

Abstract
It is only after recent advances in cryo-electron microscopy that it is now possible to describe at high-resolution structures of large macromolecules that do not crystalize. Purified 30S subunits interconvert between an “active” and “inactive” conformation. The active conformation was described by crystallography in the early 2000s, but the structure of the inactive form at high resolution remains unsolved. Here we used cryo-electron microscopy to obtain the structure of the inactive conformation of the 30S subunit to 3.6 Å resolution and study its motions. In the inactive conformation, an alternative base-pairing of three nucleotides causes the region of helix 44, forming the decoding center to adopt an unlatched conformation and the 3′ end of the 16S rRNA positions similarly to the mRNA during translation. Incubation of inactive 30S subunits at 42°C reverts these structural changes. The air–water interface to which ribosome subunits are exposed during sample preparation also peel off some ribosomal proteins. Extended exposures to low magnesium concentrations make the ribosomal particles more susceptible to the air–water interface causing the unfolding of large rRNA structural domains. Overall, this study provides new insights about the conformational space explored by the 30S ribosomal subunit when the ribosomal particles are free in solution.
Keywords: ribosome structure, 30S subunit, cryo-electron microscopy
INTRODUCTION
Ribosomes in bacteria undergo constant conformational changes that are essential for the translation process. These include from small-scale base flipping events at the decoding center to much larger-scale motions of ribosomal subunit domains induced by mRNA, tRNA, and translation factor binding (Frank 2017). Similarly, the ribosome assembly process involves constant conformational changes as the rRNA folds and ribosomal proteins are incorporated into the assembling particle (Mulder et al. 2010; Sashital et al. 2014; Razi et al. 2017a). Until recently, high-resolution structural information about these conformational states was solely contributed through X-ray crystallography. Consequently, only those states that could be stabilized in a crystal lattice were accessible providing only a reduced breath of the conformational heterogeneity existing in these processes and potentially masking important details about local and global conformational dynamics.
Today, structural biology is in the midst of a “resolution revolution” (Kuhlbrandt 2014). Because of continuous advances, cryo-electron microscopy (cryo-EM) can now routinely contribute high-resolution models of large macromolecular machines with dynamic composition and conformations that have remained impervious to crystallization (Cheng 2015; Nogales and Scheres 2015; Cheng et al. 2017). In the context of the ribosome, these cryo-EM models are illuminating new relevant transition steps in the protein translation (Hussain et al. 2016) and ribosome assembly processes (Ni et al. 2016; Nikolay et al. 2018; Razi et al. 2019; Seffouh et al. 2019).
Mainly, in the study of the ribosome assembly process, structural biologists have widely used genetic approaches to trigger the accumulation of assembly intermediates (Stokes and Brown 2015; Razi et al. 2017a). The essence of this approach consists of creating single deletion or depletion strains for one of the assembly factors to disable or slow down the ribosome biogenesis process (Daigle and Brown 2004). Invariably, these methods produce a heterogeneous mixture of immature ribosomal particles from which it is not possible to grow crystals. However, structural characterization of these assembly intermediates using cryo-EM combined with image classification approaches has shown to be a powerful approach to identify the role of protein factors in assisting specific steps in the ribosome assembly process (Jomaa et al. 2011; Guo et al. 2013; Leong et al. 2013; Razi et al. 2019).
Inferring the role of assembly factors from the structural deficiencies observed in the assembling particles accumulating in the null or depleted cells requires the existence of a common standard or reference structure that is used in the comparative analysis. Previous publications have typically used the mature 30S subunit structure obtained by crystallographic approaches as the reference structure (Wimberly et al. 2000). However, the mature 30S subunit not constrained on a crystal lattice exists in more than one conformation. Five decades ago, Elson and colleagues (Zamir et al. 1969, 1971) reported that purified 30S subunits readily interconvert between “active” and “inactive” conformations. Later, Noller's group determined, using chemical probing, that transition between both states involves structural changes in the neck and decoding center regions of the 16S rRNA (Moazed et al. 1986). This conformational variability of the mature 30S subunit is not restricted to the bacterial ribosome. The eukaryotic 40S subunit also seems to sample multiple conformations (Swiatkowska et al. 2012). More recently, it was found using RNA SHAPE (selective 2-hydroxyl acylation analyzed by primer extension) that in exponentially growing Escherichia coli cells, 16S rRNA mainly adopts the inactive conformation in free mature 30S subunits and the active conformation in translating 70S ribosomes (McGinnis and Weeks 2014; McGinnis et al. 2015). The reactivity patterns on the 30S subunit associated with the 50S subunits are entirely consistent with the RNA secondary structure exhibited by the 30S subunit in the crystal structure, suggesting that both approaches describe the same structure. However, the high-resolution structure of the inactive conformation observed in the free 30S subunits has never been obtained by X-ray crystallography or cryo-EM.
To visualize the 3D structure of the inactive conformation of the 30S subunit, and potentially other alternative conformations of the mature 30S subunits adopted outside the constraints of a crystal lattice, we exposed purified 30S subunits to buffer conditions that recall those in ribosome purification approaches (Daigle and Brown 2004; Jomaa et al. 2011). Cryo-EM revealed that the mature 30S subunits in solution adopt a variety of conformations. Magnesium concentration in the purification buffers had a large effect. Exposure to low magnesium concentrations switched the decoding center of the 30S subunit to a drastically different conformation from that observed in the crystal structure. Incubation of the purified 30S subunits at 42°C induced the decoding center to switch back to the canonical conformation. Our experiments also showed how the air–water interface to which the 30S subunits are exposed during the vitrification process in cryo-EM could also induce structural variability by causing the loss of r-proteins. Extended exposure to low magnesium concentration increased the ribosomal particles’ susceptibility to the air–water interface, causing complete unfolding of the head domain and partial unfolding of the platform domain. Studying the motion of these ribosomal particles revealed that the most predominant motions exhibited by the head domain in the free 30S subunits are similar to those observed in the 70S ribosome during protein translation.
RESULTS
High-resolution cryo-EM structure of the 30S subunit inactive conformation
In actively growing E. coli cells, 2%–5% of the existing 30S subunits remain dissociated without forming 70S ribosomes (Leong et al. 2013; Thurlow et al. 2016). Consequently, 30S subunit purification protocols typically include a step that exposes ribosomes temporarily to buffers containing 1–2 mM concentration of magnesium ions (low magnesium). This condition induces the ribosome's dissociation into its two integrating subunits, the 30S and the 50S subunits. This is mainly triggered by a structural change in the 30S subunit termed “inactivation,” as this conformational change interferes with tRNA binding in its P site (Zamir et al. 1969; Moazed et al. 1986).
To obtain the cryo-EM structure of the inactive conformation of the 30S subunit at high resolution, we purified 30S subunits from actively growing E. coli cells using a protocol that exposed the ribosomes to 1.1 mM magnesium acetate. However, at the end of the purification, the buffer in the fractions containing purified 30S subunits was exchanged and the concentration of magnesium acetate raised to 10 mM. The obtained sample (30S-inactivated-high-Mg2+) was then imaged by cryo-EM.
Using image classification approaches, we found that the purified 30S-inactivated-high-Mg2+ subunits existed in two distinct conformations that we called class A and B (Fig. 1A). The distribution of particles between these two classes was 79% and 21%, respectively. Using these particle populations, we calculated a 3.6-Å resolution cryo-EM map for class A and a 4.5-Å resolution map for class B (Supplemental Fig. S1). The structures of the body (5′ domain), platform (central domain), and head domains (3′ major domain) in both structures were identical to the canonical structure of the 30S subunit obtained by X-ray crystallography (Fig. 1A; Wimberly et al. 2000). However, the decoding region located at the convergency point of all these three domains presented important differences in both maps (Fig. 1B). In the canonical structure, helix 44 runs from the bottom of the body to the lower part of the head. The upper region of this helix near the platform domain is involved in the decoding process and is also critical in creating the subunit interface with the 50S subunit. In the map for class A, the upper domain of helix 44 adopts an alternative conformation and is not latched to the decoding center as described by the crystal structure. Instead, this entire section of the helix protrudes from the surface of the 30S subunit and distorts the interface with the 50S subunit. In class B, the whole helix 44 seemed to adopt a flexible conformation. The lower part of this helix showed a well-defined density, but the middle and upper regions exhibited a highly fragmented density, indicating this region is highly flexible.
