

Abstract
Innate immune monitoring of the cytosolic compartment for pathogen-encoded products or activities often enables the activation of a subset of caspases. In most cases, the cytosolic surveillance pathways are coupled to caspase-1 activation via canonical inflammasome complexes. A related set of caspases, caspase-11 in rodents and caspase-4 and −5 in humans, monitors the cytosol for bacterial lipopolysaccharide (LPS). Direct activation of caspase-11, −4 and −5 by intracellular LPS elicits a lytic cell death called pyroptosis, which occurs in multiple cell types. The pyroptosis is executed by a pore-forming protein gasdermin-D (GSDMD) that is activated by caspase-11, −4, or −5-mediated cleavage. In monocytes, formation of GSDMD pores can induce NLRP3 inflammasome activation for IL-1β and IL-18 maturation. Caspase-11-mediated pyroptosis in response to cytosolic LPS is critical for antibacterial defense and septic shock. Here, we review the emerging literature on cytosolic LPS sensing and its regulation and pathophysiological functions.
Germline-encoded pattern recognition receptors (PRRs) expressed by innate immune cells function to recognize microbe associated molecular patterns (MAMPs) or endogenous danger associated molecular patterns (DAMPs)1. MAMPs recognized by PRRs including LPS, lipoproteins, carbohydrates, flagellin, and nucleic acids are highly conserved and/or common among microbes, and this allows the host to sense a wide range of pathogens with a limited number of the PRRs. More than a dozen toll-like receptors (TLRs) have been characterized as PRRs for bacterial, viral and parasitic products2. In addition to TLRs, PRRs also include C-type Lectin Receptors (CLR), RIG-I-like receptors (RLR), AIM2-like receptors (ALR) as well as the relatively less characterized nucleotide-binding domain and leucine-rich repeat containing (NLR) proteins1. Among these PRRs, TLRs and CLRs are plasma or endosomal membrane-bound whereas RLRs, ALRs and NLRs are cytosolic, which together enable the innate immune system to monitor both extracellular and intracellular spaces for infectious agents.
A critical event downstream of MAMP and DAMP recognition by the PRRs is the transcriptional activation of various inflammatory mediators. TLRs and CLRs as well as certain cytosolic PRRs like the RLRs stimulate NF-κB- and interferon regulatory factor (IRF)-dependent gene transcription. An emerging and equally important response driven by many cytosolic PRRs of the ALR and NLR families is a form of proinflammatory cell death known as pyroptosis. Morphologically, pyroptosis is characterized by loss of plasma membrane integrity, cell swelling, plasma membrane rupture, and ultimately cell lysis. Pyroptosis is highly proinflammatory in nature as it is accompanied by the release of inflammatory cytokines such as interleukin-1β (IL-1β) and IL-18 as well as some DAMPs including IL-1α and HMGB13. Historically, pyroptosis refers to caspase-1-mediated monocyte death triggered by certain bacterial infections or toxin stimulation4–6. Activation of caspase-1 is now known to be mediated by the cytoplasmic multi-protein inflammasome complex, in which an NLR protein is proposed to function as a central scaffold protein7. Extensive work in the past decade or so has identified several NLR-family members (including NLRP3, NAIPs and NLRP1), AIM2, and Pyrin that function in the inflammasome pathway8,9. These cytosolic PRRs recognize a diverse spectrum of microbial products including bacterial flagellin, type III secretion system components, and toxins, as well as self or foreign double-stranded DNA and in specific cases, bacterial virulence activity to assemble various inflammasomes that activate caspase-1 directly or indirectly through an adapter protein ASC or NLRC4. The induced proteolytic activity of caspase-1, resulting from multimerization-mediated autoproteolytic processing, is responsible for the maturation of IL-1β and IL-18 as well as for the pyroptosis.
Sensing of intracellular LPS triggers pyroptosis
LPS is the major component of the outer membrane of Gram-negative bacteria. The chemical structure of LPS can be divided into three parts, the most conserved lipid A moiety, a core oligosaccharide chain and a variable polysaccharide chain known as O-antigen10. Given the abundance of LPS and its essentiality for life across bacterial species, innate immune monitoring of LPS is key to the host detection of a broad range of Gram-negative bacteria. LPS in the extracellular space is sensed by the plasma membrane-located TLR4-MD2 complex in conjunction with CD14 protein that specifically recognize the lipid A structure of LPS11. Ligated TLR4 then transduces the signal via the adapters MyD88 and TRIF to activate NF-κB- and IRF3-mediated transcription of immune system genes including cytokines and chemokines. Excessive TLR4 activation by LPS and the consequent cytokine storm was thought to underlie the endotoxic shock or sepsis. This proposal is apparently supported by the fact that Tlr4–/– mice are completely resistant to high-dose LPS injection. However, mice deficient in caspase-1 (Casp1–/–) or the related caspase-11 (Casp11–/–) located adjacent to Casp1 on the chromosome show similar resistance to LPS-induced shock12,13.
Kayagaki et al., made an important discovery clarifying why both Casp1–/– and Casp11–/– mice are similarly resistant to LPS-induced septic shock14. They first observed that the combination of LPS plus cholera toxin B (CTB) as well as certain enteric Gram-negative bacteria can trigger NLRP3 inflammasome-mediated IL-1β release by bone marrow macrophages derived from C57BL/6 but not 129 strains of mice. It turned out that Casp11 in 129 mice has a 5-bp deletion and encodes a nonfunctional protein, and this is responsible for the defective IL-1β release and pyroptosis in response to LPS plus CTB stimulation. As Casp1 knockout mice were generated originally from 129 mice-derived embryonic stem cells, they are indeed Casp1/11 double knockout, which was shown previously by anti-caspase-11 immunoblotting analyses of Casp1 knockout mice15. Subsequent mouse genetic analyses on the C57BL/6 background revealed that it is Casp11- but not Casp1-deficiency that could block high-dose LPS-induced lethality in mice14. Caspase-11 activation induces pyroptotic cell death and IL-1β release in an NLRP3 inflammasome-independent and -dependent manner, respectively. Subsequently, two independent studies discovered that caspase-11 activation responds to LPS that has gained the access to the host cytosol16,17. The fact that CTB can induce caspase-11 activation is due to the contaminant LPS that was co-internalized with CTB into the bone marrow macrophages. Surprisingly, caspase-11 activation by cytosolic LPS did not require TLR4, NLRs, or the ASC platform, which is in stark contrast to the mechanism of caspase-1 activation. This discovery raises a fundamental question: what is the receptor for intracellular LPS and which molecule is directly responsible for caspase-11 activation?
