

Abstract
Like most RNA viruses, influenza viruses generate defective viral genomes (DVGs) with large internal deletions during replication. There is accumulating evidence supporting a biological relevance of such DVGs. However, further understanding of the molecular mechanisms that underlie the production and biological activity of DVGs is conditioned upon the sensitivity and accuracy of detection methods, that is, next-generation sequencing (NGS) technologies and related bioinformatics algorithms. Although many algorithms were developed, their sensitivity and reproducibility were mostly assessed on simulated data. Here, we introduce DG-seq, a time-efficient pipeline for DVG detection and quantification, and a set of biological controls to assess the performance of not only our bioinformatics algorithm but also the upstream NGS steps. Using these tools, we provide the first rigorous comparison of the two commonly used sample processing methods for RNA-seq, with or without a PCR preamplification step. Our data show that preamplification confers a limited advantage in terms of sensitivity and introduces size- but also sequence-dependent biases in DVG quantification, thereby providing a strong rationale to favor preamplification-free methods. We further examine the features of DVGs produced by wild-type and transcription-defective (PA-K635A or PA-R638A) influenza viruses, and show an increased diversity and frequency of DVGs produced by the PA mutants compared to the wild-type virus. Finally, we demonstrate a significant enrichment in DVGs showing direct, A/T-rich sequence repeats at the deletion breakpoint sites. Our findings provide novel insights into the mechanisms of influenza virus DVG production.
Keywords: defective viral genomes, influenza, RNA-seq, amplification bias, transcription-defective mutants
INTRODUCTION
Defective viral genomes (DVGs) are generated by many RNA viruses during viral replication and have an impact on viral evolution and pathogenesis (for recent reviews, see Genoyer and López 2019; Vignuzzi and López 2019). Different types of DVGs have been observed and can be present simultaneously during infection, including DVGs with point mutations, frameshifts, deletions, insertions, and/or sequence rearrangements. Viral particles containing a DVG are unable to carry out a full replication cycle except upon coinfection with a complementing helper virus. The most commonly observed are deletion DVGs with large internal truncations that impede the production of one or several essential viral proteins, while preserving the cis-acting RNA sequences required for replication and packaging of the defective RNA. Such DVGs have the ability to interfere with the replication and production of replication-competent viruses, possibly by competing with the full-length genome for essential viral and/or host proteins.
Defective interfering viral particles were first described 60 yr ago in influenza virus stocks propagated at a high multiplicity of infection in embryonated hen eggs (von Magnus 1954). Since then interfering DVGs have been detected both during in vitro and in vivo infections with various influenza types and subtypes (Baum et al. 2010; Saira et al. 2013; Vasilijevic et al. 2017; Sheng et al. 2018; Alnaji et al. 2019; Bosma et al. 2019). In mice there is evidence that DVGs can limit influenza virus-induced pathology and trigger antiviral immunity (Tapia et al. 2013; Dimmock and Easton 2015), potentially through a preferential recognition by the cytosolic sensor RIG-I (Baum et al. 2010). Recent studies revealed that DVGs are produced during natural influenza infection in humans (Saira et al. 2013; Vasilijevic et al. 2017; Lui et al. 2019) and can be transmitted between humans (Saira et al. 2013). A reduced accumulation of DVGs was linked with a severe outcome in patients infected with the H1N1pdm09 virus (Vasilijevic et al. 2017). DVGs have therefore started to raise interest for their potential as prophylactic agents or components of attenuated vaccines (Dimmock and Easton 2014; Frensing 2015; Dimmock and Easton 2017).
The genome of influenza viruses consists of eight single-stranded RNA segments of negative polarity, which are encapsidated with nucleoproteins and associated with one copy of the viral RNA-dependent RNA polymerase. Interfering DVGs have been shown to derive predominantly from the three longest genomic segments (PB1, PB2, and PA), which encode the viral polymerase subunits (Nayak et al. 1985; Frensing 2015). The internal deletions are thought to occur during the replication process when the viral polymerase detaches from its template at one position (breakpoint start) and restarts RNA polymerization at another downstream position (breakpoint end) (Jennings et al. 1983). Evidence suggests a genetic control of DVG production as mutations in the PA subunit of the viral polymerase and the NS2/NEP or M viral protein were found to result in higher DVG levels in cell culture (Odagiri et al. 1994; Fodor et al. 2003; Perez-Cidoncha et al. 2014). Whether certain cis-acting RNA sequences in the viral genome can drive or facilitate the production of DVGs remains unknown. So far, no hotspots for the deletion breakpoints have been identified. However, accurate profiling of the multiple DVG species generated during infection still poses a number of challenges.
The advent of next-generation sequencing (NGS) has enabled to characterize the diversity of viral DVGs at an unprecedented scale. Several bioinformatics tools, such as ViReMa (Routh and Johnson 2014; Alnaji et al. 2019), DI-tector (Beauclair et al. 2018), DVG-profiler (Bosma et al. 2019), or VODKA (Sun et al. 2019) were designed in order to identify the short Illumina reads that contain DVG deletion breakpoints and are discarded by usual alignment algorithms. Two critical issues are (i) how to differentiate true deletion DVGs from artifactual deletions produced during RNA sample processing for RNA-seq, and (ii) how to accurately quantify the relative frequency of each DVG species with respect to the full-length genome. Sample processing may or may not involve PCR amplification of the reverse-transcribed viral RNAs prior to sequencing. Noticeably, most published NGS studies on influenza interfering DVGs rely on PCR amplification of the reverse-transcribed viral RNAs, while the potential artifacts and quantification biases introduced by the PCR step have not been thoroughly assessed.
Here we developed a bioinformatics pipeline for DVG detection and quantification called DG-seq. A preliminary version of this algorithm was previously used to characterize in vivo DVGs produced by Sindbis virus (Poirier et al. 2018). We demonstrate its solid performance in terms of sensitivity and reproducibility using either mixes of in vitro transcribed viral-like RNAs containing pseudo-DVGs of distinct sizes and frequencies, or viral genomic RNAs extracted from wild-type (WT) or mutant DVG-prone influenza viruses. We also provide a rigorous comparison of the two sample processing methods for RNA-seq, with or without a PCR preamplification step. We show that PCR confers increased sensitivity for the detection of DVGs only when their frequency is <10−2 and that this comes at the expense of an accurate DVG quantification due to size- but also sequence-dependent biases. Using both methods to determine the DVG landscape of WT and mutant influenza viruses, we further show that PA mutants induce more frequent and diverse DVGs than their WT counterpart. Focusing on the sequences at breakpoints, we observed a significant enrichment in deletions showing direct, A/T-rich sequence repeats at the breakpoint sites, suggesting that such repeats could be involved in the mechanism of DVG generation.
RESULTS
Fast detection and quantification of DVGs using the DG-seq pipeline
We developed a bioinformatics pipeline named DG-seq to characterize influenza virus DVGs (Fig. 1A; Supplemental Files S1, S2). Upon Illumina sequencing of viral genomic RNAs (vRNAs) extracted from A/WSN/33 virions, the sequencing data were aligned to the viral genome reference sequence using Burrows–Wheeler aligner (BWA MEM). BWA MEM is a fast alignment program that can extract chimeric reads (also termed split reads), which occur when a sequencing read aligns to two distinct sites in the reference sequence with little or no overlap. For example, if the 5′ and 3′ portion of the read maps to positions 110–160 and 1200–1270 in the reference genome, respectively, the alignment identifies that the read is spanning the junction of a deletion from position 160 to position 1200 (Fig. 1B). Upon alignment, our DG-seq algorithm extracts chimeric reads and further provides information relative to the breakpoint ends, raw read counts and normalized read counts (the latter being referred to as “Frequency determination” in Fig. 1A) for each set of deletion DVGs characterized by the exact same breakpoints.
