

Abstract
Peptidyl Arginine Deiminase 4 (PAD4/PADI4) is a posttranslational modification enzyme that converts protein arginine or mono-methylarginine to citrulline. The PAD4-mediated hypercitrullination reaction in neutrophils causes the release of nuclear chromatin to form a chromatin network termed Neutrophil Extracellular Traps (NETs). NETs were first described as antimicrobial fibers that bind and kill bacteria. However, it is not known whether PAD4 can mediate the release of chromatin DNA into the extracellular space of cancer cells. Here, we report that murine breast cancer 4T1 cells expressing high levels of PADI4 can release Cancer Extracellular Chromatin Networks (CECNs) in vitro and in vivo. Deletion of Padi4 using CRISPR/Cas9 abolished CECN formation in 4T1 cells. Padi4 deletion from 4T1 cells also reduced the rate of tumor growth in an allograft model, and decreased lung metastasis by 4T1 breast cancers. DNase I treatment, which degrades extracellular DNA including CECNs, also reduced breast to lung metastasis of Padi4 wild type 4T1 cells in allograft experiments in the Padi4 knockout mice. We further demonstrated that DNase I treatment in this mouse model did not alter circulating tumor cells but decreased metastasis through steps after intravasation. Taken together, our genetic studies show that PAD4 plays a cell autonomous role in cancer metastasis, thus revealing a novel strategy for preventing cancer metastasis by inhibiting cancer cell endogenous PAD4.
Keywords: PAD4, Hypercitrullination, Cancer extracellular chromatin network, Breast cancer, Lung metastasis
Graphical Abstract
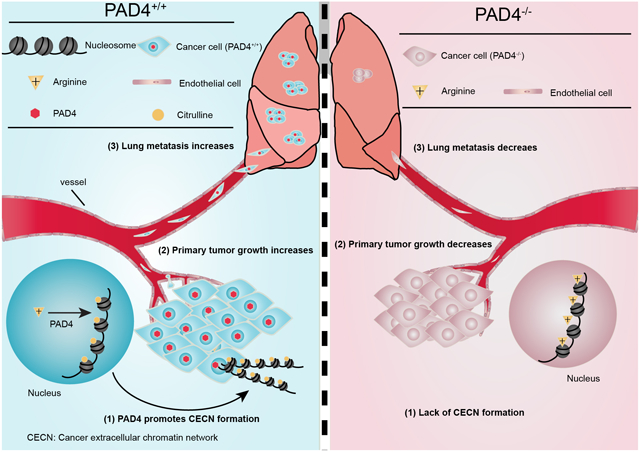
Introduction
Breast cancer is the most common cancer and the second leading cause of cancer deaths in women in the United States (1). Along with the aggressive growth of primary tumor mass, the development of distant metastases is a major cause of death from breast cancer. Due to the heterogeneity and lack of well-defined molecular targets, patients with metastatic breast cancer have a poor 5-year survival rate and finding a method to limit metastasis remains a great challenge.
Neutrophils are the predominant circulating leukocytes and can promote metastasis through different mechanisms (2–5). Recently, a novel structure formed by neutrophils, termed Neutrophil Extracellular Traps (NETs), was found to play a role in promoting metastasis (6–8). NETs were first discovered as a defense mechanism utilized by neutrophils to immobilize and kill invading microbes (9). During the past several years, a link between excessive NETs and autoimmunity has been proposed (10). NETs also form during fibrosis, ischemic stroke, preeclampsia, thrombosis, cancers and other autoimmune diseases (11–14). In the context of tumorigenesis, tumors secrete chemokines such as CXCL1 and GCSF to recruit and prime neutrophils to form NETs (6,15,16). In turn, NETs facilitate cancer metastasis by forming Circulating Tumor Cell (CTC) clusters, enhancing tumor cell adhesion to the endothelium, and potentially increasing tumor cell extravasation (7,17,18). These studies link NETs to the pathomechanism of metastatic cancer in the context of systemic inflammation and highlight the importance of developing a therapy for inhibiting NETs.
Peptidyl Arginine Deiminase 4 (PAD4 or PADI4)-catalyzed histone hypercitrullination mediates chromatin decondensation thereby leading to the formation of NETs (19,20). PAD4 is a neutrophil-enriched nuclear enzyme that converts arginine and mono-methylarginine on histones to citrulline in a calcium-dependent manner (21,22). Extracellular Trap (ET) structures are also formed by other myeloid cells like macrophages and eosinophils, and overexpression of PAD4 is sufficient to form NET-like structures in non-hematopoietic cells such as osteosarcoma U2OS cells that normally do not form ETs (20,23–25). These findings prompt us to hypothesize that PAD4-mediated hypercitrullination is able to drive ET formation, regardless of cell type.
Pathology studies detected significant expression of PAD4 in many tumor tissues derived from the colon, duodenum, esophagus, fallopian tube, gall gladder, lung, ovary, parotid, pancreas, prostate, rectum, small intestine, stomach, thyroid and uterus (26,27). Because PAD4 is also expressed in cancer cells, we investigated whether PAD4 might induce extrusion of chromatin in cancer cells and play a role in cancer progression or metastasis. Here, we report the formation of CECN both in vitro and in vivo in a PAD4-dependent manner using the murine breast cancer 4T1 model. We found that cancer cell endogenous PAD4 plays a significant role in metastasis. In cancer cells without PAD4 expression, tumor metastasis to the lung was significantly decreased. Our results support that PAD4 in cancer cells offers a new target for prevention, diagnosis, and treatment of cancer metastasis.
Materials and Methods
Cell culture and transfections
4T1 and 67NR cells were obtained from Dr. Andrea Mastro (The Pennsylvania State University, University Park, PA). Cell lines were expanded in our lab and stored in liquid nitrogen to ensure that cells used for experiments were passaged less than three times. No further genomic authentication was performed but cell lines were tested biannually for identity by appearance and growth curve analysis and validated to be mycoplasma free with PCR mycoplasma detection kit (TaKaRa) according to the manufacturer’s instruction. To establish a stable Padi4 knockout in 4T1, cells were transfected with Padi4 CRISPR/Cas9 knockout plasmid pSpCas9(BB)-2A-Puro (Addgene) using lipofectamine 2000 reagent (Invitrogen) per manufacturer’s instructions. Two different gRNAs of Padi4 were designed and purchased from IDT: AAGGCGCGGTGATCCACGTG (gRNA 1) and AAGGGCTACACAACCTTCGG (gRNA 2). gRNA 1 targets exon 1 and gRNA 2 targets exon 2. Transfected cells were selected using puromycin. Stable Padi4 knockout monoclones were identified from single-cell colonies and Padi4 knockout was determined after propagation. Genomic amplicons of the target region were subcloned into a plasmid for transformation and four individual colonies from each knockout were genotyped by sequencing. The Padi4 CRISPR-Ex1–1-C11 clone is from gRNA1 and Padi4 CRISPR-Ex2–6-E2 is from gRNA2 screening. For Padi4 knockout rescue experiments, PAD4 was overexpressed in Padi4 CRISPR cells by transient transfection with pSG5-hPAD4. Cells were then treated with 4 μM A23187 for 8 h.
Animals
BALB/c and Rag2−/−; γc−/− mice were purchased from the Jackson Laboratory. Padi4 knockout mice were originally generated in the C57BL/6 background by our laboratory (20) and backcrossed by Dr. Denisa Wagner (Harvard Medical School, Boston, MA) to BALB/c mice. All procedures were approved by the Institutional Animal Care and Use Committee and were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals.
Protein extraction and Western blot
Western blotting using the rabbit α-PAD4 (custom), mouse α-α tubulin (Sigma), rabbit α-histone H3 (Abcam) and rabbit α-H3Cit (Abcam) antibodies was performed essentially as described previously (22,23). The custom α-PAD4 was a rabbit polyclonal antibody made against a human GST-PAD4 fusion protein, and has a similar reactivity to the conserved human and mouse PAD4 protein (20,28).
Immunostaining and microscopy
Antibody staining of cells was performed using standard protocols (22). For tissue immunostaining, tumors and lungs were harvested from euthanized animals, snap frozen in OCT, cryosectioned and fixed in zinc fixative (100 mM Tris-HCl containing 37 mM zinc chloride, 23 mM zinc acetate, and 3.2 mM calcium acetate). After fixation, sections were washed with PBST three times 10 min each. Following the third wash, tissues were blocked in 2% BSA in PBST for at least 30 min at RT. Primary antibodies were diluted in PBST supplemented with 2% BSA and 5% normal goat serum. The following primary antibodies were used: rabbit α-H3Cit (Abcam), rabbit α-H3Cit26 (custom), rabbit α-H4Cit3 (Millipore), rabbit α-histone H3 (Abcam), rabbit α-H4Arg3Me (Abcam), mouse α-H4K16Ac (Millipore), rabbit α-HP1α (Active Motif), mouse α-HP1β (Active Motif), mouse α-HP1γ (Active Motif) and mouse α-CD45 (eBioscience). The custom α-H3Cit26 antibody was generated by immunizing rabbits with a synthetic peptide QLATKAA-Cit-KSVPATG corresponding to histone H3 19–33 amino acids with the Cit26 residue. The specificity of the antibody was confirmed using the same sequence without modification (histone H3 residues 19–33 QLATKAARKSVPATG). The nuclei were stained by Hoechst 33258 (Sigma). The primary antibodies were detected by fluorescent-dye conjugated secondary antibodies from Jackson ImmunoResearch Laboratories. For TUNEL staining, sections were stained with cell death detection kit (Sigma) based on the manufacturer’s instructions. Images were collected using a Carl Zeiss Axioskop 40 microscope. Images were processed with Adobe Photoshop and Illustrator.
Micrococcal nuclease (MNase) digestion of 4T1 cells after A23187 treatment
After cells were treated with 4 μM A23187 overnight in 60 mm culture dish, old medium was aspirated and cells were rinsed with PBS once to remove dead cells and any extracellular materials. MNase (0.5 U) in 500 μl Locke’s solution (10 mM Hepes-HCl pH 7.3, 150 mM NaCl, 5 mM KCl, 2 mM CaCl2, and 0.1% glucose) was added to cells for 5 min at room temperature to digest CECN. Reactions were stopped by adding EDTA to a final concentration of 10 mM. Then the supernatant was used for DNA precipitation and Western blot.
CECN formation assays
4T1 cells were stimulated with 4 μM A23187 (Sigma) for 8 h, with 200 μM H2O2 (Sigma) for 4 h or with 300 ng/ml rHMGB1 for 24 h, at 37°C and 5% CO2 in DMEM supplemented with 10% FBS and 1% Penicillin/Streptomycin (P/S). For PAD inhibition with YW3–56, 4T1 cells were incubated with 30 μM YW3–56 for 15 min before A23187 treatment. For HL60 lysis and Conditioned Medium (CM) treatment, HL60 cells were first differentiated with DMSO as previously described (22). Differentiated HL60 cells were then sonicated in medium to expose cell contents and cultured with 4T1 cells for one day. For HL60 Conditioned Medium (CM) treatment, conditioned medium from differentiated HL60 cells was used to culture 4T1 cells for one day before assays.
