

Abstract
Various amyloidogenic proteins have been suggested to be involved in the onset and progression of neurodegenerative diseases (ND) such as Alzheimer's disease (AD) and Parkinson's disease (PD). Particularly, the aggregation of misfolded amyloid-β and hyperphosphorylated tau and α-synuclein are linked to the pathogenesis of AD and PD, respectively. In order to care the diseases, multiple small molecules have been developed to regulate the aggregation pathways of these amyloid proteins. In addition to controlling the aggregation of amyloidogenic proteins, maintaining the levels of the proteins in the brain by amyloid degrading enzymes (ADE; neprilysin (NEP), insulin-degrading enzyme (IDE), asparagine endopeptidase (AEP), and ADAM10) is also essential to cure AD and PD. Therefore, numerous biological molecules and chemical agents have been investigated as either inducer or inhibitor against the levels and activities of ADE. Although the side effect of enhancing the activity of ADE could occur, the removal of amyloidogenic proteins could result in a relatively good strategy to treat AD and PD. Furthermore, since the causes of ND are diverse, various multifunctional (multitarget) chemical agents have been designed to control the actions of multiple risk factors of ND, including amyloidogenic proteins, metal ions, and reactive oxygen species. Many of them, however, were invented without considerations of regulating ADE levels and actions. Incorporation of previously created molecules with the chemical agents handling ADE could be a promising way to treat AD and PD. This review introduces the ADE and molecules capable of modulating the activity and expression of ADE.
1. Introduction
With the increase of population, particularly aged people, curing various neurodegenerative diseases (ND), whose symptoms are memory loss and cognitive impairment through the loss of neuronal function, has emerged as a new social and economic issue [1, 2]. Alzheimer's disease (AD) and Parkinson's disease (PD) are the most common cases of ND which could be caused by multiple factors, including (i) the accumulation of misfolded amyloidogenic proteins such as amyloid-β (Aβ), tau, and α-synuclein (α-syn), (ii) cholinergic deficit, (iii) metal ion dyshomeostasis, and (iv) oxidative stress induced by overproduced reactive oxygen species (ROS) [2, 3]. Particularly, the aggregates of the amyloidogenic proteins existed in both intracellular and extracellular regions have been a subject of intensive research topic for understanding of the pathology of AD and PD. Among the protein aggregates, soluble oligomer species have been informed to be the most neurotoxic species [4, 5].
AD is the most common form (ca. 60–80%) of dementia. It has been reported that 47 million people suffered from the disease worldwide, and this number is expected to increase to 75 million by 2030 [6]. The significant hallmarks of AD are (i) the shrinkage of the hippocampus and cortex in the brain, (ii) senile plaques mainly composed of Aβ aggregates, (iii) neurofibrillary tangles (NFT) containing the aggregates of hyperphosphorylated tau (ptau), (iv) miscompartmentalization of metal ions, and (v) oxidative stress [7–9]. Based on various previous studies, among those hallmarks, Aβ has been suggested to be a major risk factor of the onset and progression of AD. Therefore, the production, aggregation, and clearance of Aβ were considered to be related to the pathogenesis of AD, and multiple strategies have been tried to treat AD by (i) inhibiting β- and γ-secretases which are responsible for the generation of Aβ by cleaving amyloid precursor protein (APP) [10, 11], (ii) modulating Aβ aggregation [2], and (iii) enhancing the removal of Aβ [12–15]. Multiple studies revealed that the late-onset AD has been suggested to be associated with the reduction of Aβ clearance [16]. The age-/pathology-related decrease of the concentration and/or activity of Aβ-degrading enzymes (i.e., neprilysin (NEP) and insulin-degrading enzyme (IDE)) could be the triggers of the onset and progression of AD [17].
In addition to Aβ, ptau has been suggested as another potent major cause of AD [18]. Tau expressed in neurons exhibited a high level in neuronal axons. Hyperphosphorylation of tau makes NFTs that eventually form plaques in the brain. Tau plays important roles for neuronal trafficking and keeping the structure of synapse by stabilizing microtubules and sustaining dendrite structure; however, when the protein is hyperphosphorylated and/or fragmentized, it loses the original functions causing neuronal damages [18, 19]. Recent studies performed with transgenic mice revealed that the neurotoxicity might come from ptau oligomers in the early stage of AD [20].
The second most common form of ND is PD (ca. 6 million people were affected by the disease in 2015) which is characterized by the loss of dopaminergic neurons with the symptoms of shaking, rigidity, and slowness of movement [3, 21–23]. Besides physical disorders, emotional problems also occur including depression and anxiety [23]. One of the major reasons for the onset and progression of PD is the loss of dopaminergic neurons caused by the aggregation of α-Syn, forming Lewy bodies, in the brain. Then, the damaged neurons undergo cell death leading to the loss of astrocytes and an increase in the number of microglia, particularly in the substantia nigra [22–24]. Thus, α-Syn has been considered as the main target for curing PD [25–27].
α-Syn is a soluble 14 kDa protein. It exists at presynaptic terminals as a controller of synaptic vesicle trafficking [25]. The aggregation pathways of misfolded α-Syn are similar to those of Aβ: (i) containing lag phase, elongation phase, and steady state and (ii) forming aggregates by interacting through the central hydrophobic region composed of 11 amino acids [28–30]. Unlike monomeric α-Syn, its aggregates observed in various spots in cells indicating α-Syn species could damage both intracellular and extracellular environments. The oligomeric species have been reported to show neurotoxicity by disrupting the lipid membrane as annular Aβ oligomers [31, 32].
To succeed in treating ND, it is necessary to understand the process of generation, aggregation, and degradation of amyloidogenic proteins (i.e., Aβ, tau, and α-Syn; Table 1). In order to reduce neurotoxicity caused by the aggregates of these proteins, degrading amyloidogenic proteins could be potent therapeutic methods along with controlling the production or aggregation pathways of amyloidogenic proteins (Figure 1). In this review, we introduce the multiple ADE which are related to multiple ND and their regulators, both inducers and inhibitors toward the expression and/or activity of enzymes.
Table 1.
The correlation between the activity of ADE and related ND. Different from NEP, IDE, and ADAM10, inhibition of AEP could be a strategy to care AD and PD.
| Related neurodegenerative diseases | Enzymes | Results of enhanced enzyme activity | Amyloid aggregation |
|---|---|---|---|
| AD | NEP | Aβ degradation ↑ | Aβ aggregation ↓ |
| AD | IDE | Aβ degradation ↑ | Aβ aggregation ↓ |
| AD, PD | AEP | Tau/α-Syn fragmentation ↑ | Tau/α-Syn aggregation ↑ |
| AD | ADAM10 | Aβ generation ↓ | Aβ aggregation ↓ |
Figure 1.
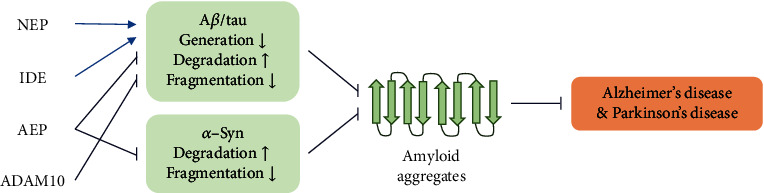
Brief mechanistic scheme of the pathogenesis of AD and PD with an aspect of amyloidogenic proteins and ADE. Regulating the degradation/fragmentation of amyloidogenic proteins by ADE could reduce their aggregation resulting in less risk of onset and/or progression of AD and PD.
2. Enzymes Related to AD and PD
2.1. Neprilysin (NEP)
NEP, known as CALLA or CD10 in the M13 subfamily of type II integral membrane endopeptidases containing 749 amino acids, is predominantly membrane-bound zinc-dependent metallopeptidase with a broad tissue distribution, including the central nervous system, kidney, and vascular endothelium [33–35]. NEP is produced in Golgi and expressed by smooth muscle cells within larger arterioles in the cerebral cortex and leptomeninges in the human brain [35, 36]. It consists of a membrane-spanning helix in the N-terminal region and C-terminal domain with a zinc-binding site, 583HEXXH587 [37].
In the neutral range of pH, NEP shows the maximum activity [38] and it cleaves the peptide bonds between hydrophobic amino acids, especially phenylalanine and leucine from the small peptides (<50 amino acids) on hydrophobic residues [37, 39]. Hence, the hydrophobic Aβ peptides could be an ideal substrate along with enkephalins, substance P, endothelin, and bradykinin [15, 34]. The active site of NEP is sterically hindered to have high selectivity of substrates. For the monomeric Aβ, multiple peptide bonds formed between (i) E3 and F4, (ii) G9 and Y10, (iii) F19 and F20, (iv) A30 and I31, and (v) G33 and L34 could be cleaved [40]. NEP has been reported to degrade oligomeric Aβ species as well [41].
In the mouse brain, the Aβ levels were shown to increase upon disruption of NEP expression [42]. Lower levels of NEP in cerebrospinal fluid were observed particularly in the prodromal phase and early AD stage [43]. Moreover, inactivation of NEP in the hAPP mouse model caused impaired synaptic plasticity and cognitive performance [42, 44], and high levels of NEP were shown to be colocalized in amyloid plaques [45]. Therefore, NEP has been suggested to be associated with the deposition of Aβ aggregates leading to the onset and progression of AD [46–50]. Based on in vivo studies, the upregulation of NEP could be a promising tactic to treat AD not only based on Aβ metabolism but also through other mechanisms (e.g., generating neuropeptide Y fragments) [48–51].
2.2. Insulin-Degrading Enzyme (IDE)
IDE is a 113 kDa zinc-dependent metallopeptidase involved in the catabolism of insulin and Aβ [52, 53]. IDE is mostly located in the cytosol; however, it could be found in mitochondria, peroxisomes, plasma membrane, and cerebrospinal fluid as well [54]. It has a 108HXXEH112 motif for zinc-binding and enzymatic processes. IDE contains 2 similar-sized domains, IDE-N and IDE-C, connected by a loop with 28 amino acids and many hydrogen bonds between two domains keeping the catalytic site closed which is located in IDE-N [55–57]. E111 acts as a base to activate the catalytic water for the hydrolysis of substrates [57]. Thus, IDE-N itself could show the proteolytic activity while the IDE-C domain itself could not have the enzyme activity [58]. The inner side of IDE-N is neutral or negatively charged while the IDE-C domain is positive in charge assisting IDE selects or excludes the substrates based on the charge [56]. Unlike NEP, IDE degrades the substrates containing β-structures causing the formation of toxic oligomeric species, subsequently leading to ND, specifically [17, 59].
Since IDE is located in mitochondria and peroxisomes, it could regulate Aβ levels at the intracellular region as well as cover the damages from Aβ aggregates [60–62]. At neutral pH, IDE shows optimal activity to cleave Aβ [63]. IDE is the major enzyme for the removal and cleavage of Aβ in hippocampal lysates, cytoplasm, and cerebrospinal fluid [64, 65]. In addition to degrading Aβ species, the inactive form of IDE could inhibit the fibrillogenesis of Aβ by acting as a chaperone [66].
