

Abstract
Purpose:
Identifying cancers with high PI3K pathway activity is critical for treatment selection and eligibility into clinical trials of PI3K inhibitors. Assessments of tumor signaling pathway activity need to consider intratumoral heterogeneity and multiple regulatory nodes.
Methods:
We established a novel, mechanistically informed approach to assessing tumor signaling pathways by quantifying single cell-level multiplex immunofluorescence using custom algorithms. In a proof-of-concept study, we stained archival formalin-fixed, paraffin-embedded (FFPE) tissue from patients with primary prostate cancer in two prospective cohort studies, the Health Professionals Follow-up Study and the Physicians’ Health Study. PTEN, stathmin, and phospho-S6 were quantified on 14 tissue microarrays as indicators of PI3K activation to derive cell-level PI3K scores.
Results:
In 1,001 men, 988,254 tumor cells were assessed (median, 743 per tumor; interquartile range, 290 to 1377). PI3K scores were higher in tumors with PTEN loss scored by a pathologist, higher Gleason grade, and a new, validated bulk PI3K transcriptional signature. Unsupervised machine-learning approaches resulted in similar clustering. Within-tumor heterogeneity in cell-level PI3K scores was high. During long-term follow-up (median, 15.3 years), rates of progression to metastases and death from prostate cancer were twice as high in the highest quartile of PI3K activation compared to the lowest quartile (hazard ratio, 2.04; 95% confidence interval, 1.13 to 3.68).
Conclusion:
Our novel pathway-focused approach to quantifying single cell-level immunofluorescence in FFPE tissue identifies prostate tumors with PI3K pathway activation that are more aggressive and may respond to pathway inhibitors.
Keywords: pathway activation, single-cell, PI3K, multiplex immunofluorescence
Introduction
Identifying tumors with activation of oncogenic signaling pathways is challenging because of the complexity of pathway regulation, downstream effectors, and genomic aberrations that result in functional activation of a pathway. The feasibility of assessing pathway activation in single cells, for example on a transcriptional level, has been amply demonstrated (1). However, biomarker assessments of tissue from cancer patients continue to rely on bulk assessments, and single-cell approaches are typically not integrated into scalable workflows that preserve spatial information—i.e., the histological structure of the tissue. Being able to leverage single cell-level information on pathway activation in cancer tissue obtained in routine clinical workflows holds the promise to more accurately identify patients whose tumors have activation of oncogenic signaling pathways and who might thus have greater benefit from therapeutic interventions targeting these pathways.
A prime example is the phosphoinositide 3-kinase (PI3K) pathway. This key oncogenic signaling pathway is activated in most cancers and drives malignant progression through various downstream mechanisms, including Akt/mammalian target of rapamycin (mTOR) signaling (2,3). More reliably identifying cancers with activation of the PI3K pathway would be paramount for the successful clinical use of PI3K pathway inhibitors. The promise of such a targeted therapy approach has been demonstrated in patients with advanced breast cancer who were eligible for a phase 3 trial if their tumors had a PIK3CA mutation. Treatment with PI3Kα-specific inhibitor prolonged progression-free survival (4). Trials that did not select tumors for PI3K alterations, such as most trials including patients with advanced prostate cancer, demonstrated little activity (5–8). Using an immunohistochemical assessment of PTEN protein expression across the entire tumor as a surrogate of PI3K pathway activation (9,10), a phase 2 trial suggested higher activity of an Akt inhibitor in tumors with PTEN loss than in those with intact PTEN (11). However, it may not be sufficient to define PI3K pathway activation by assessing solely PTEN protein expression with its intrinsically heterogeneous expression patterns, by assessing only DNA-level PI3K pathway alterations (12), or by assessing transcriptional output of just a single downstream node such as FOXO (13).
A more comprehensive assessment of PI3K activity, and of other signaling pathways, should ideally use protein-level information and account both for alternative mechanisms of pathway activation (14) and for between-cell heterogeneity within a tumor from subclonal pathway activation (15). Using the PI3K pathway in prostate cancer as a proof of concept, we piloted and validated a novel approach for determining pathway activation in tumors by quantifying multiple nodes of the pathway activation at a single-cell level using multiplex immunofluorescence (Fig. 1A). We defined a mechanistically informed tumor-level measure of PI3K activation that we applied to archival tissue from two prospective cohort studies of patients with prostate cancer and long-term follow-up for clinically meaningful outcomes.
Figure 1. Methods overview and virtual tissue microarrays for visualization of multiplex immunofluorescence results.
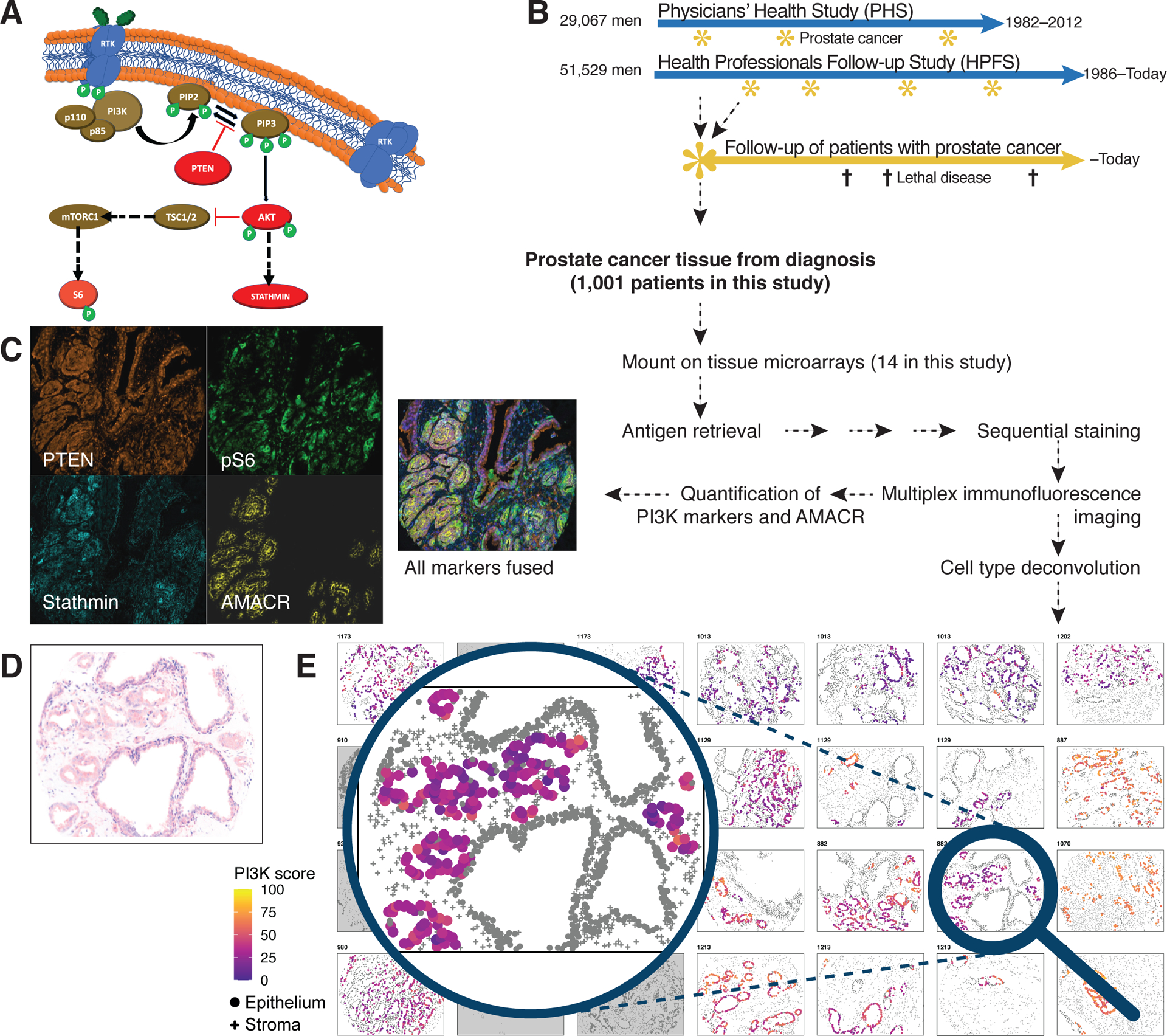
(A) An overview of the PI3K pathway, PTEN, stathmin, and phospho-S6 (pS6).
(B) Design of the prospective prostate cancer cohorts within the Health Professionals Follow-up Study (HPFS) and the Physicians’ Health Study (PHS).
(C)Example raw images from immunofluorescence imaging for PTEN, stathmin, and pS6 as markers of PI3K pathway activity and of AMACR for tumor masking, all from the same tumor core, as well as a fused image of all fluorescence channels.
(D)Hematoxylin–eosin appearance of the core highlighted from the virtual tissue microarray in (E).
(E)Virtual tissue microarray, an algorithm-based reconstruction of the multiplex immunofluorescence data. Cell type assignments from histology-based machine learning (round, epithelial cells; crosses, non-epithelial cells) and tumor cell recognition based on AMACR (colored, tumor cells; gray, non-tumor cells). Cell colors of tumor cells indicate PI3K activity scores.
