

Abstract
During zebrafish development, an early type of rRNA is gradually replaced by a late type that is substantially different in sequence. We applied RiboMeth-seq to rRNA from developmental stages for profiling of 2′-O-Me, to learn if changes in methylation pattern were a component of the shift. We compiled a catalog of 2′-O-Me sites and cognate box C/D guide RNAs comprising 98 high-confidence sites, including 10 sites that were not known from other vertebrates, one of which was specific to late-type rRNA. We identified a subset of sites that changed in methylation status during development and found that some of these could be explained by availability of their cognate SNORDs. Sites that changed during development were enriched in the novel sites revealed in zebrafish. We propose that the early type of rRNA is a specialized form and that its structure and ribose methylation pattern may be an adaptation to features of development, including translation of specific maternal mRNAs.
Keywords: box C/D snoRNA, zebrafish development, RiboMeth-seq, ribose methylation, ribosomal RNA
INTRODUCTION
Zebrafish (Danio rerio) is a well-established animal model for studying aspects of biology, in particular developmental biology. This is due to optical transparency of zebrafish embryos that facilitate easy and reliable identification of developmental stages, short generation time (∼3–4 mo), and year-round spawning (Dooley and Zon 2000; Segner 2009). Many transcriptomics and epitranscriptomics studies favor zebrafish as a model since genomic tools and whole-genome data are readily accessible. Zebrafish belongs to Teleostei infraclass, which evolved ∼340 million years ago from a common ancestor (Amores et al. 2011). In Teleostei, the genome has undergone two rounds of whole-genome duplication (Meyer and Schartl 1999; Wolfe 2000). Many human and zebrafish genes are orthologous to each other with ∼65% of zebrafish genes containing at least one human orthologue and ∼70% of human genes containing at least one zebrafish orthologue. Therefore, studies using zebrafish genetics are widely used to understand development, human diseases, and metabolism (Vilella et al. 2009; Howe et al. 2013).
Synthesis of cellular proteins is conducted by ribosomes that are ribonucleoprotein particles (RNPs). In higher eukaryotes, they are composed of ∼80 ribosomal proteins and four species of ribosomal RNA (rRNA). rRNA is the most abundant RNA in terms of mass and has a high nucleotide modification rate (∼2%) compared to mRNA. rRNA in eukaryotes is arranged into two subunits: a small subunit (SSU), which functions as the decoding center and facilitates the translocation of the tRNA/mRNA pair through the ribosome and a large subunit (LSU), which contains the peptidyltransferase center responsible for catalyzing peptide bond formation. During ribosome biogenesis, two pre-rRNAs are transcribed—one common to 18S (SSU), 5.8S, and 28S (LSU) rRNA, and one exclusively for 5S rRNA. Ribosome assembly is facilitated by ∼200 assembly factors. In addition, it involves a large number of small nucleolar RNPs in which small nucleolar RNAs (snoRNAs) are responsible for guiding enzymes to introduce nucleotide modifications, with pseudouridine (Ψ) and 2′-O-methyl (2′-O-Me) as the most abundant (Watkins and Bohnsack 2012). Ribose methylation is introduced by the generic methyltransferase, Fibrillarin, guided to the target RNA by box C/D snoRNAs (SNORDs) through base-pairing with the target. The methyl is introduced at the nucleotide base paired to the fifth nucleotide upstream of box D (“+5”) (Cavaille et al. 1996; Kiss-Laszlo et al. 1996). 2′-O-Me is believed to be important for ribosome biogenesis and translational fidelity. It provides a potential layer of regulation and thus supports the notion of specialized ribosomes (Gilbert 2011; Xue and Barna 2012; Shi and Barna 2015; Guo 2018; Ferretti and Karbstein 2019). The study of ribosome heterogeneity due to ribose methylation has become feasible by advancements in sequencing-based profiling methods (Krogh and Nielsen 2019) and was recently demonstrated in cancer cell lines (Krogh et al. 2016) and during mouse development (Hebras et al. 2020).
A recent study by Locati et al. (2017) revealed that zebrafish express two different sets of rRNA from clusters at two distinct genomic loci (Fig. 1A, left). The two types of rRNA were referred to as maternal- and somatic-type rRNAs, respectively, and they were transcribed, processed and modified (Fig. 1A, middle) at different levels throughout development (Fig. 1A, right). In the absence of direct evidence that the somatic type arises exclusively from somatic cell lineages (germ cell lineage has not been investigated) and the lack of confirmation that the maternal type is maternal-specific (the absence of de novo zygotic transcription has not been demonstrated), we have chosen to name these subtypes of rRNA in the context of developmental advancement: early- and late-rRNA, respectively. The primary sequences of the two subtypes are considerably different. Early-SSU rRNA is 1939 nt whereas the late form is 1889 nt, and the calculated similarity using BioEdit based on ClustalW alignment is 91.3%. Early-LSU rRNA is 4270 nt and late-LSU rRNA is 4106 nt, and they are 87.0% similar. These differences imply that the variable segments in rRNA display structural differences between the two subtypes. The separate chromosomal locations of the gene clusters, the structural differences, and their distinct expression patterns during development suggest that the two subtypes of rRNA have unique properties.
FIGURE 1.

Expression and modification by ribose methylation of early- and late-rRNA types of rRNA in zebrafish. (A) Schematic illustration of genomic localization, 2′-O-Me modification, and the prevalence of expressed early- and late-rRNA. The rRNA biogenesis symbolized by the arrow involves multiple steps and several types of modifications, but only ribose methylation, relevant for the present study, is illustrated. Here, an example of a canonical interaction between a box C/D guide RNA (SNORD) and its rRNA target is shown with indication of the methyl group introduced in red. (B) Bright field microscope images of the developmental stages analyzed in the present study: (Unf. Egg) unfertilized egg, the 32-cell stage, the 12-somite stage, and (PM) protruding-mouth stage. (C) Estimated relative expression (±SEM) of early- and late-SSU rRNA (left) and LSU rRNA (right) based on SNP analysis of reads from RiboMeth-seq. The result is based on the analysis of four diagnostic nucleotides in SSU and seven diagnostic nucleotides in LSU, respectively. n = 3. (D) Venn-diagrams depicting the conservation of ribose-methylated sites in SSU, LSU, and 5.8S between human rRNA and zebrafish early- and late-rRNA subtypes, respectively.
Here, we address the extent to which the differences between the two types of rRNA extend to their modification patterns, specifically with respect to 2′-O-Me sites. We used RiboMeth-seq (Birkedal et al. 2015), a sequencing-based method for mapping and estimation of modification stoichiometry, applied to zebrafish whole-cell RNA purified across selected developmental stages from unfertilized eggs to an adult sample. The method also provided low-coverage information on the expression of SNORDs that we supplemented by dedicated small RNA-seq of selected developmental stages. We identified 98 high-confidence 2′-O-Me sites, one of which was specific to late-rRNA. Several sites showed hypomethylation in early developmental stages in contrast to the adult stage, where most sites appeared close to fully methylated. Strikingly, sites that were specific to zebrafish compared to human were overrepresented among these hypomethylated sites. Structural and phylogenetic considerations suggest that early-rRNA is more divergent than late-rRNA, and it thus appears that rRNA sequence and structure, as well as ribose methylations are components that contribute to ribosome heterogeneity that may underlie specialization of ribosomes in eggs and early stages compared to adult zebrafish.
RESULTS
Zebrafish rRNA has 98 high-confidence 2′-O-Me sites, 10 of which are novel compared to human, including a late-rRNA specific site
To map the 2′-O-Me landscape in zebrafish early- and late-rRNA, RiboMeth-seq was applied to four developmental stages: unfertilized eggs, the 32-cell, 12-somite, and protruding-mouth, as well as tail from 1-yr old (adult) fish (Fig. 1B). Initially, the reads obtained from RiboMeth-seq were used for scoring the percentage of early- and late-rRNA in individual samples by mapping reads to the sequences of both types of rRNA in parallel and using a set of validated single-nucleotide differences to calculate the relative expression of the two. To minimize bias in the estimation, we chose four isolated (>30 nt distance to other variants) positions (T24, A55, C1633, and C1728) in SSU and seven positions (C1218, G1351, T1523, C2096, A2201, C3196, and C3780) in LSU (early-rRNA numbering). The single-nucleotide polymorphism (SNP) analyses of the two parallel mappings yielded complementary results at each selected position, that is, the sum of the matched and the sum of the expected mismatched reads were essentially similar. The analysis revealed similar results for SSU and LSU, with almost exclusive expression of early-rRNA in unfertilized egg and 32-cell stage, ∼20% of late-rRNA in the 12-somite stage, ∼75% in the protruding-mouth stage, and almost exclusive expression of late-rRNA in adult tail (Fig. 1C). These results are consistent with previous observations (Locati et al. 2017).