FIGURE 1.

Detection and quantification of DVGs using DG-seq. (A) Schematic representation of the DG-seq pipeline. (B) DG-seq can detect deletion DVGs. (C) Distribution of the number of reads supporting a given deletion (with unique breakpoint start and end) upon analysis of in vitro transcribed full-length pseudo-vRNAs. For instance, in RT-seq samples most deletions (>650) are covered by one read, about 100 are covered by two reads, etc. The 99th percentile was found identical in RT-seq and RT-PCR-seq samples. (D) Some DVG junctions cannot be identified because of too short sequences on one side of the junction. (E) Typical coverage per site depending on DVG frequency.
To determine the background count threshold for the DG-seq pipeline, we produced a set of eight full-length in vitro transcribed (IVT) influenza vRNAs. Five equimolar mixes of IVT vRNAs were reverse-transcribed and the cDNA was submitted to Illumina sequencing directly (RT-seq, three mixes), or the cDNA was PCR-amplified before sequencing (RT-PCR-seq, two mixes). Upon DG-seq analysis of the IVT vRNA mixes, most deletions were identified based on a very low read count of 1 or 2 reads. Considering collectively all deletions identified in the 5 IVT mixes, 99% had counts <14, that is, that the 99th percentile of the raw counts (referred to as Raw99) of the pooled data was 14, a good candidate for background noise threshold (Fig. 1C; Supplemental Table S1). To assess the robustness of this estimation, we repeated this experiment independently (four equimolar mixes of IVT RNA, two run in RT-seq and two in RT-PCR-seq). When Raw99 was computed separately for each deletion identified per mix and per segment, the distribution of Raw99 values was comparable in both experiments (Supplemental Fig. S1A). Considering collectively all deletions identified in the four IVT mixes of Expt B, we found a Raw99 of 13 counts, in line with the results from Expt A, suggesting that this threshold, in our experimental conditions, is not dependent on the experiment or the sequencing run. Notably, Raw99 values were not dependent on the method used (RT-seq or RT-PCR-seq, Supplemental Fig. S1B). Additionally, Raw99 was found to be poorly correlated to coverage depth (r = 0.2, P = 0.1, Supplemental Fig. S1C), while its normalized counterpart (referred to as Normalized99) was significantly negatively correlated to coverage depth (r = −0.56, P < 0.0001; Supplemental Fig. S1D), providing a rationale for using raw counts rather than normalized counts to determine background threshold. Together, these results suggest that the 99th percentile of raw counts of control IVT mixes is a statistic that is robust to experimental run, sequencing method and coverage depth, and identify 14 as an appropriate background threshold value in our conditions. Therefore, in subsequent analyses, we filtered out DVGs with a read count <14, as well as DVGs with a deletion of <10 nt (“Noise filtering” in Fig. 1A) in order to detect specific and biologically relevant DVGs.
The analysis of aligned chimeric reads (150 nt long) revealed that the mapped portions on each side of the breaking point were 30–120 nt long. This suggests that, for example, a read whose 5′ breakpoint is 20 nt downstream from its 5′ end will be mapped on the right side but not on the left side of the breakpoint, and therefore will not be assigned as a chimeric read (Fig. 1D). To compensate for the undetected (30 + 30)/150 = 60/150 = 2/5 of total DVGs, the read counts were multiplied by 5/3. In addition, read counts need to be normalized with regard to variations in the sequencing depth. Since the coverage can be variable along the vRNA sequence (typically it will show a central drop when a high-frequency DVG is present, Fig. 1E), normalizing over the total number of reads mapping to the vRNA is biased. Therefore, we normalized DVG read counts to the maximum coverage per position (or, rather, the mean of the 2% highest values, to avoid outliers), generally found within the packaging signals located at the 5′ and 3′ ends of vRNAs. Normalized counts are referred to below as frequencies.
Reproducibility of DVG analysis using the DG-seq pipeline
To assess the reproducibility of DVG analysis using the DG-seq pipeline, vRNAs were extracted from three stocks of recombinant A/WSN/33 virus: the wild-type (WT) virus and two PA mutants (K635A and R638A), produced by reverse genetics and submitted to one round of amplification on MDCK cells upon plaque purification. The K635A and R638A mutations in PA were shown to weaken the interaction between the influenza polymerase and the carboxy-terminal domain of cellular RNA polymerase II largest subunit (Lukarska et al. 2017), and in an independent study the PA-R638A mutation was shown to promote the generation of DVGs (Fodor et al. 2003). On each viral stock, two independent RNA extractions and independent RNA processing for RT-seq or RT-PCR-seq were performed (referred to as Expts 1 and 2).
DG-seq analysis revealed respectively for the RT-seq and RT-PCR-seq samples a total of 85 and 222 DVGs with internal deletions (at this stage the DVGs were not differentiated whether they derived from the WT or mutant A/WSN/33 virus), with a good reproducibility between the two experiments (r = 0.96 and r = 0.97, respectively) (Fig. 2A,B; Supplemental Table S2). The proportion of DVGs detected above background (i.e., with a read count ≥14) in both experiments was 56% and 66% upon RT-seq and RT-PCR-seq, respectively (Fig. 2A,B, pink dots and Venn diagrams). Noticeably, all DVGs detected above background in one experiment were also identified in the second independent experiment, albeit for some of them with a read count <14 due to differences in the sequencing depth (undetected, or UD, among the red and blue dots in Fig. 2A,B). DVGs that were detected above background in a single RNA experiment were mostly detected in Expt 1, and were found at lower frequencies than most of the DVGs detected in both experiments (Fig. 2A,B, red dots compared to pink dots). When the analysis was restricted to DVGs with a frequency >10−3, the proportion detected in both experiments was 78% and 83% upon RT-seq and RT-PCR-seq, respectively, indicating an excellent reproducibility.
FIGURE 2.

Reproducibility of DVG analysis using the DG-seq pipeline. (A,B) Expt 1 and 2 correspond to two independent purifications of genomic vRNAs from the same three viral stocks (WT and mutant A/WSN/33 viruses), followed by independent processing for RT-seq (A) or RT-PCR-seq (B) and analysis through the DG-seq pipeline. Each dot represents a distinct DVG identified with a read count above the background noise threshold, that is, ≥14 in both (pink dots) or in at least one of the experiments (red and blue dots). The frequency measured in Expt 2 is plotted as a function of the frequency measured in Expt 1, with a logarithmic scale (UD, undetected, i.e., read count = 0). Venn diagrams represent the numbers of DVGs identified above background in both or in one of the experiments. (C) For the subset of DVGs identified in Expt 1, the frequency measured upon RT-PCR-seq is plotted as a function of the frequency measured upon RT-seq, with a logarithmic scale (UD, undetected, i.e., read count = 0). Each dot represents a distinct DVG identified with a read count above the background noise threshold, that is, ≥14 with both (pink dots) or with at least one of the methods (red and blue dots). The Venn diagrams represent the numbers of DVGs identified above background with both or with one of the methods. r values indicated in A–C are Pearson correlation coefficients. (D) Same graph as in C, but with DVGs colored according to DVG size.
