simulated maps can guide the deformed model to the refined models with around 2-Å C4′ RMSD to the target structure. The incorporation of the molecular energy prevents nonphysical conformations. The EM weight of 200 was also applied in the following HNMMC simulations of different molecules.
Refine the specificity domain (S-domain) of Bacillus subtilis ribonuclease P into the experimental cryo-EM map
The S-domain of the Bacillus subtilis ribonuclease P is a 154-nt RNA that can bind pre-tRNA directly (Qin et al. 2001). The folding behavior of the S-domain in solution has been previously characterized (Baird et al. 2005, 2007, 2010a). The conformation of a folding intermediate of the S-domain at high Mg2+ concentration was captured using single-particle cryo-EM (Baird et al. 2010b). Compared to the native state (PDB 1NBS), this folding intermediate only forms secondary structures and short-range tertiary interactions, implying that the long-range tertiary interactions are formed in sequential steps during RNA folding (Baird et al. 2010b). In this example, we used HNMMC to automatically refine the native structure of the S-domain into its cryo-EM map of a folding intermediate at 15-Å resolution (EMD-5242).
In the native state, the core of the S-domain has a T-loop motif (three-way junction). Helix P10.1 connects with two coaxially stacked short helices P10 and P11 (Supplemental Fig. 2A; Krasilnikov et al. 2003). P10 forms a four-way junction with helices P7, P8, P9. P11 connects with P12 through two large internal loops, J11/12. Because it is a three-way junction, we can first define the DOFs of S-domain as three natural moves: (i) P7/P8/P9, (ii) P10.1; and (iii) P11/J11/12/P12 (blue, green, and red, respectively, in DOF 1, Fig. 2A). P10 and the loops at the junction are defined as flexible regions (gray in DOF 1, Fig. 2A; Supplemental Fig. 2C). For finer adjustment of the model inside the density, we also set the second DOF defining each helix as one natural move and the large internal loops J11/12 as another natural move (DOF 2, Fig. 2A; Supplemental Fig. 2C).
FIGURE 2.

HNMMC refinement of the native structure of the ribonuclease P S-domain against the cryo-EM map of the folding intermediate. (A) The flow chart of the refinement protocol of the S-domain. DOF 1 is applied in the first step to generate 10,000 conformations without the restraint from the cryo-EM density. DOF 2 with two levels is applied in the last step for a detailed refinement. (B) The conformational difference between the 500 representative conformations from clustering the 10,000 conformations. The scatter plot shows the differences among the 500 models by multidimensional scaling computed using the matrix of the pairwise C4′ RMSD among the 500 conformations. Each point represents one conformation. The 18 unique conformations with high cross-correlation to the target map are marked as solid purple or red dots, with the red dot representing the conformation with the highest cross-correlation. (C) The heat map of the normalized cross-correlation of the 500 representative conformations to the target map. The 18 unique conformations with high cross-correlations are marked as stars. The red star at the top-left corner indicates the conformation with the highest cross-correlation.
To avoid bias from users, we customized the protocol, which automatically refines the native structure of the S-domain against the cryo-EM map of its folding intermediate (Fig. 2A). To accomplish that, one needs to first register the model and the map in roughly matched orientations. However, due to the large conformational difference between the initial model (native) and the cryo-EM map (folding intermediate), with the best possible fitting cross-correlation of only 0.43, such a direct registration is not reliable. Therefore, we used the following strategy: (i) apply a large-scale MC sampling (10,000 decoys) to broadly search the probable conformations without the EM energy; (ii) cluster all 10,000 decoys into 500 models; (iii) dock the 500 clustered models to the map by Situs (Wriggers 2012); (iv) pick the representative models with a unique conformation and a high docking score; and (v) refine these representative models into the map locally using HNMMC in MOSAICS-EM.
To aid the selection of representative models, multidimensional scaling analysis by the R package (R Core Team 2015) was applied to visualize the conformational differences of the 500 clustered models (Fig. 2B). In addition, the docking scores (or normalized cross-correlation between the model and the cryo-EM map) of the 500 models were used to generate a heat map (Fig. 2C) on top of the plot of the multidimensional scaling. In total, 18 representative models (solid circles and stars in Fig. 2B,C, respectively; Supplemental Table 2) were selected and refined into the cryo-EM map with the finer DOF, DOF 2 (Supplemental Movie S2). The refined model with the highest cross-correlation (0.88) to the map was chosen as the best-refined model, which is in agreement to the model manually built and validated previously (Baird et al. 2010b). The two long helical arms (P11/J11/12/P12 and P10.1) form a collinear arrangement in both our model and the previous model and the orientations of the short arm (P7/P8/P9) are also the same.
Model the large-scale conformational flexibility of the “handle” in Mycobacterium tuberculosis (Mtb) ribosomes
We then expanded our application to refine the flexible region of rRNA inside the Mtb ribosome. Different from other bacterial ribosomes, the 23S rRNA in the large subunit (50S) of the Mtb ribosome has a 100 nt-long helical expansion segment H54a, termed “handle” (Yang et al. 2017). The handle undergoes a large-scale conformational change when the small subunit (30S) associates with the 50S to form the Mtb ribosome (70S). In the 70S, the handle serves as an inter-subunit bridge of the 50S and the 30S and swings between the L1 stalk of the 50S and the mRNA exit site. Therefore, the handle may modulate the translation in the Mtb and affect the association of the 50S and 30S subunits (Yang et al. 2017).
To more quantitatively measure the flexibility of the handle in the Mtb 70S, HNMMC was applied to refine its model from the bent state in the 50S into the extended states in the 70S, captured by 40 classified cryo-EM maps (Yang et al. 2017). The initial model of the handle with its connected three-way junction (rRNA fragment 1540–1659 and 1814–1828) was cut from the high-resolution structure of Mtb 50S (PDB 5V7Q) (Yang et al. 2017). The target maps with 40 unique conformations of the handle were segmented from the 40 classified maps of the Mtb 70S (downloaded as a bundle through EMD-8648) (Supplemental Fig. 3A; Yang et al. 2017). The handle consists of several short helices and motifs, such as the sarcin-ricin motif and the tetra-loop (magenta, Fig. 3). Seven levels of natural moves were defined to describe its bending and extending motions (Fig. 3A).