FIGURE 1.

Cryo-EM structure of the 30S-inactivated-high-Mg2+ particle. (A) Front (top row) and back view (bottom row) of the cryo-EM maps obtained for the two subpopulations found for the 30S-inactivated-high-Mg2+ particle. The maps are shown side-by-side with the 30S subunit structure obtained by X-ray crystallography (30S canonical structure). This structure was obtained by generating a density map from PDB file 4V4Q and low-pass filtering this structure to 4 Å. The rRNA is displayed in light gray, the r-proteins in green, and helix 44 in goldenrod orange. The r-proteins uS2, uS7, and bS21 for which representative densities do not appear in the cryo-EM maps of the 30S-inactivated-high-Mg2+ particle are shown in blue in the map of the 30S subunit obtained by X-ray crystallography. These proteins and other landmarks of the 30S subunit are labeled. (B) Zoomed-in view of the decoding region of the cryo-EM maps obtained for the two subpopulations found for the 30S-inactivated-high-Mg2+ particle and the structure of the 30S subunit obtained by X-ray crystallography. The area visualized in this panel is indicated as a frame in panel A.
To quantitatively measure the structural differences in the 30S-inactivated-high-Mg2+ class A, we produced a molecular model from the cryo-EM map and subsequently calculated a temperature map to measure the deviation of this structure from the conformation of the 16S rRNA in the canonical structure (Fig. 2A). The body and platform regions were highly similar and the rRNA in these structural motifs mostly overlapped with the canonical structure. In contrast, the position of the head and upper domain of helix 44 and contacting region in helix 28 diverged significantly. The head domain was tilted backward by 16°, opening up the decoding region (Fig. 2B). The upper domain of helix 44 and bottom part of helix 28 adopted a drastically different conformation that was stabilized by another arrangement in the base paring of the rRNA (Fig. 2C). The transition between both conformations involves nucleotides 1532–1534 at the 3′ end of the 16S rRNA. These 3 nt are not forming any base-pairing in the conventional structure. However, in class A, they approach helix 28 and unfold its bottom part (region formed by nucleotides 1391–1396 and 921–925) and base pair with nucleotides 921–923 in that region. This transition also causes the partial unfolding of the top of helix 44 formed in the canonical structure by nucleotides 1397–1407 and 1494–1503, as well as the positioning of the 3′ end of the 16S rRNA (distal to nucleotide 1534) in a conformation similar to that adopted by mRNA during translation. In this conformational transition, helix 45 slightly shifts in position but remains folded (Supplemental Movie 1).
FIGURE 2.

Molecular model of the 30S-inactivated-high-Mg2+ particle. (A) Temperature map of the 30S-inactivated-high-Mg2+ class A. The rRNA is colored according to the r.m.s.d. deviation (Å) with respect to the structure of the 30S subunit obtained by X-ray crystallography (PDB ID 4V4Q). (B) Top view of the head of the cryo-EM map of the 30S-inactivated-high-Mg2+ class A. The positions of helices 33, 39, and 42 in this structure (navy blue) and in the crystal structure of the 30S subunit (orange) are shown to illustrate the backward tilting of the head by 16° in the structure of the 30S-inactivated-high-Mg2+ class A. (C) Secondary (top panels) and tertiary (bottom panels) structures of helices 28, 44, and 45 of the 16S rRNA in the 30S subunit structure obtained by X-ray crystallography and in the molecular model derived from the cryo-EM map of the 30S-inactivated-high-Mg2+ class A. The nucleotides 1532–1534 at the 3′ end of the 16S rRNA involved in the conformational transition between both structures are highlighted in red. The regions of helices 44 and 28 that become unfolded during the conformational transition are colored in yellow in the canonical 3D structure of the 30S subunit. The molecular model of the cryo-EM map of the 30S-inactivated-high-Mg2+ class A is shown overlapped with the density of the obtained cryo-EM map. In the case of the structure of the 30S subunit obtained by X-ray crystallography, a density map was generated from the atomic coordinates and overlapped with the molecular model.
Taken together, these structures indicate that 30S subunits purified through approaches that expose them to low magnesium concentration switch from the conventional structure shown by X-ray crystallography to an inactive conformation that exhibits drastic structural differences in the decoding region. The inactive conformation is stabilized by an alternative base-pairing of the nucleotides in the 3′ end of the 16S rRNA molecule.
Exposure of the 30S subunits to the air–water interface in the cryo-EM grid causes the loss of r-protein uS2
Previously reported cryo-EM structures of mature 30S subunits (Datta et al. 2007; Razi et al. 2019) showed either fragmented or absent densities for r-proteins uS7 and bS21, suggesting that they dissociated or are intrinsically flexible when the particle is not constrained in a crystal lattice. Consistently, the densities corresponding to these two r-proteins in the cryo-EM maps obtained for the 30S-inactivated-high-Mg2+ subunit class A and B were also not present (Fig. 1A). Surprisingly, we also found the density representing uS2 to be completely absent from our cryo-EM maps. In the 30S subunit, uS2 binds in the solvent face (convex face) of the subunit, stably anchoring its two domains to the 16S rRNA and typically is fully visible in previously obtained X-ray (Wimberly et al. 2000; Schureck et al. 2016) and cryo-EM (Razi et al. 2017b) structures.
In cryo-EM, to preserve the specimen in their hydrated state, one spread the sample in a thin layer of buffer solution supported in the cryo-EM grid right before plunging the grid into liquid ethane to freeze the liquid layer into vitreous ice. In the vitrification device used in our experiments, the time that elapses between the blotting of the grid to form the thin layer and the vitrification is typically 1 sec. In this time, ribosomal particles collide with the air–water interface between 100 and 1000 times (Noble et al. 2018). These interactions have the potential to cause damages in the specimen.
To investigate whether the ribosomal's repeated interaction with the air–water interface was causing the loss of uS2, we repeated the imaging of the 30S-inactivated-high-Mg2+ particles by adding an extra thin layer of continuous carbon to the grids (Fig. 3A). We hypothesized that their exposure to the air–water interface would be reduced by adsorbing the particles to the support film. We collected a cryo-EM data set from these grids, and particle images were subjected to a similar image classification workflow. We found that particles were present mainly as one class with helix 44 in an identical conformation to the 30S-inactivated-high-Mg2+ class A particles structure. The cryo-EM map obtained from these particles (30S-inactivated-carbon-high-Mg2+) was refined to 3.8 Å (Fig. 3B; Supplemental Fig. S1). More importantly, the density representing uS2 was visible in the cryo-EM map.
FIGURE 3.

Cryo-EM structure of the 30S-inactivated-high-Mg2+ particle in grids containing a continuous layer of carbon. (A) Two representative electron micrographs containing 30S-inactivated-high-Mg2+ particles. The micrographs were obtained from EM grids without (left panel) and with (right panel) an extra layer of a continuous carbon. The inset shows the power spectra from each micrograph. The presence of a constant layer carbon makes the Thon rings in the power spectra more prominent and the background of the micrograph more prominent. (B) Front and back view of the cryo-EM map obtained for the 30S-inactivated-high-Mg2+ particle from grids containing a continuous carbon layer. The rRNA is shown in light gray, and the r-proteins are shown in green except uS2 that is colored in blue. (C) Temperature map of the 30S-inactivated-high-Mg2+ molecular model obtained from grids containing a continuous carbon layer. The rRNA is colored according to the r.m.s.d. deviation (Å) with respect to the structure 30S-inactivated-high-Mg2+ class A obtained from grids without a continuous carbon layer.