Methods
Patient cohorts
Men included in this study were diagnosed with non-metastatic prostate cancer during prospective follow-up of two well-defined cohort studies of participants across the United States, the Health Professionals Follow-up Study (HPFS) and the Physicians’ Health Study (PHS). The HPFS is an ongoing cohort study that enrolled 51,529 male health professionals from all 50 U.S. states who were 40–75 years old at baseline in 1986 (16). Participants have been reporting detailed lifestyle, dietary, and medical information through biennial questionnaires. The PHS I and II were randomized-controlled trials of aspirin and vitamin supplements for cancer and cardiovascular prevention, enrolling 29,071 male physicians who were 40–84 years old at baseline in 1982 (17,18). Participants were followed as a prospective cohort after discontinuation of randomized treatment assignments.
New cancer diagnoses reported by the medical professionals in both cohorts were verified and patients were prospectively followed through detailed biennial questionnaires, contact to treating physicians, systematic review of medical records (including for development of metastases), and detailed ascertainment of death causes (19). The clinical outcome was lethal disease (metastases or prostate cancer-specific death).
The research, conducted in accordance with the U.S. Common Rule, was approved by institutional review boards at Harvard T.H. Chan School of Public Health and Partners Healthcare and those of participating registries as required. All participants provided written informed consent.
Tumor specimens, tissue microarrays, and biomarkers
Within the prostate cancer cohorts in HPFS and PHS, a tumor biorepository was developed. Archival formalin-fixed paraffin-embedded tissue from cancer diagnosis was retrieved from treating hospitals and centrally reviewed by genitourinary pathologists, including standardized Gleason grading (20). Tissue microarrays (TMAs) were constructed from three or more 0.6 mm cores of the primary tumor nodule or the nodule with the highest Gleason grade.
TMAs were used for assessment of the PI3K markers PTEN, pS6, and stathmin. Downstream of Akt, a phosphoprotein difficult to assess in archival samples, the ribosomal protein S6 (pS6) is activated via mTOR. pS6 is responsive to PI3K inhibition (21,22). Stathmin is a microtubule-regulating phosphoprotein regulated by PI3K inhibitors (23). We previously validated individual assessments of pS6 and stathmin in archival prostate cancer tissue (14).
Using regular immunohistochemistry, we also used a genomically validated core-level assessment of PTEN loss (9,24), an activator of PI3K signaling (25). Whole-transcriptome expression profiling was performed on a subset of cases using the Affymetrix GeneChip Human Gene 1.0 ST array (Gene Expression Omnibus: GSE62872) (26).
Multiplex immunofluorescence
For fluorescence immunohistochemistry, a multiplexed tyramide signal amplification method was performed on 4-μm sections of the TMAs. Slides were deparaffinized using xylene, a graded ethanol series, 10% neutral buffered formalin for 10 minutes, and washing with distilled water and Tris-buffered saline/polysorbate 5 min thrice each. For antigen retrieval, the tissue was microwaved in citrate buffer at full power for 45 sec and at 20% power for 15 min, followed by washing. Next, the tissue was blocked using protein block (X9090, Dako) and the primary antibody (Supplementary Table 1) was applied for 30 min, followed by washing. The secondary polymer horseradish peroxidase-bound antibody against the primary species was applied for 10 min, followed by washing. Finally, the fluorescent dye-coupled tyramide signal amplification reagents (Perkin Elmer) were applied for 10 min, resulting in stable covalent bounds, followed by washing. The steps of antigen retrieval and blocking were repeated before staining each additional marker. Single-staining slides were included in each antibody cycle as controls.
Development of mechanistically informed PI3K activation scores
TMAs were scanned using a Vectra slide scanner (version 2.0.8, PerkinElmer). InForm software (version 2.1.1, PerkinElmer) was used to unmix signals and analyze the spectral images. Each single stained control slide was imaged to create spectral controls for generating the spectral library. Cores without tumor tissue were excluded based on review of adjacent sections of the same TMAs. To separate epithelial cells from stromal cells, a histology-based machine-learning image segmentation algorithm for tissue and cells provided in the software was trained on representative areas by a pathologist. The resulting segmentation masks were iteratively reviewed for quality and manually edited for further accuracy.
Further analyses were performed using custom algorithms. Analyses of PI3K activation were restricted to cytoplasmic intensity values on tumor cells. Tumors with fewer than 10 tumor cells across all cores were not included. Cell type data were merged with immunofluorescence data at the same coordinates, allowing for ±2 pixels of imprecision between coordinates of cell type and immunofluorescence; tumor cores had a diameter of >1000 pixels.
To reduce batch effects, cell-level immunofluorescence intensities were quantile-normalized across TMAs (27). To give equal weight to all three markers (PTEN, pS6, and stathmin), intensities were transformed into 33 ranks (i.e., each marker for each cell was assigned an intensity from 0 to 33). Cell-level PI3K scores were generated by summing rank-normalized pS6 and stathmin values and subtracting PTEN values, given that PTEN inhibits the PI3K pathway. This summary score is conceptually consistent with our previous approach to assessing PI3K activation using tumor-level averages (14). Scaling to a normal (Gaussian) distribution instead of using ranks had similar results but gave extreme values undue influence. Tumor-level scores of PI3K activation were defined as the median of cell-level PI3K scores across cells on all tumor cores of one tumor (i.e., one patient). For comparison, we formulated a modified tumor-level H-score based on grouping cell-level scores into quartiles across all tumors: H = 100 · Σ(quartiles indices – 1) / cell count; range, 0–300.
Intratumoral heterogeneity in PI3K scores was defined as the proportion of cells within each tumor that did not belong to the quartile of PI3K scores that the tumor was assigned to based on its median PI3K score. For comparison, Shannon’s entropy of quartiles of cell-level PI3K scores was calculated using a maximum likelihood estimator. Spatial clustering of similar PI3K scores within individual cores was assessed using Moran’s I based on Euclidean distances and Getis–Ord global G (28,29), with cells in clusters required to have pairwise distances of less than ~5% of the core span. Spatial analyses used pixel-level cell positions, avoiding grid-based artifacts of lattice-based analyses. To assess whether a small subset of cells with high PI3K scores was sufficient for associations with transcriptomic measures and with lethal disease, the tumor-level score was defined as the median of the up to 100 cells with the highest cell-level PI3K scores, rather than the median of all cells since some tumors had fewer than 100 tumor cells. Methods for unsupervised machine learning-based analyses are described in the Supplementary Text.
Validation of PI3K scores using indicators of PI3K pathway activation on bulk tumors
Validity of PI3K scores was assessed by comparing scores with a core-level PTEN immunohistochemistry (as above) and with transcriptomic signatures of PI3K activation. A signature of PI3K activation specific to primary prostate cancer was developed using The Cancer Genome Atlas (TCGA) (30), restricted to genes that had sufficient variance in the Affymetrix array. Among TCGA tumors with high-quality mRNA (n = 333), genes associated with PTEN copy number loss or mutations in PIK3CA, PIK3CB, or PTEN (ordinal), based on modeling log(RNA expression) using linear regression while adjusting for TMPRSS2:ERG fusion status (binary) (31) and Gleason grade (categorical).
For comparison, as a signature of PI3K inhibition, we used the top 100 genes from a published signature developed in Connectivity Map, which compares treating a library of cell lines with PI3K inhibitors (wortmannin, LY-294002) to treating them with other small molecule inhibitors (32). We also used a transcriptome signature of PTEN protein loss in breast cancer (33). For all three signatures, summary scores were built as más-o-menos scores based on normalized expression levels, subtracting sums of genes known to be downregulated in PI3K-activated tumors from sums of upregulated genes (34).
PI3K scores and lethal disease over long-term follow-up
To assess the strength of association between PI3K scores and rates of lethal disease, Cox regression for time to lethal disease was performed, using time since cancer diagnosis as the timescale. Scores were analyzed both using restricted cubic spline models with four knots to assess for non-linear associations (35) and using categorization in quartiles. Scaled Schoenfeld residuals did not suggest non-proportional hazards. Absolute differences in survival were estimated using restricted mean survival times (36). Effect measure modification on the multiplicative scale by Gleason grade (14), heterogeneity, and spatial autocorrelation/clustering of PI3K scores was assessed using likelihood ratio tests.
Results
Patients, tumors, and cell types
To assess PI3K activity on a single-cell level in archival tumor tissue, we developed a scalable workflow (Fig. 1B–E). Prostate cancer tissue from 1001 men was analyzed on 14 tissue microarrays (TMAs). Median age at prostate cancer diagnosis was 66 years (interquartile range [IQR], 62 to 70 years; Table 1). Most men had clinically localized disease (cT1/T2, 96%) at diagnosis.
Table 1.
