

ABSTRACT
Simulating a viral infection in tumor cells is an attractive concept to eliminate tumor cells. We previously reported the molecular design and the in vitro potency of recombinant monoclonal antibodies fused to a virus-derived peptide MHC class I complex that bypass the peptide processing and MHC loading pathway and directly displays a viral peptide in an MHC class I complex on the tumor cell surface. Here, we show that a vaccination-induced single peptide-specific CD8 T cell response was sufficient to eliminate B16 melanoma tumor cells in vivo in a fully immunocompetent, syngeneic mouse tumor model when mice were treated with mouse pMHCI-IgGs fusion proteins targeting the mouse fibroblast activation protein. Tumor growth of small, established B16 lung metastases could be controlled. The pMHCI-IgG had similar potency as an analogous pan-CD3 T-cell bispecific antibody. In contrast to growth control of small tumors, none of the compounds controlled larger solid tumors of MC38 cancer cells, despite penetration of pMHCI-IgGs into the tumor tissue and clear attraction and activation of antigen-specific CD8 T cells inside the tumor. pMHCI-IgG can have a similar potency as classical pan-T-cell recruiting molecules. The results also highlight the need to better understand immune suppression in advanced solid tumors.
KEYWORDS: Cancer immunotherapy, CMV, anti-viral CD 8 T cells, single peptide vaccination, MHCI restricted T-cell activation, antibody fusion, major histocompatibility class I, targeted T-cell recruiter, viral mimicry on cancer cells, tumor cell elimination, B16 lung metastases
Introduction
Immunotherapy is changing the way cancer is treated. Novel immunotherapeutic concepts are at various development stages, with some already approved.1–4 Most immunotherapies, including immune checkpoint blockade (e.g., anti-PD-1 and anti-CTLA-4), cytokine therapy, co-stimulatory therapies, modification of the tumor microenvironment, vaccines, and oncolytic viruses, rely largely on the endogenous antigenicity of tumor cells. In contrast, adoptive transfer of chimeric antigen receptor-expressing T cells (CAR-T cells), and T cell-recruiting bispecific antibodies (TCBs), which activate via CD3, do not rely on neoantigens. These latter immunotherapies bypass the major histocompatibility complex (MHC)/peptide recognition normally required for T cell activation. Despite the efficacy of CAR-T cells and TCBs, they frequently unleash severe adverse effects, likely due to the strong and extensive T cell activation and related cytokine release.1–4
An alternative method of activating T cells to eliminate tumor cells is to use only antigen-specific T cells instead of all T cells. For viral infections, potent antigens are well described and can be used as antigenic peptides in recombinant peptide MHC class I complexes to engage preexisting effector cells. For that purpose, pMHCI-IgG molecules have been designed.5–7 These chimeric proteins contain a monoclonal antibody (mAb), specific for a tumor-associated antigen, linked to a single-chain peptide-MHC I molecule (pMHCI) presenting an antigenic, mostly immunodominant viral peptide. The pMHCI-IgGs are named according to the peptide in the MHC class I complex and the antibody target (e.g., M38-αFAP). The molecules mimic an infection in the tumor cells via a viral antigen presented on MHC I, which activates virus-specific CD8 T cells. Similar to CAR-T cell and TCB immunotherapies, tumor cells are flagged for CD8 T cell killing, regardless of whether or not they process and present endogenous MHC I.
The T cell population specific for cytomegalovirus (CMV) is constant over many years in infected humans and continuously re-supplied due to the chronic nature of CMV infection. CMV infection is highly prevalent, and the number of CD8 T cells directed against a single antigenic peptide is high (0.5% to 20% in patients).8 Compared to pan-T cell recruiters, such as CAR-T cells and TCBs, these anti-viral T cell populations would be expected to release low levels of only Th1 cytokines. Indeed, we detected lower levels of cytokines in vitro, and only Th1 but not Th2 cytokines, following pMHCI-IgG activation compared to pan-T cell recruiters, yet retained complete tumor cell killing.7 Also, the IgG Fc confers favorable pharmacokinetic properties, such as long half-life and favorable distribution.7
The pMHCI-IgG treatment requires a prior infection of the patient with CMV and a match of the highly polymorphic MHC allotype.9 A human therapeutic pMHCI-IgG would be applicable, for example, to CMV-infected individuals who express HLA-A*0201, limiting the patient population to about 25% to 30%, who are both CMV-positive and express HLA-A*0201. Individuals with the correct HLA allele, but negative for CMV, would need to be vaccinated against the peptide contained within the pMHCI-IgG for the therapy to be effective. Here we report a syngeneic, fully immunocompetent mouse model resembling the anticipated situation with CMV-negative patients. Mice were primed with a mouse cytomegalovirus (MCMV) peptide coupled to a DC-targeted XCR1 mAb and received a peptide-cytokine boost to elicit robust antigen-specific CD8 T cell responses. We engineered pMHCI-IgGs and TCBs to target mouse fibroblast activation protein (mFAP) and directly compared their activity against B16 melanoma and MC38 colon carcinoma in fully syngeneic mice. We found that pMHCI-IgGs could target tumor cells in vivo and conferred a viral antigenicity to them, which activated vaccine-induced anti-viral CD8 T cells. The pMHCI-IgGs and TCBs both controlled tumor growth in a metastatic lung model. However, treatment of established solid tumors was ineffective. Despite clear activation of CD8 T cells inside the tumor, the activated CD8 T cells were quickly suppressed.
Results
Generation of peptide-specific CD8 T cells by vaccination in C57BL/6N mice
Vaccination consistently primed 1–4% M38-H-2Kb-specific CD8 T cells, characterized as CD44+/CD62L-/CD127+ effector memory T cells 3 weeks after vaccination. M38-specific CD8 T cells averaged 15% at the day 9 peak, although individual mice reached up to 60%, and reached an equilibrium of ~2.5% by day 40 (a typical example is shown in Figure S1).
pMHCI-IgG and TCB molecules activated CD8 T cells in vitro in a peptide-specific and concentration-dependent manner
The ability of pMHCI-IgGs and TCBs (Figure 1a) to activate CD8 T cells was tested in an in vitro interferon (IFN)-γ activation assay (Figure 1b). MC38-FAP colorectal cancer cells were incubated with splenocytes from vaccinated mice in the presence of M38-αFAP fusion molecules, IE3-αFAP control fusion molecules, or the TCB-αFAP molecule. In the IFN-γ activation assay, 66% of CD3+ cells were CD8 T cells, and 62% of these were M38-H-2Kb-specific. The M38-αFAP triggered IFN-γ expression in 6% of all CD8 T cells, which represented 10% of the M38-H-2Kb-specific CD8 T cell population. In comparison, the TCB-αFAP, recruiting effector cells via CD3, activated about 10% of all CD8 T. M38 peptide-loaded target cells mediated activation of about 12% of the entire CD8 T cell population. The pMHCI-IgG containing the control peptide IE3, or only tumor cells and splenocytes, mediated no IFN-γ activation of CD8 T cells, indicating that activation of effector cells was strictly peptide- and construct-specific. We found that peptide-loading of tumor cells achieved the maximum of IFN-γ activation only at higher concentrations (5 nM) compared to pMHCI-IgG treated tumor cells, which reached maximal IFN-γ activation at lower concentrations (1 nM).
Figure 1.
