

Abstract
Small cell lung cancer (SCLC) remains a recalcitrant disease where limited therapeutic options have not improved overall survival and approved targeted therapies are lacking. Amplification of the tyrosine kinase receptor FGFR1 (fibroblast growth factor receptor 1) is one of the few actionable alterations found in the SCLC genome. However, efforts to develop targeted therapies for FGFR1-amplified SCLC are hindered by critical gaps in knowledge around the molecular origins and mediators of FGFR1-driven signalling as well as the physiological impact of targeting FGFR1. Here we show that increased FGFR1 promotes tumorigenic progression in precancerous neuroendocrine cells and is required for SCLC development in vivo. Notably, Fgfr1 knockout suppressed tumor development in a mouse model lacking the retinoblastoma-like protein 2 (Rbl2) tumor suppressor gene but did not affect a model with wild-type Rbl2. In support of a functional interaction between these two genes, loss of RBL2 induced FGFR1 expression and restoration of RBL2 repressed it, suggesting a novel role for RBL2 as a regulator of FGFR1 in SCLC. Additionally, FGFR1 activated phospholipase C gamma 1 (PLCG1) while chemical inhibition of PLCG1 suppressed SCLC growth, implicating PLCG1 as an effector of FGFR1 signaling in SCLC. Collectively, this study uncovers mechanisms underlying FGFR1-driven SCLC that involve RBL2 upstream and PLCG1 downstream, thus providing potential biomarkers for anti-FGFR1 therapy.
Keywords: FGFR1, RBL2, PLCG1, SCLC
Introduction
SCLC accounts for approximately 13% of all lung cancers and remains a recalcitrant disease. The standard chemotherapy of cisplatin and etoposide has not significantly improved overall survival in SCLC patients and the development of targeted therapies has been challenging due to the paucity of clinically viable targets (1). SCLC is mostly driven by loss of tumor suppressors, including near universal inactivation of RB and p53 and frequent loss of other tumor suppressors such as the RB family members RBL1 and RBL2 (coded by the RBL1 and RBL2 and also known as p107 and p130, respectively) (2, 3). Unfortunately, these loss-of-function alterations are not directly actionable. Although oncogenic alterations are less frequent in SCLC, gene amplifications of FGFR1 are detected in approximately 6% of SCLC tumors and high FGFR1 expression is also detected in a subset of tumors without the gene amplification (2–6). Importantly, an FGFR-selective tyrosine kinase inhibitor (TKI) can inhibit the growth of SCLC cell lines with high FGFR1 expression (7–9). However, FGFR1 amplification and high FGFR1 levels alone do not guarantee sensitivity to FGFR inhibitors (7–9). Intracellular mediators of FGFR1 signaling as well as other genetic alterations may modulate sensitivity to treatment, but these mechanisms remain poorly defined. For example, while the MEK-ERK pathway mediates oncogenic FGFR1 signaling in some SCLC cell lines, a hyperactive RAF-MEK-ERK pathway is tumor suppressive in others (10, 11). In addition, while AKT, STAT, and PLCG1 act as transducers downstream of FGFR1 during organ development, little is known of their roles in SCLC (12). Importantly, physiological significance of FGFR1, either amplified or nonamplified, in SCLC has not formally been determined using autochthonous model. The oncogenic role has also recently been challenged by a study in which a constitutively active form of FGFR1 suppressed SCLC development in Rb1/Trp53-mutant genetically engineered mouse model (Rb1/Trp53-GEMM) (13). Thus, there are critical gaps in knowledge that need to be addressed not only to target FGFR1 in SCLC, but also to identify SCLC patients in ongoing clinical trials who would benefit from anti-FGFR1 therapies.
Here, we characterize FGFR1 in SCLC using Rb1/Trp53-GEMM and its variants in which adenoviral Cre (Ad-Cre)-mediated deletion of Rb1 and Trp53 results in lung tumors recapitulating major histopathological features of human SCLC (14,15). We also introduce genetic and chemical perturbations in primary tumor cells (mSCLC) as well as precancerous neuroendocrine cells (preSC) derived from the Rb1/Trp53-GEMM and that can transform into SCLC upon activation of oncogenic drivers (16). Our data show that FGFR1 is required for the development and growth of Rbl2 loss-driven SCLC, and that this dependency is due to the induction of FGFR1 expression. Furthermore, FGFR1 promotes tumor development and growth via activation of PLCG1 and alteration in neural differentiation. Our findings provide novel insight into the mechanism of SCLC progression and potential biomarkers for identifying patients appropriate for anti-FGFR1 therapies.
Materials and Methods
Plasmids and chemicals
pCW57.1, pLKO-puro, pL-CRISPR.EFS.tRFP, psPAX2 and pMD2.G plasmids were obtained from Addgene (#41393, #21915, #57819, #12259, #12260; the gifts from David Root, Dmitri Wiederschain, Benjami Ebert, Didier Trono). Full-length cDNAs of Fgfr1 and Rbl2 were cloned using total RNAs from murine SCLC cells and verified by Sanger sequencing before cloning into pCW57.1. The sequences of oligo nucleotides for cloning Fgfr1 and Rbl2, shRNAs for pLKO-puro, and gRNAs for pL-CRISPR.EFS.tRFP are listed in Supplementary Materials and Methods. A small molecule inhibitor of PLCG1 and its structural analog, U73122 and U73343, were obtained from Cayman Chemical. A tyrosine kinase inhibitor, PD173074, was obtained from Sigma.
Cell culture
Murine SCLC cells (mSCLC) and precancerous cells (preSC) were derived from lung tumors and early-stage neuroendocrine lesions, respectively, developed in the Rb1/Trp53-GEMM (16). Human SCLC cell lines, including H69, H82, H209, H524, were the gifts of Adi Gazdar and John Minna (UT Southwestern), were authenticated by profiling patterns of seventeen short tandem repeats (ATCC, 135-XV and 200FTA), and were tested negative for Mycoplasma using PlasmoTest-Mycoplasma Detection Kit (InvivoGen, rep-pt1). Experiments were performed within first few passages after thawing frozen cell stocks. All human and mouse cells were cultured in RPMI1640 media (Invitrogen) and 293T cells were cultured in DMEM media (Corning), both supplemented with 10% bovine growth serum (GE Healthcare) and 1% penicillin-streptomycin (Invitrogen). For lentivirus production, we transfected lentiviral plasmids with packaging plasmids in 293T cells using polyethylenimine (Sigma), harvested supernatants containing viral particles 48 and 72 hours later, filtered through 0.45μm PVDF filter before adding to culture of target cells in the presence of 5μg/ml polybrene (Sigma). Puromycin (Thermo Fisher) was used to select stably transduced cells following lentiviral infection. To induce FGFR1 expression in Fgfr1-preSC, cells were treated with doxycycline (0.2ug/mL). CRISPR-mediated knockout of Rbl2 was achieved by transient transfection of Cas9 and gRNA in a single vector using Lipofectamine 2000 (Invitrogen). Forty-eight hours later, transfected cells were enriched by FACS sorting for red fluorescent protein expressed from the same vector. Sanger sequencing verified mutation in the target sequence in Rbl2 gene and immunoblot validated loss of RBL2. MTT assay was performed to measure cell viability using thiazolyl blue tetrazolium bromide (Sigma). Cells were seeded at 1 × 104 per well in 96-well plates at day 0, and MTT reagents were added on day 4. O.D values were determined at a wavelength of 590 nm using an ELISA reader (Thermo Scientific) The percentage survival was determined as the ratio of treated cells versus vehicle control after background subtraction. Soft-agar assay was performed by seeding 1×104 cells per well in 12-well plates as previously described (16). Images of wells are acquired using Olympus MVX10 scope, and colonies from the whole field of image were counted using the imaging software NIS-Elements Basic Research (Nikon). All of the cell culture experiments were performed in triplicates and repeated for a minimum of two biological replicates.