FIGURE 3.

HNMMC refinement of the handle in Mtb 50S rRNA. (A) Seven levels of the DOF to describe possible motions of the handle. For each level, the red fragment can move as a group while the black fragment is fixed. The magenta labels the sarcin-ricin motif (close to the three-way junction) and the tetra-loop (at the tip of the handle). Combining these seven levels can effectively describe the bending of the handle at each junction. Note that each nucleotide is still allowed to fluctuate around its position within the red fragment for subtle conformational variations. (B) The initial model of the handle obtained from the Mtb 50S is docked against the cryo-EM density of the handle from the Mtb 70S. (C) The HNMMC refined model of the handle.
HNMMC refinements against the 40 maps improved the cross-correlations between the models of the handle and their corresponding maps from 0.2 ∼ 0.4 to 0.6 ∼ 0.7 (Supplemental Table 3). For the segment between the three-way junction and the sarcin-ricin motif, limited movement is observed when the entire handle extends from the conformation in the 50S to the one in the 70S. However, for the segments from the sarcin-ricin motif to the tip of the handle, it swings around 35° when forming the 70S from the 50S (Fig. 3B,C; Supplemental Movie S3). Moreover, the handle is also flexible in the 70S. Comparing these 40 refined models, the tip of the handle can travel around 60 Å in the short axis and 70 Å in the long axis (Supplemental Fig. 3). While the solvent exclusion volume and solvent-accessible area of a single conformation of the handle is around 71.9 nm3 and 156.1 nm2, respectively, the 40 conformations of the handle lead to a much larger solvent exclusion volume and solvent-accessible area of 265.4 nm3 and 301.6 nm2, respectively. Such increased solvent accessibility may help the handle efficiently interact with potential regulatory subunits or nonribosomal factors (Polikanov et al. 2012; Li et al. 2018; Mishra et al. 2018). Such a hypothesis is yet to be tested.
Building atomic models of viral gRNA in the virion
Single-stranded (ss) RNA viruses use RNA as their genetic material and infect all domains of life, including animals, plants, and bacteria. Recently, it has been revealed by cryo-EM that the ssRNA bacteriophages, such as Qβ and MS2, have a major 3D conformation of the genomic RNA (gRNA) among all viral particles (Gorzelnik et al. 2016; Koning et al. 2016; Zhong et al. 2016; Cui et al. 2017; Dai et al. 2017). The gRNA presents stem–loops, termed the “packaging signals” (Dykeman et al. 2013; Rolfsson et al. 2016), which form stable interactions with the phage capsid proteins, potentially providing a roadmap for capsid assembly.
In a previous cryo-EM study of MS2 (Dai et al. 2017), a few gRNA stem–loops or packaging signals directly interacting with the viral capsid were solved to high resolutions. However, much of the gRNA densities were only resolved to intermediate resolutions or missing, due to the internal flexibility of the RNA within the capsid. Based on the cryo-EM map and the secondary structure prediction, approximately 80% of the gRNA backbone was manually traced, presenting long RNA helices connected by complex junctions. Although the backbone model shows the organization of the gRNA, the lack of atomic details in many regions prevents the understanding of the accurate RNA/RNA or RNA/protein interactions that can affect the gRNA packing and releasing. Such an atomic model of the gRNA was difficult to obtain due to the lower resolution and missing density throughout the cryo-EM map. Recently, a method has been developed in Rosetta to build atomic models of RNA by assembling fragments of the RNA model into cryo-EM densities (Kappel et al. 2018) and applied to build 10 gRNA fragments inside the MS2 capsid. For each region, this modeling procedure (known as De novo Ribonucleoprotein modeling in Real-space through Assembly of Fragments Together with Electron density in Rosetta or DRRAFTER) started from at least one ideal RNA helix as an anchor point and was used to build the models, which connected the remaining helices with the ideal RNA helices, into their corresponding densities at intermediate resolutions.
Here, we first focused on the gRNA fragment 2394–2604, which was not modeled by the DRRAFTER (Kappel et al. 2018). The density of this region shows a four-way junction motif. The initial model of the gRNA fragment 2394–2604 was generated using RNA De novo in Rosetta (Das et al. 2010; Cheng et al. 2015) based on its secondary structure (Fig. 4A). One stem–loop (corresponding to the gRNA fragment 2468–2481) has high affinity with the viral capsid, therefore, was resolved to high resolution previously (PDB 5TC1) (Dai et al. 2017). The rest of the gRNA in this fragment does not directly interact with the capsid and is flexible, thus was resolved at a lower local resolution. Therefore, in our modeling, the high-resolution stem–loop (gRNA fragment 2468–2481) was used as a single fixed anchor while the remaining gRNAs were automatically fitted into the density through two runs of the refinement by four levels of natural moves (Fig. 4A), keeping the correct geometry of the stem–loops.
FIGURE 4.