To quantitatively assess the conformational differences between the 16S rRNA in the structure obtained from grids having or lacking the extra carbon layer, we produced a molecular model from the cryo-EM map obtained from grids containing the extra carbon layer. We then calculated a temperature map (Fig. 3C) with respect to the 30S-inactivated-high-Mg2+ class A particles that were imaged without an extra layer of carbon on the grids. We found that the conformation of the 16S rRNA in both structures was very similar, including the alternative folding adopted by the upper domain of helix 44 and the 3′ end of the rRNA molecule.
These results revealed that the air–water interface caused uS2 to fall off. They also demonstrated that the noncanonical folding observed for helix 44 and the 3′ end of the 16S rRNA was induced by the exposure of the ribosomal particles to low magnesium concentrations, and it occurs independently of the presence of uS2.
The transition from the inactive to the active state reverts the decoding center to the standard conformation
Elson and colleagues (Zamir et al. 1969, 1971) described that incubation of inactive 30S subunits at 42°C in the presence of 10–20 mM Mg2+ reverts the structural changes induced by low magnesium concentration to an “active” conformation, in which the 30S subunits regain their ability to bind tRNA.
To structurally describe the conformational changes that this incubation triggers, purified 30S-inactivated-high-Mg2+ ribosomal particles were incubated at 42°C and imaged by cryo-EM. These particles were called 30S-activated-high-Mg2+. Image classification revealed that the activation treatment had transformed all the 30S subunits to adopt a single conformation (Fig. 4A) that closely resembled the structure of the 30S subunit as described by X-ray crystallography (Fig. 1; Wimberly et al. 2000). The structure refined to a resolution of 3.6 Å (Supplemental Fig. S1) and allowed us to derive a molecular model from the cryo-EM map (Fig. 4B). We then used this model to calculate a temperature map that compared this structure with the crystallographic structure. We observed that helix 44 and rRNA forming the decoding center closely overlapped, indicating that the 42°C incubation treatment reverted the conformation of the functional domain induced by low magnesium concentration to that observed in the 70S ribosome and the 30S subunit structure produced by X-ray crystallography (Fig. 4C).
FIGURE 4.
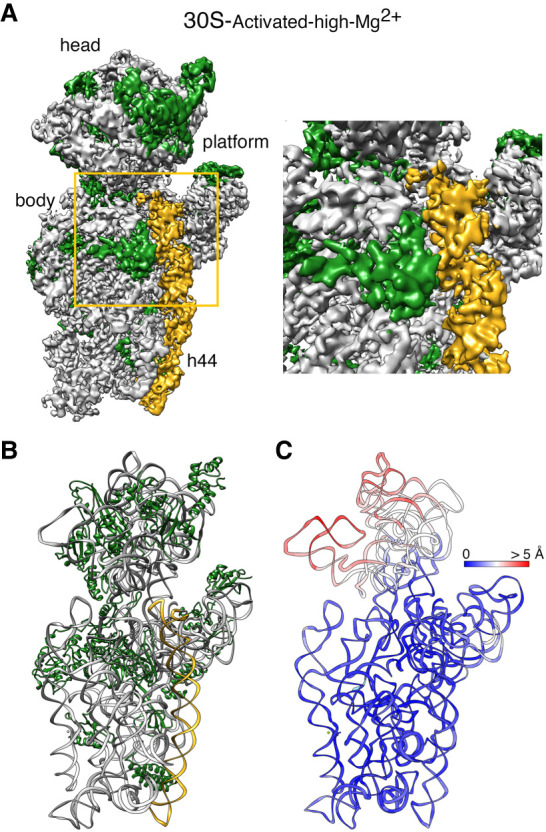
Cryo-EM structure of the 30S-activated-high-Mg2+ particle. (A) Front view (left panel) of the cryo-EM map obtained for the 30S-activated-high-Mg2+ particle. The framed area is shown as a zoomed-in view in the right panel. The main landmarks of the 30S subunit are labeled. The rRNA is shown in light gray, and the r-proteins are shown in green. Helix 44 is shown in goldenrod orange. (B) Molecular model of the 30S-activated-high-Mg2+ particle. The rRNA and the r-proteins are colored as in panel A. (C) Temperature map of the 30S-activated-high-Mg2+ molecular model. The rRNA is colored according to the r.m.s.d. deviation (Å) with respect to the structure of the 30S subunit obtained by X-ray crystallography (PDB ID 4V4Q).
Extended exposure to low magnesium concentration makes the 30S subunit more susceptible to the air–water interface
Next, we inquired about the effect of extended exposure of the 30S subunits to a low concentration of magnesium ions. The purified 30S subunits were maintained in buffer containing 1.1 mM magnesium acetate before imaging them by cryo-EM. Similar classification approaches to those followed in the previous samples revealed that the 30S ribosomal subunits coexisted under low magnesium concentrations in two distinct conformations (Fig. 5A). One of the classes represented 64% of the population, and these particles generated a cryo-EM map that refined to 3.4 Å resolution (30S-inactivated-low-Mg2+ class A) (Supplemental Fig. S2). The remaining particles generated a different cryo-EM map that refined to 3.9 Å resolution (30S-inactivated-low-Mg2+ class B) (Supplemental Fig. S2). Both classes significantly diverged from the structure considered as a mature 30S subunit and contained structural features only previously observed in structures of immature 30S subunits (Jomaa et al. 2011; Guo et al. 2013; Leong et al. 2013; Razi et al. 2019). Density representing the entirety of helix 44 was missing in the two cryo-EM maps (Fig. 5A).
FIGURE 5.

Cryo-EM structure of 30S-inactivated-low-Mg2+ particle. (A) Front (top panels) and back (bottom panels) views of the two conformers, class A and B of the 30S-inactivated-low-Mg2+ particle. The rRNA is shown in light gray, and the r-proteins are shown in green. Helix 44 is shown in goldenrod orange. These structures are shown side-by-side with the 30S subunit structure obtained by X-ray crystallography (PDB file 4V4Q). In this structure, the rRNA is displayed in light gray, the r-proteins in green, and helix 44 in goldenrod orange. The r-proteins uS2, bS6, uS7, uS11, uS18, and bS21 for which a representative density does not appear in the cryo-EM maps of the 30S-inactivated-low-Mg2+ particle are indicated using a color different than green. These proteins and other landmarks of the 30S subunit are labeled. (B) Zoomed-in view of the decoding and platform region of the cryo-EM maps obtained for the 30S-inactivated-low-Mg2+ class A and B using the same color coding as in (A). (C) Overlap of helix 44 and platform region of the molecular model derived from the cryo-EM structure of the 30S-inactivated-low-Mg2+ class A and the corresponding region from the structure of the 30S subunit obtained by X-ray crystallography. Parts of the structure present in the cryo-EM structure of the 30S-inactivated-low-Mg2+ class A are displayed in navy blue and red. The atomic model of the X-ray structure with all the elements of the complete 30S subunit structure is displayed in light gray. (D) Temperature maps of the 30S-inactivated-low-Mg2+ class A and B molecular models. The rRNA is colored according to the r.m.s.d. deviation (Å) with respect to the structure of the 30S subunit obtained by X-ray crystallography (PDB ID 4V4Q). (E) Front (left panel) and back (right panel) views of the 30S-inactivated-low-Mg2+ particle imaged in grids with an extra layer of continuous carbon. The r-proteins and rRNA helices missing in the same sample imaged in grids without the extra thin layer of carbon but present in this cryo-EM map are colored as in the structure of the 30S subunit obtained by X-ray crystallography shown in panel A.
Similarly, helices 23 and 24 in the platform region exhibited highly fragmented densities. The r-proteins bound to this region (bS6, uS11, bS21, and bS18) were also not observed in the cryo-EM map (Fig. 5B,C). The EM grids used to image this sample did not contain an additional layer of continuous carbon and, consequently, uS2 was missing from the cryo-EM maps of both classes (Fig. 5A, lower panel). The main difference observed between class A and B was in the head domain, which was completely missing in the cryo-EM map for class B. Density for all regions of this domain was present in the map obtained for class A, except for r-protein uS7 that is similar to other structures or the 30S subunit obtained in solution, maybe adopting a flexible conformation (Fig. 5A, top panel).