Tumor induction and allograft
Rb1lox/lox; Trp53lox/lox (Rb1/Trp53-GEMM) and Rb1lox/lox; Trp53lox/lox; Rbl2lox/lox (Rb1/Trp53/Rbl2-GEMM) have been previously described (14, 16). Mouse strains with Fgfr1lox and Fgfr2lox alleles have been previously described (17, 18). For tumor induction, lungs of 10-week-old mice, including both male and female mice and littermates, were infected with adenoviral Cre via intratracheal instillation as previously described and mice were aged 6 months (19). For allograft experiment, we inject 5.0×105 control or Fgfr1-preSC in the flanks of nude mice (Foxn1nu/nu; Envigo) and 1.0×106 Fgfr1lox/lox Rb1/Trp53/Rbl2 murine cells infected with Ad-Cre in the flanks of B6/129S F1 hybrid mice (Jackson Laboratory). To induce FGFR1 expression in Fgfr1-preSC following implantation, mice were fed doxycycline diet (625mg/kg, Envigo). Mice were maintained according to guidelines from the National Institutes of Health. Animal procedures were approved by the Institutional Animal Care and Use Committee at the University of Virginia.
Histology, immunohistochemistry, and immunoblot
Mouse tissues were fixed in 4% paraformaldehyde in phosphate-buffered saline before paraffin embedding. Five micrometer-thick tissue sections were stained with hematoxylin and eosin staining and immunostaining as previously described (16). For quantification of pHH3-positive cells, tumors of similar size and area were included. For immunoblot analysis, protein was extracted from mouse tumor tissues and human and murine cell lines in RIPA buffer. All primary and secondary antibodies used are listed in Supplementary Methods. Macroscopic images of lung sections were acquired using Olympus MVX10. Images of H&E and immunostained tissues were acquired using Nikon Eclipse Ni-U microscope. Image analysis and automated quantification were performed using NIS-Elements Basic Research (Nikon)
RNA sequencing and analysis
Sequencing libraries were generated by the Genome Analysis and Technology Core at University of Virginia using oligo dT-purified mRNA from 500ng of total RNA and the NEB Next Ultra RNA library preparation kit (New England Biolabs), and 50-bp single-end sequencing was performed on Illumina NextSeq 500 platform (Illumina). The sequencing data have been deposited to the SRA Database (https://www.ncbi.nlm.nih.gov/sra, identifier: PRJNA564798). Reads were mapped to the Mus musculus genome assembly GRCm38 (mm10) using TopHat and counts of reads map to each gene were obtained using HTSeq (20, 21). Differentially expressed genes regulated by FGFR1 were identified using DESeq2 package (22). A regularized log-transformation was applied to the read count data of all sequenced samples and resultant data were used for gene ontology (GO) term analysis at DAVID Bioinformatics Resources (https://david.ncifcrf.gov/home.jsp).
Statistical analysis
Using GraphPad Prism, the results were presented as the mean ± standard deviation. and evaluated using an unpaired two-tailed Student’s t-test. p<0.05 was considered statistically significant.
Results
Ectopic FGFR1 promotes progression of precancerous cells to SCLC
Fgfr1 amplification has not been detected in lung tumors in Rb1/Trp53-GEMM (3, 23). Instead, Fgfr1 mRNA levels were uniformly higher in mouse SCLC cells derived from the GEMM than precancerous cells (preSC) (Fig. 1A). Similarly, FGFR1 protein levels were markedly higher in tumor cells than preSC (Fig. 1A). In contrast, the levels of Fgfr2 and Fgfr4, which are implicated in other types of cancers (12), were generally low and did not show tumor-specific increase (Fig. 1A). Additionally, we guaged how widespread FGFR1 expression and activity are in human SCLC by examining a few human SCLC cells, which are commonly used and known as neuroendocrine subtypes with expression of the MYC family members (16, 24). While FGFR1 levels were heterogeneous across human SCLC lines, intriguingly, cell lines that are known to have high MYCL (H209 and H2141) showed markedly higher levels of phosphorylated FGFR1, indicative of protein activation, than high MYC or MYCN-expressing lines (H524, H82, H2171, and H69) (Supplementary Fig. 1A). These findings led us to hypothesize that FGFR1 promote SCLC development in Rb1/Trp53-GEMM and plays a role in a subset of human SCLC.