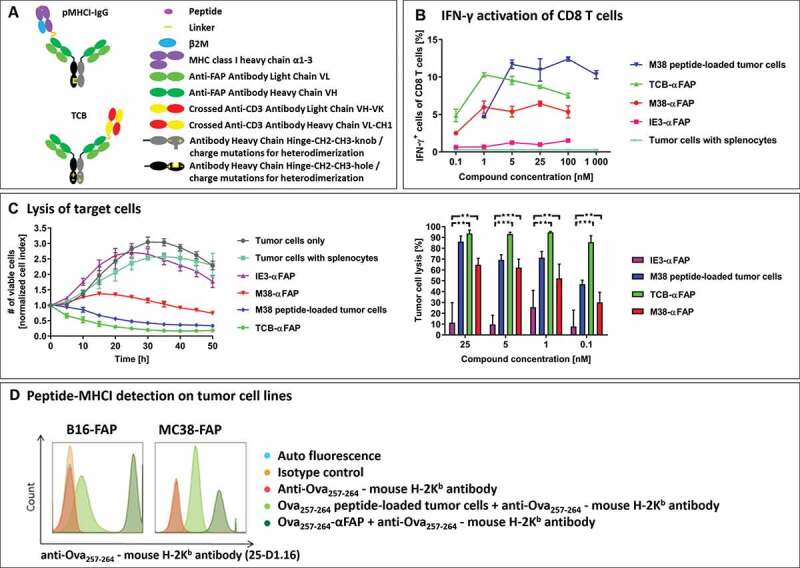
(a) Schematic design of the pMHCI-IgG (top) and the TCB fusion proteins (bottom). Both formats consist of a full monoclonal antibody in an IgG format (light green: antibody light chain, dark green: antibody heavy chain variable domain and CH1, gray/black: constant domain of the antibody heavy chain in the knob and hole format or with charge mutations to drive heterodimerization). A single pMHCI complex consisting of the antigenic peptide (magenta), MHC class I heavy chain lacking the transmembrane domain (purple) and beta-2-microglobulin (blue) or a single anti-CD3 binding CrossMab Fab (crossed heavy and light chain in yellow and red) are fused to one of the antibody heavy chains. (b) Frequency of IFN-γ expressing CD8 T cells measured in flow cytometry after exposure to compound-treated target cells. Incubation of MC38-FAP tumor cells loaded with MCMV-derived M38 peptide (blue), T cell bispecific TCB-αFAP (green), pMHCI-IgG M38-αFAP (red) or pMHCI-IgG IE3-αFAP (pink) and unloaded tumor cells (cyan) with splenocytes from M38 vaccinated mice. Splenocytes were isolated nine days after start of vaccination. All graphs show mean of replicates (n = 2) with error bars indicating standard deviation. (c) Specific tumor cell lysis mediated by compounds after incubation with splenocytes from M38 vaccinated mice. Number of viable cells is measured with xCELLigence as electrical impedance mediated by adherent cells on the culture dish bottom. Color code: IE3-αFAP (purple), M38 peptide (blue), TCB-αFAP (green), M38-αFAP (red). Kinetics of cell lysis at a compound concentration of 25 nM is shown (left). Tumor cell elimination after 40 hours is shown for all concentrations tested (right). All graphs show mean of replicates (n = 3) with error bars indicating standard deviation. Two-sided t-test for significance evaluation was applied. P-values from 0.01 to 0.05 were considered as significant (*), p-values from 0.001 to < 0.01 were considered as very significant (**) and p-values < 0.001 were considered as extremely significant (***). (d) Relative expression of endogenous MHC class I compared to pMHCI-IgG delivered recombinant MHC class I. FAP expressing B16 and MC38 tumor cells were loaded with Ova257-264 peptide (light green) or pMHCI-IgG Ova257-264-αFAP (dark green) and SIINFEKL peptide-MHC class I complexes were detected with the monoclonal antibody anti-Ova257-264 – mouse H-2Kb antibody (25-D1.16) in flow cytometry
Tumor cells are eliminated in vitro by pMHCI-IgG and TCB through redirection of CD8 T cells
pMHCI-IgG and TCB molecules were able to induce a concentration-dependent and target-specific lysis of tumor cells (Figure 1c). MC38-FAP cells were co-cultured with splenocytes from vaccinated mice in the presence of either M38-αFAP, IE3-αFAP control or TCB-αFAP. Tumor cells loaded with M38 peptide were used as a positive control. The M38-specific T cell effector to MC38-FAP target cell ratio (E:T ratio) was 0.25:1. The TCB-αFAP led to a rapid elimination of target cells and achieved a tumor cell lysis of 93% after 40 hours of incubation at compound concentrations of 1, 5, and 25 nM. Tumor cells loaded with M38 peptide were eliminated with slower kinetics. The M38-αFAP showed a delayed onset of cell killing, as previously observed.7 Sixty-three percent of tumor cells could be eliminated at compound concentrations of 5 and 25 nM after 40 hours. The kinetics of continuous cell killing showed that cell elimination was maintained after 40 hrs and that complete elimination of tumors may be achieved at later time points. IE3-αFAP, containing an irrelevant control peptide, did not trigger cell killing even at high concentrations. The expression of endogenous MHC class I is lower than the recombinant peptide-carrying MHC class I delivered via the antibody fusion protein in M38 and B16 cells, recombinantly expressing FAP (Figure 1d). Both formats used here for the pMHCI-IgG as well as the TCB had expected in vitro potencies in the low nM range for the pMHCI-IgG and pM range for the TCB.
pMHCI-IgG and TCB protect mice from experimental lung metastases in vivo
Mice were vaccinated to induce an endogenous CD8 T cell response specific for the M38 epitope of MCMV (Figure S1, Figure 2a, preventive setting). At the peak of the immune response, when M38-specific CD8 T cells averaged 3.2% in peripheral blood (range 1.4%-5.7%), mice were treated with pMHCI-IgGs or TCBs. After 24 hours, mice were challenged intravenously (i.v.) with 2 × 105 B16-FAP melanoma cells. Two days after tumor cell injection, a second treatment with M38-αFAP or TCB-αFAP was administered. On day 21 after injection of tumor cells, the lung metastatic burden was assessed by counting visible metastases on the lung surface and quantitation of the melanoma marker TRP-2 expression by RT-qPCR (Figure 2b, and supplement Figure S2a). Vaccination alone induced a marked reduction of the average metastatic burden, from 254 metastases without vaccination to 58 with vaccination. TRP-2 expression in the lung was reduced from 6506 without vaccination to 1571 with vaccination. The treatment with either M38-αFAP or TCB-αFAP eliminated almost all metastases in all mice (number of metastasis 1 and 0, respectively, and TRP-2 expression level 2 and 1, respectively). The TCB-αFAP also eliminated all metastases in the non-vaccinated mice. The TRP-2 expression level quantified by RT-qPCR was in line with the metastasis count for all treatment groups (Figure 2b, supplement Figure S2a).
Figure 2.
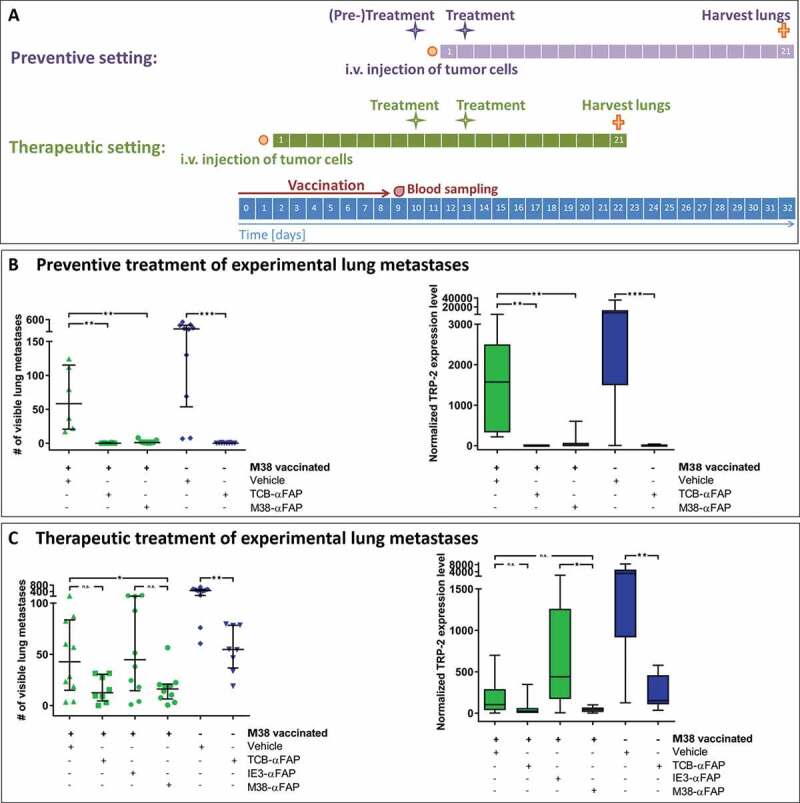
Experimental lung metastasis model
(a) Schedule for the preventive (purple) and therapeutic (green) treatment setting: The overall timeline is depicted in blue. Intravenous injections of the compounds are shown as purple or green stars in each setting. The duration of the vaccination is shown as a red arrow. Blood sampling is illustrated with a red drop. Orange points indicate intravenous injection of 2 × 105 B16-FAP melanoma cells. Orange crosses demonstrate euthanasia of mice and isolation of lungs for assessment of metastatic burden. (b) Metastatic burden after preventive treatment. Visible lung metastases were counted after harvest of lungs 21 days after injection of the tumor cells. M38 vaccinated (green) and non-vaccinated (blue) mice were either treated twice with vehicle (PBS, green triangles/blue hashes), TCB-αFAP (green squares/blue inverted triangles) or M38-αFAP (green dots) 24 hours before injection of tumor cells and 2 days after. All graphs show median per group (n = 6–10) with error bars indicating interquartile range of counted lung metastasis (left). All graphs show box plots with median of qPCR quantification of melanoma cell marker TRP-2 with whiskers indicating maximum and minimum value (right). (c) Metastatic burden after therapeutic treatment. Visible lung metastases were counted after harvest of lungs on day 21 of metastasis growth (left). TRP-2 expression was quantified by qPCR (right). M38 vaccinated (green) and non-vaccinated (blue) mice were treated twice with either vehicle (PBS, green triangles/blue hashes), TCB-αFAP (green squares/blue inverted triangles), IE3-αFAP (green hexagons) or M38-αFAP (green dots). Treatment started 9 days after injection of B16-FAP melanoma cells. All graphs show median per group (n = 8–10) with error bars indicating interquartile range of counted lung metastasis (left). All graphs show box plots with median of qPCR quantification of TRP-2 with whiskers indicating maximum and minimum value (right). For statistical analysis, Wilcoxon-test was used. P-values from 0.01 to 0.05 were considered as significant (*), p-values from 0.001 to <0.01 were considered as very significant (**) and p-values < 0.001 were considered as extremely significant (***).
Therapeutic treatment of established lung metastases with pMHCI-IgG and TCB molecules reduces tumor burden
M38 peptide-vaccinated mice were injected i.v. with 2 × 105 B16-FAP melanoma cells. After 9 days, when lung metastases were already established, mice were treated twice at an interval of 3 days, with M38-αFAP or TCB-αFAP, during the CD8 T cell peak with an average of 11.8% M38-H-2Kb-specific CD8 T cells (Figure 2a, therapeutic setting, range 5%-21%). Twenty-one days after injection of tumor cells, lungs were removed and metastasis burden was assessed (Figure 2c, supplement Figure S2b).
Both the therapeutic pMHCI-IgG and TCB treatment equally reduced already established lung metastases. The vaccinated control group averaged 43 metastases per lung and a median TRP-2 expression level of 103, while pMHCI-IgG-treated lungs averaged 16 metastases and a median TRP-2 expression level of 43, and TCB-treated lungs averaged 13 metastases and a median TRP-2 expression level of 26. M38-vaccinated mice treated with control IE3-αFAP had a similar number of metastases as M38-vaccinated mice not treated with pMHCI-IgG, indicating that killing mediated by pMHCI-IgGs is antigen-specific. Also in this experiment, immunization alone had a marked effect on metastasis growth. Without vaccination, the number of metastases averaged 463, and with vaccination, it was reduced to 43. Vaccination also contributed to the efficacy of the TCB. The effect of the vaccination was independent of the peptide used for vaccination. We observed the same impact on tumor burden when we vaccinated with ovalbumin-derived peptide (data not shown). Treatment with a pMHCI-IgG carrying a peptide not used in the vaccination (IE3-αFAP, Figure 2c) did not add any anti-tumor potency. The non-vaccinated TCB treatment group had a higher tumor burden than the vaccinated vehicle group and the vaccinated TCB treatment group showing least tumor burden (Figure 2c, supplement Figure S2b). The experimental lung metastases sustained the mFAP expression in vivo (Figure 3). The experiment demonstrated that with both pMHCI-IgG and TCB treatment, growth of experimental metastases could be delayed and reduced when B16 tumor cells have already settled in the lung.