We next focused on the 226 DVGs detected above background in Expt 1 and compared the frequency obtained using RT-seq versus RT-PCR-seq (Fig. 2C). A moderate correlation coefficient was observed (r = 0.7) and overall only 35% of the DVGs were detected with both methods (Fig. 2C, pink dots and Venn diagram). Importantly, some (mostly small-sized) DVGs detected above background with frequencies up to 10−2 in RT-PCR-seq remained undetected in RT-seq (blue dots and UD in Fig. 2C,D). Overall, our data indicate that the DG-seq pipeline allows a robust and very reproducible detection of DVGs, but suggests that RT-seq and RT-PCR-seq induce different biases in DVG frequency estimation.
Comparison of RT-seq and RT-PCR-seq using synthetic pseudo-DVG RNAs
To rigorously assess and compare the sensitivity and biases of the RT-seq and RT-PCR-seq methods for the detection of DVGs, we used three synthetic in vitro transcribed RNAs that mimic DVGs from the PB2 vRNA ranging from 424 to 1184 nt in size (Fig. 3A). To reveal sequence-dependent biases, we designed an additional DVG derived from the PB1 vRNA with almost the same size as the shortest PB2 pseudo-DVG (425 nt) (Fig. 3A). Each synthetic pseudo-DVG RNA was mixed at different molar ratios (1:1, 1:9, 1:99, 1:999, corresponding to frequencies of 0.5, 0.1, 0.01, and 0.001, respectively) with the corresponding synthetic full-length vRNA. Final mixes containing an equimolar ratio of in vitro transcribed RNAs corresponding to the eight full-length genomic segments were processed for RT-seq or RT-PCR-seq and analyzed with the DG-seq pipeline (Supplemental Table S1).
FIGURE 3.

Comparison of RT-seq and RT-PCR-seq using synthetic pseudo-DVG RNAs. (A) Schematic representation of the synthetic RNAs corresponding to PB2- and PB1-derived pseudo-DVGs. (B–E) The synthetic RNAs shown in A were added to an equimolar mix of eight synthetic full-length pseudo-vRNAs, using different molar ratios between the pseudo-DVG and the corresponding full-length vRNA. The final mixes were processed for RT-seq (B,C) or RT-PCR-seq (D,E) and analyzed with the DG-seq pipeline. The measured frequency of each synthetic pseudo-DVG is plotted as a function of the actual frequency with a linear (B,D) or a logarithmic (C,E) scale. Open circles represent read counts below the background noise threshold (i.e., <14).
Upon RT-seq or RT-PCR-seq, all pseudo-DVGs could be detected when their actual frequency was 0.5 or 0.1 or 0.01 (closed circles in Fig. 3B–E), except for the PB2-424-nt-long DVG which was not detected above background upon RT-seq when present at a frequency of 0.01 (Fig. 3C, open circles indicate a read count <14). When pseudo-DVGs were present at a lower frequency of 0.001, they systematically remained undetected above background noise upon RT-seq (open circles in Fig. 3C); in contrast, upon RT-PCR-seq, all but the PB1-425-nt-long DVGs were detected above background noise (Fig. 3E), indicating that RT-PCR-seq is more sensitive than RT-seq to detect low frequency DVGs in the 424–1184 nt range.
Upon RT-seq, the measured frequencies were systematically lower than the actual frequencies (Fig. 3B,C). For instance, at actual frequencies of 0.5 to 0.01, the underestimation rates were 1.3- to 2.7-fold and 1.5- to sixfold for the 1184- and 765-nt-long DVGs, respectively (Fig. 3B,C). The underestimations were more pronounced for the smaller 424- and 425-nt-long DVGs, which were detected at an ∼10- to 30-fold lower frequency than their actual frequency (Fig. 3B,C). The lower detection efficiency of shorter DVGs might be partly due to the specific range of fragment size retained during Illumina library preparation (400–700 nt, see Materials and Methods), as the proportion of 400–700-nt-long fragments that contain a deletion breakpoint is expected to be lower than for longer DVGs. Importantly, the measured frequencies were very similar for the 424- and 425-nt-long DVG, indicating that the bias observed upon RT-seq is sequence-independent.
In contrast, the measured frequencies upon RT-PCR-seq were up to 12-fold higher compared to the actual frequencies, and they differed more substantially from one DVG to another (Fig. 3D,E). Notably, the measured frequency for the largest, 1184-nt-long, DVG laid between the measured frequencies for the 765- and 424-nt-long DVGs, suggesting that the frequency bias was not only size-dependent (in which case the bias would have increased or decreased with size, and the largest pseudo-DVGs would have shown the smallest or largest bias). In support of this hypothesis, the PB2-424- and PB1-425-nt-long DVGs showed very different biases: for instance, when the actual frequency was 0.1 they were detected at a frequency of 0.6 and 0.25, respectively (Fig. 3D–E). These data indicate that PCR amplification, in addition to the expected size-dependent bias, induces a sequence-dependent bias, which prevents accurate estimation of DVG frequencies either in absolute or in relative terms.
Characterization of DVGs in WT and transcription-defective PA mutant viruses
We next characterized separately the DVGs found upon DG-seq analysis of vRNAs purified from the WT, PA-K635A and PA-R638A viral stocks in Expt 1 (Supplemental Table S3). As shown in Figure 4A,B, DVGs were overall more frequent and diverse for the mutants (red and orange) than for the WT (blue), and more markedly so for the PA-K635A mutant (red). The data obtained upon RT-seq (left panels) and RT-PCR-seq (right panels) showed the same trend, although the measured DVG frequencies were higher upon RT-PCR-seq. The segments showing the highest frequency (Fig. 4A) and diversity (Fig. 4B) of DVGs were the polymerase gene segments PB2, PB1, and PA, in agreement with previously published observations (Nayak et al. 1985; Frensing 2015). The PA-K635A mutant was the only virus for which, notably, DVGs were detected from every single segment upon RT-seq, with an overall frequency around 0.1 for the PB1, PB2, and PA segments, 0.01 for the HA, NP, and NA segments, and 0.001 for the M and NS segments (Fig. 4A, red bars). By comparison, only PB1- and PA-derived DVGs were detected for the WT virus, with an overall frequency of 0.01 and 0.003, respectively (Fig. 4A, blue bars). DVGs derived from the PB1, PB2, and PA segments showed mostly large deletions (i.e., >25% of the total segment length) whereas DVGs derived from other segments showed a higher proportion of small deletions, as illustrated in Figure 4B by their position relative to the diagonal line (representing a zero distance between the breakpoint start and end positions).
FIGURE 4.

DVG landscapes for the WT and transcription-deficient A/WSN/33 viruses. The DVGs identified upon DG-seq analysis of vRNAs purified from the WT, PA-K635A and PA-R638A viral stocks in Expt 1 were characterized separately for each virus (while they were analyzed all together in Fig. 2C). (A) The total DVG frequency is indicated for each genomic segment. (B) 2D plots indicate, for each identified DVG represented by a dot, the breakpoint start and end positions and the frequency (size of the dot). The axes scale is adapted according to the size of each segment.
No hotspots for the breakpoint start or end position were identified (Fig. 4B), and, accordingly, only few DVGs with the exact same breakpoint start and end positions were found in distinct viral stocks (Supplemental Table S3). Therefore, we assessed whether breakpoint sites displayed a particular nucleotide environment. To this end, we selected DVGs whose breakpoint start and end could be mapped unambiguously to unique positions in the reference A/WSN/33 sequences (61 DVGs out of 226, see next paragraph), and calculated the frequency of each nucleotide at each position upstream of the breakpoint start (−10 to −1) and downstream from the breakpoint end (+1 to +10). No conserved motif was observed (Fig. 5A,B). We next defined the distribution of A, T, G, and C nucleotides within the −10 to +10 windows surrounding a breakpoint and found no significant difference when compared to their overall distribution in the A/WSN/33 genome (Fig. 5C). Finally, we tested whether the observed breakpoint sites were leading preferentially to in-frame or out-of-frame deletions. As shown in Figure 5D, the three types of frameshifts possibly induced by internal deletions (+0, +1, and +2) were equally distributed, irrespective of the virus under study and whether the RT-seq or RT-PCR-seq method was used.