MTT assay
The in vitro growth curves of 4T1 wild type and Padi4 knockout cells were analyzed using the MTT assay. Cells were seeded in 24-well plates at 4000 cells/well, for 24 h, 48 h, 72 h, or 96 h after the initial seeding. MTT reagent was added to a final concentration of 0.5 mg/ml. After 2 h incubation at 37 °C, the culture medium was removed and 1 ml DMSO was added to each well, and plates were agitated for 10 min. Spectrophotometric absorbance at 570 nm was measured. The Population Doubling = log10(ODday n/ODday 0)/log102.
Tumorigenesis studies
A total of 106 4T1 cells with different genotypes suspended in 100 μl of DMEM medium without FBS and P/S were injected into the inguinal mammary fat pad areas of 6- to 8-week-old female BALB/c and Rag2−/−; γc−/− mice. Tumor sizes were measured every three days using a caliper. Tumor volume was calculated using the equation (L × W2) × 0.4, where “L” = length and “W” = width. Tumor weights were taken at termination point of each study. To produce experimental lung metastases, a total of 106 4T1 cells with different genotypes were intravenously (i.v.) injected into BALB/c mice. Lungs were harvested on day 14 after tumor cell injection.
Histology
Animal specimens (tumors and lungs) were collected, fixed in 3.7% buffered formalin, embedded in paraffin, and stained with hematoxylin and eosin (H&E) according to standard protocols (29) or based on the manufacturer’s instructions. For lung metastasis quantification, lungs were fixed in Bouin’s solution (75 ml saturated picric acid, 25 ml formalin and 5 ml glacial acetic acid) and the number of macroscopically detectable foci was counted manually on fixed lungs.
In vivo treatment with DNase I and YW4–03
To test the effect of DNase I (Thermo Scientific) on spontaneous lung metastasis, daily intraperitoneal (i.p.) injections with DNase I (75 U per mouse) were initiated one week after tumor implantation and continued until termination point. For PAD inhibitor YW4–03 in vivo treatment, the first dose of intratumoral (i.t.) injection of YW4–03 (25 mg/kg) or vehicle (10% DMSO in PBS) was given one week after tumor implantation and then daily doses were administered. Tumor growth and lung metastatic burden were quantified as described above.
Colony formation assay
For circulating tumor cell quantification, 300 μl whole blood was collected from size-matched (1.5 cm in diameter in the longest dimension) tumor-bearing mice. Blood cells were washed once with 1 × Hank’s Balanced Salt Solution (HBSS) and then cultured in DMEM supplemented with 10% FBS, 1 × P/S, and 60 μM 6-thioguanine for 12 days (30). Cells were then fixed by adding 5 ml methanol and stained with 0.03% methylene blue for quantification.
To quantify the number of the Padi4 knockout and parental cells in the lungs, lungs from wild type BALB/c mice were harvested 4 hours after i.v. injection of 106 4T1 wild type or knockout cells. Harvested lungs were minced into small pieces with scissors and digested with 5 ml collagenase IV (1 mg/ml in 1 × HBSS) at 4 °C for 75 min on a rotating wheel. After completion of the enzyme digestion, HBSS was added to bring the volume of the sample to 10 ml. Each sample was filtered with a 70-μm nylon cell strainer, centrifuged to collect the cell pellet, and washed twice with HBSS. Cell pellets were cultured with complete DMEM medium supplemented with 60 μM 6-thioguanine in 37 °C incubator, 5% CO2 for 9 days. After culture, the dishes were stained by methylene blue similarly as blood colony formation assay.
Statistical analysis
For all numerical analyses, results are expressed as mean ± standard deviation (SD) or standard error of the mean (SEM). Group comparisons were performed using Student’s t-test. Values of p<0.05 were considered statistically significant. These analyses were carried out using the GraphPad Prism 5 software (GraphPad Software).
Results
PAD4 gene amplification and overexpression in breast cancer
To determine whether PAD4 expression was associated with breast cancer in humans, we interrogated the Tissue Cancer Genome Atlas and Oncomine database (31). In a meta-analysis of gene expression profiling, increased PAD4 expression was significantly associated with breast carcinoma compared with normal (Fig. 1A). To investigate the functional role of PAD4 in breast cancer, we studied this gene in the highly metastatic mammary carcinoma 4T1 cells (32), which express a much higher level of PAD4 than other cancer cell lines (Fig. 1B). To quantify the amount of PADI4 in 4T1 cells, we performed Western blot analyses with a titration of purified PAD4 protein along with the total extract from 4T1 cells (Fig. 1C). The PADI4 protein from 5 × 104 4T1 cells is roughly comparable to 100 ng purified PAD4 protein, which corresponds to ~16 million molecules of PAD4 protein per cell (Fig. 1C).
Figure 1. PAD4 expression in human and mouse mammary tumors.
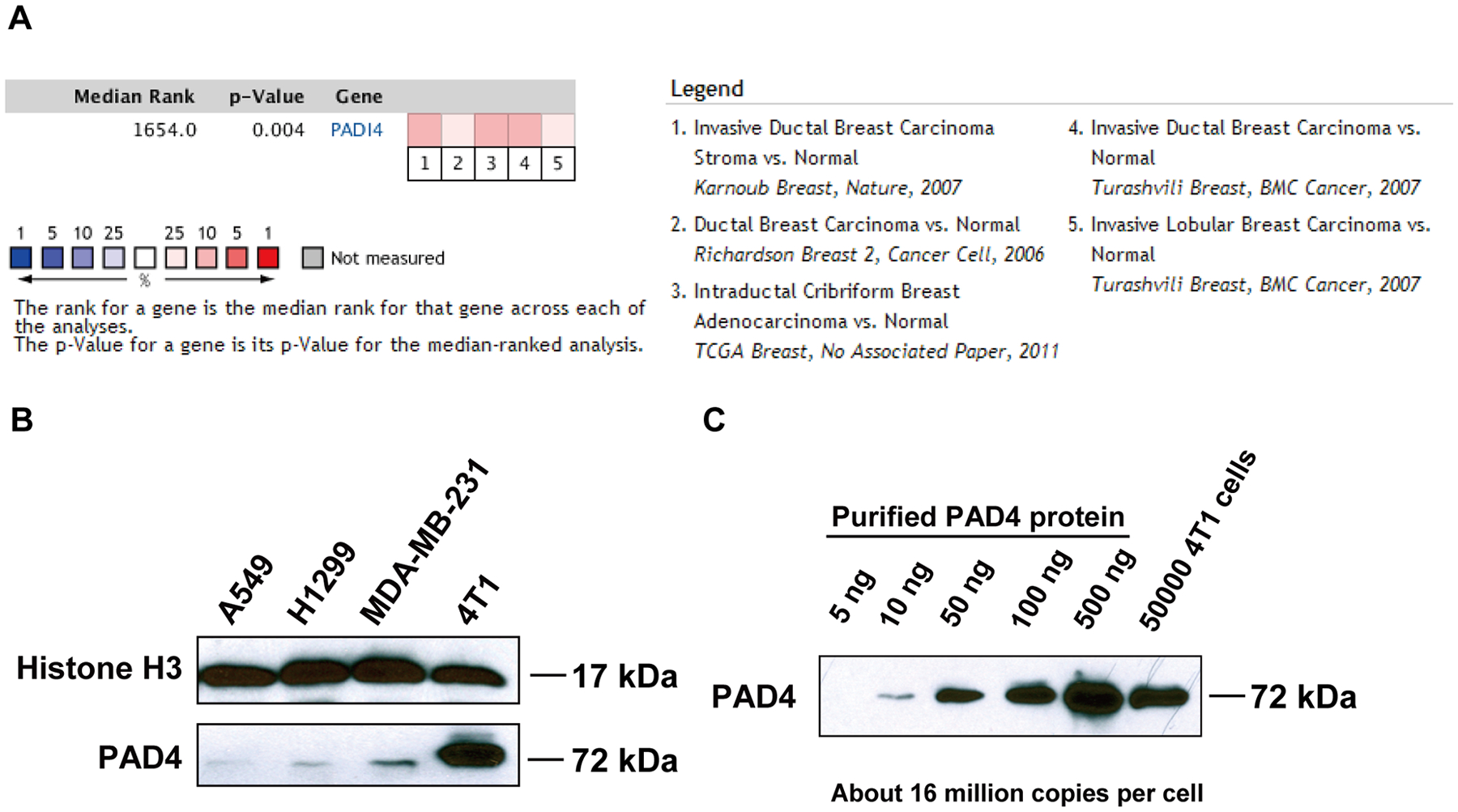
(A) Meta-analysis of gene expression profiling for PAD4 where the colored squares indicate the median rank for PAD4 across each analysis. PAD4 ranks in the top 10% to 25% in all 5 analyses. (B) Relative protein expression of PAD4 assessed in cancer cell lines by Western blot with antibody against PAD4. (C) Quantification of PADI4 in 4T1 cells by comparison to known amounts of purified PAD4 protein.
Histone hypercitrullination mediates chromatin decondensation and Cancer Extracellular Chromatin Network formation
The high level of PADI4 expressed in 4T1 cells prompted us to further explore its function in this triple negative breast cancer cell line. PAD4 is a calcium dependent enzyme. Therefore, we performed Western blot experiments after calcium ionophore treatment, and found that histone H3 citrullination detected with the H3Cit antibody (made against a histone H3 N-terminal peptide containing Cit2, Cit8, and Cit17) increased significantly after calcium ionophore treatment (Fig. 2A; lane 2). In addition, pan-PAD inhibitor YW3–56 pre-treatment for 15 min before calcium ionophore treatment decreased the level of citrullination induced by calcium ionophore, suggesting that PAD activity is involved in the elevated histone citrullination (Fig. 2A; lane 3). To test how histone citrullination activity affects chromatin structure, we analyzed cell morphology with immunostaining. Strikingly, activation of PAD activity induced 4T1 cells to rupture and release extensive web-like chromatin fibers into the extracellular space (Fig. 2B and 3A). In contrast, Padi4 deletion by CRISPR/Cas9 fully abolished histone citrullination and chromatin fiber release into the extracellular space (see below in Fig. 3D), indicating that PAD4 activity is directly involved in this process. Furthermore, we found that H3Cit staining was greatly increased at areas of highly decondensed chromatin (Fig. 2B; denoted by arrows), with a structure that is reminiscent of Neutrophil Extracellular Traps (NETs). We will refer this cancer extracellular chromatin network as CECN. In cells without calcium treatment, only sporadic hypercitrullinated cells, but not extracellular chromatin fibers, can be detected (Fig. 2B; upper panel and 3A).