Figure 3.
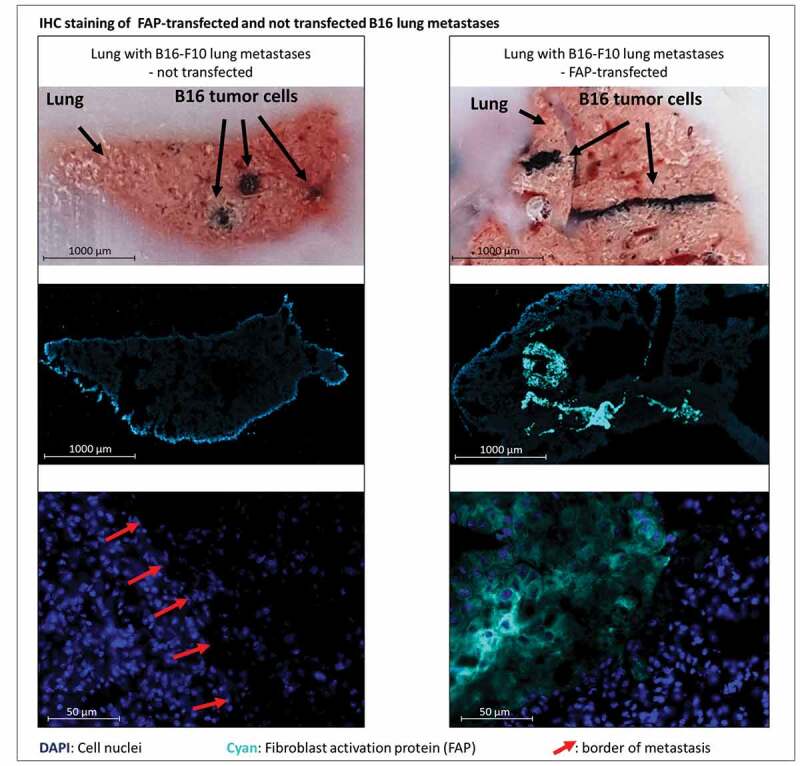
Bright field and immunofluorescent staining of lungs containing metastases derived from either murine FAP-transfected (right) or non-transfected (left) B16 melanoma cells at day 21 after injection of tumor cells. Pigmented B16 cells can be recognized in the bright field (top row). Murine FAP in cyan blue, cell nuclei in blue (DAPI). In the top row cut ends of cryo-section tissue blocks are depicted so that metastases can be localized in the corresponding stained cryo-sections below (middle row). The close-up images (bottom row) show normal lung tissue and parts of metastases. Red arrows indicate borders of metastases
pMHCI-IgG or TCB treatment does not mediate tumor growth inhibition in advanced, solid MC38 colorectal cancer cell model
MC38-FAP colorectal cancer cells were injected subcutaneously into the flank of mice to induce solid tumors. When tumors reached an average volume of 75 mm3, the M38-XCR1 vaccination was performed (Figure 4). Animals were assigned to treatment groups to have an average blood level of about 5% of M38-H-2Kb-specific CD8 T cells (average and standard deviation each group, n = 8: Vehicle group: 4.8% ± 2.0, TCB-αFAP: 5.0% ± 4.3, IE3-αFAP: 5.5% ± 2.8, M38-αFAP: 4.2 ± 2.8) and an average tumor volume of about 100 mm3 (average and standard deviation each group, n = 8: Vehicle group: 96.5 mm3 ± 29.0, TCB-αFAP: 96.7 mm3 ± 28.8, IE3-αFAP: 97.7 mm3 ± 29,0, M38-αFAP: 96.8 mm3 ± 28.7) at start of treatment. The existence of some tumor-free mice in all groups, including the phosphate-buffered saline (PBS)-treated control group, indicates that the XCR1-targeted vaccination alone had a marked influence on tumor take and induced tumor regression in some cases. However, the remaining subcutaneous tumors grew similarly in all groups and tumor volumes of vehicle and treatment groups showed no significant differences. Neither the pMHCI-IgG nor the TCB could induce tumor growth inhibition in this experimental setting (Figure 4b). In all treated groups, tumors either disappeared to undetectable sizes or showed similar growth compared to the control groups, leading to high variability and non-significant differences between groups.
Figure 4.
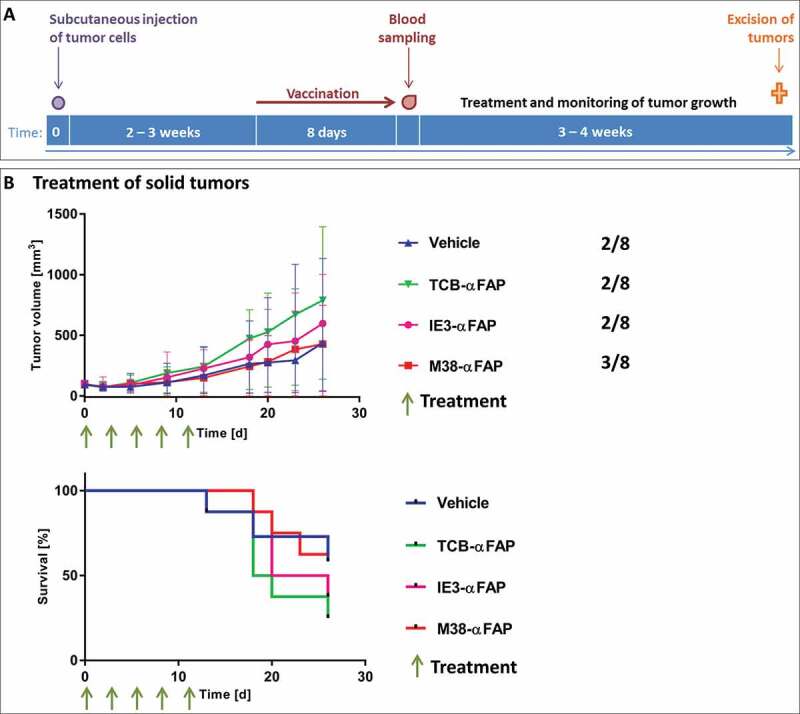
Solid tumor model
(a) Schedule for the subcutaneous MC38 tumor model. The purple dot indicates the subcutaneous inoculation of 106 MC38-FAP colorectal cancer cells. Duration of the vaccination is depicted with a red arrow. One day after finalization of vaccination, blood was taken and the amount of M38-specific T cells was determined (red drop). Green stars show time points of treatments. The orange cross demonstrates euthanasia of mice and harvest of tumors. (b) Top: Growth kinetics of solid subcutaneous MC38 tumors after treatment with TCB-αFAP (green), M38-αFAP (red), IE3-MHCI-IgG (pink) or vehicle treatment (blue). Treatments started at an average tumor volume of 97 mm3 and were administered i.v. q3dx5 as shown by green arrows. Tumor-free mice per group are indicated on the right. All graphs show median per group (n = 8) with error bars indicating interquartile range. Bottom: Time-to-event analysis of solid subcutaneous MC38 tumors after treatment with TCB-αFAP (green), M38-αFAP (red), IE3-αFAP (pink), or vehicle treatment (blue). Treatments were administered i.v. q3dx5 as shown by green arrows. Group size was n = 8. A tumor volume > 500 mm3 was defined as event.
pMHCI-IgG fusion molecules penetrate into the tumor tissue
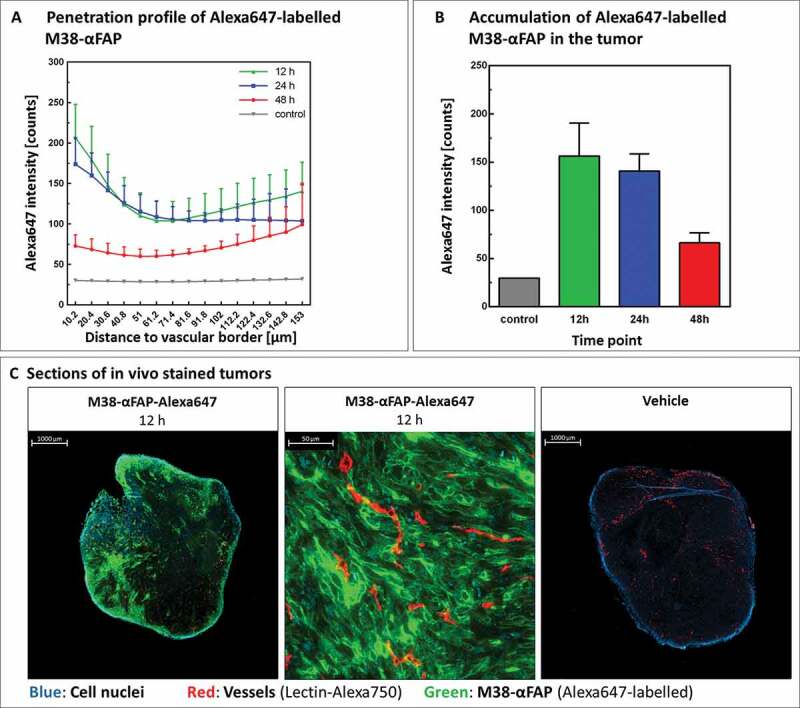
Maintenance of mFAP expression on B16 and MC38 tumor cells grown in vivo was confirmed by immunohistochemistry (Figures 3, 5). We also confirmed penetration of the pMHCI-IgG test molecules in the tumor by 3D-LSFM analysis (Figure 6). After 12 hours, the A647-labeled M38-αFAP fusion molecule penetrated into the solid MC38 tumor (Figure 6). The distribution pattern was spotty. The highest amounts of labeled fusion molecules were found in marginal regions and in areas where vessel density was high (Figure 6a). After 24 and 48 hours, decreasing quantities of the molecule were visible in the tissue due to clearance from tumor, as expected based on the known half-life of pMHCI-IgG in mice.7 At larger distances to vessels, the amount of M38-αFAPs increased again. These findings indicate some accumulation of molecules in non-vascularized, necrotic areas of the tumor with less clearance. We did not test the penetration of the TCB fusion proteins here, as they were based on the same targeting antibody and had a very similar molecular size. The results of the T cell infiltration into the tumor mass, however, indicated that the TCBs reached the inside of the tumor as well (see below and Figure 7). In summary, we observed penetration of the M38-αFAP into the tumor mass and enrichment on the cell surface of tumor cells.