FIGURE 5.

DVG breakpoints. (A–C) Nucleotidic composition of the sequences adjacent to the DVG breakpoints. Analysis was performed by combining DVGs obtained from all segments for the WT and mutant viruses. (A,B) The frequency of each nucleotide found both upstream and downstream from the breakpoints is shown. Negative and positive numbers denote positions upstream of the breakpoint start and downstream from the breakpoint end, respectively. As a reference, the overall frequency of each nucleotide in the IAV genome is indicated by the dashed horizontal lines. (C) Distribution of each nucleotide when grouping positions −10 through +10, compared with the null distribution consisting of IAV reference sequences through all segments. Significance was determined using χ2 goodness-of-fit test. (D) Pie charts showing the relative proportion of DVGs found with each frameshift, depending on the method used. Significance was determined using χ2 goodness-of-fit test.
Direct sequence repeats adjacent to DVG breakpoints
Close analysis of the DVG sequences revealed in some instances the presence of short direct repeats flanking each side of the internal deletion, which hinder the precise delineation of breakpoint start and end positions. In the particular example shown in Figure 6A, due to the presence of a short direct repeat of 3 nt (TAA) on each side of the deletion, there are four possible alignments of the DVG sequence with respect to the reference sequence, and therefore an uncertainty n = 3 compared to DVGs for which the alignment is unambiguous. The DG-seq algorithm was designed so that the uncertainty value is provided in the output file for each DVG (n = 0 to 12 among the 226 DVGs detected in Expt 1, Supplemental Table S3). In cases where the uncertainty was >0, the breakpoint start position was arbitrarily mapped to the nucleotide preceding the direct repeat on the 5′ side of the deletion (bottom alignment in Fig. 6A).
FIGURE 6.
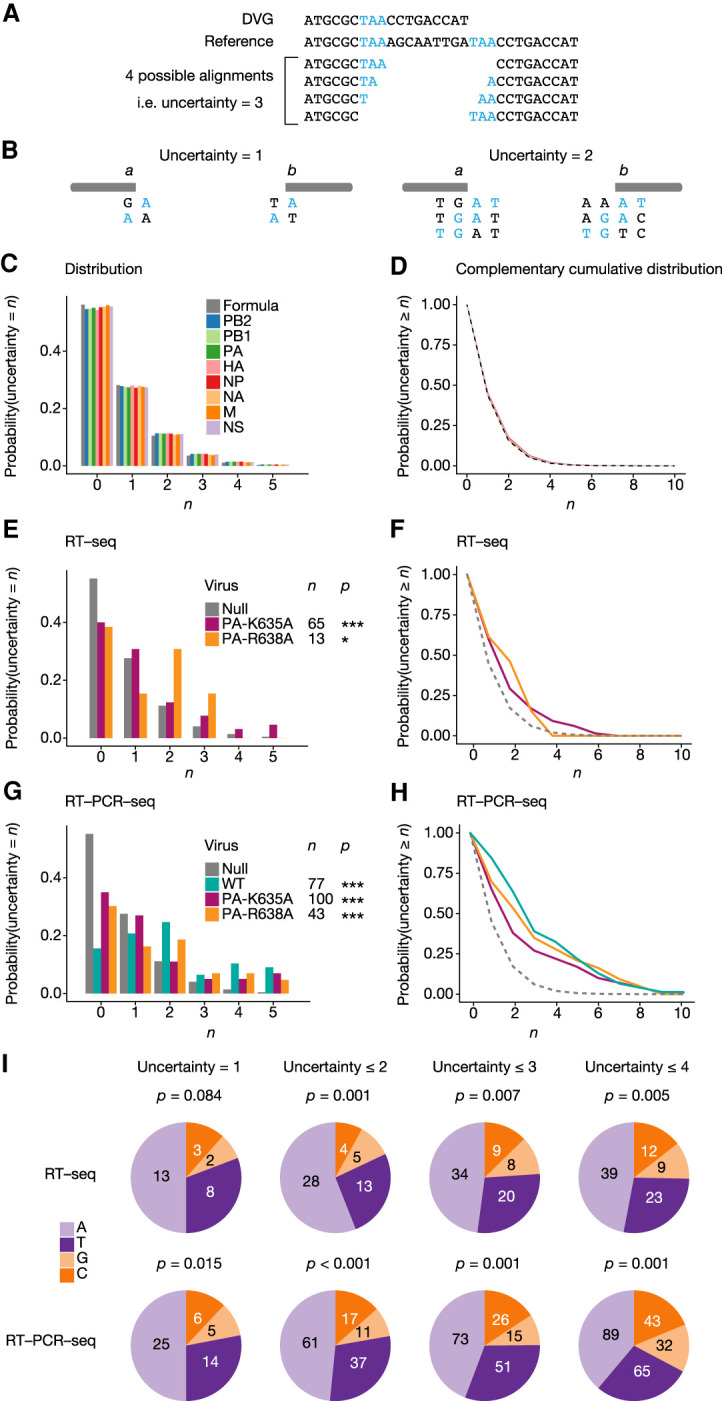
Direct sequence repeats adjacent to DVG breakpoints. (A) Example of a DVG with a direct sequence repeat of 3 nt adjacent to the breaking point (indicated in blue). (B) Conditions for a jump from position a to position b to create a DVG with an uncertainty ≥1 (left diagram) or ≥2 (right diagram). (C,D) Distribution function (C) and complementary cumulative distribution function (D) of the uncertainty value for DVGs with random breakpoints, either computed from a random sequence (gray bars and line) or computed from the sequence of each segment (colored bars and line). (E–H) Distribution function (E,G) and complementary cumulative distribution function (F,H) of uncertainties of DVGs obtained for each indicated virus upon RT-seq (E,F) or RT-PCR-seq (G,H) analysis, compared with random DVGs computed for each sequence. (*) P < 0.05, (***) P < 0.001. Significance was determined using χ2 goodness-of-fit test, followed by multiple testing correction using Holm's method. (I) Distribution of each nucleotide within uncertainty sequences in DVGs with uncertainty = 1, ≤2, ≤3, or ≤4. Significance was determined using χ2 goodness-of-fit test, with the null distribution being computed from the A/WSN/33 full-length genome reference sequences, as shown in Figure 5C.
To assess whether our data set was enriched with DVGs showing an uncertainty >0, we determined the probability at which such uncertainties would appear if the polymerase jumps were occurring randomly. Suppose the polymerase jumps from position a to position b. There is uncertainty ≥1 if (and only if) either positions a + 1 and b show the same nucleotide [denoted N(a + 1) = N(b)], or positions a and b − 1 show the same nucleotide [i.e., N(a) = N(b − 1)] (Fig. 6B, left diagram). There is an uncertainty ≥2 if (and only if) positions either (i) N(a + 1) = N(b) and N(a + 2) = N(b + 1), or (ii) N(a) = N(b − 1) and N(a + 1) = N(b), or (iii) N(a − 1) = N(b − 2) and N(a) = N(b − 1) (Fig. 6B, right diagram). Likewise, it is possible to define the necessary and sufficient conditions for an uncertainty ≥n to exist, for any value of n (details of the calculations are provided in the Materials and Methods section). From those equations we derived, in the case of polymerase jumps occurring randomly, the distribution (for n = 0 to 5, Fig. 6C, gray bars) and complementary cumulative distribution of uncertainty values (Fig. 6D, gray dashed line).
