

Abstract
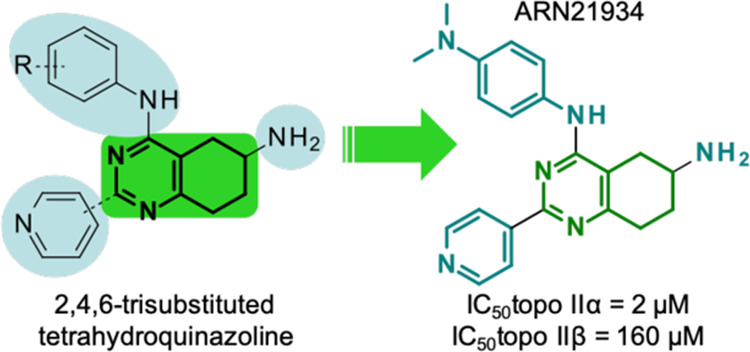
We disclose a novel class of 6-amino-tetrahydroquinazoline derivatives that inhibit human topoisomerase II (topoII), a validated target of anticancer drugs. In contrast to topoII-targeted drugs currently in clinical use, these compounds do not act as topoII poisons that enhance enzyme-mediated DNA cleavage, a mechanism that is linked to the development of secondary leukemias. Instead, these tetrahydroquinazolines block the topoII function with no evidence of DNA intercalation. We identified a potent lead compound [compound 14 (ARN-21934) IC50 = 2 μM for inhibition of DNA relaxation, as compared to an IC50 = 120 μM for the anticancer drug etoposide] with excellent metabolic stability and solubility. This new compound also shows ~100-fold selectivity for topoIIα over topoβ, a broad antiproliferative activity toward cultured human cancer cells, a favorable in vivo pharmacokinetic profile, and the ability to penetrate the blood–brain barrier. Thus, ARN-21934 is a highly promising lead for the development of novel and potentially safer topoII-targeted anticancer drugs.
Introduction
Human topoisomerase II (topoII) is a crucial enzyme that controls the topology of DNA in cells and regulates vital cellular processes such as DNA replication, transcription, recombination, and chromosome segregation.1−5 In addition to its important cellular functions, topoII is a validated target for the treatment of cancer,1,6,7 and several topoII-targeted drugs are in clinical use.1,2 Notably, all of these drugs act by trapping the covalent enzyme/DNA cleavage complex that is formed during DNA topology modifications.6−8 As a result, topoII generates double-stranded breaks in the genome. Although effective against cancer, this mechanism of action, referred to as topoII poisoning, is linked to severe side effects including the development of secondary leukemias in patients.6−10 These unwanted effects represent timely and urgent issues that have motivated the recent renaissance of research to seek new and safer topoII-targeted drugs.4,11−13
To address this issue, we have previously reported synthesis and modeling of new small molecules that act as topoII poisons.14−20 Here, we outline the discovery of novel 6-amino-tetrahydroquinazoline derivatives that are potent topoII inhibitors and that, notably, do not act as topoII poisons. Remarkably, these molecules preferentially inhibit the α isoform of human topoII, over the β one.21,22 Furthermore, they have broad antiproliferative activity against cultured human cancer cells and display a favorable in vivo pharmacokinetic (PK) profile. These findings highlight the potential of these tetrahydroquinazoline derivatives for the development of novel anticancer agents (Figure 1).
Figure 1.
Virtual screening of the D3 library of compounds and the experimental test of the most promising hits resulted in the identification of a new type of topoII inhibitor consisting of bicyclic pyrimidines derivatives, which was used to build a structure–activity relationship, from compound 1 (IC50 of 160 μM) to the lead compound 14 (IC50 of 2 μM, ARN-21934).
Results and Discussion
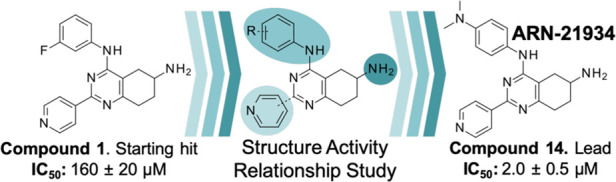
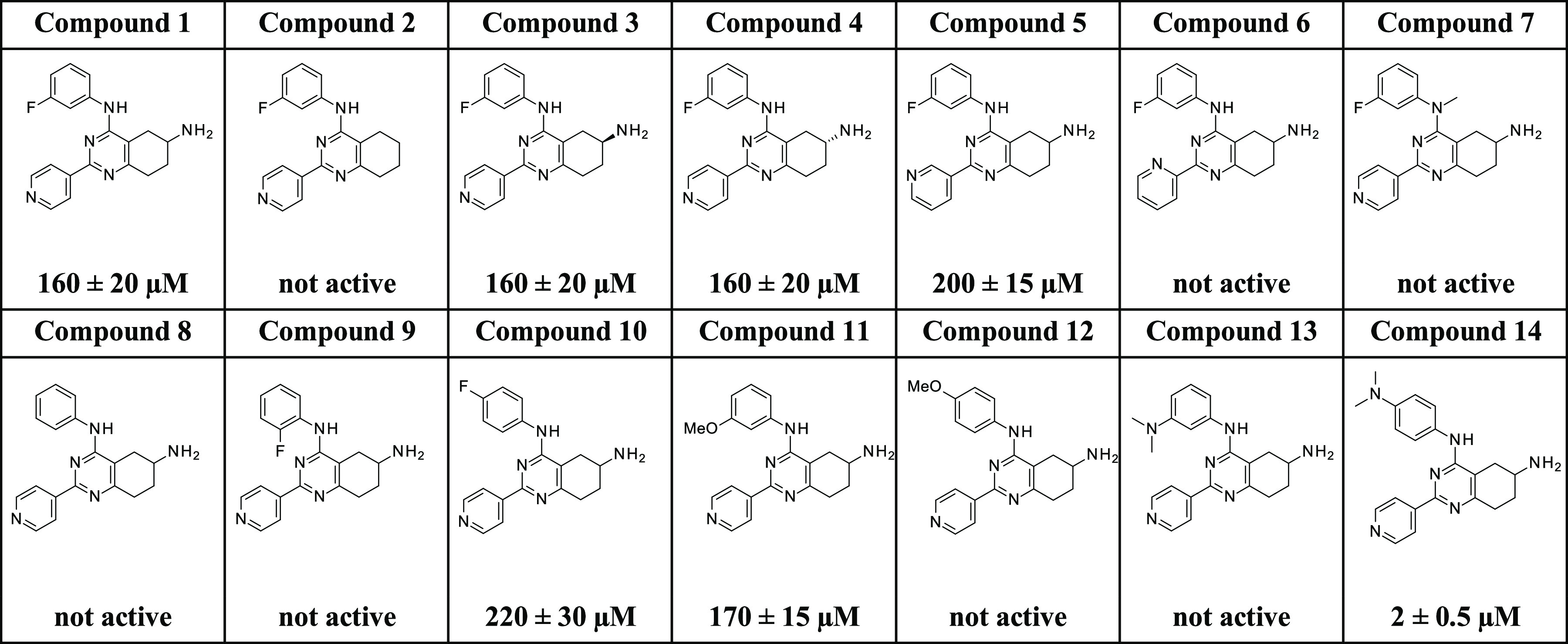
A virtual screening of an internal library of small compounds docked against the cleavage complex returned promising hits against human topoIIα. From these, ∼70 compounds were measured for their ability to inhibit human topoIIα in a DNA relaxation assay. We identified the small molecule with formula N4-(3-fluorophenyl)-2-(4-pyridyl)-5,6,7,8-tetrahydroquinazoline-4,6-di-amine (compound 1, Table 2), as a novel inhibitor with an IC50 of 160 μM.
Table 2. Topoisomerase IIα Inhibitiona.
Values shown correspond to the IC50 ± standard deviation (SD).
Notably, the core of the starting hit 1 is based on the privileged scaffold of functionalized quinazolines (Figure 2), which display a large spectrum of biological activities, including antibacterial, antifungal, anticonvulsant, anti-inflammatory, antihuman immunodeficiency virus (anti-HIV), anticancer, and analgesic effects.23 Furthermore, we measured the chemical similarity (atom-pair Tanimoto coefficients) between the proposed quinazoline scaffold and known families of topoIIα inhibitors (Figure 2). These results confirmed that partially saturated quinazolines occupy a novel structural niche in the context of topoIIα inhibition.
Figure 2.
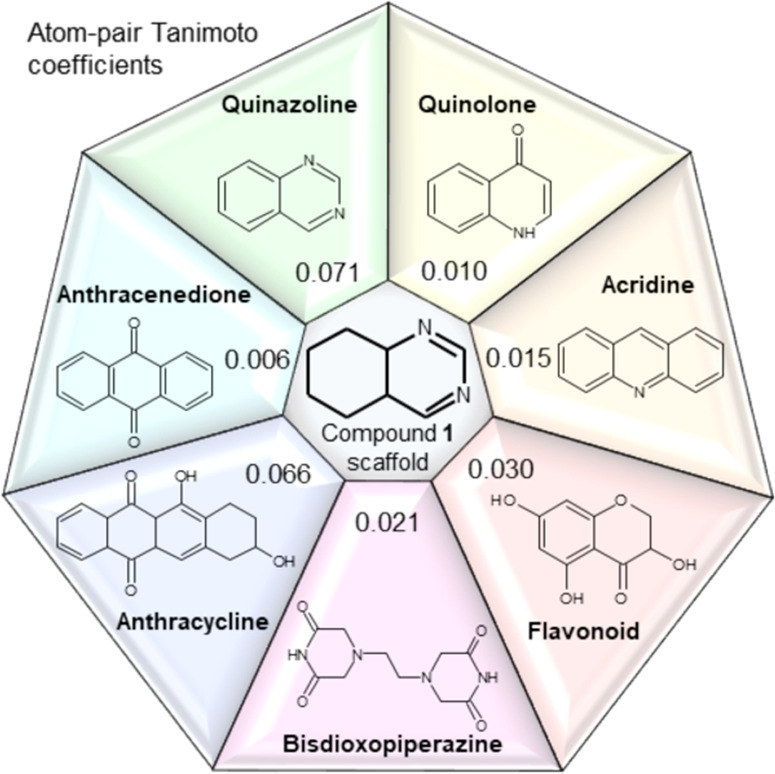
Chemical similarity between chemotypes commonly employed to target topoisomerase IIα. The values in each box correspond to the atom-pair Tanimoto coefficient calculated between the partially saturated quinazoline scaffold of compound 1 and seven other substructures, namely, quinazoline, quinolone, acridine, flavonoid, bisdioxopiperazine, anthracycline, and anthracenedione.
Compound 1 includes a partially saturated quinazoline heterocycle bearing a pyridine, an aniline, and an amino group in positions 2, 4, and 6, respectively (Figure 1). Thus, we started a focused structure–activity relationship (SAR) study and explored an initial set of 13 close analogs that were synthesized and tested against topoIIα-catalyzed DNA relaxation (summarized in Scheme 1b–d and Table 1). This systematic exploration of 1 led to six additional new active compounds. In particular, we identified compound 14 (named ARN-21934, Figure 1), which has an IC50 of 2 μM (Figure 1). Thus, 14 is ∼80-fold more potent than the starting hit 1 and is more potent than the anticancer drug etoposide (∼120 μM), which was used as a reference compound in our topoIIα inhibition assay.11,24
Scheme 1.
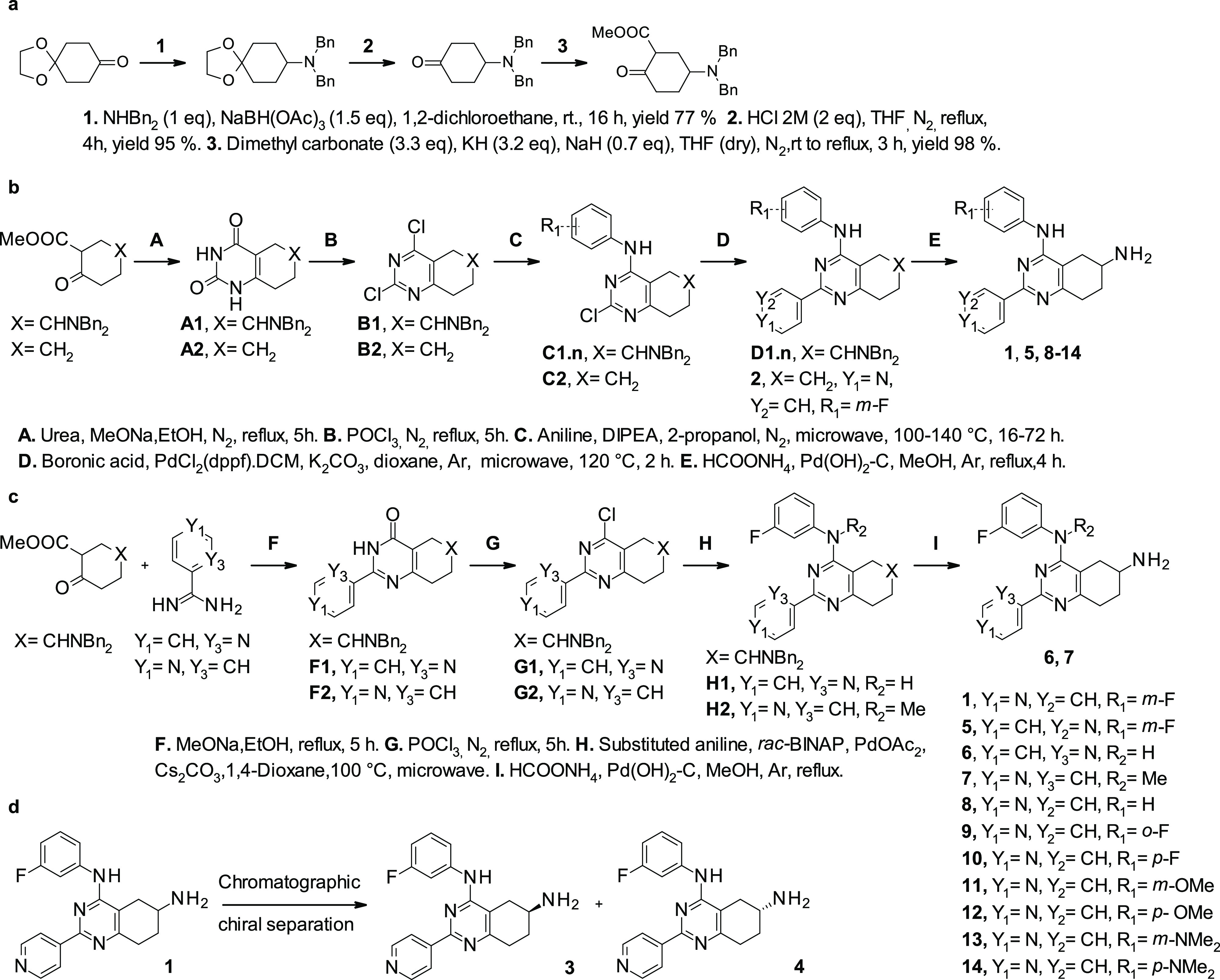
Table 1. Structures of Synthesized Intermediates C1.n and D1.n.

In view of its potency, we further characterized 14 at the cellular level, analyzed its solubility and metabolic stability, and evaluated its pharmacokinetic properties in mice. We show that 14 represents a new, isoform-specific, and highly promising topoIIα lead inhibitor that may be an excellent starting point for a full-fledged future anticancer drug discovery program. First, we investigated the chiral center at 6 position in 1. We removed the 6-amino group in 1 using the procedure in Scheme 1b, obtaining 2. This, however, led to an inactive compound, suggesting that the amino group is essential for topoIIα inhibition. We then determined that the pure enantiomers 3 and 4, obtained with a semipreparative chiral high-performance liquid chromatography (HPLC) purification (Scheme 1d; see the Experimental Section for detailed chromatographic conditions), were equipotent to the starting racemic mixture in 1. This finding suggests that position 6 stereoconfiguration does not significantly affect topoIIα inhibition, although the poor inhibitory activities of compounds 1, 3, and 4 suggest that further investigation on this point is needed.
