

Abstract
R-loops are a prevalent class of non-B DNA structures that have been associated with both positive and negative cellular outcomes. DNA:RNA immunoprecipitation (DRIP) approaches based on the anti-DNA:RNA hybrid S9.6 antibody revealed that R-loops form dynamically over conserved genic hotspots. We have developed an orthogonal approach that queries R-loops via the presence of long stretches of single-stranded DNA on their looped-out strand. Nondenaturing sodium bisulfite treatment catalyzes the conversion of unpaired cytosines to uracils, creating permanent genetic tags for the position of an R-loop. Long-read, single-molecule PacBio sequencing allows the identification of R-loop ‘footprints’ at near nucleotide resolution in a strand-specific manner on long single DNA molecules and at ultra-deep coverage. Single-molecule R-loop footprinting coupled with PacBio sequencing (SMRF-seq) revealed a strong agreement between S9.6-based and bisulfite-based R-loop mapping and confirmed that R-loops form over genic hotspots, including gene bodies and terminal gene regions. Based on the largest single-molecule R-loop dataset to date, we show that individual R-loops form nonrandomly, defining discrete sets of overlapping molecular clusters that pileup through larger R-loop zones. R-loops most often map to intronic regions and their individual start and stop positions do not match with intron-exon boundaries, reinforcing the model that they form cotranscriptionally from unspliced transcripts. SMRF-seq further established that R-loop distribution patterns are not simply driven by intrinsic DNA sequence features but most likely also reflect DNA topological constraints. Overall, DRIP-based and SMRF-based approaches independently provide a complementary and congruent view of R-loop distribution, consolidating our understanding of the principles underlying R-loop formation.
Keywords: R-loops, DNA:RNA immunoprecipitation, SMRT-sequencing, Nondenaturing bisulfite conversion, S9.6 antibody
Introduction
R-loops are three-stranded nucleic acid structures that primarily form during transcription as a result of the nascent RNA annealing to the template DNA strand [1]. The formation of this RNA:DNA hybrid causes the nontemplate DNA strand to loop out in a single-stranded state. R-loops have been comprehensively mapped at genome scale [2,3] using the S9.6 antibody to immunoprecipitate the RNA:DNA hybrid moiety of the structure [4,5]. DNA:RNA immunoprecipitation (DRIP)–based studies revealed that R-loops are prevalent and dynamic non-B DNA structures that form over specific and conserved genic hotspots during transcription [3]. An accumulating body of data has linked R-loops to both physiological and pathological outcomes [6,7]. On one hand, R-loops have been linked with efficient transcription termination [3,8], chromatin patterning [3,9], and the regulation of gene expression [10,11]. On the other hand, deregulated R-loop formation has been implicated in genome instability arising from transcription-replication conflicts caused by dysfunctions in a variety of nuclear factors, including RNA processing enzymes [1,12-16]. Deregulated R-loop formation has also been implicated in a number of human diseases, including certain cancers and neurological disorders [17].
Although DRIP-based methods provide robust and specific information on the global distribution of R-loops in a genome, they suffer from a few important limitations. First, DRIP-based methods only provide a population average view of R-loop distribution; the exact positions or lengths of individual R-loops cannot be deduced from the data. This limitation is further compounded by the fact that short-read sequencing technologies are not well suited to readout typically long R-loops. Developing single-molecule R-loop profiling methods capable of measuring entire R-loop structures on long DNA fragments is therefore desirable. Second, DRIP-based methods are exclusively dependent on the S9.6 antibody. Although S9.6 possesses subnanomolar affinity to RNA:DNA hybrids, it binds to double-stranded RNA (dsRNA) with significant residual affinity [4,18]. This affinity complicates the use of S9.6 in R-loop profiling when dsRNA are produced [18] even though adequate ribonuclease pretreatments can be used to mitigate these issues. Furthermore, the report that binding sites for an inactive ribonuclease H reporter protein (dRNase H1)are often discordant with S9.6-based R-loop maps over gene body and gene terminal regions [10] raised the need for additional orthogonal R-loop profiling methodologies. A method that enables the characterization of R-loops from start to end on single DNA molecules at near nucleotide resolution in a strand-specific manner and at high depth, independently of S9.6 enrichment would set a strong benchmark for the field.
Before the development of S9.6-based DRIP methods [2,8], endogenous R-loops were queried not through their RNA:DNA hybrid moiety but through their association with long stretches of single-stranded DNA (ssDNA) on the displaced nontemplate strand [19]. This method took advantage of the fact that sodium bisulfite is an exquisitely sensitive probe of ssDNA and efficiently triggers the deamination of exposed cytosines (C) to uracils (U) [20]. When nucleic acids extracted from cells are treated with sodium bisulfite in a nondenaturing manner, intrinsically single-stranded regions undergo C to U conversion. These patches of uracils serve as permanent genetic tags for the position of a given R-loop (Fig. S1). After PCR amplification, uracils are converted to thymines (T) and the resulting amplicons are cloned and sequenced individually using Sanger sequencing. Mapping patches of C to T conversion permits single-molecule R-loop footprinting [2,19,21]. As with S9.6-based methods, pretreatment of nucleic acids with purified ribonuclease H (RNase H), an enzyme that specifically degrades RNA strands in the context of RNA:DNA hybrids [22], serves as a robust specificity control. The application of this method to R-loops induced at class-switch regions in murine B cells has provided numerous insights into the mechanisms of R-loop formation in mammalian genomes [19,21,23,24]. One drawback of this method, however, is that it suffers from low-throughput and requires laborious, time-consuming and cost-intensive cloning, sequencing, and data analysis steps. Here, we present a novel adaption of R-loop footprinting that permits single-molecule R-loop detection at near nucleotide resolution in a strand-specific manner on long amplicons and at ultra-high coverage. This method and its accompanying computational analysis and data visualization pipeline enables deep, cost-effective, targeted R-loop profiling at a range of genomic loci, under any condition and in any genome.
Results
Nondenaturing bisulfite R-loop footprinting coupled with high-throughput single-molecule sequencing
Nondenaturing bisulfite treatment allows R-loop mapping by promoting the deamination of intrinsically single-stranded cytosines to uracils on the looped-out DNA strand of an R-loop [19]. To achieve orders of magnitude higher throughput than possible using conventional sequencing methods, Pacific Biosciences (PacBio) libraries were built directly from PCR amplicons and sequenced using single-molecule, real-time sequencing (SMRT-seq; Fig. S1). SMRT-seq is well suited for this purpose as it allows sequencing through kilobases-long GC-rich amplicons [25] and delivers high-quality sequencing reads on 50,000 single DNA molecules per SMRT cell on a PacBio RSII instrument. Given this throughput, multiple independent genomic regions can be tested for R-loop formation in one SMRT cell without any prior enrichment. In the resulting method, single-molecule R-loop footprinting coupled with PacBio sequencing (SMRF-seq), C to T conversion patterns expected from true R-loops must satisfy the following predictions: (1) R-loops can be detected upon amplification of regions with native primers without any prior enrichment with the S9.6 antibody; (2) R-loop C to T conversion tracts can only be observed on the looped-outstrand, noton the RNA-paired strand; and (3) C to T conversion tracts on the looped-out strand are lost upon RNase H treatment of the genomic DNA before bisulfite treatment.
Accurately extracting C to T conversion information from SMRF-seq runs required the development of a new computational pipeline. In brief, high-quality circular consensus sequences (CCSs) were built and mapped against reference amplicons using the bisulfite mapping-enabled pipeline Bismark [26]. Strand assignments were based on the patterns of C to T or G to A conversions as described [19] (Fig. S1). Long, R-loop-associated C to T conversion footprints were then called using a threshold-based method in which a sliding window encompassing 20 consecutive cytosines were analyzed for their conversion status and a minimum user-imposed conversion threshold was applied (see Methods for details). The imposition of a minimal window size facilitated our ability to distinguish R-loop footprints from small-scale events of DNA breathing and from PacBio sequencing errors. Note that patterns of bisulfite conversion were only analyzed for non-CpG dinucleotides because methylation of CpG sites blurs our ability to interrogate the ssDNA status of these sites. Non-CpG methylation is present only at low levels in the human Ntera2 cell line [3].
S9.6-independent, strand-specific R-loop footprints in the human genome
As a proof of concept, we investigated R-loop formation at five loci (FUS, RPL13A, SNRNP70, RPS24, and PIN4) that exhibited prominent R-loop peaks previously identified using S9.6-based methods [3]. PIN4 represents an example of a GC-skewed promoter, whereas FUS, RPL13A, and SNRNP70 represent examples of gene body/early terminal R-loops and RPS24 represents a terminal R-loop peak. We used SMRF-seq to analyze these regions in the absence of any S9.6 pre-enrichment.
In all cases, prominent R-loop footprints were observed specifically on the nontemplate strand for transcription. Using FUS as a representative example, a total of 226 footprints were called on this strand out of 5,111 molecules sequenced (Fig. 1). Thus the proportion of R-loop–carrying molecules was 4.42% which is in close agreement with the range of R-loop frequencies measured by DRIP-qPCR [3]. Only three footprints were detected out of 14,489 molecules sequenced on the template strand, showing that bisulfite-reactive ssDNA was essentially confined to the non-template strand. Importantly, the collection of footprints observed at FUS was in strong agreement with bulk S9.6-based high-resolution strand-specific R-loop profiles obtained by DRIPc-seq [3] independently from the same cells. SMRF-seq delineated three regions where footprints were more likely to pileup; these three regions matched remarkably well with the corresponding subpeaks in DRIPc-seq data (Fig. 1). As a result, aggregate bisulfite conversion profiles over R-loop peaks were highly congruent with DRIPc-seq. Similar results were observed for all other tested loci (Fig. S2). This demonstrates that strand-specific R-loop footprints can be readily recovered at a range of R-loop-prone loci in the human genome using SMRF-seq. Furthermore, it shows that nondenaturing bisulfite footprinting-based and orthogonal S9.6-based approaches report on R-loop formation with strong agreement in terms of location and strandedness. Unlike population-based approaches, however, SMRF-seq allows us to peer into the collection of individual R-loops that together give rise to R-loop hotspots.