Characteristics of patients and tumors from the prospective prostate cancer cohorts within the Health Professionals Follow-up Study (HPFS) and the Physicians’ Health Study (PHS), 1982–2009, by quartiles of the PI3K score.
| By quartiles of PI3K score | |||||
|---|---|---|---|---|---|
| Overall | 1 (Lowest) | 2 | 3 | 4 (Highest) | |
| n | 1001 | 263 | 259 | 237 | 242 |
| PI3K score | 52 (40, 61) | 33 (28, 37) | 47 (44, 50) | 57 (55, 59) | 69 (65, 73) |
| Score of top 50 cells | 66 (57, 75) | 52 (40, 60) | 63 (59, 68) | 70 (64, 75) | 80 (74, 84) |
| Heterogeneity (%) | 48 (31, 58) | 35 (22, 49) | 59 (52, 67) | 55 (47, 62) | 31 (18, 44) |
| Cohort | |||||
| HPFS | 679 (68%) | 176 (67%) | 179 (69%) | 152 (64%) | 172 (71%) |
| PHS | 322 (32%) | 87 (33%) | 80 (31%) | 85 (36%) | 70 (29%) |
| Age at cancer diagnosis | 66.0 (62.0, 69.5) |
65.9 (62.0, 70.0) |
65.0 (61.0, 69.0) |
66.0 (63.0, 69.7) |
66.0 (62.3, 69.7) |
| Year of cancer diagnosis | |||||
| 1982–1989 | 65 (6%) | 18 (7%) | 12 (5%) | 13 (5%) | 22 (9%) |
| 1990–1993 | 215 (21%) | 57 (22%) | 59 (23%) | 50 (21%) | 49 (20%) |
| 1994–2000 | 412 (41%) | 107 (41%) | 101 (39%) | 104 (44%) | 100 (41%) |
| 2001–2009 | 309 (31%) | 81 (31%) | 87 (34%) | 70 (30%) | 71 (29%) |
| cTNM | |||||
| T1/T2 | 942 (96%) | 247 (97%) | 246 (95%) | 225 (96%) | 224 (95%) |
| T3 | 30 (3%) | 7 (3%) | 9 (3%) | 6 (3%) | 8 (3%) |
| T4/N1 | 8 (1%) | 1 (0%) | 3 (1%) | 2 (1%) | 2 (1%) |
| Unknown | 18 | 8 | 1 | 3 | 6 |
| pTNM | |||||
| pT2 | 661 (71%) | 171 (71%) | 186 (74%) | 151 (69%) | 153 (67%) |
| pT3/T3a | 178 (19%) | 42 (18%) | 46 (18%) | 44 (20%) | 46 (20%) |
| pT3b | 64 (7%) | 20 (8%) | 11 (4%) | 13 (6%) | 20 (9%) |
| pT4/N1 | 33 (4%) | 7 (3%) | 7 (3%) | 10 (5%) | 9 (4%) |
| Unknown1 | 65 | 23 | 9 | 19 | 14 |
| Gleason score | |||||
| 6 | 173 (17%) | 49 (19%) | 52 (20%) | 41 (17%) | 31 (13%) |
| 3+4 | 370 (37%) | 99 (38%) | 108 (42%) | 78 (33%) | 85 (35%) |
| 4+3 | 245 (24%) | 64 (24%) | 60 (23%) | 60 (25%) | 61 (25%) |
| 8 | 82 (8%) | 15 (6%) | 12 (5%) | 26 (11%) | 29 (12%) |
| 9–10 | 131 (13%) | 36 (14%) | 27 (10%) | 32 (14%) | 36 (15%) |
| PSA at diagnosis2 [mg/dl] | 7 (5, 10) | 7 (5, 10) | 7 (5, 10) | 6 (5, 10) | 6 (4, 10) |
| Unknown | 89 | 24 | 19 | 16 | 30 |
| Tumor cell count per tumor | 743 (290, 1377) | 740 (271, 1344) | 972 (400, 1536) | 667 (254, 1371) | 670 (211, 1334) |
| PTEN by immunohistochemistry | |||||
| Intact | 625 (74%) | 204 (91%) | 179 (80%) | 137 (70%) | 105 (53%) |
| Heterogeneous loss | 84 (10%) | 14 (6%) | 25 (11%) | 22 (11%) | 23 (12%) |
| Complete loss | 133 (16%) | 6 (3%) | 19 (9%) | 37 (19%) | 71 (36%) |
| Unknown | 159 | 39 | 36 | 41 | 43 |
All data are given as median (interquartile range) or as count (percent).
Includes transurethral resection of the prostate.
Prostate-specific antigen.
With tissue image segmentation, we detected a median of 3376 cells in each tumor (IQR, 1951 to 4426; total, 3.4 million) when combining all available 0.6-mm cores per tumor (median number of cores per tumor, 3; IQR, 2–3). Of these cells, a median of 743 cells per tumor were alpha-methylacyl-CoA racemase (AMACR)-positive epithelial cells (tumor cells; IQR, 290 to 1377; total, 988,254), 512 were AMACR-negative epithelial cells (benign epithelial cells; IQR, 262 to 941), and 1598 were non-epithelial cells (e.g., stroma; IQR, 1009 to 2269; Fig. 2A).
Figure 2. Cell counts and construct validity of the PI3K scores.
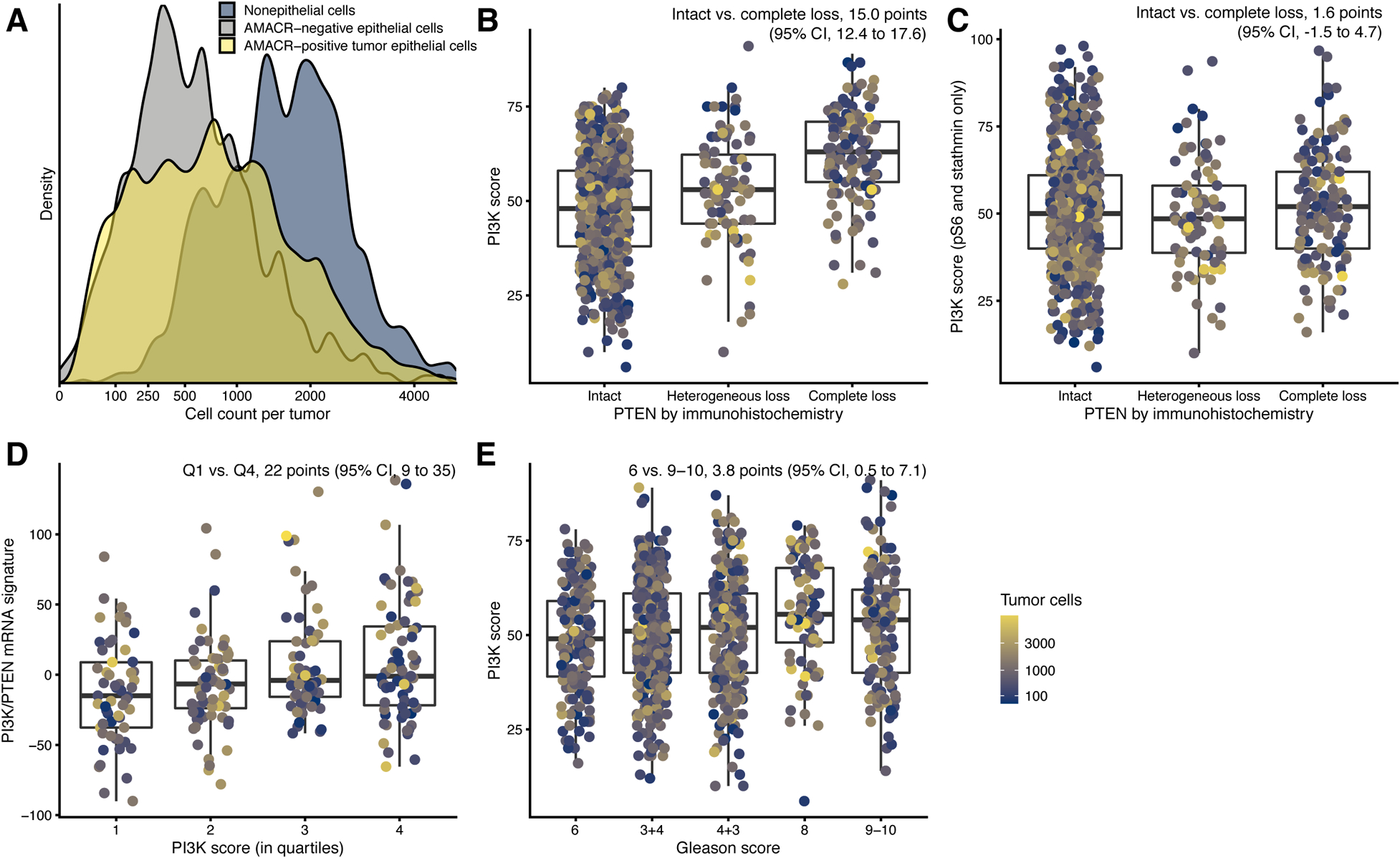
(A) Distribution of cell counts per tumor by cell type: tumor epithelial cells (yellow), non-tumor epithelial cells (gray), and non-epithelial cells (blue), from all cores of each patient.
