

Abstract
The recent success of immunotherapies has highlighted the power of leveraging the immune system in the fight against cancer. In order for most immune-based therapies to succeed, T cell subsets with the correct tumor-targeting specificities must be mobilized. When such specificities are lacking, providing the immune system with tumor antigen material for processing and presentation is a common strategy for stimulating antigen-specific T cell populations. While straightforward in principle, experience has shown that manipulation of the antigen presentation process can be incredibly complex, necessitating sophisticated strategies that are difficult to translate. Herein, we report on the design of a biomimetic nanoparticle platform that can be used to directly stimulate T cells without the need for professional antigen-presenting cells. The nanoparticles are fabricated using a cell membrane coating derived from cancer cells engineered to express a costimulatory marker. Combined with the peptide epitopes naturally presented on the membrane surface, the final formulation contains the necessary signals to promote tumor antigen-specific immune responses, priming T cells that can be used to control tumor growth. The reported approach represents an emerging strategy that can be used to develop multi-antigenic, personalized cancer immunotherapies.
Keywords: artificial antigen presentation, genetic engineering, cell membrane-coated nanoparticle, anticancer vaccine, immunotherapy
Graphical Abstract
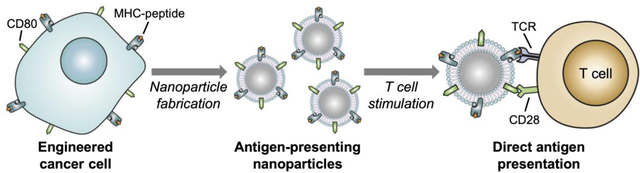
Cancer cells are genetically engineered to express a co-stimulatory marker that enables them to directly present their own antigens to the immune system under an immunostimulatory context. Cell membrane-coated nanoparticles sourced from these modified cancer cells are able to elicit antitumor immunity in vivo while bypassing the need for traditional cell-mediated antigen presentation. The reported approach may ultimately be used in the design of personalized artificial antigen presentation platforms capable of mobilizing immune cell subsets against patient-specific cancer antigens.
It is understood now that cancer pathogenesis carries a significant immunological component,[1] and tumors can utilize a number of mechanisms in order to achieve immune escape.[2] Cancer immunotherapies leverage this knowledge and seek to manipulate various aspects of the immune process in order to promote tumor destruction.[3] For example, approaches such as anticancer vaccination help to train T cells with the appropriate antigen specificities,[4] while others such as checkpoint blockade therapies work by removing the mechanisms of inhibition on existing immune cell populations.[5] Each approach has its own benefits and challenges, although even the most promising immunotherapeutic modalities only work for a subset of patients, in large part due to the large heterogeneity among cancers.[6] Currently, most of the success in the clinic has been biased towards treatments that augment or supplement the effector stage of immunity,[7, 8] perhaps because the manipulation of these downstream immune processes is more straightforward with fewer variables to consider. There is strong evidence, however, that combinatorial treatments affecting multiple aspects of immunity can promote strong antitumor immune responses,[9, 10] and it is thus imperative that the development of immunotherapies continues along multiple fronts.
Vaccines are designed to generate antigen-specific immune responses and have historically been attractive due to their ease of use and potential for broad applicability.[11] Unfortunately, vaccines formulated against cancers are notoriously difficult to develop.[12, 13] This stems largely from the fact that tumor antigens are inherently lowly immunogenic, as they are usually based on normal antigens that are subtly mutated or differentially upregulated. To address this issue, vaccines must be formulated with potent immunological adjuvants, often in the form of toll-like receptor agonists, in order to boost the immune response.[14] Delivery of antigen and adjuvant to professional antigen-presenting cells (APCs) results in the presentation of peptide epitopes in the context of major histocompatibility complex (MHC)-I, along with costimulatory markers such as CD80 or CD86.[15] Together, these signals are necessary in order to promote activation of the cognate T cells that can target and destroy tumor cells expressing the corresponding antigen epitope. Manipulation of the antigen presentation process is not straightforward, and lack of potency is a common issue for anticancer vaccine formulations.[16] This is particularly true for most vaccines that are administered parenterally, where efficient delivery to the correct APC subsets is a major challenge.[17] These difficulties have necessitated the use of production workflows that require significant time and effort,[18] greatly limiting their translational potential.
Researchers have sought to gain more control over the antigen presentation process by engineering artificial APCs (aAPCs) that can replace the function of their endogenous counterparts.[19] aAPCs can be cell-based, whereby living cells are engineered to express the appropriate MHC, as well as a costimulatory marker.[20] These modified cells have been shown to successfully engage with and activate T cells, with potential utility for adoptive cell therapy applications. More recently, there has been significant interest in developing particulate aAPCs.[21] For these synthetic platforms, the requisite biological signals, including peptide-loaded MHC multimer complexes and CD28 agonists, are conjugated onto the surface of synthetic microparticles or nanoparticles. In particular, nanoscale aAPC systems may hold significant potential given their ability to be used for in vivo applications,[22, 23] offering significant advantages such as enhanced lymphatic transport after subcutaneous administration.[24] They could ultimately be employed as a means for directly stimulating T cells that reside in the body, obviating the need for ex vivo stimulation. As aAPCs bypass the need for traditional antigen processing and presentation, such platforms could also replace vaccines for certain applications. Combined with sophisticated strategies for neoantigen identification,[25] the technology could also be amenable to a high level of personalization in the future. Overall, aAPC platforms represent powerful tools for the direct activation of T cells, particularly when individual epitopes of interest can be identified.
Almost all cells in the body express MHC-I, which allows internal monitoring by the immune system in the case of infection or aberrancy. This includes cancer cells, which often employ strategies to subvert immune detection by manipulating their MHC expression.[26] It should therefore be possible to leverage this antigen presentation machinery for the direct stimulation of cancer-targeting T cell populations,[27] given that the correct signals are provided. Here, we first engineer a model cancer cell line to express the costimulatory marker CD80, enabling it to present its own antigens in an immunostimulatory context. The membrane from these engineered cells is then collected and coated onto a nanoparticulate substrate,[28] resulting in a biomimetic nanoformulation capable of direct antigen presentation to cancer-specific T cells (Figure 1). The anticancer utility of these engineered cell membrane-coated antigen-presenting nanoparticles is evaluated both in vitro and in animal models of disease. The described platform combines the advantages of natural cell-based aAPCs with those of synthetic nanoscale APCs into a single construct that is well-suited for in vivo use. These biomimetic antigen-presenting nanoparticles have the potential to promote multi-specific T cell activation and may ultimately be used for personalized therapies.
Figure 1.
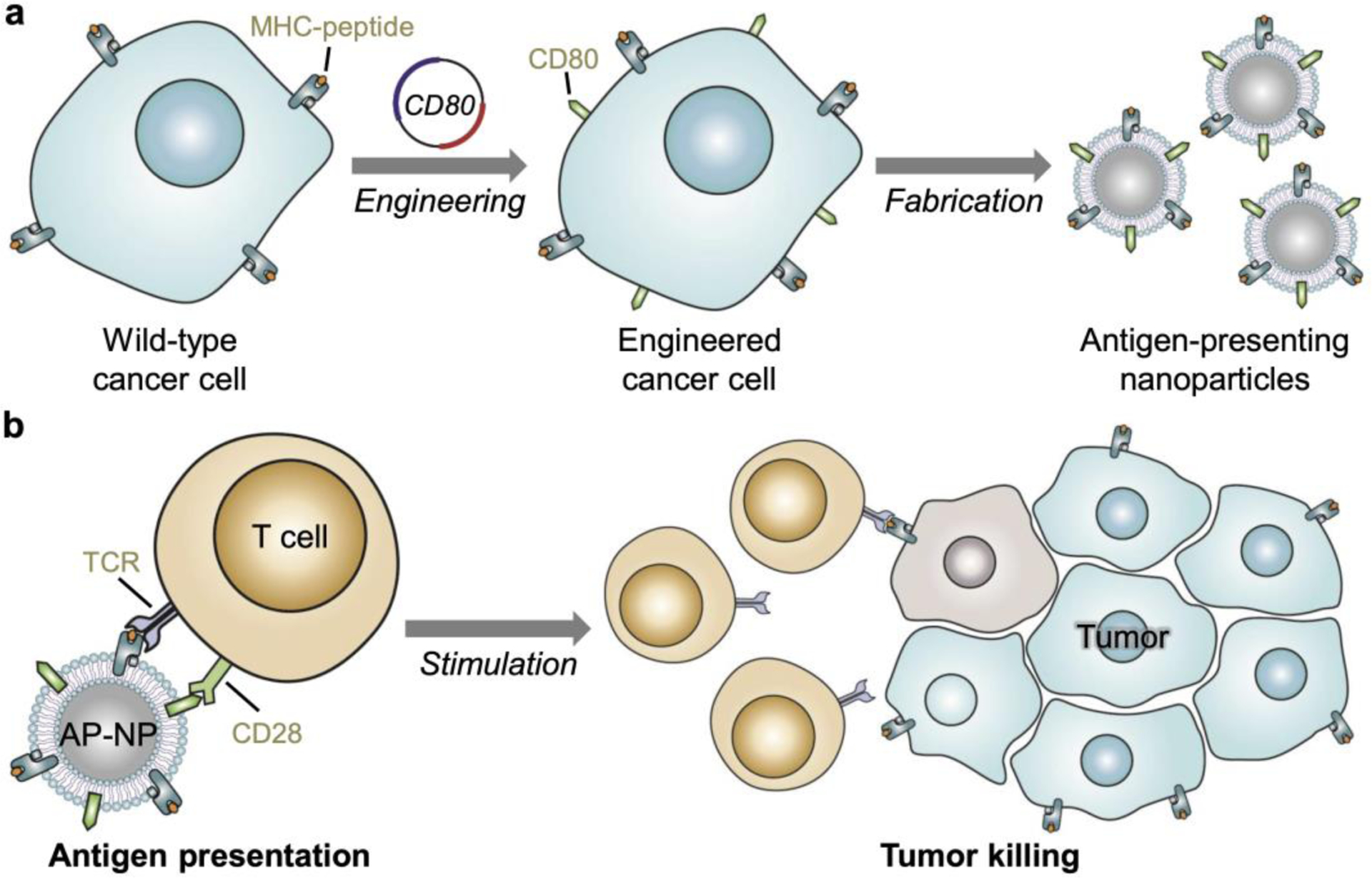
Schematic of engineered cell membrane-coated nanoparticles for direct antigen presentation. a) Wild-type cancer cells, which naturally present their own antigens via MHC-I, are engineered to express CD80, a co-stimulatory signal. The plasma membrane from these cells is then derived and coated onto polymeric nanoparticle cores. b) The resulting antigen-presenting nanoparticles (AP-NPs) can directly stimulate tumor antigen-specific T cells through engagement of the cognate T cell receptor (TCR) and CD28. Upon activation, the T cells are capable of controlling tumor growth by killing cancer cells that express the same antigens.