A surprising discovery of caspase-11, −4 and −5 as the intracellular LPS receptors
The domain structure of caspase-11 suggests that it, like caspase-9 and caspase-1, belongs to the group of initiator caspases. Caspase-9 is activated by the CARD-containing APAF-1 protein in the apoptosome complex, and caspase-1 is also activated by ASC or another CARD-containing protein in the inflammasome complex. This knowledge, together with the fact that caspase-11 activation triggers pyroptotic cell death, lead to a proposal of a noncanonical inflammasome mediating caspase-11 activation. However, unlike caspase-9 and caspase-1 whose activation can be induced by over-expression of the relevant CARD-containing proteins18,19, co-expression of 18 different CARD-containing proteins with caspase-11 in mammalian cells did not cause caspase-11 activation and pyroptosis20. Genome-wide loss-of-function screens21–23 or selected screens of the CARD or death-domain superfamily of proteins performed in several laboratories (personal communications) also failed to identify any caspase-11 activator, the central component in the presumed noncanonical inflammasome complex that may also be responsible for sensing intracellular LPS.
Clues into the mechanism of caspase-11 activation by intracellular LPS come from serendipitous observations20. It was observed that full-length caspase-11 protein, unlike most other initiator caspases, could be readily expressed and purified from Escherichia coli or insect cells with a high yield. Further, the bacteria-purified caspase-11 has a different mobility from the insect cell-derived protein on the native gel but not on the denaturing SDS gel. The former shows a much larger size than caspase-11 purified from insect cells in gel filtration analysis. Static light scattering and analytic ultracentrifugation analyses revealed a monomeric and an oligomeric state for the insect cell- and bacteria-purified caspase-11 protein, respectively20. This suggests that certain bacterial agents might induce the oligomerization of caspase-11. Consistent with the previous observation that caspase-11-dependent pyroptosis or caspase-11-dependent NLRP3-mediated IL-1β production is induced by many Gram-negative bacteria such as E. coli, Citrobacter rodentium, Vibrio cholerae, Legionella pneumophila, Salmonella enterica Typhimurium, and Burkholderia14,24–29 but not any Gram-positive bacteria, and thereby LPS was identified to be the responsible bacterial molecule. LPS incubation is sufficient to induce oligomerization of insect cell-purified caspase-11, converting its monomeric state into a large oligomer reminiscent of bacteria-purified caspase-11. A series of further biochemical assays demonstrate that the CARD domain of caspase-11 can bind directly to the lipid A moiety of LPS (Fig. 1). Mutagenesis analyses reveal that the CARD of caspase-11 alone is sufficient for LPS binding, which is mediated by several positively charged motifs in the CARD. Caspase-11 binding to LPS has a high affinity comparable to that of the TLR4-MD2 complex to LPS20. LPS binding to the CARD of caspase-11 not only triggers its oligomerization but also catalytic activation, agreeing with the model that initiator caspases adopt proximity-induced activation. Not only caspase-11 but also its human orthologs caspase-4 and −5 can bind LPS directly and become proteolytically activated to trigger pyroptosis20. Thus, caspase-11, −4 and −5 do not require an NLR-type of scaffold for their activation but instead themselves serve as the functional receptors for intracellular LPS (Fig. 1).
Figure 1. Cytosolic LPS sensing by the non-canonical inflammasome.
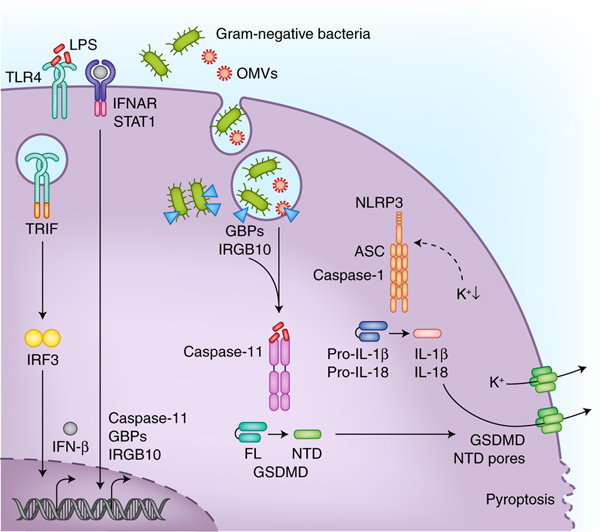
LPS that gains access to the cytosol is sensed by a subfamily of caspases namely caspase-11 in mice and caspase-4 and caspase-5 in humans. The coordinated actions of Guanylate-binding proteins (GBPs) and immunity-related GTPase family member b10 (IRGB10) facilitate the release of LPS from bacteria in the phagosomal vacuole or those that have invaded the cytosol. Outer membrane vesicles (OMVs) secreted by bacteria also enable the cytosolic localization of LPS during infections. The type I interferon signaling initiated downstream of TLR4-TRIF recognition of LPS ensures adequate expression of non-canonical inflammasome components such as caspase-11, GBPs, and IRGB10. Active caspase-11 and caspase-4 cleave gasdermin-D (GSDMD) to liberate its N terminal domain (NTD), which migrates to the plasma membrane forming pores with an inner diameter of about 18 nm. In monocytes, the NLRP3 inflammasome is also activated following GSDMD activation most likely due to the dissipation of intracellular potassium levels through the pores. Accumulation of GSDMD pores on the plasma membrane eventually leads to pyroptotic cell death which occurs in different cell types.
Caspase-1 in the canonical inflammasome pathway has a similar CARD but it does not bind LPS. Close examination of the amino acid sequences reveals some charged residues unique to the CARDs of caspase-4, −5 and −11, which is required for LPS binding. Interestingly, the predicted isoelectric points of the CARDs of caspase-4, −5, and −11 are basic (> 8) whereas that of caspase-1 is approximately 620. This may suggest or explain the binding between the CARDs of caspase-4, −5, and −11 and the acidic phosphate of the lipid A backbone in LPS. Lipid A contains six acyl chains attached to two phosphorylated glucosamines with an β (1→6) linkage. The biosynthetic precursor of lipid A is lipid IVa that has four acyl chains. Caspase-11 and caspase-4 can bind lipid IVa but this binding is nonproductive for caspase-11 activation20. A study has revealed a different situation for caspase-4 that appears to be capable of responding to tetra-acylated LPS from Francisella novicida as well as the synthetic lipid IVa30, suggesting that the human system has a broader reactivity than the mice.