(B) Tumor-level PTEN status based on a genomically validated immunohistochemistry and PI3K scores.
(C)Tumor-level PTEN status and immunofluorescence scores consisting of pS6 and stathmin.
(D) PI3K scores and the newly developed prostate cancer PI3K/PTEN signature.
(E)Gleason grade and PI3K scores.
In panels B–E, individual data points are shown, with boxes indicating interquartile ranges and the central line the median. Whiskers extend 1.5 interquartile ranges beyond the inner quartiles. In B–G and F–H, color encodes the number of tumor cells per tumor from blue (low) to yellow (high).
Validation of mechanistically informed PI3K activation scores
To assess PI3K pathway activation comprehensively at the tumor level, we calculated mechanistically informed PI3K activation scores that considered protein expression of pS6, stathmin, and PTEN (Fig. 1A) (14). Cell-level immunofluorescence intensities for PTEN, stathmin, and pS6 were corrected for batch effects across TMAs, rank-normalized, and summarized in cell-level PI3K scores. Higher pS6 and stathmin values contributed positively and higher PTEN values contributed negatively to our main measure of PI3K activity, the cell-level PI3K score, which could vary from 1 (lowest) to 100 points (highest). Tumor-level scores of PI3K activation were defined as the median of cell-level PI3K scores across cells on all tumor cores from the same tumor (i.e., one patient); scores were highly correlated with modified H-scores (Fig. S1).
We compared cell-level PI3K scores against other, independently generated indicators of PI3K activity derived from bulk assessments of the tumor. As expected, we observed strong differences in tumor-level PI3K scores between tumors with present vs. absent PTEN expression based on eye-scored immunohistochemical PTEN staining (Fig. 2B). Tumors scored by expert pathologists as “PTEN null,” i.e., those with PTEN loss in all cores (n = 133), had 15.0 point-higher tumor-level PI3K scores (95% CI, 12.4 to 17.6) than those with PTEN present in all cores (n = 625); tumors scored as “heterogeneous loss,” i.e. PTEN loss in just some cores (n = 84), had 5.6 points higher tumor-level PI3K scores (95% CI, 2.4 to 8.7; Fig. 2A).
Importantly, there was no strong association between PTEN status by eye-scoring and our tumor-level PI3K scores that were based on pS6 and stathmin but not on cell-level PTEN (Fig. 2C), suggesting that PTEN alone may not fully identify downstream activation of PI3K signaling, as indicated by high pS6 and stathmin.
As additional measure of PI3K activation, we assessed transcriptional consequences of PI3K activation. First, we used the TCGA to derive a transcriptional signature of PI3K activation, consisting of transcripts specific to tumors with DNA-level alterations that are known to result in PI3K activation in prostate cancer, and we validated this signature in tumors from HPFS/PHS (Table S2; Supplementary Text). Applying this new PI3K transcriptomic signature, we found that tumors in the highest quartile of the tumor-level PI3K score had higher transcriptional scores than tumors in the lowest quartile (Fig. 2D). Second, complementing this transcriptional signature of PI3K activation, we used a transcriptional signature of PI3K inhibition. This signature, derived from comparing cell lines treated with PI3K inhibitors versus other small molecules (32) (Supplementary Text), also tended to differ between tumors with low and high tumor-level PI3K scores (Fig. S2G). Taken together, these observations support the validity of the immunofluorescence-based PI3K scores. However, the observed associations between measures derived from cell-level measurements and the bulk transcriptional measures were modest, highlighting the potential importance of obtaining cell-level data. Tumors with higher Gleason grades had higher tumor-level PI3K scores (Fig. 2E), consistent with the association between PTEN loss and higher tumor dedifferentiation (24). Tumor-level PI3K scores, however, were not associated with the tumor proliferation index Ki-67 (Fig. S3A).
PI3K scores and lethal disease over long-term follow-up
To assess how PI3K activation is related to lethal prostate cancer, we followed the 1,101 patients for up to 28 years after cancer diagnosis (median, 15.3 years). During the 14,785 person-years of follow up for metastasis-free survival, 87 lethal events occurred (metastases or death from prostate cancer). Patients with the highest 25% of tumor-level PI3K scores (highest quartile) had rates of lethal disease twice as high as those in the lowest 25% of scores (unadjusted hazard ratio [HR], 2.04; 95% CI, 1.13 to 3.68; Fig. 3A; Table 2), an association that was only modestly attenuated after adjusting for age at cancer diagnosis and Gleason grade. Over a 20-year period after cancer diagnosis, patients in the highest quartile had a 10-months shorter time to metastases or death from prostate cancer than those in the lowest quartile (95% CI, 2 to 19 months). There was no strong evidence that the association between PI3K scores and lethal disease differed by Gleason scores (pinteraction, 0.33; Table 2).
Figure 3. PI3K activation and lethal prostate cancer.
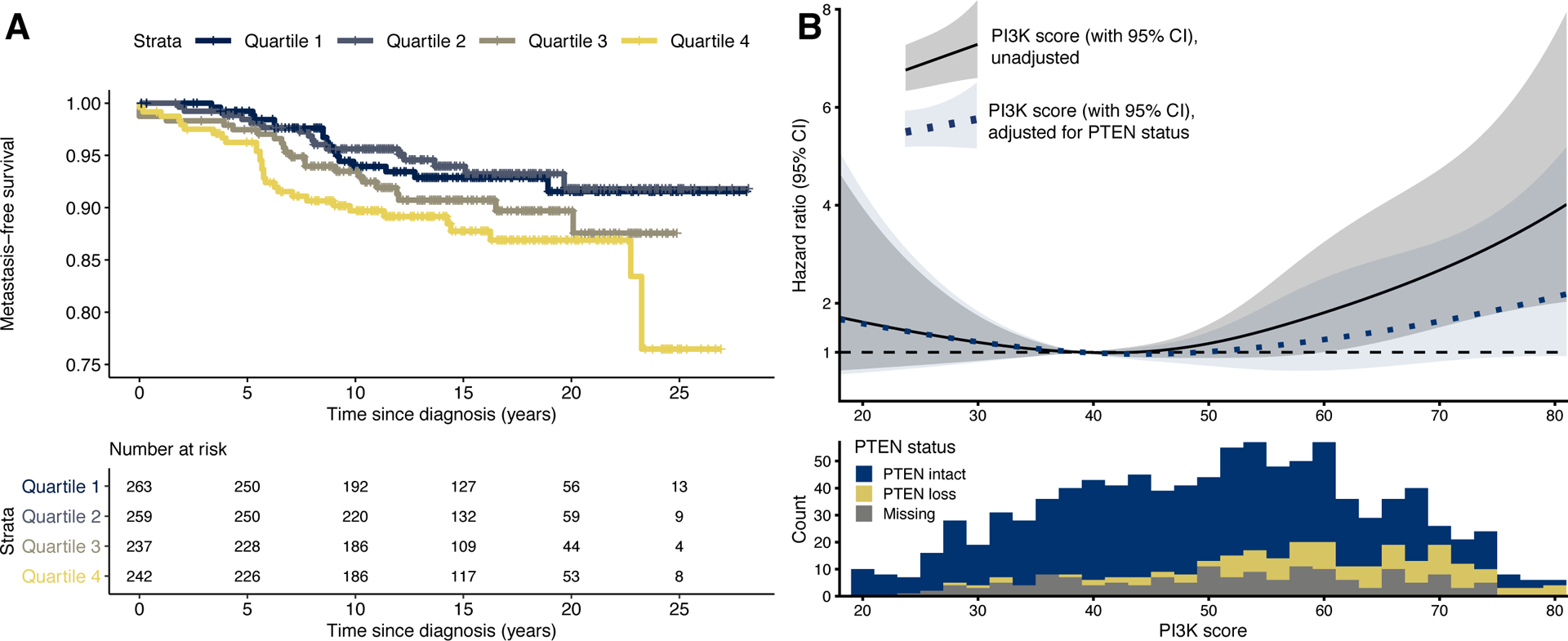
(A) Kaplan-Meier plot for PI3K scores in quartiles and time from cancer diagnosis to lethal disease. For adjusted estimates, see Table 2.
(B) Upper panel: PI3K scores (x axis), modelled continuously using restricted cubic splines, and hazard ratios for lethal disease (y axis). The solid line with gray 95% CI bands is from an unadjusted model; the dotted line with blue 95% CI bands is from a model adjusted for tumor-level PTEN status by immunohistochemistry. The reference value (hazard ratio, 1) is set to the 25th percentile of the PI3K score, a value of 40. Lower panel: Distribution of PI3K scores, according to tumor-level PTEN status by immunohistochemistry.
Table 2.