Fig. 1. Increased FGFR1 promotes SCLC development.
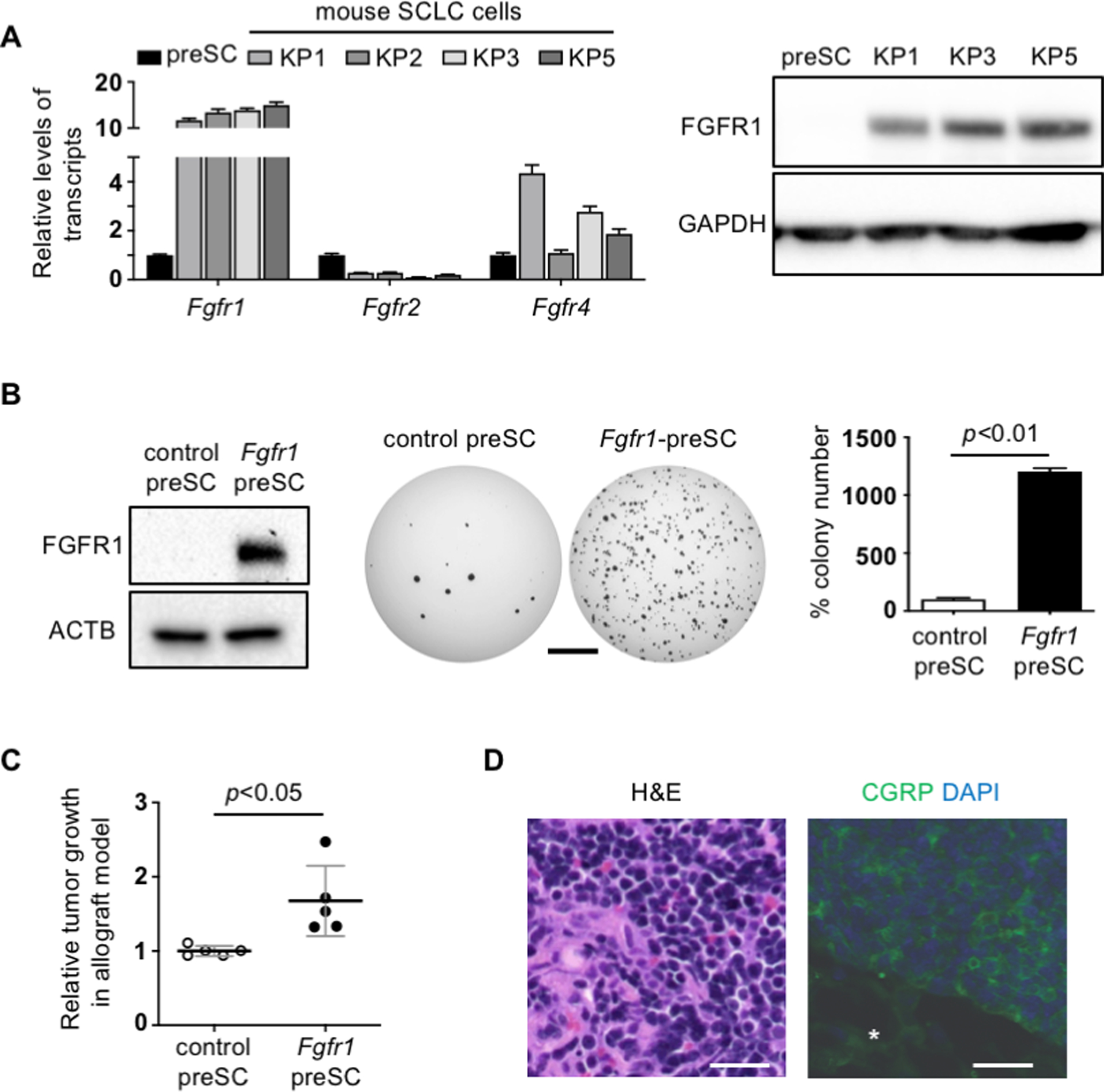
(A) RT-qPCR for Fgfr1 mRNA normalized to Gapdh (n=3 replicates per cell type) and immunoblot for FGFR1 in preSC and mSCLC. GAPDH blot verifies equal loading of total proteins. (B) Left, expression of FGFR1 in preSC infected with empty vector (control preSC) or FGFR1 expression construct (Fgfr1-preSC); ACTB blot verifies equal loading of total proteins. Middle, representative images of soft agar colonies formed by control and Fgfr1-preSC. Right, quantification of colonies >0.2mm in diameter (unpaired t-test n=3 replicates per cell type). (C) Plot of quantifying development of subcutaneous tumors >1.5cm in diameter (unpaired t-test n=5 mice per cell type); Relative tumor growth represents tumor weight (g) divided by latency (days after allograft). (D) Representative image of hematoxylin-eosin (H&E) stained section of Fgfr1-preSC-derived subcutaneous tumors and, right, of immunostaining for calcitonin gene-related peptide (CGRP, green). DAPI stains for nuclei (blue). Asterisk indicates non-tumor area. Statistical tests performed using unpaired t-test (ns: not significant). Scale bars: B, 5mm; D, 100μm. Error bars represent standard deviation.
To begin determining the functional impact of these alterations on tumor development, we expressed FGFR1 in preSC using a lentiviral vector and tested the transduced cells for tumorigenic potential. Compared with control preSC infected with empty vector, Fgfr1-preSC gave rise to more colonies in soft agar and more rapid development of subcutaneous tumors in immunocompromised nude mice (Fig. 1B, C). These subcutaneous tumors displayed histological features of SCLC including distinct small-cell morphology with scanty cytoplasm and positive staining for the neuroendocrine marker calcitonin gene-related peptide (CGRP) (Fig. 1D). These data suggest that FGFR1 overexpression is sufficient to promote tumorigenic transformation of precancerous precursors.
Deletion of FGFR1 suppresses Rbl2-deficient SCLC
The physiological significance of FGFR1 in SCLC remains unknown. To determine whether FGFR1 is necessary for tumor development, we deleted Fgfr1 in Rb1/Trp53/Fgfr1lox/lox via intratracheal instillation of Ad-Cre. Unexpectedly, we did not find a significant difference in tumor burden (tumor area/lung area) between Fgfr1Δ/Δ and Fgfr1+/+ Rb1/Trp53 mice (Fig. 2A), inconsistent with our results demonstrating a tumor-promoting role for FGFR1 in preSC (Fig. 1). We thus speculated whether a moderate impact of FGFR1 loss was not detectable in our assays, given the variability in tumor number and latency associated with Rb1/Trp53 mice (14). To exclude this possibility, we performed a similar experiment using Rb1/Trp53/Rbl2-GEMM in which tumor development is initiated by the loss of Rbl2 (a member of RB family tumor suppressor genes) in addition to Rb1 and Trp53 (25). This model develops more than a dozen tumors with latency of 6 months as opposed to 1–3 tumors with latency of 9–12 months in Rb1/Trp53-GEMM, thus providing a robust system for measuring the effect of genetic or pharmacological perturbations (26, 27). Intriguingly, Fgfr1Δ/Δ Rb1/Trp53/Rbl2 mice had significantly less tumor burden than Fgfr1+/+ Rb1/Trp53/Rbl2 mice (Fig. 2B). Lung tumors in Fgfr1Δ/Δ Rb1/Trp53/Rbl2 mice showed significantly less staining for phosphorylated histone H3 (pHH3), a marker for mitotic cell, than tumors in Fgfr1+/+ Rb1/Trp53/Rbl2 mice (Fig. 2C). These findings suggest that loss of FGFR1 suppresses tumor development in part through reduced proliferation. Additionally, to determine whether this impact on tumor cells is driven specifically by FGFR1, we used similar methods to examine the impact of deleting Fgfr2 in the Rb1/Trp53/Rbl2-GEMM. FGFR2 is expressed in the lung epithelium, and its amplification or deregulation has been implicated in the growth and survival of numerous tumor types (28–30). However, loss of Fgfr2 did not result in decreased tumor burden (Fig. 2D). Taken together, these findings suggest that Rbl2 loss-driven tumor development depends specifically on FGFR1 signaling.
Fig. 2. Deletion of Fgfr1 suppresses SCLC development in vivo.
