

Abstract
Long noncoding RNAs (lncRNAs) play essential roles in normal physiology and in disease but their mechanisms of action can be challenging to identify. For mechanistic studies, it is often useful to know a lncRNA’s intracellular abundance, i.e., approximately how many molecules of the lncRNA are present in a typical cell of a cell-type of interest. At least two approaches have been used to approximate lncRNA intracellular abundance: single-molecule sensitivity RNA fluorescence in situ hybridization (smFISH) and single-gene, calibrated reverse-transcription followed by quantitative PCR (RT-qPCR). However, like all experimental approaches, these methods have their limitations. smFISH, when analyzed using diffraction-limited microscopy, can underestimate intracellular abundance, especially for lncRNAs that accumulate in focused subcellular regions. Calibrated RT-qPCR may return inaccurate estimates of abundance because individual PCR amplicons spaced across the length of a transcript can vary in their efficiency of reverse transcription. Here, we describe a sequencing-based approach that is straightforward, orthogonal to smFISH and RT-qPCR, and can be used to approximate the intracellular abundance for most expressed long RNAs (lncRNAs and mRNAs) in a cell type of interest. Firstly, the average weight of total RNA per cell for the cell type of interest is estimated by replicate rounds of RNA purification from a known number of cells. Secondly, an rRNA-depletion RNA-Seq protocol is performed after adding spike-in control RNAs to a known quantity of total cellular RNA. Lastly, by comparing read counts per transcript to a standard curve derived from the spiked-in RNAs, the intracellular abundance for each transcript is estimated. The sequencing-based approach provides a powerful complement to existing methods, particularly in situations where it is desirable to quantify the abundance of multiple lncRNAs and/or mRNAs simultaneously.
Keywords: RNA-Seq, Ribosomal RNA depletion, lncRNA, Xist, ERCC Spike-In RNAs, Transcriptome, RNA FISH, smFISH
Background
Long noncoding RNAs (lncRNAs) play essential roles in biology but their mechanisms of action can be difficult to determine (Kopp and Mendell, 2018; Gil and Ulitsky, 2020). Relative to protein-coding genes, lncRNAs can evolve rapidly and are not constrained by codon usage ( Cabili et al., 2011 ; Kutter et al., 2012 ; Necsulea et al., 2014 ; Schuler et al., 2014 ; Washietl et al., 2014 ; Hezroni et al., 2015 ; Chen et al., 2016 ; Ulitsky, 2016). Accordingly, they often lack easily identifiable domains that might otherwise provide insight into their molecular actions. Thus, for a given lncRNA, initial footholds into its molecular mechanism are often gained by examining its sub-cellular localization and its intracellular abundance (i.e., on average, how many molecules of the lncRNA are present in a single cell of a cell type of interest).
To these ends, single-molecule sensitivity RNA fluorescence in situ hybridization (smFISH) has been a boon, providing a convenient and cost-effective way to simultaneously investigate lncRNA sub-cellular localization and intracellular abundance ( Cabili et al., 2015 ; Tsanov et al., 2016 ; Raj and Rinn, 2019). Nevertheless, while smFISH offers unparalleled benefits in regards to visualizing lncRNA sub-cellular distribution, it does have limitations in regards to estimating lncRNA intracellular abundance. In the simplest form of smFISH, the number of FISH puncta per cell is used as a proxy for a lncRNA’s intracellular abundance. However, especially when smFISH is performed using diffraction-limited microscopy, separate lncRNA molecules that are located in spatial proximity may not be individually resolved; instead, such lncRNAs may appear as single puncta. Thus, particularly for those lncRNAs that accumulate to high concentrations in specific subcellular regions (Chujo and Hirose, 2017; Ninomiya and Hirose, 2020), smFISH may underestimate intracellular abundance. This potential limitation can be overcome by performing smFISH using super-resolution microscopy and carefully quantifying signal intensity within individual puncta ( Cerase et al., 2014 ; Smeets et al., 2014 ; Sunwoo et al., 2015 ). However, this latter approach requires high-end equipment and expertise that may not be readily accessible.
Here, we describe a sequencing-based approach that is orthogonal to smFISH and can provide estimates for the intracellular abundance of lncRNAs as well as mRNAs in cultured cells (Figure 1; Schertzer et al., 2019 ). The approach relies on RNA-Seq and requires minimal expertise beyond the ability to follow standard protocols in molecular biology and bioinformatics. The approach allows for the simultaneous quantitation of the intracellular abundance of all long RNAs (lncRNAs and mRNAs) that are expressed in a cell type of interest.
Figure 1. Overview of sequencing-based approach to quantify lncRNA and mRNA intracellular abundance.

The sequencing-based approach is also likely to yield estimates of intracellular abundance that are more robust than those produced by single-gene approaches that rely on calibrated reverse transcription followed by gene-specific quantitative PCR (RT-qPCR [ Schwaber et al., 2019 ]). A primary reason for this is because different qPCR amplicons spaced across the length of a transcript may vary dramatically in their efficiency of reverse transcription. In contrast, in RNA-seq, local, intra-transcript variations in reverse transcription efficiency are inherently averaged, owing to the chemical fragmentation of RNA that occurs just prior to reverse transcription of the RNA into cDNA ( Hrdlickova et al., 2017 ). Moreover, in the sequencing-based approach, the use of ERCC Spike-In RNAs, which harbor diverse GC-contents, lengths, and abundances, obviates the need to explicitly estimate reverse transcription efficiency and instead allows transcript abundance to be estimated by comparison to a standard curve ( Jiang et al., 2011 ).
Potential sources of errors in the sequencing-based approach include (1) errors associated with read alignment, which are most relevant if the lncRNA of interest contains sequence that is highly repetitive relative to other positions in the genome, (2) errors associated with transcript isoform uncertainty, which may arise if the predominant isoform of the lncRNA of interest is misannotated in the cell type of interest, and (3) errors associated with inaccurate or variable estimates of the total weight of RNA in the cell type of interest. Nevertheless, recently, the sequencing-based approach and smFISH performed using super-resolution microscopy have been shown to arrive at similar estimates of abundance for the lncRNA Xist ( Smeets et al., 2014 ; Sunwoo et al., 2015 ; Schertzer et al., 2019 ), lending confidence that both approaches provide reasonable estimates of RNA abundance. The sequencing-based approach is additionally useful because in a single experiment, it can be used to estimate the abundance of all expressed mRNAs and lncRNAs simultaneously.