Figure 2. Activation of PADI4 leads to hypercitrullination and CECN formation in 4T1 cells in vitro.
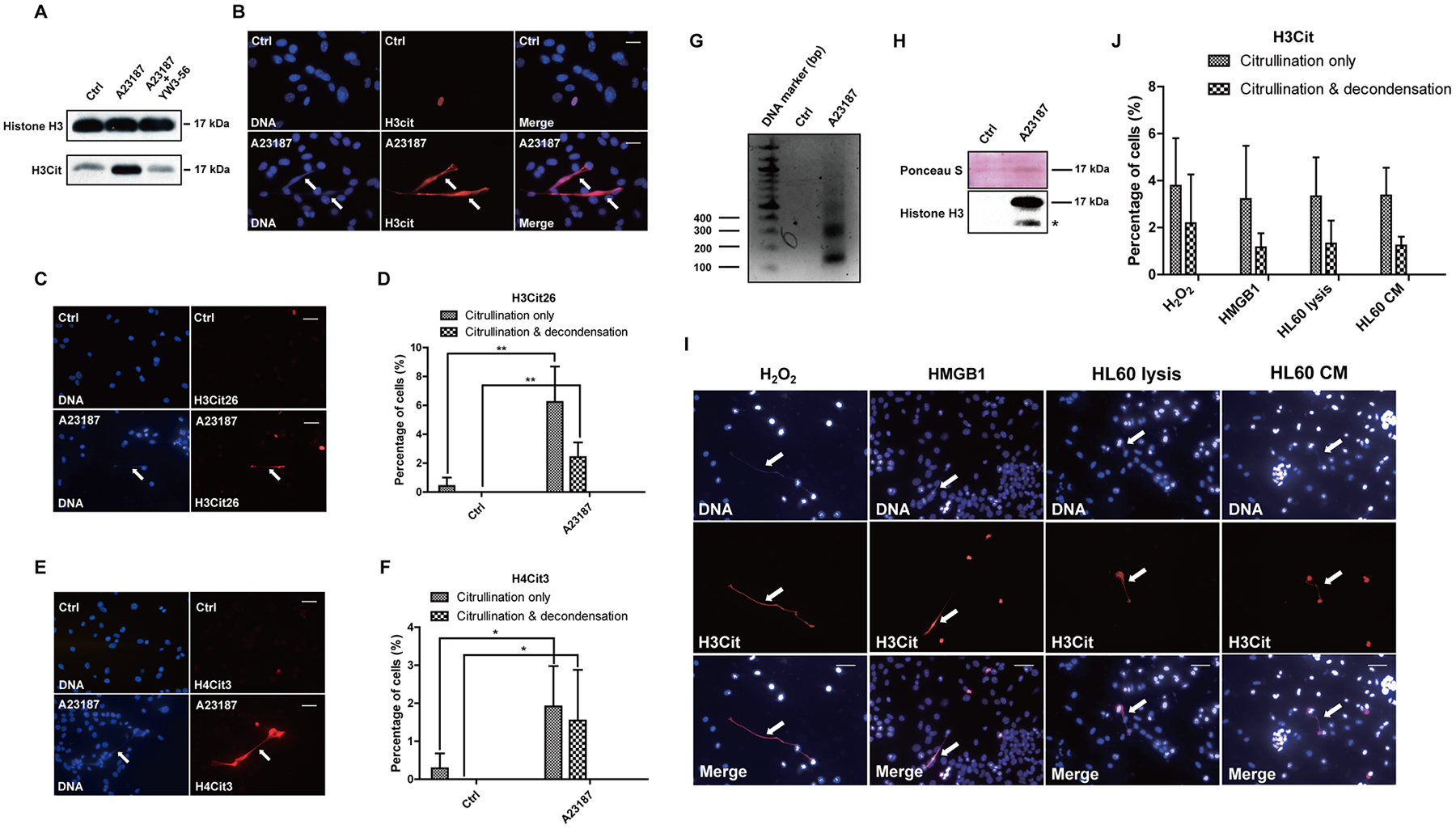
(A) H3Cit protein levels were determined by Western blot in 4T1 cells that were untreated or treated with calcium ionophore, or calcium ionophore and the PAD inhibitor YW3–56. The results are representatives of three experiments. (B) Representative immunofluorescence images of 4T1 cells showing CECN formation after 8 h of calcium ionophore treatment (magnification 40 ×) with staining for H3Cit (red), nuclei (blue). Arrows denote released chromatin DNA from 4T1 cells. Scale Bars: 25 μm. (C) Increase in the H3Cit26 level on the decondensed chromatin (denoted by arrow) stained by H3Cit26 antibody (red), nuclei (blue). Scale bars: 50 μm. (D) Quantification of H3Cit26 staining. Mean ± SD is shown (n = 4 or 5 fields from 2 independent experiments). (E) Increase in the H4Cit3 level on the decondensed chromatin (denoted by arrow) stained by H4Cit3 antibody (red), nuclei (blue). Scale bars: 50 μm. (F) Quantification of H4Cit3 staining. Mean ± SD is shown (n = 4 or 5 fields from 2 independent experiments). (G–H) Extracellular MNase digestion of 4T1 cells after control and calcium ionophore treatment. The presence of mono- and poly-nucleosome-sized fragments was detected by agarose gel (G). Histone H3 was detected by Western blot (H, * denotes potential degradation). (I) Immunofluorescence images revealed CECN release from 4T1 cells in response to H2O2, HMGB1 and in the presence of cell lysis and conditioned medium from differentiated HL60 granulocytic cells. Arrows denote released chromatin DNA from 4T1 cells. H3Cit (red), nuclei (blue). Scale Bars: 50 μm. (J) Quantification of hypercitrullination and CECN formation in 4T1 cell culture after different stimuli. Mean ± SD is shown (n > 4 fields from at least 2 independent experiments).
Figure 3. Formation of CECN is dependent on PAD4.
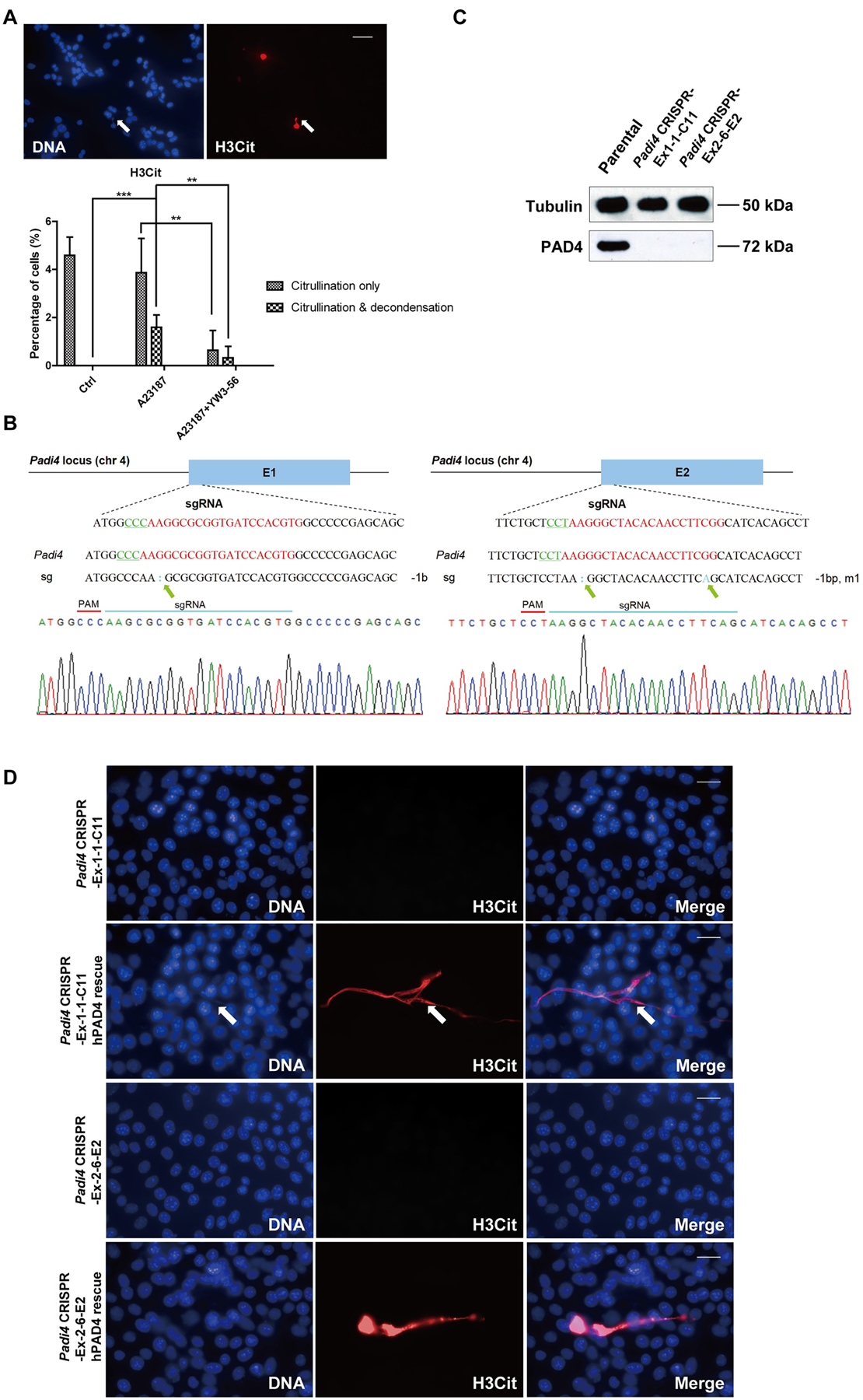
(A) Treatment of 4T1 cells with the PAD inhibitor, YW3–56, reduced both hypercitrullination and the formation of CECN. Cells were treated for 15 min with YW3–56 followed by 8 h with A23187. Upper panels: arrow denotes inhibition of CECN formation. H3Cit (red), nuclei (blue). Scale Bar: 50 μm. Lower panels: Quantification of percentage of cells with citrullinated nuclei and/or decondensed chromatin. Data are shown as mean ± SD (n = 8–10 fields from three independent experiments). **p<0.01, ***p<0.001. (B) Chromatograms from Sanger sequencing of the region encompassing mutations on exon 1 (left) and exon 2 (right). Green arrows denote mutations. (C) Knockout of the Padi4 gene in 4T1 cells via CRISPR/Cas9 was verified at protein level by Western blot. (D) Transient transfection of hPAD4 into Padi4 CRISPR cells before 8 h 4 μM A23187 treatment rescued the formation of CECN in 4T1 cells. H3Cit (red), nuclei (blue). Scale Bars: 25 μm. Data shown are representative results from 3 independent transfection experiments.