Figure 5.
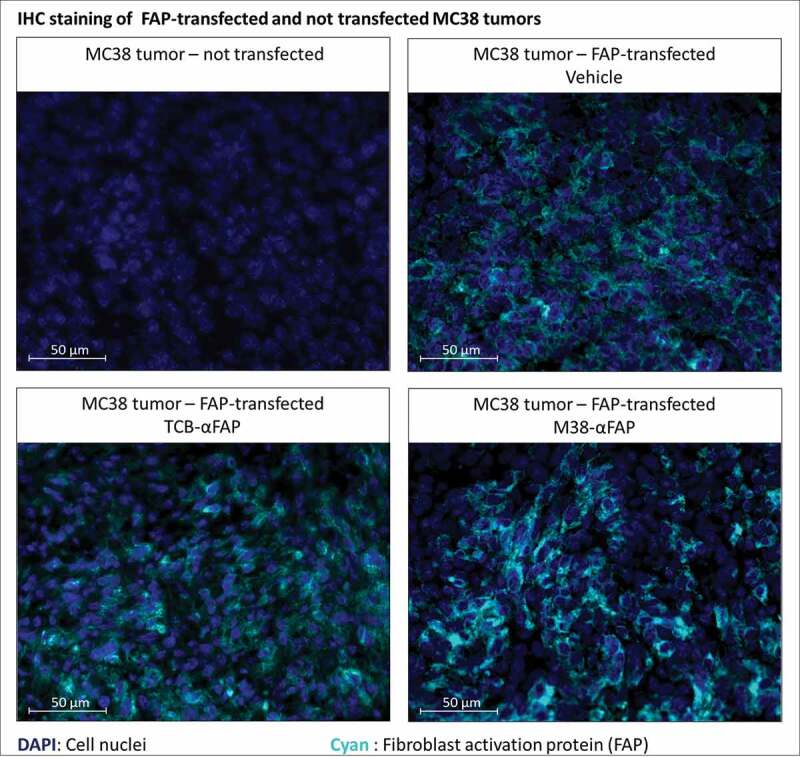
Immunofluorescent staining of murine FAP in solid tumors derived from either murine FAP-transfected (upper right, lower left, lower right) or non-transfected (upper left) MC38 colorectal cancer cells. Murine FAP-transfected tumors were treated with the TCB (lower left), the pMHCI-IgG (lower right) or untreated (upper right). Cell nuclei are stained with DAPI and depicted in blue. Murine FAP located on the cell surface is depicted in cyan blue
Figure 6.
Tumor penetration and accumulation of pMHCI-IgG molecules
(a) Penetration profile of Alexa647-labeled M38-αFAP molecule. Tumors were excised 12 hours (green), 24 hours (blue), and 48 hours (red) after intravenous injection of compound or vehicle (gray). Amount of M38-αFAP molecules in the tumor is depicted in relation to distance to vessel borders. All graphs show mean of different animals per group (n = 4) with error bars indicating standard deviation. (b) Accumulation of Alexa647-labeled M38-αFAP molecules in the tumor. Total amounts of M38-αFAP molecule present in the tumor 12 (green), 24 (blue), and 48 (red) hours after injection of molecule or vehicle (gray) are shown. All graphs show mean of different animals per group (n = 4) with error bars indicating standard deviation. (c) Sections of in vivo stained tumors excised 12 hours after injection of Alexa647-labeled M38-αFAP molecule (left) or vehicle (right). Close-up image of tumor tissue with high penetration of Alexa647-labeled M38-αFAP molecules shows binding of labeled molecule to tumor cells (middle). Color code: Cell nuclei (blue), vessels (red), M38-αFAP (green).
Figure 7.
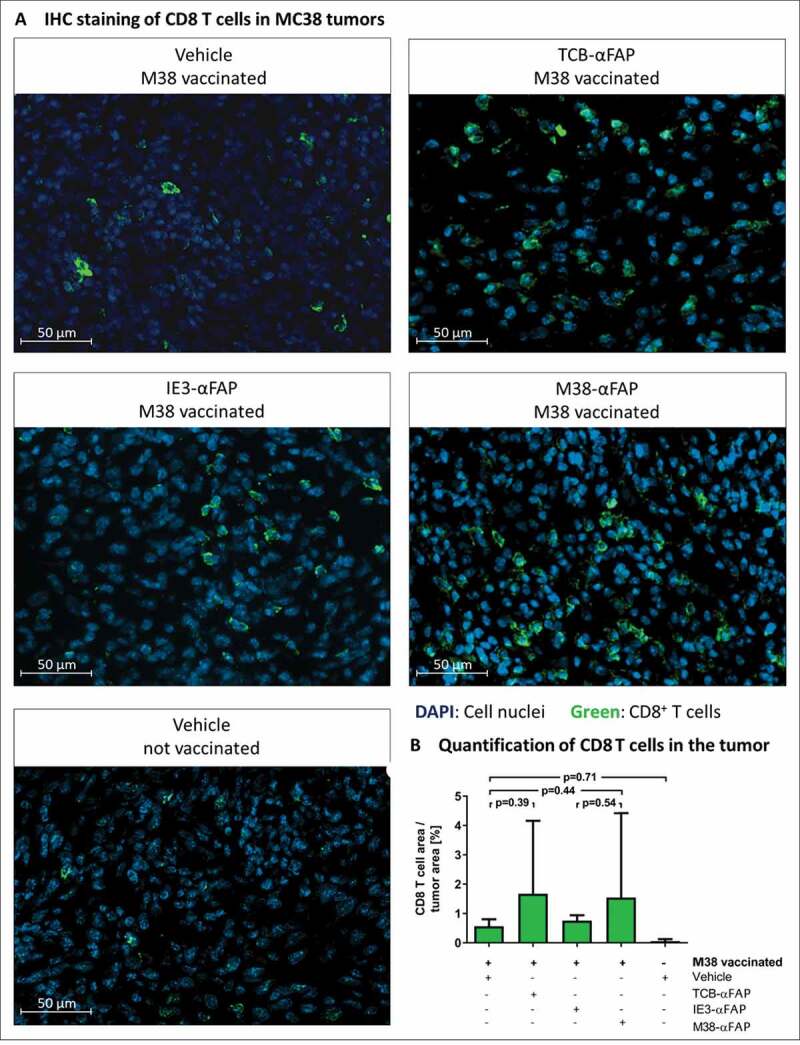
CD8 T cell infiltration in solid MC38-FAP tumors after a single treatment with compounds
(a) CD8 T cells were stained by fluorescently labeled anti-mouse CD8a antibody (green) on histological sections of the MC38-FAP tumors. Cell nuclei were stained with DAPI (blue). M38 vaccination with vehicle treatment (A: upper left), M38 vaccination with TCB-αFAP treatment (A: upper right), M38 vaccination with IE3-αFAP treatment (A: middle left), M38 vaccination with M38-αFAP treatment (A: middle right), and non-vaccinated and non-treated (A: lower left) are shown. (b) Quantification of CD8 T cells in solid MC38-FAP tumors after a single treatment with compounds. Area covered by CD8 T cells in relation to tumor area is depicted for all groups. All graphs show mean of different animals per group (n = 4) with error bars indicating standard deviation.
Vaccination-induced CD8 T cells infiltrate MC38 tumors after pMHCI-IgG and TCB treatment
Vaccinated, tumor-bearing mice were treated with M38-αFAP, control IE3-αFAP, TCB-αFAP molecules, or vehicle. Twenty-four hours after treatment, tumors were excised and analyzed by immunohistochemistry for CD8 T cells (Figure 7a). Tumor samples from non-vaccinated animals showed only poor CD8 T cell infiltration (Figure 7b). Vaccination alone, without additional treatment with pMHCI-IgG or TCB, increased the number of CD8 T cells in the tumor about ten-fold. Treatment with control IE3-αFAP did not increase the number of CD8 T cells, compared to vaccination without pMHCI-IgG treatment. Only treatment with M38-αFAP, or TCB-αFAP molecules, further increased the number of tumor-infiltrating CD8 T cells (Figure 7b).