Materials and Reagents
-
For Cell Counting
Disposable Borosilicate Glass Pasteur Pipets (Fisher Scientific, catalog number: 13-678-20D), sterilize before use
5 ml/10 ml sterile serological pipets (e.g., Genesee Scientific, catalog numbers: 12-102, 12-104)
15 ml/50 ml conical bottom centrifuge tubes (e.g., Corning, catalog numbers: 05-538-59A, 05-526B)
Glass coverslip (e.g., Fischer Scientific, Hausser Hemacytometer Cover Glass, catalog number: 02-671-53)
Kimwipes (Fisher Scientific, catalog number: 06-666)
6 cm tissue culture dishes (e.g., Genesee Scientific, catalog number: 25-260)
Mammalian cell type of interest (e.g., mouse trophoblast stem cells [ Calabrese et al., 2012 ])
Cell Culture Media supplemented with 10% serum (e.g., DMEM supplemented with 10% FBS; DMEM, Thermo Fisher Scientific, catalog number: 11995065; FBS, VWR, catalog number: 97068-085)
Sterile 1x PBS (e.g., Corning, catalog number: 21-040-CM)
0.25% Trypsin-EDTA (e.g., GIBCO, catalog number: 25200-072)
Trypan Blue Solution, 0.4% (Thermo Fisher Scientific, catalog number: 15-250-06)
70% ethanol, stored at room temperature
-
For RNA Purification
RNase Zap (Thermo Fisher Scientific, catalog number: AM9780)
P20/200/1000 Barrier pipette tips (e.g., Olympus brand tips, Genesee Scientific, catalog numbers: 23-404, 24-412, 24-430)
1.7 ml microcentrifuge tubes (e.g., Genesee Scientific, catalog number: 22-282)
TRIzol Reagent (Thermo Fisher Scientific, catalog number: 15596018)
Chloroform (Fisher Scientific, catalog number: BP1145-1)
Isopropanol (Fisher Scientific, catalog number: BP2618-1)
Linear Acrylamide (Thermo Fisher Scientific, catalog number: AM9520)
RNase-free water (e.g., we use deionized 18.2 MΩ water produced from a Synergy Water Purification System, Millipore, catalog number: SYNS0HFWW)
80% ethanol made with RNase-free water, stored at -20 °C
LE Agarose (e.g., Genesee Scientific, catalog number: 20-102QD)
Ethidium Bromide 1% Solution (Fisher Scientific, catalog number: BP1302-10)
50x TAE Buffer (e.g., Thermo Fisher Scientific, catalog number: B49)
Agarose gel loading buffer and dye (e.g., NEB, catalog number: B7024S)
1 Kb Plus DNA Ladder (Thermo Fisher Scientific, catalog number: 10787026)
-
For RNA-Seq
KAPA RNA HyperPrep Kits with RiboErase (Roche/KAPA Biosystems, catalog number: KK8560)
SeqCap Adapter Kit (see Note 1; Roche, catalog number: 714153000)
ERCC RNA Spike-In Mix 1 (Thermo Fisher Scientific, catalog number: 4456740)
Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, catalogue number: Q32854)
ERCC annotation files (ERCC92.fa, ERCC92.gtf, and ERCC_Controls_Analysis.txt found in the ERCC RNA Spike-In product page, Thermo Fisher Scientific, catalog number: 4456740)
Transcriptome annotation gtf file (e.g., downloaded from Illumina’s iGenomes site: https://support.illumina.com/sequencing/sequencing_software/igenome.html)
Appropriate genome sequence (e.g., downloaded from Illumina’s iGenomes site: https://support.illumina.com/sequencing/sequencing_software/igenome.html)
Equipment
-
For Cell Counting
Tissue Culture Incubator (e.g., Forma Series II 3110 Water-Jacketed CO2 Incubator, Thermo Fisher Scientific, catalog number: 3110)
Pipet Aid (e.g., Drummond, catalog number: DP-101)
P2/P20/200/1000 micropipettes (e.g., Research Plus 4-pack, Eppendorf, catalog number: EPPR4330)
Centrifuge for 15 ml/50 ml conical tubes (e.g., Eppendorf, model: 5810, catalog number: 022628157)
Phase Hemocytometer (e.g., Fischer Scientific, Hausser Bright-Line, catalog number: 02-671-6)
Hand Tally Counter (e.g., VWR, catalog number: 23609-102)
Inverted microscope with 10x objective (e.g., Zeiss Primovert, catalog number: 491206-0004-000)
-
For RNA Purification
Safety goggles (e.g. Thermo Fisher Scientific, catalog number: 19-053-950)
Microcentrifuge, stored at 4 °C (e.g., 5424 Microcentrifuge, Eppendorf, catalog number: 5424)
Mini Centrifuge with 1.7 ml tube rotor and PCR strip tube rotors (e.g., Genesee Scientific MyFuge Mini, catalog number: 31-500)
Nanodrop spectrophotometer (e.g., Thermo Fisher Scientific, catalog number: ND-LITE)
Mini Gel Electrophoresis System (e.g., Thermo Fisher Scientific, catalog number: B1A)
Agarose Gel Imaging System (e.g., Bio-Rad, Chemidoc MP, catalog number: 12003154)
-
For RNA-Seq
Thermal Cycler (e.g., Bio-Rad, model: C1000 Touch, catalog number: 1851148)
Magnetic bead stand (e.g., DynaMag-2 Magnet, Thermo Fisher Scientific, catalog number: 12321D)
Qubit Fluorometer (Thermo Fisher Scientific, catalog number: Q33238)
Mini Gel Electrophoresis System (e.g., Thermo Fisher Scientific, catalog number: B1A)
Agarose Gel Imaging System (e.g., Bio-Rad, Chemidoc MP, catalog number: 12003154)
Access to an Illumina sequencing instrument (e.g., Illumina, model: NextSeq500, catalog number: SY-415-1001)
Software
(Optional) SRA toolkit (Leinonen et al., 2011); https://ncbi.github.io/sra-tools/
STAR aligner ( Dobin et al., 2013 ); https://github.com/alexdobin/STAR
featureCounts from Subread package ( Liao et al., 2014 ); http://subread.sourceforge.net
Samtools ( Li et al., 2009 ); https://samtools.github.io
Microsoft Excel or equivalent, or Rstudio (RStudio_Team, 2015); https://rstudio.com
Procedure
-
Calculate the average amount of RNA per cell (see Note 2)
Prior to initiating this portion of the protocol, ensure that you have bench space, pipettes, and pipette tips that are clean and suitable for working with RNA (see Note 3).
Culture cells in a 6 cm dish until they are 60% to 80% confluent.
Preheat cell culture media and 0.25% trypsin-EDTA to 37 °C; once warmed, clean the bottles containing media, trypsin-EDTA, and 1x PBS with 70% ethanol and place them in a biological-safety cabinet.
Create a 0.125% trypsin solution by diluting the 0.25% trypsin-EDTA solution with an equal volume of 1x PBS.
Remove cultured cells from the incubator and place them in a biological-safety cabinet.
Aspirate the media from the cells using a disposable glass Pasteur pipet (or equivalent).
Wash cells on the plate by gently adding 4 ml of 1x PBS and then aspirating it.
-
Add 2 ml of 0.125% Trypsin solution to the cells and let the cell plate stand in the biological-safety cabinet at room temperature for 3 min.
Note: Dissociation protocols may differ for your cell type.
Using a clean 5 ml serological pipet attached to a pipet aid, pipet the trypsinized cell solution up and down against the plate to obtain a single-cell suspension.