To engineer cancer cells capable of displaying their own antigens under a stimulatory context, the wild-type B16-F10 (B16-WT) murine melanoma cell line was selected as the foundation. These cells are syngeneic to C57BL/6 mice and can readily be used to generate immunocompetent tumor models.[29] It has been confirmed that these cells express a certain degree of MHC class I,[30] which can present peptide epitopes from endogenous antigens to CD8+ T cells. For this study, the B16-WT cells were modified to overexpress two different genes. The first was a cytosolic form of ovalbumin (OVA),[31] which was selected as a model antigen given the wide range of immunological tools available to help facilitate its study. The second was the costimulatory marker CD80, which engages the CD28 receptor found on T cells.[32] When the two signals are presented together, CD80 and a peptide-MHC complex are generally sufficient to promote the activation of the cognate T cells. Stable cell lines were developed for both single knock-in clones, denoted B16-CD80 and B16-OVA, and a double knock-in clone, denoted B16-CD80/OVA. From western blotting analysis, it was confirmed that both B16-OVA and B16-CD80/OVA expressed the OVA protein, while no signal was detected for either B16-WT or B16-CD80 (Figure 2a). Flow cytometric analysis for CD80 confirmed that both B16-CD80 and B16-CD80/OVA cells displayed the costimulatory marker, whereas B16-WT and B16-OVA cells had little to no expression (Figure 2b). For the OVA-expressing clones, it was further confirmed that they were properly displaying the OVA-derived SIINFEKL peptide bound to H-2Kb (Kb-SIINFEKL) MHC-I (Figure 2c). This indicated that the cytoplasmic OVA was being properly restricted by endogenous cellular machinery for presentation to the immune system. When plotting CD80 expression versus Kb-SIINFEKL presentation, it was evident that the B16-CD80/OVA clone was positive for both signals required to activate anti-OVA cytotoxic T cell responses. (Figure 2d)
Figure 2.
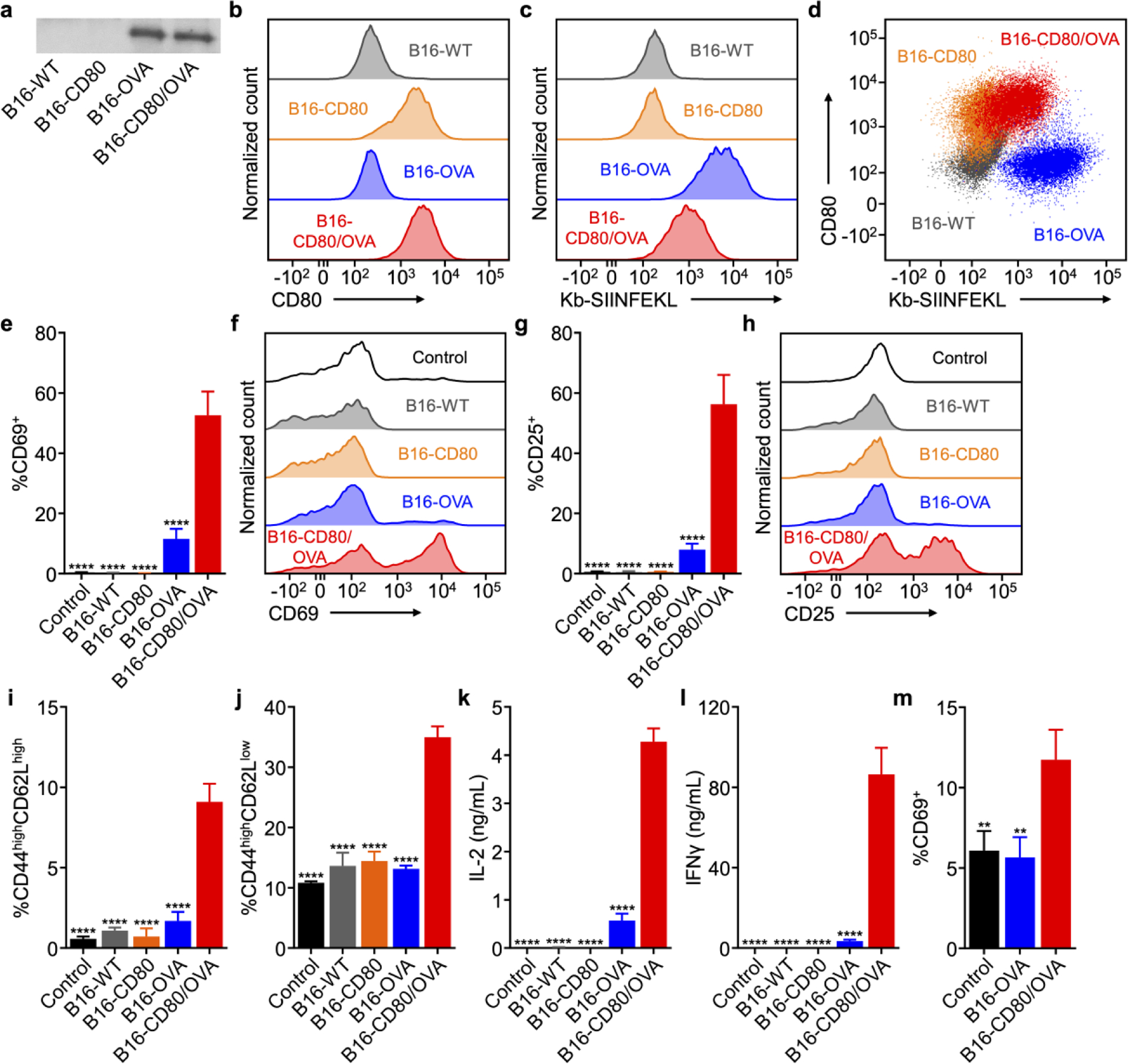
Characterization and biological activity of engineered cancer cells capable of direct antigen presentation. a) Western blot probing for OVA on B16-CD80/OVA cells and control cells. b,c) Expression of CD80 (b) and presentation of an MHC-I-restricted OVA peptide (Kb-SIINFEKL, c) by B16-CD80/OVA cells and control cells. d) Co-expression of CD80 and Kb-SIINFEKL by B16-CD80/OVA cells and control cells. e-h) Expression of CD69 (e,f) and CD25 (g,h) by OT-I CD8+ T cells incubated with B16-CD80/OVA cells and control cells for 24 h (n = 3, mean + SD). i,j) Frequency of memory phenotypes CD44highCD62Lhigh (i) and CD44highCD62Llow (j) among OT-I CD8+ T cells incubated with B16-CD80/OVA cells and control cells for 24 h (n = 3, mean + SD). k,l) Secretion of IL-2 (k) and IFNγ (l) by OT-I CD8+ T cells incubated with B16-CD80/OVA cells and control cells for 24 h (n = 3, mean + SD). m) Expression of CD69 by CD8+ T cells in a population of pmel-1 splenocytes incubated with B16- OVA or B16-CD80/OVA cells for 2 days (n = 3, mean + SD). **p < 0.01, ****p < 0.0001 (compared to B16-CD80/OVA); one-way ANOVA.