Although the level of IDE was increased in the hippocampus in the AD-affected brain, the activity of IDE was decreased with aging and at the early stages of AD [63, 67]. Colocalization of Aβ plaques and IDE was observed in the brain suggesting that IDE could be buried in the plaques or oxidized; thus, IDE may lose its amyloid-degrading function [68]. It could lead to a lack of Aβ clearance and protein aggregation resulting in neuronal damages. Also, IDE knockout animals presented the relatively high levels of Aβ [69, 70]. These experimental results proposed that the Aβ cleavage activity of IDE should be maintained to treat ND.
2.3. Asparagine Endopeptidase (AEP)
Asparagine endopeptidase (AEP) is a cysteine protease that is also called legumain. Plants legumain was discovered first; then, mammalian legumain was cloned [71, 72]. It cleaves substrates after asparagine residues and sometimes aspartic acid; however, glycosylated asparagines are not cleaved by AEP [73, 74]. It is a member of the peptidase family C13 and has structural homology with other proteases such as caspases and separases [75]. Since legumain is a type I transmembrane protein, it has a signal peptide. Activation of proAEP (56 kDa) requires autocatalytic removal of propeptides at N- and C-terminal (V18-D25 and D324-Y433) regions under acidic condition. Then, the cleavage of these two propeptides and further process produce 46 kDa of active proAEP and 36 kDa of active AEP. Cysteine protease domain (G26-N323) includes catalytic triad (N42-H148-C189) and a few N-glycosylation sites [76, 77]. The optimal pH for enzyme activity is ca. at 5.8; however, it permanently denatures above pH 7.0 [71]. The active site of AEP is blocked by its autoinhibitory C-terminal prodomain which makes the substrate difficult to access.
Moreover, AEP cleaves APP and tau in the brain with δ-secretase. Increased AEP level was observed from AD patients' brain samples [78, 79]. Aging is related to an increased AEP level accompanied by an elevated level of fragmented APP. Healthy people have uncleaved APP while AD patients have fragmented APP in the brain [79]. In mouse experiments, general symptoms of emotional problems such as anxiety and depression were reduced after the deletion of AEP knockout. In addition, spatial cognition and learning ability were also improved in AEP knockout mice compared with wild-type mice [80].
Tau and hyperphosphorylated tau can be cleaved by AEP. Cleaved tau (usually tau1-368 and tau1-255) has strong toxicity in cultured neurons since it favors aggregation [78, 81]. In transgenic mouse experiments, the pH of the brain cortex in tau P301S-expressing mice was lower than the control nontransgenic mice. Likely, AD patients have a slightly acidic environment in the brain than healthy people [82–84]. Low pH might cause activation of AEP in AD patients' brain. AEP also contributes to the splicing process of tau. In AD patients, SRPK2 which is important in pre-mRNA splicing is abnormally activated in tauopathies. Failure of tau exon 10 pre-mRNA splicing regulation results in imbalances in 3R- and 4R-tau. AEP is known to cleave SRPK2 leading to increase kinase activity. Transgenic mice with SRPK2 mutant that cannot be cleaved by AEP showed the elevated synaptic functions and spatial memory while those have truncated SRPK2 underwent 3R- and 4R-tau imbalance leading to accelerate cognitive decline [85, 86]. Tau cleavage by AEP also can be increased by BDNF deprivation. Resulting tau1-368 fragment blocks neurotrophic signals on TrkB receptors, eventually leading to cell death [87].
α-Syn is also cleaved by AEP, and the fragment of α-Syn (cleaved at 103 position) is prone to be aggregated. α-Syn cleavage by AEP is age dependent. In the PD animal model and PD patients' brain, aggregated α-Syn fragments exhibit high neurotoxicity resulting in neuronal loss and motor impairments. The wild-type and inactive AEP mutant, however, did not cleave α-Syn, and little pathogenic effects were observed [22, 23]. Oxidative stress could induce the upregulation of AEP leading to cleave α-Syn and neurotoxicity. Based on the previous studies, the cleaved α-Syn and activated AEP level showed a close correlation [88, 89]. Therefore, inhibition of AEP activity with synthetic or natural small molecules shed light on the treatment of AD and other ND.
2.4. ADAM10
The ADAM family is a zinc-dependent transmembrane and secreted metalloprotease. It usually consists of ca. 750 amino acids as a signal peptide-prodomain-metalloprotease-like domain-disintegrin-like domain-cysteine-rich domain-EGF-like domain-transmembrane domain-cytoplasmic tail. The ADAM family is involved in cell adhesion, proteolytic processing of many receptors, and signaling molecules [90]. ADAM10 is synthesized in the ER and transported to Golgi where it is matured and activated by removal of prodomain. The size of the matured ADAM10, not activated, still containing prodomain, is 68 kDa in Golgi. [91]. ADAM10 forms a dimer, and the C-terminal motif becomes an ordered domain. The zinc-coordinating catalytic core of the ADAM family is HEXGHXXGXXHD, and the active site of ADAM10 in human, rat, and bovine is HEVGHNFGSPHD. Three histidines are responsible for zinc binding, glycine next to phenylalanine allows a turn, and glutamic acid acts as a catalytic residue. Point mutation E384A loses catalytic activity and results in a decrease of soluble APPα (sAPPα) production which has a neuroprotective function [92]. The disintegrin-like domain is a ligand for integrin binding; however, this domain is not necessary for ADAM10 protease activity [92]. Although the exact mechanism of action has not been known, it is plausible that ADAM10 has a similar catalytic mechanism to other zinc proteases due to the structural similarity of active sites [92].
Aβ can be produced when APP undergoes proteolytic cleavage by β- and γ-secretases. In contrast, α-secretase like ADAM10 does not generate toxic Aβ after cleavage. Instead, the cleavage of APP by ADAM10 produces sAPPα. Thus, unlike AEP, inhibition of ADAM10 elevates the neurotoxic Aβ level in the brain and upregulation of ADAM 10 could be a promising way of treatment [93, 94]. Activated ADAM10 proteins were mostly localized on the plasma membrane while proADAM10 still remains in Golgi. This observation indicates there are two pathways to cleave APP: (1) on the cell surface and (2) along the secretory pathway [95].
ADAM10 also plays an important role in regulating synaptic proteins. Neuronal surface ADAM10 undergoes endocytosis by interacting with AP2. In AD patients, the ADAM10/AP2 association was elevated in the hippocampus. ADAM10 activity at the surface decreased upon association with AP2 and endocytosis by long-term potentiation (LTP) in hippocampal neurons [96]. In the ADAM10 knockout mouse model, decreased ADAM10 level resulted in altered network activities in the hippocampus and impaired synaptic function. Consequently, decreased neuromotor abilities and reduced learning abilities were observed. Therefore, reduced ADAM10 might affect the shedding of surface proteins to induce postsynaptic defects [97].
3. Regulators toward the Enzymes Related to AD and PD
3.1. Regulators of NEP
NEP is one of the major enzymes for Aβ clearance; thus, it has been proposed to be involved in the onset and progression of AD and could be a therapeutic target for caring the disease [15, 98]. Several chemicals have been applied to regulate the activity and expression of NEP. NNC26-9100 indirectly increases the activity of NEP in cortical tissue [99]. Also, it could decrease APP expression in both cortical and hippocampal tissues causing the reduction of Aβ production. Moreover, NNC26-9100 inhibits the formation of Aβ42 trimers within both extracellular and intracellular cortical fractions. Consequently, the upregulation of NEP expression and activity could lower Aβ42 levels and improve memory in an AD transgenic mouse model [99, 100].
In addition, some histone deacetylase (HDAC) inhibitors' potentials as NEP inducers have been investigated [101, 102]. Valproic acid could raise the expression and activity of NEP in SHSY-5Y cells as well as the cortex and hippocampus of rats [101, 102]. It could recover cognitive deficits caused by hypoxia in rats [102]. Another HDAC inhibitor, Trichostatin A also presented its ability to enhance the expression of NEP in SHSY-5Y cells [101]. Imatinib, known as Gleevec, an anticancer agent, is also proposed to elevate mRNA and protein levels of NEP in multiple cell lines [103]. Recently, 5-hydroxyindoleacetic acid (5-HIAA) also induces the levels of NEP in neuroblastoma cells and in vivo [104]. These molecules have potentials to control the Aβ level in the brain and treat AD.
On the other hand, multiple chemical agents such as phosphoramidon and tiorphan have been reported as NEP inhibitors [40, 105]. They directly interact with NEP at the active site preventing the binding of substrates to NEP [105]. Moreover, chemicals with structural modifications of phosphoramidon and tiorphan, benzimidazole, and imidazo{4,5-c}pyridine scaffold presented relatively good inhibiting ability against NEP with an IC50 value of ca. 0.2-2.0 μM [106]. MCB3937 is also suggested as NEP inhibitors with a nanomolar range of IC50 value [34]. It could interact with the zinc-binding site of NEP, 583HEXXH587, and 646EXXXD650, reducing the activity of the enzyme [34]. Another NEP inhibitor is LCZ696 which is comprised of structural moieties of valsartan and AHU377, a prodrug which metabolizes to NEP inhibitor, known as LBQ657 [107]. The IC50 values of valsartan and LBQ657 against NEP are 2.4 nM and 2.3 nM, respectively [33, 108].
As we introduced above, various molecules have been investigated in order to regulate the activity and expression of NEP. Previously, NNC26-9100, HDAC inhibitors (i.e., valproic acid and trichostatin A), imatinib, and 5-HIAA have been reported as inducers of NEP activity while phosphoramidon, tiorphan, MCB3937, LCZ696, valsartan, and LBQ657 were presented as inhibitors of NEP activity. Based on the structures and functions of these molecules, many regulators against NEP activity could be developed.
3.2. Controlling Agents toward IDE
The activity of IDE also could be regulated by various substances. Multiple short peptides, including dynorphin B-9 and A-17, have been reported to induce the activity of IDE [109]. Particularly, dynorphin B-9 increased the Aβ cleavage rate of IDE by ca. 2.5-fold whereas the cleavage rate of other substrates (i.e., insulin) was not affected [109]. Bradykinin plays a role as an inducer of IDE activity as well. Upon treatment of bradykinin, IDE presents in dimer form which is more active than tetramer form for cleaving various substrates including Aβ [17, 109]. Another short peptide, somatostatin, has been shown to increase the cleavage activity of IDE [52]. Two additional exosites have different roles according to the size and binding mode of the substrate to the IDE catalytic cleft: (i) one exosite regulates only the interaction of IDE with larger substrates (i.e., insulin and Aβ40) in a differing based on their various modes of binding to the enzyme; (ii) the other exosite is involved in the regulation of enzymatic processing by IDE of all substrates (peptides containing 10-25 amino acids) through the alteration of an open-close equilibrium [52]. Somatostatin could bind to both exosites with higher binding affinity and enhance the enzyme activity [52]. In addition to short peptides, D3, D4, and D6 developed by Cakir and coworkers presented the possibilities of increasing the activity of IDE toward Aβ degradation through unknown mechanisms [53].