The molecular model derived from the 30S-inactivated-low-Mg2+ class A, and B cryo-EM maps were used to calculate a temperature map (Fig. 5D) to compare the conformation of the 16S rRNA in these structures with that of the crystallographic structure (Wimberly et al. 2000). The body and platform regions present in the cryo-EM map of the 30S-inactivated-low-Mg2+ class A showed a close overlap. However, we found that the head domain was tilted backward in class A, similarly to what we observed in the structures derived from the 30S-inactivated-high-Mg2+ particles.
We also imaged this sample in grids containing an extra thin layer of continuous carbon. This experiment allowed us to determine whether the observed depletions of r-proteins (uS2, bS6, uS11, bS21, and bS18) and unfolding of rRNA domains in these structures was caused by the exposure of the particles to the low magnesium concentrations or because the buffer conditions made the particles easily damaged by the air–water interface. Three-dimensional classification revealed the existence of only one class of particles in the images (30S-inactivated-carbon-low-Mg2+). From these particles, we generated a cryo-EM map that refined to 3.2 Å resolution (Fig. 5E; Supplemental Fig. S2). The cryo-EM map resembled the 30S-inactivated-low-Mg2+ class A structure, as the entirety density for helix 44 was also missing. However, the platform did not exhibit any severe distortion with helices 23 and 24 and r-proteins bS6, uS11, and bS18 were fully represented in the cryo-EM map. Besides, r-protein uS2 was also fully described. Importantly, we did not observe any particles that resemble the 30S-inactivated-low-Mg2+ class B, which lacked the entire head domain.
These structures suggested that continued exposure of the mature 30S subunit to low magnesium concentrations partially destabilizes large structural motifs of the ribosomal subunit, including the central (platform), 3′ major (head), and 3′ minor (helix 44) domain and makes them more susceptible to unfolding when exposed to the air–water interface.
Motions exhibited by the free 30S subunits
We noticed that in all the cryo-EM maps obtained for the 30S subunit, density was clear and mostly complete for the body and platform domain but was fuzzier and fragmented in the head region (Fig. 6). We interpreted the partial fragmentation of these densities as an indication of the motions that the head region experiences when the 30S subunits are in solution. Using multibody refinement (Nakane et al. 2018), we investigated these motions. To this end, we split the cryo-EM maps into three bodies corresponding to three of the major domains of the 30S subunit: body, platform, and head. After multibody refinement, we compared the resulting maps obtained for the head domains with those from the consensus refinement. Visual inspection of their central section (Fig. 6) showed a small improvement, particularly in the region furthest away from the map's center.
FIGURE 6.
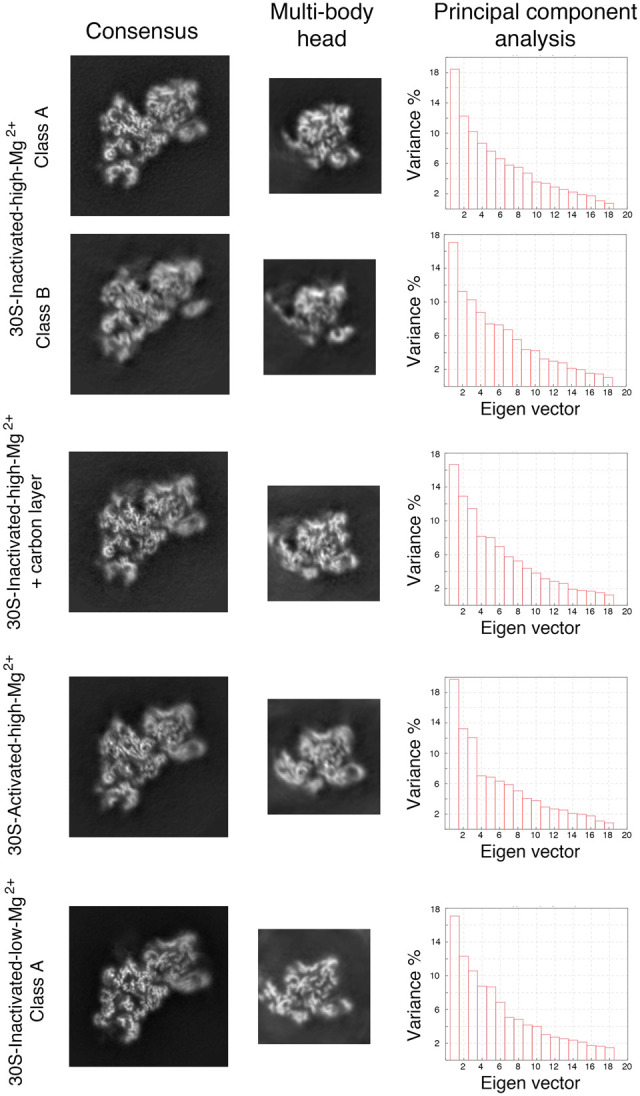
Multibody refinement analysis of the cryo-EM structures. The data set for each type of 30S particle was analyzed using multibody refinement to visualize their motions. The left and middle panels show, respectively, the central sections of the cryo-EM maps of the obtained 30S structures after the consensus refinement and of the head after multibody refinement. The high-resolution features of the density maps are apparent in the body region. Still, they are slightly blurred in the head domain in the central sections of the maps obtained through consensus refinement. High-resolution features of the head become more apparent in the maps of this region obtained through multibody refinement. The right panels show the principal component analysis of the different structures using the multibody refinement routine in RELION 3.0. This analysis indicated that between 38% and 43% of the variance is due to the movements of the head with respect to the body and platform.
More importantly, this approach performed a principal component analysis of the variance in the rotations and translations of the three bodies and allowed us to generate movies describing the most important motions in the different 30S subunit populations. In all cases, this analysis revealed that between 38% and 43% of the variance in the rotations and translations of the three bodies is explained by the first three eigenvectors (Fig. 6). Movies of the reconstructed body densities repositioned along these three eigenvectors revealed that they correspond to motions of the head with respect to the body and platform. In one of these motions, the head rotates around an axis longitudinal to the particle's longest dimension. The other two motions are back and forward tilting movements of the head with respect to the body. Supplemental Movies 2–6 show the main motion (represented by the first eigenvector) for each one of the main 30S structures obtained in this study. Overall, these results indicated that the head domain's most predominant motions in the free 30S subunits are similar to those observed in the 70S ribosome during protein translation (Noeske and Cate 2012).
DISCUSSION
The transition of the decoding center in the 30S subunit from the active to the inactive state was one of the first conformational rearrangements discovered in ribosomes (Zamir et al. 1969, 1971; Moazed et al. 1986). Chemical probing experiments at the time revealed that the adoption of the inactive conformation involved large-scale structural changes in the neck region and the fraction of helix 44 engaged in the formation of the decoding center. A precise structure could not be derived at the time from the data. However, there is a remarkable agreement between the cryo-EM structure of the inactive conformation of the 30S subunit described here and the probing data from the Noller group (Moazed et al. 1986). In particular, the decreased reactivity of nucleotides 1533–1536 suggested by the probing data correlates well with our finding of these nucleotides being involved in the base-pairing with nucleotides 921–923. Similarly, the enhanced reactivity found for nucleotides 924–926 and 1394–1397 suggested these nucleotides become unpaired, which is also observed in the cryo-EM structure.