Compounds 5 and 8–14 were then obtained using a five-step synthetic route (Scheme 1b), which was characterized by the initial formation of the bicyclic scaffold, followed by the selective introduction of different anilines at position 4, via aromatic nucleophilic substitutions. Different pyridines were then introduced using microwave-assisted Suzuki coupling conditions in position 2. Compounds 6 and 7 were synthesized with an alternative procedure (Scheme 1c). The bicyclic heterocycle was formed functionalized in 2 position, and, in a second step, corresponding anilines were introduced via microwave-assisted Buchwald coupling conditions.
TopoIIα inhibition results (Table 2) indicate that the replacement of the pyridine substituent at 2 position in 1 with the corresponding 3-pyridyl group, as in 5, was partially tolerated, with an IC50 of 200 μM. Alternatively, the compound with a 2-pyridyl substituent, which generated 6, was inactive. We therefore decided to maintain the initial pyridine ring at position 2 of the active scaffold. At this point, we investigated the effect of substituents at the 4-aniline position, generating an additional eight compounds (7–14). The introduction of a methyl group in the 2-aniline nitrogen, 7, and removal of the fluorine substituent, 8, led to an inactive compound. While a fluorine substituent in the ortho position generated an inactive compound, 9, a para-fluorine substitution was tolerated, leading to an IC50 of 220 μM, in 10. However, alternative H-bond acceptor groups did not retain this activity pattern if moved from the initial meta-position as in hit 1. For example, a methoxy substituent in the meta-position, 11, produced an IC50 of 170 μM but led to an inactive compound for the dimethylamino derivative (compound 13). In contrast, when replacing the fluorine atom in the para position, the methoxy substituent in 12 led to a complete loss of activity, while the dimethylamino one in 14 led to a significant boost of the inhibitory activity (IC50 of 2 μM). These results suggest a steric effect of aniline substituents on topoIIα inhibition.
We further characterized 14 for the mechanism by which it inhibited topoII (see Figures S2–S5, Supporting Information (SI)). First, we evaluated its ability to act as a topoII poison, like etoposide, using a DNA cleavage assay (see the SI). Surprisingly, the compound displayed no ability to increase levels of enzyme-mediated DNA cleavage, although docking suggested a favorable fit of this scaffold into the cleavage site (see Figure S6, SI). We then evaluated the ability of 14 to intercalate into DNA in the absence of enzyme using an unwinding assay. In contrast to quinazolines that intercalate into DNA,2514 did not show this behavior (Figure 3). This finding confirms that this tetrahydroquinazoline derivative can be classified as a topoII inhibitor. Thus, 14 acts differently from all of the currently approved topoII-targeted drugs, which act as poisons.6−8,26
Figure 3.

Compound 14 does not intercalate into DNA in the concentration range in which it inhibits topoIIα. A topoisomerase I-DNA unwinding assay was used to monitor intercalation. An ethidium bromide-strained agarose gel is shown. A relaxed DNA control (DNA) is included. The positions of nicked and supercoiled DNA are indicated. Relaxed DNA bands display an electrophoretic mobility between the nicked (more relaxed) and supercoiled (less relaxed) bands. Intercalation is indicated by the shift in the position of the plasmid from relaxed to negatively supercoiled. The effects of 10 μM ethidium bromide (EtBr, a strong intercalator) and 100 μM etoposide (a nonintercalator) on the DNA unwinding assay are shown as controls. The gel is representative of at least three independent experiments.
Most topoisomerase II-targeted agents display a similar affinity for topoIIα and topoIIβ.11,24 Remarkably, 14 was much more potent against the α isoform (Figure 4). Whereas the IC50 value for the inhibition of DNA relaxation by topoIIα was ∼2 μM, the value for inhibition of DNA relaxation by topoIIβ was ∼200 μM. Thus, 14 is ∼100-fold more specific for topoIIα over topoIIβ. Although additional studies will need to be carried out to determine the mechanistic basis for the actions of 14 against topoIIα, the compound is likely to interact at the DNA cleavage/ligation active site of the enzyme, as it displays little ability to inhibit topoIIα-catalyzed adenosine 5′-triphosphate (ATP) hydrolysis in the presence of DNA and none in the absence of the nucleic acid substrate (see Figures S1–S4, SI).
Figure 4.
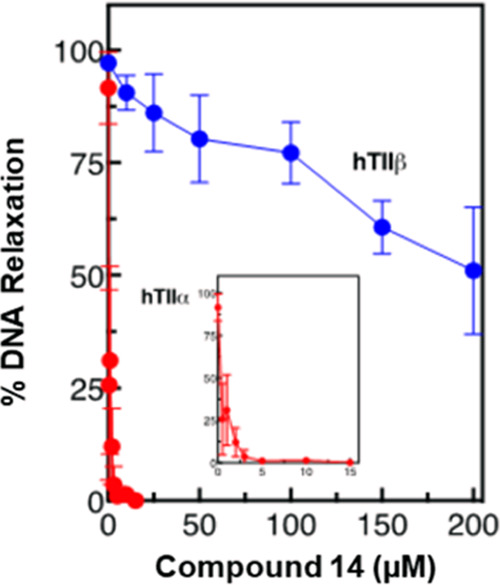
Inhibition of DNA relaxation catalyzed by human type II topoisomerases by 14. The inhibition of DNA relaxation by human topoIIα (hTIIα, red) and topoIIβ (hTIIβ, blue) is shown. The inset shows an expanded scale for results with topoIIα. Error bars represent the standard deviations of three independent experiments.
The solubility and metabolic stability of compounds 1, 5, 10, 11, and 14 were also evaluated. While the initial hit 1 showed a moderate kinetic solubility of 126 μM (measured in phosphate-buffered saline (PBS), 2.5% dimethyl sulfoxide (DMSO)), all of the following derivatives showed an excellent kinetic solubility (>250 μM; see Table S4, SI). In terms of stability, all of the compounds displayed an excellent profile both in mouse plasma (t1/2 > 120 min) and in mouse liver microsomes (t1/2 > 60 min). Taken together, these data confirm a favorable druglike profile of these tetrahydroquinazoline derivatives.
Lastly, the lead compound 14 was tested at 2 μM for off-target activity (both as agonist and as antagonist) on a set of 47 proteins for safety profiling. These include the G protein-coupled receptor (GPCR), nonkinase and kinase enzymes, transporters, ion channels (including the human ether-à-go-go-related gene (hERG) channel), and nuclear receptors. The compound showed no noteworthy activity toward these unwanted targets (see the SI); only a mild inhibitory effect against cyclooxygenase-1 (COX-1) was observed with IC50 4–4.5 μM, and it was thus much less potent than the reference COX-1 inhibitor indomethacin (IC50 0.051 μM; see the SI). The mechanistic implications of mild COX-1 inhibition remain to be investigated.
Encouraged by these results, we also evaluated the antiproliferative activity of the compounds in a small panel of human cancer cell lines (Table 3). We found an antiproliferative activity with IC50 values in the low micromolar range against melanoma (A375 and G-361), breast (MCF7), endometrial (HeLa), lung (A549), and androgen-independent prostate (DU145) cancer cell lines. In some cases, the antiproliferative activity was comparable to that of etoposide, used as the reference compound, while in others, such as lung and endometrial cancer cells, it was less sensitive (IC50 values ∼20 μM). It is notable that the most potent compound in vitro, 14, did not induce the phosphorylation of H2AX (see Figure S1, SI), which further indicates that the measured cytotoxicity is most likely due to cellular inhibition of topoII, rather than poisoning. Reassuringly, also the enantiomers 3 and 4 were equally active compared to the racemic mixture 1, reinforcing the evidence of no/little influence on cytotoxic mechanisms and activity, of the chiral center in position 6 of the tetrahydroquinazoline scaffold.
Table 3. Cell Proliferation Results in Various Cancer Cell Linesa.
| cell line | 1 | 3 | 4 | 10 | 11 | 14 | etoposide |
|---|---|---|---|---|---|---|---|
| DU145 | 15.1 ± 5.5 | 17.9 ± 7.4 | 11.1 ± 6.5 | 10.5 ± 6.0 | 8.8 ± 1.5 | 11.5 ± 1.3 | 0.5 ± 0.2 |
| A549 | 19.4 ± 1.8 | 23.3 ± 2.3 | 17.8 ± 0.6 | 22.9 ± 6.5 | 18.0 ± 0.1 | 17.1 ± 8.7 | 2.4 ± 2.0 |
| HeLa | 17.0 ± 1.9 | 17.1 ± 3.2 | 21.8 ± 4.6 | 20.8 ± 4.5 | 17.2 ± 2.6 | 38.2 ± 9.1 | 1.2 ± 0.4 |
| MCF7 | 13.9 ± 2.3 | 14.2 ± 2.5 | 16.2 ± 2.6 | 16.0 ± 0.5 | 15.8 ± 1.4 | 15.8 ± 1.3 | 8.9 ± 1.6 |
| A375 | 9.9 ± 0.1 | 10.7 ± 0.1 | 10.8 ± 0.1 | 12.3 ± 1.8 | 15.1 ± 4.8 | 12.6 ± 0.7 | 0.5 ± 0.1 |
| G-361 | 1.4 ± 0.4 | 1.6 ± 0.5 | 1.8 ± 0.6 | 3.0 ± 0.7 | 6.6 ± 3.1 | 8.1 ± 2.9 | 3.8 ± 3.0 |
A549, lung adenocarcinoma; DU145, androgen-independent prostate cancer; MCF7, breast adenocarcinoma; HeLa, cervical carcinoma; A375, human melanoma; and G-361, human melanoma. Values shown correspond to the IC50 (μM) ± SD of at least two independent experiments.
Finally, we evaluated the pharmacokinetic (PK) profile of 14 in mice (Table 4 and Figure 5). We found that 14 quickly enters the bloodstream after a single intraperitoneal (I.P. 10 mg/kg) administration, reaching a maximal plasma concentration of 0.68 μg/mL after 15 min. The half-life was 149 min in circulation, still being present in plasma 360 min after injection. The compound also showed good clearance values (0.116 L/(min kg)). Finally, 14 was able to reach the brain, with a maximum concentration of compound at 60 min, and was still present in the brain 360 min after injection. This favorable pharmacokinetic profile confirmed 14 as a highly promising lead compound for future efficacy studies in animal models for cancer, including brain tumors.
Table 4. Pharmacokinetic Parameters of 14a.
| Cmax | 0.68 μg/mL | t1/2 | 149 min |
| tmax | 15 min | VD | 24.9 L |
| AUC | 73.1 μg min/mL | CL | 0.116 L/min |
Maximum plasma concentration (Cmax); time to reach maximum concentration (tmax); area under the curve (AUC); half-life (t1/2); volume of distribution (VD); and clearance (CL).
Figure 5.

Pharmacokinetic profile of compound 14 (ARN-21934) in mouse plasma (top) and brain (down). Strain: C57B6/J. Route of administration: I.P. Dose: 10 mg/kg.
Conclusions
In summary, we have identified a class of potent topoII inhibitors that are based on a tetrahydroquinazoline core. We show that these compounds act as potent topoII inhibitors, with no accumulation of the covalent enzyme/DNA cleavage complex. Therefore, these compounds act as topoII inhibitors as opposed to poisons. Focused SAR studies showed that 2-pyridine, 4-substituted aniline, and 6-amino substitutions are mandatory for activity. Indeed, the introduction of a dimethylamino group significantly boosts topoII inhibition and yields 14, which is highly selective for topoIIα over topoIIβ. Notably, these tetrahydroquinazoline derivatives inhibit cell proliferation in various cancer cell lines, especially G-361 melanoma cells. These few novel prototype compounds also show high solubility and a favorable metabolic stability profile. In addition, compound 14 (ARN-21934) displays favorable pharmacokinetics in vivo, including brain penetration. Thus, our results support ARN-21934 for further lead optimization studies to develop novel topoII-targeted therapeutic agents that may lack the development of secondary leukemias associated with topoII poisoning. Further expansion of this chemical class will be reported in due course.
Experimental Section
Chemistry
All of the commercially available reagents and solvents were used as purchased from vendors without further purification. Dry solvents were purchased from Sigma-Aldrich. Automated column chromatography purifications were done using a Teledyne ISCO apparatus (CombiFlash Rf) with prepacked silica gel columns of different sizes (from 4 to 120 g) and mixtures of increasing polarity of cyclohexane and ethyl acetate (EtOAc), cyclohexane and tert-butyl methyl ether (TBME), or dichloromethane (DCM) and ethanol (EtOH). NMR experiments were run on a Bruker Avance III 400 system (400.13 MHz for 1H, and 100.62 MHz for 13C), equipped with a broadband Inverse (BBI) probe, and Z-gradients. Spectra were acquired at 300 K, using deuterated dimethyl sulfoxide (DMSO-d6), deuterated methanol (CD3OD), or deuterated chloroform (CDCl3) as solvents. For 1H NMR, data are reported as follows: chemical shift, multiplicity (s, singlet; d, doublet; dd, doublet of doublets; dt, doublet of triplets; td, triplet of doublets; t, triplet; q, quartet; m, multiplet), coupling constants (Hz), and integration. Ultraperformance liquid chromatography (UPLC)/mass spectroscopy (MS) analyses were run on a Waters ACQUITY UPLC/MS system consisting of a single quadrupole detector (SQD) mass spectrometer equipped with an electrospray ionization (ESI) interface and a photodiode array detector. The photodiode array (PDA) range was 210–400 nm. Analyses were performed on an ACQUITY UPLC BEH C18 column (50 × 2.1 mm2 ID, particle size 1.7 μm) with a VanGuard BEH C18 precolumn (5 × 2.1 mm2 ID, particle size 1.7 μm). The mobile phase was 10 mM NH4OAc in H2O at pH 5 adjusted with CH3COOH (A) and 10 mM NH4OAc in CH3CN–H2O (95:5) at pH 5.0 (B). Two types of gradients were applied depending on the analysis, gradient 1 (5–95% mobile phase B in 2.5 min) or gradient 2 (50–100% mobile phase B in 2.5 min). Electrospray ionization in positive and negative modes was applied. Unless otherwise indicated, all LC/MS instruments, columns, and software were from Waters Inc. (Milford, MA). An LC/MS quality check (QC) was performed for all compounds before testing. A 10 mM stock solution of the compound was prepared in DMSO-d6, diluted 20-fold with ACN–H2O (1:1), and analyzed on a Waters ACQUITY UPLC/MS system consisting of a single quadrupole detector (SQD) mass spectrometer equipped with an electrospray ionization interface and a photodiode array detector. Electrospray ionization in positive and negative modes was applied in the mass scan range 100–500 Da. The PDA range was 210–400 nm. The analyses were run on an ACQUITY UPLC BEH C18 column (100 × 2.1 mm2 ID, particle size 1.7 μm) with a VanGuard BEH C18 precolumn (5 × 2.1 mm2 ID, particle size 1.7 μm). The mobile phase was 10 mM NH4OAc in H2O at pH 5 adjusted with AcOH (A) and 10 mM NH4OAc in ACN–H2O (95:5) at pH 5 (B) with 0.5 mL/min as the flow rate. A linear gradient was applied: 0–0.2 min: 10% B, 0.2–6.2 min: 10–90% B, 6.2–6.3 min: 90–100%, 6.3–7.0 min: 100% B. All tested compounds showed ≥95% purity by NMR and UPLC/MS analyses.
General Procedure Reaction A (Scheme 1a): Pyrimidone Fused Ring Formation
A suspension of the corresponding cyclic 2-oxo ethylester (1 mmol) in EtOH (4.5 mL), urea (5 mmol), and sodium methoxide (4.5 mmol) was stirred at reflux temperature for 16 h. Afterward, the reaction crude was concentrated to dryness at low pressure, resulting in a solid triturated in water (0.5 mL), ice-cooled, pH-adjusted to 8–9 with concentrated HCl, and filtrated. The resulting solid was then rinsed with methanol (0.5 mL) and diethyl ether (0.5 mL), yielding the title compound.