In order to inhibit the activity of IDE, several chemicals have been invented with different modes of action. Firstly, the reported high-affinity IDE inhibitor is Li-1. It binds to the catalytic site of IDE with 2 nM as binding affinity and decreases the activity of IDE [110]. Another inhibitor binding to the catalytic cleft to directly interfering the binding of substrates is BDM44768, 6bk, and NTE-1. 6bk and NTE-1 bind to IDE at a different spot from Li-1 or BDM44768; 6bk and quinolone-2 moiety of NTE-1 bind to the pocket located at the outside of the catalytic site, and dipeptide aniline amide analog of NTE-1 binds to N-terminal anchoring exosite interfering the actions of IDE [111–113]. BDM41367 suppresses the activity of IDE by binding to both the N-terminal anchoring exosite and catalytic site [114]. ML345, containing a thiol group, presented different mechanisms to reduce the cleavage rate of IDE. It interacts with zinc and forms a covalent bond with C819 [115, 116].
To sum up, some biological molecules as short peptide (i.e., dynorphin B-9 and A-17, bradykinin, somatostatin, D3, D4, and D6 (by Cakir and coworkers)) could increase the degrading rate of IDE toward Aβ. Several chemical agents such as Li-1, BDM44768, 6bk, NTE-1, BDM41367, and ML345, however, could inhibit the activity of IDE. Previously presented various regulators against IDE could be the first set of library to develop the potent treatment of AD.
3.3. Inducers and Inhibitors of AEP
Short peptides such as legumain stabilization and activity modulation (LSAM) domain and αvβ3 integrin could enhance the activity of AEP. LSAM domain known as the prodomain of AEP blocks substrate binding before activation. This prodomain has a helical structure and two independent peptides. One is an activation peptide (AP, K287 to N323), and the other is a LSAM domain. LSAM domain remains even after AP is cleaved and released from protease at neutral pH via electrostatic interaction. AEP without LSAM domain has a lower melting temperature than AEP with LSAM domain [77, 117]. Another short peptide, αvβ3 integrin, can directly interact with AEP, and after forming a complex, the optimal pH for AEP activity is increased from 5.5 to 6.0. It indicates that αvβ3 binding could induce conformational stabilization of AEP accompanied by deprotonated C189. αvβ3 does not directly interact with the AEP active site; however, AEP docks to the αvβ3 RGD-binding site (allosteric effect) [117]. Based on the immunoanalysis, AEP was mostly found in lysosome and endosome as well as cell surface where αvβ3 integrin was localized [118]. Naturally occurred polysaccharides with negative charges, glycosaminoglycans (GAGs), could induce the activity of AEP as well. At low pH, proAEP undergoes autocatalytic activation and this process can be accelerated by some GAGs (such as C4S (chondroitin 4-sulfate), C6S (chondroitin 6-sulfate), C4,6S (chondroitin 4,5-sulfate), heparin, and heparin sulfate) with concentration and time dependencies at pH 4.0 [119–121].
On the other hand, various molecules, both short peptides and chemical agents, have been investigated as potent inhibitors against AEP. AENK (Fmoc-Ala-Glu-Asn-Lys-NH2) is known to inhibit AEP specifically by blocking the proteolysis of a Dnase inhibitor, SET, both in vitro and in vivo [74, 122]. AENK selectively inhibits AEP's cleavage activity of α-Syn and tau in a dose-dependent manner [22, 78]. Also, SRPK2, which is abnormally activated in tauopathy in AD, is fragmented by AEP and this proteolytic cleavage is inhibited by AENK as well [85]. In addition, Ac-YVAD-CHO (acetyl-Tyr-Val-Ala-Asp-aldehyde) could act as an AEP inhibitor as well as a reversible caspase inhibitor. Ac-ESEN-CHO (acetyl-Glu-Ser-Glu-Asn-aldehyde) is also a specific AEP inhibitor. These two short peptides suppressed AEP activity in the plant leaves [123, 124]. Cystatins consist of two groups of cysteine protease inhibitors (type 1 and type 2) that have conserved a sequence as G9, Q53, V55, and G57. Among cystatins, only cystatin C, E/M, and F (belong to type 2) have inhibitory ability against AEP. Cystatin C inhibits AEP almost completely in a dose-dependent manner. Cystatin C, E/M, and F have AEP inhibitory sites that have chemically similar residues in a loop composed of four amino acids (i.e., SNDM, SNSI, and TNDM for cystatin C, E/M, and F, respectively) [77, 121, 125–130].
Additionally, proteins from fungus could interfere with the actions of AEP. Clitocybe nebularis and Macrolepiota procera express similar proteins (ca. 20% identical sequence) with 16.8 kDa and 19 kDa, clitocypin and macrocypin, respectively, which could inhibit AEP [131, 132]. Among the five subgroups of macrocypins, only macrocypins 1 and 3 could inhibit AEP [77, 133, 134]. The C-terminal prodomain of AEP itself blocks its active site as autoinhibition, before maturation. Even after maturation, the inhibitory function is still available if the cleaved prodomain is added to the mature AEP [135]. The activated AEP returns back to the inactivated form by autoligation around pH 7.5 because autocatalytic reaction site N323 occurs reversibly [77, 136, 137].
Along with short peptides, various chemical agents also have been revealed as a potent regulator toward AEP. A nonnatural amino acid-based inhibitor Li-1 was synthesized from aza-peptidyl epoxide. It has ca. nanomolar range of IC50 and highly selective against AEP, but not against cathepsins [138]. Compound 9, developed by Xu and coworkers, is an aza-Asn epoxide that derived irreversible cysteine protease inhibitor. It showed a concentration-dependent inhibition effect, and the optimal concentration was determined as 1 μM [139, 140].
R13 is a 7,8-dihydroxyflavone (7,8-DHF)-based prodrug. Although it showed a significant therapeutic effect against AD as a TrkB agonist, 7,8-DHF has two drawbacks: poor oral bioavailability and pharmacokinetic profile. R13 is the most prominent derivative stemmed from the structure of 7,8-DHF to prevent Aβ deposition in AD mice [141]. Another small molecule, MV026630, is an acyloxymethylketone derivative presenting AEP inhibition. It reversibly inhibits AEP and able to be absorbed by living cells [142]. Compound 11 designed by Zhang and coworkers was selected as an AEP inhibitor by intensive high throughput screening in vitro and in vivo. Compound 11 could interact with both the active site and the allosteric site of AEP leading to inhibit the enzyme activity. Upon treatment of compound 11 to 5xFAD mouse models, both tau and Aβ cleavage were reduced [143].
A thyrotropin-releasing hormone (TRH) analog, taltirelin, can be applied orally. Taltirelin has 10-100 times enhanced effect on the central nervous system (CNS), and this effect lasts longer than TRH. AD and PD patients have a higher concentration of APP, α-Syn, and tau fragments than healthy condition. TRH and its analog, taltirelin, lower the phosphorylation level of tau by inhibiting AEP activation. PD mouse research proved that taltirelin can be used to downregulate tau and α-Syn-related pathology [144–146]. The AEP activity from bovine kidney and pig was inhibited by N-ethylmaleimide, p-chloromercuribenzene-sulfonic acid, mercury, and copper as well [71, 147].
In summary, the LSAM domain (i.e., αvβ3 integrin) and GAGs which are negatively charged glycosaminoglycans could accelerate the activity of AEP. In contrast, (i) tetrapeptide including AENK, Ac-YVAD-CHO, and Ac-ESEN-CHO, (ii) short peptides (i.e., cystatin C, E/M, and F), and (iii) some fungal proteins such as clitocypin and macrocypin could suppress AEP activity. In addition, AEP activity can be self-inhibited by the C-terminal prodomain. Synthetic compounds such as Li-1, compound 9 (by Xu and coworkers), R13, MV026630, compound 11 (by Zhang and coworkers), and taltirelin act as AEP inhibitors as well. The development of both biological and chemical agents as regulators of AEP could be a key to care AD and/or PD.
3.4. Regulating Molecules of ADAM10
Multiple biological molecules such as hormones and transcription factors including XBP-1 (X-box binding protein-1), SOX-2- ((sex-determining region Y-) related high mobility group (HMG) box 2), and PAX2 (paired box gene 2) and chemical agents have been developed to enhance the expression and/or activity of ADAM10. XBP-1 regulates the unfolded protein response pathway. In the transgenic mouse model, ADAM10 expression was 2-fold increased compared to nontransgenic mouse. It was also shown that insulin induces translocation of XBP-1 to the nucleus leading to enhancement of ADAM10 transcription [148]. SOX-2 exhibits a low level in AD patients' brain. SOX-2 is an ADAM10 activator in the nonamyloidogenic processing of βAPP by ADAM10. HEK293 cells transiently transfected with Sox showed an increased ADAM10 level compared to control with empty plasmid [149]. In addition, a hormone, melatonin, synthesized from tryptophan, could be related to the expression and activity of ADAM10. Its level becomes lower as aging, especially in AD patients. In vitro study revealed that melatonin upregulated cleavage of APP by ADAM10 in neuronal and nonneuronal cells. Furthermore, mouse embryonic fibroblasts which have no ADAM10 gene showed reduced melatonin-stimulating function and sAPPα secretion [150, 151].
Also, various small molecules have been reported to upregulate the activity and level of ADAM10. Bryostatin-1, a macrolide lactone and a PKC activator, enhanced the generation of soluble APP in vitro studies even at sub-nM concentration via increasing the activity of ADAM10. In the transgenic mouse model, both Aβ40 and Aβ42 levels in the brain were decreased upon the treatment of bryostatin-1. It had several clinical trials for the treatment of cancers as well as AD [152, 153]. Retinoic acid and its derivative, acitretin, could be an example to elevate the ADAM10 level. 1 μM of retinoic acid treatment to SH-SY5Y cells for 4 days results in an increase of ADAM10 mRNA level. Retinoic acid could bind to the 302 and 303 nucleotides before the translation initiation site of the ADAM10 gene [154]. Besides, in the experiments with synthetic retinoids, acitretin exhibited significant enhancement of nonamyloidogenic processing of APP with an EC50 of 1.5 μM [155, 156]. Am80 is a synthetic retinoid that acts as an agonist for retinoic acid receptors (RAR), RARα, and RARβ. Am80 can increase the ADAM10 transcription level which leads to cleavage of APP. In mouse experiment, mRNA expression and ADAM10 expression were increased upon Am80 administration. Am80 has been approved for the treatment of acute promyelocytic leukemia in Japan. Am80 was also under clinical trial for the treatment of AD from 2010 [157, 158]. Phlogacantholide C induced ADAM10 transcriptional activity and increased ADAM10 expression level. Consequently, the secretion of ADAM10 that induced APP fragments was elevated [153, 159].
A natural product, resveratrol, with a polyphenol framework found in grape skin, peanut, and pomegranates, has been reported to be applied for the treatment of ND to enhance ADAM10 expression indirectly. In mouse experiment, 4 to 5 days of resveratrol administration diminished plaque formation [153, 160]. Also, gemfibrozil and etazolate could induce the levels of ADAM10. Peroxisome proliferator-activated receptor peroxisome proliferator-activated receptor-α (PPARα) regulates genes related to fatty acid transport and catabolism and can upregulate ADAM10 expression to increase proteolytic cleavage of APP. Gemfibrozil, a PPARα agonist, enhanced ADAM10 expression in isolated mouse hippocampal neurons at 25 μM [93, 153]. Etazolate is a GABAA receptor which has an important role in neurotransmission modulator. In rat cortical neuron experiments, concentration-dependent activation of ADAM10 by etazolate was observed in the range of 0.2 to 20 μM. In vivo studies with guinea pigs revealed that soluble APP level was increased in the brain. In addition, etazolate could prevent the neurotoxicity of Aβ on cortical neurons [153, 161].