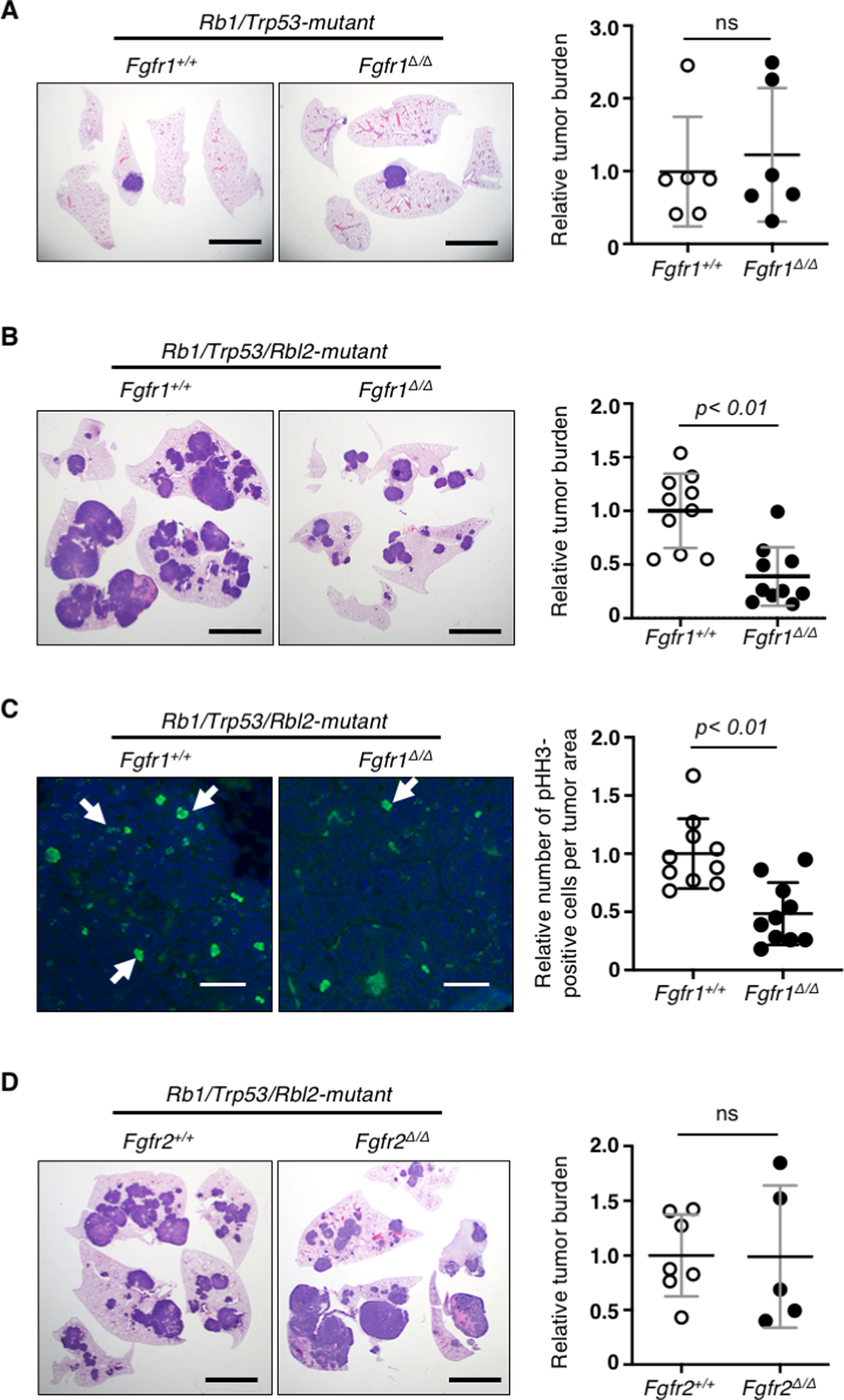
Left, representative images of H&E stained or immunostained sections of the lungs from different SCLC GEMMs and right, quantification of tumor burden (tumor area/lung area) or phosphorylated histone H3 (pHH3)-positive cells per tumor area in (A) Fgfr1+/+ vs. Fgfr1Δ/Δ Rb1/Trp53 (n=6 and n=6, respectively), (B, C) Fgfr1+/+ vs. Fgfr1Δ/Δ Rb1/Trp53/Rbl2 (n=10 and n=10, respectively); in panel C, pHH3 stain in green and DAPI in blue. Arrows indicate pHH3-positive nuclei, (D) Fgfr2+/+ vs. Fgfr2Δ/Δ Rb1/Trp53/Rbl2 n=7 and n=5, respectively). Statistical tests performed using unpaired t-test (ns: not significant). Error bars represent standard deviation. Scale bars: A, B, D, 5mm; C, 50μm.
To determine whether the decrease in tumor burden is truly due to loss of FGFR1, we validated the recombination of floxed alleles using genotyping PCR and immunoblot of primary cells from four randomly selected tumors developed in Fgfr1lox/lox Rb1/Trp53/Rbl2 mice infected with Ad-Cre. FGFR1 was not detected in primary cells from three of the four tumors, as expected (Supplementary Fig. 2). However, FGFR1 expression was present in primary cells from the remaining tumor and was modestly reduced compared to that in Fgfr1+/+ cells, indicating incomplete recombination of Fgfr1lox/lox alleles (Fig. 3A; Supplementary Fig. 2). Incomplete recombination of a floxed gene in full-blown tumors often suggests that the gene is required for tumor development (14).
Fig. 3. FGFR1 is critical for continued growth of tumor cells.
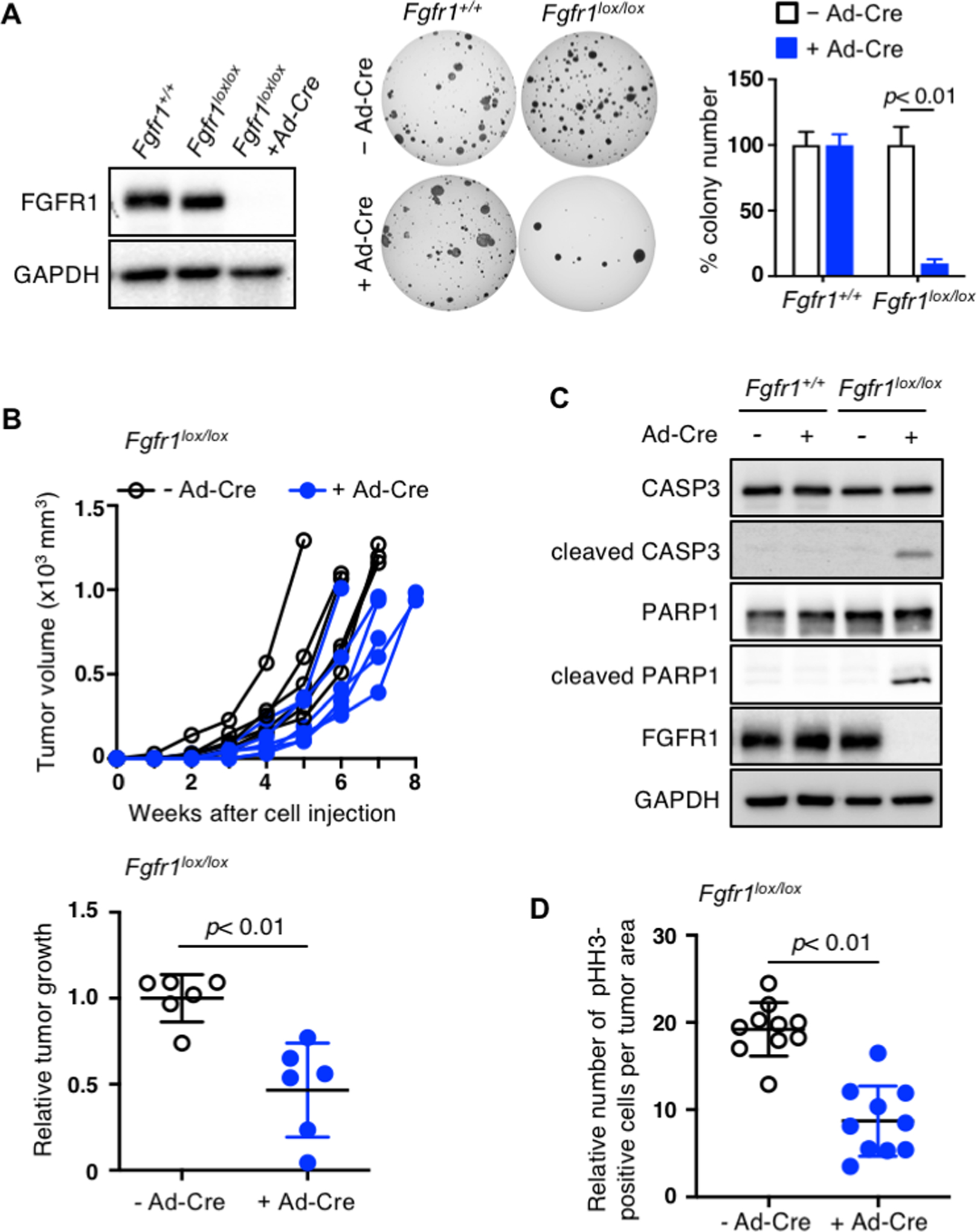
(A) Left, immunoblot for FGFR1 in primary cells derived from the lung tumors in Fgfr1+/+ and Fgfr1lox/lox Rb1/Trp53/Rbl2-GEMM infected with Ad-Cre. The cells from one of the Fgfr1lox/lox mice show little reduction of FGFR1, indicating incomplete recombination of floxed Fgfr1 alleles. Following Ad-Cre infection, these cells completely lose FGFR1 expression. Middle, representative images of soft agar colonies formed by Fgfr1+/+ and Fgfr1lox/lox Rb1/Trp53/Rbl2 cells in soft agar 3 weeks following Ad-Cre infection. Right, quantification of colonies >0.2mm in diameter (n=3 replicates per cell type). (B) Top, volumes of tumors (n=6) generated from subcutaneous injection of Fgfr1lox/lox Rb1/Trp53/Rbl2 cells with or without Ad-Cre infection. Bottom, quantification of tumor development of subcutaneous tumors >1.5cm in diameter; relative tumor growth represent tumor weight (g) divided by latency (days after allograft). (C) Immunoblot for cleaved/total CASP3 and PARP1 in Fgfr1+/+ and Fgfr1lox/lox Rb1/Trp53/Rbl2 cells with or without Ad-Cre infection. Cleaved CASP3 is a marker of apoptosis and FGFR1 blot verifies loss of FGFR1 expression in Ad-Cre infected Fgfr1lox/lox cells. GADPH blot verifies equal loading of total proteins. (D) Quantification of pHH3 staining in allograft tumors derived from Fgfr1lox/lox Rb1/Trp53/Rbl2 cells with or without Ad-Cre infection. Statistical tests performed using unpaired t-test. Error bars represent standard deviation.