Fig. 1. Single-molecule R-loop footprints at the human FUS locus.
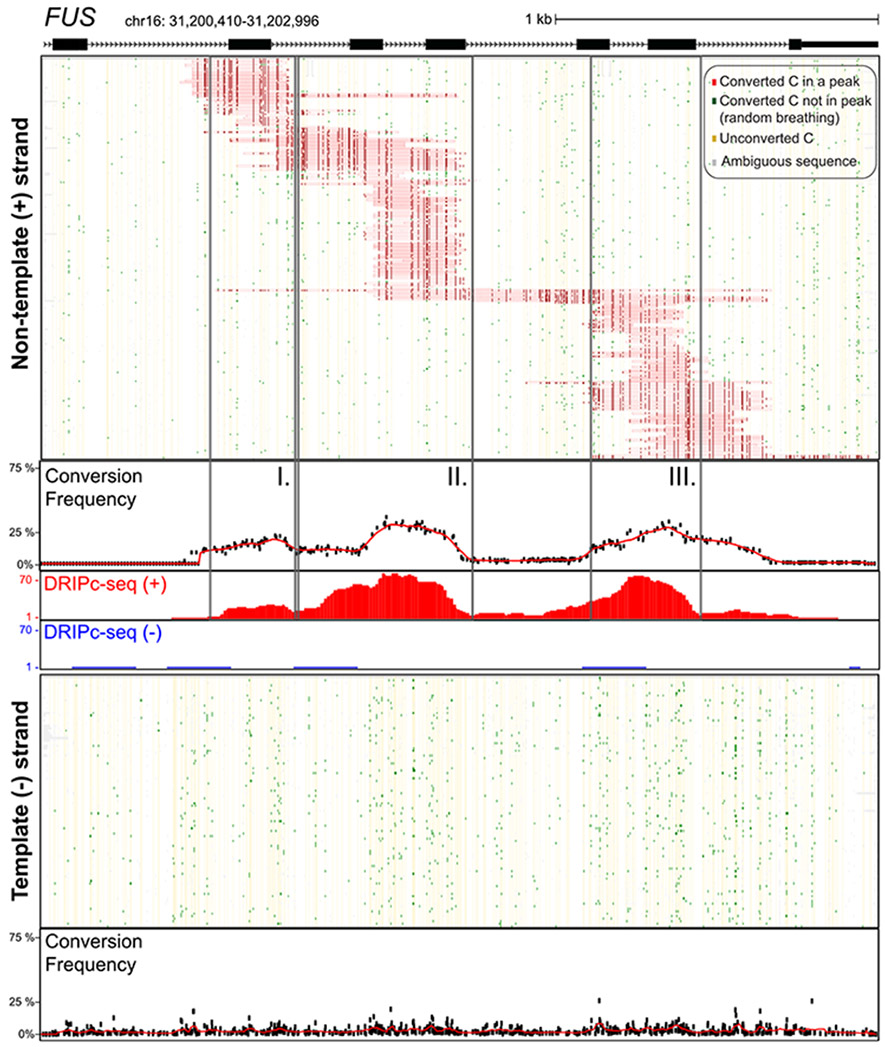
The structure of FUS over the amplicon is shown at top with exons as boxes; coordinates for the amplicon (hg19) along with a scale bar are also displayed. Two hundred twenty-six independent molecules carrying R-loop footprints on the nontemplate strand are shown at top. Each horizontal line corresponds to one DNA molecule. The status of each cytosine along the sequence is color-coded with footprints highlighted in red (see inset for color codes). These footprints were obtained using SMRF-seq with native PCR primers independently of any S9.6 enrichment. The aggregate C to T conversion frequency calculated over R-loop–containing molecules is shown along with the bulk S9.6-based R-loop signals obtained independently from DRIPc-seq (red indicates signal on the (+) strand; blue on the (−) strand). Vertical boxes highlight three regions where R-loop footprints pileup. One hundred independent molecules recovered from the template strand were randomly sampled from the 14,489 sequenced and are displayed below along with the aggregate C to T conversion frequency for that strand.
DRIP enriches but does not disturb genuine R-loop footprints
Given the overall agreement between aggregate SMRF-seq data and DRIPc-seq profiles, we asked whether the location or relative distribution of footprints would be modified by prior S9.6 enrichment. We therefore conducted DRIP followed by SMRF-seq and analyzed the same five loci previously described. As expected, the proportion of footprint-containing molecules was five- to ten-fold higher after DRIP enrichment at most loci (Table 1). Post-DRIP footprints were consistent with those observed without enrichment: they were strand-specific and their distribution in terms of aggregate profiles was consistent with and without S9.6 enrichment as shown for RPS24 and FUS (Fig. 2; Pearson's R were 0.99 and 0.94, respectively) and other loci (Fig. S3). When analyzed in terms of location and lengths, R-loop footprints with and without S9.6 enrichment were again mostly consistent across loci although minor differences were noted for individual loci (Fig. S4). For the SNRPN70 locus, one class of R-loops appeared differentially represented with and without S9.6 enrichment, leading to a discrepancy on location and lengths, but it is unclear if this difference was biologically significant or resulted from possible undersampling of the R-loop distribution. Overall, S9.6-based DRIP allowed the efficient enrichment of footprints without significantly disturbing their properties.
Table 1.
Summary of SMRF-seq data comparing footprinted sites with or without S9.6 antibody enrichment. Total number of independent reads and R-loop footprints (peaks) are shown for each tested locus and for each strand. The percentages of R-loop peaks are indicated at right in each case.
| Gene | Strand | No DRIP enrichment |
Post-DRIP |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (+) strand |
(−) strand |
(+) strand |
(−) strand |
||||||||||
| Total reads |
R-loop peaks |
% peaks |
Total reads |
R-loop peaks |
% peaks |
Total reads |
R-loop peaks |
% peaks |
Total reads |
R-loop peaks |
% peaks |
||
| CALM3 | + | 15121 | 66 | 0.4 | 1692 | 0 | 0.0 | 2729 | 351 | 12.9 | 776 | 10 | 1.3 |
| FUS | + | 5111 | 226 | 4.4 | 14489 | 3 | 0.0 | 2744 | 1457 | 53.1 | 3807 | 0 | 0.0 |
| PIN4 | + | 1912 | 110 | 5.8 | 6001 | 1 | 0.0 | 926 | 590 | 63.7 | 4851 | 0 | 0.0 |
| RPL13A | + | 4885 | 300 | 6.1 | 5654 | 14 | 0.2 | 869 | 180 | 20.7 | 1297 | 0 | 0.0 |
| RPS24 | + | 2373 | 258 | 10.9 | 5327 | 1 | 0.0 | 1992 | 1194 | 59.9 | 3968 | 0 | 0.0 |
| SNRPN70 | + | 931 | 318 | 34.2 | 3070 | 19 | 0.6 | 1994 | 1007 | 50.5 | 3073 | 5 | 0.2 |
| 30333 | 1278 | 5.9 | 11254 | 4779 | 51.8 | ||||||||
Fig. 2. Comparison of native and DRIP-enriched SMRF-seq profiles.

(A) SMRF-seq profiles are shown for the nontemplate strand of the FUS amplicon without and with S.6 enrichment (top and bottom, respectively). Two hundred twenty-six molecules are shown in both panels corresponding to the totality of footprints in the absence of S9.6 and to a random subsample after S9.6 enrichment. The plots below depict the aggregate C to T conversion frequencies observed for each sample over R-loop-containing molecules. (B) Similar data as in previous panel for the RPS24 locus. Two hundred fifty-eight independent molecules are shown in both panels corresponding to the totality of footprints in the absence of S9.6 and to a random subsample after S9.6 enrichment.
Given that S9.6 enrichment permits a tenfold higher recovery and thus coverage, we expanded the number of loci studied using DRIP followed by SMRF-seq to a total of 24 R-loop–prone single loci (Table S1), representing a total of 78.7 kb of genomic space. These loci were chosen on the basis that they: (i) displayed clear R-loop peaks based on available S9.6-based DRIPc-seq data; and (ii) covered all genic compartments including promoter, terminal, and gene body regions as defined earlier [3]. In this manner, S9.6-based mapping results could be thoroughly tested using an independent bisulfite-based approach over a range of loci and genic locations. A total of 10,429 high-confidence R-loop footprints were generated, representing the most comprehensive characterization of R-loop formation with near nucleotide, strand-specific, and single-molecule resolution. As expected, SMRF-seq footprints were overall highly strand-specific and distributed on the nontemplate strand for transcription (Table S1). In a few instances (KAT5, RPL4, and WDR3), footprints were observed on both strands. In all three cases, the corresponding portions of these loci could be transcribed on both strands as a result of convergent or divergent transcription units, and footprints were in fact stranded with respect to transcription (Fig. S5).
To determine if the ssDNA conversion footprints were caused by an RNA:DNA hybrid, we treated an S9.6-enriched population of DNA molecules with purified RNase H prior to non-denaturing sodium bisulfite treatment. RNase H treatment caused a 98.2% reduction in the proportion of footprint-carrying molecules (Table 2). The few remaining footprint-carrying molecules displayed short, mostly random footprints that are consistent with incomplete RNase H digestion. Thus, the sensitivity of the nontemplate DNA strand to bisulfite was dependent on the formation of an RNA:DNA hybrid necessarily involving the bisulfite-inaccessible template DNA strand. Overall, SMRF-seq allows the effective characterization of single-molecule R-loop footprints at high resolution and ultra-deep coverage. In every case, R-loop footprints were codirectional with transcription; they were enriched with the S9.6 antibody and were RNase H-sensitive, as expected from the formation of genuine cotranscriptional R-loops.
Table 2.