Along with inducers of ADAM10 expression and activity, various biological molecules and chemicals have been suggested as inhibitors against ADAM10. Firstly, ADAM10 itself exists as an inactive form, zymogen, before the autocatalytic cleavage process. For its activation, removal of ADAM10 prodomain is required and the cleaved prodomain could inhibit the activity of ADAM10 with IC50 of 48 nM thorough an unknown mechanism [162, 163]. Also, fish oils, particularly unsaturated fatty acids such as DHA and EPA, which is known to be good for cardiovascular health, could reduce the lease of ADAM10 substrates from endothelial cells. The exact mechanism, however, has not been revealed yet [164, 165].
In addition to biomolecules, multiple chemicals have been examined as potent inhibitors of ADAM10. Low-density lipoprotein receptor-related protein 1 (LRP1) is responsible to transport Aβ across the blood-brain barrier (BBB) resulting in the deposition of Aβ in the brain leading to AD. In activity tests of purified ADAM10 with GI254023X and GW280623, both compounds inhibited ADAM10 most completely. Long-term treatment of ADAM10 with GI254023X in AD mouse increased the Aβ level in the periphery plasma due to reduced LRP1 shedding in the brain [166]. CID 3117694 is a non-zinc-binding selective inhibitor of ADAM10 with IC50 of 6.5 μM. In cell-based assays, CID 3117694 showed a time-dependent ADAM10 inhibition function. It has similar IC50 for several ADAM10 substrate concentrations meaning it is a noncompetitive inhibitor [163].
Hydroxamate derivatives have shown their inhibiting ability against ADAM10 as well. LT4, MN8, and CAM29 are hydroxamate derivatives which were developed for selectively inhibiting ADAM10. IC50 of LT4 and MN8 against ADAM10 are 40 nM and 9.2 nM, respectively [167]. LT4 (10 μM) also could inhibit ADAM10 sheddase activity carried by ExoV purified from L428 or L540 cells [168]. CAM29 has an IC50 of 20 nM against ADAM10 [169]. Similar to LT4, CAM29 could inhibit ADAM10 sheddase activity carried by ExoV purified from L428 or L540 cells. In addition, CD30 shedding in Hodgkin lymphoma cells was reduced by LT4 and CAM29 [168].
Although INCB3619 has been developed by Incyte Corporation as an ADAM17 inhibitor, it also inhibits that ADAM10 with IC50 for ADAM10 and ADAM17 are 0.022 μM and 0.014 μM, respectively [170]. Upon addition to A549 cells, INCB3619 showed ADAM10 inhibition function leading to deactivation of heregulin and HER3 autocrine signaling [171]. Unlike INCB3619, INCB8765 is an ADAM10 selective inhibitor with 97 nM as IC50 against ADAM10 [171].
Compound 1 and compound 2 were synthesized and examined as selective ADAM10 inhibitors by Mahasenan and colleagues [172]. Compound 1, however, had low potency and selectivity against ADAM10 while compound 2 has better selectivity toward ADAM10. The phenyl piperidine group in compound 2 is suitable to occupy the shallow cavity in ADAM10 [172]. Naturally occurring molecules such as rapamycin and triptolide also presented inhibiting activity against ADAM10. Rapamycin is a widely used drug to prevent rejection in organ transplantation. Recently, rapamycin was reported to increase Aβ generation in N2a cells. In addition, the production of sAPPα was decreased while β-CFT production was increased upon the treatment of rapamycin. Two weeks' application of rapamycin (3 mg/kg/day) to Aβ overexpressing transgenic mice resulted in elevated Aβ level accompanied by decreased sAPPα due to the inhibition of ADAM10 activity [165, 173]. Triptolide could be found from a Chinese herb, Tripterygium wilfordii. Through affinity chromatography and mass spectrometric analysis, only ca. nanomolar concentration of triptolide can downregulate ADAM10 expression in U937 and MCF-7 cells [174].
In short, ADAM10 activity could be elevated by biological molecules such as XBP-1, SOX-2, PAX2, and melatonin. Small molecules such as bryostatin-1, retinoic acid, acitretin, Am80, and phlogacantholide C and multiple natural products (i.e., resveratrol, gemfibrozil, and etazolate) have been reported as upregulators of ADAM10. On the other hand, biological molecules including ADAM10 prodomain, fish oils (DHA and EPA), and various chemical agents have been known as ADAM10 inhibitors. Synthetic molecules (i.e., GI254023X, GW280623, CID 3117694, LT4, MN8, CAM29, INCB3619, INCB8765, compound 1, and compound 2 (by Mahasenan and colleagues)) and naturally occurring molecules like rapamycin and triptolide showed inhibitory effect against ADAM10. With the development of these regulators, AD could be treated by regulating the activity of ADAM10.
4. Conclusions
In order to reduce the social and economic burden, therapeutic methods to ND, such as AD and PD, should be developed [1]. Since the presence and aggregation of amyloidogenic proteins, Aβ, tau, and α-Syn, could be major causes of the onset and progression of AD and PD [2], it is necessary to control the production, aggregation, and degrading process of the proteins to care the diseases. Based on the published reports we summarized above, regulating the activity/expression of ADE by short peptide and/or chemical agents could be a promising strategy to treat AD and PD; modulation of the expression and/or activity of NEP, IDE, AEP, and ADAM10 presented the clearance of amyloid deposits and improvement of cognitive deficits in vivo. The negative effect caused by inducing the activity of ADE could occur; however, the clearance of amyloidogenic proteins may result in a relatively good way to care AD and PD. In addition, it is essential to consider multiple risk factors due to the causes of the diseases that vary. Therefore, recently, various multifunctional (multitarget) small molecules have been invented to control the actions of those amyloidogenic proteins with other risk factors of AD and PD (i.e., metal ions and reactive oxygen species) [2], but most of them do not contain the capability to adjust the actions of ADE. A combination of previously developed molecules with the chemical agents which can upregulate the expression and/or activity of ADE could be the key to the success of treatment of ND, particularly AD and PD.
Acknowledgments
This work was supported by the research grant of the Kongju National University in 2020 (to H.J.L.) and the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (2019R1F1A1059679 (to N.K.); 2020R1G1A100399311 (to H.J.L.)).
Conflicts of Interest
The authors declare that there is no conflict of interest regarding the publication of this paper.
References
- 1.Derrick J. S., Lim M. H. Tools of the trade: investigations into design strategies of small molecules to target components in Alzheimer’s disease. Chembiochem. 2015;16(6):887–898. doi: 10.1002/cbic.201402718. [DOI] [PubMed] [Google Scholar]
- 2.Savelieff M. G., Nam G., Kang J., Lee H. J., Lee M., Lim M. H. Development of multifunctional molecules as potential therapeutic candidates for Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis in the last decade. Chemical Reviews. 2018;119(2):1221–1322. doi: 10.1021/acs.chemrev.8b00138. [DOI] [PubMed] [Google Scholar]
- 3.Kalia L. V., Lang A. E. Parkinson’s disease. Lancet. 2015;386(9996):896–912. doi: 10.1016/S0140-6736(14)61393-3. [DOI] [PubMed] [Google Scholar]
- 4.Kayed R., Head E., Thompson J. L., et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300(5618):486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 5.Haass C., Selkoe D. J. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid β-peptide. Nature Reviews Molecular Cell Biology. 2007;8(2):101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 6.Alzheimer’s Association. 2018 Alzheimer’s disease facts and figures. Alzheimer's & Dementia. 2018;14(3):367–429. [Google Scholar]
- 7.Frisoni G. B., Laakso M. P., Beltramello A., et al. Hippocampal and entorhinal cortex atrophy in frontotemporal dementia and Alzheimer’s disease. Neurology. 1999;52(1):91–100. doi: 10.1212/WNL.52.1.91. [DOI] [PubMed] [Google Scholar]
- 8.Hardy J. J., Selkoe D. J. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 9.Grundke-Iqbal I., Iqbal K., Tung Y. C., Quinlan M., Wisniewski H. M., Binder L. I. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proceedings of the National Academic of Sciences of the United States of America. 1986;83(13):4913–4917. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Strooper B., Annaert W., Cupers P., et al. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature. 1999;398(6727):518–522. doi: 10.1038/19083. [DOI] [PubMed] [Google Scholar]
- 11.Panza F., Solfrizzi V., Frisardi V., et al. Disease-modifying approach to the treatment of Alzheimer’s disease. Drugs & Aging. 2009;26(7):537–555. doi: 10.2165/11315770-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 12.Leissring M. A., Lu A., Condron M. M., et al. Kinetics of amyloid beta-protein degradation determined by novel fluorescence- and fluorescence polarization-based assays. Journal of Biological Chemistry. 2003;278(39):37314–37320. doi: 10.1074/jbc.M305627200. [DOI] [PubMed] [Google Scholar]
- 13.Marr R. A., Guan H., Rockenstein E., et al. Neprilysin regulates amyloid beta peptide levels. Journal of Molecular Neuroscience. 2004;22(1-2):5–12. doi: 10.1385/JMN:22:1-2:5. [DOI] [PubMed] [Google Scholar]
- 14.Hemming M. L., Patterson M., Reske-Nielsen C., Lin L., Isacson O., Selkoem D. J. Reducing amyloid plaque burden via ex vivo gene delivery of an Aβ-degrading protease: a novel therapeutic approach to Alzheimer disease. PLoS Medicine. 2007;4(8, article e262) doi: 10.1371/journal.pmed.0040262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nalivaeva N. N., Turner A. J. Targeting amyloid clearance in Alzheimer’s disease as a therapeutic strategy. British Journal of Pharmacology. 2019;176(18):3447–3463. doi: 10.1111/bph.14593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mawuenyega K. G., Sigurdson W., Ovod V., et al. Decreased clearance of CNS-Amyloid in Alzheimer's disease. Science. 2010;330(6012):p. 1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kurochkin I. V., Guarnera E., Berezovsky I. N. Insulin-degrading enzyme in the fight against Alzheimer’s disease. Trends in Pharmacological Sciences. 2018;39(1):49–58. doi: 10.1016/j.tips.2017.10.008. [DOI] [PubMed] [Google Scholar]
- 18.Goedert M., Spillantini M. G., Jakes R., Rutherford D., Crowther R. A. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron. 1989;3(4):519–526. doi: 10.1016/0896-6273(89)90210-9. [DOI] [PubMed] [Google Scholar]
- 19.Ballatore C., Lee V. M., Trojanowski J. Q. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nature Reviews Neuroscience. 2007;8(9):663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- 20.Ward S. M., Himmelstein D. S., Lancia J. K., Binder L. I. Tau oligomers and tau toxicity in neurodegenerative disease. Biochemical Society Transactions. 2012;40(4):667–671. doi: 10.1042/BST20120134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.GBD 2015 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016;388(10053):1545–1602. doi: 10.1016/S0140-6736(16)31678-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Z., Kang S. S., Liu X., et al. Asparagine endopeptidase cleaves α-synuclein and mediates pathologic activities in Parkinson’s disease. Nature Sturctural & Molecular Biology. 2017;24(8):632–642. doi: 10.1038/nsmb.3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ahn E. H., Kang S. S., Liu X., et al. Initiation of Parkinson’s disease from gut to brain by δ-secretase. Cell Research. 2020;30(1):70–87. doi: 10.1038/s41422-019-0241-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maragakis N. J., Rothstein J. D. Mechanisms of disease: astrocytes in neurodegenerative disease. Nature Clinical Practice Neurology. 2006;2(12):679–689. doi: 10.1038/ncpneuro0355. [DOI] [PubMed] [Google Scholar]