To further validate the oncogenic role of FGFR1, we deleted any residual floxed alleles in Fgfr1lox/lox cells using Ad-Cre infection in culture and examined the impact of FGFR1 loss on tumor growth (Fig. 3A). These Ad-Cre infected cells completely lost FGFR1, gave rise to fewer colonies in soft agar, and formed smaller subcutaneous tumors than uninfected control cells or Ad-Cre-infected Fgfr1+/+ cells (Fig. 3A, B). Immunoblot assays showed that levels of cleaved CASP3 and PARP1 indicative of apoptosis markedly increased in Ad-Cre-infected Fgfr1lox/lox cells relative to uninfected control and Ad-Cre-infected Fgfr1+/+ cells (Fig. 3C). Similar to primary tumors with the same genotype, allograft tumors derived from Ad-Cre-infected Fgfr1lox/lox cells had significantly fewer pHH3-positive cells per tumor area than those derived from uninfected cells (Fig. 3D; Supplementary Fig. 3). These findings suggest that FGFR1 is important not only for tumor development but also for the continued growth of tumor cells.
Loss of RBL2 induces FGFR1 and the receptor dependency in SCLC
To our knowledge, functional relationships between RBL2 and FGFR1 have never been documented in cancer. However, RBL2 was found to repress Fgfr1 expression during muscle cell differentiation by co-binding with E2F4 at the Fgfr1 promoter (31). We thus surmised that Rbl2 loss may amplify FGFR1 dependency in SCLC by increasing FGFR1 expression levels. We first determined whether RBL2 loss induces FGFR1 expression in preSC. We found that Rbl2-KO preSC generated using CRIPSR showed a drastic reduction in RBL2 level but did not induce FGFR1 expression (Fig. 4A). Notably, however, subcutaneous tumors derived from Rbl2-KO preSC showed a significant increase in FGFR1 compared to those derived from Rbl2-WT preSC (Fig. 4A). This increase FGFR1 expression in Rbl2-KO preSC coincided with a drastic increase in Fgfr1 transcript level (Fig. 4A), and restoration RBL2 expression markedly reduced FGFR1 expression (Fig. 4B). Taken together, our findings demonstrate an inverse relationship between RBL2 and FGFR1 expression in SCLC.
Fig. 4. RBL2 loss induces FGFR1 expression and dependency.
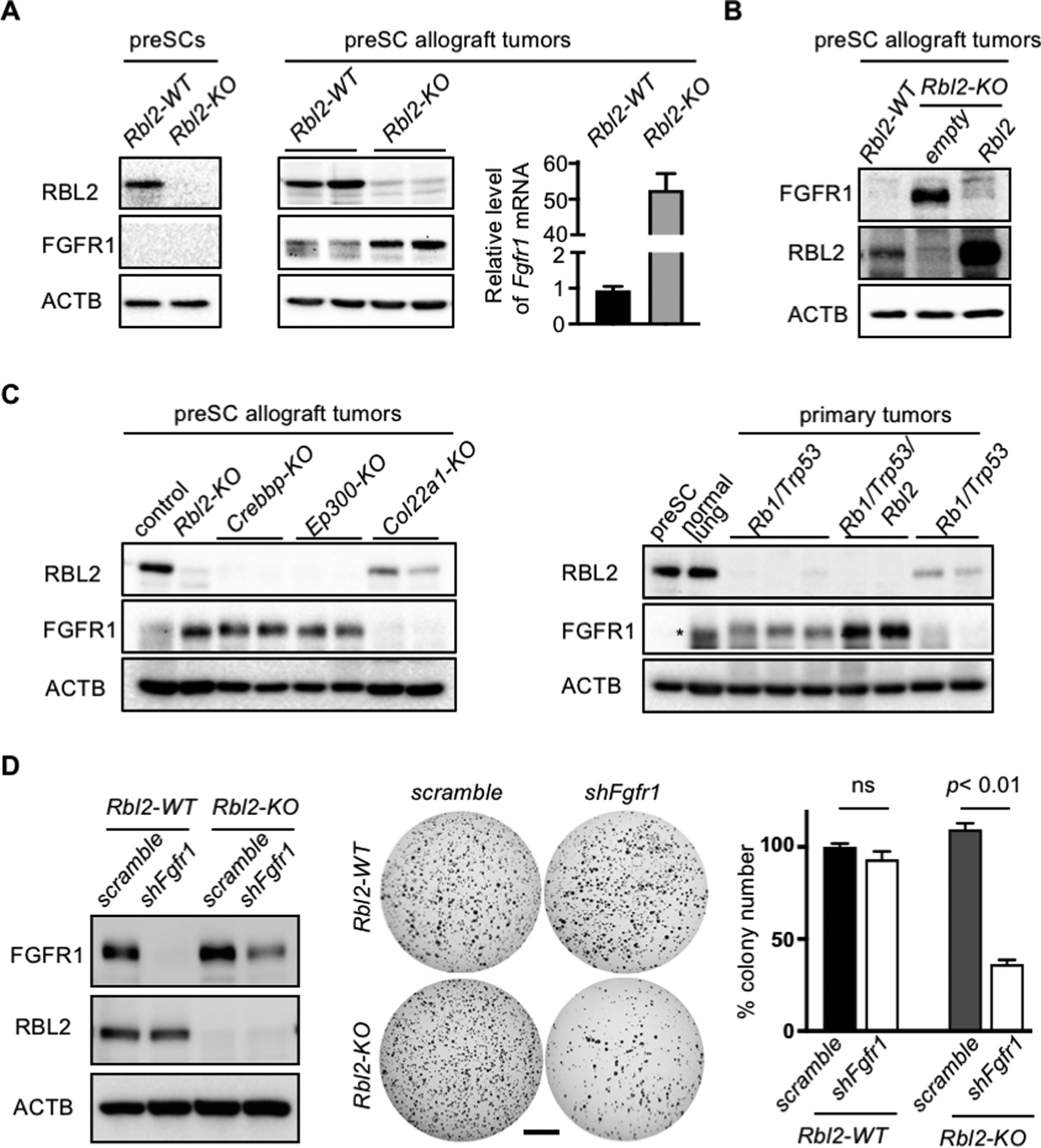
(A) Immunoblots for FGFR1 and RBL2 in control and Rbl2-KO preSC (left) and in preSC-derived subcutaneous tumors. Right, RT-qPCR for Fgfr1 mRNA normalized to Gapdh. (B) Immunoblots for FGFR1 and RBL2. Rbl2-KO preSCs were infected with empty or Rbl2 lentiviral vector. (C) Immunoblots for FGFR1 and RBL2 in the subcutaneous tumors generated from control and various targeted preSCs (left) and in primary lung tumors developed in Rb1/Trp53-mutant and Rb1/Trp53/Rbl2-mutant GEMMs (right). Asterisk indicates a non-specific band. (D) Left, immunoblots for FGFR1 and RBL2 in Rbl2-WT and Rbl2-KO mSCLC cells with or without Fgfr1 knockdown. Middle, representative images of soft agar colonies. Right, quantification of colonies >0.2mm in diameter (n=3 per cell type). ACTB blot verifies equal loading of total proteins. Statistical tests performed using unpaired t-test (ns: not significant). Error bar represents standard deviation. Scale bar: 5mm.