GSDMD is the executioner of caspase-11 and caspase-1-mediated pyroptosis
Caspase-4, −5 and −11 activation by cytosolic LPS as well as caspase-1 by canonical inflammasomes results in pyroptotic cell death31. The molecular mechanism by which these so-called inflammatory caspases execute pyroptosis was elusive for decades until 2015. Two independent unbiased genetic screens, namely the whole-genome CRISPR-Cas9 screen in macrophages22 and ethyl-N-nitrosourea mutagenesis-based forward genetic screen in mice21, identified gasdermin-D (GSDMD) as the pyroptosis executioner protein. GSDMD is a 55 kDa protein belonging to the gasdermin family of unknown function32. GSDMD contains conserved N- and C-terminal domains, and the N-terminal domain (NTD) has the membrane pore forming activity33–36. In the resting state, the C terminal domain (CTD) binds the NTD and keeps the protein in an autoinhibitory state22,34. This property is common to several members of the gasdermin family including GSDMA, GSDMB, GSDMC and GSDME; the NTDs of all the family members share a similar intrinsic membrane pore-forming activity22,34, which redefines the concept of pyroptosis as gasdermin-mediated programmed necrosis31,37. Crystal structures of GSDMA334 and GSDMD38,39 reveal the detailed mechanism for the autoinhibition by the globular helical CTD that contacts the NTD through two interfaces. Mechanistically, activated caspase-11, −4 and −5 as well as caspase-1 cleave GSDMD at an aspartate residue in the linker between NTD and CTD, producing a noncovalent NTD+CTD complex. The NTD has a high affinity for membrane phospholipids such as phosphoinositides and cardiolipin that are negatively charged in their head groups, and the binding stimulates oligomerization-mediated pore formation on the membrane. Phosphoinositides, particularly PI(4,5)P2, is abundant in the inner leaflet of the plasma, consistent with which the NTD of GSDMD was found to translocate to the plasma membrane to induce pyroptotic bubble formation and membrane lysis34. Thus, GSDMD is the final executioner protein for intracellular LPS-induced caspase-11, −4 and −5-mediated pyroptosis owing to its membrane pore-forming activity (Fig. 1).
Consistent with that phosphoinositides and cardiolipin are not present in the outer leaflet of the plasma membrane, activated GSDMD can only target the inner leaflet of the plasma membrane and therefore kill mammalian cells from the inside but not the outside. Activated GSDMD (and also other gasdermins) can efficiently form pores on cardiolipin-containing membranes in vitro, suggesting that bacteria and mitochondria may also be targets of GSDMD35,40. However, this targeting would require the stripping of the outer membranes of Gram-negative bacteria and mitochondria. Further studies are needed to nail down a physiological context where activated GSDMD could access the inner membrane of Gram-negative bacteria and mitochondria. A subsequent study was able to extract GSDMA3 pores from artificial membranes and determine the atomic structure of the entire gasdermin pore by cryo-electron microscopy41. The structure shows a single ring of 27 or 28-fold symmetry. The NTD of GSDMA3 undergoes a radical conformational change in response to lipid binding, which causes its disassociation from the inhibitory CTD and oligomerization into the ring architecture. The conformational change also generates a long four-stranded β-sheet responsible insertion of the ring into the membrane. High-resolution atomic force microscopy (AFM) analyses reveal the dynamic pore-formation process, in which the NTD protomer is inserted into the membrane followed by assembly of the arc- or slit-shaped oligomer prior to growth into the large and complete ring structure42. This mechanism should apply to the entire gasdermin family including GSDMD given their similar biochemical and structural properties.
GSDMD pore causes NLRP3 activation and IL-1β release in macrophages
The gasdermin pore has an inner and an outer diameter of 18 and 28 nm, respectively41, and formation of the pore in the plasma membrane naturally triggers a cascade of events such as ion flux and cell swelling that culminates in cell lysis. Given its large size, the GSDMD pores should allow the release of IL-1 cytokines as well as certain endogenous DAMPs from pyroptotic cells, two well-known events downstream of caspase-1-mediated pyroptosis. Genetic studies in cell culture systems show that the absence of GSDMD completely diminishes mature IL-1β release without affecting IL-1β processing by active caspase-121,22,43. The GSDMD pore is also responsible for IL-1β release even in the absence of cell lysis like in neutrophils or when cell lysis is blocked by an osmotic protectant, or when the amounts of pores are insufficient to lyse the cells such as in the case of infection with a Staphylococcus aureus mutant lacking the gene encoding O-acetyltransferase A or treatment with the oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine (oxPAPC)44,45. These studies have also shown that the GSDMD pores formed on liposome membranes can release IL-1β from the liposome lumen even when the liposomes are not ruptured yet. Two additional studies report that knockout of Gsdmd in mice can completely block all the inflammatory pathologies resulting from knocking-in NLRP3 or MEFV disease-causing mutations into the mice46,47. These genetic and biochemical evidences strongly suggest that the GSDMD pore indeed serves as a conduit for IL-1β release downstream of the caspase-1 inflammasome.
The intracellular LPS-induced pyroptosis was originally discovered in mouse macrophages, where it also leads to NLRP3-mediated IL-1β release14. Because of this early observation and also impacted by the study of the canonical caspase-1 inflammasome pathway, most studies on LPS-induced noncanonical inflammasome activation were conducted in macrophages with IL-1β release as an indicator of caspase-11 activation although caspase-11 does not directly process pro-IL-1β. Notably, two important properties about intracellular LPS sensing have become increasingly appreciated. First, caspase-11 (as well as caspase-4) and GSDMD are widely expressed in many nonmonocytic cells including most epithelial and endothelial cells as well as keratinocytes20,32,48. Indeed, the name of gasdermin comes from its high expression in the gastrointestinal tissue and skin system. Several types of epithelial cells that are positive for caspase-11 and GSDMD expression are shown to be sensitive to intracellular LPS stimulation in vitro and in vivo20,48–50. Importantly but often not noticeable, these epithelial cells do not have a functional NLRP3 pathway and therefore caspase-11 activation by intracellular LPS only causes pyroptosis without IL-1β release in these cells. Secondly, following the identification of GSDMD, it has been increasingly clear that intracellular LPS-induced NLRP3 activation in macrophages is completely diminished by the absence of GSDMD21. This is due to the potassium efflux caused by the GSDMD pores formed on the plasma membrane51, which agrees with the notion that plasma membrane disruption and potassium efflux are strong stimuli for NLRP3 inflammasome activation. Taken together, GSDMD-mediated membrane pore formation and pyroptotic death are the direct and universal event upon caspase-11 recognition of intracellular LPS while the NLRP3-dependent IL-1β response results from formation of GSDMD pores and only occurs in certain cell types like macrophage and dendritic cells (Fig. 1).