- 25.Bendor J. T., Logan T. P., Edwards R. H. The function of α-synuclein. Neuron. 2013;79(6):1044–1066. doi: 10.1016/j.neuron.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Recasens A., Dehay B., Bové J., et al. Lewy body extracts from Parkinson disease brains trigger α-synuclein pathology and neurodegeneration in mice and monkeys. Annals of Neurology. 2014;75(3):351–362. doi: 10.1002/ana.24066. [DOI] [PubMed] [Google Scholar]
- 27.Kubo S. I. Membrane lipids as therapeutic targets for Parkinson’s disease: a possible link between Lewy pathology and membrane lipids. Expert Opinion on Therapeutic Targets. 2015;20(11):1301–1310. doi: 10.1517/14728222.2016.1086340. [DOI] [PubMed] [Google Scholar]
- 28.Meuvis J., Gerard M., Desender L., Baekelandt V., Engelborghs Y. The conformation and the aggregation kinetics of α-synuclein depend on the proline residues in its C-terminal region. Biochemistry. 2010;49(43):9345–9352. doi: 10.1021/bi1010927. [DOI] [PubMed] [Google Scholar]
- 29.Giasson B. I., Murray I. V., Trojanowski J. Q., Lee V. M. A hydrophobic stretch of 12 amino acid residues in the middle of α-synuclein is essential for filament assembly. Journal of Biological Chemistry. 2001;276(4):2380–2386. doi: 10.1074/jbc.M008919200. [DOI] [PubMed] [Google Scholar]
- 30.Rodriguez J. A., Ivanova M. I., Sawaya M. R., et al. Structure of the toxic core of α-synuclein from invisible crystals. Nature. 2015;525(7570):486–490. doi: 10.1038/nature15368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wong Y. C., Krainc D. α-Synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nature Medicine. 2017;23(2):1–13. doi: 10.1038/nm.4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kayed R., Pensalfini A., Margol L., et al. Annular protofibrils are a structurally and functionally distinct type of amyloid oligomer. Journal of Biological Chemistry. 2009;284(7):4230–4237. doi: 10.1074/jbc.M808591200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Campbell D. J. Long-term neprilysin inhibition - implications for ARNIs. Nature Reviews Cardiology. 2017;14(3):171–186. doi: 10.1038/nrcardio.2016.200. [DOI] [PubMed] [Google Scholar]
- 34.Oefner C., Pierau S., Schulz H., Dale G. E. Structural studies of a bifunctional inhibitor of neprilysin and DPP-IV. Acta Crystallographica Section D. 2007;63, Part 9:975–981. doi: 10.1107/S0907444907036281. [DOI] [PubMed] [Google Scholar]
- 35.Baranello R., Bharani K., Padmaraju V., et al. Amyloid-β protein clearance and degradation (ABCD) pathways and their role in Alzheimer’s disease. Current Alzheimer Research. 2015;12(1):32–46. doi: 10.2174/1567205012666141218140953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miners J. S., Van Helmond Z., Chalmers K., Wilcock G., Love S., Kehoe P. G. Decreased expression and activity of neprilysin in Alzheimer disease are associated with cerebral amyloid angiopathy. Journal of Neuropathology & Experimental Neurology. 2006;65(10):1012–1021. doi: 10.1097/01.jnen.0000240463.87886.9a. [DOI] [PubMed] [Google Scholar]
- 37.Feygina E. E., Katrukha A. G., Semenov A. G. Neutral endopeptidase (neprilysin) in therapy and diagnostics: yin and yang. Biochemistry (Mosc) 2019;84(11):1346–1358. doi: 10.1134/S0006297919110105. [DOI] [PubMed] [Google Scholar]
- 38.Wang S., Wang R., Chen L., Bennett D. A., Dickson D. W., Wang D. S. Expression and functional profiling of neprilysin, insulin-degrading enzyme, and endothelin-converting enzyme in prospectively studied elderly and Alzheimer’s brain. Journal of Neurochemistry. 2010;115(1):47–57. doi: 10.1111/j.1471-4159.2010.06899.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shipp M. A., Tarr G. E., Chen C. Y., et al. CD10/neutral endopeptidase 24.11 hydrolyzes bombesin-like peptides and regulates the growth of small cell carcinomas of the lung. Proceedings of the National Academic of Sciences of the United States of America. 1991;88(23):10662–10666. doi: 10.1073/pnas.88.23.10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carson J. A., Turner A. J. Beta-amyloid catabolism: roles for neprilysin (NEP) and other metallopeptidases? Journal of Neurochemistry. 2002;81(1):1–8. doi: 10.1046/j.1471-4159.2002.00855.x. [DOI] [PubMed] [Google Scholar]
- 41.Kanemitsu H., Tomiyama T., Mori H. Human neprilysin is capable of degrading amyloid β peptide not only in the monomeric form but also the pathological oligomeric form. Neuroscience Letters. 2003;350(2):113–116. doi: 10.1016/S0304-3940(03)00898-X. [DOI] [PubMed] [Google Scholar]
- 42.Huang S. M., Mouri A., Kokubo H., et al. Neprilysin-sensitive synapse-associated amyloid-β peptide oligomers impair neuronal plasticity and cognitive function. Journal of Biological Chemistry. 2006;281(26):17941–17951. doi: 10.1074/jbc.M601372200. [DOI] [PubMed] [Google Scholar]
- 43.Huang J. Y., Hafez D. M., James B. D., Bennett D. A., Marr R. A. Altered NEP2 expression and activity in mild cognitive impairment and Alzheimer’s disease. Journal of Alzheimer’s Disease. 2012;28(2):433–441. doi: 10.3233/JAD-2011-111307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Farris W., Schütz S. G., Cirrito J. R., et al. Loss of neprilysin function promotes amyloid plaque formation and causes cerebral amyloid angiopathy. American Journal of Pathology. 2007;171(1):241–251. doi: 10.2353/ajpath.2007.070105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Apelt J., Ach K., Schliebs R. Aging-related down-regulation of neprilysin, a putative β-amyloid-degrading enzyme, in transgenic Tg2576 Alzheimer-like mouse brain is accompanied by an astroglial upregulation in the vicinity of β-amyloid plaques. Neuroscience Letters. 2003;339(3):183–186. doi: 10.1016/s0304-3940(03)00030-2. [DOI] [PubMed] [Google Scholar]
- 46.Iwata N., Tsubuki S., Takaki Y., et al. Metabolic regulation of brain Abeta by neprilysin. Science. 2001;292(5521):1550–1552. doi: 10.1126/science.1059946. [DOI] [PubMed] [Google Scholar]
- 47.Huttenrauch M., Baches S., Gerth J., Bayer T. A., Weggen S., Wirths O. Neprilysin deficiency alters the neuropathological and behavioral phenotype in the 5XFAD mouse model of Alzheimer’s disease. Journal of Alzheimer’s Disease. 2015;44(4):1291–1302. doi: 10.3233/JAD-142463. [DOI] [PubMed] [Google Scholar]
- 48.Saito T., Iwata N., Tsubuki S., et al. Somatostatin regulates brain amyloid β peptide Aβ42 through modulation of proteolytic degradation. Nature Medicine. 2005;11(4):434–439. doi: 10.1038/nm1206. [DOI] [PubMed] [Google Scholar]
- 49.Iwata N., Mizukami H., Shirotani K., et al. Presynaptic localization of neprilysin contributes to efficient clearance of Amyloid-Peptide in mouse brain. Journal of Neuroscience. 2004;24(4):991–998. doi: 10.1523/JNEUROSCI.4792-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Iwata N., Sekiguchi M., Hattori Y., et al. Global brain delivery of neprilysin gene by intravascular administration of AAV vector in mice. Scientific Reports. 2013;3(1, article 1472) doi: 10.1038/srep01472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rose J. B., Crews L., Rockenstein E., et al. Neuropeptide Y fragments derived from neprilysin processing are neuroprotective in a transgenic model of Alzheimer’s disease. Journal of Neuroscience. 2009;29(4):1115–1125. doi: 10.1523/JNEUROSCI.4220-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tundo G. R., di Muzio E., Ciaccio C., et al. Multiple allosteric sites are involved in the modulation of insulin-degrading-enzyme activity by somatostatin. The FEBS Journal. 2016;283(20):3755–3770. doi: 10.1111/febs.13841. [DOI] [PubMed] [Google Scholar]
- 53.Çakir B., Dağliyan O., Dağyildiz E., et al. Structure based discovery of small molecules to regulate the activity of human insulin degrading enzyme. PLoS One. 2012;7(2, article e31787) doi: 10.1371/journal.pone.0031787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tundo G. R., Sbardella D., Ciaccio C., et al. Multiple functions of insulin-degrading enzyme: a metabolic crosslight? Critical Reviews in Biochemistry and Molecular Biology. 2017;52(5):554–582. doi: 10.1080/10409238.2017.1337707. [DOI] [PubMed] [Google Scholar]
- 55.Malito E., Hulse R. E., Tang W. J. Amyloid β-degrading cryptidases: insulin degrading enzyme, presequence peptidase, and neprilysin. Cellular and Molecular Life Sciences. 2008;65(16):2574–2585. doi: 10.1007/s00018-008-8112-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shen Y., Joachimiak A., Rosner M. R., Tang W. J. Structures of human insulin-degrading enzyme reveal a new substrate recognition mechanism. Nature. 2006;443(7113):870–874. doi: 10.1038/nature05143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Im H., Manolopoulou M., Malito E., et al. Structure of substrate-free human insulin-degrading enzyme (IDE) and biophysical analysis of ATP-induced conformational switch of IDE. Journal of Biological Chemistry. 2007;282(35):25453–25463. doi: 10.1074/jbc.M701590200. [DOI] [PubMed] [Google Scholar]
- 58.Li P., Kuo W. L., Yousef M., Rosner M. R., Tang W. J. The C-terminal domain of human insulin degrading enzyme is required for dimerization and substrate recognition. Biochemical and Biophysical Research Communications. 2006;343(4):1032–1037. doi: 10.1016/j.bbrc.2006.03.083. [DOI] [PubMed] [Google Scholar]
- 59.Kurochkin I. V., Goto S. Alzheimer’s β-amyloid peptide specifically interacts with and is degraded by insulin degrading enzyme. FEBS Letters. 1994;345(1):33–37. doi: 10.1016/0014-5793(94)00387-4. [DOI] [PubMed] [Google Scholar]
- 60.Qiu W. Q., Walsh D. M., Ye Z., et al. Insulin-degrading enzyme regulates extracellular levels of amyloid β-protein by degradation. Journal of Biological Chemistry. 1998;273(49):32730–32738. doi: 10.1074/jbc.273.49.32730. [DOI] [PubMed] [Google Scholar]
- 61.Leal M. C., Magnani N., Villordo S., et al. Transcriptional regulation of insulin-degrading enzyme modulates mitochondrial amyloid β (Aβ) peptide catabolism and functionality. Journal of Biological Chemistry. 2013;288(18):12920–12931. doi: 10.1074/jbc.M112.424820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Perez A., Morelli L., Cresto J. C., Castano E. M. Degradation of soluble amyloid β-peptides 1-40, 1-42, and the Dutch variant 1-40Q by insulin degrading enzyme from Alzheimer disease and control brains. Neurochemical Research. 2000;25(2):247–255. doi: 10.1023/A:1007527721160. [DOI] [PubMed] [Google Scholar]
- 63.Stargardt A., Gillis J., Kamphuis W., et al. Reduced amyloid-β degradation in early Alzheimer's disease but not in the APPswePS1dE9 and 3xTg-AD mouse models. Aging Cell. 2013;12(3):499–507. doi: 10.1111/acel.12074. [DOI] [PubMed] [Google Scholar]
- 64.Portelius E., Mattsson N., Pannee J., et al. Ex vivo 18O-labeling mass spectrometry identifies a peripheral amyloid β clearance pathway. Molecular Neurodegeneration. 2017;12(1, article 18) doi: 10.1186/s13024-017-0152-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kurochkin I. V. Insulin-degrading enzyme: embarking on amyloid destruction. Trends in Biochemical Sciences. 2001;26(7):421–425. doi: 10.1016/S0968-0004(01)01876-X. [DOI] [PubMed] [Google Scholar]
- 66.de Tullio M. B., Castelletto V., Hamley I. W., Martino Adami P. V., Morelli L., Castano E. M. Proteolytically inactive insulin-degrading enzyme inhibits amyloid formation yielding non-neurotoxic Aβ peptide aggregates. PLoS One. 2013;8(4, article e59113) doi: 10.1371/journal.pone.0059113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhao Z., Xiang Z., Haroutunian V., Buxbaum J. D., Stetka B., Pasinetti G. M. Insulin degrading enzyme activity selectively decreases in the hippocampal formation of cases at high risk to develop Alzheimer’s disease. Neurobiology Aging. 2007;28(6):824–830. doi: 10.1016/j.neurobiolaging.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 68.Shinall H., Song E. S., Hersh L. B. Susceptibility of amyloid beta peptide degrading enzymes to oxidative damage: a potential Alzheimer’s disease spiral. Biochemistry. 2005;44(46):15345–15350. doi: 10.1021/bi050650l. [DOI] [PubMed] [Google Scholar]