Next, we selected the reference sequences of either early- or late-rRNA based on the main subtype in each sample (Fig. 1C). RiboMeth-seq scores (RMS-scores) were calculated based on early-rRNA in unfertilized egg, 32-cell, and 12-somite stages, and on late-rRNA in the protruding-mouth stage and adult tail. Importantly, the majority (87/98) of 2′-O-Me sites was in regions of the rRNA conserved between early- and late-rRNA in the scoring interval of six nucleotides on either side of the queried position and mapping the reads to either of the sequences turned out not to change the calculated RMS-scores significantly.
We defined high-confidence 2′-O-Me sites as sites with RMS-score >0.75, additionally confirmed with presence of primer extension stop signal at limiting dNTP concentration and/or presence of a plausibly assigned SNORD. In total, 104 2′-O-Me sites had RMS-score >0.75. A subset of 24 sites including those with relatively low or variable scores (within or between stages) was then subjected to the primer extension method (Supplemental Fig. S1). 20 sites were confirmed (Table 1) and four sites were excluded due to lack of primer extension signal (Supplemental Table S1). Another two of these sites were excluded because we were unable to assign a plausible SNORD (Supplemental Table S1). The resulting number of high-confidence sites total 98 with 35 located in SSU, 60 in LSU, and three in 5.8S (Fig. 1D; Supplemental Table S2). Strikingly, all sites were found in both early- and late-rRNA, except LSU-C3916, which was late-rRNA specific. This implies that the mechanism for instalment of ribose methylation functions equally well when rRNA is expressed in the context of amplified, extrachromosomal rDNA (early-rRNA) and from chromosomally integrated rDNA copies (Locati et al. 2017). Our analysis does not rule out the existence of additional sites in specialized tissues or sites with low methylation stoichiometry.
TABLE 1.
Summary of methylated sites supported by primer extension analysis
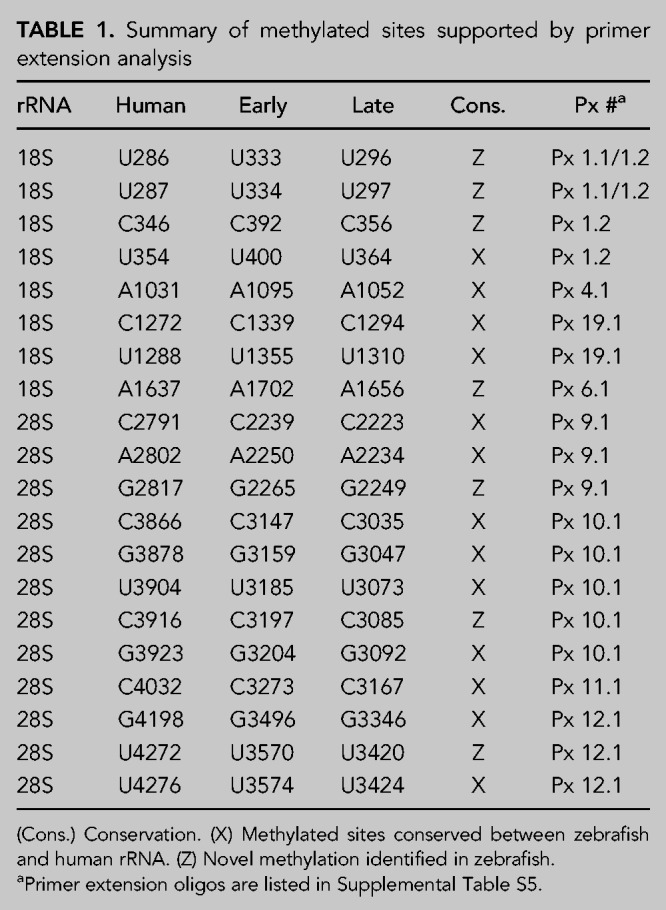
To allow easy comparison with human rRNA, we annotated zebrafish 2′-O-Me sites according to their corresponding human nucleotide counterparts using the reference sequence from the snoRNA-LBME-db (Lestrade and Weber 2006; for zebrafish nucleotide numbering, see Supplemental Tables S1–S3). In comparison with human, 10/98 sites were only found in zebrafish (five in SSU, one in 5.8S, and four in LSU [Fig. 1D; Supplemental Table S3). Conversely, 21 sites in human rRNA were not detected in zebrafish (Fig. 1D). The absence of methylation at these sites in zebrafish is consistent with sequence divergence at target sites (12/21) and absence of cognate SNORDs (17/21) as evidenced by our small RNA-seq and consistent with the small nucleolar RNA orthogonal database (snoopy; Yoshihama et al. 2013). For the remaining four sites (SSU-A159, SSU-C174, LSU-A2388, and LSU-C2811), the corresponding SNORDs were likely conserved due to a functional second antisense element.
The methylation stoichiometry at a subset of sites change during development
The RiboMeth-seq method has proven to yield estimates of methylation stoichiometry that are consistent with mass spectrometry and RP-HPLC (Krogh and Nielsen 2019). Applications of RiboMeth-seq previously revealed that cultured cells have a high proportion of sites (∼1/3) that are fractionally methylated (Krogh et al. 2016; Erales et al. 2017) in contrast to cells in differentiated tissues that are fully or close to fully methylated at almost all rRNA sites. Interestingly, a recent analysis in mice showed that several sites (∼25/108) were increasingly methylated during development (Hebras et al. 2020). Figure 2A shows the methylation profile across zebrafish developmental stages. In adult tail samples, the vast majority (76/98) of methylated sites were fully or close to fully methylated as defined by an RMS-score >0.90 with 16 sites scoring in the 0.80–0.90 range and only six sites with a score <0.80 (Fig. 2A; Supplemental Data 1). Samples from embryonic developmental stages showed significantly lower degrees of methylation at 12 sites (Fig. 2A,B). By comparing these to RiboMeth-seq of mouse tissues (Hebras et al. 2020), 4/12 sites (SSU-Um354, SSU-Gm436, 5.8S-Um14, and LSU-Gm3923) behaved similarly between the two species by showing an increasing level of methylation during development. Interestingly, zebrafish novel sites were overrepresented among the remainder of the varying sites (5/8 sites). Hence, half of the zebrafish novel sites displayed varying methylation levels during development. It should be noted that RiboMeth-seq profiling has high background levels and thus may underestimate effects in the low-scoring range.
FIGURE 2.

Ribosome-wide profiling of 2′-O-Me in zebrafish developmental stages and adult fish. (A) Graph depicting fraction methylated at all methylated sites in five developmental stages. Sites (x-axis) are numbered according to human rRNA to allow comparisons and novel sites found in this study are highlighted in bold. The corresponding nucleotide positions for zebrafish early- and late-rRNA can be found in Supplemental Table S2. Data from different stages are indicated by color and asterisks indicate the levels of statistical significance when comparing the sample in question to all other samples. Error bars indicate the standard deviation, n = 3. (B) Examples of differentially methylated sites between the five stages analyzed. Zebrafish novel methylations are highlighted in bold. Asterisks and error bars were used in the same way as in A, although coloring was omitted.
Unsupervised hierarchal clustering analysis of the RMS-scores across all samples showed the 12-somite and the 32-cell stages to group closest to unfertilized egg, whereas the protruding-mouth stage grouped together with the adult tail sample (Supplemental Fig. S2). The clustering was mainly driven by two sets of sites changing the most: SSU-U354, -C1272, LSU-C3916, and -A4560 for the first group, and SSU-C346, -G436, LSU-G2817, -G3923, -U4272, 5.8S-U8, and -U14, for the second group (Supplemental Fig. S2). Thus, it appeared that the methylation pattern at a subset of sites followed the developmental timeline and the transition from high levels of early-rRNA during the initial onset of development to high levels of late-rRNA later in development and in adult fish.