PI3K score and lethal prostate cancer over long-term follow-up, Health Professionals Follow-up Study (HPFS) and the Physicians’ Health Study (PHS), 1982–2016.
| Quartile of PI3K score | 1st lowest | 2nd | 3rd | 4th highest | |
|---|---|---|---|---|---|
| Absolute estimates | |||||
| Person-time [years] | 3,880 | 3,957 | 3,435 | 3,513 | |
| Lethal events | 17 | 16 | 23 | 31 | |
| Rate [(1,000- · year)−1] | 4.4 | 4.0 | 6.7 | 8.8 | |
| RMST1 (95% CI) [years] | 19.1 (18.7–19.5) | 19.2 (18.8–19.6) | 18.7 (18.2–19.3) | 18.3 (17.7–18.9) | |
| Hazard ratio (95% CI) | Per 10 points2 | ||||
| HPFS and PHS combined | |||||
| Unadjusted | 1 (ref.) | 0.93 (0.47–1.84) | 1.54 (0.82–2.88) | 2.04 (1.13–3.68) | 1.26 (1.07–1.48) |
| + age, grade3 | 1(ref.) | 1.12 (0.57–2.23) | 1.56 (0.83–2.94) | 1.85 (1.02–3.35) | 1.20 (0.02–1.41) |
| + age, grade, PTEN3 | 1(ref.) | 0.99 (0.47–2.10) | 1.22 (0.58–2.57) | 1.27 (0.61–2.64) | 1.08 (0.88–1.32) |
| + age, grade, PTEN, stage3 | 1 (ref.) | 1.13 (0.53–2.42) | 1.50 (0.70–3.19) | 1.51 (0.70–3.25) | 1.13 (0.92–1.39) |
| By cohort3 | |||||
| HPFS | 1 (ref.) | 1.05 (0.46–2.39) | 1.54 (0.70–3.39) | 1.55 (0.73–3.32) | 1.15 (0.94–1.41) |
| PHS | 1 (ref.) | 0.70 (0.20–2.46) | 1.54 (0.55–4.33) | 3.17 (1.22–8.25) | 1.49(1.12–1.98) |
| By Gleason grade3 | |||||
| Low, scores 6–7 | 1(ref.) | 0.69 (0.28–1.71) | 0.87 (0.35–2.18) | 1.19 (0.52–2.75) | 1.06 (0.83–1.35) |
| High, scores 8–10 | 1 (ref.) | 1.80 (0.62–5.18) | 2.46 (0.96–6.35) | 3.06 (1.23–7.62) | 1.34 (1.07–1.68) |
Restricted mean survival time (RMST) over the first 20 years of follow-up for metastasis-free survival, unadjusted.
Per 10-point increase in PI3K scores, from models using quartile medians as the exposure.
Covariate coding: age at cancer diagnosis (linear); Gleason grade (categorical: 5–6; 3+4; 4+3; 8; 9–10); pathological stage (categorical: T1/T2; T3; T4/N1; missing); PTEN status by immunohistochemistry (binary). Stratified models are otherwise unadjusted.
Individually, tumor-level PTEN levels were associated with lethal disease (Table S3). Tumor-level pS6 levels were not associated with lethal disease (Table S3), including when allowing for non-linear associations (Fig. S4B), and the association of tumor-level stathmin levels and lethal disease was unclear (Table S3). Consequently, a two-marker PI3K score consisting of PTEN and stathmin levels without pS6 was strongly associated with lethal disease (Table S3; Fig. S4C). Interestingly, this two-marker tumor-level PI3K score, when compared with the same indicators from bulk tumor assessments, had similar associations with these other indicators of PI3K pathway activation as the main (three-marker) PI3K score (Fig. S4D–F). The two-marker score remained associated with lethal disease when adjusting for Gleason grade and PTEN scoring from classic immunohistochemistry (HR, 2.59; 95% CI, 1.19 to 5.65; Table S3, Fig. S4C), while the main (three-marker) tumor-level PI3K score did not (HR, 1.27; 95% CI, 0.61 to 2.64; Table 2). Taken together, these results suggest that combining cell-level PTEN and stathmin data from multiplex immunofluorescence, but not pS6, incrementally adds to prognostication of lethal disease beyond tumor-level PTEN immunohistochemistry.
Single-cell PI3K scores as a window into intratumoral heterogeneity in pathway activation
Thus far, the above tumor-level PI3K scores were used to obtain a single estimate of PI3K activity per tumor. However, since this does not consider intratumoral heterogeneity, we next asked what proportion of cells in a tumor were misclassified when just considering the median as the tumor-level summary. Having grouped tumor-level medians in four quartiles, in half of all tumors, at least 48% of cells belonged to a different quartile than the tumor as a whole based on its median value (median heterogeneity, 48%; IQR, 31% to 58%; Fig. 4A). This measure of intratumoral heterogeneity was correlated with Shannon’s entropy (Fig. S5A). As expected, “extreme” tumors with either very low or very high tumor-level PI3K scores were those with little heterogeneity and thus uniformly extreme cell-level scores (Fig. 4B). Likewise, tumors eye-scored as “PTEN null” by immunohistochemistry had less heterogeneity (Fig. S5B). Heterogeneity increased slightly with the number of tumor cells (Fig. S5C).
Figure 4. Intratumoral heterogeneity and machine learning.
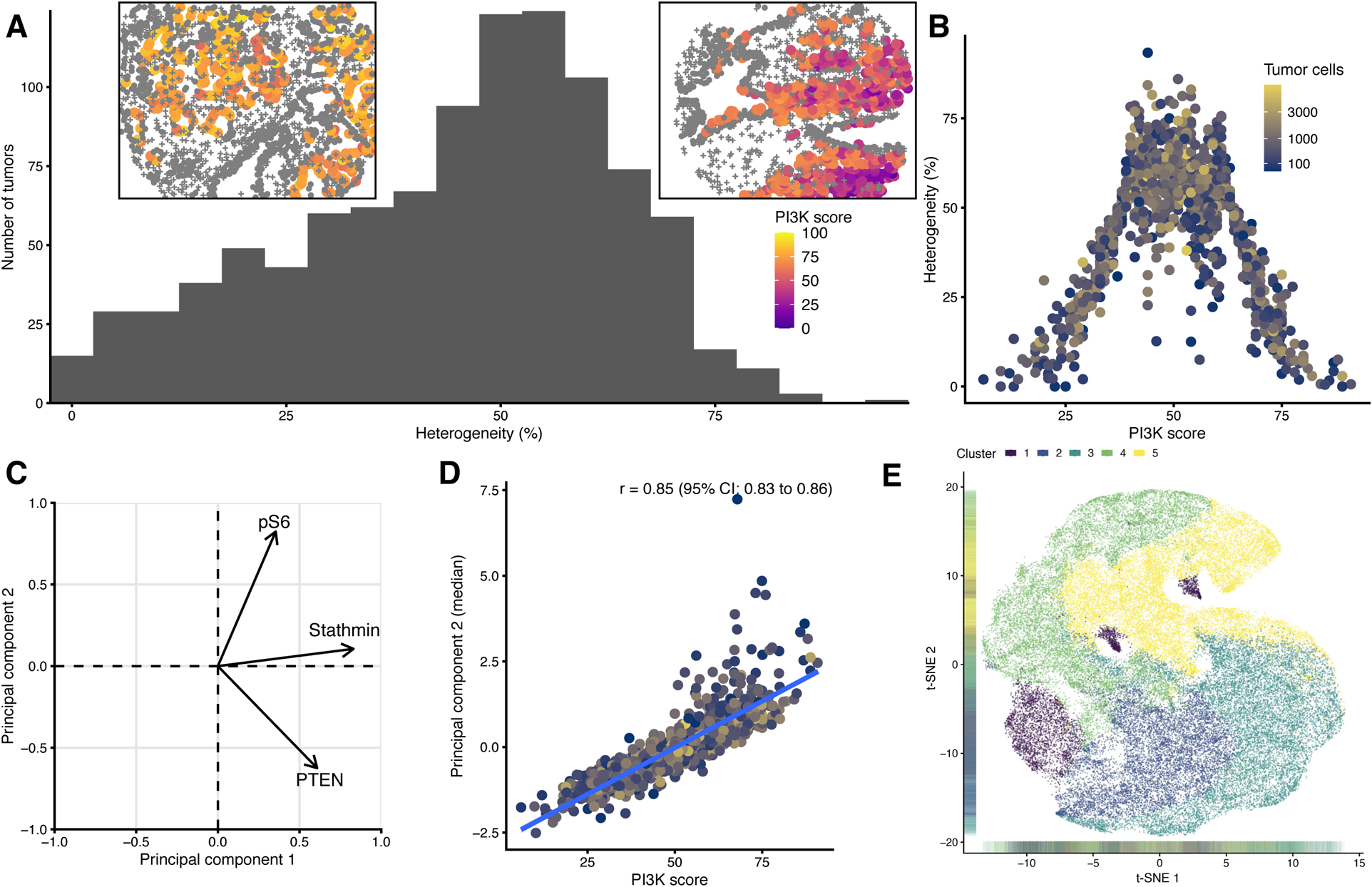
(A) Within-tumor heterogeneity. The heterogeneity is expressed as the proportion of cells that do not belong into the quartile of cell-level PI3K scores that each tumor is assigned to based on its median. Left inset: An example core with low heterogeneity. Right inset: An example core with high heterogeneity, including areas of low PI3K activity (blue) and high PI3K activity (red/yellow).