Intracellular LPS sensing in host defense and sepsis
Caspase-11 activation by cytosolic LPS is an important event during bacterial infections, which contributes to antibacterial host defense in an increasing number of bacterial infections52. Mice lacking Casp11 are extremely susceptible to even a mild infection with the cytosol-invasive bacteria Burkholderia thailandensis and B. pseudomallei whereas wild-type mice withstand much higher doses of these bacteria28,53. Interestingly, caspase-11-driven pyroptosis of pulmonary epithelial cells contribute to the antimicrobial defense against Burkholderia50. Similarly, caspase-11 was found to mediate host neutrophilic response and bacterial clearance during pulmonary infections with Klebsiella pneumoniae and Acinetobacter baumannii54,55. Particularly, in K. pneumoniae infection, caspase-11 promotes neutrophil recruitment and bacterial clearance via IL-1α rather than IL-1β. Caspase-11 activation also induces the shedding of intestinal epithelial cells infected with S. enterica Typhimurium48. Consistent with the fact that pyroptosis is the dominant response downstream of caspase-11 activation14,20, which is executed by GSDMD21,22, studies have suggested that LPS-induced GSDMD-mediated pyroptosis plays a critical role in host defenses against relevant bacterial infections. It was found that viable bacteria are trapped within the membrane debris of pyroptotic cells following GSDMD activation, a structure called a pore-induced intracellular trap (PIT)56. The PIT not only functions to immobilize the bacteria and prevent their dissemination but also can coordinates other innate immune mechanisms to recruit neutrophil for bacterial clearance. Another mechanism has been uncovered wherein intracellular LPS sensing can contribute to host defense57; GSDMD activation by caspase-11 in neutrophils infected with a Salmonella ΔsifA mutant (lost the membrane of the Salmonella-containing vacuole) or Citrobacter rodentium stimulates the formation of neutrophil extracellular traps (NETs) and cell death (NETosis). Caspase-11 and GSDMD mediate nuclear membrane permeabilization, histone degradation, nuclear delobulation, and DNA extrusion in this process. Importantly, the blockade of caspase-11 and GSDMD-dependent NET formation with deoxyribonuclease I (DNase I) during Salmonella ΔsifA infection promotes bacterial replication and dissemination57. In summary, a pivotal role for LPS-induced caspase-11-mediated pyroptosis in antibacterial immunity is emerging, which likely involves multifarious mechanisms that coordinate together to clear the infection.
Although IL-1β, IL-18, and pyroptosis are all important mechanisms contributing to pathogen clearance and antimicrobial immunity, each of them has the differential capacity to damage tissues and compromise the survival of the host if left unchecked52. Sepsis is one of the diseases resulting from inflammasome activation gone-awry. Intracellular LPS sensing by caspase-11 appears to play a central role in sepsis pathogenesis; mice deficient in Casp11 or Gsdmd are protected from death in an LPS infusion as well as cecal ligation puncture (CLP)-induced polymicrobial sepsis models14,21,58. In contrast, the absence of Nlrp3 or Il1b confers little protection from high-dose LPS-induced lethality. This is consistent with the fact that NLRP3 inflammasome activation downstream of caspase-11 activation only occurs in certain cell types whereas GSDMD-mediated pyroptosis is the universal dominant response. Mechanistically, caspase-11-mediated pyroptosis in endothelial cells appears to contribute to lung injury and lethal manifestation in high-dose LPS-induced CLP-induced polymicrobial sepsis58. Meanwhile, another study suggests that caspase-11 collaborates with caspase-8 in intestinal epithelia to induce tissue damage and executes LPS shock59. The role of caspase-8 in the latter study was determined under the receptor-interacting protein kinase 3 (RIPK3)-deficient or RIPK3 kinase activity-deficient background. Thus, the functional mechanism of caspase-8 in LPS shock remains to be further clarified. As mentioned earlier in this article, Tlr4 deletion in mice leads to a complete resistance to high-dose LPS-induced shock, and this effect can be recapitulated by eritoran, a TLR4-specific inhibitor. Unfortunately, eritoran fails to reduce deaths from sepsis in human clinical trials60. The failure can now be well explained as caspase-11 expression in mice requires priming by TLR-mediated transcription (Fig. 1); importantly, mice primed by a TLR3 agonist can bypass the requirement of TLR4 pathway for LPS-induced lethality while the caspase-11-GSDMD axis remains indispensable16,17. However, the situation in humans is different they express two caspase-11 homologs (caspase-4 and −5) and the expression of caspase-4 appears to be constitutive and does not require TLR-mediated transcriptional priming20,61. Thus, it is possible that blocking intracellular LPS induced cytotoxic responses with caspase-4 or GSDMD inhibitors may be a better therapeutic option to reduce sepsis-associated morbidities and mortalities in humans.
Factors governing LPS access to the cytosol for caspase-11 activation
LPS accessing the cytosol and the resulting caspase-11 activation occur not only during infections with cytosolic bacteria but also non-cytosolic bacteria that reside or get killed in the phagosomes as well27. In the absence of bacterial contact with the host cells, extracellular vesicles generated by the bacteria play a critical role in the cytosolic access of LPS62. Bacterial outer membrane vesicles (OMVs) are membrane-bound structures released during bacterial growth as well as upon exposure to adverse conditions such as antibiotics and antimicrobial peptides63,64. LPS is an abundant cargo of OMVs, and OMVs released by bacteria are capable of delivering LPS into the cytosol and triggering caspase-11-mediated pyroptosis. Bacterial mutants defective for OMV production are compromised in their ability to release LPS into the cytosol and activate caspase-11, indicating a key role of OMVs in cytosolic access of LPS during bacterial infections62. OMVs are taken up by the host cells via clathrin-mediated endocytosis and OMV-bound LPS is released into the cytosol from early endocytic compartments. OMV-mediated delivery of LPS into the host cell cytosol for caspase-11 or caspase-4 or −5 activation has been observed with a number of Gram-negative bacteria65–71.