Summary of SMRF-seq data comparing footprinted sites with or without RNase H pretreatment. The total number of independent reads and R-loop footprints (peaks) are shown broken down by strand for each locus. The overall percentages of R-loop footprints with and without RNase H pretreatment are shown below.
| Gene | Strand | No RNase H |
RNase H-treated |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Total (+) strand reads |
(+) strand peaks |
Total (−) strand reads |
(−) strand peaks |
Total (+) strand reads |
(+) strand peaks |
Total (−) strand reads |
(−) strand peaks |
||
| CALM3 | + | 1598 | 199 | 355 | 6 | 1685 | 36 | 173 | 0 |
| FUS | + | 2037 | 1084 | 2488 | 0 | 1052 | 0 | 2500 | 5 |
| SNRPN70 | + | 1450 | 767 | 2060 | 5 | 1092 | 0 | 1778 | 0 |
| MRFAP1L1 | − | 1833 | 0 | 762 | 491 | 1249 | 0 | 1018 | 0 |
| Total stranded peaks | 2541 | Total stranded peaks | 36 | ||||||
| Total stranded reads | 5847 | Total stranded reads | 4847 | ||||||
| % peaks | 43.46% | % peaks | 0.74% | ||||||
SMRF-seq, single-molecule R-loop footprinting coupled with PacBio sequencing; RNase, ribonuclease H.
R-loops typically extend for a few hundred base pairs but can reach kilobase lengths
SMRF-seq data allowed us to precisely determine the length distribution of individual R-loops over a large collection of footprints and loci. Peak lengths varied between loci (Fig. 3A) but in the majority of cases, they ranged from 100 bp, corresponding to our minimal length threshold up to 450 bp, corresponding to about three nucleosome length equivalents. When analyzed collectively, median R-loop lengths were longer for promoters (347 bp) than gene bodies (329 bp) and terminal regions (264 bp) (Fig. 3B). For a few individual loci, particularly promoters, median R-loop lengths could be significantly larger. For GADD45A, which we analyzed almost along its entire length, the R-loop median length was 700 bp, with structures covering promoter and gene body, as predicted by DRIPc-seq (Fig. S6A). Similarly, for the PIN4 promoter, the median R-loop length was 800 bp (Fig. S6B). Thus, some loci appear capable of giving rise to longer R-loop structures. For the majority of loci surveyed, a small subset of kilobase-length R-loops were recovered, contributing to a long tail in the length distribution (Fig. 3). In some instances, these structures exceeded 2 kb, with the longest contiguous footprints in the dataset reaching 2.7 kb for the PIN4 locus, equivalent to over 15 nucleosome lengths. This indicates that R-loops can infrequently extend to great lengths and remain stable enough to be detected in our assay.
Fig. 3. R-loop length distribution from DRIP-enriched SMRF-seq profiles.

(A) Peak lengths are shown as violin plots for each locus. (B) Aggregate lengths for promoter (n = 9), gene body (n = 7), and terminal (n = 8) regions are displayed as box plots and the median and maximal values are indicated below.
R-loops form overlapping molecular clusters spread over larger R-loop-prone zones
As was already evident for the FUS locus (Fig. 1) and was confirmed for all loci (Fig. 2, S2, S6), R-loops formed nonrandomly over series of molecular clusters. These clusters possessed distinct starts and stops and often showed overlapping patterns of distribution. Such overlaps were caused by the existence of multiple preferred initiation sites and by a tendency for structures to extend until variable termination hotspots once initiated. The observation that R-loops aggregate to form overlapping molecular clusters provides a clear rationale for how structures of median lengths 300 bp can, together, create large multi-kilobase size R-loop zones, as detected in population-average DRIPc-seq data [3].
Using bisulfite modification followed by DRIP and sequencing (bisDRIP-seq), Dumelie et al. (2017) reported that R-loops are restricted to the first exon of highly transcribed genes and that splicing tightly controlled R-loop boundaries, or alternatively, that R-loops formed from spliced transcripts [27]. These findings could not be verified here. When compared with fine gene structure annotations, R-loop footprints in most instances mapped to intronic sequences and often spread across intron-exon boundaries (Figs. 1 and 2, S2, S6). This argues strongly that R-loops are primarily formed on pre-mRNA before processing of the nascent transcript by splicing factors. Detailed analysis of R-loop initiation and termination regions did not reveal any correlation with splicing junctions, further suggesting that splicing does not constrain R-loop boundaries. Similarly, R-loop footprints were not limited to the first exons of genes and were readily observed over gene body and terminal regions, in accord with DRIPc-seq data. Even in instances where R-loops formed predominantly in the vicinity of the first exon, structures often spread into the first intron (Fig. S6C,D). We suspect that the discrepancies between the two studies arose from technical issues that limited the sensitivity of the bisDRIP-seq method. A comparison of R-loop maps produced by bisDRIP-seq and other DRIP-seq studies from independent labs [3,13] showed that bisDRIP-seq maps stand far apart from other datasets (Fig. S6E). bisDRIP-seq signal was heavily skewed toward promoter regions and failed to detect gene body and terminal genic R-loops. We speculate that the vigorous and lengthy shaking included in the initial step of the bisDRIP procedure [27] may have led to nicking or fragmentation of the displaced strand. This in turn, may have affected the recovery of R-loops, particularly longer R-loops, in addition to preventing the amplification and recovery of the nontemplate DNA strand in bisulfite-based applications.
R-loop forming sites are often not bound by inactive RNase H1
We took advantage of a ChIP-seq dataset for inactive RNase H1 (RChIP-seq) [10] to determine if this protein endowed with clear biochemical activity against RNA:DNA hybrids was bound to R-loop sites detected by both DRIPc-seq and SMRF-seq. RChIP signal was low to none over all gene body and gene terminal regions examined here, including all four regions examined by SMRF-seq with and without S9.6 enrichment (Fig. S7A). Thus, despite evidence of high, stranded, RNase H-sensitive DRIPc-seq signal and a deep collection of stranded, RNase H-sensitive R-loop footprints overlapping this S9.6-based signal (Figs. 1 and 2, S2, Table 1), little to no dRNase H1 binding could be detected. At promoter regions, a stronger agreement existed between datasets, although as reported [10], dRNase H1 tended to bind closer to core promoter regions and define shorter peaks than those defined by direct R-loop mapping. Thus, at some loci, dRNase H1 binding was confined to the promoter-proximal portion of the peak highlighted independently by both S9.6 mapping and bisulfite probing (Fig. S7B – GADD45A; Fig. S6A). Promoter-distal peaks highlighted independently by both DRIPc-seq and SMRF-seq were somehow not accessible to dRNase H1 binding. At other promoter loci (Fig. S7B – MALAT1), peaks did not overlap, with RChIP signal highlighting the downstream flanks of the main core promoter while DRIPc-seq and SMRF-seq both picked up a region located 1 kb further downstream, overlapping with strong GC skew. Overall, while only a limited number of loci have been analyzed by SMRF-seq, the available evidence suggests a disconnect between sites of dRNase H1 binding and sites of R-loop formation as measured by both DRIPc-seq and SMRF-seq.
RNA polymerase I-mediated R-loops form over ribosomal RNA genes
rDNA gene arrays represent the most abundantly transcribed regions of the human genome and have historically been considered a prominent source of R-loops in E. coli, yeast, and human cells [28-30]. To date, however, RNA Polymerase I-driven R-loops over rRNA genes have never been characterized at the single-molecule level. Here, we used SMRF-seq with and without S9.6 enrichment to characterize R-loops over a ~2 kb amplicon covering the 18S RNA gene. As observed for RNA polymerase II-driven genes, R-loops defined a range of discrete overlapping molecular clusters over the region (Fig. 4A). Thus, the formation of such clusters is observed for both RNA Pol I and RNA Pol II-driven R-loops, as well as R-loops generated in vitro by the bacteriophage T3 RNA polymerase [31]. The 18S RNA R-loop lengths were within the range measured for protein-coding genes with evidence for kilobase-length structures (Fig. 4B). A small but measurable proportion of reads (~4%) carried two ssDNA footprints separated by a gap (Fig. 4A). These events could reflect that two transcription complexes were engaged over the region, each one leading to one event of R-loop formation. This could be consistent with the high rates of transcription typically observed at rDNA regions. Alternatively, we cannot rule out that these multipatch structures could result from the partial collapse of an original larger R-loop. The overwhelming majority of structures profiled from RNA polymerase II-driven genes only carried one R-loop per molecule (~99%). Overall, SMRF-seq confirmed R-loop formation over rRNA genes mediated by RNA polymerase I.
Fig. 4. RNA polymerase I-mediated R-loop formation over the 18S region.

Black boxes highlight the gene arrangement over the rDNA region (top). The DRIPc-seq profile over this region is shown below for both positive and negative strands in red and blue, respectively. R-loop footprints collected with SMRF-seq over the 18S regions are shown for the nontemplate strand. Color codes are as described in Fig. 1. The aggregate C to T conversion frequency over R-loop containing molecules is graphed at the bottom. Possible “double R-loops” are highlighted on panel A by black arrows. (B) Peak length distribution for nontemplate strand 18S R-loops.
Short template strand ssDNA patches are found at R-loop junctions
R-loops are bounded by a pair of junctions between the structure per se and the surrounding dsDNA. It is possible that short patches of ssDNA exist on the template DNA strand at these junctions but this possibility has not been examined owing to the lack of deep coverage datasets. Using our peak calling algorithm, we could not identify C to T conversion tracks on the template strand even when lowering the minimal length threshold to 10 bp, suggesting that these patches, if they exist, are of limited size. Nonetheless, the identification of well-populated R-loop clusters enabled us to ask whether a subset of molecules carried ssDNA patches around these clusters' edges on the template strand (see Methods for details). In almost every single case analyzed, spanning numerous loci and clusters, we were able to identify a significant portion of template strand molecules carrying such junctional ssDNA patches (Fig. 5). Analysis of these junctions showed that they carried from one to three highly reactive cytosines, suggesting that junctions are short. Similar ssDNA patches could not be identified when the positions of the nontemplate strand clusters were shuffled (p < 0.05), establishing that these patches were unique to the edges of actual R-loop clusters. In some instances, a peak of ssDNA reactivity was observed on the template strand upstream of the annotated start of the R-loop cluster on the non-template strand (Fig. 5, top, PIN4 locus). Such events likely reflect that the R-loop cluster in fact initiated at this junction but could not be detected on the nontemplate strand owing to a lack of cytosines. Thus, template strand reactivity may in some instances allow a more precise annotation of R-loop boundaries.