Transfer the trypsinized cell solution to a 15 ml conical tube that contains 8 ml of culture media with 10% serum.
Invert the 15 ml conical tube 10-15 times to obtain a homogenous solution.
Using a P20 micropipette, remove 12 μl of cell suspension and carefully pipette it into the bottom of a 1.7 ml microcentrifuge tube.
Repeat Steps A11 and A12 one more time to obtain two replicates for cell counting.
Add 12 μl of Trypan Blue solution to each of the 12 μl cell suspensions and mix by pipetting, taking care to keep the Trypan Blue/cell solution at the bottom of the tube.
Separately, spin down the remainder of the trypsinized cell solution (~10 ml; in the 15 ml conical) in a centrifuge for 5 min at 1,000 rpm (~200 × g).
During the spin, clean the hemocytometer and glass coverslip with a Kimwipe sprayed with 70% ethanol, to remove any particulates.
Add 12 μl of the Trypan Blue cell suspension to the hemocytometer, ensuring that the solution distributes evenly underneath the coverslip.
Under a 20x objective, count the non-blue cells within each of the four hemocytometer quandrants, keeping track of the counts in each quadrant with a hand tally counter.
-
Calculate the number of cells per ml using the equation below, then average the cell-count-per-ml between replicates:
Replicate 1, # of cells per ml = [(sum of counts in all 4 quadrants)/4]*2*104
Replicate 2, # of cells per ml = [(sum of counts in all 4 quadrants)/4]*2*104
Average # of cells per ml = (Replicate 1 counts + Replicate 2 counts)/2
After the spin from Step A15 has completed, remove the trypsin-containing media taking care not to disturb the cell pellet, add 10 ml of 1x PBS, mix by pipeting, and spin down the PBS/cell solution in a centrifuge for 5 min at 1,250 rpm (~300 × g).
Remove the PBS, replace it with another 10 ml of 1x PBS, mix by pipetting, and spin down the cell solution in a centrifuge for 5 min at 1,250 rpm (~300 × g).
Remove all traces of PBS and add 1ml of TRIzol to the cell pellet (see Note 4).
Using a P1000, pipette up and down ~15x to lyse the cells and maximize the efficiency of RNA extraction.
Place the cell/TRIzol mixture into a -80 °C freezer.
Repeat Steps A1-A24 at least three times, ideally on separate days, to obtain biological replicates.
Remove the TRIzol suspensions from the freezer and let them thaw at room temperature.
Once thawed, let the TRIzol suspensions sit for 5 min at room temperature to help ribonucleoprotein complexes dissociate (see Note 5).
Add 0.2 ml of chloroform to the TRIzol suspension, vortex vigorously for ~20 s, and let the sample stand at room temperature for another 2 min.
Centrifuge the sample for 15 min at 12,000 × g at 4 °C.
Transfer the aqueous phase containing the RNA to a new 1.7 ml tube (~0.5 ml).
Add 10 μl of linear acrylamide to the extracted aqueous phase and vortex vigorously.
Add 0.5 ml of isopropanol to the aqueous phase (amount roughly equal to the volume of the aqueous phase), and vortex vigorously.
Incubate for ≥1 h at -20 °C (this step differs from the manufacturer’s instructions for purification of RNA from TRIzol).
Centrifuge the sample at top speed for 30 min in a microcentrifuge at 4 °C (> 12,000 × g; see Note 6).
Using a P1000, remove the water/isopropanol solution, being mindful not to remove the RNA pellet, which should be located below the hinge of the microcentrifuge tube.
To the precipitated RNA pellet, gently add 1 ml of an ice-cold mixture of 80% ethanol and 20% RNase-free water.
Using a P1000, remove the 80% ethanol, being mindful not to remove the RNA pellet.
Pulse-spin the 1.7 ml tube in a mini-centrifuge to bring the residual ethanol from the sides of the tube down to the bottom.
Using a P200 pipette and tip, remove the remaining 80% ethanol.
Repeat Steps A38-A39 until no 80% ethanol remains.
Re-suspend the pellet in 30 μl of RNase-free water.
Let the RNA-containing solution stand for 1 h at room temperature with intermittent mixing (every 15 min) by flicking or vortexing and then pulse-spinning the tube, or by pipetting the solution up and down (see Note 7).
-
After the RNA has dissolved, quantify the concentration of RNA using a Nanodrop spectrophotometer.
An ideal ratio of absorbance at 260 nm and 280 nm for RNA is between ~1.8 and ~2.
If the RNA is contaminated with residual ethanol, phenol, or guanidine, or if the RNA is not completely dissolved, the 260/280 ratio will be lower, usually < 1.6.
See Thermo Fisher's technical notes on NanoDrop Spectrophotometers for more information.
Next, determine whether the purified RNA is intact. To do this, set up a gel electrophoresis apparatus and prepare a 1% agarose/0.0001% ethidium bromide gel with 1x TAE buffer. Submerge the agarose gel in 1x TAE buffer.
In a 1.7 ml tube, mix ~250-500 ng of RNA with an appropriate amount of glycerol-based agarose gel loading buffer.
In separate lanes of the agarose gel, load the RNA/gel loading mixture as well as 0.5-1 μg of DNA ladder (the latter sample provides a size reference).
Run the samples in 1x TAE about ~8 cm through the agarose gel, at a voltage of 5 V/cm of distance between electrodes.
Take a picture of the gel on an appropriate gel imaging system. Intact RNA purified from a typical mammalian cell should yield two distinct bands running at apparent sizes of ~1,500 and ~750 nucleotides relative to the DNA ladder, which correspond to the 28s and 18s rRNA species, respectively (Figure 2; see Note 8).
-
If the RNA is intact, proceed to calculate the weight of RNA per cell:
RNA-per-cell = [concentration of RNA in g/L]*[volume in L of water used to resuspend RNA (e.g., 30 x 10-6 L)]/[the number of cells lysed in TRIzol]
Calculate the average of the RNA-per-cell numbers obtained from biological replicate RNA preparations (see Note 9).
-
Prepare cDNA libraries for RNA-Seq (see Notes 10, 11, 12, and 13)
Ensure that cDNA libraries are prepared from two or more biological replicate RNA preparations (see Note 14).
For each sample to be sequenced, aliquot 1 μg of total cellular RNA in a total volume of 8 μl of RNase-free water.
To each sample add 2 μl of a fresh 1:100 dilution of ERCC RNA Spike-In Mix #1 (see Note 15).
-
Starting with the mixture of ERCC Spike-in RNA and 1 μg of total cellular RNA, follow the manufacturer’s instructions for cDNA library preparation. We follow the instructions essentially as they are written in the user technical datasheet from KAPA (Note 16). The following are inputs required from the user:
From Section 6 of the technical datasheet (“RNA Elution, Fragmentation and Priming”), we select the fragmentation conditions of 6 min at 94 °C, which will generate 200-300 nucleotide-long RNA fragments.