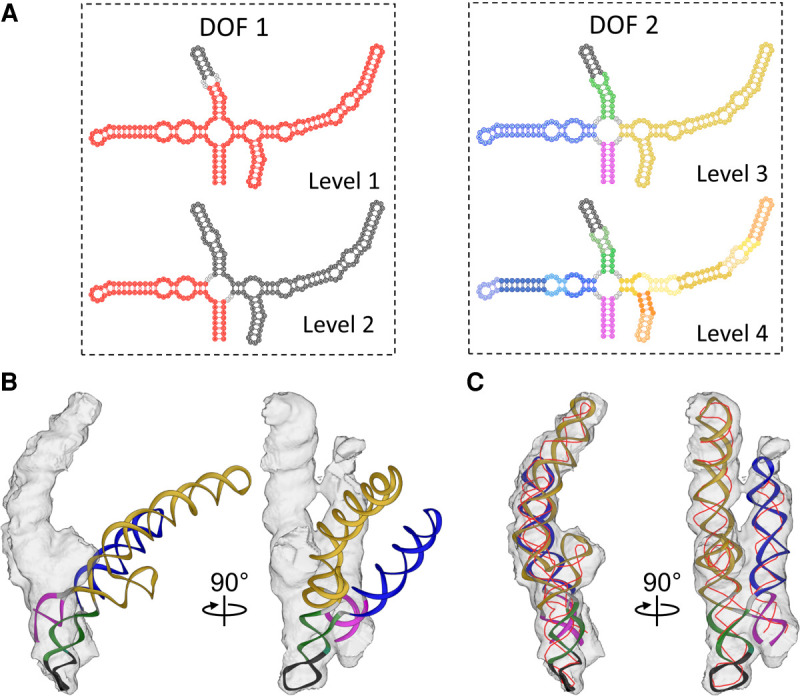
HNMMC refinement of the gRNA fragment 2394–2604 inside MS2. (A) The definition of DOFs of the gRNA fragment 2394–2604. The red fragment in Levels 1 and 2 of DOF 1 is the natural move that moves as a group. For Levels 3 and 4 of DOF 2, the different colors represent different natural moves. The black region at all levels is fixed. In Levels 3 and 4, the black region is the gRNA fragment 2468–2481 with a high-resolution EM structure. The gray color represents unpaired RNA loops that can also move. (B) The initial model of the gRNA fragment 2394–2604 and the corresponding cryo-EM density. The helical stems are colored as Level 3. (C) HNMMC refined model of the gRNA fragment 2394–2604 against the target cryo-EM density. The thin red line is the backbone trace from the previous published result (Dai et al. 2017).
HNMMC refinement increased the cross-correlation of the model and the density from 0.18 to 0.73 (Fig. 4B,C; Supplemental Movie S4). The refined model is mostly consistent with the manually traced backbone model (thin red line, Fig. 4C; Dai et al. 2017), which forms a parallel coaxial stacking (cross) of four helical stems. However, compared to the backbone model, our second helical stem (blue in Level 3, Fig. 4A) is 15-Å longer (around eight nucleotides) at the tip. In the map of the entire gRNA, the density of this helix connects with another density, suggesting its interaction with another helical stem. As expected, in the connecting area, an additional density can be found for these eight nucleotides.
Using the same strategy above, in total, we built 17 fragments of the MS2 gRNA, which were connected to establish an atomic model of the entire gRNA with all the 3569 nt. The model was then minimized using Phenix (Liebschner et al. 2019) and further refined against the cryo-EM map by 20 independent runs of HNMMC refinement with each RNA helix defined as one natural move (Fig. 5A). The 20 refined models have improved fitting to the map and maintained secondary structures in the flexible fragments with weak cryo-EM densities (Supplemental Movie S5). The average C4′ RMSD among the 20 refined models is 2.89 ± 0.24 Å and the average cross-correlation between the map and the model is 0.523 ± 0.043. As expected, comparing these 20 models shows larger variations among the refined models in regions with weaker cryo-EM densities (Fig. 5B).
FIGURE 5.

HNMMC refined models of the complete MS2 gRNA. (A) The ensemble of 20 refined models colored according to the genes in the schematic diagram at the bottom (MP: maturation protein, CP: coat protein, Rep: replicase, Lys: lysis protein). The lysis gene is not colored on the ribbon models due to the overlap with the genes for the coat protein and the replicase. (B) The conformational variation among the 20 refined models at each nucleotide colored by the average C4′ RMSD among the 20 models. The color scale from white to red denotes low to high RMSD.
To further understand the structural variations in the MS2 gRNA, a new cryo-EM data set of MS2 virions are collected and processed. Focused classifications (Bai et al. 2015) around the region with weak RNA density identifies two dominant conformations (named C1 and C2) of the gRNA, existing in 45.2% and 54.8% of the total number of virions, respectively. After the 3D refinement, the final resolutions of the two cryo-EM maps are both 7.4 Å (Supplemental Fig. 4). The atomic models of the gRNA of C1 and C2 are built and refined using HNMMC in MOSAICS-EM (Fig. 6; Supplemental Fig. 5). The two models show differences in the flexible RNA fragments, especially of the gRNA fragment 614–879, 1643–1700, 1992–2125, and 2922–3087. The C4′ RMSD of these four flexible fragments between two models are 32.58, 19.26, 24.23, and 37.06 Å, respectively, while the C4′ RMSD of the rest of the gRNAs is only 4.71 Å. In C1, two coat-protein dimers, D1 and D2 (transparent green and blue densities, Fig. 6) interact with the stem–loops of the gRNA fragment 1643–1700 (green ribbon, Fig. 6) and fragment 1992–2125 (blue ribbon, Fig. 6), respectively. However, in C2, D1 switches its interaction to a stem–loop from the gRNA fragment 2922–3087 (red ribbon, Fig. 6) and D2 interacts with the stem–loop of gRNA fragment 1643–1700 (green ribbon, Fig. 6). Moreover, in C2, due to the switch of gRNA fragment 2922–3087 (red ribbon, Fig. 6) to interact with D2, the interior space of the virion, which was originally occupied by this fragment in C1 is freed to accommodate another gRNA fragment 614–879 (yellow ribbon, Fig. 6). Such a rearrangement of the gRNA inside the capsid leads to a denser RNA packaging toward the maturation protein in C2 as compared to C1. Direct morphing between these two gRNA conformations shows collisions among themselves (Supplemental Movie S6), indicating that the gRNAs are folded into such conformations during its condensation before the capsid is formed. This result is a piece of direct evidence that the gRNA of an RNA virus can be packed in multiple conformations through different RNA folding pathways.
FIGURE 6.