Fig. 5. R-loop clusters are flanked by short template–strand junction “breathing”.

Each panel displays the C to T conversion frequencies per residue over a specific R-loop cluster at a given locus (indicated at left). The conversion frequencies on top (red) were derived from the nontemplate (displaced) strand. The number of molecules that define this cluster out of the total number of converted nontemplate strands are indicated at right. The conversion frequencies on the bottom (blue) were derived from template strand (RNA-bound) molecules carrying properly positioned template-strand junction spikes. The number of molecules carrying such junctions out of the total converted for that strand is shown at right. Background C to T conversion frequencies measured across each amplicon were first subtracted. Patches of ssDNA, reflected in a local spike-in C to T conversion, are observed on the template strand at the edges of most clusters. SMRF-seq data were obtained after S9.6 enrichment to increase depth of coverage.
It has been suggested that the time between cell lysis, DNA extraction and processing, and bisulfite probing could cause R-loop patterns to shift [27]. To test this, we modified our procedure so that bisulfite probing was conducted a mere 25 min after cell lysis without any DNA extraction or fragmentation. Loci of interest were PCR amplified immediately after nondenaturing bisulfite treatment without S9.6 enrichment. Overall, the conversion patterns observed after direct conversion were similar to those obtained after delayed conversion (Fig. S8A), suggesting that R-loop distributions did not significantly shift during DNA extraction. To further test this, we used in vitro transcription assays and profiled R-loop positions using SMRF-seq either immediately at the end of a 20 min reaction, after nucleic acid clean-up, or after further incubation of the plasmid DNA for 15, 30, 60, and 120 min at 37°C. No noticeable shift in R-loop boundaries or cluster distributions could be detected (Fig. S8B), suggesting that if R-loop junctions shift at all, shifting occurs fast.
R-loop cluster boundaries can only be partially accounted for by DNA sequence transitions
Our deep single-molecule footprint collection and the observation that R-loops are distributed among well-delineated clusters allowed us to query whether specific sequence transitions occur at the boundaries of R-loop footprints. GC skew, purine skew, and GC content have all been implicated in R-loop formation and/or elongation [2,21,32] and were accordingly analyzed. An unbiased analysis of sequence motifs (k-mers) was also conducted. For this, we devised three 100 bp windows located immediately before, across, or inside the upstream R-loop footprint boundary and measured these DNA sequence parameters across the entire collection of footprints (Fig. 6A). Three additional 100 bp windows located inside, across, and outside of the downstream R-loop footprint boundary were also used to measure sequence transitions at the end of R-loops. As shown for promoter regions, totaling 3,943 individual footprints, GC skew rose from near zero upstream of footprints to positive values inside footprints (Fig. 6B). An increase in AT skew and therefore a trend towards more purine-rich RNA sequences in R-loops was also observed (Fig. 6C, S7), consistent with the thermodynamic favorability of G/A-rich RNA:DNA hybrid sequences [33] and with R-loop mapping data from mammals to plants [2,3,10,32,34]. At the distal edge of footprints, reverse transitions back to the local average were not as prominent especially for GC skew. Only GC content showed a clear downward trend, attributable in this case to the fact that downstream R-loop boundaries are further away from core GC-rich CpG island promoters (Fig. 6D). Promoter R-loop footprints are hence characterized by upstream trends toward higher G/A skew and reduced GC content downstream, consistent with prior R-loop footprinting data [21].
Fig. 6. Analyzing sequence transitions at R-loop edges.
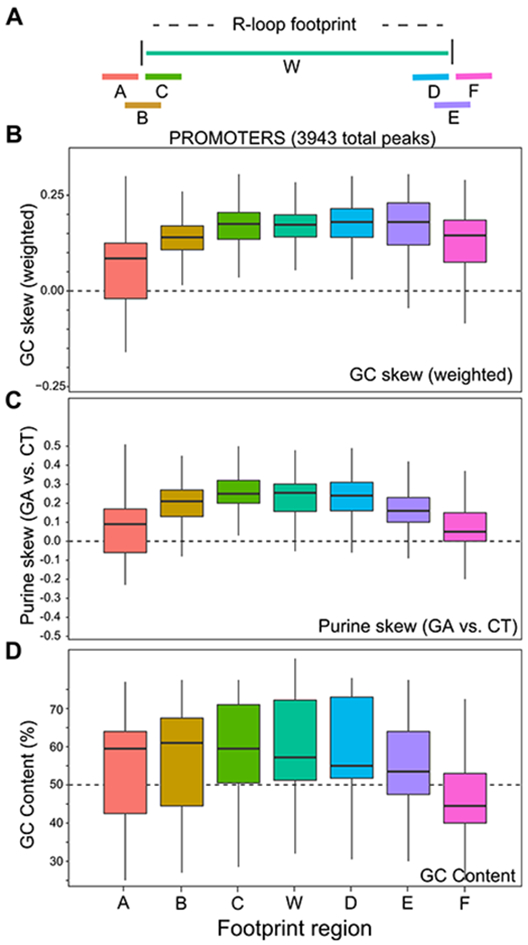
(A) Schematic of the windows used for DNA sequence analysis; each window was 100 bp long. (B-D) Box plots depicting variation in GC skew, purine skew and GC content for a broad range of promoter R-loop footprints across windows spanning R-loop proximal and distal ends.
Despite these overall trends, we noticed that even within the promoter dataset, some loci showed more pronounced DNA sequence boundary transitions than others. At the EEF2 locus, strong reciprocal shifts in GC skew were observed at both proximal and distal R-loop edges (Fig. S9A), consistent with a major role for DNA sequence in driving structure formation. At the PIN4 locus, however, GC skew overall was highly positive but did not show clear transitions at R-loop boundaries (Fig. S9B). Thus, boundaries are not strictly defined by DNA sequence parameters. For gene body and terminal regions, GC skew, GC content and purine skew transitions were consistently observed especially at the proximal edge of R-loop footprints but the trends were weaker than those described at promoters (Fig. S9C,D). Once again, some loci showed clear expected trends, whereas others did not, resulting in high variability. In addition, no significantly over-represented k-mer could be identified in any dataset other than a general tendency toward GA-rich sequences inside R-loops. Overall, this analysis revealed that while R-loop footprints delineate broadly favorable DNA sequences, the transitions into and out of an R-loop footprint are not necessarily accompanied by systematic and predictable shifts in DNA sequence properties or with any specific motif. Indeed, the R-loop DB advanced sequence-based R-loop prediction software [35] failed to predict some prominent R-loop forming regions detected both by DRIP-based methods and SMRF-seq (Fig. S10). Our data further suggest that the extent to which a set of R-loop footprints displays expected R-loop favorable DNA sequence characteristics varies from locus to locus.
One way to understand R-loop distribution patterns builds on recent advances demonstrating the key role of the interplay between DNA sequence and negative DNA superhelicity in driving R-loop formation [31]. R-loops were proposed to form because they lower the energy of the DNA fiber via the relaxation of the negative superhelical stresses and the formation of favorable RNA:DNA hybrids. Factoring in DNA topological constraints lessens the requirements for DNA sequence to drive R-loop formation and the resulting model predicted in vitro R-loop positions with high accuracy [31]. To test whether this approach can predict the position of genomic R-loops, we used the R-looper software [31] to mathematically compute the probability of R-loop formation over amplicons analyzed by SMRF-seq. A negative superhelicity of −3.5%, which is compatible with that generated by transcription [36,37], was assumed. Areas with significant R-loop probabilities were called from the probability distribution and the position of these predicted regions was compared with actual footprints in terms of direct intersect or distance. As a control, we shuffled the probability peaks 125 times independently. A similar approach was taken using predictions from R-loop DB. Both approaches performed well over promoter regions, which often possess strong favorable sequence signatures, with predictions being 2.8 to 8.8 higher (R-looper) and 2.9 to 6.1 times (R-loop DB) more likely to match an R-loop footprint than expected by chance (p < 0.008) (Table 3). Predictions remained strong over gene body and terminal region using the topology-enabled R-looper tool, with odds of intersecting actual R-loop 6.8 to 15.9 times higher than at random. The sequence-based R-loop DB tool, however, performed poorly on a number of these loci, lowering its overall predictive power. These results suggest that R-loops cannot simply be predicted using DNA sequence characteristics alone. Instead, factoring in DNA topology significantly boosted our ability to predict R-loops especially outside of promoter regions.
Table 3.