CD8 T cells present in MC38 tumors show an effector cell phenotype, but are not able to provide effector functions
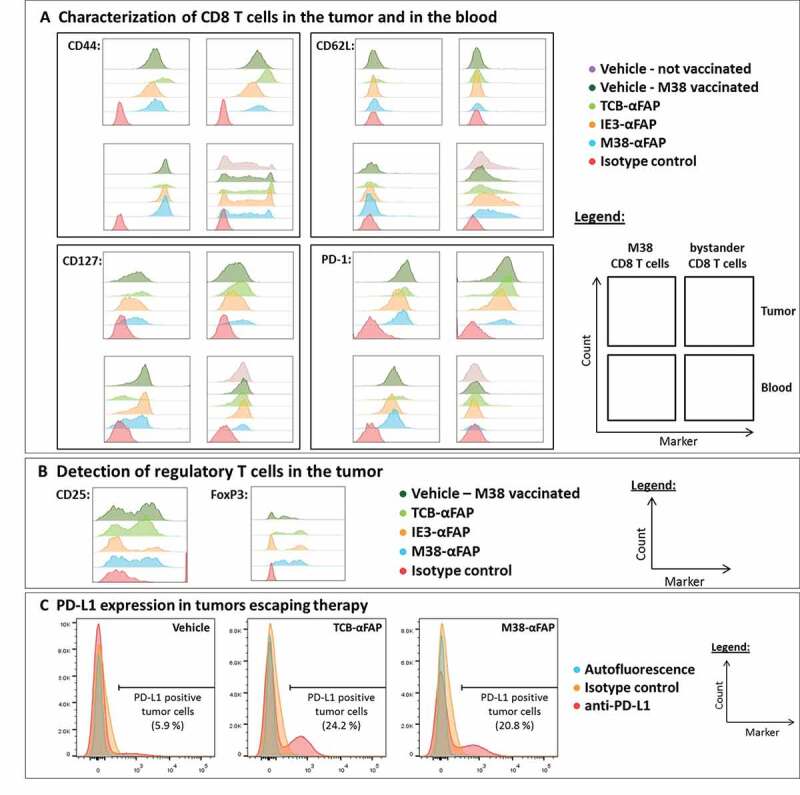
CD8 T cells from the blood and tumor were analyzed by flow cytometry after the second treatment (Figure 8). In non-vaccinated vehicle-treated mice, the phenotype of CD8 T cells could only be characterized in the blood, due to insufficient CD8 T cell numbers in the tumor. Vaccination alone increased the number of T cells in the tumor. All CD8 T cells in the tumor expressed the trafficking molecule CD44. Upon treatment with M38-αFAP or TCB-αFAP, but not with control IE3-αFAP, both M38-specific and bystander CD8 T cells expressed upregulated levels of CD44 in the tumor but not in the blood. For the non-M38-specific CD8 T cells, CD44 was not expressed. CD62L was absent in tumor and expressed only on nonspecific CD8 T cells in blood. CD127 was slightly higher on CD8 T cells in tumor in treated groups. In blood, CD127 was reduced by treatment. Expression of surface markers in M38-specific CD8 T cells generated by vaccination was characterized as effector memory T cells (CD44+, CD62L-, and CD127+) both in the tumor and in the blood (Figure 8a). The non-M38-specific CD8 T cells were effector memory T cells (CD44+ CD62L-CD127+) in the tumor and either memory or naïve T cells (CD44+ or CD44- and CD62L+CD127+) or T cells in the transition phase in the blood.
Figure 8.
Flow cytometry analysis of T cells and tumor cells from M38 vaccinated and pMHCI-IgG or TCB-treated mice
(a) CD44 (top left), CD62L (top right), CD127 (bottom left), and PD-1 (bottom right) expression of M38-specific (left column each marker) and nonspecific (right column each marker) CD8 T cells isolated from solid MC38-FAP tumors (top row each marker) and peripheral blood (bottom row each marker) 48 hours after the second treatment. Color code for treatment: Not vaccinated/vehicle (purple – markers only detectable for unspecific CD8 T cells in the blood, no T cells detectable inside the tumor mass), M38 vaccinated/vehicle (dark green), M38 vaccinated/TCB-αFAP (light green), M38 vaccinated/IE3-αFAP (orange), M38 vaccinated/M38-αFAP (blue), isotype control antibody (red). (b) Flow cytometry analysis for CD25 (left) and FoxP3 (right) of CD4 T cells in tumor samples 48 hours after the second treatment. Color code for treatment: M38 vaccinated/vehicle (dark green), M38 vaccinated/TCB-αFAP (light green), M38 vaccinated/IE3-αFAP (orange), M38 vaccinated/M38-αFAP (blue), isotype control antibody (red). (c) Flow cytometry analysis of tumors, which escaped pMHCI-IgG or TCB therapy. Tumors of the M38 vaccinated and vehicle-treated (left), M38 vaccinated and TCB-αFAP treated (middle), and M38 vaccinated and M38-αFAP treated (right) groups were analyzed for PD-L1 expression on the tumor cell surface. Color code: Autofluorescence of cells (blue), isotype control antibody (orange), anti-PD-L1 antibody (red).
Immune suppression mechanism are found upon treatment in the tumor
Regulatory T cells (Tregs; CD4+ CD25+ FoxP3+) were present in the tumors (Figure 8b). Treatment with the M38-αFAP or the TCB-αFAP, but not with the control IE3-αFAP, increased Tregs in the tumor. The M38-specific CD8 T cells expressed PD-1 both in the tumor and at a smaller level in the blood, too. Upon pMHCI-IgG or TCB treatment, PD-1 expression increased slightly. In the tumor, expression of PD-1 could be found on all CD8 T cells, regardless of whether they were M38-specific or nonspecific. In the blood, non-M38-specific CD8 T cells did not express PD-1. Tumors that escaped pMHCI-IgG or TCB therapy were excised 31 days after the last treatment and checked for PD-L1 expression. Treatment induced long-lasting expression of PD-L1 on tumor cells (Figure 8c). While tumors of the vehicle group showed only 5–6% PD-L1+ tumor cells, both pMHCI-IgG and TCB treatment resulted in PD-L1 expression on over 20% of tumor cells (Figure 8c). In summary, vaccination was required to achieve detectable numbers of CD8 T cells in the solid tumor. Antigen-specific CD8 T cells had an effector or effector-memory phenotype in the blood, while bystander CD8 T cells had an effector phenotype in the tumor and effector or naïve phenotype in the blood. T cell activation was increased for all CD8 T cells in the tumor, in particular after M38-αFAP and TCB-αFAP treatment. In the blood, only M38-specific CD8 T cells were activated as an effect of the peptide-specific vaccination. Treatment did not affect specific or unspecific CD8 T cells in the periphery. In solid tumors, we found higher PD-1 expression on specific CD8 T cells and nonspecific CD8 T cells, higher expression of PD-L1 on the tumor cells, and more Tregs in the tumor, especially upon M38-αFAP and TCB-αFAP treatment. The resistance to the therapy was not due to a loss of antibody binding to the targeted tumor cells, as the tumor cells maintained the expression of mFAP in vivo (Figure 5).
Discussion
Tumor-infiltrating lymphocytes include not only tumor-reactive T cells but also bystander T cells of irrelevant specificity. Recruiting these bystander T cells to target the tumor has the potential to vastly increase the total size and quality of the anti-tumor T cell response. Here we vaccinated mice with synthetic M38 peptide from MCMV to activate antigen-specific CD8 T cells, and compared pMHCI-IgG molecules to TCB fusion proteins for their ability to impair tumor growth. We found that pMHCI-IgG molecules displayed similar potency as TCB molecules in a metastatic setting. However, neither molecule protected mice against solid tumors. Resistance to therapy in larger tumors was not caused by a loss of the antibody tumor target, the lack of penetration of the therapeutic molecule into the tumor mass or the lack of effector T cells attracted to the tumor.
Some current immunotherapies, including anti-PD-1 and anti-CTLA-4 blockade, as well as adoptive T cell therapy, rely on CD8 T cell recognition of neoantigens. Many tumors, though are poorly immunogenic, have few neoantigens, and/or lose MHC expression. Therefore, we chose an approach that, similar to CAR T cells and TCBs, bypasses peptide-MHC recognition by CD8 T cells and targets tumor-associated antigens on the tumor cell surface. Although CAR T cells and TCBs have proven effective in some circumstances, these immunotherapies can also unleash severe adverse effects, such as cytokine release syndrome and neurotoxicity, as observed in recent clinical trials.10
Use of an antiviral immune defense to treat cancer was proposed in the past5,6 and has been receiving more attention recently.11,12 Tumor-infiltrating T cells are not limited in their specificity to tumor antigens, but also recognize a wide range of epitopes unrelated to cancer, such as those from Epstein–Barr, influenza, or cytomegalovirus (CMV).13 Use of the antiviral immune defense, in particular the immunologic memory of chronic viral infections such as CMV, to treat cancer may help to overcome some of the current challenges.
We recently developed full mAb-pMHCI fusion proteins in a novel format, pMHCI-IgGs, which can mimic a viral infection of tumor cells.7,14 The activation of CMV-specific CD8 effector T cells was efficacious in vitro with peripheral blood mononuclear cells from chronically infected donors at picomolar concentrations. Further, there was a reduced in vitro release of cytokines compared to classical anti-CD3-based TCBs.7 The human models published so far for pMHCI-IgG complexes are limited to immune-deficient mouse models in which both human tumor cells and human effector cells have been transplanted.6,15,16 Syngeneic mouse model systems have only been used in effector T cells have been derived from a T cell receptor transgenic mouse specific for ovalbumin.17
Here, we developed a fully immunocompetent mouse model and compared the potency of mouse-specific pMHCI-IgGs to TCBs, both directed against the same surface antigen on the tumor cells. We vaccinated the mice to trigger a peptide antigen-specific CD8 T cell response and engineered mouse surrogate therapeutic pMHCI-IgG and TCB fusion proteins consisting of mouse antibody sequences and MCMV-derived peptides and mouse MHC class I for the pMHCI-IgG treated tumors. The biochemical properties of the therapeutic fusion proteins, such as purity, aggregation, stability, and binding affinities were comparable to their human equivalents. As expected, target cell killing induced by pMHCI-IgGs was strictly peptide-specific, and pMHCI-IgGs carrying a non-relevant peptide did not trigger any target cell elimination in vivo, also not when they were combined with the vaccination procedure which triggered a significant immune stimulation. The in vitro potency, however, was reduced compared to the human counterparts for the pMHCI-IgGs. Only half of the antigen-specific CD8 T cells induced by the vaccination were capable of recognizing the recombinant pMHCI-IgG construct. This was not observed before for the human constructs carrying the HCMV-derived pp65 peptide.14 We also excluded that there were not enough peptide MHC class I complexes delivered to the tumor cells after M38-αFAP treatment, as we found more peptide MHC class I complexes on the MC38 tumor cell surface after targeting with pMHCI-IgG compared to endogenous MHC class I expression after peptide loading. One possibility is that structural differences of the recombinant pMHCI complex, compared to the natural pMHCI complex, may prevent some of the polyclonal T cell receptors from binding.