Next, the biological activity of both membrane-bound signals was evaluated to confirm that the engineered cells could promote the activation of antigen-specific T cells. Splenocytes derived from OT-I transgenic mice, whose CD8+ T cells predominantly express the T cell receptor against Kb-SIINFEKL,[33] were incubated with each of the B16-F10 variants. After 24 h of incubation, the CD8+ T cells in the mixed-cell population were immunophenotyped using flow cytometry (Figure S1a–f, Supporting Information). Upregulation of CD69 is a hallmark of T cell activation,[34] and the marker was highly expressed on CD8+ T cells cocultured with B16-CD80/OVA cells. In contrast, B16-OVA cells produced a modest response, while both B16-WT and B16-CD80 cells had low expression. A similar trend was observed for CD25, an interleukin-2 (IL-2) receptor that aids in the clonal expansion of T cells.[35] In terms of memory T cells,[36] only the B16-CD80/OVA cells were able to increase the proportion of CD8+ T cells with the CD44highCD62Lhigh central memory phenotype, while the effect on the CD44highCD62Llow effector memory phenotype was less pronounced. In terms of cytokine secretion, IL-2 and interferon-γ (IFNγ), both of which are essential for the survival and function of CD8+ T cells,[37] were only found in high concentrations in the media of splenocytes cocultured with B16-CD80/OVA cells (Figure S1g,h, Supporting Information). The various engineered B16-F10 cells were then incubated and tested in a similar manner with purified OT-I CD8+ cells, which should have a higher proportion of OVA-specific cytotoxic T cells (Figure 2e–l). The overall trends observed for this set of studies was largely consistent with the mixed splenocyte study, with the exception that the B16-CD80/OVA cells were able to clearly enhance the population of T cells with the CD44highCD62Llow effector memory phenotype. It was also confirmed that the CD80-modified cells could stimulate pmel-1 CD8+ T cells (Figure 2m), which are specific against the native melanoma antigen gp100 present on B16-WT.[38] Overall, this data confirmed the successful engineering of cancer cells capable of presenting their own antigens to activate specific T cell subsets.
The use of modified cancer cells in vivo would likely be accompanied with numerous safety concerns and face significant hurdles in terms of clinical translation. Thus, we sought to generate a nanoformulation safe for in vivo use by leveraging cell membrane coating nanotechnology, which is a streamlined approach for the top-down fabrication of highly functional nanoparticles that mimic many of the functions of living cells.[28] These biomimetic nanoparticles have been used in a number of different applications, including drug delivery,[39–41] detoxification,[42–44] and vaccine design.[45, 46] In terms of immune modulation, a major advantage of the technology is that it can help to stabilize membrane vesicles and decrease their size, thus enhancing in vivo transport upon subcutaneous administration.[47] Having confirmed the functionality of both surface-bound signals on the engineered B16-CD80/OVA cells, their membrane was then derived using a procedure involving cellular disruption and differential centrifugation. The purified membrane was collected and coated onto the surface of preformed polymeric nanoparticle cores by a sonication process.[48] It should be noted that the nanoparticle core serves an important function to stabilize the membrane coating and prevent unwanted fusion,[45] thus facilitating enhanced lymphatic transport.[47] The preparation of B16-CD80/OVA cell membrane-coated nanoparticles (denoted [CD80/OVA]NPs) was optimized by synthesizing the nanoparticles in water with varying weight ratios of the engineered cell membrane to polymer, followed by adjusting the resulting formulations to isotonic phosphate buffered saline (PBS) to match physiological conditions (Figure 3a). Without any membrane coating, the charge screening effect resulting from the presence of ions in the buffer caused the bare polymeric cores to aggregate significantly. This effect lessened at higher coating ratios, suggesting progressively better surface coverage. Based on the data, it was determined that the optimal membrane-to-core weight ratio was 1:2, and this [CD80/OVA]NP formulation was used for all subsequent studies.
Figure 3.
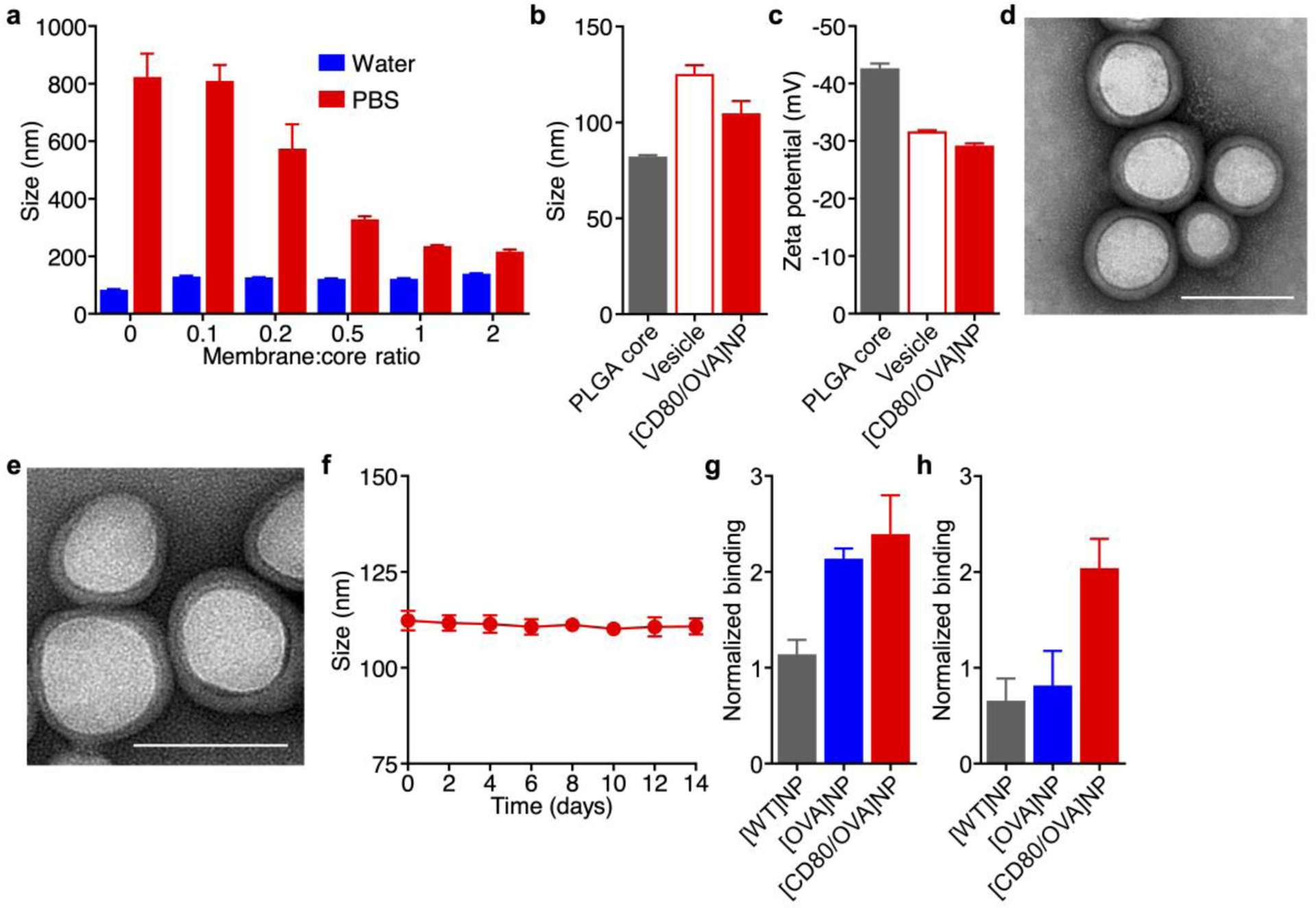
Fabrication and characterization of engineered antigen-presenting nanoparticles. a) Size of [CD80/OVA]NPs at different membrane to core weight ratios when suspended in water or PBS (n = 3; mean + SD). b,c) Hydrodynamic diameter (b) and surface zeta potential (c) of bare PLGA cores, B16-CD80/OVA membrane vesicles, and [CD80/OVA]NPs (n = 3; mean + SD). d,e) Transmission electron microscope images of [CD80/OVA]NPs immediately after synthesis (d) and after 1 week of storage (e). Scale bars = 100 nm. f) Size of [CD80/OVA]NPs over 2 weeks (n = 3; mean ± SD). g,h) Relative binding of antibodies against Kb-SIINFEKL (g) and CD80 (h) to [WT]NPs, [OVA]NPs, and [CD80/OVA]NPs (n = 3; mean + SD).
When measured by dynamic light scattering, the final [CD80/OVA]NPs were approximately 100 nm in size after coating, which was in between the sizes of the bare polymeric cores and the membrane vesicles (Figure 3b). Zeta potential measurements revealed that the surface charge of the membrane-coated nanoparticles was near the value of pure membrane vesicles, which were less negative than the highly charged polymeric cores (Figure 3c). While both these pieces of data suggested successful membrane coating, the nanoparticles were further observed under transmission electron microscopy after negative staining (Figure 3d). The imaging revealed a narrow size distribution, as well as a characteristic core–shell structure that further confirmed the membrane coating. The physical appearance of the nanoparticles remained unchanged after 1 week of storage in solution (Figure 3e). To further test their stability in solution over time, the [CD80/OVA]NPs were suspended in isotonic sucrose, and they exhibited no increase in size during a 2-week observation period (Figure 3f). Finally, to verify that the nanoparticles retained the signals required for T cell activation, immunofluorescence pull-down assays were conducted to confirm the presence of intact CD80 and Kb-SIINFEKL (Figure 3g,h). The experiment showed that nanoparticles made using plasma membrane from the B16-CD80/OVA double knock-in clone ([CD80/OVA]NPs), expressed both markers, whereas nanoparticles fabricated from the B16-OVA single knock-in clone (denoted [OVA]NPs), was high only in Kb-SIINFEKL. As expected, nanoparticles made from B16-WT cells (denoted [WT]NPs), yielded baseline signals for both markers. While the amount of both the CD80 and Kb-SIINFEKL per [CD80/OVA]NP was dictated largely by the optimal membrane coating ratio, it is possible to modulate the density of each marker by sourcing the membrane from B16-CD80/OVA clones with different levels of expression.