Because the genome of influenza virus is A/T-rich (Fancher and Hu 2011) and contains secondary structure-forming sequences (e.g., the partially complementary 12 and 13 nt at the 3′ and 5′ end of each vRNA), we reasoned that the formula generated from a random sequence may not accurately account for the null hypothesis of polymerase jumps occurring randomly within the viral RNA sequences. Therefore, we computed the distributions and complementary cumulative distribution of uncertainty values using the A/WSN/33 reference sequence, for each genomic segment. The distributions were found to be very close to that obtained with a random sequence (Fig. 6C,D, colored bars and lines). The mean distribution over all segments was subsequently used to represent the null hypothesis.
We then examined the actual distribution of uncertainty values for DVGs detected upon RT-seq with the two PA mutant viruses (Fig. 6E,F) and upon RT-PCR-seq with the WT and PA mutant viruses (Fig. 6G,H). The low number of DVGs detected upon RT-seq with the WT virus (<10) did not provide sufficient statistical power to perform this type of analysis. For each of the five experimental samples, we consistently found that the observed probability of DVGs with an uncertainty >0 was significantly higher than under the null hypothesis (colored lines compared to the gray dotted line in Fig. 6F,H). Remarkably, 13 out of 14 DVGs that were detected independently with the exact same breakpoints in two or more distinct viral stocks showed an uncertainty ≥4 (Supplemental Table S3, P < 0.001). Finally, we assessed whether the direct repeat sequences in DVGs with an uncertainty >0 showed a nucleotide composition bias. Indeed, compared to the A/WSN/33 full length genome used as a reference for the null hypothesis (Fig. 5C), the direct repeat sequences present in DVGs with an uncertainty = 1, ≤2, ≤3 or ≤4 (all viruses and segments combined) appeared to be significantly enriched in A and T nucleotides (Fig. 6I).
Comparative assessment of the DG-seq pipeline
A widely used algorithm for the analysis of DVGs is ViReMa (Viral Recombinant Mapper; Routh and Johnson 2014), which relies on iterative alignment while DG-seq relies on single alignment. We assessed how DG-seq compares to the ViReMa-based pipeline recently published by Alnaji et al. (2019) for the detection of influenza DVGs. The RNA-seq data set from Alnaji et al. relative to four DVG-enriched viral stocks, was retrieved online and analyzed with DG-seq. As shown in Supplemental Figure S2A (to be compared with Fig. 9B in Alnaji et al. 2019), the numbers of distinct DVGs detected within each genome segment were similar between DG-seq and the ViReMa-based pipeline. The specific locations of DVG junctions found in the PB1 and PA segments also showed a similar profile (Supplemental Fig. S2B compared to Fig. 9C in Alnaji et al. 2019). Conversely, we analyzed our RNA-seq data sets with the ViReMa-based pipeline developed by Alnaji et al. (https://github.com/BROOKELAB/Influenza-virus-DI-identification-pipeline). Upon analysis of the vRNAs purified from the WT, PA-K635A, and PA-R638A viral stocks in Expt 1, 90%–100% of the DVGs detected with the ViReMa-based pipeline were also detected with DG-seq (Fig. 7A, left panels), whereas in most samples the ViReMa-based pipeline detected no more than 50%–70% of the DVGs detected with DG-seq (Fig. 7A, right panels). One and three DVGs were detected exclusively with ViReMA upon RT-seq and RT-PCRseq, respectively, compared to 21 and 45 DVGs detected exclusively with DG-seq in the same conditions (Supplemental Fig. S3). As the read count threshold differed between the two methods, and to exclude the possibility that the extra-DVGs detected with DG-seq were merely noise, the comparison between the two pipelines was extended to mixes of in vitro transcribed viral-like RNAs spiked with pseudo-DVGs at frequencies of 0.5, 0.1, 0.01, or 0.001. As shown in Figure 7B, one out of the seven low-frequency DVG-samples that gave rise to lower than threshold read counts with ViReMA was detected with higher than threshold read counts with DG-seq. For the remaining samples, the read counts were overall higher with DG-seq compared to ViReMa, regardless of the size of the DVGs or the method used (RT-seq or RT-PCR-seq), therefore confirming a slight benefit of DG-seq in terms of sensitivity.
FIGURE 7.

Comparison of the DG-seq and ViReMa pipelines. (A) RNA-seq data corresponding to RT-seq and RT-PCR-seq on the WT, PA-K635A, and PA-R638A viral stocks from Expt 1 were analyzed using the ViReMa pipeline. For each sample, DVGs identified upon DG-seq analysis were compared to DVGs detected with the ViReMa pipeline. The percentage of PA, PB1, and PB2 DVGs detected with DG-seq with respect to (wrt) ViReMa (left panel) and conversely (right panel) are shown. The comparison was made using background thresholds specific for each pipeline (read counts ≥14 for DG-seq; ≥30 (PA) or 20 (PB1, PB2) for ViReMa). (B) RNA-seq data corresponding to RT-seq and RT-PCR-seq on the mixes of in vitro transcribed vRNAs spiked with pseudo-DVGs at frequencies of 0.5, 0.1, 0.01, or 0.001 (as described in Fig. 3) were analyzed using the ViReMa pipeline. The read counts of each synthetic pseudo-DVG obtained with ViReMa is plotted against the DG-seq read counts. The background thresholds specific for each pipeline are indicated by dotted lines (read counts ≥14 for DG-seq; ≥20 for ViReMa). (UD) Undetected.
DISCUSSION
Accurate quantification of viral DVGs from RNA-seq data remains a challenge. PCR amplification biases, when cDNAs prepared from viral RNAs are amplified before sequencing, have not been thoroughly evaluated. Here we examined the accuracy and reproducibility of RNA-seq with or without preamplification of the cDNA for the detection and quantification of influenza DVGs, and assessed the performance of our novel bioinformatics algorithm DG-seq. We used two types of RNA samples: two independent RNA extractions from influenza virus stocks (WT and DVG-prone viruses), and mixes of in vitro transcribed viral-like RNAs mimicking the presence of DVGs of distinct sizes and frequencies instead of simulated data like in previous studies (Routh and Johnson 2014; Beauclair et al. 2018; Alnaji et al. 2019; Bosma et al. 2019; Sun et al. 2019). We quantified the background level, reproducibility, sensitivity and accuracy in frequency determination of the whole pipeline, including all wet and dry lab steps involved.
Although both RNA-seq methods showed the same background noise level (99% of DVGs found upon analysis of synthetic full-length viral-like RNAs had counts <14, whatever the method used), RNA-seq data obtained from the synthetic RNA mixes containing pseudo-DVGs showed a higher sensitivity of the preamplification-based over preamplification-free RNA-seq method, with a possible detection of frequencies down to 10−3 for the former compared to 10−2 for the latter. However, preamplification-based RNA-seq resulted in over-estimation of frequencies up to 12-fold, and more importantly this estimation bias was not only size-dependent but also sequence-dependent, as two DVGs with near-identical sizes but distinct sequences showed highly different frequency estimation biases. On the contrary, preamplification-free RNA-seq analysis of the same two DVGs showed no sequence-dependent frequency estimation bias.