Humans chronically infected with CMV have pp65-peptide-specific CD8 effector T cells in the blood at frequencies ranging from 0.1% to 10%. These numbers are sufficient to eliminate CMV-infected cells throughout the body18,19 and can kill tumor cells in vitro.7 In cancer patients, the frequency of CMV-specific CD8 T cells can be even higher, up to 20%.8 We applied a vaccination protocol to generate a peptide-specific CD8 T cell response that was similar to the frequencies seen in humans. The range of M38 peptide-specific CD8 T cells tested here was 3.2% to 12%, of which roughly half were activated with the M38-pMHCI-IgGs, so 1.6% to 6% functional CD8 effector T cells.
Pre-treating mice with pMHCI-IgGs prevented i.v.-injected B16 melanoma cells from seeding the lungs, proving that the pMHCI-IgGs are functional in vivo when tumor cells are still in the blood. Delaying pMHCI-IgG treatment until 9 days after tumor injection reduced, but did not fully eliminate, tumor growth in the lungs. The pMHCI-IgG and the TCB had a similar potency, but neither the pMHCI-IgG nor the TCB were sufficient to completely eliminate these established B16 lung tumors. B16 tumor cells are known to be immune suppressive,20,21 in particular when they have established a suppressive tumor microenvironment in a solid tumor. Although in vitro the TCBs could activate at least twice as many T cells as the pMHCI-IgGs, they were not more potent here, indicating that, at least in these conditions, an increase in effector cells cannot overcome the immune suppression in established tumors. This became even more obvious when larger solid tumors of MC38 cells were treated. Neither the pMHCI-IgG nor the TCB could control solid MC38 tumors, despite the penetration of the therapeutic molecules into the tumor mass, persistent expression of the tumor target mFAP, and the attraction/retention of an increased number of CD8 T cells inside the tumor. Our results show that the therapeutic molecules rapidly lost the effector potency, which may not be overcome easily just by a larger number of T cells.
The analysis of the T cells resident in the tumor mass indicated that without vaccination there were very few T cells inside the tumor. Vaccination alone had a vaccine antigen-independent impact on tumor growth, either indicating a potential activation of innate immune responses or preexisting T cells against natural tumor antigens. Vaccination did not unspecifically activate T cells, as we did not observe any anti-tumor potency with pMHCI-IgGs carrying a peptide unrelated to the vaccination as tested in the B16 lung metastasis model (e.g., IE3-αFAP). However, vaccination alone was not sufficient to control tumor cells in any setting. Treatment of tumor-bearing mice with pMHCI-IgGs led to antigen-specific activation of CD8 T cells inside the tumor that triggered known immune suppression responses, such as the induction of PD-L1 expression in the tumor cells. In addition, an increased number of Tregs was observed, indicating that these and potentially additional immune suppression mechanism can easily shut off functional CD8 T cells that have been re-targeted to tumors by pMHCI-IgGs or TCBs.
The vaccination model was used here as a surrogate for the patient situation, where most but not all individuals are CMV-infected, and vaccination would enable treatment of CMV-negative patients. Perhaps not surprisingly, under the immune suppressive conditions in solid tumors, additional combination treatment regimens will be required to further increase the effectiveness of pMHCI-IgG treatment.
Materials and methods
Cell lines
B16-F10 cells (ATCC) and MC38 cells (City of Hope National Medical Center) were stably transfected to express mFAP under selection with puromycin. B16-F10 cells were grown in RPMI supplemented with 10% fetal bovine serum and MC38 cells in DMEM supplemented with 10% fetal bovine serum, L-glutamine, sodium pyruvate, and non-essential amino acids. Cell lines were verified as mouse cell lines (MALDI-TOF) and as pathogen-free directly before use.
Protein production and analytics
28H1 is a non-internalizing anti-FAP mAb with picomolar affinity for mFAP.22,23 The pMHCI-IgG and the TCB are based on the 28H1 antibody. Both molecular designs have been described previously.3,14,24 In brief, pMHCI–IgG fusion molecules consisted of peptide (MCMV-derived M38 (SSPPMFRV) or IE3 (RALEYKNL) or ovalbumin257-264 (SIINFEKL)), first linker (GCGGS(G4S)2), murine β-2-microglobulin, second linker ((G4S)4), α1–3 domains of H-2Kb mouse MHC class I complex carrying a Y84C mutation, third linker (GS) followed by the antibody heavy chain variable domain and an engineered effector-function-free mouse IgG2c constant domain (L234A, L235A and D270A, P329G).25,26 The two different heavy chains were engineered to facilitate heterodimerization via knob-into-hole mutations.27 The pMHCI-fused heavy chain carried the “hole” mutations (T366S, M368A, Y407V), while the unfused heavy chain carried the “knob” mutation (T366W).
The TCB consisted of the 28H1 antibody and the anti-mouse CD3ε antibody 145–2 C11 in a modified, effector-function-free mouse IgG1 format (D270A, P329G).28,29 Proper assembling of light chains was forced by introducing a CH1-Cκ crossover in the CD3ε binding Fab arm in the CrossMAb format.24 K392D and K409D charge variants triggered heterodimerization of the heavy chains.30 The second antibody heavy chain consisted of the variable domain of the 28H1 heavy chain followed by the antibody heavy chain constant domain mouse IgG1 with the D265A, P329G mutation and corresponding E356K and N399K charge variants to facilitate heterodimerization.30 Protein production and quality testing was performed as described previously.3,14 In brief, the protein quality (integrity and purity) was tested by SDS-PAGE (LabChip), size exclusion chromatography, Protein A HPLC, protein identity and proper formation of all disulfide bridges by mass spectroscopy. Analytical results are shown exemplarily for the pMHCI-IgG fusion protein M38-αFAP in the supplement (Figure S3). The pMHCI-IgG was monomeric and free of aggregates or fragments (below 5%). Binding to target cells was confirmed, as well as proper confirmation of the MHC complex by MHC-specific antibodies. All fusion proteins contained endotoxin levels below 2 EU per animal when injected into mice.
In vitro T cell activation assay (induction of IFN-γ expression) and cytotoxicity assays
Target cells were cultured for 2 days before splenocytes from vaccinated mice and test compounds were added. Effector cell (M38 antigen-specific CD8 T cells) to target cell (MC38-FAP tumor cells) ratio was 1:1 for the IFN-γ activation assay and 0.25:1 for the cytotoxicity assay. After 8 hours, cells were analyzed for intracellular IFN-γ (XMG1.2, BioLegend) by FACS. Specific target cell elimination was tested in the real-time xCELLigence (Roche) cell analyzer as described previously.7 Spontaneous release was measured from target cells with target and effector cells only. Specific lysis (%) was calculated as [(cell index spontaneous release – cell index specimen)/(cell index spontaneous release)] x 100.
Peptide vaccination
Vaccination was performed by targeting peptide to XCR1+ dendritic cells, as described previously.23 The MCMV M38 peptide (SSPPMFRV) was fused to the C-terminus of the anti-XCR1 antibody MARX10 by enzymatic sortase coupling.24,25 C57BL/6 N mice were immunized (i.v.) with 5 µg of anti-XCR1-M38 and 10 µg of poly(I:C) (InvivoGen). Five days later, antigen-specific CD8 T cells were amplified by administration of autologous peptide antigen-presenting cells and IL-2 cytokine receptor stimulation was started on day six (data not shown).
Flow cytometry analysis of specific CD8 T cells and tumor cells
Heparinized blood was lysed with RBC Lysis Buffer (BioLegend). Subcutaneous MC38 tumors were excised, cut into small pieces and digested in RPMI containing 1 mg/ml Dispase (StemCell Technologies), 0.8 mg/ml Collagenase D (Roche), and 0.01 mg/ml DNase (StemCell Technologies) for 20 minutes at 37°C. The digest was filtered with a 70 µm cell strainer. Fcγ receptors IIa and III on white blood cells or tumor cells were blocked with 1.0 µg of TruStain fcX antibody (BioLegend). Antigen-specific CD8 T cells were labeled with M38-Kb MHC Dextramers (Immudex) and afterward labeled with fluorescent CD8a (53–6.7, BioLegend), CD4 (GK1.5, BioLegend), CD3ε (145–2 C11, BioLegend) and CD45 (30-F11, BioLegend). Dead cells were excluded with DAPI. CD8 T cells and tumor cells (CD45-cells) were phenotyped for PD-1 (29 F.1A12, BioLegend), CD44 (IM7, BioLegend), CD62L (MEL-14, BioLegend), CD127 (A7R34, BioLegend), CD25 (PC61, BioLegend) and PD-L1 (10 F.9G2, BioLegend). For intracellular staining of FoxP3, Fixation Buffer (BioLegend), Intracellular Staining Perm Wash Buffer (BioLegend), and anti-FoxP3 (MF-14, BioLegend) were used. SIINFEKL peptide-MHC class I complexes were detected with the monoclonal anti-Ova257-264 – mouse H-2Kb antibody (25-D1.16, BioLegend).
Experimental lung metastasis in vivo model
C57BL/6 N mice (7–8 weeks) were obtained from Charles River Laboratories. To induce experimental lung metastasis,31 mice were injected i.v. with 2 × 105 B16-FAP melanoma cells. For generation of effector cells, mice were immunized with the XCR1-targeted vaccination. In the prophylactic setting, mice were pretreated with the therapeutic proteins 24 hours before tumor challenge and three days later after challenge. In the therapeutic setting, melanoma cells were allowed to grow in the lungs for 9 days after i.v. injection. Mice were treated twice with the compounds at an interval of 3 days at 5 mg/kg for the pMHCI-IgG and 2 mg/kg for the TCB. The doses were chosen based on the pharmacokinetic properties and the different in vitro potencies of both compounds. As shown in Figure 1c, the in vitro potency for the TCB is higher than for the pMHCI-IgG (about 90% of potency for the TCB at 100 pM, while the pMHCI-IgG 90% potency was higher than 5 nM). The pharmacokinetic properties either measured as tissue exposure (pMHCI-IgG, Figure 6b) or blood samples (TCB, Suppl Figure S4) indicated a similar half-life of more than 24 hrs. The doses ensured maximal potency in vivo in both the preventive and therapeutic settings. After 21 days, lungs were assessed for metastatic burden by visual quantification and qPCR.