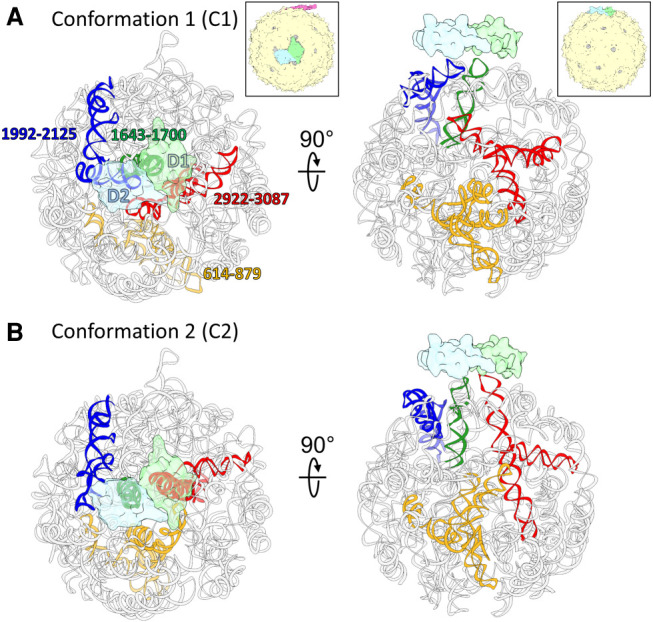
Two conformations of the gRNA inside MS2. (A,B) Two dominant gRNA conformations of the MS2 (C1,C2) are modeled based on the two cryo-EM maps. The gRNA fragments with large differences between the two models are colored yellow (gRNA fragment 614–879), green (gRNA fragment 1643–1700), blue (gRNA fragment 1992–2125), and red (gRNA fragment 2922–3087). Two copies of the coat protein dimers, which switch interacting stem–loop partners, are outlined by transparent green (Dimer 1: D1) and transparent blue (Dimer 2: D2). The insets of the capsids (magenta: maturation protein, green: D1, blue: D2, yellow: rest of the coat protein shell) show the viewing orientation for the corresponding columns and the location of the two dimers on the capsid.
DISCUSSION
In this study, we developed the molecular refinement tool, HNMMC, which combines MC simulation and customized hierarchical natural moves to efficiently refine RNA molecules into cryo-EM maps. Our energy term combines both EM energy and a molecular energy to improve the fitting to the cryo-EM map while maintaining the proper stereochemistry of the model. With the simulated density map, HNMMC successfully refined a deformed tRNA model to the correct conformation. For RNA molecules with a large conformational change, HNMMC can perform large-scale conformational sampling and then locally refine the selected candidates to the target map. Following this strategy, the structure of the folding intermediate of the ribonuclease P S-domain is automatically built from the structure of the native state. Moreover, HNMMC can be easily applied to model large RNA systems, such as ribosomes or gRNA of a virus. The customized hierarchical natural moves preserve the relatively rigid regions and allow more efficient traversing through the conformational-searching space to focus on major movements inside large molecules. For instance, two different conformations of the entire gRNA inside MS2 are built through fragments and refined into their cryo-EM maps using HNMMC.
The first step of running HNMMC is to define natural moves, which represent the potential motions of a molecule. For RNA, the natural moves can be defined using the information of the secondary structures or tertiary interactions (base pairs, motifs, etc.) from the existing high-resolution structure or computational predictions. However, in some RNA, mispredictions of the secondary structures could occur (Lorenz et al. 2011), leading to inaccuracy in the defined DOFs. In HNMMC refinement, such wrongly defined DOFs can easily cause unrealistic geometries in the RNA tertiary structures and/or low cross-correlation between the density map and the refined model. Therefore, a bad refined model is a good indicator of potential errors in the secondary structures. On the other hand, the collective motion of residues within one natural move restrains these residues around the initial geometry. In that case, when an EM density is noisy, incomplete or discontinuous, a restraint will not only keep the correct geometry of the grouped residues but also help anchor the entire region into the density without being biased by the artifacts in the density. For a map at a low resolution, the HNMMC refinement may provide multiple final models with similar conformations. To select the best-refined model, the cross-correlation score between the model and the map is helpful, but a final visual inspection is still important to identify if the selected model has wrong placements of helices or minor conformational errors that are inconsistent with experimental evidence if available.
Finally, combining the MC optimization and the hierarchical natural moves reduces the computational time required for modeling. Compared to molecular dynamics based flexible fitting (MDFF) (McGreevy et al. 2016), for example, HNMMC requires only 0.5 h of CPU time as compared to 1.5 h in MDFF to refine the model of the Mtb ribosomal handle into its EM map (Supplemental Fig. 6). Admittedly, HNMMC requires more memory (400 MB vs. 50 MB) and the speed of MDFF can be accelerated by GPU, which has not been utilized in HNMMC.
MATERIALS AND METHODS
Multiscale natural move representation of macromolecules
The goal of the HNMMC refinement is to change the conformation of the starting model to maximize its fit against the target EM density. To exhaustively search the possible conformations of an RNA, the set of natural moves for the MC simulation can be constructed as the following: (i) group nucleic acid residues into several segments of collective motions (natural moves) hierarchically based on their rigid or semi-rigid natural organizations, such as base-paired helical strands and specific motifs; (ii) define the rotational and translational DOF of each segment; (iii) maintain the continuity between each segment by the recursive stochastic chain-closure algorithm (Minary and Levitt 2010), ensuring the proper geometry of the connections and sufficient freedom in the flexible regions; and (iv) move base pairs and individual nucleotides in each segment. Moreover, natural moves can be combined into different levels to more efficiently represent global (fewer DOFs) and local movements (more DOFs) of an RNA, respectively. A common strategy is to use fewer DOFs at the beginning of the refinement to capture larger-scale movements of RNA domains and then switch to more DOFs at the later stage of the refinement to model subtle fluctuations within an RNA.
Temperature-modulated MC simulation
For the energy function of the MC simulation, besides the EM energy (EEM) defined by the negative cross-correlation between the target EM density and the density converted from the model, here, we also introduce a molecular energy (Emol) to prevent collisions within the model and improbable conformations. Emol can be calculated by an empirical physics-based force field, Amber99-bs0 (parmbsc0) (Perez et al. 2007). Therefore, during each step of the MC simulation, the generated conformation is evaluated by a total energy function (Etotal), which combines EEM and Emol. The total energy function is derived as Etotal = Emol + ωEEM. A weight (ω) at the EM energy is used to control the contribution arising from the experimental EM data. All the HNMMC refinements were performed with the simulated annealing optimization. A modulated temperature profile was applied to facilitate the exploration of different conformations and help the structure efficiently escape from local energy minima (Supplemental Fig. 1). The protocol is implemented in the software package, MOSAICS-EM (Zhang et al. 2012).