Studies have uncovered a central role for type I interferon signaling in translocation and sensing of LPS in the cytosol. The cytosolic receptor for LPS, caspase-11, itself is an interferon-stimulated gene; caspase-11, unlike human caspase-4, is weakly expressed in resting cells and its optimal expression during infections with Gram-negative bacteria is mediated directly by the NF-κB signaling downstream of the TLRs and indirectly by the TLR4-TRIF pathway through type I interferons25–27,59 (Fig. 1). Interestingly, TLR-controlled expression of caspase-11 is positively regulated by the Cpb1–C3–C3aR complement pathway in a cell autonomous and non–cell autonomous manner23. In addition, another set of interferon inducible proteins such as guanylate binding proteins (GBPs) and interferon response gene B10 (IRGB10) also play an important role in innate immune surveillance of cytosolic LPS. GBPs are a family of 65–73 kDa GTPases with seven and eleven members in humans and mice, respectively72. Though originally discovered to have antiviral functions, GBPs play strong roles during infections with intracellular protozoan and bacterial pathogens73,74. Different GBPs are recruited to the pathogen-containing vacuoles during infections with Toxoplasma gondii, Mycobacterium spp. and Listeria monocytogenes or to the cytosolic Shigella flexneri73–77. GBPs contribute to cell autonomous defense against intracellular pathogens via several mechanisms such as limiting intracellular bacterial survival and motility. Bacteria can counter the antibacterial actions of GBPs by delivering a ubiquitin ligase effector IpaH9.8 to target GBPs for proteasomal degradation75,76. In addition to their roles in cell autonomous antibacterial defense, GBPs have also been shown to be involved in noncanonical inflammasome activation. Caspase-11-mediated cell death and the downstream IL-1 response to various bacteria such as S. enterica Typhimurium, L. pneumophila, and E. coli were attenuated in macrophages derived from Gbpchr3−/− mice lacking Gbp1, Gbp2, Gbp3, Gbp5, and Gbp778,79. OMV activation of caspase-11 was similarly reduced in macrophages from Gbpchr3−/− mice. OMV-induced IL-1β and IL-18 release as well as lethality were also attenuated in Gbpchr3−/− mice67,68. While a role for GBPs in caspase-11 activation has been established, the underlying mechanism seems less clear; it appears that GBPs are recruited either to bacterial membranes or to the ruptured bacteria-containing vacuoles, where they act to extract or expose lipid A buried in bacterial membranes for caspase-11 recognition67,68,78–81 (Fig. 1). It has also been suggested that GBPs can recruit another interferon-inducible protein, IRGB10, to intracellular bacteria to execute the disruption of bacterial cell membrane and the liberation of bacterial ligands for the inflammasome recognition82.
Another report shows an important role for HMGB1 in enabling cytosolic access of LPS83. HMGB1 is an alarmin released downstream of inflammasome activation and pyroptosis20,84. HMGB1 is released by hepatocytes upon TLR4 stimulation by LPS, the HMGB1 then binds LPS and triggers its uptake by macrophages via the receptor RAGE. Subsequently, LPS-bound HMGB1 destabilizes the endosomal membrane, leading to the release of LPS into the cytosol. Supporting the notion that hepatocytes are the major source of HMGB1 released during endotoxemia, mice with conditional deletion of Hmgb1 in hepatocytes are protected from lethal endotoxic shock due to a reduced inflammasome response to LPS83. It is interesting to note that the opposite response to lethal-dose LPS injection has been observed in mice with Hmgb1 deleted in myeloid cells85. Another cytokine-like secreted protein has also been implicated in the cytosolic access of LPS. Secretoglobin 3A2 (SCGB3A2), a member of the secretoglobin family of proteins predominantly secreted by the airway epithelium, binds and chaperones LPS to the cytosol through its receptor syndecan-1 in Lewis lung carcinoma cells and RAW macrophages, leading to caspase-11-dependent pyroptosis in vitro86. Follow-up studies are needed to determine or confirm the role of these candidate LPS-entry pathways in LPS-induced septic shock. Alternatively, cytosolic entry of LPS during endotoxemia might be mediated by another high-efficient LPS transporter that is absent in commonly assayed cell culture systems.
Negative regulation of the pyroptotic response to cytosolic LPS
Intracellular LPS-elicited release of IL-1β and IL-18 as well as pyroptosis play protective roles during bacterial infections. But uncontrolled pyroptotic response has harmful consequences for the host causing organ damage and death. Host mechanisms that serve as a brake to keep pyroptosis in check are just beginning to be understood. Glutathione peroxidase-4 (GPX4) is an antioxidant enzyme belonging to the family of glutathione peroxidases. Glutathione peroxidase-4 (GPX4) negatively regulates GSDMD-mediated pyroptosis by reducing phospholipid hydroperoxides and limiting oxidative damage to lipids during pyroptotic cascade87,88 (Fig. 2). In the absence of GPX4, the elevated production of phospholipid peroxidation products has the ability to enhance cell lytic activity of GSDMD. Consequently, mice lacking GPX4 in the myeloid compartment display excessive organ damage and enhanced susceptibility to polymicrobial sepsis. oxPAPC has been shown to competitively inhibit caspase-11 binding of LPS and the subsequent inflammatory response89. In contrast, oxPAPC has also been shown to bind caspase-11 and activate IL-1 responses but not pyroptosis in dendric cells90. Stearoyl lysophosphatidylcholine (LPC), a major component of oxidized low-density lipoproteins, can block caspase-11 binding of LPS, reduce cellular responses to cytosolic LPS, and importantly improve the survival of mice during lethal endotoxin shock91. Thus, the effect of oxidized phospholipids on caspase-11 appears to be dose-, cell type- and context-dependent (Fig. 2). Endosomal sorting complexes required for transport (ESCRT)-III has been shown to limit GSDMD-mediated pyroptosis92. The plasma membrane perforation by GSDMD causes Calcium influx, which then recruits ESCRT-III complexes to the plasma membrane. ESCRT-III complexes repair the plasma membrane perforation and therefore negatively regulate the pyroptotic lysis (Fig. 2).