With the successful fabrication of the [CD80/OVA]NP formulation, its biological activity was assessed in vitro to determine if it could activate antigen-specific T cells. The nanoparticles were incubated with OT-I splenocytes, and the effect on various CD8+ T cell phenotypes, including CD69+, CD25+, central memory, and effector memory, was evaluated. A similar trend was observed as compared with whole cells, where only [CD80/OVA]NPs were able to elicit the phenotypic changes (Figure S2a–f, Supporting Information). Incubation with [OVA]NPs and [WT]NPs had minimal impact on OT-I CD8+ T cell activation state. In terms of cytokine secretion, the results were striking, as signals for IL-2 and IFNγ were barely detectable for all sample groups other than the [CD80/OVA]NPs (Figure S2g,h, Supporting Information). The advantages were largely consistent when incubated with purified OT-I CD8+ cells (Figure 4a–h), and again the double knock-in [CD80/OVA]NP formulation was able to positively modulate the CD44highCD62Llow effector memory phenotype under this experimental setup. It was confirmed that the activity of the nanoparticles was largely retained even after storage in solution for 1 week (Figure 4i,j). When looking at their ability to promote antigen-specific T cell proliferation, [CD80/OVA]NPs were able to induce a significant amount of cell division as demonstrated by a dye dilution assay, whereas all of the control samples had a minimal impact on the state of the cells (Figure 4k). This effect was shown to be dependent on nanoparticle concentration, and [CD80/OVA]NPs at 100 μg/mL caused a majority of the T cells to experience proliferation (Figure 4l). The T cell activation properties of the nanoparticles was also confirmed by quantifying expansion, where the [CD80/OVA]NP-treated cells multiplied by nearly 9-fold in 4 days, whereas cell counts for all other groups dropped below the initial value at the beginning of the experiment (Figure 4m). OT-I CD8+ cells activated using [CD80/OVA]NPs were able to preferentially kill cellular targets expressing the model antigen (Figure 4n). Overall, it is quite notable that the [CD80/OVA]NPs had significantly enhanced biological activity compared with [OVA]NPs, as this demonstrated that the T cell activation was not simply due to the introduction of OVA into the system. It was also confirmed that the CD80-modified nanoparticles could stimulate pmel-1 CD8+ T cells specific against the melanoma antigen gp100 (Figure 4o).
Figure 4.
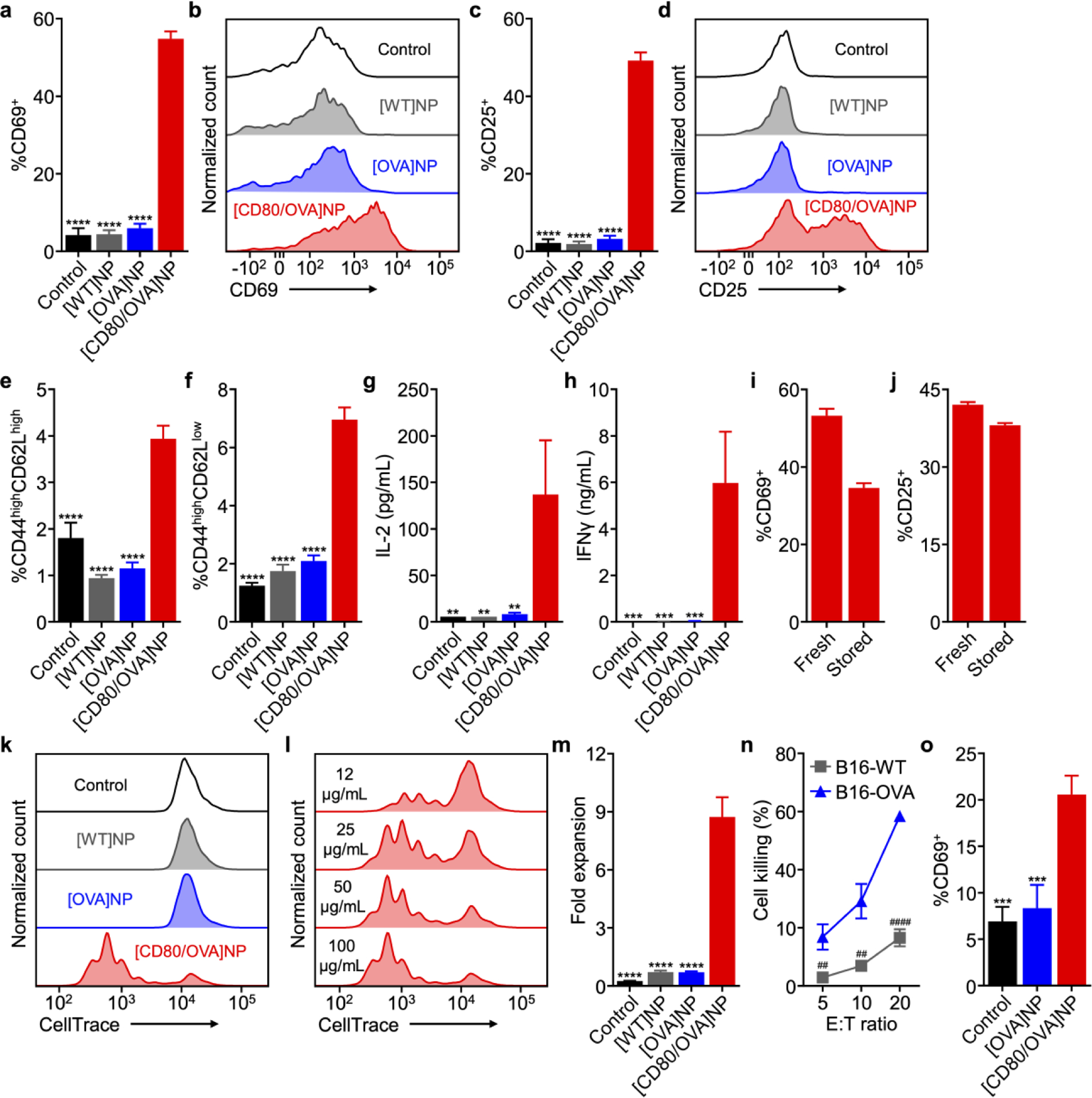
Biological activity of engineered antigen-presenting nanoparticles. a-d) Expression of CD69 (a,b) and CD25 (c,d) by OT-I CD8+ T cells incubated with [CD80/OVA]NPs and control nanoparticles for 3 days (n = 3, mean + SD). e,f) Frequency of memory phenotypes CD44highCD62Lhigh (e) and CD44highCD62Llow (f) among OT-I CD8+ T cells incubated with [CD80/OVA]NPs and control nanoparticles for 3 days (n = 3, mean + SD). g,h) Secretion of IL-2 (g) and IFNγ (h) by OT-I CD8+ T cells incubated with [CD80/OVA]NPs and control nanoparticles for 3 days (n = 3, mean + SD). i,j) Expression of CD69 (i) and CD25 (j) by CD8+ T cells in a population of OT-I splenocytes after 3 days of incubation with [CD80/OVA]NPs either freshly made or stored for 1 week (n = 3, mean + SD). k,l) Fluorescent signal dilution of CD8+ T cells in a population of OT-I splenocytes labeled with CellTrace Violet after incubation with [CD80/OVA]NPs and control nanoparticles (k) or [CD80/OVA]NPs at various concentrations (l) for 3 days. m) Fold expansion of CD8+ T cells in a population of OT-I splenocytes after incubation with [CD80/OVA]NPs and control nanoparticles for 4 days (n = 3, mean + SD). n) Cell killing by OT-I CD8+ cells activated by [CD80/OVA]NPs for 3 days and then incubated with B16-OVA or B16-WT cells at various effector to target (E:T) ratios for 18 h (n = 3, mean ± SD). o) Expression of CD69 by CD8+ T cells in a population of pmel-1 splenocytes incubated with [OVA]NPs or [CD80/OVA]NPs for 3 days (n = 3, mean + SD). **p < 0.01, ***p < 0.001, ****p < 0.0001 (compared to [CD80/OVA]NP); one-way ANOVA. ##p < 0.01, ####p < 0.0001; Student’s t-test.
After confirming the activity of the antigen-presenting nanoparticles in vitro, we next performed a set of in vivo characterizations. To evaluate their transport characteristics, fluorescently labeled [CD80/OVA]NPs were subcutaneously administered into OT-I mice, and the draining lymph nodes were collected at various time points for histological analysis (Figure 5a). At the time of injection, it could be seen that the lymph node was absent any nanoparticle signal, while CD8+ T cells were dispersed within various regions of the lymph node. At 12 h post-injection, the nanoparticle signal started to strengthen along the periphery of the lymph node. Finally, after 24 h there was a significant amount of nanoparticle fluorescence that could be visualized beyond the edges of the lymph node, and this signal was found to be adjacent to a significant number of CD8+ T cells. To evaluate the biological activity of the nanoparticles after in vivo delivery, [CD80/OVA]NPs were administered to C57BL/6 mice that had been adoptively transferred with OT-I splenocytes. The CD69 activation marker was found to be significantly upregulated on adoptively transferred CD8+ T cells in mice that were administered with [CD80/OVA]NPs, whereas those treated with [WT]NPs or [OVA]NPs had CD69 levels consistent with baseline (Figure 5b). This trend was also seen when looking at cytokine secretion, where cells derived from the lymph nodes of [CD80/OVA]NP-treated mice secreted significantly higher levels of IFNγ as compared to the control groups (Figure 5c).
Figure 5.