General Procedure Reaction B (Scheme 1a): Pyrimidone Fused Ring Chlorination
A suspension of the corresponding pyrimidone fused ring obtained from general method A (1 mmol) in POCl3 (1.5 mL) was stirred at 120 °C under a N2 atmosphere for 5 h (total reaction crude solution observed). POCl3 was then evaporated at low pressure. The resulting residue was dissolved in dichloromethane (3 mL) and poured onto ice-cold NaHCO3 saturated solution (18 mL), and the aqueous pH was adjusted to 7–8 with NaHCO3 (no gas evolution was observed after addition). The organic layer was separated, dried over Na2SO4, and concentrated to dryness at low pressure. The final normal-phase chromatography purification yielded the title compound.
General Procedure Reaction C (Scheme 1a): Aniline Introduction
A suspension of dichlorinated pyrimidinic fused ring obtained from general method B (1 mmol), the corresponding aniline (1.1 mmol), and diisopropylethylamine (5 mmol) in 2-propanol (2 mL) was stirred in a CEM microwave apparatus at 100–160 °C (depending on the corresponding aniline) until reaction completion or no crude evolution was observed. Then, the reaction crude was concentrated to dryness at low pressure, solved in dichloromethane (20 mL), extracted with NaHCO3 saturated solution (20 mL), dried over Na2SO4, and concentrated to dryness at low pressure. The final normal phase purification yielded the title compound.
General Procedure Reaction D (Scheme 1a): Suzuki Coupling Reaction
A suspension of the compound obtained from general method C (1 mmol), the corresponding boronic acid (1.2 mmol), the PdCl2(dppf) dichloromethane complex (0.1 mmol), and K2CO3 2 M solution (2 mmol) in 1,4-dioxane (10 mL) was stirred in a CEM microwave apparatus at 120 °C for 2 h. The resulting crude was portioned between dichloromethane (25 mL) and NaHCO3 saturated solution (25 mL), and the organic layer was dried over Na2SO4 and concentrated to dryness at low pressure. The final normal phase purification yielded the title compound.
General Procedure Reaction E (Scheme 1a): Benzyl Group Removal
Under a N2 atmosphere, a suspension of the compound to be deprotected (1 mmol), ammonium formate (4 mmol), and Pd(OH)2/C (20% of the starting material weight) was stirred at reflux temperature until reaction completion. The catalyst was filtered off through a celite coarse patch, and the resulting filtrate was concentrated to dryness at low pressure. The final normal phase purification yielded the title compound.
General Procedure Reaction F (Scheme 1b): 2-Pyrido-4-pyrimidone Fused Ring Formation
A suspension of methyl 5-(dibenzylamino)-2-oxo-cyclohexanecarboxylate (1 mmol) in ethanol (4.5 mL), the corresponding pyridinecarboxamidine hydrochloride (5 mmol), and sodium methoxide (5.5 mmol) was stirred at reflux temperature for 16 h. Afterward, the reaction crude was concentrated to dryness at low pressure, triturated in water (4.5 mL), and filtered. The resulting solid was then rinsed with MeOH (0.5 mL), yielding the title compound.
General Procedure Reaction G (Scheme 1b): 2-Pyrido-4-pyrimidone Fused Ring Chlorination
A suspension of the corresponding pyrimidone fused ring obtained from general procedure reaction F (1 mmol) in POCl3 (1.5 mL) was stirred at 120 °C under a N2 atmosphere until the total solution was observed (around 4 h). POCl3 was then evaporated at low pressure, and the resulting residue was dissolved in dichloromethane (15 mL) and poured onto ice-cold NaHCO3 saturated solution (15 mL). The organic layer was separated, dried over Na2SO4, and concentrated to dryness at low pressure. Purification by normal phase chromatography finally yielded the title compound.
General Procedure Reaction H (Scheme 1b): Buchwald Coupling Reaction
A mixture of Pd(OAc)2 (0.10 mmol) and rac-BINAP (0.10 mmol) in 1,4-dioxane (5 mL) was stirred under Ar flushing for 10 min. Then, the corresponding 2-chloro-4-pyrido-pyrimidine fused ring obtained from general procedure reaction G (1 mmol), the corresponding substituted aniline (1.2 mmol), and Cs2CO3 (1.4 mmol) were stepwise added. The reaction mixture was stirred in a CEM microwave apparatus at 100 °C until reaction completion, filtrated through a celite coarse patch, rinsed with DCM, and concentrated to dryness at low pressure. The final normal phase purification yielded the title compound.
General Procedure Reaction I (Scheme 1b): Benzyl Group Removal
Under a N2 atmosphere, a suspension of the compound to be deprotected obtained from general procedure reaction H (1 mmol), ammonium formate (4 mmol), and Pd(OH)2/C (20% of the starting material weight) was stirred at reflux temperature until reaction completion. The catalyst was filtered off through a celite coarse patch, and the resulting filtrate was concentrated to dryness at low pressure. The final normal phase purification yielded the title compound.
Synthesis of Compound 1: N4-(3-Fluorophenyl)-2-(4-pyridyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine
Step 1: Synthesis of N,N-Dibenzyl-1,4-dioxaspiro[4.5]decan-8-amine (Reaction 1, Scheme 1a)
A solution of 1,4-cyclohexanedionemonoethyleneacetal (5000 mg, 31.05 mmol) in dry 1,2-dichloroethane (129 mL), dibenzylamine (6.3 mL, 31.05 mmol), and acetic acid (1.8 mL, 31.05 mmol) was stirred for 15 min at room temperature. Then, sodium triacetoxyborohydride (10 391.9 mg, 46.58 mmol) was portionwise added and the reaction mixture was stirred at room temperature for 16 h. Afterward, it was diluted with DCM (100 mL) and extracted with NaHCO3 10% solution (100 mL), and the aqueous layer was extracted twice with DCM (2 × 100 mL). The combined organic layers were dried over Na2SO4 and concentrated to dryness at low pressure. Purification by typical silica gel flash chromatography (cyclohexane/TBME from 100/0 to 90/10) afforded the pure title compound (8070 mg, yield 77%) as a white solid. Rt = 2.15 min (gradient 1); MS (ESI) m/z: 338.3 [M – H]+, [M – H]+ calcd: 338.5. 1H NMR (400 MHz, CDCl3) δ 7.39–7.33 (m, 4H), 7.28 (dd, J = 8.5, 7.0 Hz, 4H), 7.24–7.16 (m, 2H), 3.96–3.87 (m, 4H), 3.64 (s, 4H), 2.57 (tt, J = 11.5, 3.6 Hz, 1H), 1.90–1.81 (m, 2H), 1.81–1.72 (m, 2H), 1.67 (td, J = 12.5, 3.5 Hz, 2H), 1.45 (td, J = 13.0, 4.3 Hz, 2H).
Step 2: Synthesis of 4-(Dibenzylamino)cyclohexanone (Reaction 2, Scheme 1a)
A solution of 2 M HCl (62 mL, 124.70 mmol) was added to an N,N-dibenzyl-1,4-dioxaspiro[4.5]decan-8-amine (8070 mg, 23.19 mmol) solution in tetrahydrofuran (62 mL). The reaction mixture was stirred under a N2 atmosphere at reflux temperature for 4 h, cooled in an ice/water bath, basified with NaOH 5 M solution, and extracted with EtOAc (3 × 50 mL). The combined organic layers were dried over Na2SO4 and concentrated to dryness at low pressure. Purification by typical silica gel flash chromatography (cyclohexane/TBME from 100/0 to 90/10) afforded the pure title compound (6464 mg, yield 95%). Rt = 1.83 min (gradient 1); MS (ESI) m/z: 294.2 [M – H]+, [M – H]+ calcd: 294.2. 1H NMR (400 MHz, CDCl3) δ 7.40–7.35 (m, 4H), 7.34–7.28 (m, 4H), 7.26–7.20 (m, 2H), 3.66 (s, 4H), 3.02 (tt, J = 11.5, 3.4 Hz, 1H), 2.43 (p, J = 2.4 Hz, 1H), 2.39 (p, J = 2.4 Hz, 1H), 2.31–2.22 (m, 2H), 2.20–2.14 (m, 2H), 1.88–1.78 (m, 2H).
Step 3: Synthesis of Methyl 5-(Dibenzylamino)-2-oxo-cyclohexanecarboxylate (Reaction 3, Scheme 1a)
Under a N2 atmosphere, a solution of 4-(dibenzylamino)cyclohexanone (6200 mg, 20.50 mmol) in tetrahydrofuran (2.5 mL) was dropwise added to a suspension of KH (5261.3 mg, 65.59 mmol) and NaH (414.2 mg, 16.40 mmol) in dry tetrahydrofuran (256 mL) at room temperature. The reaction mixture was stirred for 30 min, and dimethyl carbonate was added (5.9 mL, 69.08 mmol), stirred at reflux temperature for 3 h, cooled to room temperature, and added to a cold NaHCO3 saturated solution (100 mL). The organic layer was separated, and the aqueous one was extracted with ethyl acetate (100 mL). The combined organic layers were dried over Na2SO4 and concentrated to dryness at low pressure. Purification by typical silica gel flash chromatography (cyclohexane/TBME from 100/0 to 90/10) afforded the pure title compound (7058 mg, yield 98%). Retention time 2.54 min (gradient 1); MS (ESI) m/z: 352.2 [M – H]+, [M – H]+ calcd: 352.2. 1H NMR (400 MHz, CDCl3) δ 12.06 (s, 1H), 7.42–7.34 (m, 4H), 7.29 (dd, J = 8.3, 6.7 Hz, 4H), 7.24–7.18 (m, 2H), 3.76 (s, 3H), 3.72 (d, J = 14.0 Hz, 2H), 3.62 (d, J = 14.0 Hz, 2H), 2.88–2.73 (m, 1H), 2.54–2.43 (m, 1H), 2.43–2.18 (m, 3H), 2.05–1.94 (m, 1H), 1.74–1.57 (m, 1H).
Step 4: Synthesis of 6-(Dibenzylamino)-5,6,7,8-tetrahydro-1H-quinazoline-2,4-dione (Compound A1, Scheme 1b)
This compound was obtained using 5-(dibenzylamino)-2-oxo-cyclohexanecarboxylate (7040.0 mg, 20.03 mmol) following the general procedure A previously described, affording a brown solid (5865 mg, yield 81%). Rt = 0.94 min (gradient 1); MS (ESI) m/z: 362.1 [M – H]+, [M – H]+ calcd: 362.4. 1H NMR (400 MHz, DMSO-d6) δ 10.85 (br s, 1H), 10.56 (br s, 1H), 7.36 (d, J = 7.2 Hz, 4H), 7.30 (t, J = 7.2 Hz, 4H), 7.20 (t, J = 7.2 Hz, 2H), 3.67 (d, J = 14.2 Hz, 2H), 3.59 (d, J = 14.2 Hz, 2H), 2.76–2.61 (m, 1H), 2.48–2.37 (m, 2H), 2.37–2.22 (m, 1H), 2.25–2.11 (m, 1H), 2.06–1.93 (m, 1H), 1.64 (qd, J = 12.1, 5.6 Hz, 1H).
Step 5: Synthesis of N,N-Dibenzyl-2,4-dichloro-5,6,7,8-tetrahydroquinazolin-6-amine (Compound B1, Scheme 1b)
This compound was obtained using compound A1 (5862 mg, 16.22 mmol) following the general procedure B previously described and affording a white solid (5685 mg, yield 88%). Rt = 2.60 min (gradient 2); MS (ESI) m/z: 398.2/400.2/402.2 [M – H]+, [M – H]+ calcd: 398.2/400.2/402.2. 1H NMR (400 MHz, CDCl3) δ 7.41 (d, J = 7.4 Hz, 4H), 7.32 (t, J = 7.4 Hz, 4H), 7.23 (d, J = 7.4 Hz, 2H), 3.82 (d, J = 13.7 Hz, 2H), 3.69 (d, J = 13.7 Hz, 2H), 3.19–3.00 (m, 1H), 3.02–2.86 (m, 1H), 2.87–2.65 (m, 2H), 2.42–2.12 (m, 1H), 2.01–1.56 (m, 2H).
Step 6: Synthesis of N6,N6-Dibenzyl-2-chloro-N4-(3-fluorophenyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine (Compound C1.1, Scheme 1b)
This compound was obtained using compound B1 (400 mg, 1.0 mmol) and 3-fluoroaniline (0.12 mL, 1.21 mmol) following the general procedure C previously described at 100 °C for 72 h. Final normal phase purification (cyclohexane/TBME from 100/0 to 80/20) afforded the pure title compound (152 mg, yield 32%). Rt = 2.61 min (gradient 2); MS (ESI) m/z: 471.1/473.1 [M – H]+, [M – H]+ calcd: 471.2/473.2. 1H NMR (400 MHz, CDCl3) δ 7.56 (dt, J = 11.0, 2.3 Hz, 1H), 7.45–7.38 (m, 4H), 7.37–7.28 (m, 5H), 7.30–7.19 (m, 3H), 6.82 (tdd, J = 8.2, 2.5, 1.2 Hz, 1H), 6.38 (s, 1H), 3.86 (d, J = 14.1 Hz, 2H), 3.67 (d, J = 14.0 Hz, 2H), 3.17–3.05 (m, 1H), 2.95 (ddd, J = 18.2, 5.0, 2.4 Hz, 1H), 2.71 (ddd, J = 18.1, 12.1, 5.6 Hz, 1H), 2.55–2.46 (m, 2H), 2.32–2.21 (m, 1H), 1.78 (qd, J = 12.3, 5.1 Hz, 1H).
Step 7: N6,N6-Dibenzyl-N4-(3-fluorophenyl)-2-(4-pyridyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine (Compound D1.1, Scheme 1b)
This compound was obtained using compound C1.1 (150 mg, 0.32 mmol) and pyridine-4-boronic acid (52 mg, 0.38 mmol) following the general procedure D previously described. Final normal phase purification (cyclohexane/TBME from 80/20 to 50/20) afforded the pure title compound (157 mg, yield 96%). Rt = 2.74 min (gradient 2); MS (ESI) m/z: 516.4 [M – H]+, [M – H]+ calcd: 516.2. 1H NMR (400 MHz, DMSO-d6) δ 8.72 (s, 1H), 8.70–8.67 (m, 2H), 8.10–8.05 (m, 2H), 7.73 (dt, J = 12.0, 2.3 Hz, 1H), 7.63 (ddd, J = 8.2, 2.0, 0.9 Hz, 1H), 7.48–7.39 (m, 5H), 7.32 (dd, J = 8.3, 6.9 Hz, 4H), 7.25–7.17 (m, 2H), 6.92 (tdd, J = 8.4, 2.5, 0.9 Hz, 1H), 3.80 (d, J = 14.2 Hz, 2H), 3.70 (d, J = 14.2 Hz, 2H), 3.05–2.84 (m, 3H), 2.84–2.63 (m, 2H), 2.16 (d, J = 12.3 Hz, 1H), 1.85 (tt, J = 12.0, 6.2 Hz, 1H).