From Section 12 of the technical datasheet (“Library Amplification”), we perform PCR amplification of our final cDNA library using only half of our purified cDNA library (i.e., 10 μl of cDNA library in a 50 μl PCR reaction), rather than all 20 μl of library in the 50 μl reaction (see Note 17).
From cDNA libraries prepared using 1 μg of total RNA, using half of the purified cDNA library, we typically perform 11 cycles of PCR for the final amplification step.
Using a Qubit fluorometer, quantify the concentration of DNA in the PCR-amplified, purified cDNA library (see Note 18).
Prepare a 1% agarose/0.0001% ethidium bromide gel with 1x TAE buffer. Submerge the agarose gel in 1x TAE buffer.
Run 2 μl (1/10th) of the amplified cDNA library ~6 cm into the agarose gel, alongside of 250 ng of 1 Kb Plus DNA Ladder.
Image the agarose gel and estimate the average size in base pairs of each prepared cDNA library (see Note 19).
-
Calculate the molarity of the purified cDNA library:
The average molecular weight of a DNA base-pair is 650 Daltons, or 650 g/mole.
-
Using the average length determined in Step B8 above, calculate the molar weight of the cDNA library:
[cDNA library g/mole] = [average_length] * 650g/mole
-
Now, using the DNA concentration determined in Step B5 above, calculate the molarity of the cDNA library (see Note 20):
[cDNA library moles/Liter] = [cDNA library concentration in ng/μl])*( [1 x 10-9 g]/[1 x 10-6 L])*(1/[cDNA library g/mole])
Pool together the cDNA libraries to be sequenced in a way that will ensure an equimolar amount of each library is present in the pool and that each library will be sequenced to a depth of > 20 million reads (see Notes 21 and 22).
Sequence the pooled cDNA libraries on an Illumina platform (see Note 23).
Figure 2. RNA run on a 1% agarose gel pre-stained with ethidium bromide.
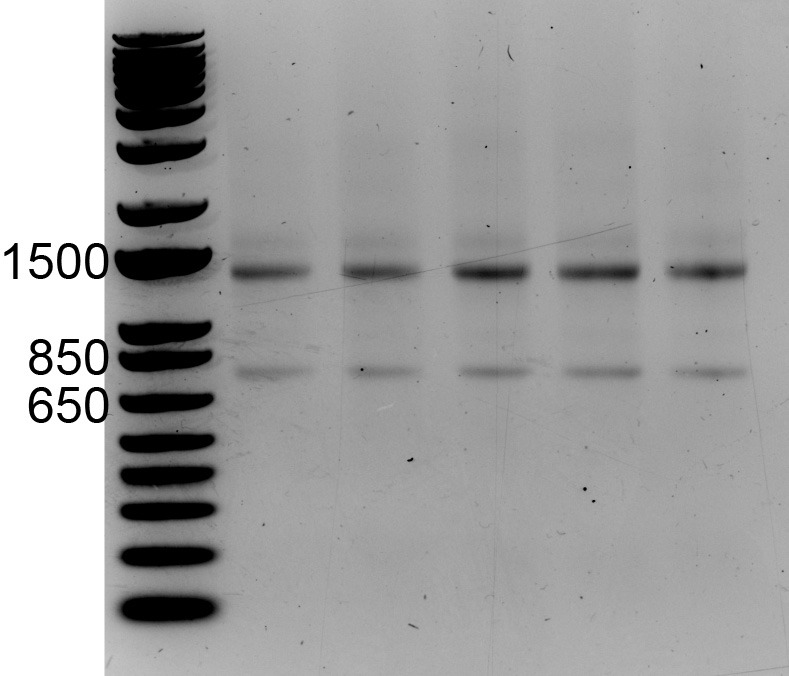
Relative to the DNA size ladder, the 28S and 18S rRNA species migrate at approximate sizes of ~1,400 and ~750 nucleotides, respectively.
Data analysis
Note: See Note 24.
-
Align data and obtain read counts (see Note 25)
-
Within a UNIX command terminal, create a master directory to perform the sequence alignment, filtering, and read-counting (e.g., ./ercc_mpc_analysis).
mkdir ./ercc_mpc_analysis
-
Obtain an RNA-Seq fastq file from the Illumina sequencing run (e.g. rnaseq_file.fastq), and move this file to the master directory (see Note 26).
mv rnaseq_file.fastq ./ercc_mpc_analysis
Download the appropriate genome fasta file (e.g., genome.fa) and gene-gtf file (e.g., genes.gtf) for your cell type and place them in the master directory (./ercc_mpc_analysis) (see Note 27).
-
Download the ERCC Spike-In RNA annotation files from the ERCC RNA Spike-In product page on Thermo Fisher’s website and place them in the master directory -- ./ercc_mpc_analysis (see Note 28). File names are:
ERCC Controls Analysis: ERCC RNA Spike-In Control Mixes (e.g., ERCC92_conc.txt)
ERCC92.fa & ERCC92.gtf sequence and annotation files (.zip)
-
Within the master directory (./ercc_mpc_analysis), create a new directory to store the genome index that will be built by STAR
mkdir ./GenomeDir/
-
Build a STAR genome-index that includes both the genome and ERCC reference sequences; the index will be created and stored in ./GenomeDir/.
STAR --runThreadN 8
--runMode genomeGenerate
--genomeDir ./GenomeDir
--genomeFastaFiles genome.fa ERCC92.fa
--sjdbGTFfile genes.gtf ERCC92.gtf
-
Use STAR to align an RNA-Seq fastq file (e.g., rnaseq_file.fastq) to the genome index. The alignments will be saved to a file with the appendix “Aligned.out.sam” (e.g., rnaseq_file_out_Aligned.out.sam).
STAR --runThreadN 12
--genomeDir ./GenomeDir
--readFilesIn rnaseq_file.fastq
--outFileNamePrefix rnaseq_file_out_
-
Use samtools to filter the “Aligned.out.sam” file for mapping quality of > 30 (this step selects for uniquely mapped reads). In this example, the filtered file is named “rnaseq_file_out _q30.sam”.
samtools view -Shq 30
rnaseq_file_out_Aligned.out.sam > rnaseq_file_out _q30.sam
-
Use featureCounts in the Subread package to count the number of reads that align to each ERCC Spike-In transcript (Note 29). In this example, the file containing ERCC counts is named “ercc_featureCounts_output.txt”.
featureCounts
-s 2
-a ERCC92.gtf
-o ercc_featureCounts_output.txt
rnaseq_file_out_q30.sam
-
Use featureCounts in the Subread package to count the number of reads that align to each genic transcript in the genes.gtf file. In this example, the file containing ERCC counts is named “mm9_genes_featureCounts_output.txt”.
featureCounts
-s 2
-a genes.gtf
-o mm9_genes_featureCounts_output.txt
rnaseq_file_out_q30.sam
-
-
Create a standard curve that relates ERCC Spike-In RNA-Seq read counts to the absolute amount of each ERCC transcript added to the RNA just prior to preparing the cDNA library for RNA-Seq. We recommend using Excel or Rstudio for these calculations. Templates and examples can be found here. Figure 3 below shows a standard curve derived from a single RNA-Seq replicate ( Schertzer et al., 2019 ).