To determine whether this inverse relationship between RBL2 and FGFR1 expression exists across SCLC cells harboring different driver mutations, we examined the subcutaneous tumors derived from variants of mutant preSC in which Crebbp, Ep300, or Col22a1 was mutated using CRISPR. Loss-of-function mutations in these genes are among the most frequent alterations found in the SCLC genome and have been shown to promote malignant progression of preSC (3, 32). Similar to Rbl2-KO tumors, Crebbp-KO or Ep300-KO tumors expressed FGFR1 but lacked RBL2, whereas Col22a1-KO tumor showed the opposite expression patterns (Fig. 4C). In all tumors, FGFR1 protein expression was consistent with the levels of Fgfr1 transcript (Supplementary Fig. 4). Additionally, a survey of multiple primary tumors developed in Rb1/Trp53-GEMM and Rb1/Trp53/Rbl2-GEMM showed a mutual exclusivity between RBL2 and FGFR1 expression (Fig. 4C).
Next, to begin to test whether RBL2 loss renders FGFR1 dependency in SCLC cells, we knocked out Rbl2 using CRISPR/Cas9. Rbl2 knockout increased FGFR1 level in mSCLC cells but did not increase the ability of these cells to form colonies in soft agar compared with Rbl2-WT cells (Fig. 4D). Notably, subsequent shRNA-mediated knockdown of Fgfr1 decreased the colony-forming ability of Rbl2-KO cells but not Rbl2-WT cells, compared with the respective control cells expressing scrambled shRNA (Fig. 4D). Taken together, these findings suggest that RBL2 represses Fgfr1 in SCLC and RBL2 loss induces FGFR1 expression, resulting in dependency on FGFR1 signaling.
FGFR1-dependent SCLC cells require activation of PLCG1
To begin to identify intracellular mediators of FGFR1-driven SCLC development, we examined the phosphorylation status of ERK1/2, AKT1, STAT1, and PLCG1, major transducers downstream of FGFR1 during organ development (12). Phosphorylation of AKT1 and PLCG1 increased in Fgfr1-preSC relative to control preSC while that of ERK1/2 and STAT did not change, suggesting lack of activation of ERK1/2 and STAT by FGFR1 (Fig. 5A). This finding was intriguing as FGFR1 signaling in SCLC had been primarily linked to MEK-ERK pathway (33, 34). We also examined the same downstream factors in human SCLC lines with varying levels of FGFR1, including H82, H209, and H524. Lentiviral shRNA-mediated knockdown of FGFR1 significantly reduced growth of H82 and H209 cells in soft agar, induced cleavage of CASP3 indicative of apoptosis, and reduced phosphorylation of PLCG1 (Fig. 5B–D). However, FGFR1 knockdown did not affect the growth of H524 colonies and PLCG1 phosphorylation (Fig. 5B, D). In all cell lines, phosphorylation levels of ERK1/2, AKT1, and STAT1 did not change regardless of FGFR1 expression, further demonstrating a specific functional relationship between FGFR1 and PLCG1 in SCLC (Fig. 5D).
Fig. 5. FGFR1-dependent SCLC cells activated PLCG1 require activation of PLCG1.