Structural comparison suggests that rRNA predominantly expressed early in development is derived
The core structure of rRNA is highly conserved. However, rRNA from different species shows considerable differences in more peripheral structural elements, in particular the expansion segments (Gerbi 1996; Ramesh and Woolford 2016). At the sequence level, early-rRNA was found to be 86.5% (SSU) and 69.9% (LSU) similar to human, whereas late-rRNA was slightly more similar, with 90.0% (SSU) and 70.6% (LSU). For a detailed comparison of human and zebrafish rRNA, see (Locati et al. 2017). Because one of the functions that have been ascribed to SNORDs is to assist in rRNA folding, we set out to specifically compare the secondary structures of zebrafish and human rRNA and relate this to 2′-O-Me sites. Structures were drawn based on human rRNA from the RiboVision database (Bernier et al. 2014) aided by sequence alignments (Fig. 3; Supplemental Data 2, 3). Compared to human rRNA, the zebrafish base-pairing scheme differed the most in expansion segments, typically with human rRNA having the longest extensions, as expected (Fig. 3A; Supplemental Figs. S3–S6). As an example, zebrafish LSU expansion segment 7 in H25 lacked ∼400 nt, primarily with shorter helixes 25ES7a, “b”, “d”, “e” and entirely lacking “f”, “g”, and “h” (Fig. 3A). In contrast to regions that lacked 2′-O-Me sites, other structural elements where early- and late-rRNA structures differed had strikingly placed zebrafish-specific 2′-O-Me sites. In the structure comprising H75-79, H78ES30 was shorter and H79ES31b was considerably longer in early-rRNA than the human counterpart, whereas these structural elements were much more similar to human in late-rRNA (Fig. 3B). The late-rRNA specific methylation LSU-C3916 was found at the three-way junction that organizes these helices (Supplemental Fig. S6) suggesting a role in the folding or conformational flexibility of late-rRNA in zebrafish. In early-SSU rRNA, the “a” part of the expansion segment in h9 (9es3a) was extended by a 28 nt long GC-rich sequence with ∼14 nt on either side of the loop compared to the human counterpart (Fig. 3C; Supplemental Figs. S3, S4). In contrast, this helix was only extended by three G–C pairs in late-SSU rRNA immediately adjacent to the loop and thus almost identical to the human structure. Furthermore, the “b” and “c” parts of 9es3b-c in early-SSU rRNA had an insertion of a 14 nt G-rich long sequence compared to the human counterpart, and was rearranged so the C-rich 5′ part of the helix was swapped with the G-rich 3′ part. In late-SSU rRNA, the insertion of the G-rich sequence was only 6 nt-long and the rearrangement seemed less dramatic (Fig. 3C; Supplemental Figs. S3, S4). Zebrafish methylation sites SSU-U286 and -U287 were found in an internal loop at the base of 9es3b-c suggesting their involvement in folding of this expansion segment, to avoid interference with the folding of the conserved core. Altogether, the sequence and structure comparison revealed that late-rRNA resembled human rRNA the most.
FIGURE 3.

Structural comparison of zebrafish early- and late-rRNA with human rRNA. The scaling compared to Supplemental Figures S3–S5 is indicated. (A) Helix 25 in LSU and expansion segment 25ES7a–h showing the deletion of 25ESf, “g,” and “h” and shortening of 25ESa, “b,” “d,” and “e.” (B) Helixes 75-79 and the two expansion segments 78ES30 and 79ES31 in Domain V in LSU with deletion and reduction of 78ES30 in early- and late-LSU, respectively, and elongation of 79ES31b in early LSU. (C) Part of the 5′ domain in SSU focusing on helix 9es3-ac. Segments of high (black) and low (red) degree of conservation, helix numbering (blue), conserved (M; black), and novel zebrafish 2′-O-Me sites (M; blue) are indicated.
Assigned SNORDs conform to the SNORD-target RNA interaction rules
Two sets of sequencing data were used to study the SNORD guides, a low-coverage data set that came along with the RiboMeth-seq analysis, and a dedicated small RNA-seq data set for which we used RNA isolated from early developmental stages. SNORDs were identified by running the FASTA file generated from the sequencing through snoScan (Schattner et al. 2005) or by manual searching for antisense elements matching a known methylation site. A plausible SNORD conforming to the SNORD-target RNA interaction rules could be assigned to 93/96 sites (Supplemental Table S4; Supplemental Data 4), not considering the two methylations in the A-loop of LSU rRNA known to be installed by a stand-alone methyltransferase (Lapeyre and Purushothaman 2004) and allowing +5 and +6 double modifications at three sites (SSU-U287, LSU-G2351, and LSU-G4198). Thus, there are currently three novel methylation sites (SSU-A1322, SSU-A1637, and LSU-G2817) lacking an assigned SNORD (Supplemental Table S4). This set of SNORDs constitutes a minimal set of expressed SNORDs that could account for the observed methylations.
The ribose methylation system in zebrafish appeared to follow strict rules sufficient to explain the experimental observations. First, the majority of SNORDs guided only a single methylation with only three SNORDs using antisense elements associated with both box D and box D′ (Fig. 4A) and one SNORD (SNORD30) guiding two sites (SSU-A1383 and LSU-A3804). Second, the rules for base-pairing interaction between the antisense element in the SNORD and the target sequence in rRNA appeared to be very strict with a minimum of nine base-pairings from position 2–10 upstream of box D (or D′) (Fig. 4B). The base-pairings were almost exclusively Watson–Crick, with few G–U pairs and rarely tolerated mismatches at the second position upstream of box D (Supplemental Fig. S7). Third, the box C and box D of almost all SNORDs complied with the consensus sequences 5′-RUGAUGA and 5′-CUGA, respectively (Fig. 4C, upper panels). Box D′ was less conserved whereas Box C′ was difficult to identify in most SNORDs and appeared almost completely degenerated (Fig. 4C, lower panels). Thus, zebrafish SNORDs and SNORD-target interactions follow the rules of other vertebrate species, such as human and mouse.
FIGURE 4.

Analysis of SNORDs identified in this study. (A) Distribution of SNORDs guiding methylations in rRNA using box D, box D′, or both. (B) Base pair interaction between the antisense element in the SNORD and the rRNA target in relation to the distance from box D (or D′). (C) Sequence logo of the box D, D′, C, and C′ of SNORDs guiding methylations in rRNA generated by WebLogo (Crooks et al. 2004). (D, upper left) sequence comparison of human and zebrafish early- and late-rRNA containing a conserved methylation at LSU-2824 with indications of identical (.), deleted (-), and methylated (m) nucleotides. (Lower left) Human and zebrafish SNORD34 and the interaction with the target sequence surrounding LSU-2824. Nucleotides that differ between the two are labeled in red. (Right) 2D structures of a conserved LSU rRNA region between human and zebrafish highlighting methylations in H47, H48, H60, and H61. Methylation at LSU-2824 is in red and the zebrafish-specific methylation in blue. (E, upper) sequence comparison of human and zebrafish early- and late-rRNA highlighting LSU-Cm3916 only found in late-rRNA. (Lower) Drawings of the SNORD base-pairings with early- and late-rRNA, respectively. The presence of a mismatch (x) with early-rRNA may prevent pairing and methylation. (Hsa) Homo sapiens, (Zbf) zebrafish.
Based on the established rules for methylation guiding, we propose that the methylation of LSU-2824, which in human is Um and in zebrafish Gm (Fig. 4D), is a remarkable example of coevolution of SNORD34 and the rRNA target involving three compensatory base pair changes in order to preserve the methylation. LSU-2824 is located in an otherwise highly conserved region of the ribosome with three additional conserved methylations, one in each of H47 and H60, and two in H61 (Fig. 4D, right). In human, there is an additional methylation 13 nucleotides upstream of LSU-2824 (LSU-C2811). In zebrafish, a G–U pair close to the base of H61 differs from a G–C pair in human (Fig. 4D, right) and the G is methylated. It would be of interest to see if these two species-specific modifications play similar roles in organizing the neighboring four-way junction. Only two other instances of compensatory base pair changes in the SNORD:target interaction were found (Supplemental Fig. S8). Another observation that is plausibly explained by applying the consensus rules is the methylation at LSU-C3916 that is exclusively found in late-rRNA (Fig. 2B). Here, we hypothesize that a C rather than an A immediately 5′ of the methylated C at position +5 prevents the formation of consensus base-pairing with SNORD202 in early-rRNA and thus the introduction of the 2′-O-Me (Fig. 4E).
SNORD expression of a subset of fractionally methylated sites is delayed and correlates with increased methylation in the course of development
We used the low-coverage RNA-seq from RiboMeth-seq to estimate SNORD levels and correlate with 2′-O-Me stoichiometry. In our hands, these estimates are consistent with RT-qPCR and Northern blot analysis, probably because they are based on sequencing of alkaline fragments that reduce the bias due to adapter ligation to the mature ends of the SNORDs. The overall SNORD levels in unfertilized eggs and the 32-cell stage were very low compared to the 12-somite and protruding-mouth stages, and the adult tail (Fig. 5A). A likely explanation is that rDNA transcription and thus rRNA biogenesis is shut-down in unfertilized eggs at the time of harvesting and that transcription of late-rRNA similarly is low in the 32-cell stage (Fig. 1C). This provides an interesting opportunity for correlating SNORD production and methylation stoichiometry during development. To this end, we identified seven 2′-O-Me sites with increasing methylation during development (methylation pattern from being fractional in the 12-somite and protruding-mouth stage to fully methylated in the adult tail sample) and their cognate SNORDs based on the results in Figure 2A,B; Supplemental Data 1. When the expression levels of these SNORDs were compared to those of the SNORDs guiding the 83 2′-O-Me sites that showed no change in methylation, we observed a significantly delayed expression of the seven SNORDs (Fig. 5B). Notably, the expression of these SNORDs was not a simple reflection of host gene expression. Three of the host genes encode additional SNORDs that followed the expression of the main group of SNORDs and guide modifications of sites that are fully methylated throughout development. The uncoupling of SNORD expression from host gene expression can, for example, occur by alternative splicing of the primary transcript and non-sense mediated decay and has previously been documented (Lykke-Andersen et al. 2014; Hebras et al. 2020).