In vivo delivery and activity of engineered antigen-presenting nanoparticles. a) Immunofluorescence images of draining lymph node sections taken from OT-I mice at different periods after administration of dye-labeled [CD80/OVA]NPs. Red: [CD80/OVA]NPs, green: CD8+ cells; scale bar = 250 μm. b) Expression of CD69 by OT-I CD8+ T cells in the draining lymph nodes 3 days after administration of [CD80/OVA]NPs or control nanoparticles into C57BL/6 mice adoptively transferred with OT-I splenocytes (n = 4, mean + SD). c) Secretion of IFNγ by draining lymph node cells 4 days after administration of [CD80/OVA]NPs or control nanoparticles into C57BL/6 mice adoptively transferred with OT-I splenocytes (n = 3, mean + SD). ***p < 0.001, ****p < 0.0001 (compared to [CD80/OVA]NP); one-way ANOVA.
The ability of the [CD80/OVA]NP formulation to control tumor growth was first tested in a prophylactic setting on an immunocompetent tumor model developed using B16-OVA cells (Figure 6a). Mice were first irradiated, followed by adoptive transfer of OT-I splenocytes. The next day, nanoparticle formulations were administered subcutaneously, and tumor cells were implanted after another 5 days. When observing tumor growth, it could be seen that both [WT]NPs and [OVA]NPs had minimal impact on the growth kinetics when compared with the control group administered with vehicle only (Figure 6b,c). On the other hand, mice treated with [CD80/OVA]NPs exhibited delayed tumor growth. This was also reflected in the survival data (Figure 6d), where the control, [WT]NP, and [OVA]NP groups had median survivals of 35, 34, and 35 days, respectively. In comparison, the [CD80/OVA]NP group had the best median survival of 44 days, with one mouse completely rejecting tumor challenge for the duration of the study. The antitumor activity was also evaluated in a more difficult to treat therapeutic scenario (Figure 6e). In this case, the tumor was implanted first, followed by irradiation for leukodepletion, adoptive transfer of OT-I CD8+ cells, and then treatment with each of the nanoformulations. The [WT]NP and [OVA]NP formulations again had minimal impact on tumor growth, while the [CD80/OVA]NP formulation was able to delay the growth kinetics (Figure 6f,g). In terms of median survival, the blank control, [WT]NP, and [OVA]NP groups had values of 29, 31, and 27 days, respectively, while the [CD80/OVA]NP group had an extended median survival of 37 days (Figure 6h). It was also confirmed that therapeutic efficacy could be achieved in the absence of leukodepletion and adoptive transfer, as [CD80/OVA]NP treatment was able to delay the growth of established B16-OVA tumors in unmanipulated mice (Figure S3a–d, Supporting Information). When benchmarked against a whole cell lysate vaccine adjuvanted with CpG 1826, the antigen-presenting nanoformulation was able to better control tumor growth and prolong survival (Figure S4a–d, Supporting Information). In this case, the improved efficacy of the [CD80/OVA]NPs may likely be attributed to their ability to present more relevant antigenic material to the immune system.[49]
Figure 6.
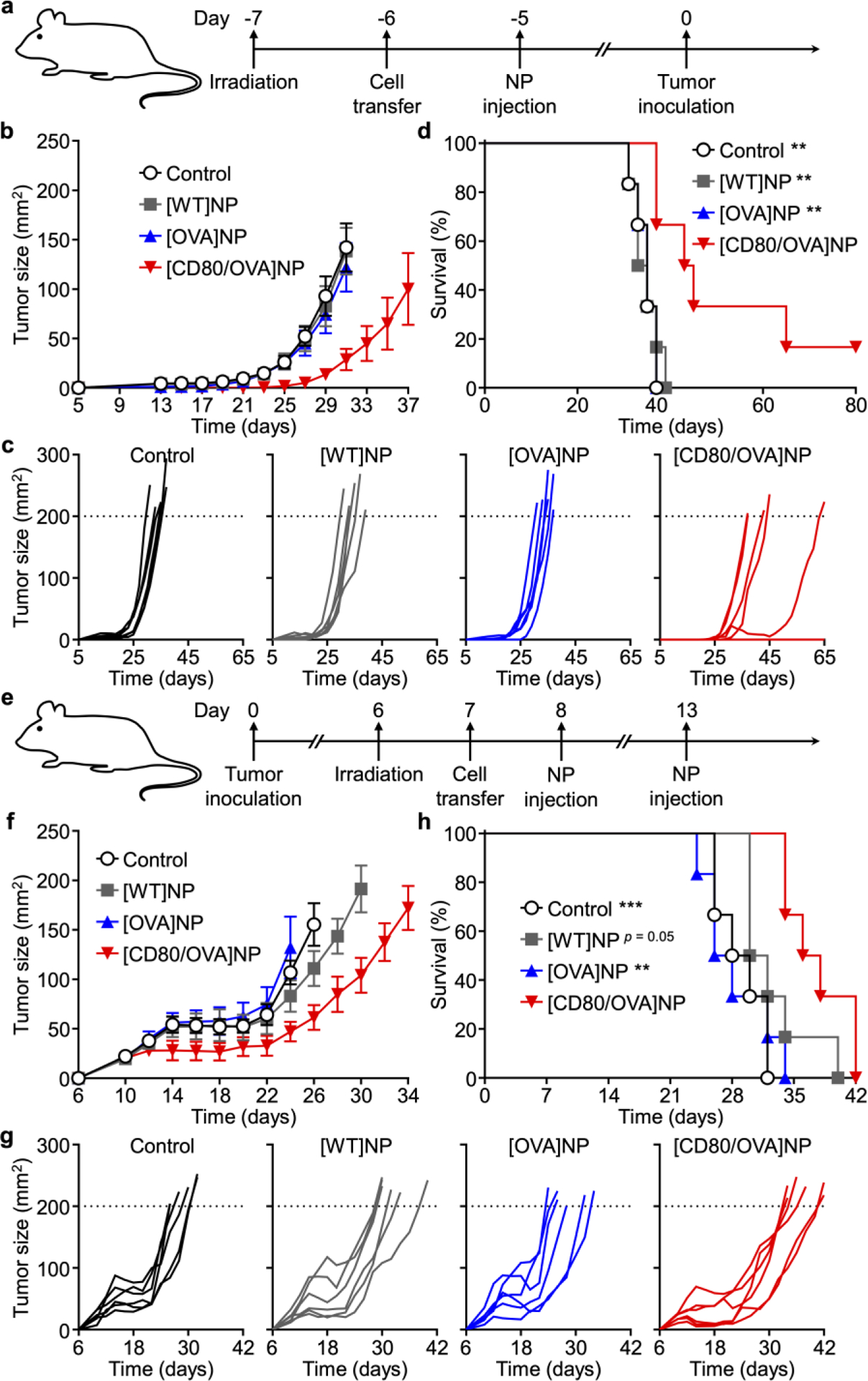
In vivo prophylactic and therapeutic efficacy. a) Experimental timeline for prophylactic efficacy study. b-d) Average tumor sizes (b), individual tumor growth kinetics (c), and survival (d) over time for the prophylactic efficacy study (n = 6; mean ± SEM). e) Experimental timeline for therapeutic efficacy study. f-h) Average tumor sizes (f), individual tumor growth kinetics (g), and survival (h) over time for the therapeutic efficacy study (n = 6; mean ± SEM). ** p < 0.01, *** p < 0.001 (compared to [CD80/OVA]NP in survival plot); log-rank test.
Throughout both the in vitro and in vivo assessment of our platform, only the experimental group expressing both CD80 and the OVA antigen was able to generate significant biological activity. This indicates that the observed effect was not simply due to endogenous processing of the antigenic material, but rather it was more likely a result of direct antigen presentation by the nanoparticles. In its current form, [CD80/OVA]NP was only able to promote a modest survival benefit, which may be attributed to the fact that the process for eliciting antitumor immunity is highly complex. The notion that effective antigen presentation alone cannot be expected to overcome the various immunosuppressive strategies employed by tumor cells is supported by the current landscape of antitumor vaccination, where durable responses are hard to achieve despite generation of T cell subsets with the correct specificities.[50] It is for this reason that researchers are actively exploring the combination of vaccines with other immunotherapies to more comprehensively activate immunity on multiple fronts,[51] and this is a strategy that will likely benefit aAPC platforms. In addition to issues posed by the tumor microenvironment, the membrane protein expression profile of the parent cells may also present its own set of challenges. The B16-F10 cell line employed in the present study is known to express low amounts of MHC-I while also expressing programmed death-ligand 1,[30, 52] and these immunosuppressive mechanisms may combine to undermine the immune-activating stimulus provided by CD80. While the main goal of the present work was to demonstrate that immune activity can be modulated via genetically engineered cell membrane-coated nanoparticles, the platform could be improved through further engineering of the cancer cells to address immune evasion mechanisms or to introduce additional immune-activating surface markers. Other avenues for improving efficacy could involving optimizing nanoparticle size to maximize lymphatic drainage[53] or to pretreat the cells with IFNγ to upregulate MHC expression.[54] Overall, the strategy outlined in this article serves as a blueprint for how to engineer complex, multimodal cell–cell interactions using biomimetic nanotechnology, and there are countless opportunities for modulating cellular function natively via their surface markers in a manner that is unique from traditional therapies.