Comparison of R-loop prediction algorithms. The ability of R-looper and R-loop DB algorithms to predict R-loop clusters was analyzed for a range of footprinted loci. Values correspond to the observed over expected ratios for direct intersect or nearest distance. Expected values were generated by shuffling predictions along the amplicon 125 times. Distances or intersects were calculated either to the start (100 bp), end (100 bp) of each footprint or with the whole footprint (middle position for distance). A value of zero indicates that no match was observed or that no prediction was made. ‘na’ indicates that this region was not computed to possible R-loops.
| R-looper |
Distance |
Intersect |
R-loop DB |
Distance |
Intersect |
Location | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Whole | Start | End | Whole | Start | End | Gene | Whole | Start | End | Whole | Start | End | |
| BTF3 | 4.7 | 4.7 | 3.7 | 12.5 | 35.9 | 15 | BTF3 | 4.7 | 4.7 | 4.1 | 5.0 | 3.0 | 4.9 | PROMOTER |
| CNBP | 1.5 | 1.7 | 1.7 | 0.0 | 0.0 | 0.0 | CNBP | 4.3 | 4.5 | 4.3 | 3.7 | 2.9 | 3.8 | PROMOTER |
| EEF2 | 2.6 | 1.7 | 3.1 | 0.1 | 0.0 | 1.0 | EEF2 | 4.7 | 4.7 | 4.3 | 8.1 | 7.4 | 7.5 | PROMOTER |
| GADD45A | 3.7 | 2.4 | 2.6 | 4 | 4.8 | 8.7 | GADD45A | 4.4 | 4.1 | 2.5 | 4.4 | 3.4 | 3.2 | PROMOTER |
| MTERFD3 | 4.2 | 2.7 | 4 | 2.4 | 0 | 2.1 | MTERFD3 | 4.8 | 4.6 | 2.1 | 4.8 | 18.9 | 0.8 | PROMOTER |
| PIN4 | 3.7 | 2.6 | 2.5 | 6.5 | 21.1 | 8.7 | PIN4 | 2.9 | 2.4 | 2.7 | 4.4 | 7.3 | 2.9 | PROMOTER |
| RPL4 | 3.2 | 3.9 | 2 | 0 | 0 | 0 | RPL4 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | PROMOTER |
| Average | 3.4 | 2.8 | 2.8 | 3.6 | 8.8 | 5.1 | Average | 3.7 | 3.6 | 2.9 | 4.3 | 6.1 | 3.3 | PROMOTER |
| CALM3 | 4.1 | 3.9 | 3.4 | 8.4 | 9 | 12.9 | CALM3 | 4.1 | 4.3 | 4.7 | 4.6 | 5.4 | 5.7 | GENEBODY |
| FUS | 4.4 | 3.9 | 4.5 | 8.8 | 19.1 | 29.2 | FUS | 3.3 | 3.2 | 3.8 | 5.0 | 2.4 | 6.3 | GENEBODY |
| KAT5 | 2.8 | 3.1 | 2.3 | 5.8 | 13.3 | 6.8 | KAT5 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | GENEBODY |
| MRFAP1L1 | 4.4 | 3.9 | 3.6 | 10 | 2.7 | 4.2 | MRFAP1L1 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | GENEBODY |
| POGK | 4.2 | 4.2 | 3 | 3.5 | 28.9 | 2 | POGK | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | GENEBODY |
| RPL13A | 4.6 | 4.2 | 4.3 | 14.2 | 20.5 | 36.3 | RPL13A | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | GENEBODY |
| RPS24 | 3.9 | 2.2 | 4.1 | 9.6 | 1 | 19.8 | RPS24 | 3.8 | 2.1 | 4.0 | 7.8 | 0.0 | 18.7 | GENEBODY |
| Average | 4.1 | 3.6 | 3.6 | 8.6 | 13.5 | 15.9 | Average | 1.6 | 1.4 | 1.8 | 2.5 | 1.1 | 4.4 | GENEBODY |
| CEP95 | 4.7 | 3.8 | 3.3 | 8.8 | 18.5 | 3.9 | CEP95 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | TERMINAL |
| MORF4L2 | 2.8 | 3 | 4 | 4.7 | 24.9 | 0 | MORF4L2 | 4.9 | 4.9 | 4.4 | 6.1 | 11.5 | 2.9 | TERMINAL |
| MYLIP | 4 | 3.7 | 4 | 6.9 | 3.6 | 12.1 | MYLIP | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | TERMINAL |
| SNRPN70 | 4 | 3.2 | 4.2 | 6.2 | 8.2 | 24 | SNRPN70 | 3.6 | 2.1 | 4.0 | 4.2 | 12.4 | 1.7 | TERMINAL |
| TRIM33 | 4.5 | 3.3 | 4.3 | 7.5 | 7.1 | 8.1 | TRIM33 | na | na | na | na | na | na | TERMINAL |
| Average | 4.0 | 3.4 | 4.0 | 6.8 | 12.5 | 9.6 | Average | 2.1 | 1.8 | 2.1 | 2.6 | 6.0 | 1.2 | TERMINAL |
All values represent observed/expected ratios.
Observed values were computed by measuring the distances or %intersects between predictions (R-looper and RloopDB) and actual SMRF-seq footprints.
SMRF-seq, single-molecule R-loop footprinting coupled with PacBio sequencing.
Expected values were computed similarly after shuffling predictions 125 times.
Distances or intersects were calculated either to the start (100 bp), end (100 bp) of each footprint or with the whole footprint (middle position for distance).
Discussion
Unlike DRIP-based approaches that query R-loops based on their RNA:DNA hybrid moiety, SMRF-seq exploits the exquisite reactivity of ssDNA to bisulfite-mediated cytosine deamination [20] to query R-loops through their displaced ssDNA strand. Compared with previous low-throughput, labor intensive, nondenaturing bisulfite mapping approaches, SMRF-seq offers orders of magnitude improvement in efficiency and permits R-loop footprinting on kilobases-long amplicons at single-molecule resolution and ultra-deep coverage. SMRF-seq can be conducted independently of S9.6 enrichment using only native PCR primers and therefore represents an unbiased orthogonal readout for R-loop formation. SMRF-seq nonetheless displays a few limitations. First, the recovery of R-loop molecules is limited by the frequency at which R-loops form in the genome. In agreement with prior DRIP-qPCR data [3], R-loop–containing molecules typically amounted to 1–10% of the total molecule pool. This can be improved about ten-fold by first enriching for R-loops using the S9.6 antibody (Table 1). We showed here that S9.6 enrichment does not significantly distort the patterns of single-molecule R-loops recovered using SMRF-seq. S9.6 pre-enrichment therefore allows to expand the throughput of the method, although it is not required. Second, SMRF-seq, such as DRIP-seq, requires initial DNA extraction and therefore samples R-loops ex vivo. One concern with ex vivo approaches is that spurious R-loops may form upon invasion of RNAs into chromosomal DNA during nucleic acid extraction. As previously pointed out [38], this is unlikely given that such strand invasion activity would require considerable energy to melt the duplex and necessitate homology recognition to both identify target sites and stabilize weak intermediates. In addition, regions susceptible to strand separation under negative superhelical constraints are primarily AT-rich [39-41]. By contrast, R-loops profiled ex vivo are predominantly found in GA-rich regions that are least susceptible to superhelical strand opening. Thus, there is a strong disconnect between loci predicted to form spurious R-loops and where R-loops are experimentally detected. Furthermore, the predicted lengths of superhelically strand-separated regions typically range from 30 to 60 bp when the median length of individual genomic R-loops recovered in SMRF-seq is 300 bp. There is therefore a strong disconnect in the predicted sizes of hypothetical spurious R-loops and observed R-loops. The possibility that an R-loop may grow from a short “seed” by spontaneous branch migration is also unlikely given strong steric issues [42]. In addition, the invading ssRNA is most likely highly folded through secondary structure formation and owing its short persistence length. Thus, extending an R-loop from a highly folded RNA will require additional energy to unfold a tight random coil and denature any secondary structures present. Such a process is expected to be highly unfavorable. In addition to these arguments, R-loop formation measured by DRIP is exquisitely sensitive to perturbations of nascent transcription, not of total RNA abundance. Inhibition of transcription elongation triggers the loss of promoter R-loops within a few minutes [3], long before steady-state mRNA pools are reduced. This data is at odds with a model of spurious R-loop formation from pools of mRNA transcripts and instead supports that they form cotranscriptionally in vivo, as is clearly know in vitro [2,43,44]. Finally, one of the key findings from this study is that R-loops repeatedly occur over molecular clusters defined by clear boundaries. It is difficult to envision how these precise clusters could result from random branch migration events following spurious RNA strand invasion events. We therefore view a de novo R-loop formation mechanism ex vivo as insufficient to explain the characteristics of the signals recovered in DRIP-seq and SMRF-seq. As acknowledged [38], one weakness of ex vivo approaches is that they could lead to an underestimation of R-loop loads owing to the dissolution of short, unstable R-loops during the DNA fragmentation step [31]. In addition, we cannot formally rule out that the precise junctions of R-loop structures may shift slightly during the process, although the available evidence demonstrates that these junctions are stable over long incubations (Fig. S8). Overall, SMRF-seq is technically straightforward and can be easily deployed in a wide range of conditions and organisms. The amplification of specific loci prior to sequencing permits lower sample sizes, expanding its possible range of application compared with DRIP-based methods. SMRF-seq will allow users to address R-loop distributions and formation mechanisms with unparalleled depth on single DNA molecules.
The dataset presented here surveyed an array of 24 R-loop-prone loci representative of the diversity of R-loop-prone loci observed previously in DRIP-based approaches. These loci include CpG island promoters that are universally understood to be favorable for R-loops and a range of gene body and terminal regions that have not necessarily been captured by other R-loop mapping strategies [10,27]. The strong agreement between SMRF-seq and DRIPc-seq at all tested loci provides strong confidence that DRIP-based methods accurately report on R-loop formation.
Based on the largest collection of R-loop footprints to date, SMRF-seq establishes that individual R-loops most often range from 200 to 500 bp in length (Fig. 3), making them an order of magnitude larger than most other non-B DNA structures such as single-stranded bubbles, B/Z transitions, or triplexes that all typically range from 20 to 50 bp [39]. For most loci, the accretion of individual R-loops defined discrete, nonrandom clusters. R-loop clusters themselves often overlapped and defined larger, kilobase-size, R-loop zones. The remarkable agreement between aggregate SMRF-seq signal and DRIPc-seq data suggest that the often kilobase-sized peaks recovered in population-average DRIP-based methods [3] are caused by the piling up of numerous smaller R-loop clusters. This provides a clear rationale for how large R-loop zones can be created by otherwise much smaller individual structures.