(B) PI3K scores and within-tumor heterogeneity. Color encodes the number of tumor cells.
(C) Loading plot from principal components analysis. Both principal components explain similar proportions of the variance, and the second principal component is positively loaded with stathmin and pS6 and negative loaded with PTEN, corresponding to the subject matter-informed approach of defining the PI3K score.
(D) Subject matter-based PI3K scores (x axis) and a machine learning-based score (principal component 2, y axis).
(E)Unsupervised clustering of cell-level PTEN, pS6, and stathmin values based on k-medoids, resulting in 5 clusters, indicated by different colors, visualized here along axes of a two-dimensional t-SNE dissimilarity decomposition.
We next assessed where cells with high cell-level PI3K scores were located in relation to their peers. We expected that tumors with higher tumor-level PI3K scores would show more clustering of PI3K-activated cells and more spatial autocorrelation. While larger tumors with more tumor cells showed more clustering (Fig. S5G), cells with high scores were no more likely to cluster together in tumors with high tumor-level PI3K scores than in those with low tumor-level PI3K scores (Fig. S5F). Moreover, in tumors with higher tumor-level PI3K scores, neighboring tumor cells tended to have more dissimilar PI3K scores, as reflected by a more negative Moran’s I for spatial autocorrelation. This association was robust to adjusting for the number of tumor cells per tumor and for Gleason grade and appeared strongest in tumors with little heterogeneity (Fig. S5H). Likewise, cell-level PI3K scores as well as cell-level PTEN expression levels were also more dissimilar in tumors eye-scored as PTEN-null than in tumors eye-scored as PTEN-intact (Fig. S5I–J). Whether clustering or spatial autocorrelation modified how tumor-level PI3K scores were associated with progression to lethal disease was unclear (Table S3). Together, our observations suggest that tumors with higher tumor-level PI3K scores had less variation in how high tumor-level PI3K scores were across the tumor. At the same time, they had more dissimilarity in cell-level PI3K scores between spatially adjacent cells.
PI3K activation scores derived through machine learning
We asked whether unsupervised machine learning approaches would reveal PI3K activity overlooked by analysis that assigned equal weights to the three markers. Results from these unsupervised analyses supported the validity of the mechanistically informed PI3K score. One factor from principal components analysis, explaining 36% of the variance in biomarker levels, was strongly correlated with our tumor-level PI3K score (Fig. 4C, D; S6). Results from a big-data implementation of k-medoids clustering were also consistent with our mechanistically informed scores, yet potentially less reproducible between patient cohorts (Fig. 4E, S7; Table S4; Supplementary Text).
Discussion
In this study, we developed and tested an approach to assessing activity of the PI3K pathway based on single cell-level data. We performed proof of concept studies in human prostate cancer using archival prostate cancer tissue from prospective cohort studies with long-term follow-up for clinically relevant outcomes. Our results demonstrate the validity of PI3K activation scores, suggest more nuanced associations with regulators of the PI3K pathway than when assessing PTEN alone, further support PI3K pathway activation as a feature of tumors with poor prognosis, and provide a window into intratumoral heterogeneity in PI3K signaling. Our results also underscore that a mechanistically grounded, interpretable approach to assessing activation of tumor signaling pathways compares favorably to solely data-driven approaches. This biomarker set should be tested for predicting susceptibility to PI3K inhibition in clinical trials and may be suitable for studying etiologic questions, such as quantifying the impact of IGF-1 (37) and of statins (38) on the risk of PI3K-activated prostate cancer. Our approach may also allow for assessing tumor signaling pathways in patient tissue more generally.
Identifying cancers with activation of the PI3K pathway is paramount for the successful clinical use of PI3K pathway inhibitors, as demonstrated in advanced breast cancer (4). Unlike in breast cancer, where PIK3CA mutations are common, a different approach to quantifying PI3K activation is required in prostate cancer with its much higher prevalence of PTEN deletions and the different impact of the PI3Kβ isoform (3,39). With stathmin and pS6, we selected protein biomarkers not in an effort to capture all potential other pathway regulators besides PTEN, but rather to obtain a reliable reflection of downstream activation. Unsurprisingly, bulk tumor assessments of PTEN predicted pS6 and stathmin levels inadequately, suggesting that its sole use as an indicator of PI3K pathway activation may be an oversimplification.
Assessment of tumor signaling pathways in patient tissues may be facilitated by single cell-based analyses. While the feasibility of multiplex immunofluorescence is clear (40), analytical approaches to leverage their full potential in large-cohort and clinical settings have lagged. A simple quantification approach could use pathology software to obtain immunofluorescence intensity as a tumor-level average, as we did in the past using classic immunohistochemistry for PI3K markers (14), or even to impose cut-offs for “high” and “low” (41,42), and then cross-classify each tumor based on multiple binary markers. Such an approach may be useful when cell populations are truly characterized by binary presence or absence of protein expression, such as subsets of immune cells defined by their surface proteins (43). Importantly, we used unprocessed numeric immunofluorescence intensity data and developed custom algorithms for quantifications and visualizations, with several benefits. First, this approach allowed us to systematically address batch effects between TMAs, a key issue in biomarker studies (44). Second, averaging or classifying each marker on a tumor level upfront, with tremendous information loss, to some extent defeats the purpose of obtaining cell-level data. We instead first calculated PI3K activity scores for each cell based on its individual markers. Doing so allowed us to identify PI3K activation more precisely as the co-occurrence of low PTEN, high pS6, and high stathmin within a cell. The high degree of heterogeneity in PI3K scores within tumors underlines that true cell-level values would have been misclassified by combining tumor averages upfront. At least on a protein level, there was considerable heterogeneity in PI3K activation, an observation that is consistent with notable heterogeneity in PTEN protein expression (10,24). Third, with cell-level PI3K scores at hand, we found that a relatively small subpopulation of cells with high PI3K score was sufficient to identify tumors with different bulk indicators of PI3K signaling and different prognosis. Perhaps counterintuitively, in tumors with high median PI3K scores, tumor cells with high values did not cluster together. Instead, they were more likely to be surrounded by cells with dissimilar PI3K scores, suggesting yet another dimension of intratumoral heterogeneity.
Alternative approaches to analyzing high-dimensional immunofluorescence data may include unsupervised machine learning-based methods (45,46). When applying such techniques, we found striking similarities between one of the principal components and our mechanistically informed PI3K scores. Unsupervised clustering also identified two groups of tumors with similar characteristics as based on mechanistically informed PI3K scores. As our study included patients from two independent prospective cohort studies, we tested the robustness of clustering by performing it independently in each cohort. Cluster coordinates were considerably different between cohorts. Even though tissues from both cohorts had been processed in parallel, the measurements include cohort-specific variation. This observation raises the question how well such highly parametrized classifications would be reproducible in independent populations and different laboratory setups. Our approach is mechanistically informed, so we conjecture it may be more robust, as it was in our study, and warrants validation in other tumor types.
A central challenge for defining PI3K activation is the absence of a gold standard. To what extent scores developed here are indicative of PI3K activation as well as predictive of response to PI3K inhibitors will need additional lines of evidence, such as assessments of our PI3K scores in pre- and post-treatment cancer tissue from clinical trials of PI3K inhibitors. Among patients with primary prostate tumors, PTEN loss is well-known to be associated with higher rates of metastases and death from prostate cancer (10,24,47). In contrast, data in advanced prostate cancer are not indicative of strong associations (12,48). Importantly, finding a similarly strong association between PI3K scores and prognosis as between PTEN status and prognosis does not mean that both perform similarly in indicating sensitivity to PI3K inhibition. Similar considerations apply to the two-marker PI3K score consisting of PTEN and stathmin only. Furthermore, validation efforts presented here are limited in that they mainly reflect PTEN copy number loss only, as one would expect in prostate cancer, and they are all based on bulk assessments of the tumor for PTEN and transcriptome signatures. Relying on TMAs, which were constructed using the highest-grade tumor nodule, potentially underestimated intratumoral heterogeneity.
A number of additional challenges are inherent to multiplex immunofluorescence. One concern is that antigens may be washed out with each additional staining step; however, this effect would be uniform across cores on all TMAs and may be less of a concern with a relatively small number of markers. Moreover, numeric values from immunofluorescence imaging are relative rather than absolute, which allows for comparisons within a study population, but makes it difficult to define reference values for routine use. Finally, we relied on the prostate cancer cell marker AMACR, whose expression can be lost in a subset of tumor cells (49), and on standard pathology software to identify cell positions and distinguish tumor cells from benign epithelium and stroma. Some of the heterogeneity in PI3K scores in tumor cells is likely due to misclassification of cell types.
In summary, we developed a novel measure of PI3K pathway activation in cancer and demonstrated feasibility in cohorts of men with prostate cancer. Our method: (a) only required routine archival FFPE specimens, (b) assessed three markers that mechanistically reflect PI3K pathway activation, (c) captured activation on a protein single-cell level, (d) was validated against external measures, and (e) was applied to tumors from two prospective cohort studies with long-term follow-up. Our analytical framework allows for quantification of single-cell data with spatial resolution from large-scale tumor biorepositories. Testing the PI3K score as a predictive biomarker should proceed using tissue from clinical trials of PI3K inhibitors as a next step. More generally, similar approaches that undertake multiplex protein quantification on a single cell-level in tumor samples from well-defined study populations should lead to insights into tumor etiology and progression. Methodological innovation to optimize quantification approaches is needed.