More recently, RNA SHAPE experiments chemically probed the RNA structure of the 30S subunits in exponentially growing E. coli cells (McGinnis and Weeks 2014; McGinnis et al. 2015). An important contribution of these experiments was in revealing that the inactive state of the 30S subunit is biologically relevant in regulating ribosome function. Based on the SHAPE data, this group also suggested an alternative base-pairing in the 16S rRNA forming the decoding center in the inactive conformation. In particular, nucleotides 1402–1408, which form an irregular helix at the top of helix 44 pairing with nucleotides 1492–1500 in the conventional structure, undergo a register shift and pair with positions 921–927 within helix 28 and nucleotides 1390–1401 form an unpaired loop. The obtained high-resolution cryo-EM map of the inactive conformation of the 30S subunit presented here diverges from the one proposed in the RNA SHAPE experiments (McGinnis et al. 2015). In the cryo-EM map obtained for the “inactive” conformation, nucleotides 1532–1534 at the 3′ end of the 16S rRNA base paired with nucleotides 921–923 in helix 28, causing the unfolding of the bottom of this helix and the top of helix 44. The newly formed base pairs force the 3′ end of the 16S rRNA in a conformation that resembles the conformation adopted by mRNA during translation.
Comparison of the previous chemical probing data (Moazed et al. 1986; McGinnis et al. 2015) with our cryo-EM results shows the clear advantage of cryo-EM for the study of the alternative conformations adopted by ribosomal particles. Whereas, in some cases, multiple structures can be potentially derived from chemical probing data, the direct observation of the structure using cryo-EM typically allows establishing unambiguously the precise nature of the conformational rearrangement that the ribosomal particle undergoes. We found this to be a likely explanation between the disagreement of the structure proposed from the SHAPE data (McGinnis et al. 2015) and that shown by cryo-EM here.
The functional importance of the “inactive” conformation of the 30S subunit has been highlighted in previous studies (Myasnikov et al. 2009; Karbstein 2013). Multiple translation initiation factors and RNases affecting 30S subunit turnover bind at sites overlapping with helices 28 and 44, suggesting that the “active–inactive” transition observed by cryo-EM could govern the accessibility of these factors and regulates translation initiation and ribosome quality control. In addition, the cryo-EM structure of the 30S-inactivated-high-Mg2+ class A provides a functional explanation for the inactivity of the ribosomal particle in this conformation. The 3′ end of 16S rRNA of the 30S subunit containing the anti-Shine–Dalgarno sequence is typically accessible in the platform of the 30S subunit and base pairs with the Shine–Dalgarno sequence in the 5′ end of the mRNA. However, in the inactive conformation, the entire 3′ end of 16S rRNA adopts a conformation similar to that adopted by mRNA during translation. In this position, the anti-Shine–Dalgarno sequence is occluded and unable to recognize the Shine–Dalgarno sequence during initiation.
Our study also characterizes the motions that the different 30S subunit subpopulations exhibit when they are in solution and not constrained in a crystal lattice. X-ray crystallographic and cryo-EM structures from many laboratories have described the large-scale conformational changes that the ribosome undergoes during protein synthesis (Noeske and Cate 2012). For example, the movement of the tRNAs from the classical A/A and P/P configurations to the hybrid A/P and P/E states is driven by a rigid body rotation of the small subunit with respect to the large subunit by 10° around an axis perpendicular to the subunit interface (ratchet-like motion) (Frank and Agrawal 2000). This movement is also accompanied by a simultaneous swiveling of the 30S subunit head domain in the direction of the E site (Spahn et al. 2004). Swiveling motions of the head also accompany the process of termination and ribosome recycling (Dunkle et al. 2011; Yokoyama et al. 2012). The head domain also exhibits other motions linked to different physiological processes, such as hibernation. The binding of ribosome modulation factor or hibernation promoting factor triggers displacement of the head away from the central protuberance (backward tilting), promoting the formation of 100S ribosome dimers (Polikanov et al. 2012). All these motions have been observed in the context of the 70S ribosome.
In our study, cryo-EM combined with multibody refinement processing approaches (Nakane et al. 2018) allowed us to investigate the most prominent motions of the different conformational subpopulations identified for free 30S subunits. Overall, we found that the free 30S subunits also exhibit the motions of swiveling and backward tilting that the head domain of the 30S subunit undergoes in the context of the 70S ribosome.
An important motivation for this study was to provide a more extensive comparative reference for those studies on the 30S subunit assembly that use single deletion or depletion strains of certain assembly factors. Drawing meaningful conclusions from the heterogeneous mixture of ribosomal particles that these strains accumulate is only possible when a comprehensive description of the conformations adopted by the mature subunit is available. Only then is it possible to separate from the observed pool of structures, those truly representing immature assembly states and derive information about the function of specific assembly factors.
Current sample preparation techniques in cryo-EM suffer from a fundamental problem. Macromolecules in solution drift during vitrification and get exposed to the air–water interface at the top and bottom of the thin films formed upon blotting. This exposure is damaging to the macromolecular assemblies and can pull their components apart and destroy them. Our results show that even for specimens considered resilient such as the ribosome or the 30S subunit, current cryo-EM sample preparation methods still cause damages. In particular, we found that r-protein uS2 is especially sensitive to the air–water interface and, in our experiment, caused the complete removal of the protein during the vitrification process. This problem is most likely exacerbated in ribosome assembly intermediates where multiple r-proteins may not be bound as tightly as in the mature subunit. Consequently, similarly to some previous studies (Jomaa et al. 2011, 2014; Guo et al. 2013; Leong et al. 2013; Li et al. 2013; Razi et al. 2017b, 2019), it may be prudent that future studies analyzing ribosome assembly intermediates by cryo-EM continue to use grids containing an extra thin layer of continuous carbon to decrease the exposure of the particles to the air–water interface. Significantly, the structures presented here will contribute to reinterpret previous cryo-EM work performed in grids without an additional layer of continuous carbon (Lopez-Alonso et al. 2017) and help to differentiate bona fide structural defects observed on heterogeneous mixtures of 30S ribosomal particles from those caused by the sample preparation technique.
Overall, this study provides new insights into the alternative conformations and motions adopted by the 30S ribosomal subunit. One of them, named by Elson and colleagues (Zamir et al. 1969, 1971) as the “inactive” conformation, was discovered over five decades ago. It has been only now, with the recent improvements in electron microscopes and direct electron detector cameras, that we have described these structures at high resolution.
MATERIALS AND METHODS
Cell strains
We used the Escherichia coli K-12 (BW25113) strain from the Keio collection (Baba et al. 2006) to produce the purified 30S subunits used in this study.
Purification of 30S ribosomal particles
To obtain purified 30S subunits, we first produced a 30 mL saturated overnight culture of E. coli K-12 strain (BW25113) in LB media and this culture was used to inoculate a 3 L culture of LB media. Cells were grown at 37°C with shaking at 225 rpm in an Innova 44 incubator shaker (New Brunswick) until they reached an OD600 of 0.6. Harvesting of the cells was done by centrifugation at 3700g for 15 min and the obtained pellets were chilled to 4°C and all the subsequent steps were done at this temperature. These pellets were resuspended in 7 mL of buffer A (20 mM Tris-HCl at pH 7.5, 10 mM magnesium acetate, 100 mM NH4Cl, 0.5 mM EDTA, 3 mM 2-mercaptoethanol, and a protease inhibitor mixture (cOmplete Protease Inhibitor Mixture Tablets; Roche) and DNase I (Roche). Cell lysis was done by passing the cell suspension through a French pressure cell at 1400 kg/cm2 three consecutive times, and the cell debris was cleared by spinning the lysate at 59,000g for 30 min. The supernatant was layered over a sucrose cushion of equal volume composed of 37.6% sucrose in buffer B (20 mM Tris-HCl pH 7.5, 10 mM magnesium acetate, 500 mM NH4Cl, 0.5 mM EDTA, and 3 mM 2-mecaptoethanol), and then spun down for 4.5 h at 321,000g. The pellet containing the ribosomal particles was resuspended in buffer C containing 10 mM Tris-HCl pH 7.5, 10 mM magnesium acetate, 500 mM NH4Cl, 0.5 mM EDTA, and 3 mM 2-mecaptoethanol and then spun for 16 h at 100,000g. This produced a pellet containing washed ribosomal particles that was resuspended in buffer F (10 mM Tris-HCl, pH 7.5, 1.1 mM magnesium acetate, 60 mM NH4Cl, 0.5 mM EDTA, and 2 mM 2-mercaptoethanol). To separate the fraction containing the 30S subunits, approximately 120 A260 units of resuspended crude ribosomes were then applied to 34 mL of 10%–30% (wt/vol) sucrose gradients prepared with buffer F. The gradient was centrifuged for 16 h at 40,000g on a Beckman Coulter SW 32 Ti rotor and fractionated using a Brandel fractionator apparatus and an AKTA Prime FPLC system (GE Healthcare). Fractions containing the 30S subunits were selected and pooled together based on the UV absorbance at A254. Subsequently, they were spun down for another 4.5 h at 321,000g on a Beckman 70 Ti rotor. The pellet was resuspended differently according to the final conditions under which we intended to study the 30S subunit. The 30S subunits under “low Mg2+ conditions” (30S-inactivated-low-Mg2+) were resuspended back in buffer F, whereas the 30S subunits under “high Mg2+ conditions” (30S-inactivated-high-Mg2+ and 30S-inactivated-carbon-high-Mg2+) were resuspended in buffer E (10 mM Tris-HCl, pH 7.5, 10 mM magnesium acetate, 60 mM NH4Cl, and 3 mM 2-mecaptoethanol). “Activated” 30S subunits (30S-activated-high-Mg2+) were resuspended in modified buffer E with a different concentration of magnesium acetate (20 mM) and stored at −80°C. To perform the activation, these 30S subunits were heated at 42°C for 20 min and then diluted by mixing equal volumes of the sample with buffer E not containing any magnesium acetate.