FIGURE 5.

SNORD expression levels during zebrafish development. (A) Relative expression levels of all SNORDs identified in this study that guides methylation in rRNA. (B) Cumulated expression of SNORDs guiding invariable 2′-O-Me sites (Invariable) and sites that exhibit increased methylation during development (Me up). This group comprised SSU-C346, -U354, -G436, LSU-C3916, -G3923, -A4560, and 5.8S-U14 and the cognate SNORDs, SNORD-200, -90, -100, -202, -111, -119, and -71. Asterisks indicate significant differences between the two groups of methylation sites within a given developmental stage. (C) Schematic of the poorly annotated FP101887 host gene with indication of 11 exons based on comparison to the longest annotated transcript that appear to encode a lncRNA. The introns host 10 SNORDs of which nine are related. Two SNORDs are given double names (see text for explanation). (D) Alignment of the SNORDs encoded by the FP101887 gene. Conserved SNORD elements are in bold and the antisense elements are underlined. Nucleotides deviating from the most abundant nucleotide at any given position in the alignment are labeled in red to highlight the relatedness of the SNORDs. (E) Expression data extracted from RiboMeth-seq analysis (low coverage RNA-seq).
Focused analysis of a host gene reveals an unanticipated multitude of SNORDs
Our SNORD search was focused on discovery of a minimal set that could explain the observed methylation patterns. However, it is known from SNORD inventories in many organisms that several SNORDs are represented by more than one family member and that SNORDs without a known target (“orphans”) as well as SNORD pseudogenes exist. Supplemental Data 4 lists additional members of the SNORD families in our minimal data set as revealed by a naïve BLAST analysis. Because methylation of LSU-C3916 was specific to late-rRNA and guided by an apparently novel SNORD (SNORD202), we conducted a thorough analysis of the host gene of this SNORD, FP101887. The annotated transcripts from the gene do not appear to encode proteins, but on the other hand, the transcripts were not detected in a survey of zebrafish lncRNA (Ulitsky et al. 2011). In addition to the SNORD202 sequence used in our minimal set, the gene has two annotated SNORD202 family members that diverge slightly in sequence, but all appear to be bona fide guide RNAs. By aligning the gene sequence and the longest of seven annotated transcripts, we derived the exon–intron structure (Fig. 5C; Supplemental Fig. S9) and searched for SNORD motifs in the intronic regions. Surprisingly, this revealed ten SNORDs, of which nine are related in sequence (Fig. 5D). Seven of these have antisense elements consistent with guiding methylations at LSU-U3904 (upstream of the D′ box) and LSU-C3916 (upstream of the D box). One has the antisense element for LSU-C3916 only, and, conversely, one has the antisense element for LSU-U3904 only. These are the two family members that we detected for our minimal set with the latter corresponding to SNORD52. All of the family members appear to be expressed at low to medium levels and all increased their expression during development as evidenced by our low-coverage RNA-seq analysis (Fig. 5E). The expression levels of individual members appeared related to the stability of the short terminal stem (Supplemental Fig. S10) formed by sequences flanking boxes C and D that pair to form a kink-turn stabilized by binding of the zebrafish homolog of the Snu13/15.5k_NHPX/L7Ae protein (Watkins and Bohnsack 2012). The antisense element associated with box D and required for guiding methylation of LSU-C3916 was not found in any of the SNORD52 homologs aligned in an evolutionary analysis of 29 vertebrate SNORDs (Kehr et al. 2014). Thus, the 3′ half of the seven double-guiding SNORDs in FP101887 and SNORD202 are formally a novel SNORD family. However, to maintain transparency in the literature, we label all of these SNORDs as SNORD52 family members. The SNORD52-unrelated SNORD encoded by the last annotated intron in FP101887 (labeled SNORDX in Fig. 5C) is among the most highly expressed SNORDs in zebrafish. It has at least three plausible targets in rRNA, but none of these were found to be methylated. Thus, it was excluded from our minimal set and awaits further analysis.
DISCUSSION
2′-O-Me sites and assigned SNORDs in the zebrafish
We applied RiboMeth-seq to whole-cell RNA from zebrafish and uncovered 98 high-confidence sites defined as methylated sites that were further supported by primer extension analysis (20 sites) and/or assignment of a SNORD guide (or an enzyme in the case of the two A-loop sites) conforming to consensus (95 sites). Apart from one site that was specific to late-rRNA, all sites were methylated in both early- and late-rRNA, albeit to varying degrees for a subset of sites. Ten of the sites were not found in human and mouse (Krogh et al. 2016; Hebras et al. 2020).
We identified 86 SNORDs responsible for 93 of the 96 RNA-guided ribose methylations. Together, these SNORDs constitute a minimal set that was sufficient to explain the observed methylation patterns in RiboMeth-seq experiments. 41 of the SNORDs were also listed in the small nucleolar RNA Orthogonal database (Yoshihama et al. 2013), and only 12 were annotated in the ZFIN database (www.zfin.org). Two of the SNORDs in our set were described separately (Makarova and Kramerov 2009). Altogether, we have updated the information on zebrafish 2′-O-Me sites in rRNA and cognate SNORDs to the level of other major model organisms. All of the SNORDs in the minimal set were encoded in intronic regions within host genes (Supplemental Data 4). The base-pairing interactions between the SNORDs and their targets almost all (89/98) conformed to strict consensus rules. The main exception was three cases of double modifications at positions +5 and +6, a phenomenon that has also been observed in several other systems. As in human (Krogh et al. 2016) and mouse (Hebras et al. 2020), but different from yeast (Birkedal et al. 2015) and many species of archaea (Dennis et al. 2015), 83/86 SNORDs appeared to guide a single methylation, predominantly using the box D′-associated antisense element (two-thirds of the cases). Five of the SNORDs appeared novel and were numbered SNORD200-204 (high numbers used in order not to interfere with human numbering). These SNORDs returned no hits when queried in the Rfam database (Kalvari et al. 2018) and we were unable to detect related SNORDs by BLAST searches with one exception. SNORD201 is related to human orphan SNORD101 in the human snoRNA atlas (Jorjani et al. 2016). The sequence similarity does not include the antisense element and thus, we maintain the SNORD201 numbering emphasizing the functional SNORD of the two until a more comprehensive phylogenetic analysis can form the basis for defining the family to which these SNORDs belong.
SNORD inventories from several organisms have revealed that many SNORDs are represented by several genomic copies that may differ in sequence and that many SNORDs without known targets (“orphans”) exist. In Supplemental data 4 we have listed 33 additional SNORDs belonging to 16 families (one to five additional copies) revealed by BLASTN analysis of the zebrafish genome. The additional copies were typically found in the same host gene as the SNORD from the minimal set. However, this is clearly not the full story as revealed by our in-depth analysis of the FP101887 host gene. Here we found a total of 10 SNORDs, seven more than annotated for the gene. Nine of these belong to the same family, with seven double guides and two in which the antisense element associated with either the D box (SNORD52) or D′ box (SNORD202) was degenerate. One possibility is that the double-guiding SNORDs gave rise to two single-guiding SNORDs following an evolutionary trend toward single-guiding SNORDs. For transparency in the literature, we assigned all of these nine SNORDs to the SNORD52 family, using double names SNORD52A/SNORD202 and SNORD52I/SNORD52 in this paper to highlight the novelty of the box D-associated antisense element guiding methylation at LSU-C3916. Clearly, a full inventory of zebrafish must await a dedicated analysis, as is the case for most other model organisms.
Early-rRNA is structurally different from late-rRNA and may have adopted functions specific to early development
Genome duplication is an important source of paralogues in evolution. However, there are only a few examples that comprise rRNA genes, which is surprising considering that sequence variants of rRNA genes have been suggested to be of functional importance (Kurylo et al. 2018; Parks et al. 2018). The few well-described examples comprise rRNA genes in the malaria parasite, Plasmodium falciparum (Gunderson et al. 1987; Mercereau-Puijalon et al. 2002), and the parasitic trypanosomatids (Liu et al. 2016; Shalev-Benami et al. 2017; Rajan et al. 2020). In the latter organisms, several studies have addressed variable ribose methylation in rRNA of the blood stream form of the parasite propagating in humans and the procyclic form propagating in the insect host (Liu et al. 2016; Shalev-Benami et al. 2017; Rajan et al. 2020). In the case of Trypanosoma brucei, the 2′-O-Me sites were recently mapped by several sequencing-based approaches, including RiboMeth-seq, and it was argued that differences in methylation could be part of adaptation to differences in nutrients and temperature in the two hosts (Rajan et al. 2020). The teleost-specific whole-genome duplications may have given rise to the two genomic rDNA clusters described in zebrafish (Locati et al. 2017). Sequence and secondary structure comparisons revealed that late-rRNA was more similar to rRNA from other vertebrates than the early-rRNA subtype (Fig. 3), suggesting that the cluster encoding early-rRNA was more diverged, and likely acquired new properties. It was recently reported that snoRNAs U3, U8, and snoZ30 also exist in early- and late-expressed versions (Pagano et al. 2020). Finally, it is possible that the genome duplication also gave rise to new SNORDs, but due to the low sequence constraints of SNORDs it was not possible to establish if the zebrafish-specific SNORDs are derived from more ancestral vertebrate SNORDs by duplication and sequence drift.