Copy and paste the contents of the ERCC92_conc.txt file (downloaded in step A4) into a new Excel spreadsheet – call this the “ERCC Mix In” spreadsheet (See Note N30). A picture of the ERCC92_conc.txt file is below (Figure 4).
Delete the final three columns of the table in the Excel spreadsheet ("concentration in Mix 2 (attomoles/ul)", "expected fold-change ratio", "log2(Mix 1/Mix 2)").
Create a new column in the table–column E, “Attomoles added”–that uses the values in column D "concentration in Mix 1 (attomoles/ul)" to calculate the number of attomoles of ERCC Spike-In RNAs added to the total RNA prior to RNA-Seq library preparation (see Note 31; Figure 5).
Create a new column–column F, “Moles”–that divides the number in column E “Attomoles added” by 1E18.
Create a new column–column G, “Molecules”–that multiplies the values in column F “Moles” by 6.022E23 (molecules per mole; Avogadro’s number).
Create a new column–column H, “log2(molecules)” – that calculates the log-base-2 of the values in column G “Molecules”.
Sort the table such that the data in column B “ERCC_ID” appear in ascending order; this will be important later (Figure 6).
In a separate Excel spreadsheet, copy and paste the contents of the ercc_featureCounts_output.txt file generated in step A9–call this the “fCounts data” spreadsheet (see Note 32).
-
Within this new spreadsheet, calculate the number of aligned reads per kilobase per million aligned reads (RPKM) for each ERCC transcript.
-
First, in your dataset of interest, find the total number of reads that aligned to the genome with a mapping quality of > 30. This can be done from the UNIX command line, using samtools view:
samtools view -c rnaseq_file_out _q30.sam > rnaseq_file_counts.txt
Next, create a new column in the Excel spreadsheet–column H, “RPM”–in which the read counts in column G are divided by aligned read count from “rnaseq_file_counts.txt” then multiplied by 1 million. This gives reads per million (RPM).
Then, create a new column in the Excel spreadsheet–column I, “RPKM”–in which the RPM value in column H is divided by the value in column F “length” (the length in nucleotides of each ERCC transcript), then multiplied by 1,000. This converts RPM into RPKM, or read counts per kilobase of transcript per million aligned reads.
Finally, create a new column in the Excel spreadsheet–column J, “log2(RPKM)”–that calculates the log-base-2 of the values in column I “RPKM” (Figure 7).
-
Ensure that the Excel spreadsheets created in step B1 and in step B8 are sorted by ERCC ID in ascending order (see Note 33).
Paste the log2(RPKM) values from the “fCounts data” spreadsheet (created in step B8) into a new column in the “ERCC Mix In” spreadsheet (created in step B1).
Remove ERCC transcripts that had zero aligned reads.
Generate a scatter plot where log2(molecules) is on the x-axis and log2(RPKM) is on the y-axis. See Figure 3 and Figure 8 below.
-
Fit a straight line to the data
In Excel, select the points on the graph, right click, and ‘Add Trendline’. In the window, select ‘Linear’ and ‘Display Equation on chart’ (Figure 9).
Expect the R2 value to be greater than 0.90 (typically, it is above 0.95).
Use the y = mx + b equation in the next section.
-
Calculate molecules per cell for the lncRNA and mRNA genes of interest.
Copy the contents of the mm9_genes_featureCounts_output.txt file (file generated in step A10) and paste them into a new Excel spreadsheet – call this the “MPC” spreadsheet (see Note 34).
Using same procedure outlined in step B9 above, convert the read counts for each gene (column G of the MPC spreadsheet) into RPM as a new column H, then into RPKM as a new column I, and then into log2(RPKM) as a new column J.
-
For each gene of interest, calculate log2(molecules) using the y = mx + b equation from step B14.
x = log2(molecules)
y = log2(RPKM) values calculated in column J
b = y-intercept from equation in step B14
m = slope from equation in step B14
Create a new column in the MPC Excel spreadsheet–column K “log2(molecules)”–which performs the following calculation:
x= (y-b)/m
Create a new column in the MPC Excel spreadsheet–column L “molecules”–in which the value in column K is converted to molecules using the exponential 2x. In Excel notation, this is performed by setting the equation in column L to “=2^column_K” (see Note 35).
-
Create a final column in the MPC Excel spreadsheet–column M “MPC”–in which the value in column L is converted to molecules-per-cell:
Divide 1 μg, the amount of total RNA used to prepare the RNA-seq library, by the weight of RNA-per-cell calculated in Step A49 of the “Procedure” section of this protocol. This value represents the approximate total number of cells used to prepare the RNA-seq library (see Note 36).
For each transcript of interest, divide the number of molecules in column L by the total number of cells used for RNA-Seq to estimate molecules of transcript per cell.
Figure 3. Representative standard curve relating RNA-Seq read counts (y-axis) to molecular abundance of the ERCC Spike-In RNAs (x-axis).
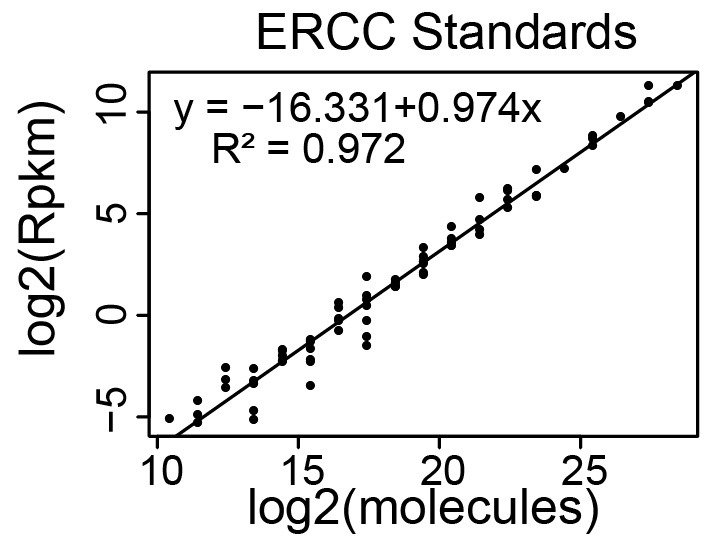
Figure 4. Screenshot of the ERCC92_conc.txt file.

Figure 5. Screenshot of an example calculation of the number of attomoles of ERCC Spike-Ins added to the RNA prior to library preparation.

Figure 6. Screenshot of sorted ERCC calculation table.

Figure 7. Screenshot of ERCC RPKM calculation.

Figure 8. Screenshot of scatter plot generation in Excel.

Figure 9. Screenshot of trendline-adding in Excel.