Supplementary Material
Translational Relevance.
The PI3K pathway is activated in a large proportion of solid cancers, including prostate cancer. Loss of its inhibitor PTEN is associated with higher risk of lethal prostate cancer. However, clinical trials of PI3K/Akt inhibitors have had mixed results in prostate cancer, possibly because patients were not selected for PI3K activation. In this study, we developed and validated a new methodology for the assessment of PI3K pathway activity in prostate cancer that (1) only required routine archival FFPE specimens, (2) assessed three markers that mechanistically reflect PI3K pathway activation, (3) captured activation on a single-cell level, (4) was validated against external measures, and (5) was applied to tumors from two prospective cohort studies with long-term follow-up. This scalable measure of PI3K activation should be explored as a predictive biomarker in clinical trials of PI3K inhibitors.
Acknowledgements
We thank the participants and staff of the Health Professionals Follow-Up Study and the Physicians’ Health Study for their valuable contributions. In particular, we would like to recognize the contributions of Liza Gazeeva, Siobhan Saint-Surin, Robert Sheahan, Betsy Frost-Hawes, Eleni Konstantis, and Sam Peisch. We thank Ericka M. Ebot, PhD MPH for expert advice. We would like to thank the following state cancer registries for their help: AL, AZ, AR, CA, CO, CT, DE, FL, GA, ID, IL, IN, IA, KY, LA, ME, MD, MA, MI, NE, NH, NJ, NY, NC, ND, OH, OK, OR, PA, RI, SC, TN, TX, VA, WA, WY. The authors assume full responsibility for analyses and interpretation of these data. This work was supported by the National Cancer Institute (R01 CA187918) and the Department of Defense (W81XWH-17-1-0483, PC180582 to M.L.). The Health Professionals Follow-up Study is supported by the National Institutes of Health (U01 CA167552). This research was funded in part by the Specialized Programs of Research Excellence program in Prostate Cancer 5P50 CA090381 and P50 CA211024, the NIH/NCI Cancer Center Support Grants P30 CA008748 and P30 CA006516, NIH/NCI grants 5R37 CA227190-02 (S.T., K.L.P., and G.P.) and R01CA131945 (M.L.), and the Prostate Cancer Foundation (M.L.). The Department of Defense supported K.H.S. (W81XWH-18-1-0330) and P.W.K. (W81XWH-14-1-0515). K.H.S., K.L.P., and L.A.M. are Prostate Cancer Foundation Young Investigators.
Footnotes
Conflicts of Interest:
P.W. Kantoff is not aware of conflicts of interest. It is his policy to disclose all relationships, which include ownership interest in Context Therapeutics LLC, DRGT, Placon, Seer Biosciences, and Tarveda Therapeutics; he is a company board member for Context Therapeutics LLC, a consultant/advisory board member for BIND Biosciences, Inc., BN Immunotherapeutics, DRGT, GE Healthcare, Janssen, Metamark, New England Research Institutes, Inc., OncoCellMDX, Progenity, Sanofi, Seer Biosciences, Tarveda Therapeutics, and Thermo Fisher, and serves on data safety monitoring boards for Genentech/Roche and Merck. No potential conflicts of interest were disclosed by other authors.
References
- 1.Levitin HM, Yuan J, Sims PA. Single-Cell Transcriptomic Analysis of Tumor Heterogeneity. Trends Cancer 2018;4(4):264–8 doi 10.1016/j.trecan.2018.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. The PI3K Pathway in Human Disease. Cell 2017;170(4):605–35 doi 10.1016/j.cell.2017.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goncalves MD, Hopkins BD, Cantley LC. Phosphatidylinositol 3-Kinase, Growth Disorders, and Cancer. N Engl J Med 2018;379(21):2052–62 doi 10.1056/NEJMra1704560. [DOI] [PubMed] [Google Scholar]
- 4.Andre F, Ciruelos E, Rubovszky G, Campone M, Loibl S, Rugo HS, et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. N Engl J Med 2019;380(20):1929–40 doi 10.1056/NEJMoa1813904. [DOI] [PubMed] [Google Scholar]
- 5.Armstrong AJ, Halabi S, Healy P, Alumkal JJ, Winters C, Kephart J, et al. Phase II trial of the PI3 kinase inhibitor buparlisib (BKM-120) with or without enzalutamide in men with metastatic castration resistant prostate cancer. Eur J Cancer 2017;81:228–36 doi 10.1016/j.ejca.2017.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Armstrong AJ, Shen T, Halabi S, Kemeny G, Bitting RL, Kartcheske P, et al. A phase II trial of temsirolimus in men with castration-resistant metastatic prostate cancer. Clin Genitourin Cancer 2013;11(4):397–406 doi 10.1016/j.clgc.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 7.Rathkopf DE, Larson SM, Anand A, Morris MJ, Slovin SF, Shaffer DR, et al. Everolimus combined with gefitinib in patients with metastatic castration-resistant prostate cancer: Phase 1/2 results and signaling pathway implications. Cancer 2015;121(21):3853–61 doi 10.1002/cncr.29578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Templeton AJ, Dutoit V, Cathomas R, Rothermundt C, Bartschi D, Droge C, et al. Phase 2 trial of single-agent everolimus in chemotherapy-naive patients with castration-resistant prostate cancer (SAKK 08/08). Eur Urol 2013;64(1):150–8 doi 10.1016/j.eururo.2013.03.040. [DOI] [PubMed] [Google Scholar]
- 9.Lotan TL, Gurel B, Sutcliffe S, Esopi D, Liu W, Xu J, et al. PTEN protein loss by immunostaining: analytic validation and prognostic indicator for a high risk surgical cohort of prostate cancer patients. Clin Cancer Res 2011;17(20):6563–73 doi 10.1158/1078-0432.CCR-11-1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jamaspishvili T, Berman DM, Ross AE, Scher HI, De Marzo AM, Squire JA, et al. Clinical implications of PTEN loss in prostate cancer. Nat Rev Urol 2018;15(4):222–34 doi 10.1038/nrurol.2018.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Bono JS, De Giorgi U, Rodrigues DN, Massard C, Bracarda S, Font A, et al. Randomized Phase II Study Evaluating Akt Blockade with Ipatasertib, in Combination with Abiraterone, in Patients with Metastatic Prostate Cancer with and without PTEN Loss. Clin Cancer Res 2019;25(3):928–36 doi 10.1158/1078-0432.CCR-18-0981. [DOI] [PubMed] [Google Scholar]
- 12.Stopsack KH, Nandakumar S, Wibmer AG, Haywood S, Weg ES, Barnett ES, et al. Oncogenic Genomic Alterations, Clinical Phenotypes, and Outcomes in Metastatic Castration-Sensitive Prostate Cancer. Clin Cancer Res 2020;26(13):3230–8 doi 10.1158/1078-0432.CCR-20-0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Ooijen H, Hornsveld M, Dam-de Veen C, Velter R, Dou M, Verhaegh W, et al. Assessment of Functional Phosphatidylinositol 3-Kinase Pathway Activity in Cancer Tissue Using Forkhead Box-O Target Gene Expression in a Knowledge-Based Computational Model. Am J Pathol 2018;188(9):1956–72 doi 10.1016/j.ajpath.2018.05.020. [DOI] [PubMed] [Google Scholar]
- 14.Martin NE, Gerke T, Sinnott JA, Stack EC, Andren O, Andersson SO, et al. Measuring PI3K Activation: Clinicopathologic, Immunohistochemical, and RNA Expression Analysis in Prostate Cancer. Mol Cancer Res 2015;13(10):1431–40 doi 10.1158/1541-7786.MCR-14-0569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McGranahan N, Favero F, de Bruin EC, Birkbak NJ, Szallasi Z, Swanton C. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci Transl Med 2015;7(283):283ra54 doi 10.1126/scitranslmed.aaa1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giovannucci E, Liu Y, Platz EA, Stampfer MJ, Willett WC. Risk factors for prostate cancer incidence and progression in the health professionals follow-up study. Int J Cancer 2007;121(7):1571–8 doi 10.1002/ijc.22788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hennekens CH, Buring JE, Manson JE, Stampfer M, Rosner B, Cook NR, et al. Lack of effect of long-term supplementation with beta carotene on the incidence of malignant neoplasms and cardiovascular disease. N Engl J Med 1996;334(18):1145–9 doi 10.1056/NEJM199605023341801. [DOI] [PubMed] [Google Scholar]
- 18.Christen WG, Gaziano JM, Hennekens CH. Design of Physicians’ Health Study II--a randomized trial of beta-carotene, vitamins E and C, and multivitamins, in prevention of cancer, cardiovascular disease, and eye disease, and review of results of completed trials. Ann Epidemiol 2000;10(2):125–34. [DOI] [PubMed] [Google Scholar]