Other potential citrullinated positions on histones include Arg26 on histone H3 and Arg3 on histone H4. We next tested the citrullination level on these two positions with α-H3Cit26 (Fig. 2C and 2D) and α-H4Cit3 antibodies (Fig. 2E and 2F) respectively. The custom H3Cit26 antibody was validated with dot blot assay (Fig. S1A). Similarly, H3Cit26 and H4Cit3 are also increased on CECN released from 4T1 cells (Fig. 2C–2F; denoted by arrows). In accordance with our previous finding that PAD4 targets histone methyl-arginine residues for citrullination (22), histone H4 Arg3 methylation was decreased on CECN (Fig. S1B). The acetylation of histone H4K16 has been correlated with chromatin decondensation (33). However, this modification was not increased at the decondensed chromatin fiber (data not shown), suggesting that the increased staining of histone citrullination is unlikely to be the result of an increase in antibody accessibility. Moreover, histone hypercitrullination after PAD4 activation excludes chromatin association of the HP1 family of proteins including HP1α,β, andγ, which are involved in the maintenance of a heterochromatin state (Fig. S1C–S1E). To further confirm the presence of extracellular chromatin structure, intact 4T1 cells with or without calcium ionophore treatment were subjected to digestion with MNase, a nuclease that cuts between nucleosomes in chromatin. Only 4T1 cells after PADI4 activation showed the presence of a nucleosome DNA ladder in the supernatant (Fig. 2G). Accordingly, histone H3 protein can only be detected from the supernatant of 4T1 cells after calcium ionophore stimulation (Fig. 2H). Together, the aforementioned immunostaining and MNase analyses indicate that histone hypercitrullination is associated with CECN formation.
Induction of hypercitrullination and CECN formation by H2O2, HMGB1 and cellular components
To evaluate whether other pathological stimuli can induce CECN formation, we treated 4T1 cancer cells with H2O2 and HMGB1. H2O2 can be generated from multiple sources like neutrophils, macrophages and tumor cells themselves in the tumor microenvironment (34–36). Histone H3 citrullination and CECN formation were detected in 4T1 cells after 4 h treatment with H2O2 (Fig. 2I and 2J). High-Mobility Group Box 1 (HMGB1) protein can be passively released or actively secreted from multiple types of immune cells and cancer cells under stress (37). We found a modest increase in extracellular DNA and H3Cit staining after exposure of 4T1 cells to HMGB1, indicating the ability of HMGB1 to induce CECN in 4T1 cells (Fig. 2I and 2J).
Neutrophils are the most abundant circulating leucocytes, which can infiltrate many types of tumors. In the tumor microenvironment, neutrophils lyse to release their cellular components and form NETs that contribute to cancer metastasis (6). To test the effect of cellular components on CECN formation, we cultured 4T1 cells with contents from lysed HL60 derived granulocytic cells or the Conditioned Medium (CM) of HL60 cells, which were centrifuged to remove cell debris. After one day’s culture, we observed extracellular DNA fibers that colocalize with H3Cit staining (Fig. 2I and 2J). Collectively, the above results from in vitro experiments identified possible pathological inducers for CECN formation.
Formation of CECN is dependent on PAD4
To address whether the observed CECN formation was dependent on PAD4 activity, we first treated the calcium ionophore stimulated 4T1 cells with pan-PAD inhibitor YW3–56 which inhibits both PAD4 and PAD2 (38). Inhibition of PAD decreased the incidence of both hypercitrullination and release of CECN (Fig. 3A). Moreover, the extent of chromatin decondensation is also reduced compared to calcium ionophore treatment alone (Fig. 3A; denoted by arrows). To test the function of PAD4 specifically, we knocked out Padi4 in tumor cells using the CRISPR/Cas9 method and selected two single-cell colonies named Padi4 CRISPR-Ex1–1-C11 and Padi4 CRIPSR-Ex2–6-E2 with Padi4 deletion in exon 1 and 2, respectively. The deletion of Padi4 was confirmed by Sanger Sequencing (Fig. 3B), Western blot (Fig. 3C) and immunostaining (Fig. S2A). We then compared a series of phenotypic features of these 4T1 Padi4 knockout clones with parental 4T1 cells and found no obvious alteration in morphology (Fig. S2C) and cell viability (Fig. S2D). Furthermore, the basal level of citrullination is absent from Padi4 knockout cells (Fig. S2B). Additionally, citrullination and the formation of CECN were not detected after calcium ionophore stimulation (Fig. 3D). To test if PAD4 can restore CECN formation, we transiently transfected Padi4 knockout cells with a human PAD4-expressing plasmid and found that overexpression of PAD4 before calcium ionophore treatment can induce the release of CECN in Padi4 knockout cells (Fig. 3D). Together, these aforementioned analyses indicate that PAD4 activity is required for histone hypercitrullination and CECN formation.
4T1 tumor cells generate hypercitrullination in allograft tumors
To investigate whether 4T1 cells can release CECN in vivo, we first compared citrullination levels from orthotopically transplanted and size-matched 4T1 wild type and Padi4 knockout tumors. As expected, PAD4 can only be detected in the parental cell line and parental tumors (Fig. 4A). In addition, H3Cit was only significantly elevated in parental tumors derived from the parental Padi4 wild type 4T1 cell line but not in tumors from the two Padi4 knockout cell lines, suggesting that histone hypercitrullination occurred in a PAD4 dependent manner in the primary tumor (Fig. 4A). To test whether hypercitrullination leads to decondensed chromatin, we first analyzed the morphology of nuclei from size-matched tumors with H&E staining. The nuclei from wild type tumors displayed a swollen pattern with diffuse DNA staining while the nuclei from Padi4 knockout tumors were compact and condensed (Fig. 4B). We then immunostained the wild type tumor sections with the H3Cit antibody and DAPI to detect potential CECN structures, and the CD45 antibody to label all the immune cells. We found potential CECN structures showing H3Cit positive, CD45 negative, along with a diffuse DNA staining pattern (Fig. 4C; denoted by arrow). In the Padi4 wild type mice, host cells could contribute to citrullination and extracellular DNA formation. To exclude the activity of PADI4 from host mice, we repeated the same set of Western blot, H&E staining and immunostaining experiments with tumors allografted on BALB/c Padi4 knockout mice. As shown in Fig. 4D to 4F, parental tumors still showed significantly higher level of H3Cit and chromatin with CECN characteristics, while citrullination was undetectable in the Padi4 CRISPR deletion tumors. Most in vitro CECN formation assays were performed within one day. However, 4T1 cells in primary tumor were exposed to sustained stimulation signals from tumor microenvironment for weeks. Thus, in vivo CECN stimulation is actually stronger than in vitro treatments. The H3Cit staining in primary tumor is area-based and has different staining patterns (Fig. 4G), with some areas positive while others negative of H3Cit staining (Fig. 4G). This is likely due to the heterogeneity of tumor microenvironment with different levels of ROS or numbers of dying cells. Collectively, these results indicate that 4T1 tumor cells are prone to elevate histone citrullination and form CECN in primary tumors.
Figure 4. CECN is present in allograft tumors.
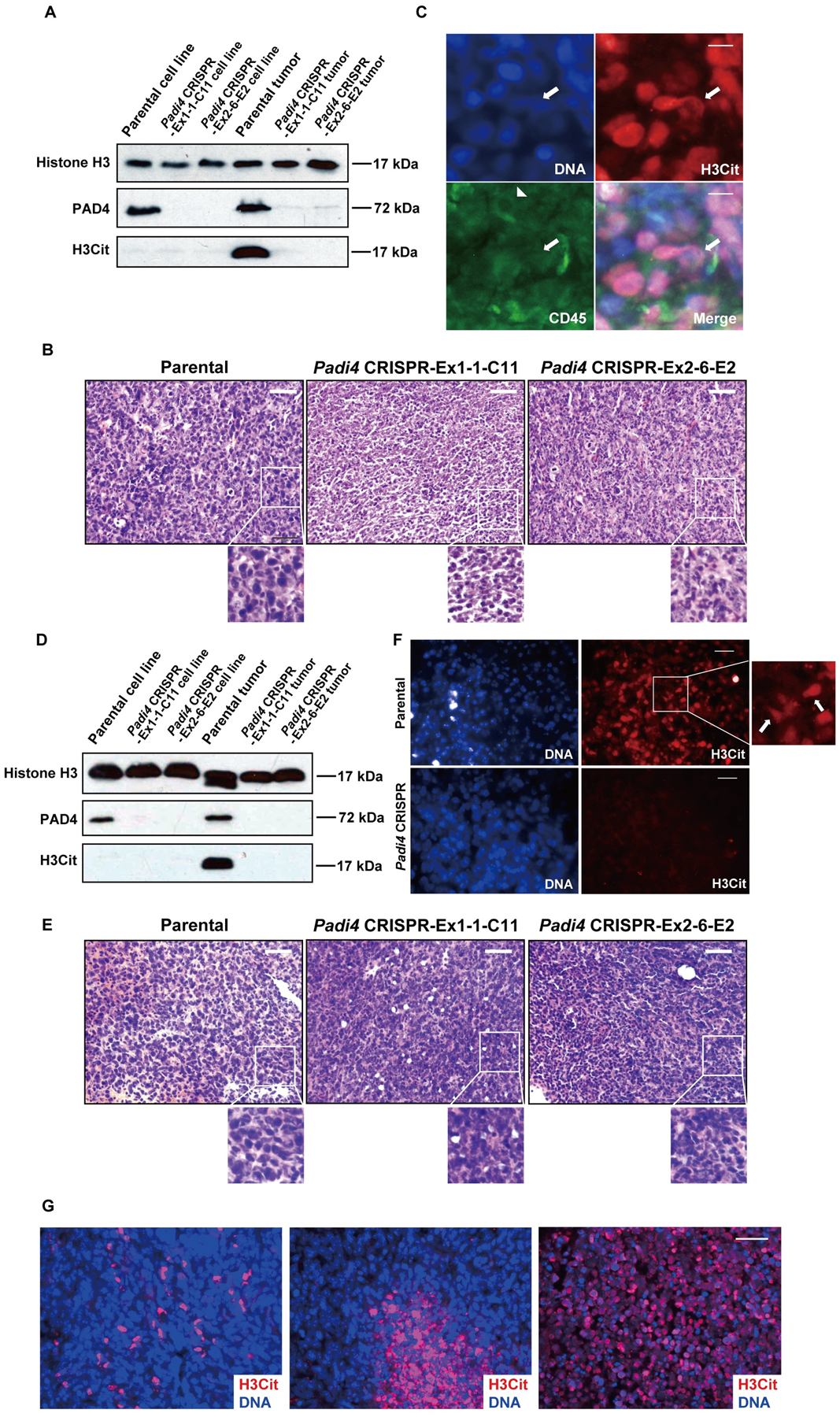
(A) PAD4 and H3Cit protein levels were determined by Western blot in 4T1 cell cultures with different genotypes (Lane 1~3) and 4T1 tumors with different genotypes growing in wild type BALB/c mice (Lane 4~6). (B) Histologic analysis of tumor sections from wild type BALB/c mice. Swollen nuclei from wild type tumor cells were detected by H&E staining. Scale Bars: 50 μm. (C) Representative immunofluorescence images of primary tumor sections from wild type BALB/c mice showing CECNs with staining for H3Cit (red), nuclei (blue), CD45 (green). Arrow denotes released chromatin from cancer cells. Arrow head denotes an immune cell. Scale Bars: 15 μm. (D) PAD4 and H3Cit protein levels were determined by Western blot in 4T1 cell cultures with different genotypes (Lane 1~3) and 4T1 tumors with different genotypes growing in BALB/c Padi4 knockout mice (Lane 4~6). (E) Histologic analysis of tumor sections from Padi4 knockout BALB/c mice. Swollen nuclei from wild type tumor cells were revealed by H&E staining. Scale Bars: 50 μm. (F) Representative immunofluorescence images of primary tumor sections from BALB/c Padi4 knockout mice showing CECNs with staining for H3Cit (red), nuclei (blue). Arrow denotes released chromatin from cancer cells. Scale Bars: 50 μm. (G) Representative immunofluorescence images of primary tumor sections showing different patterns of staining for H3Cit (red), nuclei (blue). Scale Bar: 40 μm.