Cryo-electron microscopy
Sample vitrification was performed using a Vitrobot (Thermo Fisher Scientific). For all samples, a volume of 3.6 µL of the diluted sample was applied to holey carbon grids (c-flat CF-2/2-2C-T) previously washed in chloroform for 2 h and treated with glow discharged in air at 5 mA for 15 sec right before the sample was applied. For the 30S-inactivated-carbon-high-Mg2+ and 30S-inactivated-carbon-low-Mg2+ samples, we used holey carbon grids containing an extra layer of continuous thin carbon (5–10 nm). The concentration of ribosomal particles in the solution applied to the grid varied between 200 and 300 nM depending on the sample. In the Vitrobot, each grid was blotted once for 3 sec and with a blot force +1 before they were plunged into liquid ethane. The Vitrobot was set at 25°C and 100% relative humidity.
Most of the data acquisition was performed using EPU software at FEMR-McGill using a Titan Krios microscope at 300 kV equipped with a Falcon II direct electron detector (Thermo Fisher Scientific). Movies for the 30S-inactivated-high-Mg2+, 30S-inactivated-carbon-high-Mg2+, 30S-activated-high-Mg2+, and 30S-inactivated-low-Mg2+ data sets were collected with a total dose of 50, 52, 52, and 50 e−/Å2, respectively. All data sets except for the 30S-inactivated-carbon-high-Mg2+ particles were collected as movies with seven frames acquired in 1 sec exposure at a magnification of 75,000×, producing images with a calibrated pixel size of 1.073 Å. Movies for the 30S-inactivated-carbon-high-Mg2+ particle were collected in the same manner but with 30 frames. The nominal defocus range used to collect all these data sets was between −1.25 and −2.75 µm. The data set for the and 30S-inactivated-carbon-low-Mg2+ sample was also collected in the same Titan Krios microscope but using SerialEM software (Schorb et al. 2019) and a Gatan K3 direct electron detector equipped with a Quantum LS imaging filter. Images were collected as 36-frames movies using 3.6 sec exposures in counting mode using a total dose per movie of 60 e−/A2 at a nominal magnification of 81,000× corresponding to a calibrated pixel size of 1.09 Å. Defocus range for this data set was −1 to −2.25 µm.
Image processing
Collected movies were corrected for beam-induced motion using RELION's implementation of the MotionCor2 algorithm (Zheng et al. 2017; Zivanov et al. 2018). Correction for the contrast transfer function (CTF) was done using the Gctf program (Zhang 2016). Subsequent processing steps were done using RELION 3.0 (Zivanov et al. 2018). Particle images were selected and extracted from the micrographs using auto-picking and subsequently subjected to one or more cycles of reference-free 2D classification to remove false-positive and damaged particles. This process produced clean data sets for the 30S-inactivated-high-Mg2+, 30S-inactivated-carbon-high-Mg2+, 30S-activated-high-Mg2+, 30S-inactivated-low-Mg2+, and 30S-inactivated-carbon-low-Mg2+ samples comprised of 565,255, 334,903, 407,623, 658,065, and 264,418 particles, respectively. These data sets were used for 3D classifications to separate the different conformational subpopulations existing in each sample. The initial 3D reference used for these classifications was either a 60-Å low-pass filtered map of the mature 30S subunit created from 4V4Q.pdb (Schuwirth et al. 2005) using the Xmipp program (de la Rosa-Trevin et al. 2013) or the intermediate cryo-EM maps obtained during classification. No mask was used for the 3D classifications. All 2D classification and 3D classification jobs were performed using particle images binned by 4. In each data set, maps obtained in the classification steps were visually inspected in Chimera (Pettersen et al. 2004), and particles assigned to maps representing the same conformation were pooled together and used for standard 3D auto-refinement. A soft-mask was applied in all 3D auto-refinements. These masks were created with the “relion_mask_create” command extending the binary mask by four pixels and creating a soft edge with a width of four pixels. The initial threshold for binarization of the mask varied depending on the structure. As initial map for the refinement procedures we used either a 60-Å low-pass filtered map of the mature 30S subunit created from 4V4Q.pdb (Schuwirth et al. 2005) using the Xmipp program (de la Rosa-Trevin et al. 2013) or the cryo-EM maps obtained during classification after they were rescaled back to full-size. Suitable classes were subsequently subjected to Bayesian polishing to correct for per-particle beam-induced motions, followed by CTF refinement for per-particle defocus, per-micrograph astigmatism, beam tilt, optical aberrations, and a final round of 3D auto-refinement (Zivanov et al. 2019, 2020). Sharpening of the final cryo-EM maps and the local analysis was done with RELION (Zivanov et al. 2018).
Isotropic map normalization
The cryo-EM maps for 30S-inactivated-high-Mg2+ class A and 30S-activated-high-Mg2+ were slightly affected by directional resolution anisotropy caused by the presence of preferential specimen orientations. In these cases, the underrepresented macromolecular views result on low map amplitudes along these directions causing a stretching in the reconstructed volume. In this work, we compensate the map stretching caused by preferential specimen orientations by an isotropic amplitude map normalization in Fourier space. In this approach, we normalize map amplitudes at a particular resolution along all possible map directions for resolutions higher than 9–10 Å. At these resolution ranges, respective Fourier components encode the information of secondary structures presented in the map. We hypothesized that these secondary structures should be oriented approximately random-wise. Then, the amplitudes of map Fourier components should be approximately isotropic for resolutions higher than 9–10 Å. Our map restoration method follows these steps: (i) The Fourier transform of the cryo-EM map is obtained; (ii) Starting from 9 to 10 Å, a shell of the map Fourier transform is extracted; (iii) The map amplitudes within the extracted shell and the respective q = 75% percentile value is obtained; (iv) Map amplitudes at the extracted shell are modified so their value is set to q; (v) A new shell at higher resolution is extracted, and steps (i) to (iv) are repeated; (vi) This process is iterated until the shell at highest resolution (Nyquist resolution) is processed; (vii) The inverse Fourier transform of the isotropic amplitude normalized map is computed obtaining a new corrected map. This map restoration approach transforms map Fourier amplitudes only, without modifying map phases.