SMRF-seq also establishes that genuine three-stranded R-loops, and not simply two-stranded RNA:DNA hybrids, form in the human genome. This conclusion is based on the clear evidence that the nontemplate and template strands produce highly asymmetric bisulfite sensitivity patterns: high conversion rates only exist on the nontemplate strand. This argues that while the nontemplate strand is largely single-stranded and accessible, the template strand is engaged in contiguous base-pairing interactions and chemically inaccessible (save for short conversion patches located at the edges of R-loop clusters). The observation that RNase H treatment abolishes the bisulfite reactivity of the nontemplate DNA strand indicates that the ssDNA character of that strand depends on the formation of an RNA:DNA hybrid between the template DNA strand and the nascent transcript. SMRF-seq consequently brings conclusive evidence that three strands are involved in R-loop structures. The agreement between SMRF-seq and DRIPc-seq data at all tested loci, by extension, suggests that the large majority of S9.6-based DRIP signals also correspond to three-stranded R-loop structures.
The validity of ex vivo R-loop maps as determined by DRIP- and SMRF-based approaches is underscored by a large body of work in which R-loop formation was studied using in vitro transcription assays. R-loop formation in these assays occurs cotranscriptionally in cis [2,43,44]. Every locus known to form genomic R-loops ex vivo tested so far also formed R-loops in vitro in a stranded, RNase H-sensitive, manner [2,3,19,24,45-47]. In every case where in vitro R-loop positions were analyzed at the single-molecule level after nondenaturing bisulfite probing, R-loops tended to initiate and terminate at specific positions, forming clusters [31,48-51]. Thus, a propensity toward clustering is a hallmark of both genomic R-loops mapped ex vivo ([2,19,21,23,24], this study) and cotranscriptional R-loops formed in vitro. In vitro R-loops analyzed by SMRF-seq often initiated far downstream of the transcription start site (400 to 600 bp) [31], similarly to what is commonly observed in DRIP-seq analysis of genomic promoter R-loops [3]. In addition, in vitro R-loop clusters responded to the favorability of the underlying DNA sequence and to the superhelical state of DNA, consistent with the current understanding of the physico-chemical parameters driving R-loop formation [31]. Similarly, genomic R-loops mapped ex vivo can be best understood through models that incorporate the effects of both DNA sequence and DNA topology. These lines of evidence provide independent support to the notion that R-loops observed ex vivo possess the characteristics expected of genuine cotranscriptional R-loops. A recent analysis of R-loop positions after in vitro transcription by direct visualization on single DNA molecules using atomic force microscopy was in complete agreement with SMRF-seq results [51]. The remarkable consensus between multiple orthogonal approaches provides a strong benchmark for the field.
RChIP-seq was recently used to map R-loops [10] based on the assumption that dRNase H1 can freely bind to all R-loops in cells. Gene body and terminal sites shown here to form R-loops using DRIPc-seq and SMRF-seq show little to no dRNase H1 binding. Although the sources of these discrepancies remain to be determined, we note that numerous proteins have been reported to bind R-loops in vivo [52,53], possibly limiting their accessibility to RNase H1. Alternatively, it is possible that RNase H1 is targeted to specific R-loop subsets, particularly over promoter regions. Overall, analysis of SMRF-seq footprints for more than ten thousand individual R-loops reinforces a model whereby R-loops form cotranscriptionally upon reinvasion of the nascent pre-mRNA before splicing occurs. R-loop formation was clearly observed outside first exons as shown for the FUS, SNRPN70, RPL13A, and RPS24 gene body and terminal regions that were footprinted without any S9.6 enrichment (Figs. 1 and 2 and S2). R-loop footprints were also observed at every gene body and terminal region analyzed by SMRF-seq after S9.6 enrichment, in full agreement with bulk DRIP-based findings from human cells [3,15,54]. Even in cases when R-loops occurred over or near the first exon of a gene, they were not limited to this region as evidenced by spreading downstream in the gene body (Fig. S6). Thus, R-loops are not constrained to the first exon in human cells, in agreement with findings from R-loop mapping data in mouse and Arabidopsis thaliana [3,9,32]. Furthermore, we could not obtain evidence that splicing controls R-loop boundaries. R-loops most often initiated in introns and frequently spanned (multiple) exon-intron boundaries, arguing they originated from unspliced transcripts. Similarly, we did not observe any colocalization of R-loop boundaries with splice junctions beyond what is expected by chance. Overall, the agreement between two orthogonal approaches, SMRF-seq and DRIP-based methods, strongly supports the view that R-loops can form cotranscriptionally anywhere along a gene provided R-loop permissive factors (e.g. DNA sequence and topology) exist. In that view, splicing factors and other RNA binding proteins may regulate the likelihood of interaction of the nascent pre-mRNAs with the DNA template as demonstrated in yeast [55] but not the precise patterns of R-loop formation.
One of the key findings of this study is the demonstration that genomic R-loops, similarly to cotranscriptional R-loops formed in vitro [31], aggregate over well-defined molecular clusters. SMRF-seq provided an opportunity to determine the role of DNA sequence in driving cluster formation at higher resolution than previously possible. In general, R-loop footprints coincided with R-loop favorable sequence characteristics (GA-rich), as expected. However, the transitions into and out of an R-loop were often not clear cut, suggesting that R-loops cannot be simply predicted by stereotypical sequence transitions. Patterns of R-loop formation could instead be better understood when we considered DNA topology as an additional factor driving R-loops. The relaxation of the surrounding DNA fiber by R-loop formation is indeed an important factor favoring R-loops [31], alleviating the need for favorable RNA:DNA base-pairing. If a region experiences low to mild supercoiling stress, R-loop favorable DNA sequence characteristics will play a leading role in allowing R-loops to form. This situation most likely applies to promoter regions, which carry conserved R-loop–favorable sequence characteristics [56] and experience only limited transcription-driven superhelical stress, given their location early in the transcription unit. By contrast, if a region experiences moderate to high topological stress, the role of DNA sequence favorability as a driver of R-loop stability will be reduced in favor of DNA relaxation [31,57]. Given that transcription-driven topological stress builds up with the length of DNA transcribed [58], the contribution of DNA topology to R-loop formation may be higher in gene body and terminal regions. This is consistent with reports that R-loop propensity equally correlates with gene length and gene expression [3] and that loss of topoisomerase I results in R-loop accumulation in long, highly expressed, genes for which superhelical stress dissipation is constrained [30]. We therefore suggest that loci for which prominent R-loop hotspots form over regions with moderately or poorly favorable sequence characteristics, such as gene body or terminal gene regions, may be driven primarily by the need to relax topological stresses. Importantly, these observations imply that strictly DNA sequence–based approaches are unlikely to support accurate and robust R-loop predictions. DNA topology-enabled approaches are instead required and early indications support that these approaches perform better for gene body and terminal regions (Table 3). Accurate, global R-loop prediction will require an intimate knowledge of the intensity and distribution of DNA topology.
Material and Methods
Nondenaturing single-molecule R-loop footprinting coupled to PacBio sequencing
Cell culture
NTERA-2 cells (ATCC® CRL-1973) were used for all footprinting experiments and grown under standard conditions in a humidified incubator at 5% CO2 in DMEM high glucose media supplemented with 10% FBS. Cells were grown to 80–90% confluence and split to 50% in new media and harvested 16 h later.
Nucleic acid isolation, fragmentation, and immunoprecipitation
DNA extraction was carried out as described for standard DRIP-seq protocol [38] except that the proteinase K incubation was shortened to 2 h instead of overnight. Restriction enzymes (REs) were selected to fragment genomic DNA around target regions so that fragments of not more than 15 kb were generated. We used 80–120 units total of REs and incubated overnight at 37°C followed by a DNA purification step using 1X AMPure beads. This digested template was either sodium bisulfite treated directly to footprint R-loops or after immunoprecipitation using the anti-RNA:DNA hybrid S9.6 antibody [5] as per the standard DRIP protocol [38].
In vitro transcription assays
In vitro transcription assays were conducted as described previously on the supercoiled R-loop-prone pFC8 plasmid [2]. Reactions were incubated for 20 min and nucleic acids were subsequently purified by phenol and chloroform extraction followed by ethanol precipitation. DNAs were resuspended in TE and further incubated at 37°C for up to 2 h. Aliquots were withdrawn for R-loop characterization using SMRF-seq immediately after transcription, after nucleic acids clean up, and 15, 30, 60, and 120 min after incubation.
Nondenaturing bisulfite conversion
Samples were treated with sodium bisulfite in accordance with the Zymo EZ DNA Methylation-Lightning kit (PN D5030) following the manufacturer's instructions except for a few critical modifications to permit the probing of intrinsically single-stranded cytosines under nondenaturing conditions. In brief, the DNA was not denatured before bisulfite treatment and treatment with the conversion reagent was performed at 37°C for 2 h with gentle rotation. Desulfonation and sample recovery were performed as instructed. As a positive control, a linearized denatured plasmid was spiked-in with genomic samples and treated accordingly. All recovered DNA molecules were heavily converted and the overall C to T conversion efficiency was 86% (Fig. S11). This indicates that bisulfite treatment under these conditions was efficient and suitable for R-loop detection.
Direct nondenaturing bisulfite conversion was performed based on procedure B in the Zymo EZ DNA Methylation-Direct kit (PN D5020) with a few modifications. Briefly, samples containing approximately 30,000 cells were pelleted and resuspended in 1 mL 1X DPBS, lysed with 1X final concentration of M-Digestion buffer, and incubated with Proteinase K (included in the kit) for 25 min at 37°C. The digested sample was then directly treated with sodium bisulfite at 37°C for 3.5 h with gentle rotation. The following steps were performed as per manufacturer's instructions.
Site-specific PCR amplification
We identified a test set of 24 R-loop forming regions (Table S1). Amplicons ranging from 2 to 4 kb were designed to capture not only the presumptive R-loop peak but also non–R-loop flanks. Primers were designed using Primer3 (bioinfo.ut.ee/primer3-0.4.0) (Table S2). PCR reactions using ThermoFisher PhusionU DNA polymerase (ThermoFisher PN F555S) were optimized to produce long-range, high fidelity single-band products (conditions available upon request). Standard PCR reaction included 1X PCR buffer, 0.2 mM dNTPs, 0.8 μM each forward and reverse primers, 5–30 ng DNA template, 0.02 U/μL Phusion U DNA polymerase, 1 M Betaine (optional), and PCR grade water to a 30 μL final volume. PCR cycling was as follows: (1) Initial denaturing at 98°C for 30 s; (2) 30–35 cycles of: (2a) denature at 98°C for 10 s, (2b) anneal at optimized temperature for 30 s, (2c) extend at 72°C for 2.5 min; (3) Final extension at 72°C for 5 min; (4) Hold at 4°C infinitely. All PCR products were purified using 1X Ampure beads.