Step 8: N4-(3-Fluorophenyl)-2-(4-pyridyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine (Compound 1, Scheme 1b)
This compound was obtained using compound D1.1 (154.3 mg, 0.29 mmol) following the general procedure E previously described for 4 h. Final normal phase purification (DCM/DCM:NH3 1 M MeOH 4:1 from 90/10 to 60/40) afforded the pure title compound as a solid (62.9 mg, yield 64%). Rt = 1.53 min (gradient 1); MS (ESI) m/z: 336.2 [M – H]+, [M – H]+ calcd: 336.2. HRMS m/z: 336.1623, calcd for C19H19FN5+: 336.1624. QC analysis: Rt = 2.58 min, m/z: 336.10 [M – H]+, UV (215 nm): 98%. 1H NMR (400 MHz, DMSO-d6) δ 8.73 (s, 1H), 8.72–8.67 (m, 2H), 8.16–8.05 (m, 2H), 7.75 (dt, J = 12.1, 2.3 Hz, 1H), 7.64 (dd, J = 8.0, 1.9 Hz, 1H), 7.41 (td, J = 8.2, 6.9 Hz, 1H), 6.90 (td, J = 8.4, 2.6 Hz, 1H), 3.27 (s, 3H), 2.89 (dddd, J = 19.8, 15.1, 8.6, 5.4 Hz, 3H), 2.46–2.35 (m, 1H), 2.09–1.89 (m, 1H), 1.70 (dtd, J = 12.6, 9.5, 5.9 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 163.08 (C), 162.05 (CF, d, JCF = 240.7 Hz), 158.61 (C), 157.26 (C), 150.24 (CH), 145.23 (C), 141.69 (C, d, JCF = 11.1 Hz), 129.86 (CH, d, JCF = 9.6 Hz), 121.29 (CH), 117.23 (CH, d, JCF = 1.5 Hz), 112.76 (C), 109.11 (CH, d, JCF = 21.0 Hz), 108.11 (CH, d, JCF = 25.9 Hz), 46.01 (CH), 31.84 (CH2), 30.60 (CH2), 30.35 (CH2).
Synthesis of Compound 2: N-(2-Fluorophenyl)-2-(4-pyridyl)-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-amine
Step 1: Synthesis of 5,6,7,8-Tetrahydro-1H-quinazoline-2,4-dione (Compound A2, Scheme 1b)
This compound was obtained using ethyl 2-oxocyclohexanecarboxylate (10 000.0 mg, 55.81 mmol) following the general procedure A previously described and affording a brown solid (8440 mg, yield 91%). Rt = 1.01 min (gradient 1); MS (ESI) m/z: 167.1 [M – H]+, [M – H]+ calcd: 167.1. 1H NMR (400 MHz, DMSO-d6) δ 10.71 (s, 2H), 2.29 (t, J = 6.1 Hz, 2H), 2.13 (t, J = 6.0 Hz, 2H), 1.61 (dddd, J = 17.5, 9.3, 7.6, 4.6 Hz, 4H).
Step 2: Synthesis of 2,4-Dichloro-5,6,7,8-tetrahydroquinazoline (Compound B2, Scheme 1a)
This compound was obtained using compound A2 (3000 mg, 18.05 mmol) following the general procedure B previously described and affording a pure white solid (2055 mg, yield 56%). Rt = 2.21 min (gradient 1); MS (ESI) m/z: 203.1/205.1/207.1 [M – H]+, [M – H]+ calcd: 203.1/205.1/207.1. 1H NMR (400 MHz, CDCl3) δ 2.88 (ddt, J = 5.4, 4.1, 2.5 Hz, 2H), 2.73 (ddt, J = 6.6, 4.6, 2.3 Hz, 2H), 1.88 (h, J = 3.8, 3.3 Hz, 4H).
Step 3: Synthesis of 2-Chloro-N-(3-fluorophenyl)-5,6,7,8-tetrahydroquinazolin-4-amine (Compound C2, Scheme 1b)
This compound was obtained using compound B2 (300 mg, 1.48 mmol) and 3-fluoroaniline (0.16 mL, 1.21 mmol) following the general procedure C previously described at 120 °C for 72 h. Final normal phase purification (cyclohexane/EtOAc from 100/0 to 80/20) afforded the pure title compound (115 mg, yield 28%). Rt = 2.32 min (gradient 1); MS (ESI) m/z: 278.1/280.1 [M – H]+, [M – H]+ calcd: 278.1/280.1. 1H NMR (400 MHz, DMSO-d6) δ 8.76 (s, 1H), 7.61 (dt, J = 11.9, 2.3 Hz, 1H), 7.48 (ddd, J = 8.2, 2.0, 0.9 Hz, 1H), 7.37 (m, 1H), 6.91 (tdd, J = 8.5, 2.6, 0.9 Hz, 1H), 2.69–2.60 (m, 2H), 2.58–2.52 (m, 2H), 1.86–1.69 (m, 4H).
Step 4: Synthesis of N-(2-Fluorophenyl)-2-(4-pyridyl)-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-amine (Compound 2, Scheme 1b)
This compound was obtained using compound C2 (110 mg, 0.40 mmol) and pyridine-4-boronic acid (64.9 mg, 0.48 mmol) following the general procedure D previously described. Final normal phase purification (cyclohexane/EtOAc from 80/20 to 60/40) afforded the pure title compound (65 mg, yield 51%). Rt = 2.41 min (gradient 1); MS (ESI) m/z: 321.1 [M – H]+, [M – H]+ calcd: 321.1. HRMS m/z: 321.1509, calcd for C19H18FN4+: 321.1515. QC analysis: Rt = 4.82 min, m/z: 321.79 [M – H]+, UV (215 nm): >99.5%. 1H NMR (400 MHz, DMSO-d6) δ 8.72–8.69 (m, 2H), 8.62 (s, 1H), 8.15–8.08 (m, 2H), 7.77 (dt, J = 12.1, 2.3 Hz, 1H), 7.64 (dd, J = 8.1, 1.8 Hz, 1H), 7.41 (td, J = 8.2, 6.9 Hz, 1H), 6.90 (td, J = 8.4, 2.6 Hz, 1H), 2.79 (t, J = 5.9 Hz, 2H), 2.64 (t, J = 5.8 Hz, 2H), 1.94–1.77 (m, 4H). 13C NMR (151 MHz, DMSO-d6) δ 163.53 (C), 162.03 (CF, d, JCF = 240.9 Hz), 158.42 (C), 157.12 (C), 150.26 (CH), 145.21 (C), 141.64 (C, d, JCF = 11.0 Hz), 129.88 (CH, d, JCF = 9.2 Hz), 121.25 (CH), 117.25 (CH, d, JCF = 2.1 Hz), 114.08 (C), 109.14 (CH, d, JCF = 21.0 Hz), 108.12 (CH, d, JCF = 26.1 Hz), 31.98 (CH2), 22.59 (CH2), 21.62 (CH2).
Chiral Chromatographic Separation of Compound 3Scheme 1d ((6R)-N4-(3-Fluorophenyl)-2-(4-pyridyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine) and Compound 4Scheme 1d ((6S)-N4-(3-Fluorophenyl)-2-(4-pyridyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine)
Chiral semipreparative separation of both enantiomers by HPLC was run on a Waters Alliance HPLC instrument consisting of an e2695 Separation Module and a 2998 photodiode array detector. The PDA range was 210–400 nm. The separation was performed isocratically on a Chiralcel OD-H column (250 × 10 mm2 ID, particle size: 5 μm) using 0.1% triethylamine (TEA) heptane–EtOH (95:5) as the mobile phase at 5 mL/min. Postanalysis of each isolated enantiomer was performed on a Chiralcel OD-H column (250 × 4.6 mm2 ID, particle size: 5 μm) using 0.1% TEA heptane–EtOH (95:5) as the mobile phase at 1 mL/min. The chiral structure of enantiomers 3 and 4 was arbitrarily assigned.
(6R)-N4-(3-Fluorophenyl)-2-(4-pyridyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine (3, Scheme 1d)
Enantiomeric purity of 95.2% enantiomeric excess (ee) was obtained at 290 nm of the first eluting enantiomer at Rt = 63.3 min in a Chiralcel OD-H column. Rt = 1.53 min (gradient 1); MS (ESI) m/z: 336.2 [M – H]+, [M – H]+ calcd: 336.2. HRMS m/z: 336.1623, calcd for C19H19FN5+: 336.1624. QC analysis: Rt = 2.57 min, m/z: 336.11 [M – H]+, UV (215 nm): >99.5%. 1H NMR (400 MHz, DMSO-d6) δ 8.73 (s, 1H), 8.72–8.67 (m, 2H), 8.16–8.05 (m, 2H), 7.75 (dt, J = 12.1, 2.3 Hz, 1H), 7.64 (dd, J = 8.0, 1.9 Hz, 1H), 7.41 (td, J = 8.2, 6.9 Hz, 1H), 6.90 (td, J = 8.4, 2.6 Hz, 1H), 3.27 (s, 3H), 2.89 (dddd, J = 19.8, 15.1, 8.6, 5.4 Hz, 3H), 2.46–2.35 (m, 1H), 2.09–1.89 (m, 1H), 1.70 (dtd, J = 12.6, 9.5, 5.9 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 163.08 (C), 162.05 (CF, d, JCF = 240.7 Hz), 158.61 (C), 157.26 (C), 150.24 (CH), 145.23 (C), 141.69 (C, d, JCF = 11.1 Hz), 129.86 (CH, d, JCF = 9.6 Hz), 121.29 (CH), 117.23 (CH, d, JCF = 1.5 Hz), 112.76 (C), 109.11 (CH, d, JCF = 21.0 Hz), 108.11 (CH, d, JCF = 25.9 Hz), 46.01 (CH), 31.84 (CH2), 30.60 (CH2), 30.35 (CH2).
(6S)-N4-(3-Fluorophenyl)-2-(4-pyridyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine (4, Scheme 1d)
Enantiomeric purity was >99.5% ee at 290 nm of the second eluting enantiomer at Rt = 67. 6 min in the Chiralcel OD-H column. Rt = 1.53 min (gradient 1); MS (ESI) m/z: 336.2 [M – H]+, [M – H]+ calcd: 336.2. HRMS m/z: 336.1623, calcd for C19H19FN5+: 336.1624. QC analysis: Rt = 2.56 min, m/z: 336.14 [M – H]+, UV (215 nm): >99.5%. 1H NMR (400 MHz, DMSO-d6) δ 8.73 (s, 1H), 8.72–8.67 (m, 2H), 8.16–8.05 (m, 2H), 7.75 (dt, J = 12.1, 2.3 Hz, 1H), 7.64 (dd, J = 8.0, 1.9 Hz, 1H), 7.41 (td, J = 8.2, 6.9 Hz, 1H), 6.90 (td, J = 8.4, 2.6 Hz, 1H), 3.27 (s, 3H), 2.89 (dddd, J = 19.8, 15.1, 8.6, 5.4 Hz, 3H), 2.46–2.35 (m, 1H), 2.09–1.89 (m, 1H), 1.70 (dtd, J = 12.6, 9.5, 5.9 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 163.08 (C), 162.05 (CF, d, JCF = 240.7 Hz), 158.61 (C), 157.26 (C), 150.24 (CH), 145.23 (C), 141.69 (C, d, JCF = 11.1 Hz), 129.86 (CH, d, JCF = 9.6 Hz), 121.29 (CH), 117.23 (CH, d, JCF = 1.5 Hz), 112.76 (C), 109.11 (CH, d, JCF = 21.0 Hz), 108.11 (CH, d, JCF = 25.9 Hz), 46.01 (CH), 31.84 (CH2), 30.60 (CH2), 30.35 (CH2).
Synthesis of Compound 5: N4-(3-Fluorophenyl)-2-(3-pyridyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine
Step 1: Synthesis of N6,N6-Dibenzyl-N4-(3-fluorophenyl)-2-(3-pyridyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine (Compound D1.2, Scheme 1b)
This compound was obtained using compound C1.1 (55 mg, 0.12 mmol) and pyridine-3-boronic acid (19.2 mg, 0.14 mmol) following the general procedure D previously described. Final normal phase purification (cyclohexane/TBME from 80/20 to 50/50) afforded the pure title compound (58.0 mg, yield 96%). Rt = 3.0 min (gradient 2); MS (ESI) m/z: 516.4 [M – H]+, [M – H]+ calcd: 516.2. 1H NMR (400 MHz, CDCl3) δ 9.54 (dd, J = 2.2, 0.9 Hz, 1H), 8.65 (dd, J = 4.8, 1.7 Hz, 1H), 8.58 (dt, J = 7.9, 2.0 Hz, 1H), 7.68 (dt, J = 11.2, 2.3 Hz, 1H), 7.46 (d, J = 7.8 Hz, 5H), 7.41–7.29 (m, 7H), 7.26 (s, 2H), 6.81 (tdd, J = 8.3, 2.6, 1.2 Hz, 1H), 6.49 (s, 1H), 3.91 (d, J = 13.7 Hz, 2H), 3.71 (d, J = 14.0 Hz, 2H), 3.29–3.10 (m, 1H), 3.11–2.98 (m, 1H), 2.80 (ddd, J = 17.9, 12.2, 5.4 Hz, 1H), 2.65 (d, J = 8.3 Hz, 2H), 2.46–2.22 (m, 1H), 1.84 (qd, J = 12.2, 4.8 Hz, 1H).
Step 2: Synthesis of N4-(3-Fluorophenyl)-2-(3-pyridyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine (Compound 5, Scheme 1b)
This compound was obtained using compound D1.2 (58.0 mg, 0.11 mmol) following the general procedure E previously described for 4 h. Final normal phase purification (DCM/DCM:NH3 1 N MeOH 4:1 from 95/5 to 75/25) afforded the pure title compound (12.2 mg, yield 33%). Rt = 1.71 min (gradient 1); MS (ESI) m/z: 336.1 [M – H]+, [M – H]+ calcd: 336.2. HRMS m/z: 336.1620, calcd for C19H19 FN5+: 336.1624. QC analysis: Rt = 2.67 min, m/z: 336.10 [M – H]+, UV (215 nm): 95%. 1H NMR (400 MHz, CD3OD) δ 9.45–9.32 (m, 1H), 8.64 (dt, J = 8.0, 1.9 Hz, 1H), 8.58 (dd, J = 4.9, 1.7 Hz, 1H), 7.70 (dt, J = 11.7, 2.3 Hz, 1H), 7.56–7.44 (m, 2H), 7.35 (td, J = 8.2, 6.6 Hz, 1H), 6.83 (tdd, J = 8.4, 2.6, 0.9 Hz, 1H), 3.29–3.22 (m, 1H), 3.04–2.83 (m, 3H), 2.40 (dd, J = 16.3, 9.1 Hz, 1H), 2.21–2.08 (m, 1H), 1.77 (dtd, J = 12.8, 10.3, 6.2 Hz, 1H). 13C NMR (101 MHz, CD3OD) δ 164.21 (CF, d, JCF = 241.9 Hz), 163.95 (C), 159.68 (C), 150.93 (CH), 149.77 (CH), 137.16 (CH), 135.75 (C), 130.75 (CH, d, JCF = 9.3 Hz), 125.04 (CH), 118.43 (CH, d, JCF = 2.6 Hz), 112.15 (C), 110.71 (CH, d, JCF = 21.4 Hz), 109.86 (CH, d, JCF = 26.0 Hz), 47.71 (CH), 31.91 (CH2), 31.62 (CH2), 31.25 (CH2).
Synthesis of Compound 6:N4-(3-Fluorophenyl)-2-(2-pyridyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine
Step 1: Synthesis of 6-(Dibenzylamino)-2-(2-pyridyl)-5,6,7,8-tetrahydro-3H-quinazolin-4-one (Compound F1, Scheme 1c)
This compound was obtained using 5-(dibenzylamino)-2-oxo-cyclohexanecarboxylate (335.0 mg, 0.95 mmol) and pyridine-2-carboxamidine hydrochloride (774 mg, 4.77 mmol) following the general procedure F previously described and affording a pure white solid (300 mg, yield 74%). Rt = 1.89 min (gradient 2); MS (ESI) m/z: 423.5 [M – H]+, [M – H]+ calcd: 423.2. 1H NMR (400 MHz, DMSO-d6) δ 11.81 (s, 1H), 8.70 (dt, J = 4.6, 1.3 Hz, 1H), 8.23 (d, J = 7.9 Hz, 1H), 7.99 (td, J = 7.8, 1.7 Hz, 1H), 7.60 (ddd, J = 7.7, 4.7, 1.2 Hz, 1H), 7.43–7.36 (m, 4H), 7.31 (dd, J = 8.3, 6.8 Hz, 4H), 7.25–7.15 (m, 2H), 3.73 (d, J = 14.3 Hz, 2H), 3.65 (d, J = 14.2 Hz, 2H), 2.91–2.76 (m, 1H), 2.73 (s, 1H), 2.70–2.54 (m, 2H), 2.12–2.05 (m, 1H), 1.77 (qd, J = 12.0, 5.2 Hz, 1H).