Reduced growth and lung metastasis of Padi4 knockout tumors in mice
To assess the cell-autonomous role of PAD4 on tumor growth and metastasis, wild type female BALB/c mice were inoculated in the mammary gland with 4T1 wild type and Padi4 knockout cells. Although all 4T1 cell types showed similar degrees of in vitro growth rates (Fig. 5A), the allograft Padi4 knockout tumors grew significantly slower compared with wild type 4T1 tumors in wild type BALB/c mice (Fig. 5B). The reduced growth rate of Padi4 knockout tumors were also observed in Padi4 knockout BALB/c mice (Fig. 5C). Since the generation of CRISPR/Cas9 Padi4 knockout cell lines as clones could introduce some discrepancy other than Padi4 manipulation, we also tested a pan-PAD inhibitor. To inhibit PADI4 on population level in tumor cells and limit the effects of drug on host cells, we performed intratumoral injection of PAD4 inhibitor YW4–03 on wild type tumor allografted in Padi4 knockout mice thereby limiting NETosis of host neutrophils. Similarly, intratumoral PAD4 inhibitor treatment also reduced primary tumor growth rate (Fig. 5D).
Figure 5. Growth rates of primary mammary tumors formed by 4T1 cells with different Padi4 genotypes or combined with YW4–03 treatment.
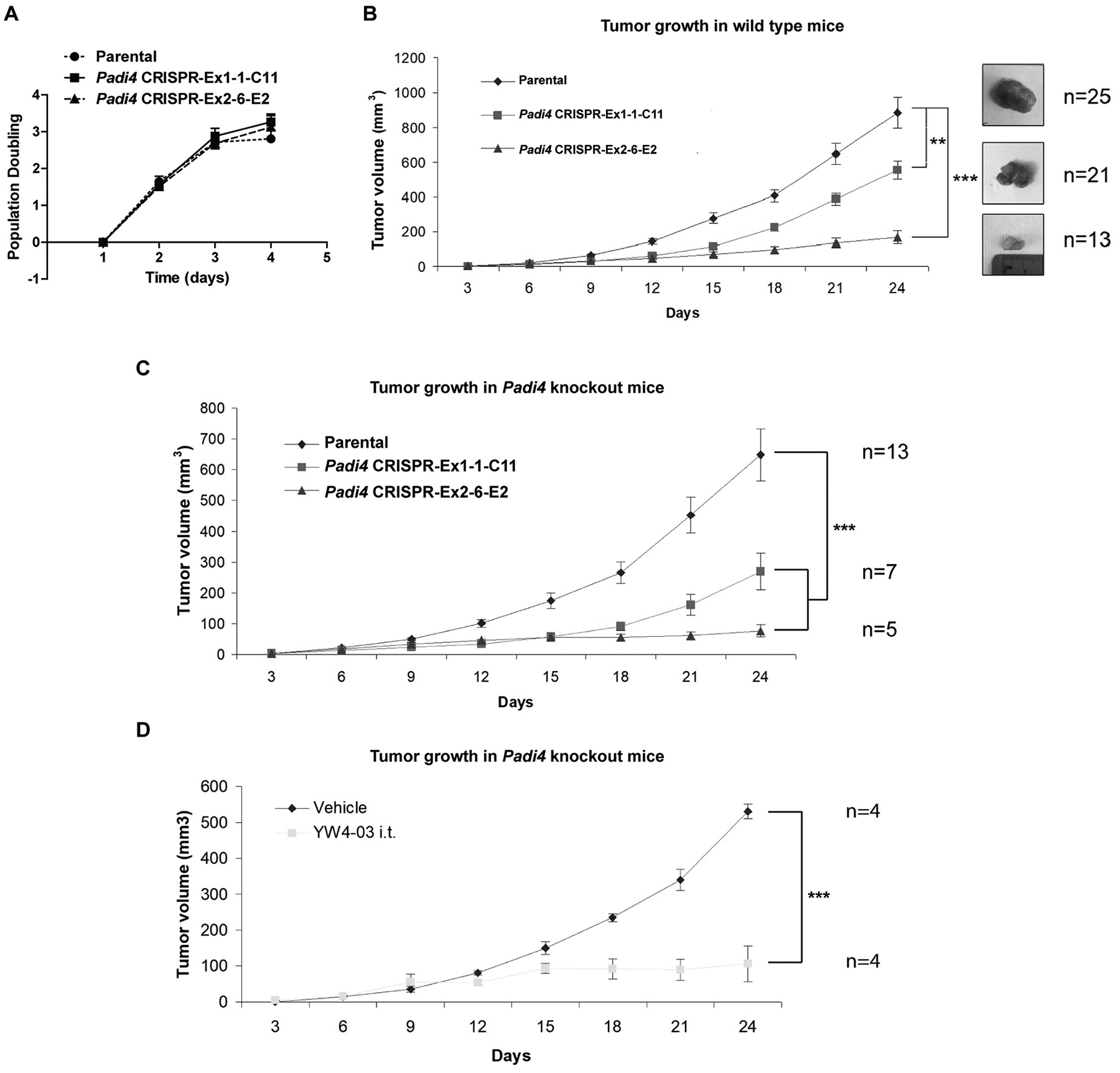
(A) Proliferation of the 4T1 cells with different Padi4 genotypes. Each data point represents the mean ± SD number of cells counted from three experiments. Each experiment includes four wells. (B) Growth rates of primary mammary tumors formed by 4T1 cells with different Padi4 genotypes. Each data point represents the mean ± SEM of indicated number of primary tumors. **p<0.01, ***p<0.001. The number of samples (n) for each genotype was denoted in the figure. (C) Growth rates of primary mammary tumors formed by 4T1 cells with different Padi4 genotypes on Padi4 knockout BALB/c mice. ***p<0.001. The number of samples (n) for each genotype was denoted in the figure. (D) Growth rates of primary mammary tumors formed by wild type 4T1 cells on Padi4 knockout BALB/c mice combined with vehicle or YW4–03 i.t. treatment. The number of samples (n) for each genotype was denoted in the figure. ***p<0.001.
To quantify cell death in the primary tumors, we performed TUNEL assay on size-matched tumors with different Padi4 genotypes at the end-point (1.5 cm in diameter in the longest dimension). Padi4 knockout 4T1 tumors showed similar proportions of apoptotic cells compared with wild type tumors, suggesting that they at least have same cell death level when tumors reach the same size (Fig. S3A and S3B).
4T1 cells are highly metastatic compared with a sibling cell line 67NR which was derived from the same mouse tumor but cannot metastasize (32). As shown in Fig. 6A, the PAD4 level is significantly higher in 4T1 cells compared with 67NR cells, prompting us to test a hypothesis that an increase in PAD4 expression can affect tumor metastasis. After the mice carrying tumors with different genotypes were euthanized at day 27 after allograft, mice bearing wild type tumors formed significantly higher number of metastatic foci on the lung surface than the two Padi4 knockout cell lines (Fig. 6B and 6C). Since the Padi4 knockout tumors are smaller than the Padi4 wild type tumors, the Padi4 knockout tumors were allowed to grow longer time to reach the same size (1.5 cm in diameter in the longest dimension) as the wild type tumors. Lung metastasis was still significantly reduced in mice bearing tumors derived from Padi4 CRISPR cancer cells even after prolonged growth (Fig. 6D–6F). Representative lung metastasis images from mice bearing tumors derived from wild type and Padi4 CRISPR cancer cells after whole mount fixation (Fig. 6D and 6E) or section staining (Fig. 6F) are shown. Typical metastatic foci were denoted by white arrows (Fig. 6F). To test the effects of PADI4 inhibition on population level, lung metastasis was also compared in the YW4–03 intratumoral treatment model using Padi4 knockout mice. Due to the sustained inhibition of tumor growth, comparison was performed at day 27 after tumor allograft. Compared with those receiving vehicle control, Padi4 knockout mice bearing tumors derived from parental wild type 4T1 cancer cells exhibited lower level of lung metastasis after YW4–03 treatment (Fig. 6G and 6H).
Figure 6. Lack of PAD4 in tumor leads to reduced lung metastasis.