Multibody refinement and motion analysis
We used the RELION 3.0 implementation (Nakane et al. 2018) to perform the multibody refinement and motion analysis. The same approach was followed for all 30S subunit populations. The consensus cryo-EM map obtained for each class was divided into three bodies corresponding to the three major domains of the subunit: body, platform, and head. The masks for the corresponding bodies were made using a 25-Å low-pass filtered version of the consensus map. We use available atomic models of the 30S subunit (PDB ID 4V4Q) to determine the boundaries between the bodies. By extending the binary map by 10 pixels and placing a soft edge with 10 pixels, all three bodies overlapped with each other. The multibody refinement job was run with downsized particle images with a box size of 218 and a pixel size of 1.496 Å. The standard deviation of the Gaussian prior on the rotations was set to 10° for all three bodies, and the standard deviations on the body translations were all set to 2 pixels. Based on the domain architecture of the 30S subunit, the head was set to rotate with respect to platform and body. Multibody refinement was started using an initial angular sampling rate of 1.8°, an initial offset range of 3 pixels and an initial offset step of 0.25 pixels.
Map analysis and atomic model building
Before starting to build the molecular models for each structure using the obtained cryo-EM maps from RELION, the connectivity of the densities of the cryo-EM maps was improved using automatic sharpening as implemented in the PHENIX suite (Adams et al. 2010). Model building of all maps started by fitting the 4V4Q.pdb (Schuwirth et al. 2005) X-ray structure of the 30S subunit into the obtained cryo-EM maps using the rigid-body docking tools in Chimera (Pettersen et al. 2004). The molecular models were then built through multiple rounds of manual model building in Coot (Emsley and Cowtan 2004; Emsley et al. 2010) and real-space refinement using Phenix (Adams et al. 2010).
Panels for figures were prepared using the PyMOL program (The PyMOL Molecular Graphics System, Version 2.3.3, Schrodinger), UCSF Chimera, and Chimera X. Figures were assembled using Photoshop (Adobe). Secondary structures of RNA were produced with DSSR (http://x3dna.org) (Lu and Olson 2008) and Varna (http://varna.lri.fr) (Darty et al. 2009).
DATA DEPOSITION
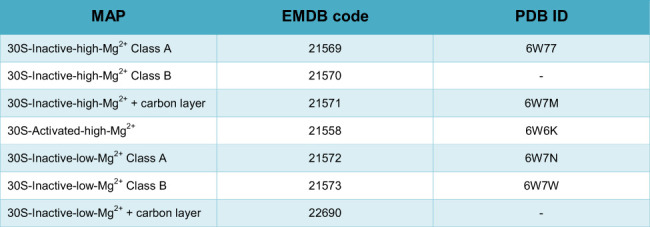
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Kelly Sears, Mike Strauss, and other staff members of the Facility for Electron Microscopy Research (FEMR) at McGill University for help with microscope operation and data collection. We acknowledge Clara Ortega for assistance with graphic design. This work was supported by grants from the Canadian Institutes of Health Research (PJT-153044) to J.O. Titan Krios cryo-EM data were collected at FEMR (McGill). FEMR is supported by the Canadian Foundation for Innovation, Quebec government, and McGill University. The funders had no role in study design, data collection, and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.075846.120.
REFERENCES
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66: 213–221. 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2: 2006.0008 10.1038/msb4100050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y. 2015. Single-particle cryo-EM at crystallographic resolution. Cell 161: 450–457. 10.1016/j.cell.2015.03.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Glaeser RM, Nogales E. 2017. How cryo-EM became so hot. Cell 171: 1229–1231. 10.1016/j.cell.2017.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle DM, Brown ED. 2004. Studies of the interaction of Escherichia coli YjeQ with the ribosome in vitro. J Bacteriol 186: 1381–1387. 10.1128/JB.186.5.1381-1387.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darty K, Denise A, Ponty Y. 2009. VARNA: interactive drawing and editing of the RNA secondary structure. Bioinformatics 25: 1974–1975. 10.1093/bioinformatics/btp250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta PP, Wilson DN, Kawazoe M, Swami NK, Kaminishi T, Sharma MR, Booth TM, Takemoto C, Fucini P, Yokoyama S, et al. 2007. Structural aspects of RbfA action during small ribosomal subunit assembly. Mol Cell 28: 434–445. 10.1016/j.molcel.2007.08.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Rosa-Trevin JM, Oton J, Marabini R, Zaldivar A, Vargas J, Carazo JM, Sorzano CO. 2013. Xmipp 3.0: an improved software suite for image processing in electron microscopy. J Struct Biol 184: 321–328. 10.1016/j.jsb.2013.09.015 [DOI] [PubMed] [Google Scholar]
- Dunkle JA, Wang L, Feldman MB, Pulk A, Chen VB, Kapral GJ, Noeske J, Richardson JS, Blanchard SC, Cate JH. 2011. Structures of the bacterial ribosome in classical and hybrid states of tRNA binding. Science 332: 981–984. 10.1126/science.1202692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60: 2126–2132. 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66: 486–501. 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank J. 2017. The translation elongation cycle-capturing multiple states by cryo-electron microscopy. Philos Trans R Soc Lond B Biol Sci 372: 20160180 10.1098/rstb.2016.0180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank J, Agrawal RK. 2000. A ratchet-like inter-subunit reorganization of the ribosome during translocation. Nature 406: 318–322. 10.1038/35018597 [DOI] [PubMed] [Google Scholar]
- Guo Q, Goto S, Chen Y, Feng B, Xu Y, Muto A, Himeno H, Deng H, Lei J, Gao N. 2013. Dissecting the in vivo assembly of the 30S ribosomal subunit reveals the role of RimM and general features of the assembly process. Nucleic Acids Res 41: 2609–2620. 10.1093/nar/gks1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain T, Llacer JL, Wimberly BT, Kieft JS, Ramakrishnan V. 2016. Large-scale movements of IF3 and tRNA during bacterial translation initiation. Cell 167: 133–1 44.e13 10.1016/j.cell.2016.08.074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jomaa A, Stewart G, Martin-Benito J, Zielke R, Campbell TL, Maddock JR, Brown ED, Ortega J. 2011. Understanding ribosome assembly: the structure of in vivo assembled immature 30S subunits revealed by cryo-electron microscopy. RNA 17: 697–709. 10.1261/rna.2509811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jomaa A, Jain N, Davis JH, Williamson JR, Britton RA, Ortega J. 2014. Functional domains of the 50S subunit mature late in the assembly process. Nucleic Acids Res 42: 3419–3435. 10.1093/nar/gkt1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbstein K. 2013. Quality control mechanisms during ribosome maturation. Trends Cell Biol 23: 242–250. 10.1016/j.tcb.2013.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlbrandt W. 2014. Biochemistry. The resolution revolution. Science 343: 1443–1444. 10.1126/science.1251652 [DOI] [PubMed] [Google Scholar]
- Leong V, Kent M, Jomaa A, Ortega J. 2013. Escherichia coli rimM and yjeQ null strains accumulate immature 30S subunits of similar structure and protein complement. RNA 19: 789–802. 10.1261/rna.037523.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Chen Y, Guo Q, Zhang Y, Yuan Y, Ma C, Deng H, Lei J, Gao N. 2013. Cryo-EM structures of the late-stage assembly intermediates of the bacterial 50S ribosomal subunit. Nucleic Acids Res 41: 7073–7083. 10.1093/nar/gkt423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Alonso JP, Kaminishi T, Kikuchi T, Hirata Y, Iturrioz I, Dhimole N, Schedlbauer A, Hase Y, Goto S, Kurita D, et al. 2017. RsgA couples the maturation state of the 30S ribosomal decoding center to activation of its GTPase pocket. Nucleic Acids Res 45: 6945–6959. 10.1093/nar/gkx324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu XJ, Olson WK. 2008. 3DNA: a versatile, integrated software system for the analysis, rebuilding and visualization of three-dimensional nucleic-acid structures. Nat Protoc 3: 1213–1227. 10.1038/nprot.2008.104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinnis JL, Weeks KM. 2014. Ribosome RNA assembly intermediates visualized in living cells. Biochemistry 53: 3237–3247. 10.1021/bi500198b [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinnis JL, Liu Q, Lavender CA, Devaraj A, McClory SP, Fredrick K, Weeks KM. 2015. In-cell SHAPE reveals that free 30S ribosome subunits are in the inactive state. Proc Natl Acad Sci 112: 2425–2430. 10.1073/pnas.1411514112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moazed D, Van Stolk BJ, Douthwaite S, Noller HF. 1986. Interconversion of active and inactive 30 S ribosomal subunits is accompanied by a conformational change in the decoding region of 16 S rRNA. J Mol Biol 191: 483–493. 10.1016/0022-2836(86)90143-9 [DOI] [PubMed] [Google Scholar]