Figure 2. Negative regulation of the non-canonical inflammasome.
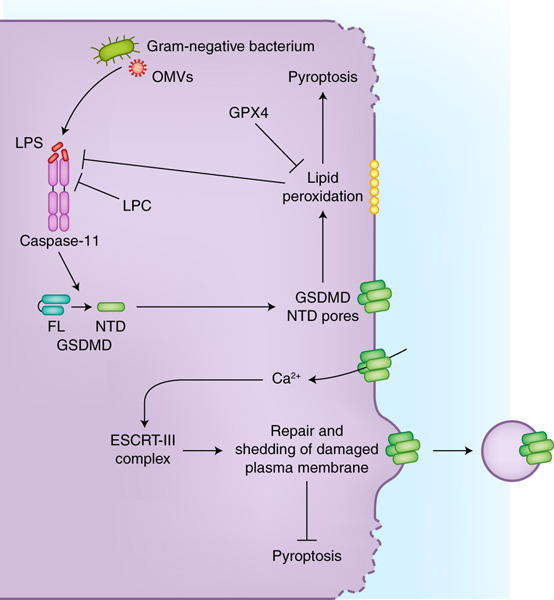
Host cells mitigate the pyroptotic cascade, driven by the non-canonical inflammasome downstream of cytosolic LPS, via a number of mechanisms. Glutathione peroxidase-4 (GPX4) reduces the lipid peroxidation that occurs during pyroptosis and thereby limits the extent of pyroptosis. Stearoyl lysophosphatidylcholine (LPC) can inhibit LPS binding by caspase-11. Furthermore, calcium influx elicited by the GSDMD pores leads to the activation of ESCRT-III complex, which repairs plasma membrane containing pores, thereby attenuating pyroptotic cell rupture.
Conclusions and future directions
Intensive research efforts have discovered a new mechanism of innate immune sensing of LPS. This mode of surveillance for intracellular LPS by caspase-11 in mice and caspase-4 and −5 in humans elicits lytic cell death via the pore forming protein GSDMD, which indirectly triggers the maturation of IL-1β and IL-18 in certain cell types. Studies have revealed a critical role of this intracellular LPS sensing pathway not only in antibacterial defense but also in sepsis pathogenesis as well. While remarkable progress has been made in this area, several pressing questions remain. First, the structure of caspase-11 and caspase-4 with LPS needs to be resolved to gain insights into the molecular basis of how LPS is specifically recognized by these caspases and how the binding induces their activation. Second, further insights into the precise mechanism underlying LPS translocation from endosomes to the cytosol during infections as well as LPS transport into the cytosol during endotoxic shock is also needed. Third, certain bacteria have been found to evade caspase-11-mediated detection by synthesizing tetra-acylated LPS16 or counteracting the caspase-11 pathway through secreted effector protein during infection93. Such mechanisms might be exploited by other pathogens, which should be taken into consideration in future studies determining the contribution of the intracellular LPS sensing pathway for defenses against particular bacterial pathogens. Finally, the relevance of caspase-4 and caspase-5-based recognition of LPS to human infectious diseases as well as human sepsis remains to be further explored. Although caspase-4 appears to be widely expressed in various cell types and tissues, information about caspase-5 expression and function is scarce in the literature. Recently, a new study has uncovered that ADP-heptose, a precursor sugar molecule essential for LPS biosynthesis, is directly recognized by host alpha-kinase 1 (ALPK1), which in turn results in potent activation of the NF-κB proinflammatory response94. Given that ADP-heptose is an abundant metabolite universal to Gram-negative bacteria, future studies are needed to understand how ALPK1 might coordinate with caspase-11, −4 or −5 in antibacterial defense and whether or not its activation by ADP-heptose also contributes to Gram-negative bacteria-induced sepsis, particularly in humans.
Acknowledgements
We thank J. Snyder for editing the manuscript. Research in the Shao laboratory is supported by the Basic Science Center Project of the National Natural Science Foundation of China (81788101) and National Key Research and Development Program of China (2016YFA0501500 and 2017YFA0505900). Research in the Rathinam laboratory is supported by the National Institutes of Health (R01AI119015 and R21AI 135528).
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Takeuchi O & Akira S Pattern recognition receptors and inflammation. Cell 140, 805–20 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Pandey S, Kawai T & Akira S Microbial sensing by Toll-like receptors and intracellular nucleic acid sensors. Cold Spring Harb Perspect Biol 7, a016246 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Man SM, Karki R & Kanneganti TD Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol Rev 277, 61–75 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Friedlander AM Macrophages are sensitive to anthrax lethal toxin through an acid-dependent process. J Biol Chem 261, 7123–6 (1986). [PubMed] [Google Scholar]
- 5.Zychlinsky A, Prevost MC & Sansonetti PJ Shigella flexneri induces apoptosis in infected macrophages. Nature 358, 167–9 (1992). [DOI] [PubMed] [Google Scholar]
- 6.Fink SL, Bergsbaken T & Cookson BT Anthrax lethal toxin and Salmonella elicit the common cell death pathway of caspase-1-dependent pyroptosis via distinct mechanisms. Proc Natl Acad Sci U S A 105, 4312–7 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martinon F, Burns K & Tschopp J The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 10, 417–26 (2002). [DOI] [PubMed] [Google Scholar]
- 8.Zhao Y & Shao F Diverse mechanisms for inflammasome sensing of cytosolic bacteria and bacterial virulence. Curr Opin Microbiol 29, 37–42 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Broz P & Dixit VM Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol 16, 407–20 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Raetz CR & Whitfield C Lipopolysaccharide endotoxins. Annu Rev Biochem 71, 635–700 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park BS et al. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 458, 1191–5 (2009). [DOI] [PubMed] [Google Scholar]
- 12.Li P et al. Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock. Cell 80, 401–11 (1995). [DOI] [PubMed] [Google Scholar]
- 13.Wang S et al. Murine caspase-11, an ICE-interacting protease, is essential for the activation of ICE. Cell 92, 501–9 (1998). [DOI] [PubMed] [Google Scholar]
- 14.Kayagaki N et al. Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–21 (2011). [DOI] [PubMed] [Google Scholar]
- 15.Kang SJ et al. Dual role of caspase-11 in mediating activation of caspase-1 and caspase-3 under pathological conditions. J Cell Biol 149, 613–22 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hagar JA, Powell DA, Aachoui Y, Ernst RK & Miao EA Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science 341, 1250–3 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kayagaki N et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341, 1246–9 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Perkins C, Kim CN, Fang G & Bhalla KN Overexpression of Apaf-1 promotes apoptosis of untreated and paclitaxel- or etoposide-treated HL-60 cells. Cancer Res 58, 4561–6 (1998). [PubMed] [Google Scholar]
- 19.Srinivasula SM et al. The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. J Biol Chem 277, 21119–22 (2002). [DOI] [PubMed] [Google Scholar]