The quantification of DVGs obtained from viral stocks in two independent replicates demonstrated the robustness of the DG-seq bioinformatic pipeline. Indeed, for each RNA-seq method, the high reproducibility pattern (with a regression slope very close to 1) confirmed our detection limit (14 counts) as being stringent enough, in addition to validating our normalization method. The much poorer correlation between the preamplification-based and preamplification-free quantification of DVGs is due to the different nature of size-dependent biases in both methods, and more importantly to the existence of a strong sequence-dependent bias in the preamplification-based RNA-seq method. Notably, many DVGs that were detected above background in RT-PCR-seq, some with frequencies up to 10−2, remained completely undetected in RT-seq (with counts = 0). Since RT-seq can detect frequencies lower than 10−3 with counts >0, the actual frequency of these DVGs are likely to be lower than 10−3, and the sequence-dependent bias observed with pseudo-DVGs suggest they might diverge erratically from the estimated frequencies. In contrast, our data demonstrate that for the detection of DVGs at an actual frequency ≥10−2, preamplification-free RNA-seq is as sensitive as preamplification-based RNA-seq and provides a more quantitative determination of DVG landscapes. In conclusion, unless there is a need for the detection of DVGs present at a frequency lower than 10−2, and/or the initial viral input is too low, the preamplification-free method is clearly preferable.
After direct comparison of DG-seq with the ViReMa algorithm, which relies on multiple realignments (Routh and Johnson 2014), DG-seq appeared to provide very similar DVG information and even showed a slight benefit over ViReMa in terms of sensitivity. Advantageous features of the DG-seq pipeline are a substantially reduced requirement for time since it relies on a fast single-alignment method and does not require multiple realignments per read: BWA MEM takes 40 to 90 sec to align one sample, to which 1–30 sec must be added for DG-seq, compared to 2–15 min with the ViReMa pipeline), and easier handling for noninformatics personnel. Both pipelines can be used to analyze RNA-seq data produced from other viruses than IAVs. The primary endpoint of our study was to compare RNA-seq methods, but the biological control tools we developed could be used to further compare alignment procedures, as well as to determine parameter values of complex algorithms to maximize their reproducibility and sensitivity on real biological controls rather than on simulated data sets.
In the present study, rather than performing high-MOI passages to obtain DVG-enriched influenza virus stocks, we chose to use two PA mutants of A/WSN/33 which, based on published data (Fodor et al. 2003; Lukarska et al. 2017), were expected to produce high amounts of DVGs. The WT and mutant viruses were rescued by reverse genetics, plaque-purified and amplified once on MDCK in parallel, using the exact same procedure. We confirmed and extended the initial observations by Fodor et al. (2003) on the PA-R638A mutant, as we found an increased diversity and frequency of DVGs produced by the PA-R638A and also to a larger extent by the PA-K635A mutant virus compared to the wild-type. As these two mutations specifically induce a strong defect in the transcription of viral messenger RNAs, one may speculate that low levels of viral proteins, in particular the nucleoprotein that encapsidates viral genomic RNAs and the complementary cRNAs intermediates that serve as template for genome replication, may favor the production of DVGs. Although we focused on deletion DVGs, DG-seq (along with other algorithm such as DI-tector) can theoretically detect DVGs that show insertions as well as copy-back or snap-back DVGs (as the breakpoint end need not be matched downstream from the breakpoint start in the reference sequence), which are commonly found in paramyxoviruses (Vignuzzi and López 2019). However, insertion, copy-back and snap-back influenza DVGs were not detected above background levels in our viral RNA samples, in agreement with previous reports (Nayak et al. 1985; Frensing 2015; Vignuzzi and López 2019).
Upon preamplification-free RNA-seq of the WT virus, DVGs with large internal deletions were exclusively detected in polymerase segments, while with the PA mutants they were also detected in the HA, NA, and/or NP segments. The internal deletions systematically preserved the 5′ and 3′ ends essential for the packaging of genomic segments but showed no hotspots, in agreement with previous characterizations of influenza DVGs (Nayak et al. 1985; Frensing 2015). Our systematic analysis of the 20 nt surrounding the DVG junction site did not reveal any conserved motif, enrichment in a particular type of nucleotide, or preference for in-frame deletions. Remarkably, however, we found a significant enrichment in DVGs showing direct sequence repeats at their junction site, mostly short repeats up to 12 nt in length. Building on the fact that DG-seq provides for each DVG an uncertainty value which corresponds to the length in nucleotides of the direct repeat at the junction site, we showed that in all conditions examined the observed frequency of DVGs with an uncertainty >0 was significantly higher than under the null hypothesis, consisting in random deletion DVGs derived from the A/WSN/33 reference sequence. In addition, we found that the direct repeat sequences present in DVGs with an uncertainty = 1, ≤2, ≤3 or ≤4 were significantly enriched in A and T nucleotides.
The presence of direct repeats at DVG junction sites was observed by others with the A/PR/8/34 virus (Jennings et al. 1983) and more recently with H1N1pdm09 (Saira et al. 2013; Alnaji et al. 2019) and H7N9 (Lui et al. 2019) viruses, but to our knowledge we are the first to demonstrate a significant association between the two features. The fact that Alnaji et al. did not find such a significant association may lie in the fact that they grouped uncertainties = 0 and = 1 when computing their statistics (Alnaji et al. 2019), thereby undermining their power to detect a significant enrichment in uncertainties >0. Differences in the method used to obtain DVG-enriched viral stocks (high MOI passaging versus DVG-prone mutations) could also contribute to our discrepant findings. Most interestingly, we found that among the few DVGs that were detected, with the exact same breakpoints, in two or more distinct stocks, almost all (13 out of 14) showed direct repeat sequences of ≥4 nt. This highly significant enrichment strongly suggests that, in addition to a genetic control exerted through the viral polymerase subunits (Fodor et al. 2003; Vasilijevic et al. 2017; and this study) and other viral proteins (Odagiri et al. 1994; Perez-Cidoncha et al. 2014), the presence of A/T-rich direct repeats in the viral genome may direct polymerase jumps and control the production of deletion DVGs. Further investigations are needed to fully understand the underlying mechanisms, which is of high interest given the potential of DVGs in influencing infection outcome and their possible use as a novel antiviral therapy approach.
MATERIALS AND METHODS
RNA extractions from influenza virus stocks
Recombinant A/WSN/33 influenza viruses (NCBI:txid382835), either wild-type or bearing single mutations in the PA polymerase subunit (PA-K635A and PA-R638A), were used. Genomic vRNAs were extracted from 150 µL of viral stocks using the QIAamp Viral RNA Mini Kit (Qiagen), eluted in 45 µL of nuclease-free water and subjected to RT-PCR (from 5 µL) or to cDNA synthesis (from 30 µL) as described below.
In vitro transcription of synthetic pseudo-vRNAs
In order to produce synthetic full-length or defective pseudo-vRNAs, overlap extension PCR (Higuchi et al. 1988) was performed using the reverse genetics plasmids pPolI-WSN-PB2 or pPolI-WSN-PB1 (Fodor et al. 1999) as templates. The first step PCRs were performed using primers complementary to the extremities of the PB2 or PB1 segment, and primers complementary to internal sequences surrounding the breakpoint start (BS) and breakpoint end (BE) of the pseudo-DVGs PB2-424 (BS:207, BE:2125), PB2-765 (BS:364, BE:1941), PB2-1184 (BS:556, BE:1714), and PB1-425 (BS: 101, BE: 2018). The second step PCRs were performed using primers complementary to the extremities of the PB2 or PB1 segment, the reverse primer being extended at the 5′ end by a modified sequence of the T7 promoter (5′-GGAAATTTAATACGACTCACTATA…-3′). The exact sequence of all primers can be provided upon request.