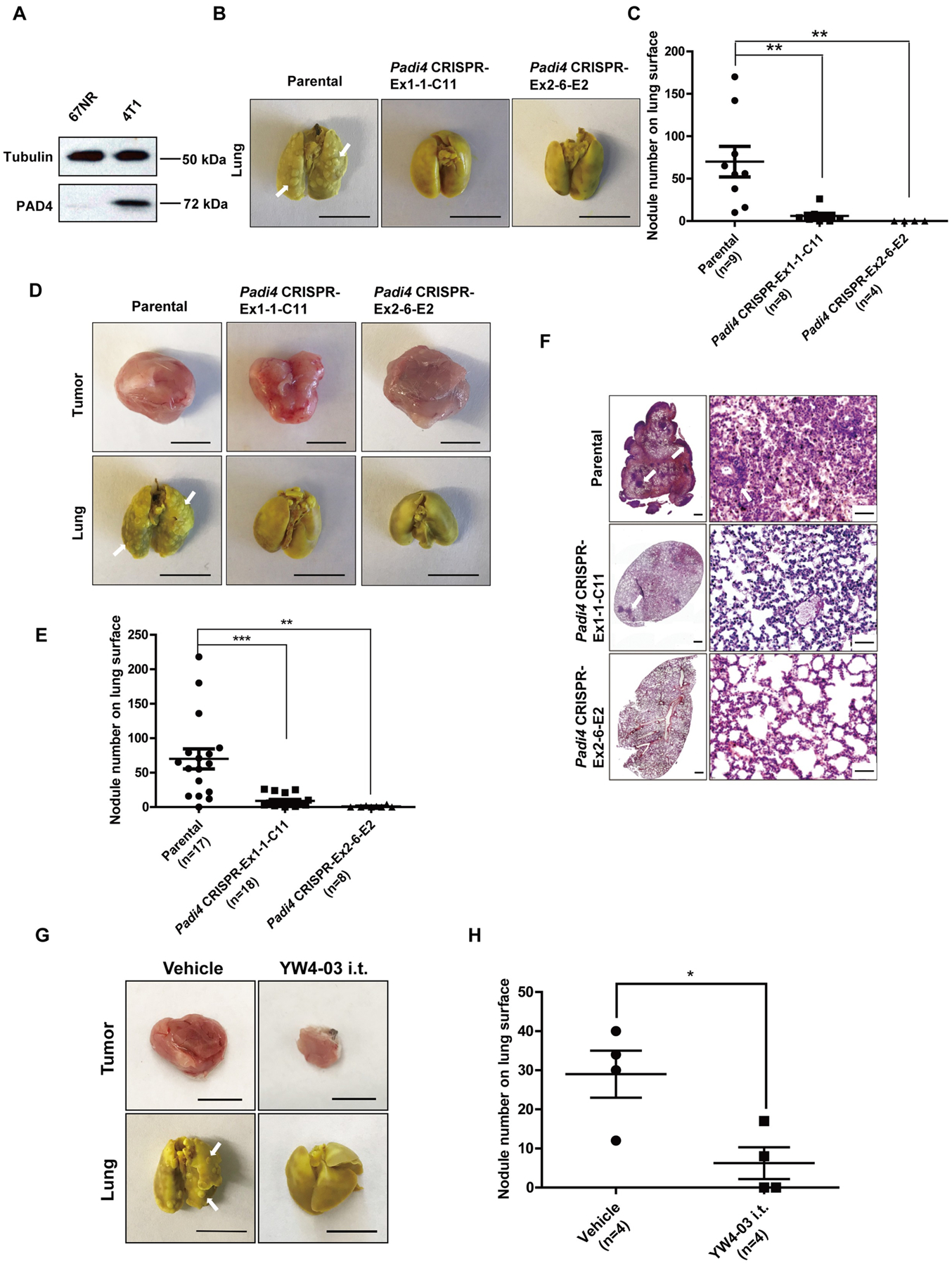
(A) Expression of PAD4 protein was examined by Western blot in two murine breast cancer cell lines. Tubulin was used as a loading control. (B) Representative images of metastasized lungs in mice bearing wild type and Padi4 knockout tumors in whole mount at day 27. Typical metastatic nodules were denoted by white arrows. Scale Bars: 1 cm. (C) Quantification of lung nodules in mice bearing wild type and Padi4 knockout tumors at day 27. Data are shown as mean ± SEM. The number of samples (n) for each genotype was denoted in the figure. **p<0.01. (D) Representative images of primary tumors and metastasized lungs in mice bearing wild type and Padi4 knockout tumors in whole mount at the end-point (1.5 cm in diameter in the longest dimension). Typical metastatic nodules were denoted by white arrows. Scale Bars: 1 cm. (E) Quantification of lung nodules in mice bearing wild type and Padi4 knockout tumors at end point (1.5 cm in diameter in the longest dimension). Data are shown as mean ± SEM. The number of samples (n) for each genotype was denoted in the figure. **p<0.01, ***p<0.001. (F) Representative H&E staining images of metastasized lungs in mice bearing wild type and Padi4 knockout tumors at the end-point (1.5 cm in diameter in the longest dimension). Scale Bars: 500 μm (left panels) and 50 μm (right panels). (G) Representative images of primary tumors and metastasized lungs in wild type tumor bearing vehicle and i.t. YW4–03 treated Padi4 knockout mice at day 27 after tumor implantation. Scale Bars: 1 cm. (H) Quantification of lung nodules in wild type tumor bearing vehicle and i.t. YW4–03 treated Padi4 knockout mice at day 27 after tumor implantation. Data are shown as mean ± SEM. The number of samples (n) for each genotype was denoted in the figure. *p<0.05
Adaptive immune cells play important roles in tumor growth and metastasis. To explore the involvement of the immune system in PAD4 mediated cancer metastasis, we transplanted 4T1 control and Padi4 knockout cells into the inguinal mammary fat pad areas of Rag2−/−; γc−/− immunodeficient mice, which lack functional T and B cells and natural killer (NK) cells (39). Although both Padi4 wild type and Padi4 knockout tumors grew faster in the Rag2−/−; γc−/− mice (data not shown), mice with tumors derived from Padi4 knockout cancer cells still showed a significant decrease in lung metastases compared with mice with tumors derived from parental wild type 4T1 cancer cells when all the tumors reach 1.2 cm in diameter in the longest dimension (Fig. S4A and S4B). This result demonstrates that tumor-derived PADI4 facilitates metastasis, at least partially, in a manner independent of the adaptive immune cells. Taken together, the above data suggest that loss of Padi4 decreases metastasis of 4T1 cell derived tumors.
Cancer cell autonomous PAD4 promotes metastasis through steps after intravasation
To further test the idea that CECNs are involved in metastasis, we used DNase I to disrupt extracellular DNA including CECN structures in the 4T1 wild type tumors growing in the Padi4 knockout mice, which lack the ability to release chromatin mediated by PADI4 from immune cells such as neutrophils (20). Although DNase I treatment did not significantly alter the growth rate of the primary tumor (Fig. S5A), lung metastasis was significantly decreased (Fig. 7A and 7B). We next tested whether PAD4 expressed in cancer cells influences intravasation by analyzing the Circulating Tumor Cells (CTCs) in blood with colony formation assays. Neither loss of Padi4 nor DNase I treatment influenced the CTC numbers from size-matched (1.5 cm in diameter in the longest dimension) tumor bearing mice, suggesting cancer cell endogenous PAD4 does not promote metastasis through the intravasation process (Fig. 7C–7F).
Figure 7. Cancer cell endogenous PAD4 promotes metastasis through steps after intravasation.
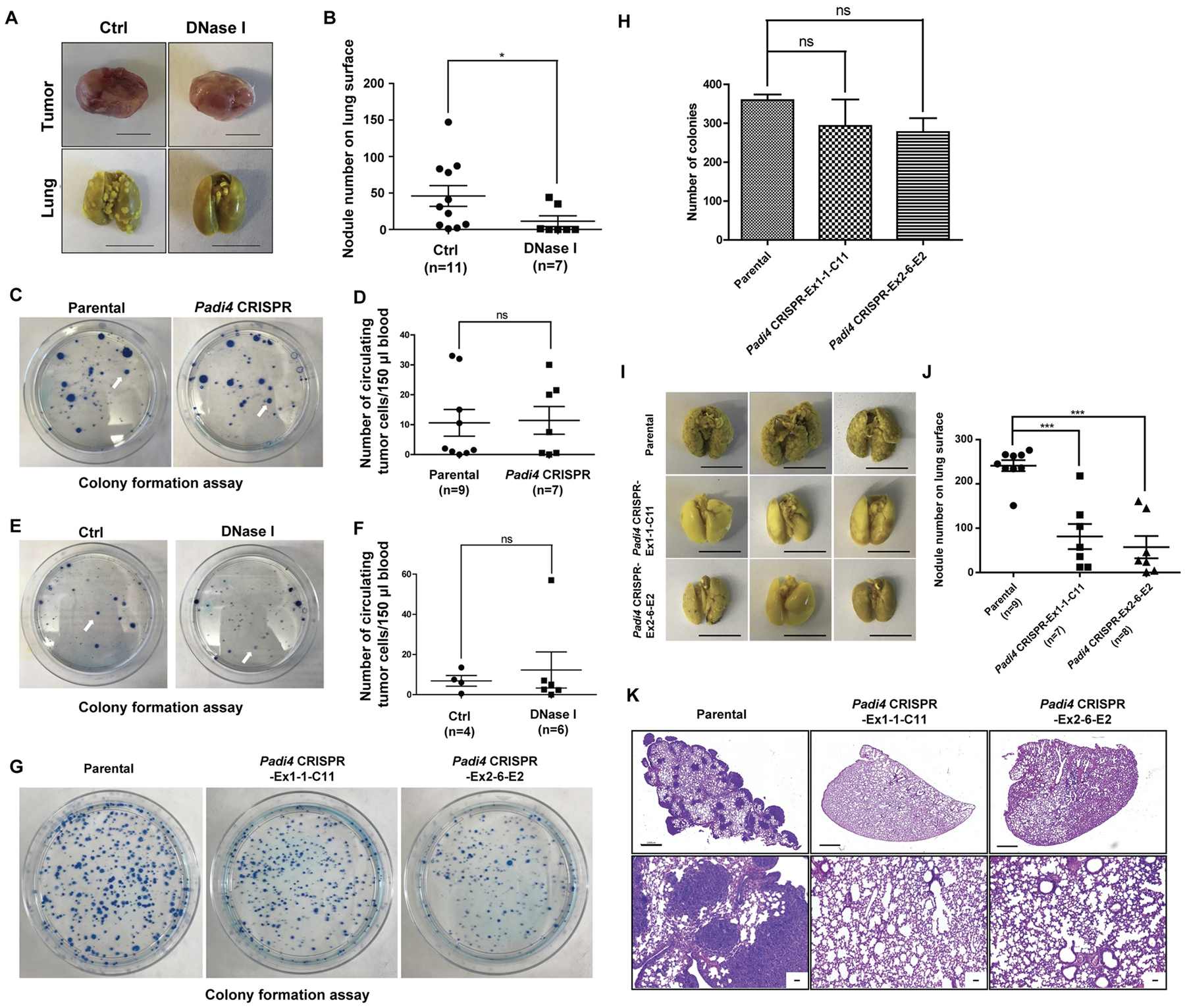
(A–B) Representative images of primary tumors and metastasized lungs (A) and quantification assays (B) in control and DNase I treated Padi4 knockout mice bearing tumors derived from parental wild type 4T1 cells at the end-point (1.5 cm in diameter in the longest dimension). Scale Bars: 1 cm. Scattered plot in (B) shows mean ± SEM. The number of samples (n) for each treatment was denoted in the figure. *p<0.05. (C–D) Colony formation assay of circulating tumor cells (CTCs) in blood from wild type mice bearing wild type and Padi4 knockout primary tumors at end point (1.5 cm in diameter in the longest dimension). Arrows denote representative colonies. Scattered plot in (D) shows mean ± SEM. The number of samples (n) for each genotype was denoted in the figure. ns, not significant. (E–F) Colony formation assay of CTCs in blood from the control and DNase I treated Padi4 knockout mice bearing Padi4 wild type 4T1 primary tumors at end point (1.5 cm in diameter in the longest dimension). Arrows denote representative colonies. Scattered plot in (F) shows mean ± SEM. The number of samples (n) for each treatment was denoted in the figure. ns, not significant. (G–H) Colony formation assay of parental wild type and Padi4 knockout 4T1 cells in lungs 4 hours after i.v. injection. Bar graph in (H) shows mean ± SEM. n = 3 for all groups. ns, not significant. (I–J) Representative images in (I) show metastasized lungs in experimental metastasis model with parental wild type and Padi4 knockout 4T1 cells at day 14 after i.v. injection. Scale Bars: 1 cm. Scattered plot in (J) shows mean ± SEM. The number of samples (n) for each genotype was denoted in the figure. ***p<0.001. (K) Representative H&E staining images of metastasized lungs in mice 14 days after i.v. injection of parental wild type and Padi4 knockout 4T1 cells. Scale Bar: 1 mm (upper panels) and 50 μm (lower panels).