Cryo-EM data collection for MS2
The RNA virus, MS2 particles were imaged under an FEI Tecnai F20 cryo-electron microscope with a field emission gun (Thermo Fisher Scientific) operated at 200 kV. Data was collected on a K2 Summit direct detection camera (Gatan) in the super-resolution mode. A nominal magnification of ×29,000 was used, yielding a sub-pixel size of 0.625 Å. A 33-frame movie stack was recorded for each exposure, with a dose rate of 8 e−/pixel/s on the camera and 0.2 sec per frame for a total exposure time of 6.6 sec.
Cryo-EM data processing for MS2
The collected super-resolution movie stacks were processed as previously described (Meng et al. 2019). First, the movie stacks were binned by 2, then aligned and summed using Unblur (Grant and Grigorieff 2015) with electron dose filtering. The defocus value of each summed micrograph was estimated using CTFFIND4 (Mindell and Grigorieff 2003). The particles were picked semi-automatically by e2boxer.py in EMAN2 (Tang et al. 2007). The particles were then downscaled by a factor of 2, yielding a pixel size of 2.5 Å, and refined by RELION (Scheres 2012). To analyze the structural difference of the gRNA inside the capsid, focused classification in RELION was used with a soft-edged mask around the flexible region of the gRNA. The orientation parameters for the 3D classification were directly taken from the consensus refinement. Four classes were requested in the 3D classification and yielded four maps representing 22.92%, 31.69%, 23.11%, and 22.28% of the total particles, respectively. Visual inspection grouped Classes 1, 4 together (45.2%) and Classes 2, 3 together (54.8%), due to the similar gRNA conformations, respectively, leading to two dominant gRNA conformations in MS2 virions. After refinement, we obtained two cryo-EM maps of MS2 with different gRNA conformations, both at 7.4-Å resolution.
DATA DEPOSITION
The software and examples can be obtained from https://cryoem.tamu.edu/software/. The cryo-EM maps of MS2 are deposited into EM-Databank with accession ID: EMD-21659 (gRNA conformation 1) and EMD-21660 (gRNA conformation 2).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge the Texas A&M High-Performance Research Computing Center for providing the computational resources, and the Microscopy and Imaging Center for cryo-EM data collection. J.Z. is supported by start-up funding from the Department of Biochemistry and Biophysics and the Center for Phage Technology, jointly sponsored by Texas AgriLife and Texas A&M University. J.Z. also acknowledges the National Institutes of Health (NIH) grants R21AI137696, P01AI095208, the National Science Foundation (NSF) grant MCB-1902392, the Welch Foundation grant #1863, and support by the Texas A&M Triads for Transformation grant, the TAMU X-Grant Program, the TAMU School of Science Strategic Transformative Research Program, and the CST*R Foundation. We thank Karl Gorzelnik for comments and editing of the paper.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.071100.119.
REFERENCES
- Bai XC, Rajendra E, Yang G, Shi Y, Scheres SH. 2015. Sampling the conformational space of the catalytic subunit of human γ-secretase. Elife 4: e11182 10.7554/eLife.11182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird NJ, Westhof E, Qin H, Pan T, Sosnick TR. 2005. Structure of a folding intermediate reveals the interplay between core and peripheral elements in RNA folding. J Mol Biol 352: 712–722. 10.1016/j.jmb.2005.07.010 [DOI] [PubMed] [Google Scholar]
- Baird NJ, Fang XW, Srividya N, Pan T, Sosnick TR. 2007. Folding of a universal ribozyme: the ribonuclease P RNA. Q Rev Biophys 40: 113–161. 10.1017/S0033583507004623 [DOI] [PubMed] [Google Scholar]
- Baird NJ, Gong H, Zaheer SS, Freed KF, Pan T, Sosnick TR. 2010a. Extended structures in RNA folding intermediates are due to nonnative interactions rather than electrostatic repulsion. J Mol Biol 397: 1298–1306. 10.1016/j.jmb.2010.02.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird NJ, Ludtke SJ, Khant H, Chiu W, Pan T, Sosnick TR. 2010b. Discrete structure of an RNA folding intermediate revealed by cryo-electron microscopy. J Am Chem Soc 132: 16352–16353. 10.1021/ja107492b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrmann E, Loerke J, Budkevich TV, Yamamoto K, Schmidt A, Penczek PA, Vos MR, Burger J, Mielke T, Scheerer P, et al. 2015. Structural snapshots of actively translating human ribosomes. Cell 161: 845–857. 10.1016/j.cell.2015.03.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannone JJ, Subramanian S, Schnare MN, Collett JR, D'Souza LM, Du Y, Feng B, Lin N, Madabusi LV, Muller KM, et al. 2002. The comparative RNA web (CRW) site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinformatics 3: 2 10.1186/1471-2105-3-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cech TR, Zaug AJ, Grabowski PJ. 1981. In vitro splicing of the ribosomal RNA precursor of Tetrahymena: involvement of a guanosine nucleotide in the excision of the intervening sequence. Cell 27: 487–496. 10.1016/0092-8674(81)90390-1 [DOI] [PubMed] [Google Scholar]
- Cheng CY, Chou FC, Das R. 2015. Modeling complex RNA tertiary folds with Rosetta. Methods Enzymol 553: 35–64. 10.1016/bs.mie.2014.10.051 [DOI] [PubMed] [Google Scholar]
- Chou FC, Sripakdeevong P, Dibrov SM, Hermann T, Das R. 2013. Correcting pervasive errors in RNA crystallography through enumerative structure prediction. Nat Methods 10: 74–76. 10.1038/nmeth.2262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick FHC. 1970. Central dogma of molecular biology. Nature 227: 561–563. 10.1038/227561a0 [DOI] [PubMed] [Google Scholar]