Temporal concurrence of the early-rRNA subtype and maternal mRNA, as well as their similar rate of decline in the course of zebrafish development, suggests a functional link. Maternal mRNA, stockpiled in an oocyte and utilized for translation well beyond the time point of zygotic genome activation, has unique features ensuring its stability and mechanisms of activation and degradation. These include short or no poly(A) tail, and the subsequent cytoplasmic polyadenylation regulates translational maturation and efficiency, transcript stability, and controls maternal-to-zygotic transition (Vasudevan et al. 2006; Subtelny et al. 2014; Winata and Korzh 2018; Winata et al. 2018; Vastenhouw et al. 2019). Also, maternal mRNA is enriched in epitranscriptomics modifications, such as N6-methyladenosine (m6A), which functions in destabilization (clearance), but also affect the translational efficiency (Huang et al. 2017; Aanes et al. 2020), and 5-methylcytosine (m5C), which enhances the stability of maternal mRNA (Yang et al. 2019). Finally, maternal mRNA contains translation-dependent information, in particular codon usage that influences translation efficiency and transcript decay (Bazzini et al. 2016; Mishima and Tomari 2016). Together, the features of maternal mRNA support the possibility of specialized translational machinery evolved in parallel. Ribosomal heterogeneity is well-established and has been associated with differential transcript preferences and translation efficiencies, although hard evidence for functionally specialized ribosomes is scarce (Genuth and Barna 2018; Guo 2018; Ferretti and Karbstein 2019).
Fractional modification at certain 2′-O-Me sites is a feature of development
The RiboMeth-seq analysis revealed that most sites were fully or close to fully methylated in adult tissues, leaving only six sites as fractionally modified (RMS-score <0.80). This is in line with our observations from human and mouse (Hebras et al. 2020) and in contrast to the early analyses of cell cultures in which one-third of sites were fractionally modified (Krogh et al. 2016). Fractional, or substoichiometric methylation, is an important phenomenon because it implies that the ribosome population is heterogeneous. However, fractional modification could be a passive phenomenon without functional consequences. One possibility is that fractional modification is caused by recently acquired SNORDs that have not yet become optimized for targeting. By comparison of fractional sites to a careful phylogenetic analysis of the origin of their cognate SNORDs there is no support in humans (Kehr et al. 2014; Krogh et al. 2016) or yeast (Birkedal et al. 2015; Canzler et al. 2018) for such an explanation. Another trivial explanation is that the methylation stoichiometry reflects SNORD availability. Although this appears to be the case for some SNORDs, there is no global correlation between SNORD levels and methylation stoichiometry in humans (Krogh et al. 2016), mouse (Hebras et al. 2020), or in the present study. Finally, methylation could reflect SNORD access and thus reflect partitioning between different rRNA folding pathways. If this was the case, the variation in methylation in studies like the present, would imply considerable changes to ribosome biogenesis during development. Based on these arguments, we propose that fractional methylation in at least some cases is a nontrivial phenomenon that should be subjected to functional studies.
An important find in the present study was that 8/98 2′-O-Me sites were fractionally modified during zebrafish development, but fully modified in the adult fish. Conversely, 2/5 sites that were fractional in adult were close to fully methylated in all investigated developmental stages. Thus, changes to the level of methylation appear to be a feature of zebrafish development similar to what was previously observed in the mouse (Hebras et al. 2020). Strikingly, species-specific sites appear particularly enriched among sites that display developmental variation. A significant fraction (6/10) of the zebrafish-specific sites found in the present study conformed to this notion. Similarly, among the 21 sites in mouse that were not found in zebrafish, 12 displayed methylation differences between developmental stages and adult mouse. These constituted a large fraction of the sites (12/20) that showed variation. Fractional methylation implies heterogeneity at the level of the ribosome and may be related to a regulatory transition, a hallmark of early development. Thus, we suggest that a subset of ribose methylations in rRNA constitute an adaptation to specific features of organismal development in vertebrates. Other studies have noted changes in SNORD levels during development, for example, in the classical developmental model Dictyostelium discoideum (Aspegren et al. 2004) and in Drosophila melanogaster (Angrisani et al. 2015). It will be of interest to study if these changes in SNORDs are paralleled by changes in ribosomal RNA methylation patterns and affect development.
The components of box C/D snoRNPs have multiple functions. Fibrillarin is the methyltransferase responsible for RNA-guided ribose methylation (Cavaille et al. 1996; Kiss-Laszlo et al. 1996), but it has additional catalytic functions, including histone methylation (Tessarz et al. 2014; Iyer-Bierhoff et al. 2018), and a key role as a structural component in nucleolar phase transitions (Feric et al. 2016). Proteins that combine with RNAs for targeting, such as Fibrillarin and box C/D RNAs, constitute powerful and highly adaptable systems. Several of the box C/D snoRNPs are involved in ribosome biogenesis without introduction of methylations, for example, U3 snoRNP that is essential for early cleavages of pre-rRNA (Beltrame and Tollervey 1995; Marmier-Gourrier et al. 2011). Those that conform to the methylation paradigm may in fact also be diverse in function. In some cases, the SNORD may carry the main function in chaperoning the folding of rRNA. In other cases, the methylation may carry the main function in stabilizing the ribosome or mediating translation. For this reason, it is important to experimentally establish the methylation pattern in key model organisms and to relate this to the specific biology of the organism. In this study, we have provided the foundation for such work in zebrafish. We suggest that some zebrafish modifications are adaptations to structural alterations in rRNA (Fig. 3) and that others may impact mRNA recruitment and translation during development. These observations open the possibility for functional studies involving manipulations of selected SNORDs using antisense oligonucleotides that interfere with host gene splicing or processing of the SNORD (e.g., using morpholinos; Higa-Nakamine et al. 2012) or by CRISPR–Cas9 KO of SNORD sequences. Both strategies are technically challenging because the SNORDs are located close to splice sites and because the effects of manipulating the SNORD sequences on the often complex splicing of the host gene transcripts are unknown.
MATERIALS AND METHODS
Zebrafish maintenance and sampling
The AB zebrafish line embryos and adults were sourced from the zebrafish facility belonging to Nord University, Norway. The experimental procedures and husbandry were performed in agreement with the Norwegian Regulation on Animal Experimentation (The Norwegian Animal Protection Act, No. 73 of 20 December 1974). This was certified by the National Animal Research Authority, Norway, General License for Fish Maintenance and Breeding no. 17. The maintenance of zebrafish was done using an Aquatic Habitats recirculating system (Pentair) and followed the standard protocol (Westerfield 2000). The fish were fed SDS zebrafish-specific diet (Special Diet Services) according to the manufacturers’ instruction.
Sampling was performed in triplicates, and the source fish originated from three different tanks to eliminate tank-specific variations. To collect unfertilized eggs, two sexually mature females were randomly chosen from each experimental tank, sedated using 50 mg/L MS-222 (Tricaine; Sigma Aldrich) buffered with equal parts of sodium bicarbonate (NaHCO3), and the unfertilized eggs were obtained by manual stripping. The eggs were then washed in ice-cold PBS, drained and immediately snap-frozen for later RNA purification (Presslauer et al. 2017). To collect 32-cell, 12-somite and protruding-mouth stages, parental fish were free-spawning in their respective tanks without prior isolating males and females. Embryos were collected within 10 min after the fertilization and further incubated in Petri dish placed in a cell culture incubator at 28.5°C. Their development was visually tracked under light microscopy. The required embryonic stages were identified according to Kimmel et al. (1995), snap-frozen in liquid nitrogen and stored at −80°C for later RNA purification. To collect samples of trunk from adult individuals (here referred to as “tail” sample), single random male and female were chosen from each experimental tank. These individuals were euthanatized with 200 mg/L MS-222 and the sample filets were dissected and snap-frozen for later RNA purification. Images of zebrafish developmental stages were taken with a Zeiss Axio Zoom V16 microscope using Zeiss Zen image analysis software.