Synthetic RNAs corresponding to the eight full-length vRNAs or the four DVGs were in vitro transcribed (IVT) using 200 ng of gel-purified PCR product and the MEGAscript Kit (Thermo Fisher Scientific). RNAs were recovered after lithium chloride precipitation and quantified using the Qubit RNA HS Assay Kit (Thermo Fisher Scientific). The eight IVT full-length vRNAs were mixed at an equimolar ratio of 5 × 10−3 pmol each and one of the synthetic DVGs was added in a 1:1, 1:9, 1:99, 1:999 molar ratio, in a total volume of 50 µL. RT-PCR (for preamplification-based RNA-seq, or RT-PCR-seq) and cDNA synthesis (for preamplication-free RNA-seq, or RT-seq) were performed on each of these IVT RNA mixes.
RT-PCR for preamplification-based RNA-seq (RT-PCR-seq)
RT-PCR reactions were performed according to a protocol adapted from Watson et al. (2013). Briefly, 5 µL of vRNAs or 5 µL of IVT RNA mixes were amplified using the Superscript One-Step RT-PCR with Platinium Taq (Thermo Fisher Scientific) and primers complementary to the extremities conserved in all viral genomic segments (U12 and U13). PCR products were purified using the Nucleospin PCR Clean-up Kit (Machery Nagel) and quantified using the Quant-iT RNA Assay Kit (Thermo Fisher Scientific).
cDNA synthesis for preamplification-free RNA-seq (RT-seq)
Briefly, 30 µL of vRNAs or 30 µL of IVT RNA mixes were purified and concentrated to 10 µL using Agencourt RNAClean XP SPRI beads (Beckman Coulter) and subjected to first strand cDNA synthesis using the Superscript III First-Strand Synthesis System for RT-PCR Kit (Thermo Fisher Scientific) and a mixture of random hexamers and U12–U13 specific primers (see above). The second cDNA strand was synthetized using 5 U of E. coli RNase H, 40 U of E. coli DNA polymerase, and 10 U of E. coli DNA ligase for 2 h at 16°C (New England Biolabs). Double-stranded cDNAs were purified using Agencourt RNAClean XP SPRI beads (Beckman Coulter) and quantified using the Quant-iT RNA Assay Kit (Thermo Fisher Scientific).
Next-generation sequencing
The Illumina library construction and sequencing were performed by the P2M platform at Institut Pasteur. In brief, the Nextera XT DNA Library Preparation kit (Illumina) was used for library construction. Upon enzymatic fragmentation, a size selection was performed using Pippin Prep (Labtech) to retain fragments between 400 to 700 nt in length. Finally, the pooled libraries were sequenced on an Illumina NextSeq 500 instrument (150 nt paired end reads). The resulting fastq files were demultiplexed with the bcl2fastq Conversion Software v2.20 (Illumina).
Alignment and identification of DVGs
Fastq files were trimmed and aligned using BWA MEM, and sorted bam files were fed to the DG-seq pipeline using R v. 3.4.3 and the Rsamtools package (Bioconductor). Reads that were not aligned or did not pass quality control (default thresholds) were discarded. DG-seq script and example file are provided (Supplemental Files S1, S2).
Reads containing an SA tag were isolated, and the following information was used to classify DVGs: CIGAR (c1), SA CIGAR (c2), 0 × 10 flag (s1, indicating whether the alignment is the reverse complement of the sequence), and SA strand argument (s2). Obtained reads were divided into three categories:
If s1 = s2 and (c1, c2) = (MH, HM) or (MS, SM), simple deletion or insertion
If s1 ≠ s2 and (c1, c2) = (MH, MH) or (MS, MS), 5′ copy-back DVG
If s1 ≠ s2 and (c1, c2) = (HM, HM) or (SM, SM), 3′ copy-back DVG.
In addition, simple deletions, detected as c1 = MDM without SA tags, were added.
Breakpoint positions were computed accordingly and filtering was performed to avoid duplicate entries. Overlapping alignments were detected as potential uncertainty junctions and the leftmost breakpoint start was used, while reads with unaligned middle sequences were discarded.
Except for background noise computation, deletions of length <10 were discarded. Read counts n were normalized as 5/3 × n/N, where N denotes the mean of the 2% highest coverage values per position.
In Figures 4–7, data from Expt 1 are shown and analyzed. For the quantitative analyses performed in Figures 5 and 6, data points consisted of unique (breakpoint start, breakpoint end) pairs.
Uncertainties
Uncertainties were computed for each start i and end j positions (with j ≥ i + 1), using the reference sequence, giving rise to a matrix M. When the uncertainty computed by length of the overlapping sequences in the two alignments were in disagreement with the uncertainty computed using the reference sequence (from M), the latter was used.
To determine the null probability for random polymerase jumps, suppose the polymerase jumps from position a to position b (Fig. 6B). There is uncertainty ≥1 if (and only if) either positions a + 1 and b share the same nucleotide, or if positions a and b − 1 share the same nucleotide. In other words,
where Si is the nucleotide at position i in the reference sequence, Ui,j the uncertainty for a jump from i to j. Similarly,
where Si→j denotes (Si,…, Sj) (Fig. 6B). Therefore,
In other words, the uncertainty is ≥n if and only if, within a window of 2n around a, one can find a sequence of size n which is exactly similar to one centered on b − 1 of the same size. So the probability that U ≥ n is the probability qn of n consecutive successes in a sequence of 2n Bernouilli trials with a probability = 1/4 of winning (the odds that two random nucleotides are equal are 1/4). Feller (2008) showed that when n → ∞,
| F |
where p = 1/4, q = 1 − p, and x is the root of f(x) = 1 − x + qpnxn+1 that is not 1/p. This asymptotical approximation is extremely good, even for small n’s.
To determine the uncertainty distribution of random sequences, either formula (F) (using the function uniroot.all of package rootSolve to determine the roots of f), or all entries of matrix M, were used.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Aartjan te Velthuis, Stephen Cusack, and the Vignuzzi and Albert laboratories, especially Laura Levi, Enzo Poirier, Rasmus Henningsson, and Matthew Albert, for helpful discussions and advice. We thank Solenne Brun and Mélanie Albert (Centre National de Référence des Virus des Infections Respiratoires, Institut Pasteur, Paris, France) for their help with sample processing for RNA-seq. This work was supported by the LabEx IBEID (grant 10-LABX-0062) to Sylvie van der Werf and the ANR (grant ANR-18-CE11-0028) to Nadia Naffakh. This work was funded in part by the DARPA INTERCEPT program managed by Dr. Jim Gimlett, Dr. Brad Ringiesen, and Dr. Seth Cohen and administered through DARPA Cooperative Agreement #HR0011-17-2-0023 to Marco Vignuzzi (the content of the information does not necessarily reflect the position or the policy of the U.S. government, and no official endorsement should be inferred). J.B. was supported by a grant from the Ecole normale supérieure and by the école doctorale Frontières du vivant—programme Bettencourt.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.077529.120.