- 20.Shi J et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514, 187–92 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Kayagaki N et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signaling. Nature 526, 666–71 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Shi J et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–5 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Napier BA et al. Complement pathway amplifies caspase-11-dependent cell death and endotoxin-induced sepsis severity. J Exp Med 213, 2365–2382 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Akhter A et al. Caspase-11 promotes the fusion of phagosomes harboring pathogenic bacteria with lysosomes by modulating actin polymerization. Immunity 37, 35–47 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Broz P et al. Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature 490, 288–91 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gurung P et al. Toll or interleukin-1 receptor (TIR) domain-containing adaptor inducing interferon-beta (TRIF)-mediated caspase-11 protease production integrates Toll-like receptor 4 (TLR4) protein- and Nlrp3 inflammasome-mediated host defense against enteropathogens. J Biol Chem 287, 34474–83 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rathinam VA et al. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell 150, 606–19 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aachoui Y et al. Caspase-11 protects against bacteria that escape the vacuole. Science 339, 975–8 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Case CL et al. Caspase-11 stimulates rapid flagellin-independent pyroptosis in response to Legionella pneumophila. Proc Natl Acad Sci U S A 110, 1851–6 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lagrange B et al. Human caspase-4 detects tetra-acylated LPS and cytosolic Francisella and functions differently from murine caspase-11. Nat Commun 9, 242 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shi J, Gao W & Shao F Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem Sci 42, 245–254 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Saeki N & Sasaki H Gasdermin Superfamily: A Novel Gene Family Functioning in Epithelial Cells in Endothelium and epithelium : composition, functions, and pathology (eds. Carrasco J & Matheus M) 193–211 (Nova Science Publishers, Hauppauge, N.Y., 2011). [Google Scholar]
- 33.Aglietti RA et al. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci U S A 113, 7858–63 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ding J et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535, 111–6 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Liu X et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sborgi L et al. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J 35, 1766–78 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547, 99–103 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Kuang S et al. Structure insight of GSDMD reveals the basis of GSDMD autoinhibition in cell pyroptosis. Proc Natl Acad Sci U S A 114, 10642–10647 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Z et al. Structures of the Gasdermin D C-Terminal Domains Reveal Mechanisms of Autoinhibition. Structure 26, 778–784 e3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Platnich JM et al. Shiga Toxin/Lipopolysaccharide Activates Caspase-4 and Gasdermin D to Trigger Mitochondrial Reactive Oxygen Species Upstream of the NLRP3 Inflammasome. Cell Rep 25, 1525–1536 e7 (2018). [DOI] [PubMed] [Google Scholar]
- 41.Ruan J, Xia S, Liu X, Lieberman J & Wu H Cryo-EM structure of the gasdermin A3 membrane pore. Nature 557, 62–67 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mulvihill E et al. Mechanism of membrane pore formation by human gasdermin-D. EMBO J 37(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.He WT et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res 25, 1285–98 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Evavold CL et al. The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity 48, 35–44 e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heilig R et al. The Gasdermin-D pore acts as a conduit for IL-1beta secretion in mice. Eur J Immunol 48, 584–592 (2018). [DOI] [PubMed] [Google Scholar]
- 46.Kanneganti A et al. GSDMD is critical for autoinflammatory pathology in a mouse model of Familial Mediterranean Fever. J Exp Med 215, 1519–1529 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xiao J et al. Gasdermin D mediates the pathogenesis of neonatal-onset multisystem inflammatory disease in mice. PLoS Biol 16, e3000047 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Knodler LA et al. Noncanonical inflammasome activation of caspase-4/caspase-11 mediates epithelial defenses against enteric bacterial pathogens. Cell Host Microbe 16, 249–256 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pallett MA et al. Bacterial virulence factor inhibits caspase-4/11 activation in intestinal epithelial cells. Mucosal Immunol 10, 602–612 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang J et al. Caspase-11-dependent pyroptosis of lung epithelial cells protects from melioidosis while caspase-1 mediates macrophage pyroptosis and production of IL-18. PLoS Pathog 14, e1007105 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ruhl S & Broz P Caspase-11 activates a canonical NLRP3 inflammasome by promoting K(+) efflux. Eur J Immunol 45, 2927–36 (2015). [DOI] [PubMed] [Google Scholar]
- 52.Russo AJ, Behl B, Banerjee I & Rathinam VAK Emerging Insights into Noncanonical Inflammasome Recognition of Microbes. J Mol Biol 430, 207–216 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Aachoui Y et al. Canonical Inflammasomes Drive IFN-gamma to Prime Caspase-11 in Defense against a Cytosol-Invasive Bacterium. Cell Host Microbe 18, 320–32 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang J et al. Caspase-11 deficiency impairs neutrophil recruitment and bacterial clearance in the early stage of pulmonary Klebsiella pneumoniae infection. Int J Med Microbiol 307, 490–496 (2017). [DOI] [PubMed] [Google Scholar]
- 55.Wang W et al. Caspase-11 Plays a Protective Role in Pulmonary Acinetobacter baumannii Infection. Infect Immun 85(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jorgensen I, Zhang Y, Krantz BA & Miao EA Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J Exp Med 213, 2113–28 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen KW et al. Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci Immunol 3(2018). [DOI] [PubMed] [Google Scholar]