Purification of whole-cell RNA
RNA samples (<50 embryos per replicate) were treated with 1 mL Qiazol (Qiagen) and an adequate amount of lysis beads, and homogenized in the Precellys 24 system (Bertin instrumentation) at 5000 RPM for 2 × 20 sec with a 20 sec gap. Unclear lysates were centrifuged at 13,500 RPM for 10 min at 4°C to remove debris. Otherwise, RNA was purified according to the manufacturer. Subsequently, RNA pellets were dried, resuspended in RNase free water, and 1 µL was used to measure quantity (average of >400 ng/µL) and quality (RIN > 9.6) using an Agilent TapeStation 2200 (Agilent). For long-term storage, RNA was repelleted and kept in 70% ethanol at −80°C. Prior to use, the integrity of the RNA was assessed on a denaturing 1% agarose gel.
RiboMeth-seq and SNP analysis
The initial mapping and quantitation of 2′-O-Me in rRNA was achieved using RiboMeth-seq on biological triplicates except for the adult tail sample which was conducted in duplicate. RiboMeth-seq was essentially performed as previously described (Birkedal et al. 2015; Krogh et al. 2016). Whole-cell RNA was subjected to partial alkaline degradation, purified on a 10% UPAG. Prior to cDNA synthesis using SuperScript IV RT (ThermoFisher Scientific), adaptor ligation was done using a tRNA ligase. The resulting libraries were sequenced on Ion PI Chips (v3) using an Ion Proton semiconductor sequencer with default analysis parameters except that –trim-adapter-cutoff=0 was added to the “Pre-BaseCaller for calibration” and “BaseCaller” to include adapters in the FASTQ file generated using the FastqCreator on the Torrent server. Reads were subsequently sorted based on barcodes in the 5′adapter (cDNA) and this adapter was removed using a Python script. Here, it is important to keep in mind that the analysis is critically dependent on recording of read ends and that the sequence primarily serves to map the read. The generic Ion P1B (3′adapter) was subsequently removed and untrimmed reads or reads shorter than 15 were discarded using cutadapt v2.0. The trimmed reads (median length of ∼34 nt with >95% of reads being between 25 and 45 nt long) were mapped separately to the zebrafish early- or late-rRNA (Locati et al. 2017) and SNORDs (Supplemental Data 5) using Bowtie2 v2.3.4.1 with -k 10. Prior to read-end counting multiple mapped reads were removed allowing only the best mapped reads using a Python script. The “fraction methylated” (RMS-score) was calculated as previously (“score C” in (Birkedal et al. 2015). Subsequently, at a few sites, the RMS-score was manually corrected based on an inherent problem that arises when the commercial RNA oligo used in the first adapter ligation is less than full-length. If the adapter is missing a nucleotide at its 5′ end and ligated to a library RNA fragment with an identical nucleotide at its 3′ end, the barcode is thus intact and the library nucleotide will be removed during data processing. Such errors are easily detected and dealt with by excluding the 3′ end data set from the calculation in the affected replicate and at the problematic site only (see Krogh et al. 2017). Corrected sites are indicated in Supplemental Data 1.
SNP analysis was performed using Samtools v1.3.1. To calculate relative expression of early- and late-rRNAs, isolated SNPs with >30 nt distance to other SNPs were picked based on differences between early- and late-rRNA described in Locati et al. (2017). In total, four positions (T24, A55, C1633, and C1728) in SSU and seven positions (C1218, G1351, T1523, C2096, A2201, C3196, and C3780) in LSU (early-rRNA numbering) were used in the analysis.
Detection of 2′-O-Me by primer extension
A subset of 2′-O-Me sites with low (RMS < 0.75) or inconsistent scores and sites without a plausible SNORD were further assessed by the high/low dNTP-concentration primer extension method (Maden 2001). Primers were designed based on the predicted 2′-O-Me sites and a list of all the primers can be found in Supplemental Table S5. Reverse transcription of 1 µg of whole-cell RNA from appropriate developmental stages were performed in 20 µL 1× RT buffer at 42°C for 60 min supplemented with 1 µL AMV RT (Promega, 20 U) at low and high dNTP concentrations (0.01 mM and 1 mM, respectively). The cDNA generated were separated on an 8% UPAG together with a sequencing ladder. Dried gels were exposed to Phosphor Imager Screens and scanned using a Typhoon Biomolecular Imager (Amersham) to visualize the radioactive signals from the probes. Images were analyzed using Fiji software.
Identification of SNORDs by small RNA-seq
Embryos from different zebrafish developmental stages (32-cell, oblong, and 15% epiboly) were collected as described above, snap-frozen in liquid nitrogen and stored at −80°C. Whole-cell RNA from each of the stages were extracted from ∼30 zebrafish embryos using QIAzol lysis reagent (QIAGEN) as described above. The quality of the RNA was assessed by an Agilent TapeStation 2200 and all samples used for library construction had a RIN value >9. One μg of whole-cell RNA from each stage was subjected to rRNA depletion using the RiboMinus Eukaryote System v2 (Invitrogen), and small RNA libraries were constructed using Ion Total RNA-seq Kit v2 (ThermoFisher Scientific) with minor modifications. In short, the rRNA-depleted RNA samples were enriched for small RNA (<200 nt) using the magnetic bead clean-up module supplied with the kit or the Monarch RNA Cleanup Kit (New England Biolabs), adapters diluted 1:2 were ligated to the RNA for 2 h and the RNA subsequently reverse transcribed using Superscript IV (ThermoFisher Scientific). The cDNA was purified using the magnetic bead clean-up module, without size-selection. Amplification of cDNA and purification were performed according to the manufacturer's instructions. Manual template preparation of libraries was carried out on the Ion OneTouch 2 System (ThermoFisher Scientific) and subsequently sequenced on Ion 540 chips, the Ion GeneStudio S5 System. Reads were automatically trimmed, low quality reads discarded using default settings on the Torrent server, and a FASTQ file generated using the FastqCreator plugin.
SNORD search and rRNA interaction prediction
The FASTQ files from the small RNA-seq of the three stages were converted to FASTA files using a Perl script, merged and subsequently used as the basis of the SNORD search and rRNA interaction prediction. Initially, SNORDs were identified by running the merged FASTA file through snoScan (Schattner et al. 2005) against zebrafish early- and late-rRNA reference sequences (Locati et al. 2017). Only the top-ranked SNORDs conforming to the consensus rules for SNORD-target rRNA interaction were picked for further analyses (Krogh et al. 2016). A few SNORDs were manually identified by searching for the predicted sequence of an antisense element based on the neighboring sequence of the 2′-O-Me site in question. Subsequently, SNORDs were assigned to the methylated sites identified by RiboMeth-seq and primer extension analysis and named in accordance with the human SNORDs (HGCN). The generated list of SNORDs with predicted rRNA interactions was aligned against the zebrafish genome (ENSEMBL, GRCz11) using BLASTN, with search sensitivity set to normal (http://www.ensembl.org/Danio_rerio/Tools/Blast) to extract genomic location, host gene information (Supplemental Table S4; Supplemental Data 4, 5), to validate the SNORD sequences, and extract information on potential homologs. By mapping reads from the RiboMeth-seq analyses against the SNORD sequences, the list of SNORDs was subsequently filtered based on expression and thus only expressed SNORDs were considered in this study (Supplemental Data 5).
Statistical analyses
The RMS-score from RiboMeth-seq results are expressed as mean ± SD. SNORD expression data as mean ± SEM. Correlations were analyzed using Spearman's Rank correlation. Statistical analyses were performed using Microsoft Excel software and GraphPad Prism 7. Comparison of two groups was analyzed by Student's unpaired t-test (two-tailed) and statistically significant differences between groups are indicated as *P < 0.05, ** P < 0.01, and *** P < 0.001. The unsupervised hierarchical cluster analysis and associated heatmap was generated in R using the pheatmap package with the complete linkage method and otherwise default settings (https://CRAN.R-project.org/package=pheatmap).
DATA DEPOSITION
Sequencing data from RiboMeth-seq and small RNA-seq are deposited at the NCBI Gene Expression Omnibus database and accessible through GSE151797.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We wish to thank Cesilie Røtnes Amundsen and Purushothaman Kathiresan, Nord University, for their assistance in quality assessments and sampling of the embryos. The study was funded by the Research Council of Norway, InnControl project (grant #275786) and The Danish Cancer Society (grant R167-A10943-17-S2; to N.K.).
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.076760.120.