To further test whether PADI4 is functioning at steps after intravasation, we performed experimental metastasis assays by intravenously injecting the same number of tumor cells into mice. At 4 hours after i.v. injection of tumor cells, we analyzed the number of Padi4 knockout and parental cells accumulated in the lung using colony formation assays. The loss of Padi4 in 4T1 cells didn’t significantly reduce the number of cancer cells accumulated compared with the Padi4 wild type cells (Fig. 7G and 7H). However, mice receiving Padi4 knockout 4T1 cells displayed large decreases in the numbers of lung metastatic nodules compared with wild type cells at day 14 after tumor cell i.v. injection (Fig. 7I and 7J). Representative sections of stained lungs from mice injected with Padi4 wild type or Padi4 knockout 4T1 cells are shown in Fig. 7K. In addition, lungs from mice receiving parental wild type tumor cells were physically larger and weighed significantly more than lungs from mice receiving Padi4 mutant cancer cells (Fig. S4C and S4D). These results suggest that PAD4 regulates the growth of tumor cells disseminated into the lung and subsequent tumor nodule formation.
Discussion
Our study provides evidence suggesting that high levels of PADI4 result in the release of Cancer Extracellular Chromatin Network (CECN) in 4T1 cells in culture and in tissue sections. Loss of Padi4 in 4T1 cells or DNase I treatment in tumor bearing Padi4 knockout host mice significantly decreased cancer metastasis in the orthotopic allograft breast cancer mouse model. In addition, we offered evidence showing that cancer cell autonomous PAD4 promotes metastasis through steps after intravasation. These results are consistent with findings dated back to the 1960s, when reports implicated extracellular DNA in cancer development (40–42). Recent studies identified NETs as a mechanism underlying the role of extracellular DNA in carcinogenesis (6–8). In addition, others have shown cancer cell-related extracellular DNA (exDNA) is involved in cell metastatic potential in vitro and that DNase I treatment significantly decreased cancer metastasis in an orthotopic xenograft cancer mouse model (43). Here, we showed that PAD4 mediated extracellular chromatin networks from cancer cells themselves are contributing to the pool of extracellular DNA.
In our experiment, the two Padi4 CRISPR cell lines exhibited the same growth kinetics as wild type cells in cell culture. However, they were growing slower than wild type tumors in the allograft model and some of the exon 2 mutant tumors were very much arrested and even completely disappeared. The difference between in vitro and in vivo kinetics is striking. One clear difference is PADI4 is only highly activated in allograft tumors in vivo, as indicated by the high level of H3Cit (Fig. 4A and 4D). The two CRISPR cell lines also showed different kinetics of growth in allograft experiments. The one base pair deletion in each cell line led to a frame shift mutation resulting in an early stop codon soon after the mutation. The exon 2 CRISPR/Cas9 cell line could encode a 49 amino acid polypeptide, while the exon 1 CRISPR/Cas9 cell line could encode a short 5 amino acid peptide. In addition, the two CRISPR/Cas9 cell lines were individually selected single cell colonies, which may introduce some discrepancy through the procedures such as transfection steps and clonal selection. In addition, since the 4T1 cell line is heterogenous, single cell clones possibly behave differently from parental cells. As an alternative method to test the role of PAD4, we treated Padi4 knockout mice bearing wild type tumors with PAD inhibitor YW4–03 (38) to inhibit PAD4 at population level and found tumors under this treatment also displayed slower growth pattern (Fig. 5D). However, we did not observe a significant difference in tumor growth in the DNase I treatment model in the Padi4 knockout mice (Fig. S5A). This indicates differential effects of the CECN in tumor growth and metastasis. PAD4 in cancer cells has functions important for primary tumor growth that cannot be targeted by digesting extracellular DNA. The change of gene expression due to Padi4 knockout led to the reduced primary tumor growth but not removal of CECN. However, we didn’t examine further how loss of PADI4 in 4T1 cancer cells reduced primary tumor growth in this study. One future direction is to elucidate the mechanism of PAD4-mediated tumor growth, progression, and metastasis in the tumor microenvironment in mouse models.
In addition, not all NETosis is dependent on PAD4 and there are other sources of extracellular DNA than CECNs and NETs. Therefore, the DNase I treatment on Padi4 knockout mice bearing wild type tumors is not specific to CECNs and can only provide us a supportive evidence for the function of CECNs. In our study, we used Padi4 knockout tumor and mice to genetically remove Padi4 mediated extracellular chromatin network formation and performed DNase I treatment to digest extracellular DNA. However, due to technical issue, it’s impossible for us to specifically block CECN formation without manipulation of Padi4 at this moment. Other techniques are required to be developed to target on CECNs specifically and elucidate the role of CECNs more precisely.
The CECN in our cell culture experiments is only found in PADI4 high 4T1 cells after stimulation. It can also be found in allograft model but never occurs in 4T1 cells in normal culture conditions. Thus, the high level of PAD4 and its activation are requirements for PAD4-mediated CECN formation. Since not all tumor types express PAD4 at a high level, the PAD4-mediated CECN likely affects a subset of metastatic cancer cells. In addition, under inflammatory conditions, NETs can contribute extracellular chromatin fibers thereby facilitating cancer cell adhesion, metastasis and awakening (6,17,44). Future work should be done to address the molecular signatures of the cancer cell types that have a high metastatic potency and are dependent on PAD4.
To date, the mechanism of NET formation has been tackled at the macroscopic level via visualization of stimulated neutrophils. From these studies, we know that there is a loss of nuclear integrity, mixing of chromatin with cellular granules and finally, disintegration of the nuclear membrane prior to chromatin release (45). However, a detailed signaling pathway has not been established yet. The similarity between PAD4 mediated NETs and CECNs prompts us to hypothesize that the mechanism should also be similar except that granules are not present in cancer cells. The involvement of reactive oxygen species (ROS), particularly H2O2, in the tumor promotion process is supported by both in vivo and in vitro studies. The association between H2O2 and tumor progression along with the fact that NETs can be induced by ROS prompted us to test the effect of H2O2 on CECN formation. HMGB1 has also been demonstrated to promote NET formation through interactions with Toll-like receptor 4 (46) and HMGB1 released during NET formation may in turn drive proliferation and migration of cancer cells (47). In addition, oxidative stress is a central regulator of HMGB1’s translocation, release and activity in inflammation and cell death (48,49), thus establishing linkage between ROS and HMGB1. In our study, we found calcium ionophore, H2O2 and HMGB1 could trigger CECN in vitro. Thus another important future direction will be to identify the physiological stimulator and its signaling pathways that trigger cellular cascade leading to CECN formation.
Our lab first identified PAD4’s function in histone hypercitrullination, its role in NETs formation and its requirement for bacterial killing mediated by these chromatin fibers (19,20,22). Meanwhile, increasing evidence also linked excessive levels of NETs to many autoimmune diseases and cancers. Above findings suggest a role for histones and their epigenetic modifications in a physiological process that was not fully recognized. Although PAD4 has been shown to be overexpressed in a variety of tumors (26,27), its function in tumor cells still remains poorly understood. Cancer cell derived PAD4 was shown to be essential for the growth of liver metastases from colorectal cancer through citrullination of a key matrix component collagen type I (50). It was also reported that PAD4 is an important mediator of the p53 signaling pathway via citrullination of histone H3 at target gene promoters (51) and cooperates with Elk-1 to activate c-Fos (52). On the contrary, loss of PAD4 from breast tumor using MCF7 cells as a model was shown to initiate early steps of metastasis through dysregulation of citrullination of nuclear GSK3β in these tumors (53). The differences in the responses of cancer cells to PAD4 could be due to the different choice of cancer cell lines. Knockdown of PAD4 in MCF7 cells showed active spreading, loss of cell-cell contact, and a fibroblast-like morphology compared to control MCF7 cells (53). However, Padi4 deletion using CRISPR/Cas9 didn’t change the morphology of 4T1 cells (Fig. S2C) and has no influence on intravasation (Fig. 7C and D) in our study. Possible relevant differences leading to the different phenotypes are that the MCF7 cells are human and estrogen-dependent and 4T1 cells are murine and triple negative breast cancer cells.
Our findings provide a mechanistic basis for the high metastatic tendency in tumors with elevated levels of PAD4. We combined biochemical and physiological approaches to establish the occurrence of PADI4-mediated CECN in 4T1 cells in vitro and in vivo. The tumor microenvironment harbors the signal to activate PADI4 and deletion of Padi4 or removal of extracellular DNA including CECN with DNase I in 4T1 cells inhibits metastatic lung colonization. How PADI4 regulates primary tumor growth warrants further exploration. PAD4’s vital role in promoting tumor growth and metastasis renders itself a tangible therapeutic target for drug development to treat highly metastatic cancers involving PAD4 as an underlying mechanism.
Supplementary Material
Implication:
This study shows that PADI4 can mediate the formation of Cancer Extracellular Chromatin Networks (CECNs) in 4T1 cells, and that endogenous PAD4 plays an essential role in breast cancer lung metastasis.
Acknowledgments
We thank Dr. Andrea Mastro for providing us the 4T1 and 67NR cells, Dr. Denisa D. Wagner for backcrossing Padi4 knockout mice to BALB/c background and Dr. Na Xiong for providing us the Rag2−/−; γc−/− mice. We also thank Dr. David S. Gilmour for comments on the manuscript. This work was supported by funding from The Pennsylvania State University and Henan University to YW; and by a grant from National Institutes of Health (NIH)/National Cancer Institute (R01 CA214865) to AT.