REFERENCES
- Alnaji FG, Holmes JR, Rendon G, Vera JC, Fields CJ, Martin BE, Brooke CB. 2019. Sequencing framework for the sensitive detection and precise mapping of defective interfering particle-associated deletions across influenza A and B viruses. J Virol 93: e00354–19 10.1128/JVI.00354-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum A, Sachidanandam R, García-Sastre A. 2010. Preference of RIG-I for short viral RNA molecules in infected cells revealed by next-generation sequencing. Proc Natl Acad Sci 107: 16303–16308. 10.1073/pnas.1005077107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauclair G, Mura M, Combredet C, Tangy F, Jouvenet N, Komarova AV. 2018. DI-tector: defective interfering viral genomes’ detector for next-generation sequencing data. RNA 24: 1285–1296. 10.1261/rna.066910.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosma TJ, Karagiannis K, Santana-Quintero L, Ilyushina N, Zagorodnyaya T, Petrovskaya S, Laassri M, Donnelly RP, Rubin S, Simonyan V, et al. 2019. Identification and quantification of defective virus genomes in high throughput sequencing data using DVG-profiler, a novel post-sequence alignment processing algorithm. PLoS One 14: e0216944 10.1371/journal.pone.0216944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmock NJ, Easton AJ. 2014. Defective interfering influenza virus RNAs: time to reevaluate their clinical potential as broad-spectrum antivirals? J Virol 88: 5217–5227. 10.1128/JVI.03193-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmock N, Easton A. 2015. Cloned defective interfering influenza RNA and a possible pan-specific treatment of respiratory virus diseases. Viruses 7: 3768–3788. 10.3390/v7072796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmock NJ, Easton AJ. 2017. Can defective interfering RNAs affect the live attenuated influenza vaccine? Lancet Infect Dis 17: 1234–1235. 10.1016/S1473-3099(17)30637-0 [DOI] [PubMed] [Google Scholar]
- Fancher KC, Hu W. 2011. Codon bias of influenza a viruses and their hosts. Am J Mol Biol 1: 174–182. 10.4236/ajmb.2011.13017 [DOI] [Google Scholar]
- Feller W. 2008. An introduction to probability theory and its applications, Vol. 1 Wiley, Hoboken, NJ. [Google Scholar]
- Fodor E, Devenish L, Engelhardt OG, Palese P, Brownlee GG, García-Sastre A. 1999. Rescue of influenza A virus from recombinant DNA. J Virol 73: 9679–9682. 10.1128/JVI.73.11.9679-9682.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fodor E, Mingay LJ, Crow M, Deng T, Brownlee GG. 2003. A single amino acid mutation in the PA subunit of the influenza virus RNA polymerase promotes the generation of defective interfering RNAs. J Virol 77: 5017–5020. 10.1128/JVI.77.8.5017-5020.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frensing T. 2015. Defective interfering viruses and their impact on vaccines and viral vectors. Biotechnol J 10: 681–689. 10.1002/biot.201400429 [DOI] [PubMed] [Google Scholar]
- Genoyer E, López CB. 2019. The impact of defective viruses on infection and immunity. Annu Rev Virol 6: 547–566. 10.1146/annurev-virology-092818-015652 [DOI] [PubMed] [Google Scholar]
- Higuchi R, Krummel B, Saiki RK. 1988. A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res 16: 7351–7367. 10.1093/nar/16.15.7351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings PA, Finch JT, Winter G, Robertson JS. 1983. Does the higher order structure of the influenza virus ribonucleoprotein guide sequence rearrangements in influenza viral RNA? Cell 34: 619–627. 10.1016/0092-8674(83)90394-X [DOI] [PubMed] [Google Scholar]
- Lui W-Y, Yuen C-K, Li C, Wong WM, Lui P-Y, Lin C-H, Chan K-H, Zhao H, Chen H, To KKW, et al. 2019. SMRT sequencing revealed the diversity and characteristics of defective interfering RNAs in influenza A (H7N9) virus infection. Emerg Microbes Infect 8: 662–674. 10.1080/22221751.2019.1611346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukarska M, Fournier G, Pflug A, Resa-Infante P, Reich S, Naffakh N, Cusack S. 2017. Structural basis of an essential interaction between influenza polymerase and Pol II CTD. Nature 541: 117–121. 10.1038/nature20594 [DOI] [PubMed] [Google Scholar]
- Nayak DP, Chambers TM, Akkina RK. 1985. Defective-interfering (DI) RNAs of influenza viruses: origin, structure, expression, and interference. In Current topics in microbiology and immunology (ed. Cooper M, et al. ), pp. 103–151. Springer, Berlin/Heidelberg. [DOI] [PubMed] [Google Scholar]
- Odagiri T, Tominaga K, Tobita K, Ohta S. 1994. An amino acid change in the non-structural NS2 protein of an influenza A virus mutant is responsible for the generation of defective interfering (DI) particles by amplifying DI RNAs and suppressing complementary RNA synthesis. J Gen Virol 75: 43–53. 10.1099/0022-1317-75-1-43 [DOI] [PubMed] [Google Scholar]
- Perez-Cidoncha M, Killip MJ, Oliveros JC, Asensio VJ, Fernandez Y, Bengoechea JA, Randall RE, Ortin J. 2014. An unbiased genetic screen reveals the polygenic nature of the influenza virus anti-interferon response. J Virol 88: 4632–4646. 10.1128/JVI.00014-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier EZ, Goic B, Tomé-Poderti L, Frangeul L, Boussier J, Gausson V, Blanc H, Vallet T, Loyd H, Levi LI, et al. 2018. Dicer-2-dependent generation of viral DNA from defective genomes of RNA viruses modulates antiviral immunity in insects. Cell Host Microbes 23: 353–365.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Routh A, Johnson JE. 2014. Discovery of functional genomic motifs in viruses with ViReMa—a Virus Recombination Mapper—for analysis of next-generation sequencing data. Nucleic Acids Res 42: e11 10.1093/nar/gkt916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saira K, Lin X, DePasse JV, Halpin R, Twaddle A, Stockwell T, Angus B, Cozzi-Lepri A, Delfino M, Dugan V, et al. 2013. Sequence analysis of in vivo defective interfering-like RNA of influenza A H1N1 pandemic virus. J Virol 87: 8064–8074. 10.1128/JVI.00240-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng Z, Liu R, Yu J, Ran Z, Newkirk SJ, An W, Li F, Wang D. 2018. Identification and characterization of viral defective RNA genomes in influenza B virus. J Gen Virol 99: 475–488. 10.1099/jgv.0.001018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Kim EJ, Felt SA, Taylor LJ, Agarwal D, Grant GR, López CB. 2019. A specific sequence in the genome of respiratory syncytial virus regulates the generation of copy-back defective viral genomes. PLoS Pathog 15: e1007707 10.1371/journal.ppat.1007707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapia K, Kim WK, Sun Y, Mercado-López X, Dunay E, Wise M, Adu M, López CB. 2013. Defective viral genomes arising in vivo provide critical danger signals for the triggering of lung antiviral immunity. PLoS Pathog 9: e1003703 10.1371/journal.ppat.1003703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasilijevic J, Zamarreño N, Oliveros JC, Rodriguez-Frandsen A, Gómez G, Rodriguez G, Pérez-Ruiz M, Rey S, Barba I, Pozo F, et al. 2017. Reduced accumulation of defective viral genomes contributes to severe outcome in influenza virus infected patients. PLoS Pathog 13: e1006650 10.1371/journal.ppat.1006650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignuzzi M, López CB. 2019. Defective viral genomes are key drivers of the virus–host interaction. Nat Microbiol 4: 1075–1087. 10.1038/s41564-019-0465-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Magnus P. 1954. Incomplete forms of influenza virus. In Advances in virus research (ed. Smith KM, Lauffer MA), Vol. 2, pp. 59–79. Academic Press, MA. [DOI] [PubMed] [Google Scholar]
- Watson SJ, Welkers MRA, Depledge DP, Coulter E, Breuer JM, de Jong MD, Kellam P. 2013. Viral population analysis and minority-variant detection using short read next-generation sequencing. Philos Trans R Soc B Biol Sci 368: 20120205 10.1098/rstb.2012.0205 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.