In vivo solid tumor model
A total of 1 x 106 MC38-FAP cells were injected subcutaneously into C57BL/6 N mice. After 2 to 3 weeks, tumors averaged 75 mm3 and mice were vaccinated with M38-XCR1. Mice were allocated into treatment groups with equivalent tumor volumes and similar frequencies of M38-specific CD8 T cells in the blood. Mice were treated with 5 mg/kg of pMHCI-IgG (i.v. q3dx5) or 2 mg/kg of TCB (i.v. q3dx5). Tumors were analyzed by fluorescence ultramicroscopy, flow cytometry, and immunohistochemistry. Tumor volume was calculated as an ellipse (tumor volume = (width2 * length)/2).
Immunohistochemistry
Frozen tumors or lungs, embedded in Richard-Allan Scientific NEG 50 Section Medium (Thermo Scientific), were cut into 14 µm sections. Tissues were fixed in 4% paraformaldehyde containing 5 mM sucrose for 10 min at room temperature and blocked with Protein Block Serum-Free (Dako) for 10 min. mFAP expression was detected with A647-labeled 28H1 (Roche) for 1 h at room temperature. CD8 T cells were stained with polyclonal rabbit anti-mouse CD8a (Synaptic Systems, 1:200) for 60 min. Sections were washed with Tween 20 containing Tris-buffered saline and subsequently incubated with A488-labeled goat anti-rabbit IgG (Invitrogen, 1:400) for 30 min. Slides were mounted with a coverslip with ProLong Gold antifade reagent containing DAPI (Life Technologies). For quantification of CD8 T cells present in the tumor, the average signal for each slide was quantified as positive stained area per five randomly selected tumor areas of 2,000 x 1,000 µm by automated analysis with the Image Intensity Threshold Tool (Roche).
3D light sheet fluorescence microscopy (3D-LSFM)
Fluorescence ultramicroscopy was performed as described before.32 Briefly, tumor-bearing mice received 5 mg/kg A647-labeled M38-αFAP fusion protein. Five minutes before euthanasia, 4 mg/kg A750-labeled Bandeiraea simplicifolia lectin (Sigma-Aldrich, fluorescence labeling in-house) was administered i.v. Explanted tumors were fixed 24 h in 10% formalin. Fixed tissues were dehydrated by incubation in a graded ethanol dilution series. For 3D-LSFM, samples were incubated at 4°C in the dark in clearing solution, consisting of one part benzyl alcohol and two parts benzyl benzoate, until optically transparent. After ultramicroscopic analysis, tumors were incubated in a graded ethanol/xylene dilution series and embedded in paraffin. 2 µm tissue slices were cut from the middle of the tumors and mounted to glass slides. After deparaffination and rehydration of tissue, slides were sealed with ProLong Gold Antifade Reagent with DAPI (Life Technologies) and scanned with a Pannoramic 250 Flash III (3DHISTECH).
Reverse-transcriptase quantitave PCR (RT-qPCR)
RT-qPCR of TRP-2 mRNA from lungs was performed according to a modified protocol.33 Lungs were fixed in RNA later (Qiagen). RNA was isolated with the MagNA Pure LC RNA Isolation Kit III (Roche) according to manufacturer recommendations. First-strand cDNA was synthesized with the iScript Select cDNA Synthesis Kit (Bio-Rad). For qPCR, the LightCycler FastStart DNA Master SYBR Green I Kit (Roche) and the LightCycler Carousel-Based System (Roche) were used. PCR primers used were for TRP-2 (forward TTAGGTCCAGGACGCCCC; reverse CTGTGCCACGTGACAAAGGC) and GAPDH (forward CAATGTGTCCGTCGTGGA; reverse GATGCCTGCTTCACCACC). CP values were used to quantify relative gene expression. TRP-2 expression was normalized to GAPDH using the following formula: Normalized TRP-2 expression = 2-ΔΔCP, ΔΔCP = ΔCPsample – ΔCPcontrol, ΔCP = CPTRP-2 – CPGAPDH.
Statistical analysis
For statistical analysis, the JMP Software (SAS Institute) was used applying the two-sided t-test for significance evaluation in the in vitro analyses and the Wilcoxon-test for significance evaluation in the in vivo analyses. P-values from 0.01 to 0.05 were considered as significant (*), p-values from 0.001 to <0.01 were considered as very significant (**) and p-values < 0.001 were considered as extremely significant (***).
Supplementary Material
Acknowledgments
The authors thank Inge van Halst for critical reading of the article, Anne-Kathrin Winter, Marco Gruendl, Carsten Wolter, and Martin Lechmann for supporting the in vivo work, Joerg Zielonka and team for engineering MC38-mFAP and B16-mFAP cell lines, and Katrin Krause for the technical support.
Funding Statement
All studies were funded by Roche Diagnostics GmbH, Roche Glycart AG, or F. Hoffmann-La Roche Ltd.
Abbreviations
- B16
Murine melanoma cell line
- C57BL6
Inbred mouse strain
- CAR
Chimeric antigen receptor
- CD
Cluster of differentiation
- CMV
Cytomegalovirus
- CTLA-4
Cytotoxic T-lymphocyte-associated protein 4
- DC
Dendritic cell
- DP47
Germline DP47 antibody
- FAP
Fibroblast activation protein
- Fc
Fragment crystallizable region
- H-2Kb
Mouse histocompatibility class I complex
- HCMV
Human cytomegalovirus
- hFAP
Human fibroblast activation protein
- HLA
Human leukocyte antigen
- HPLC
High performance liquid chromatography
- IE3
Mouse cytomegalovirus peptide epitope of the immediate early regulatory protein
- IFN
Interferon
- IgG
Immunoglobin G
- i.v.
Intravenous
- kDA
Kilodalton
- M38
Mouse cytomegalovirus peptide epitope of M38.5 protein
- M38-αFAP
peptide MHC class I – anti FAP antibody fusion protein displaying the M38 peptide epitope
- mAbs
Monoclonal antibodies
- MALDI-TOF
Matrix-assisted laser desorption/ionization – time of flight mass spectroscopy
- MARX10
Anti-X-C motif chemokine receptor 1 antibody
- MC38
C57BL6 murine colon adenocarcinoma cells
- MC38-FAP
MC38 cells expressing fibroblast activation protein
- MCMV
Mouse cytomegalovirus
- mFAP
Mouse fibroblast activation protein
- MHC
Major histocompatibility complex
- Ova
Ovalbumin
- PBS
Phosphate buffered saline
- PCR
Polymerase chain reaction
- PD-1
Programmed cell death protein 1
- PD-L1
Programmed death-ligand 1
- pMHCI
Peptide MHC class I
- pMHCI-IgG
Peptide major histocompatibility complex class I - Immunoglobin G fusion protein
- qPCR
Quantitative polymerase chain reaction
- RPMI
Roswell Park Memorial Institute Medium
- RT-PCR
Reverse transcription - polymerase chain reaction
- SEC
Size exclusion chromatography
- TCB
T cell bispecific antibody
- TCB-αFAP
T cell bispecific antibody targeting fibroblast activation protein
- Tregs
Regulatory T cells
- TRP-2
Tyrosinase-related protein-2
- XCR1
X-C motif chemokine receptor 1
- 145-2C11
Anti-mouse CD3ε monoclonal antibody
- 28H1
Anti-human fibroblast activation protein monoclonal antibody
- 3D-LSFM
Three dimensional light sheet fluorescence microscopy
Authors’ contributions
Cornelia Fischer (molecule design, protein supply, in vitro and in vivo experiments, manuscript), Michael Munks, Ann Hill, Christian Klein (design of the experiments), Robert Kroczek (design of the vaccination procedure), Martina Schmittnaegel, Sabine Imhof-Jung, Eike Hoffmann (molecule design, protein supply), Stefan Bissinger (3D-LSFM), Verena Brand (IHC), Frank Herting (design of the in vivo study), Hendrik Knoetgen (molecule design, design of the experiments, manuscript)
Competing interests
Cornelia Fischer, Martina Schmittnaegel, Sabine Imhof-Jung, Eike Hoffmann and Frank Herting are employees of Roche Diagnostics GmbH Germany, Christian Klein is employee of Roche Glycart AG, hold stock and patents with Roche, and Hendrik Knoetgen is employee of F. Hoffmann-La Roche Ltd, Switzerland. Michael Munks and Ann Hill declare no conflicts.
Declarations
Ethics approval and consent to participate
All animal experiments were carried out according to the German Animal Welfare Act guidelines.
Consent for publication
All authors gave their consent for publication.
Availability of data and material
Primary data are available upon request. Recombinant protein sequences have been published and are available upon request.