Step 2: Synthesis of N,N-Dibenzyl-4-chloro-2-(2-pyridyl)-5,6,7,8-tetrahydroquinazolin-6-amine (Compound G1, Scheme 1c)
This compound was obtained using compound F1 (300.0 mg, 0.71 mmol) following the general procedure G previously described. Normal phase purification (CHCl3/CHCl3:MeOH 4:1 from 100/0 to 50/50) afforded the pure title compound (275 mg, yield 88%). Rt = 2.29 min (gradient 2); MS (ESI) m/z: 441/443 [M – H]+, [M – H]+ calcd: 441/443. 1H NMR (400 MHz, CDCl3) δ 8.82 (ddd, J = 4.7, 1.8, 0.9 Hz, 1H), 8.44 (dt, J = 8.0, 1.1 Hz, 1H), 7.82 (td, J = 7.8, 1.8 Hz, 1H), 7.46–7.39 (m, 4H), 7.37 (ddd, J = 7.5, 4.8, 1.2 Hz, 1H), 7.35–7.28 (m, 4H), 7.25–7.20 (m, 2H), 3.83 (d, J = 14.0 Hz, 2H), 3.70 (d, J = 14.1 Hz, 2H), 3.28 (ddd, J = 18.3, 5.0, 2.5 Hz, 1H), 3.16–3.00 (m, 2H), 2.98–2.79 (m, 2H), 2.29 (ddd, J = 12.5, 5.4, 2.6 Hz, 1H), 1.85 (qd, J = 12.3, 5.0 Hz, 1H).
Step 3: Synthesis of N6,N6-Dibenzyl-N4-(3-fluorophenyl)-2-(2-pyridyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine (Compound H1, Scheme 1c)
This compound was obtained using compound G1 (135 mg, 0.31 mmol) and 3-fluoroaniline (0.036 mL, 0.37 mmol) following the general procedure H previously described for 1 h. Final normal phase purification (cyclohexane/EtOAc from 40/60 to 60/40) afforded the pure title compound (131.1 mg, yield 83%). Rt = 2.26 min (gradient 1); MS (ESI) m/z: 516.5 [M – H]+, [M – H]+ calcd: 516.2. 1H NMR (400 MHz, DMSO-d6) δ 8.69 (ddd, J = 4.7, 1.8, 0.9 Hz, 1H), 8.63 (s, 1H), 8.20 (dt, J = 8.0, 1.1 Hz, 1H), 8.06 (dt, J = 12.5, 2.3 Hz, 1H), 7.90 (td, J = 7.7, 1.8 Hz, 1H), 7.70 (ddd, J = 8.3, 2.0, 0.9 Hz, 1H), 7.48–7.41 (m, 5H), 7.38 (td, J = 8.3, 7.0 Hz, 1H), 7.32 (t, J = 7.6 Hz, 4H), 7.26–7.18 (m, 2H), 6.86 (tdd, J = 8.4, 2.6, 0.9 Hz, 1H), 3.80 (d, J = 14.2 Hz, 2H), 3.70 (d, J = 14.2 Hz, 2H), 3.06–2.86 (m, 3H), 2.85–2.69 (m, 1H), 2.25–2.10 (m, 1H), 1.85 (qd, J = 12.2, 5.0 Hz, 1H).
Step 4: Synthesis of N4-(3-Fluorophenyl)-2-(2-pyridyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine (Compound 6, Scheme 1c)
This compound was obtained using compound H1 (131 mg, 0.25 mmol) following the general procedure I previously described for 4 h. Final normal phase purification (DCM/DCM:NH3 1 N MeOH 4:1 from 90/10 to 60/40) afforded the pure title compound (51.1 mg, yield 60%). Rt = 1.57 min (gradient 1); MS (ESI) m/z: 336.1 [M – H]+, [M – H]+ calcd: 336.2. HRMS m/z: 336.1623, calcd for C19H19FN5+: 336.1624. QC analysis: Rt = 2.70 min, m/z: 336.50 [M – H]+, UV (215 nm): 97%. 1H NMR (400 MHz, DMSO-d6) δ 8.71 (ddd, J = 4.7, 1.8, 0.9 Hz, 1H), 8.58 (s, 1H), 8.23 (dt, J = 8.0, 1.1 Hz, 1H), 8.10 (dt, J = 12.6, 2.3 Hz, 1H), 7.92 (td, J = 7.7, 1.8 Hz, 1H), 7.78–7.68 (m, 1H), 7.46 (ddd, J = 7.5, 4.7, 1.2 Hz, 1H), 7.35 (td, J = 8.2, 7.0 Hz, 1H), 6.90–6.79 (m, 1H), 3.18 (tdd, J = 8.4, 5.0, 3.0 Hz, 1H), 2.96–2.71 (m, 3H), 2.35 (dd, J = 17.0, 8.3 Hz, 1H), 2.00–1.89 (m, 1H), 1.64 (dtd, J = 12.7, 9.6, 5.8 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 163.48 (C), 162.56 (d, JCF = 240.1 Hz), 159.52 (C), 159.02 (C), 155.95 (C), 149.91 (CH), 142.61 (C, d, JCF = 11.5 Hz), 137.19 (CH), 130.14 (CH, d, JCF = 9.5 Hz), 124.82 (CH), 123.27 (CH), 117.00 (CH), 112.75 (C), 108.95 (CH, d, JCF = 21.2 Hz), 108.06 (CH, d, JCF = 26.8 Hz), 46.58 (CH), 32.65 (CH2), 31.62 (CH2), 30.89 (CH2).
Synthesis of Compound 7: N4-(3-Fluorophenyl)-N4-methyl-2-(4-pyridyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine
Step 1: Synthesis of 6-(Dibenzylamino)-2-(4-pyridyl)-5,6,7,8-tetrahydro-3H-quinazolin-4-one (Compound F2, Scheme 1c)
This compound was obtained using 5-(dibenzylamino)-2-oxo-cyclohexanecarboxylate (2000.0 mg, 5.69 mmol) and pyridine-4-carboxamidine hydrochloride (4624.0 mg, 28.46 mmol) following the general procedure F previously described and affording a pure white solid (1620 mg, yield 67%). Rt = 1.34 min (gradient 2); MS (ESI) m/z: 423.5 [M – H]+, [M – H]+ calcd: 423.2. 1H NMR (400 MHz, DMSO-d6) δ 12.78 (s, 1H), 8.79–8.69 (m, 2H), 8.03–7.93 (m, 2H), 7.40 (d, J = 7.1 Hz, 4H), 7.32 (t, J = 7.5 Hz, 4H), 7.21 (t, J = 7.3 Hz, 2H), 3.74 (d, J = 14.2 Hz, 2H), 3.66 (d, J = 14.2 Hz, 2H), 2.90–2.76 (m, 1H), 2.78–2.70 (m, 1H), 2.71–2.55 (m, 2H), 2.17–2.05 (m, 1H), 1.77 (qd, J = 12.0, 5.4 Hz, 1H).
Step 2: Synthesis of N,N-Dibenzyl-4-chloro-2-(4-pyridyl)-5,6,7,8-tetrahydroquinazolin-6-amine (Compound G2, Scheme 1c)
This compound was obtained using compound F2 (1620.0 mg, 3.83 mmol) following the general procedure G previously described. Normal phase purification (CHCl3/CHCl3:MeOH 4:1 from 95/5 to 65/35) afforded the pure title compound (1488 mg, yield 88%). Rt = 2.67 min (gradient 2); MS (ESI) m/z: 441/443 [M – H]+, [M – H]+ calcd: 441/443. 1H NMR (400 MHz, DMSO-d6) δ 8.99–8.89 (m, 2H), 8.55–8.39 (m, 2H), 7.73 (s, 2H), 7.62 (d, J = 6.0 Hz, 2H), 7.51–7.26 (m, 6H), 4.72 (d, J = 13.7 Hz, 2H), 4.60 (d, J = 12.2 Hz, 2H), 3.84–3.68 (m, 1H), 3.62–3.50 (m, 1H), 3.47–3.32 (m, 1H), 3.25–3.17 (m, 1H), 3.09–2.91 (m, 1H), 2.81–2.64 (m, 1H), 2.29–2.11 (m, 1H).
Step 3: Synthesis of N6,N6-Dibenzyl-N4-(3-fluorophenyl)-N4-methyl-2-(4-pyridyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine (Compound H2, Scheme 1c)
This compound was obtained using compound G2 (150.0 mg, 0.34 mmol) and 3-fluoro-N-methylaniline (52.7 mg, 0.41 mmol) following the general procedure H previously described for 6.5 h. Final normal phase purification (cyclohexane/EtOAc from 95/5 to 70/30) afforded the pure title compound (80.7 mg, yield 46%). Rt = 1.72 min (gradient 1); MS (ESI) m/z: 530.5 [M – H]+, [M – H]+ calcd: 530.3. 1H NMR (400 MHz, DMSO-d6) δ 8.76–8.67 (m, 2H), 8.22–8.18 (m, 2H), 7.45–7.36 (m, 2H), 7.35–7.26 (m, 1H), 7.29–7.19 (m, 3H), 7.21–7.16 (m, 2H), 7.16–7.10 (m, 4H), 7.03 (td, J = 8.4, 2.5 Hz, 1H), 6.97 (dd, J = 8.0, 2.0 Hz, 1H), 3.52 (s, 3H), 3.45 (d, J = 14.2 Hz, 2H), 3.40 (d, J = 14.2 Hz, 2H), 3.07–2.90 (m, 1H), 2.80–2.60 (m, 1H), 2.13–1.90 (m, 4H), 1.67 (qd, J = 12.0, 5.6 Hz, 1H).
Step 4: Synthesis of N4-(3-Fluorophenyl)-N4-methyl-2-(4-pyridyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine (Compound 7, Scheme 1c)
This compound was obtained using compound H2 (80.0 mg, 0.25 mmol) following the general procedure I previously described for 2 h. Final normal phase purification (DCM/DCM:NH3 1 N MeOH 4:1 from 95/5 to 75/25) afforded the pure title compound (11.9 mg, yield 23%). Rt = 1.57 min (gradient 1); MS (ESI) m/z: 350.1 [M – H]+, [M – H]+ calcd: 350.4. HRMS m/z: 350.1778, calcd for C20H21FN5+: 350.1781. QC analysis: Rt = 2.77 min, m/z: 350.20 [M – H]+, UV (215 nm): >99.5%. 1H NMR (400 MHz, DMSO-d6) 8.79–8.66 (m, 2H), 8.27–8.17 (m, 2H), 7.36 (td, J = 8.2, 6.8 Hz, 1H), 7.02 (dt, J = 11.0, 2.3 Hz, 1H), 6.97 (td, J = 8.4, 2.5 Hz, 1H), 6.88–6.83 (m, 1H), 3.54 (s, 3H), 3.07–2.91 (m, 1H), 2.91–2.75 (m, 2H), 2.20–2.07 (m, 1H), 1.93–1.76 (m, 1H), 1.64 (dd, J = 16.8, 8.3 Hz, 1H), 1.51 (dtd, J = 12.7, 9.3, 6.2 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 165.74 (C), 162.94 (C), 162.69 (CF, d, JCF = 243.3 Hz), 157.78 (C), 150.26 (CH), 144.90 (C), 130.69 (CH, d, JCF = 9.6 Hz), 121.42 (CH), 118.87 (CH, d, JCF = 2.3 Hz), 117.94 (C), 110.72 (CH, d, JCF = 20.9 Hz), 110.01 (CH, d, JCF = 23.5 Hz), 45.79 (CH), 40.78 (CH3), 35.40 (CH2), 30.58 (CH2), 30.44 (CH2).
Synthesis of Compound 8: N4-Phenyl-2-(4-pyridyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine
Step 1: Synthesis of N6,N6-Dibenzyl-2-chloro-N4-phenyl-5,6,7,8-tetrahydroquinazoline-4,6-diamine (Compound C1.2, Scheme 1b)
This compound was obtained using compound B1 (200 mg, 0.500 mmol) and aniline (0.05 mL, 0.55 mmol) following the general procedure C previously described at 100 °C for 16 h. Final normal phase purification (cyclohexane/TBME from 95/5 to 75/25) afforded the pure title compound (141.7 mg, yield 62%). Rt = 2.50 min (gradient 2); MS (ESI) m/z: 455.2/457.2 [M – H]+, [M – H]+ calcd: 455.2/457.2. 1H NMR (400 MHz, CDCl3) δ 7.62–7.53 (m, 2H), 7.43 (d, J = 7.5 Hz, 4H), 7.39–7.28 (m, 6H), 7.29–7.19 (m, 2H), 7.12 (t, J = 7.6 Hz, 1H), 6.42 (s, 1H), 3.87 (d, J = 14.1 Hz, 2H), 3.67 (d, J = 14.0 Hz, 2H), 3.18–3.02 (m, 1H), 2.97–2.86 (m, 1H), 2.68 (ddd, J = 18.1, 12.1, 5.5 Hz, 1H), 2.54 (d, J = 8.1 Hz, 2H), 2.33–2.16 (m, 1H), 1.77 (qd, J = 12.3, 5.0 Hz, 1H).
Step 2: Synthesis of N6,N6-Dibenzyl-N4-phenyl-2-(4-pyridyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine (Compound D1.3, Scheme 1b)
This compound was obtained using compound C1.2 (75 mg, 0.16 mmol) and pyridine-4-boronic acid (27.0 mg, 0.20 mmol) following the general procedure D previously described. Final normal phase purification (cyclohexane/TBME from 75/25 to 45/55) afforded the pure title compound (57 mg, yield 69%). Rt = 2.72 min (gradient 2); MS (ESI) m/z: 514.4 [M – H]+, [M – H]+ calcd: 514.3. 1H NMR (400 MHz, CDCl3) δ 8.75–8.64 (m, 2H), 8.22–8.12 (m, 2H), 7.75–7.65 (m, 2H), 7.43 (ddd, J = 16.0, 7.8, 1.6 Hz, 6H), 7.33 (t, J = 7.5 Hz, 4H), 7.29–7.20 (m, 3H), 7.15 (td, J = 7.3, 1.1 Hz, 1H), 6.44 (s, 1H), 3.90 (d, J = 14.0 Hz, 2H), 3.71 (d, J = 14.0 Hz, 2H), 3.28–3.10 (m, 1H), 3.11–2.96 (m, 1H), 2.80 (ddd, J = 17.9, 12.3, 5.4 Hz, 1H), 2.64 (d, J = 8.1 Hz, 2H), 2.43–2.24 (m, 1H), 1.83 (qd, J = 14.6, 13.4, 6.1 Hz, 1H).
Step 3: Synthesis of N4-Phenyl-2-(4-pyridyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine (Compound 8, Scheme 1b)
This compound was obtained using compound D1.3 (56.6 mg, 0.11 mmol) following the general procedure E previously described for 4 h. Final normal phase purification (DCM/DCM:NH3 1 N MeOH 4:1 from 95/5 to 75/25) afforded the pure title compound (20.2 mg, yield 57%). Rt = 1.43 min (gradient 1); MS (ESI) m/z: 318.3 [M – H]+, [M – H]+ calcd: 318.2. HRMS m/z: 318.1716, calcd for C19H20N5+: 318.1719. QC analysis: Rt = 2.36 min, m/z: 318.19 [M – H]+, UV (215 nm): >99.5%. 1H NMR (400 MHz, DMSO-d6) δ 8.74–8.61 (m, 2H), 8.54 (s, 1H), 8.22–8.03 (m, 2H), 7.85–7.70 (m, 2H), 7.39 (dd, J = 8.5, 7.3 Hz, 2H), 7.14–7.03 (m, 1H), 3.24–3.10 (m, 1H), 2.98–2.72 (m, 3H), 2.34 (dd, J = 16.9, 8.2 Hz, 1H), 2.06–1.88 (m, 1H), 1.64 (dtd, J = 12.6, 9.6, 5.8 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 162.60 (C), 158.85 (C), 157.23 (C), 150.16 (CH), 145.40 (C), 139.73 (C), 128.36 (CH), 122.96 (CH), 121.91 (CH), 121.34 (CH), 112.34 (C), 46.09 (CH), 32.06 (CH2), 30.90 (CH2), 30.35 (CH2).