- Crothers DM, Cole PE, Hilbers CW, Shulman RG. 1974. The molecular mechanism of thermal unfolding of Escherichia coli formylmethionine transfer RNA. J Mol Biol 87: 63–88. 10.1016/0022-2836(74)90560-9 [DOI] [PubMed] [Google Scholar]
- Cui Z, Gorzelnik KV, Chang JY, Langlais C, Jakana J, Young R, Zhang J. 2017. Structures of Qβ virions, virus-like particles, and the Qβ-MurA complex reveal internal coat proteins and the mechanism of host lysis. Proc Natl Acad Sci 114: 11697–11702. 10.1073/pnas.1707102114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai X, Li Z, Lai M, Shu S, Du Y, Zhou ZH, Sun R. 2017. In situ structures of the genome and genome-delivery apparatus in a single-stranded RNA virus. Nature 541: 112–116. 10.1038/nature20589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danev R, Baumeister W. 2016. Cryo-EM single particle analysis with the Volta phase plate. Elife 5: e13046 10.7554/eLife.13046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danev R, Tegunov D, Baumeister W. 2017. Using the Volta phase plate with defocus for cryo-EM single particle analysis. Elife 6: e23006 10.7554/eLife.23006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R, Karanicolas J, Baker D. 2010. Atomic accuracy in predicting and designing noncanonical RNA structure. Nat Methods 7: 291–294. 10.1038/nmeth.1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dashti A, Schwander P, Langlois R, Fung R, Li W, Hosseinizadeh A, Liao HY, Pallesen J, Sharma G, Stupina VA, et al. 2014. Trajectories of the ribosome as a Brownian nanomachine. Proc Natl Acad Sci 111: 17492–17497. 10.1073/pnas.1419276111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dykeman EC, Stockley PG, Twarock R. 2013. Packaging signals in two single-stranded RNA viruses imply a conserved assembly mechanism and geometry of the packaged genome. J Mol Biol 425: 3235–3249. 10.1016/j.jmb.2013.06.005 [DOI] [PubMed] [Google Scholar]
- Eddy SR. 2004. How do RNA folding algorithms work? Nat Biotechnol 22: 1457–1458. 10.1038/nbt1104-1457 [DOI] [PubMed] [Google Scholar]
- Esteller M. 2011. Non-coding RNAs in human disease. Nat Rev Genet 12: 861–874. 10.1038/nrg3074 [DOI] [PubMed] [Google Scholar]
- Fenoglio C, Ridolfi E, Galimberti D, Scarpini E. 2013. An emerging role for long non-coding RNA dysregulation in neurological disorders. Int J Mol Sci 14: 20427–20442. 10.3390/ijms141020427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer N, Neumann P, Konevega AL, Bock LV, Ficner R, Rodnina MV, Stark H. 2015. Structure of the E. coli ribosome-EF-Tu complex at <3 Å resolution by Cs-corrected cryo-EM. Nature 520: 567–570. 10.1038/nature14275 [DOI] [PubMed] [Google Scholar]
- Garst AD, Edwards AL, Batey RT. 2011. Riboswitches: structures and mechanisms. Cold Spring Harb Perspect Biol 3: a003533 10.1101/cshperspect.a003533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopal A, Zhou ZH, Knobler CM, Gelbart WM. 2012. Visualizing large RNA molecules in solution. RNA 18: 284–299. 10.1261/rna.027557.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorzelnik KV, Cui Z, Reed CA, Jakana J, Young R, Zhang J. 2016. Asymmetric cryo-EM structure of the canonical Allolevivirus Qβ reveals a single maturation protein and the genomic ssRNA in situ. Proc Natl Acad Sci 113: 11519–11524. 10.1073/pnas.1609482113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant T, Grigorieff N. 2015. Measuring the optimal exposure for single particle cryo-EM using a 2.6 Å reconstruction of rotavirus VP6. Elife 4: e06980 10.7554/eLife.06980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigorieff N. 2007. FREALIGN: high-resolution refinement of single particle structures. J Struct Biol 157: 117–125. 10.1016/j.jsb.2006.05.004 [DOI] [PubMed] [Google Scholar]
- Guerrier-Takada C, Gardiner K, Marsh T, Pace N, Altman S. 1983. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell 35: 849–857. 10.1016/0092-8674(83)90117-4 [DOI] [PubMed] [Google Scholar]
- Ha M, Kim VN. 2014. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol 15: 509–524. 10.1038/nrm3838 [DOI] [PubMed] [Google Scholar]
- Huarte M. 2015. The emerging role of lncRNAs in cancer. Nat Med 21: 1253–1261. 10.1038/nm.3981 [DOI] [PubMed] [Google Scholar]
- Jiang J, Chan H, Cash DD, Miracco EJ, Ogorzalek Loo RR, Upton HE, Cascio D, O'Brien Johnson R, Collins K, Loo JA, et al. 2015. Structure of Tetrahymena telomerase reveals previously unknown subunits, functions, and interactions. Science 350: aab4070 10.1126/science.aab4070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jossinet F, Ludwig TE, Westhof E. 2010. Assemble: an interactive graphical tool to analyze and build RNA architectures at the 2D and 3D levels. Bioinformatics 26: 2057–2059. 10.1093/bioinformatics/btq321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappel K, Liu S, Larsen KP, Skiniotis G, Puglisi EV, Puglisi JD, Zhou ZH, Zhao R, Das R. 2018. De novo computational RNA modeling into cryo-EM maps of large ribonucleoprotein complexes. Nat Methods 15: 947–954. 10.1038/s41592-018-0172-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoshouei M, Radjainia M, Baumeister W, Danev R. 2017. Cryo-EM structure of haemoglobin at 3.2 Å determined with the Volta phase plate. Nat Commun 8: 16099 10.1038/ncomms16099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp B, Demharter S, Deane CM, Minary P. 2016. Exploring peptide/MHC detachment processes using hierarchical natural move Monte Carlo. Bioinformatics 32: 181–186. 10.1093/bioinformatics/btv502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koning RI, Gomez-Blanco J, Akopjana I, Vargas J, Kazaks A, Tars K, Carazo JM, Koster AJ. 2016. Asymmetric cryo-EM reconstruction of phage MS2 reveals genome structure in situ. Nat Commun 7: 12524 10.1038/ncomms12524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasilnikov AS, Yang X, Pan T, Mondragon A. 2003. Crystal structure of the specificity domain of ribonuclease P. Nature 421: 760–764. 10.1038/nature01386 [DOI] [PubMed] [Google Scholar]