Notes
Roche recently purchased KAPA Biosystems and discontinued their small reaction-number sequence adapter kits. The SeqCap Adapter Kit is their replacement product, but note that this kit only comes in a 96-reaction format. For less than the list-price of the SeqCap kit, users can purchase their own Illumina-compatible adapters in bulk from a commercial oligonucleotide provider. These adapters then need to be resuspended and annealed by the user. However, the advantage of purchasing adapters in bulk is that the cost per reaction is dramatically reduced. Thus, users that plan to perform many RNA-seq assays (or, for that matter, any other *–seq assay) will find that purchasing and annealing their own adapters is far more cost effective than purchasing a pre-aliquoted set of adapters. For those interested in purchasing and annealing their own adapters, we have provided instructions here. Users that plan to carry out only a small number of RNA-seq assays may find it more cost-effective to purchase their RNA-seq kits and adapters from a manufacturer that sells reagents in small-sized packages, such as NEB.
This portion of the protocol describes how to calculate the average amount of RNA per cell in a cell type of interest. The protocol is designed for cultured adherent cells but could easily be adapted to cultured suspension cells. With additional optimization, it could also be adapted to a tissue of interest. In this latter case, the user would need a method to approximate the total number of cells per mass of tissue of interest (for example, how many cells are present in one milligram of tissue?). With an accurate estimate of cell-number-per-mass-of-tissue, the user could then perform replicate rounds of RNA purification from a known mass of tissue. The yield in weight of RNA would then be divided by the number of cells that were used to obtain the RNA to derive an estimate of the amount of RNA per average cell in the tissue of interest.
To clean a bench, spray the area with a light coat of RNase Zap and wipe the solution clean with paper towels. Lightly spray the pipettes to be used for the RNA prep with RNase Zap and then wipe them clean. Use boxes of pipette tips that have not been exposed to any source of RNase. Common sources of RNase are from plasmid DNA preparation kits and human skin/saliva. On a given workday, minimize the chance of RNase contamination by performing RNA work prior to performing any plasmid DNA preparations (or any other protocol that involves an RNase) and wear gloves at all times. Moreover, although it may sound draconian, once an RNA preparation begins in earnest (Step A26, Procedure section) avoid talking, coughing, chewing gum etc. in the vicinity of open microcentrifuge tubes. We also use barrier tips on our pipettes. By taking these simple precautions, you will help to ensure the success of the protocol.
TRIzol contains phenol, which is corrosive to the eyes, skin, and respiratory tract. When working with TRIzol, users should wear safety goggles, closed-toed shoes, and a lab coat at all times and take care not to splash TRIzol on any part of their body. If users are sensitive to fumes from TRIzol, work should be performed in a fume hood. TRIzol needs to be disposed of by following safety guidelines that are appropriate to the institution.
Recently, Chujo and colleagues found that certain nuclear-retained lncRNAs are recovered at a higher efficiency when TRIzol suspensions are incubated for 10 min at 55 °C (Chujo and Hirose, 2017). In our prior study of lncRNA intracellular abundance ( Schertzer et al., 2019 ), we did not perform this 55 °C incubation step. However, we see no downside to the 55 °C incubation, and may perform it in the future.
Prior to starting the spin cycle, align the spines of each microcentrifuge tube so that the hinges are all facing outward. Aligning the spines will ensure that the RNA/acrylamide pellet in each tube is located directly below the tube hinge. Knowing where in the tube to expect the pellet helps to take some of the guesswork out of the protocol, especially when you are working with small amounts of RNA.
If mixing by pipetting, please note that the RNA pellet can sometimes stick to the inside of the pipette tip. If this scenario occurs, continue pipetting the water up and down until the pellet visibly dissolves.
RNA that has experienced varying levels of degradation will appear as a smear on the gel.
Using this procedure, our RNA-per-cell estimates for mouse embryonic stem and trophoblast stem cells arrived at 20 picograms and 30 picograms, respectively ( Calabrese et al., 2007 ; Schertzer et al., 2019 ).
To estimate intracellular abundance, we recommend using RNA-Seq library preparation protocols that purify genic transcripts away from rRNA by rRNA-depletion rather than by polyA-selection. In a pilot study, we estimated intracellular abundance using rRNA-depletion and polyA-selection for three lncRNAs of interest (Xist, Airn, and Kcnq1ot1) and found that the two protocols arrived at dramatically different estimates (not shown). Our interpretation of these pilot data is that during the polyA-selection protocol, a number of factors likely cause the efficiency of capture to vary for different polyadenylated RNAs. Variations in the extent of polyadenylation, the length of the polyA tail, the extent to which RNA base-pairing interferes with polyA capture, and the amount of internal A-rich sequence may cause certain polyadenylated transcripts to be captured with greater efficiency than others. These variations would skew estimates of intracellular abundance in ways that are difficult to predict. In contrast, variations in polyA-capture efficiency are not relevant for rRNA-depletion protocols, which deplete rRNA from total RNA preparations using oligonucleotides that are complementary to the major rRNA species. Thus, intracellular abundance may be measured more accurately by rRNA-depletion RNA-Seq than by polyA-selection RNA-seq. That being said, transcripts harboring internal homology to the rRNAs would be selectively depleted by rRNA-depletion and would require special consideration under this protocol.
To prepare samples for RNA-Seq, our lab routinely uses the RNA HyperPrep Kit with RiboErase from Kapa Biosciences/Roche. The instructions from the KAPA RNA HyperPrep Kit with RiboErase are clear and walk users in-depth through each step of the protocol, which begins with the degradation of rRNA, followed by DNase treatment, RNA fragmentation and priming, cDNA synthesis, second-strand cDNA synthesis and A-tailing, adapter ligation, and finally, library amplification. Most of these steps are followed by a purification and buffer exchange using polystyrene–magnetite beads that are provided as part of the kit. In our lab, this kit has been robust to multiple users collecting different datasets over timeframes that span multiple years. However, many other companies sell high-quality kits to prepare ribo-deplete RNA-seq libraries, including Illumina and NEB. Generally speaking, these kits should perform equivalently. For users that plan to perform only a few RNA-seq experiments, a company such as NEB may be preferable, because they sell kits in smaller sizes than KAPA.
The KAPA RNA HyperPrep protocol is a modified version of the dUTP second-strand protocol described in Parkhomchuk et al. (2009) and Levin et al. (2010) , in which stranded-ness of the RNA-seq library is maintained by performing second-strand cDNA synthesis in the presence of dUTP, followed by cDNA library amplification using a DNA polymerase that has been engineered to preferentially amplify DNA that contains deoxythymidine and not deoxyuridine. When using this kit or any other kit that employs a dUTP-based method, researchers should be aware that the stranded-ness of the amplified library is not perfect. With a low frequency, the engineered DNA polymerases will still amplify the deoxyuridine-containing second-strand. The result of this low-frequency second-strand amplification that for highly expressed genes, a “shadow” of RNA-seq signal is often visible on the strand that is opposite (i.e., antisense) to the correct strand of the gene. In practice, this shadow signal has never affected our downstream analyses, but users should be aware that it exists.
It is not uncommon to make mistakes during the first run-through of this protocol. We recommend that first-time users start by going through the entire protocol below using only one or two samples from which biological material is non-limiting. This way, users can work out the logistics of library preparation without the stress of needing the protocol to work the very first time.