Synthesis of Compound 9:N4-(2-Fluorophenyl)-2-(4-pyridyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine
Step 1: Synthesis of N6,N6-Dibenzyl-2-chloro-N4-(2-fluorophenyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine (Compound C1.3, Scheme 1b)
This compound was obtained using compound B1 (500 mg, 1.26 mmol) and 2-fluoroaniline (0.15 mL, 1.51 mmol) following the general procedure C previously described at 160 °C for 72 h. Final normal phase purification (cyclohexane/TBME from 100/0 to 80/20) afforded the title compound pure enough to be used in the next step (235 mg). Rt = 2.38 min (gradient 2); MS (ESI) m/z: 473.1/475.1 [M – H]+, [M – H]+ calcd: 473.2/475.2. 1H NMR (400 MHz, CDCl3) δ 8.39 (t, J = 8.5 Hz, 1H), 7.41 (d, J = 7.4 Hz, 4H), 7.32 (t, J = 7.5 Hz, 4H), 7.23 (t, J = 7.6 Hz, 2H), 7.18 (d, J = 8.6 Hz, 1H), 7.13 (dd, J = 9.9, 2.5 Hz, 1H), 7.10–7.04 (m, 1H), 6.63 (d, J = 3.8 Hz, 1H), 3.85 (d, J = 14.0 Hz, 2H), 3.68 (d, J = 14.0 Hz, 2H), 3.12 (q, J = 9.0 Hz, 1H), 3.03–2.89 (m, 1H), 2.72 (ddd, J = 18.2, 12.1, 5.7 Hz, 1H), 2.31–2.20 (m, 1H), 2.11–2.00 (m, 1H), 1.80 (qd, J = 12.2, 5.0 Hz, 1H), 1.31–1.21 (m, 1H).
Step 2: Synthesis of N6,N6-Dibenzyl-N4-(2-fluorophenyl)-2-(4-pyridyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine (Compound D1.4, Scheme 1b)
This compound was obtained using crude C1.3 (230 mg, 0.49 mmol) and pyridine-4-boronic acid (79.7 mg, 0.58 mmol) following the general procedure D previously described. Final normal phase purification (cyclohexane/TBME from 80:20 to 60:40) afforded the pure title compound (175 mg, overall yield 70% for steps 1 and 2). Rt = 2.63 min (gradient 2); MS (ESI) m/z: 516.4 [M – H]+, [M – H]+ calcd: 516.2. 1H NMR (400 MHz, CDCl3) δ 8.70 (dt, J = 4.8, 0.8 Hz, 2H), 8.49 (t, J = 8.2 Hz, 1H), 8.22–8.14 (m, 2H), 7.45 (d, J = 7.5 Hz, 4H), 7.33 (t, J = 7.5 Hz, 4H), 7.25–7.20 (m, 3H), 7.20–7.14 (m, 1H), 7.09 (q, J = 7.5, 7.0 Hz, 1H), 6.66 (s, 1H), 3.91 (d, J = 14.0 Hz, 2H), 3.73 (d, J = 14.0 Hz, 2H), 3.20 (s, 1H), 3.07 (dd, J = 17.1, 4.6 Hz, 1H), 2.82 (ddd, J = 17.9, 12.2, 5.4 Hz, 1H), 2.69 (d, J = 10.2 Hz, 2H), 2.33 (d, J = 12.5 Hz, 1H), 1.87 (qd, J = 12.3, 4.9 Hz, 1H).
Step 3: Synthesis of N4-(2-Fluorophenyl)-2-(4-pyridyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine (Compound 9, Scheme 1b)
This compound was obtained using compound D1.4 (170 mg, 0.33 mmol) following the general procedure E previously described for 4 h. Final normal phase purification (DCM/DCM:NH3 1 N MeOH 4:1 from 95/5 to 75/25) afforded the pure title compound (55.2 mg, yield 22%). Rt = 1.41 min (gradient 1); MS (ESI) m/z: 336.2 [M – H]+, [M – H]+ calcd: 336.2. HRMS m/z: 336.1617, calcd for C19H19FN5+: 336.1624. QC analysis: Rt = 2.29 min, m/z: 336.20 [M – H]+, UV (215 nm): 99%. 1H NMR (400 MHz, DMSO-d6) δ 8.64–8.58 (m, 2H), 8.51 (s, 1H), 7.97–7.91 (m, 2H), 7.58 (td, J = 7.6, 2.4 Hz, 1H), 7.36–7.21 (m, 4H), 3.18 (d, J = 11.0 Hz, 1H), 3.03–2.67 (m, 3H), 2.32 (dd, J = 16.9, 8.3 Hz, 1H), 2.06–1.87 (m, 1H), 1.64 (dtd, J = 12.7, 9.6, 5.7 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 163.09 (C), 157.69 (C), 150.51 (CH), 145.71 (C), 144.87 (C), 127.82 (CH, d, JCF = 2.0 Hz), 127.41 (CH, d, JCF = 3.0 Hz), 124.65 (CH, d, JCF = 3.5 Hz), 121.64 (CH), 116.12 (CH, d, JCF = 20.0 Hz), 112.52 (C), 46.48 (CH), 32.67 (CH2), 31.66 (CH2), 30.66 (CH2).
Synthesis of Compound 10: N4-(4-Fluorophenyl)-2-(4-pyridyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine
Step 1: Synthesis of N6,N6-Dibenzyl-2-chloro-N4-(4-fluorophenyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine (Compound C1.4, Scheme 1b)
This compound was obtained using compound B1 (300 mg, 0.75 mmol) and 4-fluoroaniline (0.07 mL, 0.75 mmol) following the general procedure C previously described at 100 °C for 16 h. Final normal phase purification (cyclohexane/TBME from 100/0 to 90/10) afforded the pure title compound (303 mg, yield 73%). Rt = 2.70 min (gradient 2); MS (ESI) m/z: 473.1 [M – H]+, [M – H]+ calcd: 473.2. 1H NMR (400 MHz, CDCl3) δ 7.70–7.58 (m, 3H), 7.45–7.38 (m, 4H), 7.37–7.28 (m, 6H), 7.12–7.02 (m, 2H), 3.67 (d, J = 14.0 Hz, 2H), 3.17–3.05 (m, 1H), 2.95 (ddd, J = 18.2, 5.0, 2.4 Hz, 1H), 2.71 (ddd, J = 18.1, 12.1, 5.6 Hz, 1H), 2.55–2.46 (m, 2H), 2.32–2.21 (m, 1H), 1.78 (qd, J = 12.3, 5.1 Hz, 1H).
Step 2: Synthesis of N6,N6-Dibenzyl-N4-(4-fluorophenyl)-2-(4-pyridyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine (Compound D1.5, Scheme 1b)
This compound was obtained using compound C1.4 (75 mg, 0.16 mmol) and pyridine-4-boronic acid (26.0 mg, 0.19 mmol) following the general procedure D previously described. Final normal phase purification (cyclohexane/TBME from 80/20 to 50/50) afforded the pure title compound (75.7 mg, yield 92%). Rt = 2.97 min (gradient 2); MS (ESI) m/z: 516.4 [M – H]+, [M – H]+ calcd: 516.2. 1H NMR (400 MHz, CDCl3) δ 8.74–8.62 (m, 2H), 8.21–8.05 (m, 2H), 7.69–7.55 (m, 2H), 7.50–7.39 (m, 4H), 7.38–7.29 (m, 4H), 7.26 (s, 1H), 7.16–7.05 (m, 2H), 6.41 (s, 1H), 3.91 (d, J = 14.0 Hz, 2H), 3.71 (d, J = 14.0 Hz, 2H), 3.29–3.10 (m, 1H), 3.11–2.98 (m, 1H), 2.79 (ddd, J = 17.9, 12.3, 5.5 Hz, 1H), 2.65 (d, J = 8.1 Hz, 2H), 2.43–2.22 (m, 1H), 1.95–1.81 (m, 1H).
Step 3: Synthesis of N4-(4-Fluorophenyl)-2-(4-pyridyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine (Compound 10, Scheme 1b)
This compound was obtained using compound D1.5 (75.7 mg, 0.14 mmol) following the general procedure E previously described for 4 h. Final normal phase purification (DCM/DCM:NH3 1 N MeOH 4:1 from 95/5 to 75/25) afforded the pure title compound (45.3 mg, yield 94%). Rt = 1.65 min (gradient 1); MS (ESI) m/z: 336.1 [M – H]+, [M – H]+ calcd: 336.2. HRMS m/z: 336.1619, calcd for C19H19FN5+: 336.1624. QC analysis: Rt = 2.52 min, m/z: 336.10 [M – H]+, UV (215 nm): 96%. 1H NMR (400 MHz, DMSO-d6) δ 8.67 (d, J = 5.2 Hz, 2H), 8.59 (s, 1H), 8.08 (d, J = 5.2 Hz, 2H), 7.76 (ddd, J = 9.3, 5.0, 2.3 Hz, 2H), 7.22 (t, J = 8.9 Hz, 2H), 2.93–2.71 (m, 3H), 2.31 (dd, J = 16.9, 8.1 Hz, 1H), 1.94 (ddt, J = 11.1, 7.8, 4.4 Hz, 1H), 1.63 (dtd, J = 12.4, 9.6, 5.7 Hz, 1H), 1.23 (t, J = 7.2 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 162.58 (C), 158.84 (C), 151.25 (CF, d, JCF = 1193.8 Hz), 150.16 (CH), 135.98 (C, d, JCF = 2.1 Hz), 123.91 (CH, d, JCF = 7.8 Hz), 121.33 (CH), 114.89 (CH, d, JCF = 22.2 Hz), 112.24 (C), 46.10 (CH), 32.20 (CH2), 31.09 (CH2), 30.37 (CH2).
Synthesis of Compound 11:N4-(3-Methoxyphenyl)-2-(4-pyridyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine
Step 1: Synthesis of N6,N6-Dibenzyl-2-chloro-N4-(3-methoxyphenyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine (Compound C1.5, Scheme 1b)
This compound was obtained using compound B1 (250 mg, 0.63 mmol) and 3-methoxyaniline (0.08 mL, 0.69 mmol) following the general procedure C previously described at 100 °C for 16 h. Final normal phase purification (cyclohexane/TBME from 100/0 to 90/10) afforded the pure title compound (140 mg, yield 46%). Rt = 2.43 min (gradient 2); MS (ESI) m/z: 485.2 [M – H]+, [M – H]+ calcd: 469.2. 1H NMR (400 MHz, CDCl3) δ 7.48–7.38 (m, 5H), 7.33 (t, J = 7.4 Hz, 5H), 7.24 (dd, J = 7.8, 2.5 Hz, 3H), 6.95 (d, J = 7.5 Hz, 1H), 6.35 (s, 1H), 3.86 (d, J = 14.1 Hz, 2H), 3.67 (d, J = 14.0 Hz, 2H), 3.11 (q, J = 9.0 Hz, 1H), 2.92 (ddd, J = 18.3, 5.1, 2.2 Hz, 1H), 2.69 (ddd, J = 18.1, 12.2, 5.5 Hz, 1H), 2.52 (d, J = 8.5 Hz, 2H), 2.38 (s, 3H), 2.30–2.19 (m, 1H), 1.85–1.70 (m, 1H).
Step 2: Synthesis of N6,N6-Dibenzyl-N4-(3-methoxyphenyl)-2-(4-pyridyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine (Compound D1.6, Scheme 1b)
This compound was obtained using compound C1.5 (135 mg, 0.28 mmol) and pyridine-4-boronic acid (45.6 mg, 0.33 mmol) following the general procedure D previously described. Final normal phase purification (cyclohexane/EtOAc from 75/25 to 60/40) afforded the pure title compound (132 mg, yield 90%). Rt = 2.62 min (gradient 2); MS (ESI) m/z: 528.4 [M – H]+, [M – H]+ calcd: 528.3. 1H NMR (400 MHz, DMSO-d6) δ 8.71–8.63 (m, 2H), 8.55 (s, 1H), 8.15–8.05 (m, 2H), 7.48–7.40 (m, 5H), 7.38 (ddd, J = 8.1, 2.0, 1.0 Hz, 1H), 7.31 (q, J = 7.7 Hz, 5H), 7.25–7.18 (m, 2H), 6.69 (ddd, J = 8.1, 2.5, 1.0 Hz, 1H), 3.82–3.78 (m, 5H), 3.70 (d, J = 14.2 Hz, 2H), 3.03–2.84 (m, 3H), 2.84–2.63 (m, 2H), 2.16 (d, J = 12.2 Hz, 1H), 1.84 (qd, J = 12.1, 5.1 Hz, 1H).
Step 3: Synthesis of N4-(3-Methoxyphenyl)-2-(4-pyridyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine (Compound 11, Scheme 1b)
This compound was obtained using compound D1.6 (130.0 mg, 0.25 mmol) following the general procedure E previously described for 4 h. Final normal phase purification (neutral alumina, DCM/DCM:MeOH 4:1 from 90/10 to 60/40) afforded the pure title compound (51.4 mg, yield 60%). Rt = 1.45 min (gradient 1); MS (ESI) m/z: 348.2 [M – H]+, [M – H]+ calcd: 348.2. HRMS m/z: 348.1818, calcd for C20H22N5O+: 348.1824. QC analysis: Rt = 2.44 min, m/z: 348.16 [M – H]+, UV (215 nm): >99.5%. 1H NMR (400 MHz, DMSO-d6) δ 8.80–8.63 (m, 2H), 8.48 (s, 1H), 8.30–8.02 (m, 2H), 7.50 (t, J = 2.2 Hz, 1H), 7.43 (ddd, J = 8.2, 2.0, 0.9 Hz, 1H), 7.27 (t, J = 8.1 Hz, 1H), 6.65 (ddd, J = 8.2, 2.5, 0.9 Hz, 1H), 3.78 (s, 3H), 3.17 (qd, J = 5.5, 2.2 Hz, 1H), 2.95–2.70 (m, 3H), 2.40–2.25 (m, 1H), 2.01–1.88 (m, 1H), 1.63 (dtd, J = 12.3, 9.4, 5.5 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 162.70 (C), 159.33 (C), 158.78 (C), 157.17 (C), 150.14 (CH), 145.38 (C), 141.03 (C), 129.02 (CH), 121.27 (CH), 113.82 (CH), 112.63 (C), 108.70 (CH), 106.95 (CH), 55.01 (CH3), 46.07 (CH), 32.28 (CH2), 31.13 (CH2), 30.37 (CH2).
Synthesis of Compound 12: N4-(4-Methoxyphenyl)-2-(4-pyridyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine
Step 1: Synthesis of N6,N6-Dibenzyl-2-chloro-N4-(4-methoxyphenyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine (Compound C1.6, Scheme 1b)
This compound was obtained using compound B1 (250 mg, 0.63 mmol) and 4-methoxyaniline (0.087 mL, 0.69 mmol) following the general procedure C previously described at 120 °C for 4 h. Final normal phase purification (cyclohexane/TBME from 80/20 to 60/40) afforded the pure title compound (216 mg, yield 71%). Rt = 2.30 min (gradient 2); MS (ESI) m/z: 485.3/487.3 [M – H]+, [M – H]+ calcd: 485.0/487.0. 1H NMR (400 MHz, DMSO-d6) δ 8.53 (s, 1H), 7.66–7.54 (m, 2H), 7.42–7.38 (m, 4H), 7.35 (t, J = 7.5 Hz, 4H), 7.25–7.14 (m, 2H), 7.06–6.97 (m, 2H), 3.82 (d, J = 14.2 Hz, 2H), 3.81 (s, 3H), 3.72 (d, J = 14.2 Hz, 2H), 3.05–2.83 (m, 3H), 2.73 (dt, J = 17.2, 11.2 Hz, 2H), 2.17 (d, J = 12.2 Hz, 1H), 1.85 (qd, J = 12.2, 5.1 Hz, 1H).