Supplementary data
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Galluzzi L, Martin P.. CARs on a highway with roadblocks. Oncoimmunology. 2017;6(12):e1388486. doi: 10.1080/2162402X.2017.1388486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ribas A, Dummer R, Puzanov I, VanderWalde A, Andtbacka RHI, Michielin O, Olszanski AJ, Malvehy J, Cebon J, Fernandez E, et al. Oncolytic virotherapy promotes intratumoral T cell infiltration and improves anti-PD-1 immunotherapy. Cell. 2017;170(6):1109–15. doi: 10.1016/j.cell.2017.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bacac M, Klein C, Umana P. CEA TCB: A novel head-to-tail 2:1 T cell bispecific antibody for treatment of CEA-positive solid tumors. Oncoimmunology. 2016;5(8):e1203498. doi: 10.1080/2162402X.2016.1203498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marin-Acevedo JA, Soyano AE, Dholaria B, Knutson KL, Lou Y. Cancer immunotherapy beyond immune checkpoint inhibitors. J Hematol Oncol. 2018;11(1):8. doi: 10.1186/s13045-017-0552-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cesson V, Stirnemann K, Robert B, Luescher I, Filleron T, Corradin G, Mach J-P, Donda A. Active antiviral T-lymphocyte response can be redirected against tumor cells by antitumor antibody x MHC/viral peptide conjugates. Clin Cancer Res. 2006;12(24):7422–30. doi: 10.1158/1078-0432.ccr-06-1862. [DOI] [PubMed] [Google Scholar]
- 6.Lev A, Noy R, Oved K, Novak H, Segal D, Walden P, Zehn D, Reiter Y. Tumor-specific Ab-mediated targeting of MHC-peptide complexes induces regression of human tumor xenografts in vivo. Proc Natl Acad Sci U S A. 2004;101(24):9051–56. doi: 10.1073/pnas.0403222101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmittnaegel M, Levitsky V, Hoffmann E, Georges G, Mundigl O, Klein C, Knoetgen H. Committing cytomegalovirus-specific CD8 T cells to eliminate tumor cells by bifunctional major histocompatibility class I antibody fusion molecules. Cancer Immunol Res. 2015;3(7):764–76. doi: 10.1158/2326-6066.CIR-15-0037. [DOI] [PubMed] [Google Scholar]
- 8.Khan N, Shariff N, Cobbold M, Bruton R, Ainsworth JA, Sinclair AJ, Nayak L, Moss PAH. Cytomegalovirus seropositivity drives the CD8 T cell repertoire toward greater clonality in healthy elderly individuals. J Immunol. 2002;169(4):1984–92. doi: 10.4049/jimmunol.169.4.1984. [DOI] [PubMed] [Google Scholar]
- 9.Solberg OD, Mack SJ, Lancaster AK, Single RM, Tsai Y, Sanchez-Mazas A, Thomson G. Balancing selection and heterogeneity across the classical human leukocyte antigen loci: a meta-analytic review of 497 population studies. Hum Immunol. 2008;69(7):443–64. doi: 10.1016/j.humimm.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schmidt C. The struggle to do no harm in clinical trials. Nature. 2017;552(7685):S74–S5. doi: 10.1038/d41586-017-08705-4. [DOI] [PubMed] [Google Scholar]
- 11.De Vries J, Figdor C. Immunotherapy: cancer vaccine triggers antiviral-type defences. Nature. 2016;534(7607):329–31. doi: 10.1038/nature18443. [DOI] [PubMed] [Google Scholar]
- 12.Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC, Meng M, Fritz D, Vascotto F, Hefesha H, et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature. 2016;534(7607):396–401. doi: 10.1038/nature18300. [DOI] [PubMed] [Google Scholar]
- 13.Simoni Y, Becht E, Fehlings M, Loh CY, Koo SL, Teng KWW, Yeong JPS, Nahar R, Zhang T, Kared H, et al. Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature. 2018;557:575–79. doi: 10.1038/s41586-018-0130-2. [DOI] [PubMed] [Google Scholar]
- 14.Schmittnaegel M, Hoffmann E, Imhof-Jung S, Fischer C, Drabner G, Georges G, Klein C, Knoetgen H. A new class of bifunctional major histocompatibility class I antibody fusion molecules to redirect CD8 T cells. Mol Cancer Ther. 2016;15(9):2130–42. doi: 10.1158/1535-7163.MCT-16-0207. [DOI] [PubMed] [Google Scholar]
- 15.Lev A, Novak H, Segal D, Reiter Y. Recruitment of CTL activity by tumor-specific antibody-mediated targeting of single-chain class I MHC-peptide complexes. J Immunol. 2002;169(6):2988–96. doi: 10.4049/jimmunol.169.6.2988. [DOI] [PubMed] [Google Scholar]
- 16.Novak H, Noy R, Oved K, Segal D, Wels WS, Reiter Y. Selective antibody-mediated targeting of class I MHC to EGFR-expressing tumor cells induces potent antitumor CTL activity in vitro and in vivo. Int J Cancer. 2007;120(2):329–36. doi: 10.1002/ijc.22168. [DOI] [PubMed] [Google Scholar]
- 17.King BC, Hamblin AD, Savage PM, Douglas LR, Hansen TH, French RR, Johnson PWM, Glennie MJ. Antibody-peptide-MHC fusion conjugates target non-cognate T cells to kill tumour cells. Cancer Immunol Immunother. 2013;62(6):1093–105. doi: 10.1007/s00262-013-1408-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wills MR, Carmichael AJ, Mynard K, Jin X, Weekes MP, Plachter B, Sissons JG. The human cytotoxic T-lymphocyte (CTL) response to cytomegalovirus is dominated by structural protein pp65: frequency, specificity, and T-cell receptor usage of pp65-specific CTL. J Virol. 1996;70(11):7569–79. doi: 10.1128/JVI.70.11.7569-7579.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gratama JW, Brooimans RA, van der Holt B, Sintnicolaas K, van Doornum G, Niesters HG, Löwenberg B, Cornelissen JJ. Monitoring cytomegalovirus IE-1 and pp65-specific CD4+ and CD8+ T-cell responses after allogeneic stem cell transplantation may identify patients at risk for recurrent CMV reactivations. Cytometry B Clin Cytom. 2008;74(4):211–20. doi: 10.1002/cyto.b.20420. [DOI] [PubMed] [Google Scholar]
- 20.Burnstein P, Brody NI. Suppression of the delayed type hypersensitivity response by tumor facilitating factor of B16 melanoma. A tumor factor suppresses immune responses. J Dermatol Surg Oncol. 1993;19(6):543–52. doi: 10.1111/j.1524-4725.1993.tb00388.x. [DOI] [PubMed] [Google Scholar]
- 21.Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A. 2010;107(9):4275–80. doi: 10.1073/pnas.0915174107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laverman P, van der Geest T, Terry SY, Gerrits D, Walgreen B, Helsen MM, Nayak TK, Freimoser-Grundschober A, Waldhauer I, Hosse RJ, et al. Immuno-PET and immuno-SPECT of rheumatoid arthritis with radiolabeled anti-fibroblast activation protein antibody correlates with severity of arthritis. J Nucl Med. 2015;56(5):778–83. doi: 10.2967/jnumed.114.152959. [DOI] [PubMed] [Google Scholar]
- 23.Brunker P, Wartha K, Friess T, Grau-Richards S, Waldhauer I, Koller CF, Weiser B, Majety M, Runza V, Niu H, et al. RG7386, a novel tetravalent FAP-DR5 antibody, effectively triggers FAP-dependent, avidity-driven DR5 hyperclustering and tumor cell apoptosis. Mol Cancer Ther. 2016;15(5):946–57. doi: 10.1158/1535-7163.MCT-15-0647. [DOI] [PubMed] [Google Scholar]
- 24.Schaefer W, Regula JT, Bahner M, Schanzer J, Croasdale R, Durr H, Gassner C, Georges G, Kettenberger H, Imhof-Jung S, et al. Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc Natl Acad Sci U S A. 2011;108(27):11187–92. doi: 10.1073/pnas.1019002108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arduin E, Arora S, Bamert PR, Kuiper T, Popp S, Geisse S, Grau R, Calzascia T, Zenke G, Kovarik J, et al. Highly reduced binding to high and low affinity mouse Fc gamma receptors by L234A/L235A and N297A Fc mutations engineered into mouse IgG2a. Mol Immunol. 2015;63(2):456–63. doi: 10.1016/j.molimm.2014.09.017. [DOI] [PubMed] [Google Scholar]
- 26.Baehner MJS, Kubbies M, Moessner E, Schlothauer T. Genentech, assignee. ANTIBODY Fc VARIANTS 2012.
- 27.Ridgway JB, Presta LG, Carter P. ‘Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng. 1996;9(7):617–21. doi: 10.1093/protein/9.7.617. [DOI] [PubMed] [Google Scholar]
- 28.Leo O, Foo M, Sachs DH, Samelson LE, Bluestone JA. Identification of a monoclonal antibody specific for a murine T3 polypeptide. Proc Natl Acad Sci U S A. 1987;84(5):1374–78. doi: 10.1073/pnas.84.5.1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alegre ML, Vandenabeele P, Depierreux M, Florquin S, Deschodt-Lanckman M, Flamand V, Moser M, Leo O, Urbain J, Fiers W. Cytokine release syndrome induced by the 145-2C11 anti-CD3 monoclonal antibody in mice: prevention by high doses of methylprednisolone. J Immunol. 1991;146(4):1184–91. [PubMed] [Google Scholar]
- 30.Gunasekaran K, Pentony M, Shen M, Garrett L, Forte C, Woodward A, Ng SB, Born T, Retter M, Manchulenko K, et al. Enhancing antibody Fc heterodimer formation through electrostatic steering effects: applications to bispecific molecules and monovalent IgG. J Biol Chem. 2010;285(25):19637–46. doi: 10.1074/jbc.M110.117382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fidler IJ, Nicolson GL. Organ selectivity for implantation survival and growth of B16 melanoma variant tumor lines. J Natl Cancer Inst. 1976;57(5):1199–202. doi: 10.1093/jnci/57.5.1199. [DOI] [PubMed] [Google Scholar]
- 32.Dobosz M, Ntziachristos V, Scheuer W, Strobel S. Multispectral fluorescence ultramicroscopy: three-dimensional visualization and automatic quantification of tumor morphology, drug penetration, and antiangiogenic treatment response. Neoplasia. 2014;16(1):1–13. doi: 10.1593/neo.131848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sorensen MR, Pedersen SR, Lindkvist A, Christensen JP, Thomsen AR. Quantification of B16 melanoma cells in lungs using triplex Q-PCR–a new approach to evaluate melanoma cell metastasis and tumor control. PLoS One. 2014;9(1):e87831. doi: 10.1371/journal.pone.0087831. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.