REFERENCES
- Aanes H, Engelsen D, Manaf A, Alemu EA, Vågbø CB, Martín L, Lerdrup M, Hansen K, Mathavan S, Winata C, et al. 2020. N6-methyladenosine dynamics during early vertebrate embryogenesis. bioRxiv 10.1101/528422 [DOI] [Google Scholar]
- Amores A, Catchen J, Ferrara A, Fontenot Q, Postlethwait JH. 2011. Genome evolution and meiotic maps by massively parallel DNA sequencing: spotted gar, an outgroup for the teleost genome duplication. Genetics 188: 799–808. 10.1534/genetics.111.127324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angrisani A, Tafer H, Stadler PF, Furia M. 2015. Developmentally regulated expression and expression strategies of Drosophila snoRNAs. Insect Biochem Mol Biol 61: 69–78. 10.1016/j.ibmb.2015.01.013 [DOI] [PubMed] [Google Scholar]
- Aspegren A, Hinas A, Larsson P, Larsson A, Soderbom F. 2004. Novel non-coding RNAs in Dictyostelium discoideum and their expression during development. Nucleic Acids Res 32: 4646–4656. 10.1093/nar/gkh804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzini AA, Del Viso F, Moreno-Mateos MA, Johnstone TG, Vejnar CE, Qin Y, Yao J, Khokha MK, Giraldez AJ. 2016. Codon identity regulates mRNA stability and translation efficiency during the maternal-to-zygotic transition. EMBO J 35: 2087–2103. 10.15252/embj.201694699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltrame M, Tollervey D. 1995. Base pairing between U3 and the pre-ribosomal RNA is required for 18S rRNA synthesis. EMBO J 14: 4350–4356. 10.1002/j.1460-2075.1995.tb00109.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernier CR, Petrov AS, Waterbury CC, Jett J, Li F, Freil LE, Xiong X, Wang L, Migliozzi BL, Hershkovits E, et al. 2014. RiboVision suite for visualization and analysis of ribosomes. Faraday Discuss 169: 195–207. 10.1039/C3FD00126A [DOI] [PubMed] [Google Scholar]
- Birkedal U, Christensen-Dalsgaard M, Krogh N, Sabarinathan R, Gorodkin J, Nielsen H. 2015. Profiling of ribose methylations in RNA by high-throughput sequencing. Angew Chem Int Ed Engl 54: 451–455. 10.1002/anie.201408362 [DOI] [PubMed] [Google Scholar]
- Canzler S, Stadler PF, Schor J. 2018. The fungal snoRNAome. RNA 24: 342–360. 10.1261/rna.062778.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavaille J, Nicoloso M, Bachellerie JP. 1996. Targeted ribose methylation of RNA in vivo directed by tailored antisense RNA guides. Nature 383: 732–735. 10.1038/383732a0 [DOI] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE. 2004. WebLogo: a sequence logo generator. Genome Res 14: 1188–1190. 10.1101/gr.849004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis PP, Tripp V, Lui L, Lowe T, Randau L. 2015. C/D box sRNA-guided 2′-O-methylation patterns of archaeal rRNA molecules. BMC Genomics 16: 632 10.1186/s12864-015-1839-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dooley K, Zon LI. 2000. Zebrafish: a model system for the study of human disease. Curr Opin Genet Dev 10: 252–256. 10.1016/S0959-437X(00)00074-5 [DOI] [PubMed] [Google Scholar]
- Erales J, Marchand V, Panthu B, Gillot S, Belin S, Ghayad SE, Garcia M, Laforets F, Marcel V, Baudin-Baillieu A, et al. 2017. Evidence for rRNA 2′-O-methylation plasticity: control of intrinsic translational capabilities of human ribosomes. Proc Natl Acad Sci 114: 12934–12939. 10.1073/pnas.1707674114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feric M, Vaidya N, Harmon TS, Mitrea DM, Zhu L, Richardson TM, Kriwacki RW, Pappu RV, Brangwynne CP. 2016. Coexisting liquid phases underlie nucleolar subcompartments. Cell 165: 1686–1697. 10.1016/j.cell.2016.04.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferretti MB, Karbstein K. 2019. Does functional specialization of ribosomes really exist? RNA 25: 521–538. 10.1261/rna.069823.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genuth NR, Barna M. 2018. The discovery of ribosome heterogeneity and its implications for gene regulation and organismal life. Mol Cell 71: 364–374. 10.1016/j.molcel.2018.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerbi SA. 1996. Expansion segments: regions of variable size that interrupt the universal secondary structure of ribosomal RNA. In Ribosomal RNA, structure, evolution, processing, and function in protein biosynthesis (ed. Zimmermann RA, Dahlberg AE), pp. 71–87. CRC Press, Boca Raton, FL. [Google Scholar]
- Gilbert WV. 2011. Functional specialization of ribosomes? Trends Biochem Sci 36: 127–132. 10.1016/j.tibs.2010.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunderson JH, Sogin ML, Wollett G, Hollingdale M, de la Cruz VF, Waters AP, McCutchan TF. 1987. Structurally distinct, stage-specific ribosomes occur in Plasmodium. Science 238: 933–937. 10.1126/science.3672135 [DOI] [PubMed] [Google Scholar]
- Guo H. 2018. Specialized ribosomes and the control of translation. Biochem Soc Trans 46: 855–869. 10.1042/BST20160426 [DOI] [PubMed] [Google Scholar]
- Hebras J, Krogh N, Marty V, Nielsen H, Cavaille J. 2020. Developmental changes of rRNA ribose methylations in the mouse. RNA Biol 17: 150–164. 10.1080/15476286.2019.1670598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higa-Nakamine S, Suzuki T, Uechi T, Chakraborty A, Nakajima Y, Nakamura M, Hirano N, Suzuki T, Kenmochi N. 2012. Loss of ribosomal RNA modification causes developmental defects in zebrafish. Nucleic Acids Res 40: 391–398. 10.1093/nar/gkr700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe K, Clark MD, Torroja CF, Torrance J, Berthelot C, Muffato M, Collins JE, Humphray S, McLaren K, Matthews L, et al. 2013. The zebrafish reference genome sequence and its relationship to the human genome. Nature 496: 498–503. 10.1038/nature12111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Shi J, Guo Y, Huang W, Huang S, Ming S, Wu X, Zhang R, Ding J, Zhao W, et al. 2017. A snoRNA modulates mRNA 3′ end processing and regulates the expression of a subset of mRNAs. Nucleic Acids Res 45: 8647–8660. 10.1093/nar/gkx651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer-Bierhoff A, Krogh N, Tessarz P, Ruppert T, Nielsen H, Grummt I. 2018. SIRT7-dependent deacetylation of fibrillarin controls histone H2A methylation and rRNA synthesis during the cell cycle. Cell Rep 25: 2946–2954. 10.1016/j.celrep.2018.11.051 [DOI] [PubMed] [Google Scholar]
- Jorjani H, Kehr S, Jedlinski DJ, Gumienny R, Hertel J, Stadler PF, Zavolan M, Gruber AR. 2016. An updated human snoRNAome. Nucleic Acids Res 44: 5068–5082. 10.1093/nar/gkw386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalvari I, Argasinska J, Quinones-Olvera N, Nawrocki EP, Rivas E, Eddy SR, Bateman A, Finn RD, Petrov AI. 2018. Rfam 13.0: shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Res 46: D335–D342. 10.1093/nar/gkx1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehr S, Bartschat S, Tafer H, Stadler PF, Hertel J. 2014. Matching of Soulmates: coevolution of snoRNAs and their targets. Mol Biol Evol 31: 455–467. 10.1093/molbev/mst209 [DOI] [PubMed] [Google Scholar]
- Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. 1995. Stages of embryonic development of the zebrafish. Dev Dyn 203: 253–310. 10.1002/aja.1002030302 [DOI] [PubMed] [Google Scholar]
- Kiss-Laszlo Z, Henry Y, Bachellerie JP, Caizergues-Ferrer M, Kiss T. 1996. Site-specific ribose methylation of preribosomal RNA: a novel function for small nucleolar RNAs. Cell 85: 1077–1088. 10.1016/S0092-8674(00)81308-2 [DOI] [PubMed] [Google Scholar]
- Krogh N, Nielsen H. 2019. Sequencing-based methods for detection and quantitation of ribose methylations in RNA. Methods 156: 5–15. 10.1016/j.ymeth.2018.11.017 [DOI] [PubMed] [Google Scholar]
- Krogh N, Jansson MD, Hafner SJ, Tehler D, Birkedal U, Christensen-Dalsgaard M, Lund AH, Nielsen H. 2016. Profiling of 2′-O-Me in human rRNA reveals a subset of fractionally modified positions and provides evidence for ribosome heterogeneity. Nucleic Acids Res 44: 7884–7895. 10.1093/nar/gkw482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh N, Kongsbak-Wismann M, Geisler C, Nielsen H. 2017. Substoichiometric ribose methylations in spliceosomal snRNAs. Org Biomol Chem 15: 8872–8876. 10.1039/C7OB02317K [DOI] [PubMed] [Google Scholar]