- Krawczyk K, Sim AY, Knapp B, Deane CM, Minary P. 2016. Tertiary element interaction in HIV-1 TAR. J Chem Inf Model 56: 1746–1754. 10.1021/acs.jcim.6b00152 [DOI] [PubMed] [Google Scholar]
- Li X, Mooney P, Zheng S, Booth CR, Braunfeld MB, Gubbens S, Agard DA, Cheng Y. 2013. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat Methods 10: 584–590. 10.1038/nmeth.2472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Ge X, Zhang Y, Zheng L, Sanyal S, Gao N. 2018. Cryo-EM structure of Mycobacterium smegmatis ribosome reveals two unidentified ribosomal proteins close to the functional centers. Protein Cell 9: 384–388. 10.1007/s13238-017-0456-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebschner D, Afonine PV, Baker ML, Bunkoczi G, Chen VB, Croll TI, Hintze B, Hung LW, Jain S, McCoy AJ, et al. 2019. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr D Struct Biol 75: 861–877. 10.1107/S2059798319011471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Cheng L. 2015. Cryo-EM shows the polymerase structures and a nonspooled genome within a dsRNA virus. Science 349: 1347–1350. 10.1126/science.aaa4938 [DOI] [PubMed] [Google Scholar]
- Liu Z, Gutierrez-Vargas C, Wei J, Grassucci RA, Sun M, Espina N, Madison-Antenucci S, Tong L, Frank J. 2017. Determination of the ribosome structure to a resolution of 2.5 Å by single-particle cryo-EM. Protein Sci 26: 82–92. 10.1002/pro.3068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz R, Bernhart SH, Honer Zu Siederdissen C, Tafer H, Flamm C, Stadler PF, Hofacker IL. 2011. ViennaRNA Package 2.0. Algorithms Mol Biol 6: 26 10.1186/1748-7188-6-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loughrey D, Watters KE, Settle AH, Lucks JB. 2014. SHAPE-Seq 2.0: systematic optimization and extension of high-throughput chemical probing of RNA secondary structure with next generation sequencing. Nucleic Acids Res 42: e165 10.1093/nar/gku909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Chang HY. 2016. Decoding the RNA structurome. Curr Opin Struct Biol 36: 142–148. 10.1016/j.sbi.2016.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews DH, Turner DH. 2006. Prediction of RNA secondary structure by free energy minimization. Curr Opin Struct Biol 16: 270–278. 10.1016/j.sbi.2006.05.010 [DOI] [PubMed] [Google Scholar]
- Mathews DH, Sabina J, Zuker M, Turner DH. 1999. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J Mol Biol 288: 911–940. 10.1006/jmbi.1999.2700 [DOI] [PubMed] [Google Scholar]
- Mathews DH, Moss WN, Turner DH. 2010. Folding and finding RNA secondary structure. Cold Spring Harb Perspect Biol 2: a003665 10.1101/cshperspect.a003665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGreevy R, Teo I, Singharoy A, Schulten K. 2016. Advances in the molecular dynamics flexible fitting method for cryo-EM modeling. Methods 100: 50–60. 10.1016/j.ymeth.2016.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMullan G, Faruqi AR, Clare D, Henderson R. 2014. Comparison of optimal performance at 300keV of three direct electron detectors for use in low dose electron microscopy. Ultramicroscopy 147: 156–163. 10.1016/j.ultramic.2014.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng R, Jiang M, Cui Z, Chang JY, Yang K, Jakana J, Yu X, Wang Z, Hu B, Zhang J. 2019. Structural basis for the adsorption of a single-stranded RNA bacteriophage. Nat Commun 10: 3130 10.1038/s41467-019-11126-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minary P, Levitt M. 2010. Conformational optimization with natural degrees of freedom: a novel stochastic chain closure algorithm. J Comput Biol 17: 993–1010. 10.1089/cmb.2010.0016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minary P, Levitt M. 2014. Training-free atomistic prediction of nucleosome occupancy. Proc Natl Acad Sci 111: 6293–6298. 10.1073/pnas.1404475111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mindell JA, Grigorieff N. 2003. Accurate determination of local defocus and specimen tilt in electron microscopy. J Struct Biol 142: 334–347. 10.1016/S1047-8477(03)00069-8 [DOI] [PubMed] [Google Scholar]
- Mishra S, Ahmed T, Tyagi A, Shi J, Bhushan S. 2018. Structures of Mycobacterium smegmatis 70S ribosomes in complex with HPF, tmRNA, and P-tRNA. Sci Rep 8: 13587 10.1038/s41598-018-31850-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriya T, Saur M, Stabrin M, Merino F, Voicu H, Huang Z, Penczek PA, Raunser S, Gatsogiannis C. 2017. High-resolution single particle analysis from electron cryo-microscopy images using SPHIRE. J Vis Exp 55448 10.3791/55448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TH, Galej WP, Bai XC, Savva CG, Newman AJ, Scheres SH, Nagai K. 2015. The architecture of the spliceosomal U4/U6.U5 tri-snRNP. Nature 523: 47–52. 10.1038/nature14548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onoa B, Tinoco I. 2004. RNA folding and unfolding. Curr Opin Struct Biol 14: 374–379. 10.1016/j.sbi.2004.04.001 [DOI] [PubMed] [Google Scholar]