For a robust biological replicate, we recommend using RNA prepared on different days or from different animals etc.
To minimize pipetting error, we recommend pipetting volumes of 2 μl or more. For example, instead of pipetting 1 μl of ERCC Spike-In solution to 99 μl RNase-free water, we would pipette 2 μl of Spike-In solution to 198 μl of RNase-free water.
KAPA RNA HyperPrep Kit with RiboErase; catalog number: KK8560; technical datasheet version KR1351–v2.17; this same datasheet is also included in our github page.
The reason for using only half of the library is that it preserves cDNA material in case the user needs to repeat the final PCR reaction owing to an error in PCR setup, or over- or under-amplification of the cDNA library.
After PCR amplification, clean-up, and elution in 20 μl of buffer as specified in the technical datasheet, the amount of DNA per sample should be in the range of 7-150 ng/μl. Concentrations of DNA outside of this range may be acceptable, but in the rare instances in which the concentrations of our own cDNA libraries have fallen outside of this range, we have elected to repeat the final PCR amplification using an adjusted number of PCR cycles, rather than submit the originally-amplified library for RNA-seq. The reason for this is that the user is trying to ensure that the final PCR remains within the linear range of amplification. Amplification to a concentration of 150 ng/μl may be close to the top of the linear range of the KAPA kit. Similarly, final library concentrations < 7 ng/μl may also be acceptable, but under this scenario, we have elected to repeat the final PCR using more cycle numbers rather than submit the low concentration libraries for sequencing. Most frequently, the final concentration of our amplified libraries is between 10-50 ng/μl; this is our optimal target range.
Using the conditions for cDNA library preparation specified above, users should expect the average size of amplified DNA fragment in each library to be between 300-400 nucleotides.
Here is an example with numbers: The concentration calculated in Step B5 (Prodedure section) is 25 ng/μl. The average library size estimated in Step B8 (Prodedure section) is 300 base pairs. The molarity of the library is 25 * (1 x 10-9)/(1 x 10-6)/(300 * 650) = 128 nM.
The high-throughput sequencing facility at UNC asks that users submit their pooled cDNA libraries to them at a final concentration of 15 nM. Thus, as an example, if we were hoping to include 12 separate cDNA libraries in a single pool, that would mean each library would need to be present in the pool at a final concentration of 1.25 nM, or [15 nM/12]. One easy way to create a pool of cDNA libraries at the appropriate concentration is to first create separate aliquots of each library at the final concentration of the pool–in this example, that concentration would be 15 nM. Then, equal volumes of each library can be combined to create the 15 nM pool.
In order to determine the maximum number of libraries that can be included in a pool such that that each library is still sequenced to an appropriate depth, first determine the number of sequencing reads that you expect to be returned from your run on the Illumina sequencing instrument. For example, at UNC, the average 75-cycle high-output run on a NextSeq500 Instrument will return 500 million reads. In order to obtain at least 20 million reads per cDNA library in a pool, the maximum number of libraries we should include in that pool is 500/20, or 25 libraries. In practice, we often include fewer libraries than this maximum number, which results in read-depths per library of > 20 million reads. High read-depth per sample is never a problem. Moreover, fewer than 20 million reads from a single library may also be tolerable; just note that as the number of reads decreases, so does your ability to confidently quantify the abundance lowly-expressed transcripts.
We have performed our data analyses after obtaining 75 base, single-end reads from an Illumina NextSeq500 instrument. Shorter, longer, or even paired-end reads would also be suitable.
Please see the github page associated with this protocol for example files and analysis templates in Excel and in R (https://github.com/mschertzer/ercc_analysis).
For RNA-Seq alignments we generally use STAR, but note that other aligners that support gapped-alignments should perform equivalently ( Baruzzo et al., 2017 ; Bushnell, 2010; Dobin et al., 2013 ).
If following along with the example provided on the github page, you may use the SRA toolkit to download the fastq file associated with record SRR7685881 in the NCBI Sequence Read Archive ( Leinonen et al., 2011 ).
In the example in this protocol, we use the mm9 genome.fa and genes.gtf files compiled by the UCSC Genome Browser ( Haeussler et al., 2019 ) and downloaded from Illumina’s iGenomes site: https://support.illumina.com/sequencing/sequencing_software/igenome.html.
Users can also find these files on the github page associated with this protocol (https://github.com/mschertzer/ercc_analysis).
The “-s 2” option of featureCounts is specific to libraries that are prepared using methods that generate “reverse-stranded” data, such as the KAPA RNA HyperPrep Kit with RiboErase described in this protocol.
Steps B2 through B7 (Data analysis) below have already been performed in the “ERCC Mix In” spreadsheet that is included in the “ERCC_analysis_template.xlsx” template on the github page associated with this protocol (https://github.com/mschertzer/ercc_analysis).
In Schertzer et al. (2019) , we added 2 μl of a 1:100 dilution of ERCC Mix 1 Spike-In RNA to 1 μg total RNA. Thus, to follow our example, divide the ERCC Spike-In Mix concentration in column D by 100 and then multiply by 2 to calculate the number of attomoles added as column E.
In the template “ERCC_analysis_template.xlsx” provided on the github page, this second spreadsheet is called “fCounts data”.
After sorting, the order in which each ERCC transcript appears in each spreadsheet should be identical.
For an example of MPC calculations performed over the entire transcriptome using an RNA-seq dataset from Schertzer et al. (2019) , see the MPC spreadsheet in the “ERCC vprtta analysis example.xlsx” file on the github page associated with this protocol (https://github.com/mschertzer/ercc_analysis).
The result of this calculation is an estimation of the number of RNA molecules that were present in the pool of total RNA that was used to prepare the RNA-Seq cDNA library (which in our case was 1 μg total RNA).
In Schertzer et al. (2019) , we estimated that our trophoblast stem cell line harbors 30 picograms per cell. Thus, 1 μg of RNA corresponds to 33,333 cells.
Acknowledgments
We thank Keean Braceros for proofreading this protocol and Jackson Trotman for the gel image used in Figure 2. This work was supported by the National Institutes of Health (NIH) Grant GM121806. Schertzer et al. (2019) is the original paper from which this protocol was derived.
Competing interests
The authors have no competing interests to declare.
Citation
Readers should cite both the Bio-protocol article and the original research article where this protocol was used.
References
- 1. Baruzzo G., Hayer K. E., Kim E. J., Di Camillo B., FitzGerald G. A. and Grant G. R.(2017). Simulation-based comprehensive benchmarking of RNA-seq aligners. Nat Methods 14(2): 135-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bushnell B.(2010). BBMap(sourceforge.net/projects/bbmap/).