SMRTbell library construction
We used the PacBio RSII system to achieve long-read, single-molecule resolution sequencing of R-loop footprints. We generated libraries by pooling nonoverlapping amplicons (less than 20 products per run) adding equal amounts for each. Starting with 1–2 μg of PCR products, pooled samples were concentrated using 1X Ampure bead wash. Libraries were built following the “Procedure & Checklist – 2 kb Template Preparation and Sequencing” protocol (PN 001-143-835-08) from PacBio with a few modifications. No prior DNA damage repair step was done. AMPure bead wash steps were carried out using 0.8X concentration. Ligation was carried out for 1 h at 25°C. SMRTbell libraries were quantified and size confirmation carried out by either by gel electrophoresis or Agilent Genomic's 2100 Bioanalyzer. Libraries were sequenced on a PacBio RSII instrument with 6-h movie times.
R-loop profiling using DRIPc-seq
DRIPc-seq was applied to NTERA-2 cells as described [3] except a Ribonuclease A pretreatment (10 μg/ml for 30 min at 37°C) was applied to the extracted nucleic acids before S9.6 immunoprecipitation.
Computational data processing
CCS generation. Subreads of read quality at least 90% were further processed into CCS using PacBio SMRT Analysis pipeline (ConsensusTools.sh) with a minimum pass filter of 3. Enforcing a 3x minimal pass together with additional steps in the following context allowed us to have a >95% read accuracy.
Duplicate read removal. To avoid oversampling from potential PCR duplicates, we used dedupe2.sh from package BBMap V37.90 (https://sourceforge.net/projects/bbmap/) with default parameters except for mid = 98, nam = 4, k = 31, and e = 30. An average of 43% reads (254,951 out of 592,444 total CCS reads) were removed as duplicates in the combined datasets.
Gargamel computational pipeline
The Gargamel pipeline (available at https://github.com/srhartono/footLoop) allow user to map reads, assign strand, call SMRFs as peaks of C to T conversion, perform clustering on SMRFs, and visualize the data.
Read mapping. Reads were mapped to the hg19 human genome reference focusing on their respective amplicon regions using Bismark v0.13.1 [26] including a 10 bp buffer off their beginning and end positions. Bismark default settings were used except for a slightly relaxed minimum score threshold (–score_min of L,0,−0.3 instead of L,0,−0.2). Truncated reads shorter than 95% of their expected length were discarded. Altogether, the stringent requirements imposed for circular consensus, duplicate removal, mapping, and size, ensured the selection of very high-quality reads.
Strand assignment. For each read, we assigned strand based on their conversion patterns. Reads with insufficient conversions (C- > T < 6 and G->A < 6) could not be assigned a strand. Likewise, if the number of C- > T conversions was within±10% of G- > A conversions on a read, then the strandedness was considered as unknown. Such reads represented less than ~5% of the total pool. Otherwise, reads with predominant C to T or G to A conversions were assigned as nontemplate or template strand, respectively. Ambiguous regions carrying indels owing to PacBio sequencing errors were masked (including a 5 bp buffer around the indel) so as not to distort the conversion frequency calculation. Some individual loci consistently resulted in more reads mapping to one strand than to the other, regardless of which strand carried R-loop footprints, most likely reflecting PCR strand biases. Considering all tested loci together, reads were approximately equally distributed to each strand.
Peak calling. A threshold-based sliding window method was used to call tracts of C to T conversion referred to as R-loop peaks. The windows spanned 20 consecutive cytosines and were moved across each read one cytosine at a time. For each window, we calculated the C to T conversion frequency and called a window R-loop positive if a minimum of 55% cytosines were converted. Positive windows were further extended if neighboring windows also satisfied the 55% threshold. Upon encountering a window with conversion frequency less than the threshold, the peak was terminated and its boundaries recorded. In this study, we imposed that each positive peak should be at least 100 bp in length. We tested a combination of window sizes (10, 15, 20, and 30 cytosines), conversion thresholds (35%, 55%, 65%, 70%, 75%, and 80%), and minimal peak sizes (30 and 100 bp); the results were qualitatively similar except that more positive peaks were recovered for less stringent conditions. A window size of 20 cytosines, minimal C to T conversion of 55%, and a minimal length of 100 bp permitted a good combination of specificity and sensitivity. When presenting aggregate C to T conversion frequency plots, the frequency was calculated on the subpopulation of molecules that were determined to carry an R-loop footprint. For samples directly treated with bisulfite after a short proteinase K digestion, peak calling thresholds were modified to 35% conversion frequency with 30 bp minimum peak length.
Clustering. For each gene, R-loop peaks were clustered using their start and stop coordinates using k-means clustering. K was determined automatically by minimizing intracluster distances, iterating until minimum within-group distance for each cluster was at most three.
Reproducibility and location analysis. For each locus, we combined reads from all replicates and generated clusters as described previously. For each biological replicate, we then quantified the distribution of that replicate's reads across these predefined clusters. If independent replicates are sampling from the same overall biological distribution, the expectation is that read distributions across replicates will be similar. Reproducibility was therefore measured by calculating Pearson correlation coefficients applied to the read distribution across clusters between replicates. As a control, we shuffled the position of each read around the amplicon region (keeping length information intact) and assigned the shuffled reads to the same set of predefined clusters. A shuffled read was considered to belong to a specific cluster if its start and end positions fell within±100 bp of the mean start and end of all experimental reads in that cluster. Reads that could not be assigned to any experimentally determined cluster were assigned to an extra “shuffled” cluster. The Pearson correlation coefficients between experimental and shuffled reads were calculated across clusters as described previously.
Identification of template-strand ssDNA patches at the edge of R-loop clusters
C to T conversion frequencies were measured on the template strand for more than a 20 bp window around the edges of R-loop footprint clusters. Molecules with conversion frequencies >5% were selected. To determine if these conversion events were a product of random DNA breathing or reflected the presence of a bisulfite-susceptible ssDNA patch on certain molecules, we subtracted background C to T conversion frequencies for all nontemplate strand molecules. This was performed either by subtracting the average background C to T conversion frequency across the entire amplicon or the average C to T conversion frequency for each position along the amplicon, with similar results. As a negative control, we determined if randomly assigned clusters outside of actual footprint regions would show C to T conversion patches at their edges. Shuffled clusters were not associated with significant template-strand ssDNA patches.
Supplementary Material
Acknowledgments
The authors thank members of the Chedin lab for critical feedback and useful discussions. The authors also thank Dr. Shaheen Mohammad for assistance with in vitro transcription assays and Tonia Brown for help with editing.
Funding
This work was supported by National Institutes of Health Grant [R01-GM120607 to F.C.]; supported in part by National Science Foundation Graduate Research Fellowship Grant [1650042 to M.M.]; and by National Institute of General Medical Sciences Biomolecular Technology Predoctoral Training Program Grant [T32-GM008799 to M.M.].
Footnotes
Availability of data and materials
SMRF-seq and DRIPc-seq datasets have been uploaded to the NCBI GEO databased under accession number GSE130726. The Gargamel computational pipeline for read mapping, strand assignment, peak calling, clustering, and data visualization is available from GitHub (https://github.com/srhartono/footLoop).
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflicts of interest
The authors declare no competing interests.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jmb.2020.02.014.