- 58.Cheng KT et al. Caspase-11-mediated endothelial pyroptosis underlies endotoxemia-induced lung injury. J Clin Invest 127, 4124–4135 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mandal P et al. Caspase-8 Collaborates with Caspase-11 to Drive Tissue Damage and Execution of Endotoxic Shock. Immunity 49, 42–55 e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Opal SM et al. Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: the ACCESS randomized trial. JAMA 309, 1154–62 (2013). [DOI] [PubMed] [Google Scholar]
- 61.Lin XY, Choi MS & Porter AG Expression analysis of the human caspase-1 subfamily reveals specific regulation of the CASP5 gene by lipopolysaccharide and interferon-gamma. J Biol Chem 275, 39920–6 (2000). [DOI] [PubMed] [Google Scholar]
- 62.Vanaja SK et al. Bacterial Outer Membrane Vesicles Mediate Cytosolic Localization of LPS and Caspase-11 Activation. Cell 165, 1106–1119 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kulp A & Kuehn MJ Biological functions and biogenesis of secreted bacterial outer membrane vesicles. Annu Rev Microbiol 64, 163–84 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kaparakis-Liaskos M & Ferrero RL Immune modulation by bacterial outer membrane vesicles. Nat Rev Immunol 15, 375–87 (2015). [DOI] [PubMed] [Google Scholar]
- 65.Chen S et al. Dysregulated hemolysin liberates bacterial outer membrane vesicles for cytosolic lipopolysaccharide sensing. PLoS Pathog 14, e1007240 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wacker MA, Teghanemt A, Weiss JP & Barker JH High-affinity caspase-4 binding to LPS presented as high molecular mass aggregates or in outer membrane vesicles. Innate Immun 23, 336–344 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Finethy R et al. Inflammasome Activation by Bacterial Outer Membrane Vesicles Requires Guanylate Binding Proteins. MBio 8(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Santos JC et al. LPS targets host guanylate-binding proteins to the bacterial outer membrane for non-canonical inflammasome activation. EMBO J 37(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bitto NJ et al. Membrane vesicles from Pseudomonas aeruginosa activate the noncanonical inflammasome through caspase-5 in human monocytes. Immunol Cell Biol 96, 1120–1130 (2018). [DOI] [PubMed] [Google Scholar]
- 70.Gu L et al. Toll-Like Receptor 4 Signaling Licenses the Cytosolic Transport of Lipopolysaccharide From Bacterial Outer Membrane Vesicles. Shock 51, 256–265 (2019). [DOI] [PubMed] [Google Scholar]
- 71.Cecil JD et al. Outer Membrane Vesicles Prime and Activate Macrophage Inflammasomes and Cytokine Secretion In Vitro and In Vivo. Front Immunol 8, 1017 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim BH et al. Interferon-induced guanylate-binding proteins in inflammasome activation and host defense. Nat Immunol 17, 481–9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kim BH et al. A family of IFN-gamma-inducible 65-kD GTPases protects against bacterial infection. Science 332, 717–21 (2011).21551061 [Google Scholar]
- 74.Yamamoto M et al. A cluster of interferon-gamma-inducible p65 GTPases plays a critical role in host defense against Toxoplasma gondii. Immunity 37, 302–13 (2012). [DOI] [PubMed] [Google Scholar]
- 75.Wandel MP et al. GBPs Inhibit Motility of Shigella flexneri but Are Targeted for Degradation by the Bacterial Ubiquitin Ligase IpaH9.8. Cell Host Microbe 22, 507–518 e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li P et al. Ubiquitination and degradation of GBPs by a Shigella effector to suppress host defence. Nature 551, 378–383 (2017). [DOI] [PubMed] [Google Scholar]
- 77.Pilla-Moffett D, Barber MF, Taylor GA & Coers J Interferon-Inducible GTPases in Host Resistance, Inflammation and Disease. J Mol Biol 428, 3495–513 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Meunier E et al. Caspase-11 activation requires lysis of pathogen-containing vacuoles by IFN-induced GTPases. Nature 509, 366–70 (2014). [DOI] [PubMed] [Google Scholar]
- 79.Pilla DM et al. Guanylate binding proteins promote caspase-11-dependent pyroptosis in response to cytoplasmic LPS. Proc Natl Acad Sci U S A 111, 6046–51 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Meunier E et al. Guanylate-binding proteins promote activation of the AIM2 inflammasome during infection with Francisella novicida. Nat Immunol 16, 476–484 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Man SM et al. The transcription factor IRF1 and guanylate-binding proteins target activation of the AIM2 inflammasome by Francisella infection. Nat Immunol 16, 467–75 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Man SM et al. IRGB10 Liberates Bacterial Ligands for Sensing by the AIM2 and Caspase-11-NLRP3 Inflammasomes. Cell 167, 382–396 e17 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Deng M et al. The Endotoxin Delivery Protein HMGB1 Mediates Caspase-11-Dependent Lethality in Sepsis. Immunity 49, 740–753 e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lamkanfi M et al. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J Immunol 185, 4385–92 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yanai H et al. Conditional ablation of HMGB1 in mice reveals its protective function against endotoxemia and bacterial infection. Proc Natl Acad Sci U S A 110, 20699–704 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yokoyama S et al. A novel pathway of LPS uptake through syndecan-1 leading to pyroptotic cell death. Elife 7(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kang R et al. Lipid Peroxidation Drives Gasdermin D-Mediated Pyroptosis in Lethal Polymicrobial Sepsis. Cell Host Microbe 24, 97–108 e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Russo AJ & Rathinam VAK Lipid Peroxidation Adds Fuel to Pyr(optosis). Cell Host Microbe 24, 8–9 (2018). [DOI] [PubMed] [Google Scholar]
- 89.Chu LH et al. The oxidized phospholipid oxPAPC protects from septic shock by targeting the non-canonical inflammasome in macrophages. Nat Commun 9, 996 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zanoni I et al. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science 352, 1232–6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li W et al. Stearoyl Lysophosphatidylcholine Inhibits Endotoxin-Induced Caspase-11 Activation. Shock 50, 339–345 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ruhl S et al. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 362, 956–960 (2018). [DOI] [PubMed] [Google Scholar]
- 93.Kobayashi T et al. The Shigella OspC3 effector inhibits caspase-4, antagonizes inflammatory cell death, and promotes epithelial infection. Cell Host Microbe 13, 570–583 (2013). [DOI] [PubMed] [Google Scholar]
- 94.Zhou P et al. Alpha-kinase 1 is a cytosolic innate immune receptor for bacterial ADP-heptose. Nature 561, 122–126 (2018). [DOI] [PubMed] [Google Scholar]