In conclusion, we have constructed a biomimetic nanoscale aAPC platform capable of directly activating T cells against tumor antigens based on the direct presentation of epitopes found on cancer cells. This was achieved by engineering cancer cells to express costimulatory markers in order to leverage their endogenous antigen presentation machinery. The membrane from these cells, which contained the requisite signals for T cell stimulation, was then stabilized onto a nanoparticulate substrate to enable in vivo application. It was demonstrated that the double knock-in [CD80/OVA]NP formulation was able to control tumor growth in murine models. One of the key advantages of this biomimetic approach towards antigen presentation is its ability to bridge the gap that exists between current cell-based and synthetic nanoparticle-based anticancer immunotherapies. On one hand, the non-living nature of the biomimetic antigen-presenting nanoparticles eliminates concerns associated with the derivation, manipulation, and re-administration of patient-derived cells, which should simplify manufacture and quality control. On the other hand, the biomembrane component readily enables the presentation of multiple tumor antigens without requiring the specific identification of the relevant epitopes. Further, there has been evidence suggesting that the fluidity afforded by lipid membrane structures can enhance antigen presentation efficiency.[55, 56] It is also notable that the anticancer immunity in the present study was generated in the absence of other immunostimulatory compounds, such as adjuvants, cytokines, or checkpoint blockades, which may be included in the future within the nanoparticle core to enhance treatment potency by providing additional immunological signals. With regards to clinical translation, the well-established workflows for modifying patient-derived cells in chimeric antigen receptor T cell therapy can be adapted for engineering autologous cancer cells prior to fabricating the antigen-presenting nanoparticles.[57] Ultimately, the platform represents an effective means of producing tumor-targeting immune cell subsets and could be combined with other modes of immunotherapy to produce a more comprehensive solution for generating robust antitumor responses in the clinic.
Experimental Section
Cell Culture and Engineering.
Wild-type B16-F10 (B16-WT) mouse melanoma cells (CRL-6475; American Type Culture Collection) were maintained in Dulbecco’s modified eagle medium (DMEM; Mediatech) supplemented with 10% bovine growth serum (Hyclone) and 1% penicillin-streptomycin (Gibco). To generate the B16-CD80 cell line, B16-WT cells were transfected with a plasmid encoding CD80 (pUNO1-mB7–1; InvivoGen) using lipofectamine 2000 (Life Technologies), followed by selection in media containing blastidicin (InvivoGen). Monoclonal selection was conducted by plating the blasticidin-selected cells in 96-well tissue culture plates at an average density of 0.5 cells per well, and the clone with the highest expression was expanded for further study. B16-CD80 cells were maintained in culture media supplemented with 4 μg/mL of blasticidin. To generate the B16-CD80/OVA cell line, a gene encoding for a cytoplasmic form of ovalbumin from pCI-neo-cOVA (a gift from Maria Castro; #25097; Addgene) was cloned into the pQCXIH retroviral expression vector (Clontech), and the resulting plasmid was used to transfect AmphoPhoenix cells (obtained from the National Gene Vector Biorepository). After 48 h, virus-containing cell culture supernatant was collected, mixed with polybrene (Sigma-Aldrich) at a final concentration of 4 μg/mL, and added to B16-CD80 cells. Viral transduction was facilitated by centrifuging the culture plate at 800 g for 90 min. After 48 h of transduction, selection was performed by culturing the cells in media containing hygromycin B (InvivoGen). A monoclonal cell line was obtained in a similar manner as above. B16-CD80/OVA cells were maintained in media containing 300 μg/mL hygromycin B and 4 μg/mL blasticidin. B16-OVA cells were generated using the same protocol starting with B16-WT cells and were maintained in media containing 300 μg/mL hygromycin B.
Western Blotting.
The various cell lines were collected by scraping and lysed by sonicating in water for 5 min using a Fisher FS30D bath sonicator. Each sample was diluted to 0.7 mg/mL of protein content in water based on a BCA protein assay (Pierce) and then mixed in a 3 to 1 volume ratio with NuPAGE 4× lithium dodecyl sulfate sample loading buffer (Novex). After heating for 10 min at 70 °C, the samples were loaded into 12-well Bolt 4–12% bis-tris gels (Invitrogen) and run at 165 V for 45 min in MOPS running buffer (Novex). The proteins were then transferred onto 0.45 μm nitrocellulose membrane (Pierce) in Bolt transfer buffer (Novex) at 15 V for 30 min. The blots were blocked with 5% milk (Genesee Scientific) in PBS with 0.05% Tween 20 (National Scientific), followed by incubation with a mouse anti-ovalbumin monoclonal antibody (3G2E1D9, Santa Cruz Biotechnology) as the primary immunostain and a horseradish peroxidase-conjugated secondary (Biolegend) as the secondary immunostain. Membranes were developed with ECL western blotting substrate (Pierce) in an ImageWorks Mini-Medical/90 Developer.
Surface Marker Characterization
Flow cytometry was employed to examine the MHC-mediated surface presentation of the SIINFEKL (OVA257–264) peptide and the expression of CD80 on the various cell lines. To facilitate the analysis of SIINFEKL presentation, the cells were incubated with 10 ng/mL recombinant mouse IFNγ (Biolegend) overnight to upregulate MHC-I expression.[54] Cells were collected from culture using 1 mM ethylenediaminetetraacetic acid (USB Corporation) in PBS and stained with PE-conjugated anti-mouse Kb-SIINFEKL antibody (25-D1.16, Biolegend) and Alexa647-conjugated anti-mouse CD80 antibody (16–10A1, Biolegend), or the corresponding isotype antibodies (Biolegend). Data was collected using a Becton Dickinson FACSCanto-II flow cytometer and analyzed using FlowJo.
In Vitro Biological Activity of Engineered Cells.
All animal experiments were performed in accordance with NIH guidelines and approved by the Institutional Animal Care and Use Committee (IACUC) of the University of California San Diego. C57BL/6-Tg(TcraTcrb)1100Mjb/J transgenic mice (OT-I; The Jackson Laboratory) were euthanized and their spleens were collected. To obtain single cell suspensions, each spleen was physically extruded through 70 μm nylon cell strainers (Fisher Scientific), followed by red blood cell removal using RBC lysis buffer (Biolegend) per the manufacturer’s instructions. The cells were then washed with 1× PBS and CD8+ cells were isolated out by magnetic separation using CD8a (Ly-2) MicroBeads (Miltenyi Biotec) on MACS LS separation columns (Miltenyi Biotec) per the manufacturer’s instructions. Splenocytes or isolated CD8+ cells were resuspended using warm media consisting of 500 mL Isocove’s modification of DMEM with 2 mM L-glutamine and 25 mM HEPES (Mediatech) supplemented with 50 mL USDA certified fetal bovine serum (Omega Scientific), 500 μL 55 mM β-mercaptoethanol (Gibco), 5 mL 200 mM L-glutamine (Gibco), and 5 mL penicillin-streptomycin. Either 2 × 105 CD8+ cells or 3 × 106 splenocytes were incubated with 5 × 104 of the various engineered B16 cells for 24 h. For cytokine analysis, culture supernatant was collected and assayed using mouse IFNγ and IL-2 ELISA kits (Biolegend) according to the manufacturer’s instructions. For surface marker analysis, CD8+ cells or splenocytes in culture were collected and stained with one of the two following sets of antibodies: (1) PE/Cy7-conjugated anti-mouse CD3 antibody (17A2, Biolegend), APC-conjugated anti-mouse CD8a antibody (53–6.7, Biolegend), FITC-conjugated anti-mouse/human CD44 antibody (IM7, Biolegend), and Pacific Blue-conjugated anti-mouse CD62L antibody (MEL-14, Biolegend), or (2) PE/Cy7-conjugated anti-mouse CD3 antibody, Pacific Blue-conjugated anti-mouse CD8a antibody (Biolegend), Alexa647-conjugated anti-mouse CD69 antibody (H1.2F3, Biolegend), and FITC-conjugated anti-mouse CD25 antibody (3C7, Biolegend). Data was collected using a Becton Dickinson FACSCanto-II flow cytometer and analyzed using FlowJo. All analysis was conducted on the CD3+CD8+ T cell population. To examine the ability of the engineered cells to activate T cells specific for a native antigen, splenocytes were derived from B6.Cg-Thy1a/Cy Tg(TcraTcrb)8Rest/J transgenic mice (pmel-1; The Jackson Laboratory). Prior to the study, B16-OVA or B16-CD80/OVA cells were cultured in the presence of 10 ng/mL recombinant mouse IFNγ for 1 day. The engineered cells were then rinsed before incubating with 3 × 106 pmel-1 splenocytes for 48 h. Afterwards, surface marker expression was analyzed by staining with PE/Cy7-conjugated anti-mouse CD3 antibody, Pacific Blue-conjugated anti-mouse CD8a antibody, and Alexa647-conjugated anti-mouse CD69 antibody.
Nanoparticle Preparation.
The cell membrane from B16-WT, B16-OVA, and B16-CD80/OVA cells was collected according to a previously published protocol.[46] Briefly, cells were suspended in a lysis buffer containing 30 mM Tris-HCl pH 7.0 (Quality Biological) with 0.0759 M sucrose (Sigma-Aldrich), 0.225 M D-mannitol (Sigma-Aldrich), and a cocktail of phosphatase and protease inhibitors (Sigma-Aldrich), followed by physical disruption using a Kinematica Polytron PT 10/35 probe homogenizer at 70% power for 15 passes. The membrane was separated by first centrifuging at 10,000 g and then centrifuging the resulting supernatant at 150,000 g to obtain a membrane pellet using a Beckman Coulter Optima XPN-80 ultracentrifuge. To prepare polymeric cores, 1 mL of poly(DL-lactic-co-glycolic acid) (50:50 PLGA, 0.67 dL/g, Lactel Absorbable Polymers) in acetone at 10 mg/mL was added dropwise into 1 mL of water and the mixture was placed under a vacuum aspirator to evaporate the organic solvent. Membrane coating was carried out by mixing the preformed PLGA cores with the membrane at the appropriate ratios and sonicating the mixture for 2 min in a Fisher Scientific FS30D bath sonicator.