- Mulder AM, Yoshioka C, Beck AH, Bunner AE, Milligan RA, Potter CS, Carragher B, Williamson JR. 2010. Visualizing ribosome biogenesis: parallel assembly pathways for the 30S subunit. Science 330: 673–677. 10.1126/science.1193220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myasnikov AG, Simonetti A, Marzi S, Klaholz BP. 2009. Structure-function insights into prokaryotic and eukaryotic translation initiation. Curr Opin Struct Biol 19: 300–309. 10.1016/j.sbi.2009.04.010 [DOI] [PubMed] [Google Scholar]
- Nakane T, Kimanius D, Lindahl E, Scheres SH. 2018. Characterisation of molecular motions in cryo-EM single-particle data by multi-body refinement in RELION. eLife 7: e36861 10.7554/eLife.36861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni X, Davis JH, Jain N, Razi A, Benlekbir S, McArthur AG, Rubinstein JL, Britton RA, Williamson JR, Ortega J. 2016. YphC and YsxC GTPases assist the maturation of the central protuberance, GTPase associated region and functional core of the 50S ribosomal subunit. Nucleic Acids Res 44: 8442–8455. 10.1093/nar/gkw678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolay R, Hilal T, Qin B, Mielke T, Burger J, Loerke J, Textoris-Taube K, Nierhaus KH, Spahn CMT. 2018. Structural visualization of the formation and activation of the 50S ribosomal subunit during in vitro reconstitution. Mol Cell 70: 881–893.e3. 10.1016/j.molcel.2018.05.003 [DOI] [PubMed] [Google Scholar]
- Noble AJ, Wei H, Dandey VP, Zhang Z, Tan YZ, Potter CS, Carragher B. 2018. Reducing effects of particle adsorption to the air–water interface in cryo-EM. Nat Methods 15: 793–795. 10.1038/s41592-018-0139-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noeske J, Cate JH. 2012. Structural basis for protein synthesis: snapshots of the ribosome in motion. Curr Opin Struct Biol 22: 743–749. 10.1016/j.sbi.2012.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogales E, Scheres SH. 2015. Cryo-EM: a unique tool for the visualization of macromolecular complexity. Mol Cell 58: 677–689. 10.1016/j.molcel.2015.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25: 1605–1612. 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- Polikanov YS, Blaha GM, Steitz TA. 2012. How hibernation factors RMF, HPF, and YfiA turn off protein synthesis. Science 336: 915–918. 10.1126/science.1218538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razi A, Britton RA, Ortega J. 2017a. The impact of recent improvements in cryo-electron microscopy technology on the understanding of bacterial ribosome assembly. Nucleic Acids Res 45: 1027–1040. 10.1093/nar/gkw1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razi A, Guarne A, Ortega J. 2017b. The cryo-EM structure of YjeQ bound to the 30S subunit suggests a fidelity checkpoint function for this protein in ribosome assembly. Proc Natl Acad Sci 114: E3396–E3403. 10.1073/pnas.1618016114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razi A, Davis JH, Hao Y, Jahagirdar D, Thurlow B, Basu K, Jain N, Gomez-Blanco J, Britton RA, Vargas J, et al. 2019. Role of era in assembly and homeostasis of the ribosomal small subunit. Nucleic Acids Res 47: 8301–8317. 10.1093/nar/gkz571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sashital DG, Greeman CA, Lyumkis D, Potter CS, Carragher B, Williamson JR. 2014. A combined quantitative mass spectrometry and electron microscopy analysis of ribosomal 30S subunit assembly in E. coli. eLife 3: e04491 10.7554/eLife.04491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schorb M, Haberbosch I, Hagen WJH, Schwab Y, Mastronarde DN. 2019. Software tools for automated transmission electron microscopy. Nat Methods 16: 471–477. 10.1038/s41592-019-0396-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schureck MA, Maehigashi T, Miles SJ, Marquez J, Dunham CM. 2016. mRNA bound to the 30S subunit is a HigB toxin substrate. RNA 22: 1261–1270. 10.1261/rna.056218.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuwirth BS, Borovinskaya MA, Hau CW, Zhang W, Vila-Sanjurjo A, Holton JM, Cate JH. 2005. Structures of the bacterial ribosome at 3.5 A resolution. Science 310: 827–834. 10.1126/science.1117230 [DOI] [PubMed] [Google Scholar]
- Seffouh A, Jain N, Jahagirdar D, Basu K, Razi A, Ni X, Guarne A, Britton RA, Ortega J. 2019. Structural consequences of the interaction of RbgA with a 50S ribosomal subunit assembly intermediate. Nucleic Acids Res 47: 10414–10425. 10.1093/nar/gkz770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spahn CM, Gomez-Lorenzo MG, Grassucci RA, Jorgensen R, Andersen GR, Beckmann R, Penczek PA, Ballesta JP, Frank J. 2004. Domain movements of elongation factor eEF2 and the eukaryotic 80S ribosome facilitate tRNA translocation. EMBO J 23: 1008–1019. 10.1038/sj.emboj.7600102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes JM, Brown ED. 2015. Chemical modulators of ribosome biogenesis as biological probes. Nat Chem Biol 11: 924–932. 10.1038/nchembio.1957 [DOI] [PubMed] [Google Scholar]
- Swiatkowska A, Wlotzka W, Tuck A, Barrass JD, Beggs JD, Tollervey D. 2012. Kinetic analysis of pre-ribosome structure in vivo. RNA 18: 2187–2200. 10.1261/rna.034751.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurlow B, Davis JH, Leong V, T FM, Williamson JR, Ortega J. 2016. Binding properties of YjeQ (RsgA), RbfA, RimM and Era to assembly intermediates of the 30S subunit. Nucleic Acids Res 44: 9918–9932. 10.1093/nar/gkw613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimberly BT, Brodersen DE, Clemons WM Jr, Morgan-Warren RJ, Carter AP, Vonrhein C, Hartsch T, Ramakrishnan V. 2000. Structure of the 30S ribosomal subunit. Nature 407: 327–339. 10.1038/35030006 [DOI] [PubMed] [Google Scholar]
- Yokoyama T, Shaikh TR, Iwakura N, Kaji H, Kaji A, Agrawal RK. 2012. Structural insights into initial and intermediate steps of the ribosome-recycling process. EMBO J 31: 1836–1846. 10.1038/emboj.2012.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamir A, Miskin R, Elson D. 1969. Interconversions between inactive and active forms of ribosomal subunits. FEBS Lett 3: 85–88. 10.1016/0014-5793(69)80103-1 [DOI] [PubMed] [Google Scholar]
- Zamir A, Miskin R, Elson D. 1971. Inactivation and reactivation of ribosomal subunits: amino acyl-transfer RNA binding activity of the 30 s subunit of Escherichia coli. J Mol Biol 60: 347–364. 10.1016/0022-2836(71)90299-3 [DOI] [PubMed] [Google Scholar]
- Zhang K. 2016. Gctf: real-time CTF determination and correction. J Struct Biol 193: 1–12. 10.1016/j.jsb.2015.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, Agard DA. 2017. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods 14: 331–332. 10.1038/nmeth.4193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, Scheres SH. 2018. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7: e42166 10.7554/eLife.42166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Scheres SHW. 2019. A Bayesian approach to beam-induced motion correction in cryo-EM single-particle analysis. IUCrJ 6: 5–17. 10.1107/S205225251801463X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Scheres SHW. 2020. Estimation of high-order aberrations and anisotropic magnification from cryo-EM data sets in RELION-3.1. IUCrJ 7: 253–267. 10.1107/S2052252520000081 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.