Step 2: Synthesis of N6,N6-Dibenzyl-N4-(4-methoxyphenyl)-2-(4-pyridyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine (Compound D1.7, Scheme 1b)
This compound was obtained using crude C1.6 (210 mg, 0.43 mmol) and pyridine-4-boronic acid (71.0 mg, 0.52 mmol) following the general procedure D previously described. Final normal phase purification (cyclohexane/TBME from 70/30 to 50/50) afforded the pure title compound (170 mg, overall yield 74% from step 1). Rt = 2.50 min (gradient 2); MS (ESI) m/z: 528.4 [M – H]+, [M – H]+ calcd: 528.7. 1H NMR (400 MHz, DMSO-d6) δ 8.69–8.60 (m, 2H), 8.51 (s, 1H), 8.10–8.00 (m, 2H), 7.68–7.57 (m, 2H), 7.47–7.40 (m, 4H), 7.32 (t, J = 7.5 Hz, 4H), 7.26–7.18 (m, 2H), 7.03–6.94 (m, 2H), 3.80 (d, J = 14.2 Hz, 2H), 3.79 (s, 3H), 3.70 (d, J = 14.2 Hz, 2H), 3.02–2.81 (m, 3H), 2.71 (dt, J = 17.2, 11.2 Hz, 2H), 2.15 (d, J = 12.2 Hz, 1H), 1.83 (qd, J = 12.2, 5.1 Hz, 1H).
Step 3: Synthesis of N4-(4-Methoxyphenyl)-2-(4-pyridyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine (Compound 12, Scheme 1b)
This compound was obtained using compound D1.7 (165 mg, 0.31 mmol) following the general procedure E previously described for 4 h. Final normal phase purification (DCM/DCM:NH3 1 N MeOH 4:1 from 95/5 to 75/25) afforded the pure title compound (78.8 mg, yield 76%). Rt = 1.49 min (gradient 1); MS (ESI) m/z: 348.2 [M – H]+, [M – H]+ calcd: 348.4. HRMS m/z: 348.1815, calcd for C20H22N5O+: 348.1824. QC analysis: Rt = 2.43 min, m/z: 348.09 [M – H]+, UV (215 nm): >99.5. 1H NMR (400 MHz, DMSO-d6) δ 8.71–8.61 (m, 2H), 8.43 (s, 1H), 8.14–8.02 (m, 2H), 7.69–7.59 (m, 2H), 7.00–6.92 (m, 2H), 3.77 (s, 3H), 3.22–3.10 (m, 1H), 2.93–2.68 (m, 3H), 2.29 (dd, J = 16.9, 8.3 Hz, 1H), 2.02–1.87 (m, 1H), 1.62 (dtd, J = 12.7, 9.6, 5.7 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 162.01 (C), 158.95 (C), 157.18 (C), 155.25 (C), 150.07 (CH), 145.47 (C), 132.59 (C), 123.74 (CH), 121.29 (CH), 113.50 (CH), 111.78 (C), 55.20 (CH3), 46.12 (CH), 32.10 (CH2), 31.05 (CH2), 30.30 (CH2).
Synthesis of Compound 13: N4-[3-(Dimethylamino)phenyl]-2-(4-pyridyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine
Step 1: Synthesis of N6,N6-Dibenzyl-2-chloro-N4-[3-(dimethylamino)phenyl]-5,6,7,8-tetrahydroquinazoline-4,6-diamine (Compound C1.7, Scheme 1b)
This compound was obtained using compound B1 (250 mg, 0.63 mmol) and N,N-dimethyl-1,3-phenylenediamine dihydrochloride (145.8 mg, 0.69 mmol) following the general procedure C previously described at 100 °C for 6 h. Final normal phase purification (cyclohexane/EtOAc from 100/0 to 80/20) afforded the pure title compound (175 mg, yield 56%). Rt = 2.51 min (gradient 2); MS (ESI) m/z: 498.3/500.3 [M – H]+, [M – H]+ calcd: 498.2/500.2. 1H NMR (400 MHz, DMSO-d6) δ 8.61 (s, 1H), 7.48–7.38 (m, 4H), 7.32 (dd, J = 8.3, 6.8 Hz, 4H), 7.26–7.19 (m, 2H), 7.16 (t, J = 8.1 Hz, 1H), 7.02 (t, J = 2.2 Hz, 1H), 6.92 (ddd, J = 8.0, 2.0, 0.8 Hz, 1H), 6.52 (ddd, J = 8.4, 2.6, 0.9 Hz, 1H), 3.78 (d, J = 14.2 Hz, 2H), 3.67 (d, J = 14.2 Hz, 2H), 2.91 (s, 6H), 2.99–2.84 (m, 0H), 2.75 (dd, J = 7.9, 3.6 Hz, 1H), 2.73–2.56 (m, 2H), 2.10 (d, J = 12.4 Hz, 1H), 1.79 (tt, J = 12.1, 6.0 Hz, 1H).
Step 2: Synthesis of N6,N6-Dibenzyl-N4-[3-(dimethylamino)phenyl]-2-(4-pyridyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine (Compound D1.8, Scheme 1b)
This compound was obtained using crude C1.7 (170 mg, 0.34 mmol) and pyridine-4-boronic acid (55.9 mg, 0.41 mmol) following the general procedure D previously described. Final normal phase purification (cyclohexane/EtOAc from 90/10 to 60/40) afforded the pure title compound (150 mg, overall yield 81% from step 1). 1H NMR (400 MHz, DMSO-d6) δ 8.71–8.62 (m, 2H), 8.41 (s, 1H), 8.14–8.06 (m, 2H), 7.48–7.41 (m, 4H), 7.35–7.29 (m, 4H), 7.25–7.17 (m, 4H), 7.08 (ddd, J = 7.9, 1.9, 0.9 Hz, 1H), 6.50 (ddd, J = 8.3, 2.5, 0.9 Hz, 1H), 3.80 (d, J = 14.2 Hz, 2H), 3.70 (d, J = 14.2 Hz, 2H), 2.94 (s, 6H), 3.00–2.83 (m, 2H), 2.82–2.69 (m, 1H), 2.15 (d, J = 12.4 Hz, 1H), 1.83 (qd, J = 12.0, 5.1 Hz, 1H).
Step 3: Synthesis of N4-[3-(Dimethylamino)phenyl]-2-(4-pyridyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine (Compound 13, Scheme 1b)
This compound was obtained using compound D1.8 (150 mg, 1.39 mmol) following the general procedure E previously described for 4 h. Final normal phase purification (DCM/DCM:NH3 1 N MeOH 4:1 from 95/5 to 75/25) afforded the pure title compound (83.0 mg, yield 83%). Rt = 1.57 min (gradient 1); MS (ESI) m/z: 361.2 [M – H]+, [M – H]+ calcd: 361.2. HRMS m/z: 361.2134, calcd for C21H25N6+: 361.2141. QC analysis: Rt = 2.65 min, m/z: 361.11 [M – H]+, UV (215 nm): 99%. 1H NMR (600 MHz, DMSO-d6) δ 8.70–8.65 (m, 2H), 8.30 (s, 1H), 8.19–8.09 (m, 2H), 7.28–7.21 (m, 1H), 7.19–7.12 (m, 2H), 6.51–6.44 (m, 1H), 3.17 (dddd, J = 11.1, 8.3, 5.1, 2.9 Hz, 1H), 2.92 (s, 6H), 2.88 (p, J = 2.3 Hz, 1H), 2.86 (d, J = 5.0 Hz, 1H), 2.78 (ddd, J = 17.4, 9.7, 5.9 Hz, 1H), 2.31 (dd, J = 16.8, 8.2 Hz, 1H), 1.94 (dp, J = 12.8, 4.4, 3.6 Hz, 1H), 1.71 (s, 2H), 1.67–1.57 (m, 1H). 13C NMR (151 MHz, DMSO-d6) δ 162.39 (C), 158.90 (C), 157.14 (C), 150.70 (C), 150.13 (CH), 145.48 (C), 140.44 (C), 128.64 (CH), 121.30 (CH), 112.46 (C), 110.02 (CH), 107.55 (CH), 105.90 (CH), 46.14 (CH), 40.29 (CH3), 32.33 (CH2), 31.23 (CH2), 30.38 (CH2).
Synthesis of Compound 14:N4-[4-(Dimethylamino)phenyl]-2-(4-pyridyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine
Step 1: Synthesis of N6,N6-Dibenzyl-2-chloro-N4-[4-(dimethylamino)phenyl]-5,6,7,8-tetrahydroquinazoline-4,6-diamine (Compound C1.8, Scheme 1b)
This compound was obtained using compound B1 (250 mg, 0.63 mmol) and N4,N4-dimethylbenzene-1,4-diamine (0.085 mL, 0.69 mmol) following the general procedure C previously described at 100 °C for 16 h. Final normal phase purification (cyclohexane/EtOAc from 100:0 to 70:30) afforded the pure title compound (240 mg, yield 77%). Rt = 2.44 min (gradient 2); MS (ESI) m/z: 498.1/500.1 [M – H]+, [M – H]+ calcd: 498.2/500.2. 1H NMR (400 MHz, CDCl3) δ 7.42 (d, J = 7.5 Hz, 4H), 7.34 (dt, J = 14.7, 8.1 Hz, 6H), 7.24 (t, J = 7.4 Hz, 2H), 6.72 (s, 2H), 6.28 (s, 1H), 3.84 (d, J = 14.1 Hz, 2H), 3.66 (d, J = 14.0 Hz, 2H), 3.16–3.01 (m, 1H), 3.01–2.81 (m, 1H), 2.73–2.58 (m, 1H), 2.48 (s, 2H), 2.30–2.16 (m, 1H), 1.74 (qd, J = 12.3, 5.1 Hz, 1H), 1.43 (s, 6H).
Step 2: Synthesis of N6,N6-Dibenzyl-N4-[4-(dimethylamino)phenyl]-2-(4-pyridyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine (Compound D1.9, Scheme 1b)
This compound was obtained using compound C1.8 (240 mg, 0.48 mmol) and pyridine-4-boronic acid (85.7 mg, 0.58 mmol) following the general procedure D previously described. Final normal phase purification (cyclohexane/EtOAc from 75/25 to 50/50) afforded the pure title compound (197 mg, yield 76%). Rt = 2.62 min (gradient 2); MS (ESI) m/z: 541.2 [M – H]+, [M – H]+ calcd: 541.7. 1H NMR (400 MHz, DMSO-d6) δ 8.68–8.60 (m, 2H), 8.40 (s, 1H), 8.09–8.01 (m, 2H), 7.57–7.49 (m, 2H), 7.47–7.39 (m, 4H), 7.32 (t, J = 7.5 Hz, 4H), 7.25–7.17 (m, 2H), 6.84–6.74 (m, 2H), 3.79 (d, J = 14.2 Hz, 2H), 3.69 (d, J = 14.2 Hz, 2H), 3.02–2.92 (m, 1H), 2.91 (s, 6H), 2.89–2.78 (m, 2H), 2.76–2.60 (m, 2H), 2.14 (d, J = 12.1 Hz, 1H), 1.81 (qd, J = 12.3, 5.1 Hz, 1H).
Step 3: Synthesis of N4-[4-(Dimethylamino)phenyl]-2-(4-pyridyl)-5,6,7,8-tetrahydroquinazoline-4,6-diamine (Compound 14, Scheme 1b)
This compound was obtained using compound D1.9 (197.2 mg, 0.48 mmol) following the general procedure E previously described for 5 h. Final normal phase purification (DCM/DCM:NH3 1 M MeOH 4:1 from 95/5 to 40/60) afforded the pure title compound (79.4 mg, yield 46%). Rt = 1.32 min (gradient 1); MS (ESI) m/z: 361.2 [M – H]+, [M – H]+ calcd: 361.2. HRMS m/z: 361.2137, calcd for C21H25N6+: 361.2141. QC analysis: Rt = 2.52 min, m/z: 361.17 [M – H]+, UV (215 nm): 96%. 1H NMR (400 MHz, DMSO-d6) δ 8.73–8.61 (m, 2H), 8.52 (s, 1H), 8.12–8.00 (m, 2H), 7.57–7.44 (m, 2H), 6.84–6.75 (m, 2H), 3.50–3.60 (m, 1H), 2.97 (dd, J = 17.0, 5.4 Hz, 1H), 2.91 (s, 6H), 2.86 (d, J = 6.6 Hz, 2H), 2.60 (dd, J = 16.6, 8.8 Hz, 1H), 2.17 (d, J = 19.0 Hz, 1H), 1.90 (p, J = 8.2 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 160.97 (C), 158.93 (C), 157.76 (C), 150.11 (CH), 147.27 (C), 145.34 (C), 128.75 (C), 123.86 (CH), 121.31 (CH), 112.32 (CH), 109.52 (C), 45.74 (CH), 40.50 (CH3), 29.34 (CH2), 27.82 (CH2), 26.26 (CH2).
Topoisomerase II Activity Assay
The activity of topoIIα was measured using a decatenation assay (Inspiralis) following the manufacturer’s instructions. Compounds were dissolved in DMSO and used at a concentration ranging from 200 to 1 μM. The final DMSO concentration in the assay was ≤1%. Reaction mixtures were incubated for 30 min at 37 °C and terminated with STEB buffer (40% (w/v) sucrose, 100 mM Tris–HCl, pH 8, 1 mM ethylenediaminetetraacetic acid (EDTA), 0.5 mg/ mL bromophenol blue). Reaction products were resolved by electrophoresis in 1% agarose gels containing SYBR Safe DNA stain (Invitrogen), scanned, and quantified using the ChemiDoc system (BioRad). IC50 values were obtained with GraphPad Prism software (version 5.03) using the band intensities of the dose–response gels. Values are reported as the mean ± SD of two independent experiments.
Biology: Cell Viability Assay
Human cancer cell lines A549 (lung adenocarcinoma, ATCC CCL-185), DU145 (androgen-human cancer cell lines A549 (lung adenocarcinoma, ATCC CCL-185), MCF7 (breast adenocarcinoma, ATCC HTB-22), DU145 (androgen-independent prostate cancer, ATCC HTB-81), and HeLa (cervical carcinoma, ATCC CCL-2) were obtained from ATCC. Human melanoma cell lines A375 (88113005) and G-361 (88030401) were obtained from European Collection of Authenticated Cell Cultures (ECACC). Cells were routinely grown in minimal essential medium containing Eagle’s salts and l-glutamine (A549, DU-145, MCF7, and HeLa) or Dulbecco’s Modified Minimal Essential Medium (A375 and G-361) supplemented with 10% heat-inactivated fetal bovine serum (FBS) in a humidified atmosphere of 5% CO2 at 37 °C. To assess the antiproliferative activity of the compounds, cells were seeded at a density of 2500 cells/well (HeLa), 5000 cells/well (A549, DU145, and A375) or 10000 cells/well (MCF7 and G-361) in 96-well plates, and cell viability was measured using the MTT assay. Values are reported as the mean ± SD of two independent experiments.
TopoII Inhibition Mechanism Characterization of Compound 14: Materials
Recombinant wild-type human topoisomerase IIα (topoIIα) and topoisomerase IIβ (topoIIβ) were expressed in Saccharomyces cerevisiae JEL-1Δtop1 and purified as described previously.25−27 The enzymes were stored at −80 °C as 1.5 mg/mL stocks in 50 mM Tris–HCl, pH 7.9, 0.1 mM NaEDTA, 750 mM KCl, and 40% glycerol. Negatively supercoiled pBR322 DNA was prepared from Escherichia coli using a Plasmid Mega Kit (Qiagen) as described by the manufacturer and the exonuclease was treated to remove any chromosomal DNA contaminants from the plasmid.28 Analytical-grade etoposide and ethidium bromide were purchased from Sigma-Aldrich. [γ32P]ATP (3000 Ci/mmol stock) was purchased from PerkinElmer. Compound 14 was stored at −20 °C as a 20 mM solution in dimethyl sulfoxide.