Nanoparticle Optimization and Characterization.
To optimize the nanoformulation, [CD80/OVA]NPs were fabricated at membrane to polymer weight ratios of 0, 0.1, 0.2, 0.5, 1, and 2 in water and then adjusted to 1× PBS using 20× PBS (Teknova). Nanoparticle size and surface zeta potential were measured by dynamic light scattering using a Malvern ZEN 3600 Zetasizer. To evaluate stability over time, nanoparticles were suspended in 10% sucrose (Sigma-Aldrich) solution and stored at 4 °C for two weeks; size was monitored every 2 days. For electron microscopic visualization, freshly synthesized nanoparticles or nanoparticles stored for 1 week at 4 °C in 10% sucrose were first deposited onto carbon-coated 400-square mesh copper grids (Electron Microscopy Sciences), followed by negative staining with 1 wt% uranyl acetate (Electron Microscopy Sciences). Imaging was performed on a Tecnai Spirit transmission electron microscope. The pull-down assay was conducted by first suspending nanoparticles at 4 mg/mL in 1% bovine serum albumin (BSA; Sigma-Aldrich) for 1 h at 4 °C. Then, Alexa647-conjugated anti-mouse CD80 antibody, APC-conjugated anti-mouse Kb-SIINFEKL antibody (Biolegend), or the corresponding isotype antibodies were incubated with the nanoparticles for 30 min at 4 °C. After incubation, the nanoparticles were centrifuged at 21,000 g, and the fluorescence of the supernatant was measured. Binding was calculated as (initial fluorescence – supernatant fluorescence)/(initial fluorescence) and normalized to the value for the corresponding isotype antibody.
In Vitro Biological Activity of Engineered Nanoparticles.
To evaluate nanoparticle biological activity, 8 × 105 CD8+ cells or 3 × 106 splenocytes from OT-I mice were cultured in the presence of 100 μg/mL of each nanoformulation for 3 days. CD25, CD69, CD44, and CD62L surface marker expression, as well as IFNγ and IL-2 secretion, were analyzed as described above. To measure biological functionality after storage, nanoparticles stored for 1 week at 4 °C in 10% sucrose were incubated with 8 × 105 OT-I CD8+ cells at 100 μg/mL for 3 days. Expression of CD25 and CD69 on the CD8+ T cells was measured as described above. For the cell proliferation study, 3 × 106 splenocytes derived from OT-I mice were labeled using CellTrace Violet (Invitrogen) according to the manufacturer’s instructions and incubated with the various nanoformulations at a concentration of 0, 12.5, 25, 50, and 100 μg/mL. After 3 days of incubation, the cells were collected and stained with PE/Cy7-conjugated anti-mouse CD3 antibody and FITC-conjugated anti-mouse CD8a antibody (Biolegend). Data was collected using a Becton Dickinson FACSCanto-II flow cytometer and analyzed using FlowJo. All analysis was conducted on the CD3+CD8+ T cell population. To quantify fold expansion, splenocytes derived from OT-I mice were cultured in 24-well plates at a density of 4 × 106 cells per well. Nanoparticles were added to the wells at a final concentration of 100 μg/mL. On day 4, the cells were collected and stained with PE/Cy7-conjugated anti-mouse CD3 antibody and FITC-conjugated anti-mouse CD8a antibody for enumeration by flow cytometry. Fold expansion was calculated by normalizing the total number of CD3+CD8+ T cells on day 4 to the number on day 0. To measure cell killing, CD8+ cells derived from OT-I mouse were activated in the presence of 50 μg/mL of [CD80/OVA]NPs and 20 ng/mL of recombinant mouse IL-2 (Biolegend) for 3 days. On the day before the assay, target B16-WT and B16-OVA cells were plated at a density of 1 × 104 cells per well in a 96-well plate in the presence of 10 ng/mL of recombinant mouse IFNγ. To perform the assay, the target cells were first rinsed twice with culture media, and then 5 × 104, 1 × 105, or 2 × 105 activated CD8+ cells were added, followed by incubation for 18 h at 37 °C. The cytotoxicity was measured by an LDH assay (Biolegend) per the manufacturer’s instructions (Biolegend). To examine the ability of the nanoparticles to activate immunity against native antigens, 3 × 106 pmel-1 splenocytes were cultured with 100 μg/mL of nanoformulations fabricated using membrane derived from engineered cells pretreated with 10 ng/mL recombinant mouse IFNγ for 72 h. Expression of CD69 was examined as described above.
In Vivo Delivery.
To evaluate the in vivo localization of the nanoparticles, 400 μg of [CD80/OVA]NPs labeled with 0.1 wt% 1,1’-dioctadecyl-3,3,3’,3’- tetramethylindodicarbocyanine dye (Biotium) were subcutaneously injected into the flanks of OT-I mice. The subcutaneous administration route was chosen over the intravenous route due to the propensity of the latter for generating tolerance.[58] At 0, 12, and 24 h, the inguinal lymph nodes were collected and cryosectioned, followed by fixation in 10% phosphate-buffered formalin (Fisher Chemical) for 15 min at room temperature. After fixation, the slides were blocked with 2% BSA in PBS for 30 min and stained with 7.5 μg/mL FITC-conjugated anti-mouse CD8a antibody in 2% BSA solution overnight at 4 °C. Finally, the slides were mounted in Vectashield mounting media (Vector Laboratories) and imaged on a Keyence BZ-X710 fluorescence microscope using the GFP and Cy5 filters.
In Vivo Biological Activity.
Whole-body irradiation was performed on female C57BL/6 mice (Envigo) at a dosage of 6 Gy. After 1 day, 2.5 × 107 splenocytes derived from OT-I mice were labeled with CellTrace Violet and adoptively transferred into each irradiated mouse by intravenous injection. The next day, 450 μg of the various nanoformulations in 10% sucrose was injected subcutaneously into each the neck, left flank, and right flank. After another 3 days, the inguinal and axillary lymph nodes were collected and processed into single cell suspensions, after which they were stained with PE/Cy7-conjugated anti-mouse CD3 antibody, FITC-conjugated anti-mouse CD8a antibody, and Alexa647-conjugated anti-mouse CD69 antibody. Data was collected using a Becton Dickinson FACSCanto-II flow cytometer and analyzed using FlowJo. All analysis was conducted on the CellTrace+CD3+CD8+ T cell population. To assess cytokine secretion, whole-body irradiation was performed on female C57BL/6 mice at a dosage of 6 Gy and, after 1 day, 2 × 107 splenocytes derived from OT-I mice were adoptively transferred. The next day, 450 μg of the various nanoformulations in 10% sucrose was injected subcutaneously into each the neck, left flank, and right flank. After another 4 days, the inguinal and axillary lymph nodes were collected and processed into a single cell suspension, which was cultured in 250 μL of media in a 96-well tissue culture plate. After 3 days of culture, the supernatant was collected and assayed using a mouse IFNγ ELISA kit according to the manufacturer’s instructions.
In Vivo Antitumor Efficacy.
To study prophylactic efficacy, whole-body irradiation was performed on female C57BL/6 mice at a dosage of 5.5 Gy. After 1 day, 1 × 107 splenocytes derived from OT-I mice were adoptively transferred into each mouse by intravenous injection. The following day, 400 μg of nanoparticles was subcutaneously injected into each the neck, left flank, and right flank. Tumor challenge was performed by subcutaneous administration of 4 × 105 B16-OVA cells into the lower right flank after another 5 days. To study therapeutic efficacy, female C57BL/6 mice were first challenged with 5 × 105 B16-OVA cells. Whole-body irradiation at a dosage of 5.5 Gy was performed on day 6 after tumor inoculation. On day 7, 1 × 106 CD8+ T cells derived from OT-I mice (main therapeutic study) or 1 × 107 splenocytes derived from OT-I mice (whole cell lysate study) were adoptively transferred into each mouse by intravenous injection. Treatment was administered on days 8 and 13 by subcutaneous injection of 300 μg of nanoparticles into each the neck, left flank, and right flank. Whole cell lysate was prepared by resuspending B16-OVA cells in water and subjecting them to 3 freeze-thaw cycles. Mice were administered on days 8 and 13 with lysate at a dosage where the amount of membrane material was equivalent to the [CD80/OVA]NP sample,[46] along with 10 μg of CpG 1826 (InvivoGen). To study therapeutic efficacy without irradiation and adoptive transfer, female C57BL/6 mice were first challenged with 3 × 105 B16-OVA cells. Treatment was administered on days 3 and 7 by subcutaneous injection of 300 μg of nanoparticles into each the neck, left flank, and right flank. Tumors were measured every other day and the experimental endpoint was defined as either death or tumor size greater than 200 mm2.
Supplementary Material
Acknowledgements
This work is supported by the National Institutes of Health under Award Number R01CA200574 and the Defense Threat Reduction Agency Joint Science and Technology Office for Chemical and Biological Defense under Grant Number HDTRA1-18-1-0014. Y.J. and J.Z. were supported by a National Institutes of Health 5T32CA153915 training grant from the National Cancer Institute. Support was also received from the NHLBI funded National Gene Vector Biorepository at Indiana University (Contract Number 75N92019D00018).