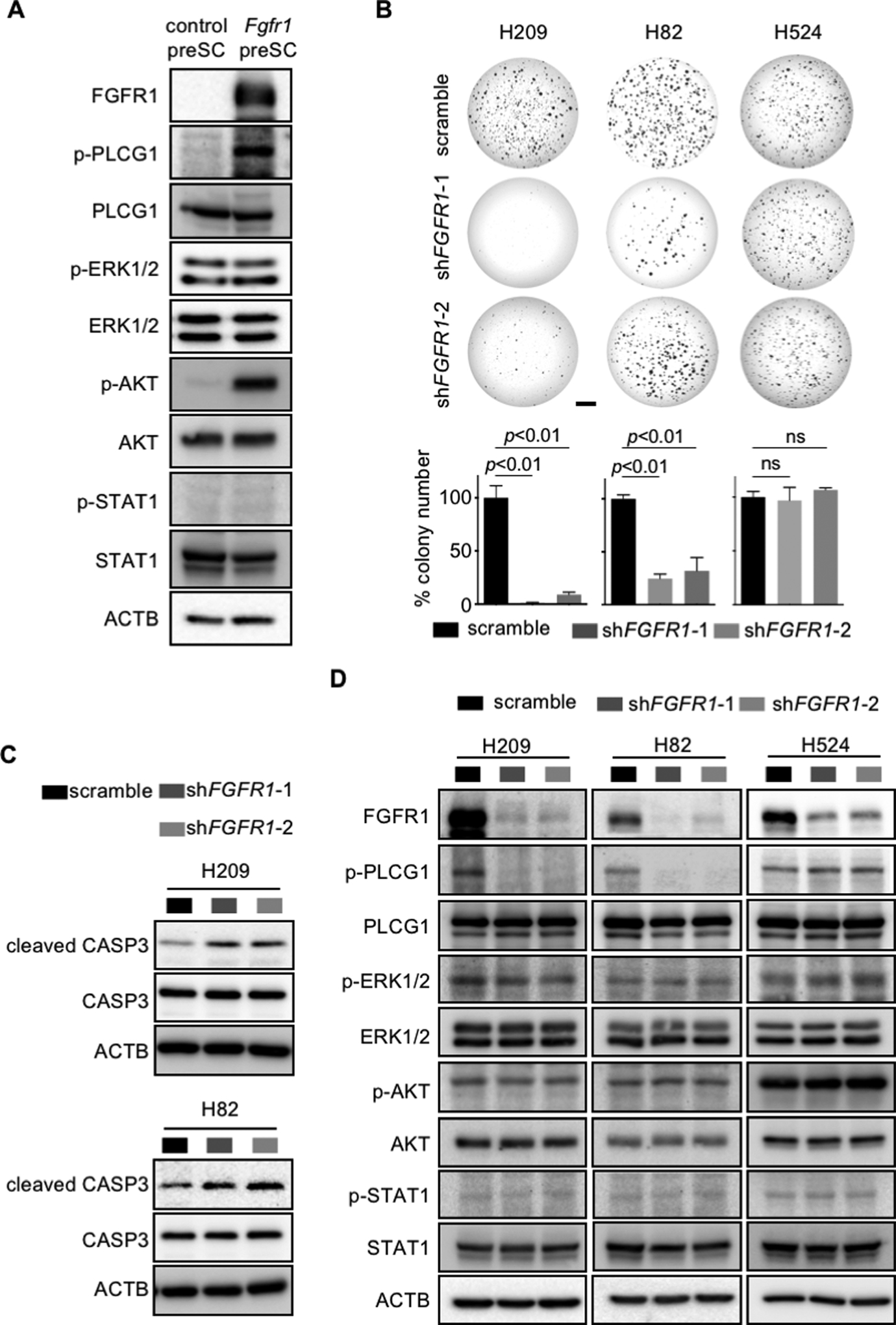
(A) Immunoblot for phosphorylated forms and respective total protein in control and Fgfr1-preSC. ACTB blot verifies equal loading of total proteins. (B) Representative images of soft agar colonies formed from human SCLC lines with FGFR1 knockdown (top) and quantification of colonies >0.2mm in diameter (bottom) (n=3 replicates per cell type). (C, D) Immunoblots for cleaved/total CASP3 and phosphorylated/total proteins of PLCG1, ERK1/2, AKT, and STAT1 in human SCLC lines with FGFR1 knockdown. Scale bar: 5mm.
To determine the significance of PLCG1 activation in SCLC cells, we tested effects of U73122, a chemical inhibitor of PLCG1, and its structural analog U73343 as an innocuous control (35). Compared with control, U73122 reduced viability of H82 and H209 cells in a concentration-dependent manner (Fig. 6A) and suppressed their ability to give rise to colonies in soft agar (Fig. 6B). However, these effects of U73122 were not seen for H524 (Fig. 6A,B), consistent with the observation that this cell line does not respond to FGFR1 knockdown in growth. Importantly, treatment of a pan-FGFR inhibitor PD173074 did not affect viability in the tested SCLC lines (Fig. 6C). Taken together, these data suggest that PLCG1 mediates FGFR1 signaling to promote tumorigenic progression of preSCs and continued growth of SCLC cells independently of ERK1/2, AKT1, and STAT1.
Fig. 6. Chemical inhibition of PLCG1 suppresses SCLC cells.
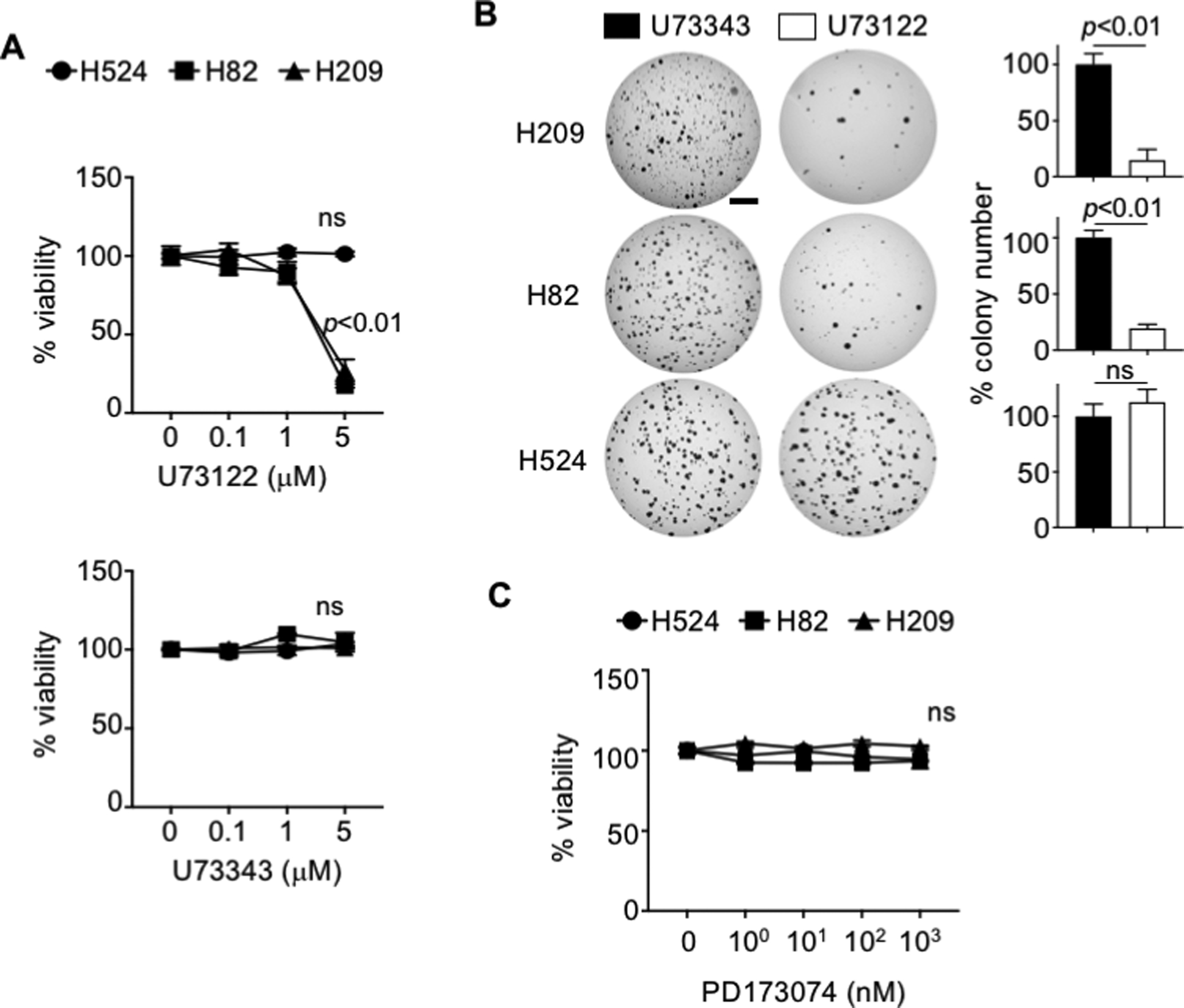
(A) Viability assays for human SCLC lines treated with U73122 (PLCG1 inhibitor) and U73343 (innocuous analog of U73122) once for 4 days, as measured by MTT assay. (B) Representative images of soft agar culture of human SCLC lines treated with 1μM U73122 and U73343 every three days for 14 days (H82 and H524) or 21 days (H209) and quantification of colonies (>0.2mm) in diameter (n=3 replicates per cell line). (C) Viability assay for human SCLC lines treated with PD173074 (pan-FGFR inhibitor) once for 4 days, as measured by MTT assay. Statistical tests performed using unpaired t-test (ns: not significant). Error bar represents standard deviation. Scale bar: 5mm.