Financial Support: This work was supported by funding from The Pennsylvania State University and Henan University to YW; and by a grant from National Institutes of Health (NIH)/National Cancer Institute (R01 CA214865) to AT.
Footnotes
Conflict of interest: The authors declare no potential conflicts of interest.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin 2018;68:7–30 [DOI] [PubMed] [Google Scholar]
- 2.Tazawa H, Okada F, Kobayashi T, Tada M, Mori Y, Une Y, et al. Infiltration of neutrophils is required for acquisition of metastatic phenotype of benign murine fibrosarcoma cells: implication of inflammation-associated carcinogenesis and tumor progression. Am J Pathol 2003;163:2221–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau CS, et al. IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis. Nature 2015;522:345–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wculek SK, Malanchi I. Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature 2015;528:413–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spiegel A, Brooks MW, Houshyar S, Reinhardt F, Ardolino M, Fessler E, et al. Neutrophils Suppress Intraluminal NK Cell-Mediated Tumor Cell Clearance and Enhance Extravasation of Disseminated Carcinoma Cells. Cancer Discov 2016;6:630–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park J, Wysocki RW, Amoozgar Z, Maiorino L, Fein MR, Jorns J, et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci Transl Med 2016;8:361ra138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cools-Lartigue J, Spicer J, McDonald B, Gowing S, Chow S, Giannias B, et al. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J Clin Invest 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Najmeh S, Cools-Lartigue J, Rayes RF, Gowing S, Vourtzoumis P, Bourdeau F, et al. Neutrophil extracellular traps sequester circulating tumor cells via beta1-integrin mediated interactions. Int J Cancer 2017;140:2321–30 [DOI] [PubMed] [Google Scholar]
- 9.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science 2004;303:1532–5 [DOI] [PubMed] [Google Scholar]
- 10.Cooper PR, Palmer LJ, Chapple IL. Neutrophil extracellular traps as a new paradigm in innate immunity: friend or foe? Periodontol 2000 2013;63:165–97 [DOI] [PubMed] [Google Scholar]
- 11.Martinod K, Witsch T, Erpenbeck L, Savchenko A, Hayashi H, Cherpokova D, et al. Peptidylarginine deiminase 4 promotes age-related organ fibrosis. J Exp Med 2017;214:439–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thalin C, Demers M, Blomgren B, Wong SL, von Arbin M, von Heijne A, et al. NETosis promotes cancer-associated arterial microthrombosis presenting as ischemic stroke with troponin elevation. Thromb Res 2016;139:56–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gupta AK, Hasler P, Holzgreve W, Gebhardt S, Hahn S. Induction of neutrophil extracellular DNA lattices by placental microparticles and IL-8 and their presence in preeclampsia. Hum Immunol 2005;66:1146–54 [DOI] [PubMed] [Google Scholar]
- 14.Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr., et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A 2010;107:15880–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Demers M, Wong SL, Martinod K, Gallant M, Cabral JE, Wang Y, et al. Priming of neutrophils toward NETosis promotes tumor growth. Oncoimmunology 2016;5:e1134073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Demers M, Krause DS, Schatzberg D, Martinod K, Voorhees JR, Fuchs TA, et al. Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proc Natl Acad Sci U S A 2012;109:13076–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoo HJ, Lee JS, Kim JE, Gu J, Koh Y, Kim I, et al. Extracellular Histone Released from Leukemic Cells Increases Their Adhesion to Endothelium and Protects them from Spontaneous and Chemotherapy-Induced Leukemic Cell Death. PLoS One 2016;11:e0163982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol 2011;187:538–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y, Li M, Stadler S, Correll S, Li P, Wang D, et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol 2009;184:205–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med 2010;207:1853–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakashima K, Hagiwara T, Yamada M. Nuclear localization of peptidylarginine deiminase V and histone deimination in granulocytes. J Biol Chem 2002;277:49562–8 [DOI] [PubMed] [Google Scholar]
- 22.Wang Y, Wysocka J, Sayegh J, Lee YH, Perlin JR, Leonelli L, et al. Human PAD4 regulates histone arginine methylation levels via demethylimination. Science 2004;306:279–83 [DOI] [PubMed] [Google Scholar]
- 23.Leshner M, Wang S, Lewis C, Zheng H, Chen XA, Santy L, et al. PAD4 mediated histone hypercitrullination induces heterochromatin decondensation and chromatin unfolding to form neutrophil extracellular trap-like structures. Front Immunol 2012;3:307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mohanan S, Horibata S, McElwee JL, Dannenberg AJ, Coonrod SA. Identification of macrophage extracellular trap-like structures in mammary gland adipose tissue: a preliminary study. Front Immunol 2013;4:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dworski R, Simon HU, Hoskins A, Yousefi S. Eosinophil and neutrophil extracellular DNA traps in human allergic asthmatic airways. J Allergy Clin Immunol 2011;127:1260–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chang X, Han J. Expression of peptidylarginine deiminase type 4 (PAD4) in various tumors. Mol Carcinog 2006;45:183–96 [DOI] [PubMed] [Google Scholar]
- 27.Chang X, Han J, Pang L, Zhao Y, Yang Y, Shen Z. Increased PADI4 expression in blood and tissues of patients with malignant tumors. BMC Cancer 2009;9:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li P, Yao H, Zhang Z, Li M, Luo Y, Thompson PR, et al. Regulation of p53 target gene expression by peptidylarginine deiminase 4. Mol Cell Biol 2008;28:4745–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fischer AH, Jacobson KA, Rose J, Zeller R. Hematoxylin and eosin staining of tissue and cell sections. CSH Protoc 2008;2008:pdb prot4986 [DOI] [PubMed] [Google Scholar]
- 30.Pulaski BA, Ostrand-Rosenberg S. Mouse 4T1 breast tumor model. Curr Protoc Immunol 2001;Chapter 20:Unit 20 2 [DOI] [PubMed] [Google Scholar]
- 31.Rhodes DR, Kalyana-Sundaram S, Mahavisno V, Varambally R, Yu J, Briggs BB, et al. Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia 2007;9:166–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aslakson CJ, Miller FR. Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res 1992;52:1399–405 [PubMed] [Google Scholar]
- 33.Robinson PJ, An W, Routh A, Martino F, Chapman L, Roeder RG, et al. 30 nm chromatin fibre decompaction requires both H4-K16 acetylation and linker histone eviction. J Mol Biol 2008;381:816–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nathan CF. Neutrophil activation on biological surfaces. Massive secretion of hydrogen peroxide in response to products of macrophages and lymphocytes. J Clin Invest 1987;80:1550–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kono K, Salazar-Onfray F, Petersson M, Hansson J, Masucci G, Wasserman K, et al. Hydrogen peroxide secreted by tumor-derived macrophages down-modulates signal-transducing zeta molecules and inhibits tumor-specific T cell-and natural killer cell-mediated cytotoxicity. Eur J Immunol 1996;26:1308–13 [DOI] [PubMed] [Google Scholar]
- 36.Yan D, Cui H, Zhu W, Talbot A, Zhang LG, Sherman JH, et al. The Strong Cell-based Hydrogen Peroxide Generation Triggered by Cold Atmospheric Plasma. Sci Rep 2017;7:10831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kang R, Zhang Q, Zeh HJ 3rd, Lotze MT, Tang D. HMGB1 in cancer: good, bad, or both? Clin Cancer Res 2013;19:4046–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Y, Li P, Wang S, Hu J, Chen XA, Wu J, et al. Anticancer peptidylarginine deiminase (PAD) inhibitors regulate the autophagy flux and the mammalian target of rapamycin complex 1 activity. J Biol Chem 2012;287:25941–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Song J, Willinger T, Rongvaux A, Eynon EE, Stevens S, Manz MG, et al. A mouse model for the human pathogen Salmonella typhi. Cell Host Microbe 2010;8:369–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sugihara S, Yamamoto T, Tanaka H, Kambara T, Hiraoka T, Miyauchi Y. Deoxyribonuclease treatment prevents blood-borne liver metastasis of cutaneously transplanted tumour cells in mice. Br J Cancer 1993;67:66–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De Lamirande G. Action of deoxyribonuclease and ribonuclease on the growth of Ehrlich ascites carcinoma in mice. Nature 1961;192:52–4 [DOI] [PubMed] [Google Scholar]
- 42.Salganik RI, Martynova RP, Matienko NA, Ronichevskaya GM. Effect of deoxyribonuclease on the course of lymphatic leukaemia in AKR mice. Nature 1967;214:100–2 [DOI] [PubMed] [Google Scholar]
- 43.Wen F, Shen A, Choi A, Gerner EW, Shi J. Extracellular DNA in pancreatic cancer promotes cell invasion and metastasis. Cancer Res 2013;73:4256–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME, et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 2018;361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Medina E. Beyond the NETs. J Innate Immun 2009;1:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tadie JM, Bae HB, Jiang S, Park DW, Bell CP, Yang H, et al. HMGB1 promotes neutrophil extracellular trap formation through interactions with Toll-like receptor 4. Am J Physiol Lung Cell Mol Physiol 2013;304:L342–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tohme S, Yazdani HO, Al-Khafaji AB, Chidi AP, Loughran P, Mowen K, et al. Neutrophil Extracellular Traps Promote the Development and Progression of Liver Metastases after Surgical Stress. Cancer Res 2016;76:1367–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tsung A, Klune JR, Zhang X, Jeyabalan G, Cao Z, Peng X, et al. HMGB1 release induced by liver ischemia involves Toll-like receptor 4 dependent reactive oxygen species production and calcium-mediated signaling. J Exp Med 2007;204:2913–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu Y, Tang D, Kang R. Oxidative stress-mediated HMGB1 biology. Front Physiol 2015;6:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yuzhalin AE, Gordon-Weeks AN, Tognoli ML, Jones K, Markelc B, Konietzny R, et al. Colorectal cancer liver metastatic growth depends on PAD4-driven citrullination of the extracellular matrix. Nat Commun 2018;9:4783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tanikawa C, Ueda K, Nakagawa H, Yoshida N, Nakamura Y, Matsuda K. Regulation of protein Citrullination through p53/PADI4 network in DNA damage response. Cancer Res 2009;69:8761–9 [DOI] [PubMed] [Google Scholar]
- 52.Zhang X, Gamble MJ, Stadler S, Cherrington BD, Causey CP, Thompson PR, et al. Genome-wide analysis reveals PADI4 cooperates with Elk-1 to activate c-Fos expression in breast cancer cells. PLoS Genet 2011;7:e1002112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stadler SC, Vincent CT, Fedorov VD, Patsialou A, Cherrington BD, Wakshlag JJ, et al. Dysregulation of PAD4-mediated citrullination of nuclear GSK3beta activates TGF-beta signaling and induces epithelial-to-mesenchymal transition in breast cancer cells. Proc Natl Acad Sci U S A 2013;110:11851–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.