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
The authors declare no conflict of interest.
References
- [1].Finn OJ, Engl N. J. Med 2008, 358, 2704. [DOI] [PubMed] [Google Scholar]
- [2].Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD, Nat. Immunol 2002, 3, 991. [DOI] [PubMed] [Google Scholar]
- [3].Martin-Liberal J, Ochoa de Olza M, Hierro C, Gros A, Rodon J, Tabernero J, Cancer Treat. Rev 2017, 54, 74. [DOI] [PubMed] [Google Scholar]
- [4].Pardoll DM, Nat. Med 1998, 4, 525. [DOI] [PubMed] [Google Scholar]
- [5].Postow MA, Callahan MK, Wolchok JD, J. Clin. Oncol 2015, 33, 1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Jamal-Hanjani M, Quezada SA, Larkin J, Swanton C, Clin. Cancer Res 2015, 21, 1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hinrichs CS, Rosenberg SA, Immunol. Rev 2014, 257, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Azoury SC, Straughan DM, Shukla V, Curr. Cancer Drug Targets 2015, 15, 452. [DOI] [PubMed] [Google Scholar]
- [9].Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, Benci JL, Xu B, Dada H, Odorizzi PM, Herati RS, Mansfield KD, Patsch D, Amaravadi RK, Schuchter LM, Ishwaran H, Mick R, Pryma DA, Xu X, Feldman MD, Gangadhar TC, Hahn SM, Wherry EJ, Vonderheide RH, Minn AJ, Nature 2015, 520, 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Fecek RJ, Storkus WJ, Immunotherapy 2016, 8, 1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Stern AM, Markel H, Health Aff. (Millwood) 2005, 24, 611. [DOI] [PubMed] [Google Scholar]
- [12].Tabi Z, Man S, Adv. Drug Deliv. Rev 2006, 58, 902. [DOI] [PubMed] [Google Scholar]
- [13].Finn OJ, Nat. Rev. Immunol 2003, 3, 630. [DOI] [PubMed] [Google Scholar]
- [14].Circelli L, Tornesello M, Buonaguro FM, Buonaguro L, Hum. Vaccin. Immunother 2017, 13, 1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Awate S, Babiuk LA, Mutwiri G, Front. Immunol 2013, 4, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Jacobs JJ, Snackey C, Geldof AA, Characiejus D, Van Moorselaar RJ, Den Otter W, Anticancer Res 2014, 34, 2689. [PubMed] [Google Scholar]
- [17].Chen P, Liu X, Sun Y, Zhou P, Wang Y, Zhang Y, Hum. Vaccin. Immunother 2016, 12, 612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Silverman E, Biotechnol. Healthc 2012, 9, 13. [PMC free article] [PubMed] [Google Scholar]
- [19].Turtle CJ, Riddell SR, Cancer J 2010, 16, 374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Butler MO, Hirano N, Immunol. Rev 2014, 257, 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Eggermont LJ, Paulis LE, Tel J, Figdor CG, Trends Biotechnol 2014, 32, 456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sunshine JC, Green JJ, Nanomedicine (Lond.) 2013, 8, 1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Rhodes KR, Green JJ, Mol. Immunol 2018, 98, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Fang RH, Zhang L, Annu. Rev. Chem. Biomol. Eng 2016, 7, 305. [DOI] [PubMed] [Google Scholar]
- [25].Hutchison S, Pritchard AL, Mamm. Genome 2018, 29, 714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Garrido F, Aptsiauri N, Doorduijn EM, Garcia Lora AM, van Hall T, Curr. Opin. Immunol 2016, 39, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chong H, Hutchinson G, Hart IR, Vile RG, Br J Cancer 1998, 78, 1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Fang RH, Kroll AV, Gao W, Zhang L, Adv. Mater 2018, 30, 1706759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Overwijk WW, Restifo NP, Curr. Protoc. Immunol 2000, 39, 20.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bohm W, Thoma S, Leithauser F, Moller P, Schirmbeck R, Reimann J, J. Immunol 1998, 161, 897. [PubMed] [Google Scholar]
- [31].Yang J, Sanderson NS, Wawrowsky K, Puntel M, Castro MG, Lowenstein PR, Proc. Natl. Acad. Sci. U. S. A 2010, 107, 4716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Schweitzer AN, Borriello F, Wong RC, Abbas AK, Sharpe AH, J. Immunol 1997, 158, 2713. [PubMed] [Google Scholar]
- [33].Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, Carbone FR, Cell 1994, 76, 17. [DOI] [PubMed] [Google Scholar]
- [34].Yokoyama WM, Koning F, Kehn PJ, Pereira GM, Stingl G, Coligan JE, Shevach EM, J. Immunol 1988, 141, 369. [PubMed] [Google Scholar]
- [35].Valenzuela J, Schmidt C, Mescher M, J. Immunol 2002, 169, 6842. [DOI] [PubMed] [Google Scholar]
- [36].Sckisel GD, Mirsoian A, Minnar CM, Crittenden M, Curti B, Chen JQ, Blazar BR, Borowsky AD, Monjazeb AM, Murphy WJ, J. Immunother. Cancer 2017, 5, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Tanchot C, Lemonnier FA, Perarnau B, Freitas AA, Rocha B, Science 1997, 276, 2057. [DOI] [PubMed] [Google Scholar]
- [38].Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, Dellemijn TA, Antony PA, Spiess PJ, Palmer DC, Heimann DM, Klebanoff CA, Yu Z, Hwang LN, Feigenbaum L, Kruisbeek AM, Rosenberg SA, Restifo NP, J. Exp. Med 2003, 198, 569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hu CM, Fang RH, Wang KC, Luk BT, Thamphiwatana S, Dehaini D, Nguyen P, Angsantikul P, Wen CH, Kroll AV, Carpenter C, Ramesh M, Qu V, Patel SH, Zhu J, Shi W, Hofman FM, Chen TC, Gao W, Zhang K, Chien S, Zhang L, Nature 2015, 526, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Hu CM, Zhang L, Aryal S, Cheung C, Fang RH, Zhang L, Proc. Natl. Acad. Sci. U. S. A 2011, 108, 10980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Fang RH, Hu CM, Luk BT, Gao W, Copp JA, Tai Y, O’Connor DE, Zhang L, Nano Lett 2014, 14, 2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Thamphiwatana S, Angsantikul P, Escajadillo T, Zhang Q, Olson J, Luk BT, Zhang S, Fang RH, Gao W, Nizet V, Zhang L, Proc. Natl. Acad. Sci. U. S. A 2017, 114, 11488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zhang Q, Dehaini D, Zhang Y, Zhou J, Chen X, Zhang L, Fang RH, Gao W, Zhang L, Nat. Nanotechnol 2018, 13, 1182. [DOI] [PubMed] [Google Scholar]
- [44].Hu CM, Fang RH, Copp J, Luk BT, Zhang L, Nat. Nanotechnol 2013, 8, 336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hu CM, Fang RH, Luk BT, Zhang L, Nat. Nanotechnol 2013, 8, 933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kroll AV, Fang RH, Jiang Y, Zhou J, Wei X, Yu CL, Gao J, Luk BT, Dehaini D, Gao W, Zhang L, Adv. Mater 2017, 29, 1703969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Gao W, Fang RH, Thamphiwatana S, Luk BT, Li J, Angsantikul P, Zhang Q, Hu CM, Zhang L, Nano Lett 2015, 15, 1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Copp JA, Fang RH, Luk BT, Hu CM, Gao W, Zhang K, Zhang L, Proc. Natl. Acad. Sci. U. S. A 2014, 111, 13481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Lokhov PG, Balashova EE, J. Cancer 2010, 1, 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].van der Burg SH, Arens R, Ossendorp F, van Hall T, Melief CJ, Nat. Rev. Cancer 2016, 16, 219. [DOI] [PubMed] [Google Scholar]
- [51].Grenier JM, Yeung ST, Khanna KM, Front. Immunol 2018, 9, 610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Juneja VR, McGuire KA, Manguso RT, LaFleur MW, Collins N, Haining WN, Freeman GJ, Sharpe AH, J. Exp. Med 2017, 214, 895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Reddy ST, van der Vlies AJ, Simeoni E, Angeli V, Randolph GJ, O’Neil CP, Lee LK, Swartz MA, Hubbell JA, Nat. Biotechnol 2007, 25, 1159. [DOI] [PubMed] [Google Scholar]
- [54].Merritt RE, Yamada RE, Crystal RG, Korst RJ, J. Thorac. Cardiovasc. Surg 2004, 127, 355. [DOI] [PubMed] [Google Scholar]
- [55].Lin JC, Chien CY, Lin CL, Yao BY, Chen YI, Liu YH, Fang ZS, Chen JY, Chen WY, Lee NN, Chen HW, Hu CJ, Nat. Commun 2019, 10, 1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Zhang Q, Wei W, Wang P, Zuo L, Li F, Xu J, Xi X, Gao X, Ma G, Xie HY, ACS Nano 2017, 11, 10724. [DOI] [PubMed] [Google Scholar]
- [57].Wang X, Riviere I, Mol. Ther. Oncolytics 2016, 3, 16015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Liblau RS, Tisch R, Shokat K, Yang X, Dumont N, Goodnow CC, McDevitt HO, Proc. Natl. Acad. Sci. U. S. A 1996, 93, 3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.