- 19.Pettersson A, Graff RE, Bauer SR, Pitt MJ, Lis RT, Stack EC, et al. The TMPRSS2:ERG rearrangement, ERG expression, and prostate cancer outcomes: a cohort study and meta-analysis. Cancer Epidemiol Biomarkers Prev 2012;21(9):1497–509 doi 10.1158/1055-9965.EPI-12-0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stark JR, Perner S, Stampfer MJ, Sinnott JA, Finn S, Eisenstein AS, et al. Gleason score and lethal prostate cancer: does 3 + 4 = 4 + 3? J Clin Oncol 2009;27(21):3459–64 doi 10.1200/JCO.2008.20.4669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Serra V, Markman B, Scaltriti M, Eichhorn PJ, Valero V, Guzman M, et al. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res 2008;68(19):8022–30 doi 10.1158/0008-5472.CAN-08-1385. [DOI] [PubMed] [Google Scholar]
- 22.Bendell JC, Rodon J, Burris HA, de Jonge M, Verweij J, Birle D, et al. Phase I, dose-escalation study of BKM120, an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol 2012;30(3):282–90 doi 10.1200/JCO.2011.36.1360. [DOI] [PubMed] [Google Scholar]
- 23.Andersen JN, Sathyanarayanan S, Di Bacco A, Chi A, Zhang T, Chen AH, et al. Pathway-based identification of biomarkers for targeted therapeutics: personalized oncology with PI3K pathway inhibitors. Sci Transl Med 2010;2(43):43ra55 doi 10.1126/scitranslmed.3001065. [DOI] [PubMed] [Google Scholar]
- 24.Ahearn TU, Pettersson A, Ebot EM, Gerke T, Graff RE, Morais CL, et al. A Prospective Investigation of PTEN Loss and ERG Expression in Lethal Prostate Cancer. J Natl Cancer Inst 2016;108(2) doi 10.1093/jnci/djv346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zu K, Martin NE, Fiorentino M, Flavin R, Lis RT, Sinnott JA, et al. Protein expression of PTEN, insulin-like growth factor I receptor (IGF-IR), and lethal prostate cancer: a prospective study. Cancer Epidemiol Biomarkers Prev 2013;22(11):1984–93 doi 10.1158/1055-9965.EPI-13-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Penney KL, Sinnott JA, Tyekucheva S, Gerke T, Shui IM, Kraft P, et al. Association of prostate cancer risk variants with gene expression in normal and tumor tissue. Cancer Epidemiol Biomarkers Prev 2015;24(1):255–60 doi 10.1158/1055-9965.EPI-14-0694-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on bias and variance. Bioinformatics 2003;19(2):185–93. [DOI] [PubMed] [Google Scholar]
- 28.Nawaz S, Yuan Y. Computational pathology: Exploring the spatial dimension of tumor ecology. Cancer Lett 2016;380(1):296–303 doi 10.1016/j.canlet.2015.11.018. [DOI] [PubMed] [Google Scholar]
- 29.Getis A, Ord JK. The Analysis of Spatial Association by Use of Distance Statistics. Geographical Analysis 1992;24(3):189–206. [Google Scholar]
- 30.Cancer Genome Atlas Research Network. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015;163(4):1011–25 doi 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berglund AE, Rounbehler RJ, Gerke T, Awasthi S, Cheng CH, Takhar M, et al. Distinct transcriptional repertoire of the androgen receptor in ETS fusion-negative prostate cancer. Prostate Cancer Prostatic Dis 2019;22(2):292–302 doi 10.1038/s41391-018-0103-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Creighton CJ, Fu X, Hennessy BT, Casa AJ, Zhang Y, Gonzalez-Angulo AM, et al. Proteomic and transcriptomic profiling reveals a link between the PI3K pathway and lower estrogen-receptor (ER) levels and activity in ER+ breast cancer. Breast Cancer Res 2010;12(3):R40 doi 10.1186/bcr2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saal LH, Johansson P, Holm K, Gruvberger-Saal SK, She QB, Maurer M, et al. Poor prognosis in carcinoma is associated with a gene expression signature of aberrant PTEN tumor suppressor pathway activity. Proc Natl Acad Sci U S A 2007;104(18):7564–9 doi 10.1073/pnas.0702507104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao SD, Parmigiani G, Huttenhower C, Waldron L. Maás-o-menos: a simple sign averaging method for discrimination in genomic data analysis. Bioinformatics 2014;30:3062–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dose-response Greenland S. and trend analysis in epidemiology: alternatives to categorical analysis. Epidemiology 1995;6(4):356–65. [DOI] [PubMed] [Google Scholar]
- 36.Uno H, Claggett B, Tian L, Inoue E, Gallo P, Miyata T, et al. Moving beyond the hazard ratio in quantifying the between-group difference in survival analysis. J Clin Oncol 2014;32(22):2380–5 doi 10.1200/JCO.2014.55.2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Travis RC, Appleby PN, Martin RM, Holly JMP, Albanes D, Black A, et al. A Meta-analysis of Individual Participant Data Reveals an Association between Circulating Levels of IGF-I and Prostate Cancer Risk. Cancer Res 2016;76(8):2288–300 doi 10.1158/0008-5472.CAN-15-1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Allott EH, Ebot EM, Stopsack KH, Gonzalez-Feliciano AG, Markt SC, Wilson KM, et al. Statin Use Is Associated with Lower Risk of PTEN-Null and Lethal Prostate Cancer. Clin Cancer Res 2020;26(5):1086–93 doi 10.1158/1078-0432.CCR-19-2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bergholz JS, Roberts TM, Zhao JJ. Isoform-Selective Phosphatidylinositol 3-Kinase Inhibition in Cancer. J Clin Oncol 2018;36(13):1339–42 doi 10.1200/JCO.2017.77.0891. [DOI] [PubMed] [Google Scholar]
- 40.Liu J, Lau SK, Varma VA, Moffitt RA, Caldwell M, Liu T, et al. Molecular mapping of tumor heterogeneity on clinical tissue specimens with multiplexed quantum dots. ACS Nano 2010;4(5):2755–65 doi 10.1021/nn100213v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hamid AA, Gray KP, Huang Y, Bowden M, Pomerantz M, Loda M, et al. Loss of PTEN Expression Detected by Fluorescence Immunohistochemistry Predicts Lethal Prostate Cancer in Men Treated with Prostatectomy. Eur Urol Oncol 2019;2(5):475–82 doi 10.1016/j.euo.2018.09.003. [DOI] [PubMed] [Google Scholar]
- 42.McKinley ET, Sui Y, Al-Kofahi Y, Millis BA, Tyska MJ, Roland JT, et al. Optimized multiplex immunofluorescence single-cell analysis reveals tuft cell heterogeneity. JCI Insight 2017;2(11) doi 10.1172/jci.insight.93487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Toki MI, Merritt CR, Wong PF, Smithy JW, Kluger HM, Syrigos KN, et al. High-Plex Predictive Marker Discovery for Melanoma Immunotherapy-Treated Patients Using Digital Spatial Profiling. Clin Cancer Res 2019;25(18):5503–12 doi 10.1158/1078-0432.CCR-19-0104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marrone MT, Joshu CE, Peskoe SB, De Marzo AM, Heaphy CM, Lupold SE, et al. Adding the Team into T1 Translational Research: A Case Study of Multidisciplinary Team Science in the Evaluation of Biomarkers of Prostate Cancer Risk and Prognosis. Clin Chem 2019;65(1):189–98 doi 10.1373/clinchem.2018.293365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin JR, Izar B, Wang S, Yapp C, Mei S, Shah PM, et al. Highly multiplexed immunofluorescence imaging of human tissues and tumors using t-CyCIF and conventional optical microscopes. Elife 2018;7 doi 10.7554/eLife.31657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gerdes MJ, Sevinsky CJ, Sood A, Adak S, Bello MO, Bordwell A, et al. Highly multiplexed single-cell analysis of formalin-fixed, paraffin-embedded cancer tissue. Proc Natl Acad Sci U S A 2013;110(29):11982–7 doi 10.1073/pnas.1300136110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leapman MS, Nguyen HG, Cowan JE, Xue L, Stohr B, Simko J, et al. Comparing Prognostic Utility of a Single-marker Immunohistochemistry Approach with Commercial Gene Expression Profiling Following Radical Prostatectomy. Eur Urol 2018;74(5):668–75 doi 10.1016/j.eururo.2018.08.020. [DOI] [PubMed] [Google Scholar]
- 48.Abida W, Cyrta J, Heller G, Prandi D, Armenia J, Coleman I, et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc Natl Acad Sci U S A 2019;116(23):11428–36 doi 10.1073/pnas.1902651116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Browne TJ, Hirsch MS, Brodsky G, Welch WR, Loda MF, Rubin MA. Prospective evaluation of AMACR (P504S) and basal cell markers in the assessment of routine prostate needle biopsy specimens. Hum Pathol 2004;35(12):1462–8 doi 10.1016/j.humpath.2004.09.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.