- 69.Farris W., Mansourian S., Chang Y., et al. Insulin-degrading enzyme regulates the levels of insulin, amyloid-protein, and the-amyloid precursor protein intracellular domain in vivo. Proceedings of the National Academic of Sciences of the United States of America. 2003;100(7):4162–4167. doi: 10.1073/pnas.0230450100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Miller B. C., Eckman E. A., Sambamurti K., et al. Amyloid-β peptide levels in brain are inversely correlated with insulysin activity levels in vivo. Proceedings of the National Academic of Sciences of the United States of America. 2003;100(10):6221–6226. doi: 10.1073/pnas.1031520100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen J. M., Dando P. M., Rawlings N. D., et al. Cloning, isolation, and characterization of mammalian legumain, an asparaginyl endopeptidase. Journal of Biological Chemistry. 1997;272(12):8090–8098. doi: 10.1074/jbc.272.12.8090. [DOI] [PubMed] [Google Scholar]
- 72.Chen J. M., Dando P. M., Stevens R. A., Fortunato M., Barrett A. J. Cloning and expression of mouse legumain, a lysosomal endopeptidase. Biochemical Journal. 1998;335(1):111–117. doi: 10.1042/bj3350111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dando P. M., Fortunato M., Smith L., Knight C. G., McKendrick J. E., Barrett A. J. Pig kidney legumain: an asparaginyl endopeptidase with restricted specificity. Biochemical Journal. 1999;339(3):743–749. doi: 10.1042/bj3390743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Manoury B., Hewitt E. W., Morrice N., Dando P. M., Barrett A. J., Watts C. An asparaginyl endopeptidase processes a microbial antigen for class II MHC presentation. Nature. 1998;396(6712):695–699. doi: 10.1038/25379. [DOI] [PubMed] [Google Scholar]
- 75.Chen J. M., Rawlings N. D., Stevens R. A., Barrett A. J. Identification of the active site of legumain links it to caspases, clostripain and gingipains in a new clan of cysteine endopeptidases. FEBS Letters. 1998;441(3):361–365. doi: 10.1016/S0014-5793(98)01574-9. [DOI] [PubMed] [Google Scholar]
- 76.Li D. N., Matthews S. P., Antoniou A. N., Mazzeo D., Watts C. Multistep autoactivation of asparaginyl endopeptidase in vitro and in vivo. Journal of Biological Chemistry. 2003;278(40):38980–38990. doi: 10.1074/jbc.M305930200. [DOI] [PubMed] [Google Scholar]
- 77.Dall E., Brandstetter H. Structure and function of legumain in health and disease. Biochimie. 2016;122(122):126–150. doi: 10.1016/j.biochi.2015.09.022. [DOI] [PubMed] [Google Scholar]
- 78.Zhang Z., Song M., Liu X., et al. Cleavage of tau by asparagine endopeptidase mediates the neurofibrillary pathology in Alzheimer’s disease. Nature Medicine. 2014;20(11):1254–1262. doi: 10.1038/nm.3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang Z., Song M., Liu X., et al. Delta-secretase cleaves amyloid precursor protein and regulates the pathogenesis in Alzheimer’s disease. Nature Communications. 2015;6(1, article 8762) doi: 10.1038/ncomms9762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gao J., Li K., Du L., Yin H., Tan X., Yang Z. Deletion of asparagine endopeptidase reduces anxiety- and depressive-like behaviors and improves abilities of spatial cognition in mice. Brain Research Bulletin. 2018;142:147–155. doi: 10.1016/j.brainresbull.2018.07.010. [DOI] [PubMed] [Google Scholar]
- 81.Basurto-Islas G., Grundke-Iqbal I., Tung Y. C., Liu F., Iqbal K. Activation of asparaginyl endopeptidase leads to tau hyperphosphorylation in Alzheimer disease. Journal of Biological Chemistry. 2013;288(24):17495–17507. doi: 10.1074/jbc.M112.446070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yates C. M., Butterworth J., Tennant M. C., Gordon A. Enzyme activities in relation to pH and lactate in postmortem brain in Alzheimer-type and other dementias. Journal of Neurochemistry. 1990;55(5):1624–1630. doi: 10.1111/j.1471-4159.1990.tb04948.x. [DOI] [PubMed] [Google Scholar]
- 83.Fang B., Wang D., Huang M., Yu G., Li H. Hypothesis on the relationship between the change in intracellular pH and incidence of sporadic Alzheimer’s disease or vascular dementia. International Journal of Neuroscience. 2010;120(9):591–595. doi: 10.3109/00207454.2010.505353. [DOI] [PubMed] [Google Scholar]
- 84.Pirchl M., Humpel C. Does acidosis in brain play a role in Alzheimer’s disease? Neuropsychiatry. 2009;23(3):187–192. [PubMed] [Google Scholar]
- 85.Wang Z. H., Liu P., Liu X., Yu S. P., Wang J. Z., Ye K. Delta-secretase (AEP) mediates tau-splicing imbalance and accelerates cognitive decline in tauopathies. Journal of Experimental Medicine. 2018;215(12):3038–3056. doi: 10.1084/jem.20180539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang Z. H., Liu P., Liu X., et al. Delta-secretase phosphorylation by SRPK2 enhances its enzymatic activity, provoking pathogenesis in Alzheimer’s disease. Molecular Cell. 2017;67(5):812–825.e5. doi: 10.1016/j.molcel.2017.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Xiang J., Wang Z. H., Ahn E. H., et al. Delta-secretase-cleaved Tau antagonizes TrkB neurotrophic signalings, mediating Alzheimer’s disease pathologies. Proceedings of the National Academic of Sciences of the United States of America. 2019;116(18):9094–9102. doi: 10.1073/pnas.1901348116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kang S. S., Ahn E. H., Zhang Z., et al. α-Synuclein stimulation of monoamine oxidase-B and legumain protease mediates the pathology of Parkinson’s disease. The EMBO Journal. 2018;37(12, article e98878) doi: 10.15252/embj.201798878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Luo S., Kang S. S., Wang Z. H., et al. Akt phosphorylates NQO1 and triggers its degradation, abolishing its antioxidative activities in Parkinson’s disease. Journal of Neuroscience. 2019;39(37):7291–7305. doi: 10.1523/JNEUROSCI.0625-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Huovila A. P., Turner A. J., Pelto-Huikko M., Karkkainen I., Ortiz R. M. Shedding light on ADAM metalloproteinases. Trends in Biochemical Sciences. 2005;30(7):413–422. doi: 10.1016/j.tibs.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 91.Endres K., Deller R. Regulation of α-secretase ADAM10 in vitro and in vivo: genetic, epigenetic, and protein-based mechanisms. Frontiers in Molecular Neuroscience. 2017;10, article 56 doi: 10.3389/fnmol.2017.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wolfsberg T. G., Primakoff P., Myles D. G., White J. M. ADAM, a novel family of membrane proteins containing a disintegrin and metalloprotease domain: multipotential functions in cell-cell and cell-matrix interactions. Journal of Cell Biology. 1995;131(2):275–278. doi: 10.1083/jcb.131.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Corbett G. T., Gonzalez F. J., Pahan K. Activation of peroxisome proliferator-activated receptor α stimulates ADAM10-mediated proteolysis of APP. Proceedings of the National Academic of Sciences of the United States of America. 2015;112(27):8445–8450. doi: 10.1073/pnas.1504890112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Marcello E., Borroni B., Pelucchi S., Gardoni F., Di Luca M. ADAM10 as a therapeutic target for brain diseases: from developmental disorders to Alzheimer’s disease. Expert Opinion on Therapeutic Targets. 2017;21(11):1017–1026. doi: 10.1080/14728222.2017.1386176. [DOI] [PubMed] [Google Scholar]
- 95.Lammich S., Kojro E., Postina R., et al. Constitutive and regulated-secretase cleavage of Alzheimer's amyloid precursor protein by a disintegrin metalloprotease. Proceedings of the National Academic of Sciences of the United States of America. 1999;96(7):3922–3927. doi: 10.1073/pnas.96.7.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Marcello E., Saraceno C., Musardo S., et al. Endocytosis of synaptic ADAM10 in neuronal plasticity and Alzheimer’s disease. Journal of Clinical Investigation. 2013;123(6):2523–2538. doi: 10.1172/JCI65401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Prox J., Bernreuther C., Altmeppen H., et al. Postnatal disruption of the disintegrin/metalloproteinase ADAM10 in brain causes epileptic seizures, learning deficits, altered spine morphology, and defective synaptic functions. Journal of Neuroscience. 2013;33(32):12915–12928. doi: 10.1523/JNEUROSCI.5910-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Iwata N., Tsubuki S., Takaki Y., et al. Identification of the major Aβ1-42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nature Medicine. 2000;6(2):143–150. doi: 10.1038/72237. [DOI] [PubMed] [Google Scholar]
- 99.Sandoval K. E., Farr S. A., Banks W. A., Crider A. M., Morley J. E., Witt K. A. Somatostatin receptor subtype-4 agonist NNC 26-9100 decreases extracellular and intracellular Aβ1-42 trimers. European Journal of Pharmacology. 2012;683(1-3):116–124. doi: 10.1016/j.ejphar.2012.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sandoval K. E., Farr S. A., Banks W. A., Crider A. M., Morley J. E., Witt K. A. Somatostatin receptor subtype-4 agonist NNC 26-9100 mitigates the effect of soluble Aβ42 oligomers via a metalloproteinase-dependent mechanism. Brain Research. 2013;1520:145–156. doi: 10.1016/j.brainres.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Belyaev N. D., Nalivaeva N. N., Makova N. Z., Turner A. J. Neprilysin gene expression requires binding of the amyloid precursor protein intracellular domain to its promoter: implications for Alzheimer disease. EMBO Reports. 2008;10(1):94–100. doi: 10.1038/embor.2008.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nalivaeva N. N., Belyaev N. D., Lewis D. I., et al. Effect of sodium valproate administration on brain neprilysin expression and memory in rats. Journal of Molecular Neuroscience. 2012;46(3):569–577. doi: 10.1007/s12031-011-9644-x. [DOI] [PubMed] [Google Scholar]
- 103.Eisele Y. S., Baumann M., Klebl B., Nordhammer C., Jucker M., Kilger E. Gleevec increases levels of the amyloid precursor protein intracellular domain and of the amyloid-β degrading enzyme neprilysin. Molecular Biology of the Cell. 2007;18(9):3591–3600. doi: 10.1091/mbc.e07-01-0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Klein C., Roussel G., Brun S., et al. 5-HIAA induces neprilysin to ameliorate pathophysiology and symptoms in a mouse model for Alzheimer’s disease. Acta Neuropathologica Communications. 2018;6(1, article 136) doi: 10.1186/s40478-018-0640-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Oefner C., D'Arcy A., Hennig M., Winkler F. K., Dale G. E. Structure of human neutral endopeptidase (neprilysin) complexed with phosphoramidon. Journal of Molecular Biology. 2000;296(2):341–349. doi: 10.1006/jmbi.1999.3492. [DOI] [PubMed] [Google Scholar]
- 106.Sahli S., Stump B., Welti T., et al. Structure-based design, synthesis, and in vitro evaluation of nonpeptidic neprilysin inhibitors. ChemBioChem. 2004;5(7):996–1000. doi: 10.1002/cbic.200400005. [DOI] [PubMed] [Google Scholar]
- 107.Gu J., Noe A., Chandra P., et al. Pharmacokinetics and pharmacodynamics of LCZ696, a novel dual-acting angiotensin receptor-neprilysin inhibitor (ARNi) The Journal of Clinical Pharmacology. 2010;50(4):401–414. doi: 10.1177/0091270009343932. [DOI] [PubMed] [Google Scholar]
- 108.Brown P. C. Center for Drug Evaluation and Research. Application Number: 207620Orig1s000. Pharmacology Review. FDA. 2015. 2020. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/207620Orig1s000PharmR.pdf.