DNA Relaxation
DNA relaxation reactions were carried out using the procedure of Fortune and Osheroff.29 Reaction mixtures contained 5 nM negatively supercoiled pBR322 DNA, 3 nM human topoIIα or 4 nM topoIIβ, 1 mM ATP, and 0–200 μM compound 14 in a total of 20 μL of 10 mM Tris–HCl, pH 7.9, 5 mM MgCl2, 175 mM KCl, 0.1 mM NaEDTA, and 2.5% (v/v) glycerol. Mixtures were incubated at 37 °C for 4 min. Reactions were stopped by the addition of 3 μL of 0.77% sodium dodecyl sulfate (SDS)–77 mM NaEDTA, pH 8.0. Samples were mixed with 2 μL of agarose loading dye [60% sucrose (w/v), 10 mM Tris–HCl, pH 7.9, 0.5% bromophenol blue, 0.5% xylene cyanol], heated for 2 min at 45 °C, and subjected to electrophoresis in a 1% agarose gel in 100 mM Tris-borate, pH 8.3, and 2 mM EDTA. Gels were stained for 30 min using 1.0 μg/mL ethidium bromide and rinsed in deionized water. DNA bands were visualized by medium-wave UV light and quantified using an α Innotech digital imaging system. DNA relaxation was monitored by the loss of supercoiled plasmid molecules.
DNA Cleavage
DNA cleavage reactions were performed using the procedure of Fortune and Osheroff.29 Reaction mixtures contained 10 nM negatively supercoiled pBR322 DNA and 150 nM human topoIIα in a final volume of 20 μL of cleavage buffer [10 mM Tris–HCl, pH 7.9, 5 mM MgCl2, 100 mM KCl, 0.1 mM NaEDTA, and 2.5% (v/v) glycerol] containing 0–200 μM compound 14. Reactions were incubated for 6 min at 37 °C, and enzyme–DNA cleavage complexes were trapped by the addition of 2 μL of 4% SDS followed by 2 μL of 250 mM NaEDTA, pH 8.0. Proteinase K (2 μL of a 0.8 mg/mL solution) was added, and samples were incubated for 30 min at 45 °C to digest the enzyme. Samples were mixed with 2 μL of agarose loading dye, heated for 2 min at 45 °C, and subjected to electrophoresis in a 1% agarose gel in 40 mM Tris-acetate, pH 8.3, and 2 mM EDTA containing 0.5 μg/mL ethidium bromide. DNA bands were visualized and quantified as described above. DNA cleavage was monitored by the conversion of supercoiled plasmid to linear molecules.
DNA Intercalation
DNA intercalation was carried out using the protocol of Fortune et al.30 Calf thymus DNA topoisomerase I (Invitrogen, 0.5 U) and 5 mM relaxed pBR322 were incubated in 10 mM Tris–HCl, pH 7.5, 10 mM KCl, 2 mM MgCl2, 0.02 mM EDTA, 0.1 mM dithiothreitol, and 6 μg/mL bovine serum albumin. Assays were carried out in the presence of 0–25 μM compound 14. Mixtures were incubated for 15 min at 37 °C. Reactions containing 10 μM ethidium bromide (a well-characterized intercalator) or 100 μM etoposide (a nonintercalative topoisomerase II poison) were used as positive or negative controls, respectively. Samples were extracted using 20 μL of phenol/chloroform/isoamyl alcohol (25:24:1), and the aqueous layer was mixed with 2 μL of agarose loading dye and heated for 5 min at 45 °C. Reactions were stopped by the addition of 3 μL of 0.77% SDS–77 mM NaEDTA, pH 8.0. Samples were mixed with 2 μL of agarose loading dye, heated for 2 min at 45 °C, and subjected to electrophoresis in a 1% agarose gel in 100 mM Tris-borate, pH 8.3, and 2 mM EDTA. Gels were stained for 30 min using 1.0 μg/mL ethidium bromide and rinsed in deionized water. DNA bands were visualized and quantified as described above. DNA intercalation was monitored by the conversion of relaxed to supercoiled plasmid molecules.
ATP Hydrolysis
ATPase assays were performed as described by Osheroff et al.31 Reaction mixtures contained 30 nM human topoIIα, 1 mM [γ32P]ATP, and 0–25 μM compound 14 in a total of 20 μL of cleavage buffer. Some assays were carried out in the presence of 5 nM negatively supercoiled pB322 DNA. Reactions were initiated by the addition of topoIIα, and mixtures were incubated at 37 °C for 8 min. Samples (2.0 μL) were removed and spotted on to polyethyleneimine-impregnated thin-layer cellulose chromatography plates. Plates were developed by chromatography in freshly prepared 400 mM NH4HCO3, and released 32P was visualized and quantified using a Bio-Rad Pharos FX Plus Molecular Imager.
Acknowledgments
M.D.V. and V.G. thank Paolo Bisignano for technical assistance.
Glossary
Abbreviations
- topoII
topoisomerase II enzyme
- topoIIα
topoisomerase II enzyme α, isoform
- hTIIα
human topoisomerase II enzyme α, isoform, topoIIβ, topoisomerase II enzyme β, isoform
- hTIIβ
human topoisomerase II enzyme β, isoform
- SD
standard deviation
- Cmax
maximum plasma concentration
- tmax
time to reach maximum concentration
- VD
volume of distribution
- CL
clearance
- ACN
acetonitrile
- EtOAc
ethyl acetate
- EtOH
ethanol
- MeOH
methanol
- SQD
single quadrupole detector
- PDA
photodiode array
- QC
quality check
- UPLC
ultraperformance liquid chromatography
- PdCl2(dppf)
[1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II)
- Pd(OH)2/C
Pd(OH)2 on activated carbon
- rac-BINAP
(±)-2,2′-bis(diphenylphosphino)-1,1′-binaphthalene
- Rt
retention time
- TEA
triethylamine
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c00774.
Author Contributions
○ J.A.O. and J.M.A. contributed equally to this work.
This work was supported in part by the Italian Association for Cancer Research (AIRC) through the “IG 23679” and the National Institutes of Health (grant R01 GM126363 to N.O.).
The authors declare the following competing financial interest(s): One patent application protecting the class of compounds disclosed in this manuscript has been filed by the following authors: Marco De Vivo, Jose Antonio Ortega, Jose M. Aren cibia, Claudia Sissi and Anna Minarini.
Supplementary Material
References
- Deweese J. E.; Osheroff M. A.; Osheroff N. DNA topology and topoisomerases: teaching a “Knotty” Subject. Biochem. Mol. Biol. Educ. 2009, 37, 2–10. 10.1002/bmb.20244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitiss J. L. DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer 2009, 9, 327–337. 10.1038/nrc2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos S. M.; Tretter E. M.; Schmidt B. H.; Berger J. M. All tangled up: how cells direct, manage and exploit topoisomerase function. Nat. Rev. Mol. Cell Biol. 2011, 12, 827–841. 10.1038/nrm3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y.; Sun Y.; Huang S. N.; Nitiss J. L. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell Biol. 2016, 17, 703–721. 10.1038/nrm.2016.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashley R. E.; Osheroff N.. Regulation of DNA Topology by Topoisomerases: Mathemtics at the Molecular Level. In Knots, Low-Dimensional Topology and Applications; Adams C. C.; Gordon C. M.; Jones V. F. R.; Kauffman L. H.; Lambropoulou S.; Millett K. C.; Przytycki J. H.; Ricca R.; Sazdanovic R., Eds.; Springer: Switzerland, 2019; pp 411–433. [Google Scholar]
- Deweese J. E.; Osheroff N. The DNA cleavage reaction of topoisomerase II: wolf in sheep’s clothing. Nucleic Acids Res. 2009, 37, 738–748. 10.1093/nar/gkn937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y. Drugging topoisomerases: lessons and challenges. ACS Chem. Biol. 2013, 8, 82–95. 10.1021/cb300648v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitiss J. L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. 10.1038/nrc2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joannides M.; Mays A. N.; Mistry A. R.; Hasan S. K.; Reiter A.; Wiemels J. L.; Felix C. A.; Coco F. L.; Osheroff N.; Solomon E.; Grimwade D. Molecular pathogenesis of secondary acute promyelocytic leukemia. Mediterr. J. Hematol. Infect. Dis. 2011, 3, e2011045 10.4084/mjhid.2011.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendleton M.; Lindsey R. H. Jr.; Felix C. A.; Grimwade D.; Osheroff N. Topoisomerase II and leukemia. Ann. N. Y. Acad. Sci. 2014, 1310, 98–110. 10.1111/nyas.12358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailly C. Contemporary challenges in the design of topoisomerase II inhibitors for cancer chemotherapy. Chem. Rev. 2012, 112, 3611–3640. 10.1021/cr200325f. [DOI] [PubMed] [Google Scholar]
- Palermo G.; Minniti E.; Greco M. L.; Riccardi L.; Simoni E.; Convertino M.; Marchetti C.; Rosini M.; Sissi C.; Minarini A.; De Vivo M. An optimized polyamine moiety boosts the potency of human type II topoisomerase poisons as quantified by comparative analysis centered on the clinical candidate F14512. Chem. Commun. 2015, 51, 14310–14313. 10.1039/C5CC05065K. [DOI] [PubMed] [Google Scholar]
- Hu W.; Huang X. S.; Wu J. F.; Yang L.; Zheng Y. T.; Shen Y. M.; Li Z. Y.; Li X. Discovery of novel topoisomerase II Inhibitors by medicinal chemistry approaches. J. Med. Chem. 2018, 61, 8947–8980. 10.1021/acs.jmedchem.7b01202. [DOI] [PubMed] [Google Scholar]
- Arencibia J. M.; Brindani N.; Franco-Ulloa S.; Nigro M.; Akkarapattiakal Kuriappan J.; Ottonello G.; Bertozzi S. M.; Summa M.; Girotto S.; Bertorelli R.; Armirotti A.; De Vivo M. Design, synthesis, dynamic docking, biochemical characterization, and in vivo pharmacokinetics studies of novel topoisomerase II poisons with promising antiproliferative activity. J. Med. Chem. 2020, 63, 3508–3521. 10.1021/acs.jmedchem.9b01760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccardi L.; Genna V.; De Vivo M. Metal–ligand interactions in drug design. Nat. Rev. Chem. 2018, 2, 100–112. 10.1038/s41570-018-0018-6. [DOI] [Google Scholar]
- Oviatt A. A.; Kuriappan J. A.; Minniti E.; Vann K. R.; Onuorah P.; Minarini A.; De Vivo M.; Osheroff N. Polyamine-containing etoposide derivatives as poisons of human type II topoisomerases: differential effects on topoisomerase IIalpha and IIbeta. Bioorg. Med. Chem. Lett. 2018, 28, 2961–2968. 10.1016/j.bmcl.2018.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega J. A.; Riccardi L.; Minniti E.; Borgogno M.; Arencibia J. M.; Greco M. L.; Minarini A.; Sissi C.; De Vivo M. Pharmacophore hybridization to discover novel topoisomerase II poisons with promising antiproliferative activity. J. Med. Chem. 2018, 61, 1375–1379. 10.1021/acs.jmedchem.7b01388. [DOI] [PubMed] [Google Scholar]
- Minniti E.; Byl J. A. W.; Riccardi L.; Sissi C.; Rosini M.; De Vivo M.; Minarini A.; Osheroff N. Novel xanthone-polyamine conjugates as catalytic inhibitors of human topoisomerase II. Bioorg. Med. Chem. Lett. 2017, 27, 4687–4693. 10.1016/j.bmcl.2017.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palermo G.; Cavalli A.; Klein M. L.; Alfonso-Prieto M.; Dal Peraro M.; De Vivo M. Catalytic metal ions and enzymatic processing of DNA and RNA. Acc. Chem. Res. 2015, 48, 220–228. 10.1021/ar500314j. [DOI] [PubMed] [Google Scholar]
- Palermo G.; Stenta M.; Cavalli A.; Dal Peraro M.; De Vivo M. Molecular simulations highlight the role of metals in catalysis and inhibition of type II topoisomerase. J. Chem. Theory Comput. 2013, 9, 857–862. 10.1021/ct300691u. [DOI] [PubMed] [Google Scholar]
- Bau J. T.; Kang Z.; Austin C. A.; Kurz E. U. Salicylate, a catalytic inhibitor of topoisomerase II, inhibits DNA cleavage and is selective for the alpha isoform. Mol. Pharmacol. 2014, 85, 198–207. 10.1124/mol.113.088963. [DOI] [PubMed] [Google Scholar]
- Kuriappan J. A.; Osheroff N.; De Vivo M. Smoothed potential MD simulations for dissociation kinetics of etoposide to unravel isoform specificity in targeting human topoisomerase II. J. Chem. Inf. Model. 2019, 59, 4007–4017. 10.1021/acs.jcim.9b00605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alagarsamy V.; Chitra K.; Saravanan G.; Solomon V. R.; Sulthana M. T.; Narendhar B. An overview of quinazolines: Pharmacological significance and recent developments. Eur. J. Med. Chem. 2018, 151, 628–685. 10.1016/j.ejmech.2018.03.076. [DOI] [PubMed] [Google Scholar]
- Wu C. C.; Li T. K.; Farh L.; Lin L. Y.; Lin T. S.; Yu Y. J.; Yen T. J.; Chiang C. W.; Chan N. L. Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science 2011, 333, 459–462. 10.1126/science.1204117. [DOI] [PubMed] [Google Scholar]
- Worland S. T.; Wang J. C. Inducible overexpression, purification, and active site mapping of DNA topoisomerase II from the yeast Saccharomyces cerevisiae. J. Biol. Chem. 1989, 264, 4412–4416. [PubMed] [Google Scholar]
- Wasserman R. A.; Austin C. A.; Fisher L. M.; Wang J. C. Use of yeast in the study of anticancer drugs targeting DNA topoisomerases: expression of a functional recombinant human DNA topoisomerase II alpha in yeast. Cancer Res. 1993, 53, 3591–3596. [PubMed] [Google Scholar]
- Kingma P. S.; Greider C. A.; Osheroff N. Spontaneous DNA lesions poison human topoisomerase IIalpha and stimulate cleavage proximal to leukemic 11q23 chromosomal breakpoints. Biochemistry 1997, 36, 5934–5939. 10.1021/bi970507v. [DOI] [PubMed] [Google Scholar]
- Minniti E.; Byl J. A. W.; Riccardi L.; Sissi C.; Rosini M.; De Vivo M.; Minarini A.; Osheroff N. Novel xanthone-polyamine conjugates as catalytic inhibitors of human topoisomerase IIalpha. Bioorg. Med. Chem. Lett. 2017, 27, 4687–4693. 10.1016/j.bmcl.2017.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortune J. M.; Osheroff N. Merbarone inhibits the catalytic activity of human topoisomerase II. J. Biol. Chem. 1998, 273, 17643–17650. 10.1074/jbc.273.28.17643. [DOI] [PubMed] [Google Scholar]
- Fortune J. M.; Velea L.; Graves D. E.; Utsugi T.; Yamada Y.; Osheroff N. DNA topoisomerases as targets for the anticancer drug TAS-103: DNA interactions and topoisomerase catalytic inhibition. Biochemistry 1999, 38, 15580–15586. 10.1021/bi991792g. [DOI] [PubMed] [Google Scholar]
- Osheroff N.; Shelton E. R.; Brutlag D. L. DNA topoisomerase II from Drosophila melanogaster. Relaxation of supercoiled DNA. J. Biol. Chem. 1983, 258, 9536–9543. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.