- Kurylo CM, Parks MM, Juette MF, Zinshteyn B, Altman RB, Thibado JK, Vincent CT, Blanchard SC. 2018. Endogenous rRNA sequence variation can regulate stress response gene expression and phenotype. Cell Rep 25: 236–248. 10.1016/j.celrep.2018.08.093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapeyre B, Purushothaman SK. 2004. Spb1p-directed formation of Gm2922 in the ribosome catalytic center occurs at a late processing stage. Mol Cell 16: 663–669. 10.1016/j.molcel.2004.10.022 [DOI] [PubMed] [Google Scholar]
- Lestrade L, Weber MJ. 2006. snoRNA-LBME-db, a comprehensive database of human H/ACA and C/D box snoRNAs. Nucleic Acids Res 34: D158–D162. 10.1093/nar/gkj002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Gutierrez-Vargas C, Wei J, Grassucci RA, Ramesh M, Espina N, Sun M, Tutuncuoglu B, Madison-Antenucci S, Woolford JL, et al. 2016. Structure and assembly model for the Trypanosoma cruzi 60S ribosomal subunit. Proc Natl Acad Sci 113: 12174–12179. 10.1073/pnas.1614594113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locati MD, Pagano JFB, Girard G, Ensink WA, van Olst M, van Leeuwen S, Nehrdich U, Spaink HP, Rauwerda H, Jonker MJ, et al. 2017. Expression of distinct maternal and somatic 5.8S, 18S, and 28S rRNA types during zebrafish development. RNA 23: 1188–1199. 10.1261/rna.061515.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykke-Andersen S, Chen Y, Ardal BR, Lilje B, Waage J, Sandelin A, Jensen TH. 2014. Human nonsense-mediated RNA decay initiates widely by endonucleolysis and targets snoRNA host genes. Genes Dev 28: 2498–2517. 10.1101/gad.246538.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maden BE. 2001. Mapping 2'-O-methyl groups in ribosomal RNA. Methods 25: 374–382. [DOI] [PubMed] [Google Scholar]
- Makarova JA, Kramerov DA. 2009. Analysis of C/D box snoRNA genes in vertebrates: the number of copies decreases in placental mammals. Genomics 94: 11–19. 10.1016/j.ygeno.2009.02.003 [DOI] [PubMed] [Google Scholar]
- Marmier-Gourrier N, Clery A, Schlotter F, Senty-Segault V, Branlant C. 2011. A second base pair interaction between U3 small nucleolar RNA and the 5′-ETS region is required for early cleavage of the yeast pre-ribosomal RNA. Nucleic Acids Res 39: 9731–9745. 10.1093/nar/gkr675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercereau-Puijalon O, Barale JC, Bischoff E. 2002. Three multigene families in Plasmodium parasites: facts and questions. Int J Parasitol 32: 1323–1344. 10.1016/S0020-7519(02)00111-X [DOI] [PubMed] [Google Scholar]
- Meyer A, Schartl M. 1999. Gene and genome duplications in vertebrates: the one-to-four (-to-eight in fish) rule and the evolution of novel gene functions. Curr Opin Cell Biol 11: 699–704. 10.1016/S0955-0674(99)00039-3 [DOI] [PubMed] [Google Scholar]
- Mishima Y, Tomari Y. 2016. Codon usage and 3′ UTR length determine maternal mRNA stability in zebrafish. Mol Cell 61: 874–885. 10.1016/j.molcel.2016.02.027 [DOI] [PubMed] [Google Scholar]
- Pagano JFB, Locati MD, Ensink WA, van Olst M, van Leeuwen S, De Leeuw WS, Nehrdich U, Spaink HP, Rauwerda H, Jonker MJ, et al. 2020. Maternal- and somatic-type snoRNA expression and processing in zebrafish development. bioRxiv 10.1101/858936 [DOI] [Google Scholar]
- Parks MM, Kurylo CM, Dass RA, Bojmar L, Lyden D, Vincent CT, Blanchard SC. 2018. Variant ribosomal RNA alleles are conserved and exhibit tissue-specific expression. Sci Adv 4: eaao0665 10.1126/sciadv.aao0665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presslauer C, Tilahun Bizuayehu T, Kopp M, Fernandes JM, Babiak I. 2017. Dynamics of miRNA transcriptome during gonadal development of zebrafish. Sci Rep 7: 43850 10.1038/srep43850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajan KS, Zhu Y, Adler K, Doniger T, Cohen-Chalamish S, Srivastava A, Shalev-Benami M, Matzov D, Unger R, Tschudi C, et al. 2020. The large repertoire of 2′-O-methylation guided by C/D snoRNAs on Trypanosoma brucei rRNA. RNA Biol 17: 1018–1039. 10.1080/15476286.2020.1750842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramesh M, Woolford JL Jr. 2016. Eukaryote-specific rRNA expansion segments function in ribosome biogenesis. RNA 22: 1153–1162. 10.1261/rna.056705.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schattner P, Brooks AN, Lowe TM. 2005. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res 33: W686–W689. 10.1093/nar/gki366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segner H. 2009. Zebrafish (Danio rerio) as a model organism for investigating endocrine disruption. Comp Biochem Physiol C Toxicol Pharmacol 149: 187–195. 10.1016/j.cbpc.2008.10.099 [DOI] [PubMed] [Google Scholar]
- Shalev-Benami M, Zhang Y, Rozenberg H, Nobe Y, Taoka M, Matzov D, Zimmerman E, Bashan A, Isobe T, Jaffe CL, et al. 2017. Atomic resolution snapshot of Leishmania ribosome inhibition by the aminoglycoside paromomycin. Nat Commun 8: 1589 10.1038/s41467-017-01664-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Z, Barna M. 2015. Translating the genome in time and space: specialized ribosomes, RNA regulons, and RNA-binding proteins. Annu Rev Cell Dev Biol 31: 31–54. 10.1146/annurev-cellbio-100814-125346 [DOI] [PubMed] [Google Scholar]
- Subtelny AO, Eichhorn SW, Chen GR, Sive H, Bartel DP. 2014. Poly(A)-tail profiling reveals an embryonic switch in translational control. Nature 508: 66–71. 10.1038/nature13007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessarz P, Santos-Rosa H, Robson SC, Sylvestersen KB, Nelson CJ, Nielsen ML, Kouzarides T. 2014. Glutamine methylation in histone H2A is an RNA-polymerase-I-dedicated modification. Nature 505: 564–568. 10.1038/nature12819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulitsky I, Shkumatava A, Jan CH, Sive H, Bartel DP. 2011. Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell 147: 1537–1550. 10.1016/j.cell.2011.11.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vastenhouw NL, Cao WX, Lipshitz HD. 2019. The maternal-to-zygotic transition revisited. Development 146: dev161471 10.1242/dev.161471 [DOI] [PubMed] [Google Scholar]
- Vasudevan S, Seli E, Steitz JA. 2006. Metazoan oocyte and early embryo development program: a progression through translation regulatory cascades. Genes Dev 20: 138–146. 10.1101/gad.1398906 [DOI] [PubMed] [Google Scholar]
- Vilella AJ, Severin J, Ureta-Vidal A, Heng L, Durbin R, Birney E. 2009. EnsemblCompara GeneTrees: complete, duplication-aware phylogenetic trees in vertebrates. Genome Res 19: 327–335. 10.1101/gr.073585.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins NJ, Bohnsack MT. 2012. The box C/D and H/ACA snoRNPs: key players in the modification, processing and the dynamic folding of ribosomal RNA. Wiley Interdiscip Rev RNA 3: 397–414. 10.1002/wrna.117 [DOI] [PubMed] [Google Scholar]
- Westerfield M. 2000. The zebrafish book. A guide for the laboratory use of zebrafish (Danio rerio). University of Oregon Press, Eugene, OR. [Google Scholar]
- Winata CL, Korzh V. 2018. The translational regulation of maternal mRNAs in time and space. FEBS Lett 592: 3007–3023. 10.1002/1873-3468.13183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winata CL, Lapinski M, Pryszcz L, Vaz C, Bin Ismail MH, Nama S, Hajan HS, Lee SGP, Korzh V, Sampath P, et al. 2018. Cytoplasmic polyadenylation-mediated translational control of maternal mRNAs directs maternal-to-zygotic transition. Development 145: dev159566 10.1242/dev.159566 [DOI] [PubMed] [Google Scholar]
- Wolfe K. 2000. Robustness–it's not where you think it is. Nat Genet 25: 3–4. 10.1038/75560 [DOI] [PubMed] [Google Scholar]
- Xue S, Barna M. 2012. Specialized ribosomes: a new frontier in gene regulation and organismal biology. Nat Rev Mol Cell Biol 13: 355–369. 10.1038/nrm3359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Wang L, Han X, Yang WL, Zhang M, Ma HL, Sun BF, Li A, Xia J, Chen J, et al. 2019. RNA 5-methylcytosine facilitates the maternal-to-zygotic transition by preventing maternal mRNA decay. Mol Cell 75: 1188–1202 e1111. 10.1016/j.molcel.2019.06.033 [DOI] [PubMed] [Google Scholar]
- Yoshihama M, Nakao A, Kenmochi N. 2013. snOPY: a small nucleolar RNA orthological gene database. BMC Res Notes 6: 426 10.1186/1756-0500-6-426 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.