- Perez A, Marchan I, Svozil D, Sponer J, Cheatham TE III, Laughton CA, Orozco M. 2007. Refinement of the AMBER force field for nucleic acids: improving the description of α/γ conformers. Biophys J 92: 3817–3829. 10.1529/biophysj.106.097782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polikanov YS, Blaha GM, Steitz TA. 2012. How hibernation factors RMF, HPF, and YfiA turn off protein synthesis. Science 336: 915–918. 10.1126/science.1218538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punjani A, Rubinstein JL, Fleet DJ, Brubaker MA. 2017. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods 14: 290–296. 10.1038/nmeth.4169 [DOI] [PubMed] [Google Scholar]
- Qin H, Sosnick TR, Pan T. 2001. Modular construction of a tertiary RNA structure: the specificity domain of the Bacillus subtilis RNase P RNA. Biochemistry 40: 11202–11210. 10.1021/bi010076n [DOI] [PubMed] [Google Scholar]
- R Core Team. 2015. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- Rolfsson O, Middleton S, Manfield IW, White SJ, Fan B, Vaughan R, Ranson NA, Dykeman E, Twarock R, Ford J, et al. 2016. Direct evidence for packaging signal-mediated assembly of bacteriophage MS2. J Mol Biol 428: 431–448. 10.1016/j.jmb.2015.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rother M, Rother K, Puton T, Bujnicki JM. 2011. ModeRNA: a tool for comparative modeling of RNA 3D structure. Nucleic Acids Res 39: 4007–4022. 10.1093/nar/gkq1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruskin RS, Yu Z, Grigorieff N. 2013. Quantitative characterization of electron detectors for transmission electron microscopy. J Struct Biol 184: 385–393. 10.1016/j.jsb.2013.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH. 2012. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 180: 519–530. 10.1016/j.jsb.2012.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schluenzen F, Tocilj A, Zarivach R, Harms J, Gluehmann M, Janell D, Bashan A, Bartels H, Agmon I, Franceschi F, et al. 2000. Structure of functionally activated small ribosomal subunit at 3.3 angstroms resolution. Cell 102: 615–623. 10.1016/S0092-8674(00)00084-2 [DOI] [PubMed] [Google Scholar]
- Seetin MG, Mathews DH. 2012. RNA structure prediction: an overview of methods. Methods Mol Biol 905: 99–122. 10.1007/978-1-61779-949-5_8 [DOI] [PubMed] [Google Scholar]
- Shalev-Benami M, Zhang Y, Matzov D, Halfon Y, Zackay A, Rozenberg H, Zimmerman E, Bashan A, Jaffe CL, Yonath A, et al. 2016. 2.8-A cryo-EM structure of the large ribosomal subunit from the eukaryotic parasite leishmania. Cell Rep 16: 288–294. 10.1016/j.celrep.2016.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Moore PB. 2000. The crystal structure of yeast phenylalanine tRNA at 1.93 A resolution: a classic structure revisited. RNA 6: 1091–1105. 10.1017/S1355838200000364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim AY, Levitt M, Minary P. 2012. Modeling and design by hierarchical natural moves. Proc Natl Acad Sci 109: 2890–2895. 10.1073/pnas.1119918109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smola MJ, Rice GM, Busan S, Siegfried NA, Weeks KM. 2015. Selective 2′-hydroxyl acylation analyzed by primer extension and mutational profiling (SHAPE-MaP) for direct, versatile and accurate RNA structure analysis. Nat Protoc 10: 1643–1669. 10.1038/nprot.2015.103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, Ludtke SJ. 2007. EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol 157: 38–46. 10.1016/j.jsb.2006.05.009 [DOI] [PubMed] [Google Scholar]
- Uchida S, Dimmeler S. 2015. Long noncoding RNAs in cardiovascular diseases. Circ Res 116: 737–750. 10.1161/CIRCRESAHA.116.302521 [DOI] [PubMed] [Google Scholar]
- Woodson SA. 2010. Compact intermediates in RNA folding. Annu Rev Biophys 39: 61–77. 10.1146/annurev.biophys.093008.131334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wriggers W. 2012. Conventions and workflows for using Situs. Acta Crystallogr D Biol Crystallogr 68: 344–351. 10.1107/S0907444911049791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan C, Hang J, Wan R, Huang M, Wong CC, Shi Y. 2015. Structure of a yeast spliceosome at 3.6-angstrom resolution. Science 349: 1182–1191. 10.1126/science.aac7629 [DOI] [PubMed] [Google Scholar]
- Yang K, Chang JY, Cui Z, Li X, Meng R, Duan L, Thongchol J, Jakana J, Huwe CM, Sacchettini JC, et al. 2017. Structural insights into species-specific features of the ribosome from the human pathogen Mycobacterium tuberculosis. Nucleic Acids Res 45: 10884–10894. 10.1093/nar/gkx785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Minary P, Levitt M. 2012. Multiscale natural moves refine macromolecules using single-particle electron microscopy projection images. Proc Natl Acad Sci 109: 9845–9850. 10.1073/pnas.1205945109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Ding K, Yu X, Chang W, Sun J, Zhou ZH. 2015. In situ structures of the segmented genome and RNA polymerase complex inside a dsRNA virus. Nature 527: 531–534. 10.1038/nature15767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Lai M, Chang W, Yu I, Ding K, Mrazek J, Ng HL, Yang OO, Maslov DA, Zhou ZH. 2016. Structures and stabilization of kinetoplastid-specific split rRNAs revealed by comparing leishmanial and human ribosomes. Nat Commun 7: 13223 10.1038/ncomms13223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Keane SC, Su Z, Irobalieva RN, Chen M, Van V, Sciandra CA, Marchant J, Heng X, Schmid MF, et al. 2018. Structure of the 30 kDa HIV-1 RNA dimerization signal by a hybrid cryo-EM, NMR, and molecular dynamics approach. Structure 26: 490–498. 10.1016/j.str.2018.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Q, Carratala A, Nazarov S, Guerrero-Ferreira RC, Piccinini L, Bachmann V, Leiman PG, Kohn T. 2016. Genetic, structural, and phenotypic properties of MS2 coliphage with resistance to ClO2 disinfection. Environ Sci Technol 50: 13520–13528. 10.1021/acs.est.6b04170 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.