- 109.Song E. S., Juliano M. A., Juliano L., Hersh L. B. Substrate activation of insulin-degrading enzyme (insulysin) Journal of Biological Chemistry. 2003;278(50):49789–49794. doi: 10.1074/jbc.M308983200. [DOI] [PubMed] [Google Scholar]
- 110.Leissring M. A., Malito E., Hedouin S., et al. Designed inhibitors of insulin-degrading enzyme regulate the catabolism and activity of insulin. PLoS One. 2010;5(5, article e10504) doi: 10.1371/journal.pone.0010504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Deprez-Poulain R., Hennuyer N., Bosc D., et al. Catalytic site inhibition of insulin-degrading enzyme by a small molecule induces glucose intolerance in mice. Nature Communications. 2015;6(1, article 8250) doi: 10.1038/ncomms9250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Costes S., Butler P. C. Insulin-degrading enzyme inhibition, a novel therapy for type 2 diabetes? Cell Metabolism. 2014;20(2):201–203. doi: 10.1016/j.cmet.2014.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Durham T. B., Toth J. L., Klimkowski V. J., et al. Dual exosite-binding inhibitors of insulin-degrading enzyme challenge its role as the primary mediator of insulin clearance in vivo. Journal of Biological Chemistry. 2015;290(33):20044–20059. doi: 10.1074/jbc.M115.638205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Charton J., Gauriot M., Guo Q., et al. Imidazole-derived 2-[N-carbamoylmethyl-alkylamino]acetic acids, substrate-dependent modulators of insulin-degrading enzyme in amyloid-β hydrolysis. European Journal of Medicinal Chemistry. 2014;79:184–193. doi: 10.1016/j.ejmech.2014.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bannister T. D., Wang H., Abdul-Hay S. O., et al. ML345, a small-molecule inhibitor of the insulin-degrading enzyme (IDE). Probe Reports from the NIH Molecular Libraries Program. Bethesda (MD): National Center for Biotechnology Information (US); 2010. [PubMed] [Google Scholar]
- 116.Abdul-Hay S. O., Bannister T. D., Wang H., et al. Selective targeting of extracellular insulin-degrading enzyme by quasi-irreversible thiol-modifying inhibitors. ACS Chemical Biology. 2015;10(12):2716–2724. doi: 10.1021/acschembio.5b00334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Dall E., Brandstetter H. Mechanistic and structural studies on legumain explain its zymogenicity, distinct activation pathways, and regulation. Proceedings of the National Academic of Sciences of the United States of America. 2013;110(27):10940–10945. doi: 10.1073/pnas.1300686110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Liu Y., Bajjuri K. M., Liu C., Sinha S. C. Targeting cell surface αvβ3 integrin increases therapeutic efficacies of a legumain protease-activated auristatin prodrug. Molecular Pharmaceutics. 2012;9(1):168–175. doi: 10.1021/mp200434n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Berven L., Johansen H. T., Solberg R., Kolset S. O., Samuelsen A. B. Autoactivation of prolegumain is accelerated by glycosaminoglycans. Biochimie. 2013;95(4):772–781. doi: 10.1016/j.biochi.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 120.Lunde N. N., Haugen M. H., Bodin Larsen K. B., et al. Glycosylation is important for legumain localization and processing to active forms but not for cystatin E/M inhibitory functions. Biochimie. 2017;139:27–37. doi: 10.1016/j.biochi.2017.05.009. [DOI] [PubMed] [Google Scholar]
- 121.Zhang Z., Tian Y., Ye K. δ-Secretase in neurodegenerative diseases: mechanisms, regulators and therapeutic opportunities. Translational Neurodegeneration. 2020;9(1) doi: 10.1186/s40035-019-0179-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Liu Z., Jang S. W., Liu X., et al. Neuroprotective actions of PIKE-L by inhibition of SET proteolytic degradation by asparagine endopeptidase. Molecular Cell. 2008;29(6):665–678. doi: 10.1016/j.molcel.2008.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Saska I., Gillon A. D., Hatsugai N., et al. An asparaginyl endopeptidase mediates in vivo protein backbone cyclization. Journal of Biological Chemistry. 2007;282(40):29721–29728. doi: 10.1074/jbc.M705185200. [DOI] [PubMed] [Google Scholar]
- 124.Harris K. S., Guarino R. F., Dissanayake R. S., et al. A suite of kinetically superior AEP ligases can cyclise an intrinsically disordered protein. Scientific Reports. 2019;9(1, article 10820) doi: 10.1038/s41598-019-47273-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Alvarez-Fernandez M., Barrett A. J., Gerhartz B., Dando P. M., Ni J., Abrahamson M. Inhibition of mammalian legumain by some cystatins is due to a novel second reactive site. Journal of Biological Chemistry. 1999;274(27):19195–19203. doi: 10.1074/jbc.274.27.19195. [DOI] [PubMed] [Google Scholar]
- 126.Turk V., Bode W. The cystatins: protein inhibitors of cysteine proteinases. FEBS Letters. 1991;285(2):213–219. doi: 10.1016/0014-5793(91)80804-C. [DOI] [PubMed] [Google Scholar]
- 127.Abrahamson M., Barrett A. J., Salvesen G., Grubb A. Isolation of six cysteine proteinase inhibitors from human urine. Their physicochemical and enzyme kinetic properties and concentrations in biological fluids. Journal of Biological Chemistry. 1986;261(24):11282–11289. [PubMed] [Google Scholar]
- 128.Turk V., Stoka V., Vasiljeva O., et al. Cysteine cathepsins: from structure, function and regulation to new frontiers. Biochimica et Biophysica Acta. 2012;1824(1):68–88. doi: 10.1016/j.bbapap.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Cheng T., Hitomi K., van Vlijmen-Willems I. M. J. J., et al. Cystatin M/E is a high affinity inhibitor of cathepsin V and cathepsin L by a reactive site that is distinct from the legumain-binding site. Journal of Biological Chemistry. 2006;281(23):15893–15899. doi: 10.1074/jbc.M600694200. [DOI] [PubMed] [Google Scholar]
- 130.Ni J., Abrahamson M., Zhang M., et al. Cystatin E is a novel human cysteine proteinase inhibitor with structural resemblance to family 2 cystatins. Journal of Biological Chemistry. 1997;272(16):10853–10858. doi: 10.1074/jbc.272.16.10853. [DOI] [PubMed] [Google Scholar]
- 131.Brzin J., Rogelj B., Popovic T., Strukelj B., Ritonja A. Clitocypin, a new type of cysteine proteinase inhibitor from fruit bodies of mushroom Clitocybe nebularis. Journal of Biological Chemistry. 2000;275(26):20104–20109. doi: 10.1074/jbc.M001392200. [DOI] [PubMed] [Google Scholar]
- 132.Sabotic J., Gaser D., Rogelj B., Gruden K., Strukelj B., Brzin J. Heterogeneity in the cysteine protease inhibitor clitocypin gene family. Biological Chemistry. 2006;387(12):1559–1566. doi: 10.1515/BC.2006.194. [DOI] [PubMed] [Google Scholar]
- 133.Sabotic J., Popovic T., Puizdar V., Brzin J. Macrocypins, a family of cysteine protease inhibitors from the basidiomycete Macrolepiota procera. FEBS Journal. 2009;276(16):4334–4345. doi: 10.1111/j.1742-4658.2009.07138.x. [DOI] [PubMed] [Google Scholar]
- 134.Renko M., Sabotic J., Mihelic M., Brzin J., Kos J., Turk D. Versatile loops in mycocypins inhibit three protease families. Journal of Biological Chemistry. 2009;285(1):308–316. doi: 10.1074/jbc.M109.043331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kuroyanagi M., Nishimura M., Hara-Nishimura I. Activation of Arabidopsis vacuolar processing enzyme by self-catalytic removal of an auto-inhibitory domain of the C-terminal propeptide. Plant and Cell Physiology. 2002;43(2):143–151. doi: 10.1093/pcp/pcf035. [DOI] [PubMed] [Google Scholar]
- 136.Dall E., Fegg J. C., Briza P., Brandstetter H. Structure and mechanism of an aspartimide-dependent peptide ligase in human legumain. Angewandte Chemie International Edition. 2015;54(10):2917–2921. doi: 10.1002/anie.201409135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zhao L., Hua T., Crowley C., et al. Structural analysis of asparaginyl endopeptidase reveals the activation mechanism and a reversible intermediate maturation stage. Cell Research. 2014;24(3):344–358. doi: 10.1038/cr.2014.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lee J., Bogyo M. Synthesis and evaluation of aza-peptidyl inhibitors of the lysosomal asparaginyl endopeptidase, legumain. Bioorganic & Medicinal Chemistry Letters. 2012;22(3):1340–1343. doi: 10.1016/j.bmcl.2011.12.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Xu C., Cao L., Liu J., et al. Suppression of asparaginyl endopeptidase inhibits polyomavirus middle T antigen-induced tumor formation and metastasis. Oncology Research. 2017;25(3):407–415. doi: 10.3727/096504016X14743350548249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yao P., Ding Y., Han Z., et al. Suppression of asparaginyl endopeptidase attenuates breast cancer-induced bone pain through inhibition of neurotrophin receptors. Molecular Pain. 2017;13:1–10. doi: 10.1177/1744806917708127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chen C., Wang Z., Zhang Z., et al. The prodrug of 7,8-dihydroxyflavone development and therapeutic efficacy for treating Alzheimer’s disease. Proceedings of the National Academic of Sciences of the United States of America. 2018;115(3):578–583. doi: 10.1073/pnas.1718683115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Loak K., Li D. N., Manoury B., et al. Novel cell-permeable acyloxymethylketone inhibitors of asparaginyl endopeptidase. Biological Chemistry. 2003;384(8):1239–1246. doi: 10.1515/BC.2003.136. [DOI] [PubMed] [Google Scholar]