- 3. Cabili M. N., Dunagin M. C., McClanahan P. D., Biaesch A., Padovan-Merhar O., Regev A., Rinn J. L. and Raj A.(2015). Localization and abundance analysis of human lncRNAs at single-cell and single-molecule resolution. Genome Biol 16: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cabili M. N., Trapnell C., Goff L., Koziol M., Tazon-Vega B., Regev A. and Rinn J. L.(2011). Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 25(18): 1915-1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Calabrese J. M., Seila A. C., Yeo G. W. and Sharp P. A.(2007). RNA sequence analysis defines Dicer's role in mouse embryonic stem cells. Proc Natl Acad Sci U S A 104(46): 18097-18102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Calabrese J. M., Sun W., Song L., Mugford J. W., Williams L., Yee D., Starmer J., Mieczkowski P., Crawford G. E. and Magnuson T.(2012). Site-specific silencing of regulatory elements as a mechanism of X inactivation. Cell 151(5): 951-963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cerase A., Smeets D., Tang Y. A., Gdula M., Kraus F., Spivakov M., Moindrot B., Leleu M., Tattermusch A., Demmerle J., Nesterova T. B., Green C., Otte A. P., Schermelleh L. and Brockdorff N.(2014). Spatial separation of Xist RNA and polycomb proteins revealed by superresolution microscopy. Proc Natl Acad Sci U S A 111(6): 2235-2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen J., Shishkin A. A., Zhu X., Kadri S., Maza I., Guttman M., Hanna J. H., Regev A. and Garber M.(2016). Evolutionary analysis across mammals reveals distinct classes of long non-coding RNAs. Genome Biol 17: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chujo T. and Hirose T.(2017). Nuclear bodies built on architectural long noncoding rnas: unifying principles of their construction and function. Mol Cells 40(12): 889-896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M. and Gingeras T. R.(2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29(1): 15-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gil N. and Ulitsky I.(2020). Regulation of gene expression by cis-acting long non-coding RNAs. Nat Rev Genet 21(2): 102-117. [DOI] [PubMed] [Google Scholar]
- 12. Haeussler M., Zweig A. S., Tyner C., Speir M. L., Rosenbloom K. R., Raney B. J., Lee C. M., Lee B. T., Hinrichs A. S., Gonzalez J. N., Gibson D., Diekhans M., Clawson H., Casper J., Barber G. P., Haussler D., Kuhn R. M. and Kent W. J.(2019). The UCSC Genome Browser database: 2019 update. Nucleic Acids Res 47(D1): D853-D858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hezroni H., Koppstein D., Schwartz M. G., Avrutin A., Bartel D. P. and Ulitsky I.(2015). Principles of long noncoding RNA evolution derived from direct comparison of transcriptomes in 17 species. Cell Rep 11(7): 1110-1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hrdlickova R., Toloue M. and Tian B.(2017). RNA-Seq methods for transcriptome analysis. Wiley Interdiscip Rev RNA 8(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jiang L., Schlesinger F., Davis C. A., Zhang Y., Li R., Salit M., Gingeras T. R. and Oliver B.(2011). Synthetic spike-in standards for RNA-seq experiments. Genome Res 21(9): 1543-1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kopp F. and Mendell J. T.(2018). Functional Classification and Experimental Dissection of Long Noncoding RNAs. Cell 172(3): 393-407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kutter C., Watt S., Stefflova K., Wilson M. D., Goncalves A., Ponting C. P., Odom D. T. and Marques A. C.(2012). Rapid turnover of long noncoding RNAs and the evolution of gene expression. PLoS Genet 8(7): e1002841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Leinonen R., Sugawara H., Shumway M. and International Nucleotide Sequence Database C.(2011). The sequence read archive. Nucleic Acids Res 39(Database issue): D19-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Levin J. Z., Yassour M., Adiconis X., Nusbaum C., Thompson D. A., Friedman N., Gnirke A. and Regev A.(2010). Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nat Methods 7(9): 709-715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. and Genome Project Data Processing S.(2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25(16): 2078-2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liao Y., Smyth G. K. and Shi W.(2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30(7): 923-930. [DOI] [PubMed] [Google Scholar]
- 22. Necsulea A., Soumillon M., Warnefors M., Liechti A., Daish T., Zeller U., Baker J. C., Grutzner F. and Kaessmann H.(2014). The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature 505(7485): 635-640. [DOI] [PubMed] [Google Scholar]
- 23. Ninomiya K. and Hirose T.(2020). Short tandem repeat-enriched architectural rnas in nuclear bodies: functions and associated diseases. Noncoding RNA 6(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Parkhomchuk D., Borodina T., Amstislavskiy V., Banaru M., Hallen L., Krobitsch S., Lehrach H. and Soldatov A.(2009). Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res 37(18): e123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Raj A. and Rinn J. L.(2019). Illuminating Genomic Dark Matter with RNA Imaging. Cold Spring Harb Perspect Biol 11(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. RStudio_Team(2015). RStudio: Integrated Development for R. RStudio: Integrated Development for R.
- 27. Schertzer M. D., Braceros K. C. A., Starmer J., Cherney R. E., Lee D. M., Salazar G., Justice M., Bischoff S. R., Cowley D. O., Ariel P., Zylka M. J., Dowen J. M., Magnuson T. and Calabrese J. M.(2019). lncRNA-Induced Spread of Polycomb Controlled by Genome Architecture, RNA Abundance, and CpG Island DNA. Mol Cell 75(3): 523-537 e510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schuler A., Ghanbarian A. T. and Hurst L. D.(2014). Purifying selection on splice-related motifs, not expression level nor RNA folding, explains nearly all constraint on human lincRNAs. Mol Biol Evol 31(12): 3164-3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schwaber J., Andersen S. and Nielsen L.(2019). Shedding light: The importance of reverse transcription efficiency standards in data interpretation. Biomol Detect Quantif 17: 100077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Smeets D., Markaki Y., Schmid V. J., Kraus F., Tattermusch A., Cerase A., Sterr M., Fiedler S., Demmerle J., Popken J., Leonhardt H., Brockdorff N., Cremer T., Schermelleh L. and Cremer M.(2014). Three-dimensional super-resolution microscopy of the inactive X chromosome territory reveals a collapse of its active nuclear compartment harboring distinct Xist RNA foci. Epigenetics Chromatin 7: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sunwoo H., Wu J. Y. and Lee J. T.(2015). The Xist RNA-PRC2 complex at 20-nm resolution reveals a low Xist stoichiometry and suggests a hit-and-run mechanism in mouse cells. Proc Natl Acad Sci U S A 112(31): E4216-4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tsanov N., Samacoits A., Chouaib R., Traboulsi A. M., Gostan T., Weber C., Zimmer C., Zibara K., Walter T., Peter M., Bertrand E. and Mueller F.(2016). smiFISH and FISH-quant- a flexible single RNA detection approach with super-resolution capability. Nucleic Acids Res 44(22): e165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ulitsky I.(2016). Evolution to the rescue: using comparative genomics to understand long non-coding RNAs. Nat Rev Genet 17(10): 601-614. [DOI] [PubMed] [Google Scholar]
- 34. Washietl S., Kellis M. and Garber M.(2014). Evolutionary dynamics and tissue specificity of human long noncoding RNAs in six mammals. Genome Res 24(4): 616-628. [DOI] [PMC free article] [PubMed] [Google Scholar]