References
- [1].Santos-Pereira JM, Aguilera A, R loops: new modulators of genome dynamics and function, Nat. Rev. Genet 16 (2015) 583–597. [DOI] [PubMed] [Google Scholar]
- [2].Ginno PA, Lott PL, Christensen HC, Korf I, Chedin F, R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters, Mol. Cell 45 (2012) 814–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Sanz LA, Hartono SR, Lim YW, Steyaert S, Rajpurkar A, Ginno PA, et al. , Prevalent, dynamic, and conserved R-loop structures associate with specific epigenomic signatures in mammals, Mol. Cell 63 (2016) 167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Phillips DD, Garboczi DN, Singh K, Hu Z, Leppla SH, Leysath CE, The sub-nanomolar binding of DNA-RNA hybrids by the single-chain Fv fragment of antibody S9.6, J. Mol. Recogn 26 (2013) 376–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Boguslawski SJ, Smith DE, Michalak MA, Mickelson KE, Yehle CO, Patterson WL, et al. , Characterization of monoclonal antibody to DNA.RNA and its application to immunodetection of hybrids, J. Immunol. Methods 89 (1986) 123–130. [DOI] [PubMed] [Google Scholar]
- [6].Skourti-Stathaki K, Proudfoot NJ, A double-edged sword: R loops as threats to genome integrity and powerful regulators of gene expression, Genes Dev. 28 (2014) 1384–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Costantino L, Koshland D, The yin and yang of R-loop biology, Curr. Opin. Cell Biol 34 (2015) 39–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Skourti-Stathaki K, Proudfoot NJ, Gromak N, Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination, Mol. Cell 42 (2011) 794–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chen PB, Chen HV, Acharya D, Rando OJ, Fazzio TG, R loops regulate promoter-proximal chromatin architecture and cellular differentiation, Nat. Struct. Mol. Biol 22 (2015) 999–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chen L, Chen JY, Zhang X, Gu Y, Xiao R, Shao C, et al. , R-ChIP using inactive RNase H reveals dynamic coupling of R-loops with transcriptional pausing at gene promoters, Mol. Cell 68 (2017) 745–757 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Boque-Sastre R, Soler M, Oliveira-Mateos C, Portela A, Moutinho C, Sayols S, et al. , Head-to-head antisense transcription and R-loop formation promotes transcriptional activation, Proc. Natl. Acad. Sci. U. S. A 112 (2015) 5785–5790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Paulsen RD, Soni DV, Wollman R, Hahn AT, Yee MC, Guan A, et al. , A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability, Mol. Cell 35 (2009) 228–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hamperl S, Bocek MJ, Saldivar JC, Swigut T, Cimprich KA, Transcription-replication conflict orientation modulates R-loop levels and activates distinct DNA damage responses, Cell 170 (2017) 774–786 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sollier J, Cimprich KA, Breaking bad: R-loops and genome integrity, Trends Cell Biol. 25 (2015) 514–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Stork CT, Bocek M, Crossley MP, Sollier J, Sanz LA, Chedin F, et al. , Co-transcriptional R-loops are the main cause of estrogen-induced DNA damage, Elife 5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Huertas P, Aguilera A, Cotranscriptionally formed DNA: RNA hybrids mediate transcription elongation impairment and transcription-associated recombination, Mol. Cell 12 (2003) 711–721. [DOI] [PubMed] [Google Scholar]
- [17].Richard P, Manley JL, R loops and links to human disease, J. Mol. Biol 429 (2017) 3168–3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hartono SR, Malapert A, Legros P, Bernard P, Chedin F, Vanoosthuyse V, The affinity of the S9.6 antibody for double-stranded RNAs impacts the accurate mapping of R-oops in fission yeast, J. Mol. Biol 430 (2018) 272–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Yu K, Chedin F, Hsieh CL, Wilson TE, Lieber MR, R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells, Nat. Immunol 4 (2003) 442–451. [DOI] [PubMed] [Google Scholar]
- [20].Clark SJ, Harrison J, Paul CL, Frommer M, High sensitivity mapping of methylated cytosines, Nucleic Acids Res. 22 (1994) 2990–2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Huang FT, Yu K, Hsieh CL, Lieber MR, Downstream boundary of chromosomal R-loops at murine switch regions: implications for the mechanism of class switch recombination, Proc. Natl. Acad. Sci. U. S. A 103 (2006) 5030–5035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Cerritelli SM, Crouch RJ, Ribonuclease H: the enzymes in eukaryotes, FEBS J. 276 (2009) 1494–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Huang FT, Yu K, Balter BB, Selsing E, Oruc Z, Khamlichi AA, et al. , Sequence dependence of chromosomal R-loops at the immunoglobulin heavy-chain Smu class switch region, Mol Cell Biol 27 (2007) 5921–5932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kao YP, Hsieh WC, Hung ST, Huang CW, Lieber MR, Huang FT, Detection and characterization of R-loops at the murine immunoglobulin Salpha region, Mol. Immunol 54 (2013) 208–216. [DOI] [PubMed] [Google Scholar]
- [25].Loomis EW, Eid JS, Peluso P, Yin J, Hickey L, Rank D, et al. , Sequencing the unsequenceable: expanded CGG-repeat alleles of the fragile X gene, Genome Res. 23 (2013) 121–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Krueger F, Andrews SR, Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications, Bioinformatics 27 (2011) 1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Dumelie JG, Jaffrey SR, Defining the location of promoter-associated R-loops at near-nucleotide resolution using bisDRIP-seq, Elife 6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Masse E, Phoenix P, Drolet M, DNA topoisomerases regulate R-loop formation during transcription of the rrnB operon in Escherichia coli, J. Biol. Chem 272 (1997) 12816–12823. [DOI] [PubMed] [Google Scholar]
- [29].El Hage A, French SL, Beyer AL, Tollervey D, Loss of Topoisomerase I leads to R-loop-mediated transcriptional blocks during ribosomal RNA synthesis, Genes Dev. 24 (2010) 1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Manzo SG, Hartono SR, Sanz LA, Marinello J, De Biasi S, Cossarizza A, et al. , DNA Topoisomerase I differentially modulates R-loops across the human genome, Genome Biol. 19 (2018) 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Stolz R, Sulthana S, Hartono SR, Malig M, Benham CJ, Chedin F, Interplay between DNA sequence and negative superhelicity drives R-loop structures, Proc. Natl. Acad. Sci. U. S. A 116 (2019) 6260–6269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Xu W, Xu H, Li K, Fan Y, Liu Y, Yang X, et al. , The R-loop is a common chromatin feature of the Arabidopsis genome, Native Plants 3 (2017) 704–714. [DOI] [PubMed] [Google Scholar]
- [33].Huppert JL, Thermodynamic prediction of RNA-DNA duplex-forming regions in the human genome, Mol. Biosyst 4 (2008) 686–691. [DOI] [PubMed] [Google Scholar]
- [34].Ginno PA, Lim YW, Lott PL, Korf I, Chedin F, GC skew at the 5' and 3' ends of human genes links R-loop formation to epigenetic regulation and transcription termination, Genome Res. 23 (2013) 1590–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Jenjaroenpun P, Wongsurawat T, Sutheeworapong S, Kuznetsov VA, R-loopDB: a database for R-loop forming sequences (RLFS) and R-loops, Nucleic Acids Res. 45 (2017) D119–D127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kouzine F, Gupta A, Baranello L, Wojtowicz D, Ben-Aissa K, Liu J, et al. , Transcription-dependent dynamic supercoiling is a short-range genomic force, Nat. Struct. Mol. Biol 20 (2013) 396–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Naughton C, Avlonitis N, Corless S, Prendergast JG, Mati IK, Eijk PP, et al. , Transcription forms and remodels supercoiling domains unfolding large-scale chromatin structures, Nat. Struct. Mol. Biol 20 (2013) 387–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Sanz LA, Chedin F, High-resolution, strand-specific R-loop mapping via S9.6-based DNA-RNA immunoprecipitation and high-throughput sequencing, Nat. Protoc 14 (2019) 1734–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kouzine F, Wojtowicz D, Baranello L, Yamane A, Nelson S, Resch W, et al. , Permanganate/S1 nuclease footprinting reveals non-B DNA structures with regulatory potential across a mammalian genome, Cell. Syst 4 (2017) 344–356 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kowalski D, Natale DA, Eddy MJ, Stable DNA unwinding, not “breathing,” accounts for single-strand-specific nuclease hypersensitivity of specific A+T-rich sequences, Proc. Natl. Acad. Sci. U. S. A 85 (1988) 9464–9468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Benham CJ, Torsional stress and local denaturation in supercoiled DNA, Proc. Natl. Acad. Sci. U. S. A 76 (1979) 3870–3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Belotserkovskii BP, Tornaletti S, D'Souza AD, Hanawalt PC, R-loop Generation during Transcription: Formation, Processing and Cellular Outcomes, DNA repair, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Drolet M, Bi X, Liu LF, Hypernegative supercoiling of the DNA template during transcription elongation in vitro, J. Biol. Chem 269 (1994) 2068–2074. [PubMed] [Google Scholar]
- [44].Duquette ML, Handa P, Vincent JA, Taylor AF, Maizels N, Intracellular transcription of G-rich DNAs induces formation of G-loops, novel structures containing G4 DNA, Genes Dev. 18 (2004) 1618–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Reaban ME, Griffin JA, Induction of RNA-stabilized DNA conformers by transcription of an immunoglobulin switch region, Nature 348 (1990) 342–344. [DOI] [PubMed] [Google Scholar]
- [46].Daniels GA, Lieber MR, RNA:DNA complex formation upon transcription of immunoglobulin switch regions: implications for the mechanism and regulation of class switch recombination, Nucleic Acids Res. 23 (1995) 5006–5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Duquette ML, Pham P, Goodman MF, Maizels N, AID binds to transcription-induced structures in c-MYC that map to regions associated with translocation and hypermutation, Oncogene 24 (2005) 5791–5798. [DOI] [PubMed] [Google Scholar]
- [48].Roy D, Lieber MR, G clustering is important for the initiation of transcription-induced R-loops in vitro, whereas high G density without clustering is sufficient thereafter, Mol Cell Biol 29 (2009) 3124–3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Roy D, Yu K, Lieber MR, Mechanism of R-loop formation at immunoglobulin class switch sequences, Mol Cell Biol 28 (2008) 50–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Roy D, Zhang Z, Lu Z, Hsieh CL, Lieber MR, Competition between the RNA transcript and the nontemplate DNA strand during R-loop formation in vitro: a nick can serve as a strong R-loop initiation site, Mol Cell Biol 30 (2010) 146–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Carrasco-Salas Y, Malapert A, Sulthana S, Molcrette B, Chazot-Franguiadakis L, Bernard P, et al. , The extruded non-template strand determines the architecture of R-loops, Nucleic Acids Res. 47 (2019) 6783–6795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Cristini A, Groh M, Kristiansen MS, Gromak N, RNA/DNA hybrid interactome identifies DXH9 as a molecular player in transcriptional termination and R-loop-associated DNA damage, Cell Rep. 23 (2018) 1891–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Wang IX, Grunseich C, Fox J, Burdick J, Zhu Z, Ravazian N, et al. , Human proteins that interact with RNA/DNA hybrids, Genome Res. 28 (2018) 1405–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Lim YW, Sanz LA, Xu X, Hartono SR, Chedin F, Genome-wide DNA hypomethylation and RNA:DNA hybrid accumulation in Aicardi-Goutieres syndrome, Elife 4 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Bonnet A, Grosso AR, Elkaoutari A, Coleno E, Presle A, Sridhara SC, et al. , Introns protect eukaryotic genomes from transcription-associated genetic instability, Mol. Cell 67 (2017) 608–621 e6. [DOI] [PubMed] [Google Scholar]
- [56].Hartono SR, Korf IF, Chedin F, GC skew is a conserved property of unmethylated CpG island promoters across vertebrates, Nucleic Acids Res. 43 (2015) 9729–9741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Baaklini I, Usongo V, Nolent F, Sanscartier P, Hraiky C, Drlica K, et al. , Hypernegative supercoiling inhibits growth by causing RNA degradation, J. Bacteriol 190 (2008) 7346–7356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Liu LF, Wang JC, Supercoiling of the DNA template during transcription, Proc. Natl. Acad. Sci. U. S. A 84 (1987) 7024–7027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.