- 143.Zhang Z., Obianyo O., Dall E., et al. Inhibition of delta-secretase improves cognitive functions in mouse models of Alzheimer’s disease. Nature Communications. 2017;8(1, article 14740) doi: 10.1038/ncomms14740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zheng C., Chen G., Tan Y., et al. TRH analog, taltirelin protects dopaminergic neurons from neurotoxicity of MPTP and rotenone. Frontiers in Cellular Neuroscience. 2018;12, article 485 doi: 10.3389/fncel.2018.00485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kinoshita K., Nagao T., Ono H. Effects of TA-0910, an orally active TRH analog, on the spinal reflex in spinal rats. Neuropharmacology. 1994;33(10):1183–1188. doi: 10.1016/S0028-3908(05)80008-5. [DOI] [PubMed] [Google Scholar]
- 146.Konoshita K., Yamamura M., Sugihara J., Suzuki M., Matsuoka Y. Taltirelin hydrate (TA-0910): an orally active thyrotropin-releasing hormone mimetic agent with multiple actions. CNS Drug Reviews. 2006;4(1):25–41. [Google Scholar]
- 147.Yamane T., Takeuchi K., Yamamoto Y., et al. Legumain from bovine kidney: its purification, molecular cloning, immunohistochemical localization and degradation of annexin II and vitamin D-binding protein. Biochimica et Biophysica Acta. 2002;1596(1):108–120. doi: 10.1016/S0167-4838(02)00209-1. [DOI] [PubMed] [Google Scholar]
- 148.Reinhardt S., Schuck F., Grösgen S., et al. Unfolded protein response signaling by transcription factor XBP-1 regulates ADAM10 and is affected in Alzheimer’s disease. FASEB Journal. 2013;28(2):978–997. doi: 10.1096/fj.13-234864. [DOI] [PubMed] [Google Scholar]
- 149.Sarlak G., Htoo H. H., Hernandez J. F., et al. Sox2 functionally interacts with βAPP, the βAPP intracellular domain and ADAM10 at a transcriptional level in human cells. Neuroscience. 2016;312:153–164. doi: 10.1016/j.neuroscience.2015.11.022. [DOI] [PubMed] [Google Scholar]
- 150.Shukla M., Htoo H. H., Wintachai P., et al. Melatonin stimulates the nonamyloidogenic processing ofβAPP through the positive transcriptional regulation of ADAM10 and ADAM17. Journal of Pineal Research. 2015;58(2):151–165. doi: 10.1111/jpi.12200. [DOI] [PubMed] [Google Scholar]
- 151.Shukla M., Govitrapong P., Boontem P., Reiter R. J., Satayavivad J. Mechanisms of melatonin in alleviating Alzheimer’s disease. Current Neuropharmacology. 2017;15(7):1010–1031. doi: 10.2174/1570159X15666170313123454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Etcheberrigaray R., Tan M., Dewachter I., et al. Therapeutic effects of PKC activators in Alzheimer’s disease transgenic mice. Proceedings of the National Academic of Sciences of the United States of America. 2004;101(30):11141–11146. doi: 10.1073/pnas.0403921101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Wetzel S., Seipold L., Saftig P. The metalloproteinase ADAM10: a useful therapeutic target? Biochimica et Biophysica Acta - Molecular Cell Research. 2017;1864(11):2071–2081. doi: 10.1016/j.bbamcr.2017.06.005. [DOI] [PubMed] [Google Scholar]
- 154.Prinzen C., Muller U., Endres K., Fahrenholz F., Postina R. Genomic structure and functional characterization of the human ADAM10 promoter. FASEB Journal. 2005;19(11):1522–1524. doi: 10.1096/fj.04-3619fje. [DOI] [PubMed] [Google Scholar]
- 155.Tippmann F., Hundt J., Schneider A., Endres K., Fahrenholz F. Up-regulation of the α-secretase ADAM10 by retinoic acid receptors and acitretin. FASEB Journal. 2009;23(6):1643–1654. doi: 10.1096/fj.08-121392. [DOI] [PubMed] [Google Scholar]
- 156.Vincent B. Regulation of the α-secretase ADAM10 at transcriptional, translational and post-translational levels. Brain Research Bulletin. 2016;126, Part 2:154–169. doi: 10.1016/j.brainresbull.2016.03.020. [DOI] [PubMed] [Google Scholar]
- 157.Fukasawa H., Nakagomi M., Yamagata N., et al. Tamibarotene: a candidate retinoid drug for Alzheimer’s disease. Biological and Pharmaceutical Bulletin. 2012;35(8):1206–1212. doi: 10.1248/bpb.b12-00314. [DOI] [PubMed] [Google Scholar]
- 158.Kitaoka K., Shimizu N., Ono K., et al. The retinoic acid receptor agonist Am80 increases hippocampal ADAM10 in aged SAMP8 mice. Neuropharmacology. 2013;72:58–65. doi: 10.1016/j.neuropharm.2013.04.009. [DOI] [PubMed] [Google Scholar]
- 159.Meineck M., Schuck F., Abdelfatah S., Efferth T., Endres K. Identification of phlogacantholide C as a novel ADAM10 enhancer from traditional Chinese medicinal plants. Medicines. 2016;3(4):p. 30. doi: 10.3390/medicines3040030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Karuppagounder S. S., Pinto J. T., Xu H., Chen H. L., Beal M. F., Gibson G. E. Dietary supplementation with resveratrol reduces plaque pathology in a transgenic model of Alzheimer's disease. Neurochemistry International. 2009;54(2):111–118. doi: 10.1016/j.neuint.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Marcade M., Bourdin J., Loiseau N., et al. Etazolate, a neuroprotective drug linking GABAAreceptor pharmacology to amyloid precursor protein processing. Journal of Neurochemistry. 2008;106(1):392–404. doi: 10.1111/j.1471-4159.2008.05396.x. [DOI] [PubMed] [Google Scholar]
- 162.Moss M. L., Bomar M., Liu Q., et al. The ADAM10 prodomain is a specific inhibitor of ADAM10 proteolytic activity and inhibits cellular shedding events. Journal of Biological Chemistry. 2007;282(49):35712–35721. doi: 10.1074/jbc.M703231200. [DOI] [PubMed] [Google Scholar]
- 163.Madoux F., Dreymuller D., Pettiloud J. P., et al. Discovery of an enzyme and substrate selective inhibitor of ADAM10 using an exosite-binding glycosylated substrate. Scientific Reports. 2016;6(1):p. 11. doi: 10.1038/s41598-016-0013-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Speck N., Brandsch C., Schmidt N., et al. The antiatherogenic effect of fish oil in male mice is associated with a diminished release of endothelial ADAM17 and ADAM10 substrates. Journal of Nutrition. 2015;145(6):1218–1226. doi: 10.3945/jn.115.211375. [DOI] [PubMed] [Google Scholar]
- 165.Kato T., Hagiyama M., Ito A. Renal ADAM10 and 17: their physiological and medical meanings. Frontiers in Cell and Developmental Biology. 2018;6 doi: 10.3389/fcell.2018.00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Shackleton B., Crawford F., Bachmeier C. Inhibition of ADAM10 promotes the clearance of Aβ across the BBB by reducing LRP1 ectodomain shedding. Fluids and Barriers of the CNS. 2016;13(1):p. 14. doi: 10.1186/s12987-016-0038-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Zocchi M. R., Camodeca C., Nuti E., et al. ADAM10 new selective inhibitors reduce NKG2D ligand release sensitizing Hodgkin lymphoma cells to NKG2D-mediated killing. Oncoimmunology. 2015;5(5, article e1123367) doi: 10.1080/2162402X.2015.1123367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Tosetti F., Venè R., Camodeca C., et al. Specific ADAM10 inhibitors localize in exosome-like vesicles released by Hodgkin lymphoma and stromal cells and prevent sheddase activity carried to bystander cells. Oncoimmunology. 2018;7(5, article e1421889) doi: 10.1080/2162402X.2017.1421889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Camodeca C., Nuti E., Tosetti F., et al. Synthesis and in vitro evaluation of ADAM10 and ADAM17 highly selective bioimaging probes. ChemMedChem. 2018;13(19):2119–2131. doi: 10.1002/cmdc.201800482. [DOI] [PubMed] [Google Scholar]
- 170.Moss M. L., Minond D. Recent advances in ADAM17 research: a promising target for cancer and inflammation. Mediators of Inflammation. 2017;2017:21. doi: 10.1155/2017/9673537.9673537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Zhou B.-B. S., Peyton M., He B., et al. Targeting ADAM-mediated ligand cleavage to inhibit HER3 and EGFR pathways in non-small cell lung cancer. Cancer Cell. 2006;10(1):39–50. doi: 10.1016/j.ccr.2006.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Mahasenan K. V., Ding D., Gao M., et al. In search of selectivity in inhibition of ADAM10. ACS Medicinal Chemistry Letters. 2018;9(7):708–713. doi: 10.1021/acsmedchemlett.8b00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Zhang S., Salemi J., Hou H., et al. Rapamycin promotes β-amyloid production via ADAM-10 inhibition. Biochemical and Biophysical Research Communications. 2010;398(3):337–341. doi: 10.1016/j.bbrc.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Soundararajan R., Sayat R., Robertson G. S., Marignani P. A. Triptolide: an inhibitor of a disintegrin and metalloproteinase 10 (ADAM10) in cancer cells. Cancer Biology & Therapy. 2009;8(21):2054–2062. doi: 10.4161/cbt.8.21.9803. [DOI] [PubMed] [Google Scholar]