To further explore the mechanism by which FGFR1 drives tumor growth, we performed RNA sequencing and analyzed a total of 1823 genes that were differentially expressed (DE) in Fgfr1-preSC (Fig. 7A; Supplementary Data 1). Gene ontology (GO) analysis of DE genes indicated enrichment of GO terms for cell cycle/mitosis and neuron differentiation and development (Fig. 7B; Supplementary Data 2). The GO terms related to neuron differentiation were further enriched in top 228 DE genes (with 1.5-fold or higher changes in expression) (Fig. 7B; Supplementary Data 2). Enrichment of these GO terms reflected the upregulation of Rhoa, Gli3, Etv4, Arx, and Lif and downregulation of Nefl, Chl1, Dcx, Dcc, Slitrk3, Nr2e1, and Bdnf (Fig. 7A). These results suggest that, in addition to promoting cell cycle, FGFR1 signaling regulates neural differentiation in cells progressing from precancerous neuroendocrine cells to neuroendocrine tumor. This observation is consistent with established roles of FGFR1 in proliferation of neural progenitor cells and neural development (36), and with recent studies showing that dysregulation in neural differentiation is a major event associated with SCLC progression (37, 38).
Fig. 7. FGFR1 induces transcriptome-wide change in the expression of genes involved in proliferation and differentiation.
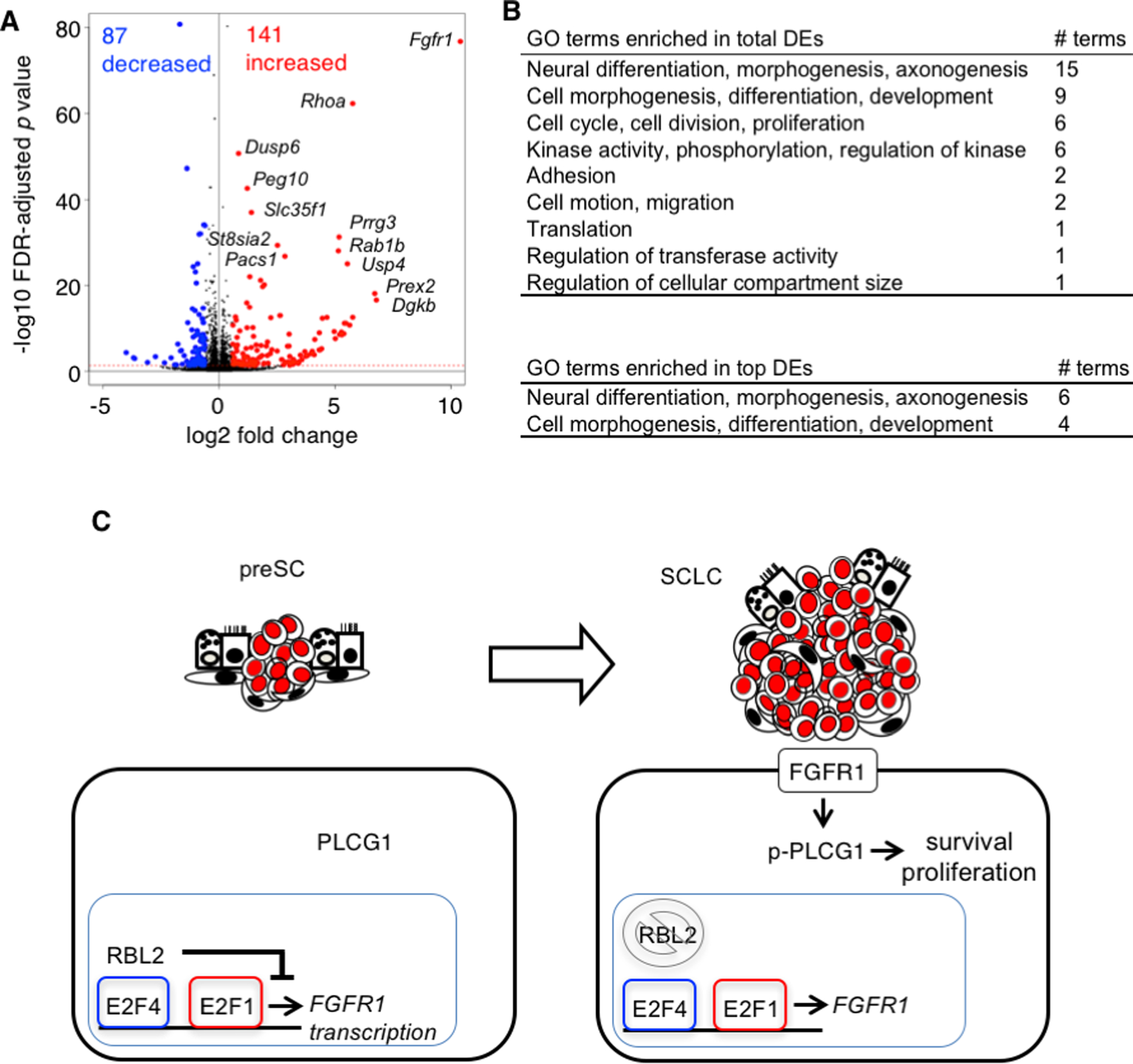
(A) Volcano plot showing differential gene expression between control and Fgfr1-preSC; changes in gene expression are significant above the red horizontal line, with an FDR-adjusted p-value < 0.05, and red and blue dots indicate genes (top DEs) with a 1.5-fold or higher increase or decrease in expression. FDR: false discovery rate. (B), Gene ontology (GO) terms enriched in genes differentially expressed (DE) in Fgfr1-preSC relative to control preSC (FDR-adjusted p-value < 0.05). Bottom, summary of GO terms enriched in top DEs. (C), A model depicting the proposed relationships among RBL2, FGFR1, and PLCG1 during early stages of SCLC development. RBL2 interacts with E2F4 to suppression FGFR1 expression. In the event of RBL2 loss or phosphorylation, E2F4 loses the repressor function and E2F1 activates FGFR1 transcription. FGFR1 then triggers phosphorylation of PLCG1 that mediates signaling for cell survival and proliferation.
Discussion
Here we report for the first time a direct role for nonamplified FGFR1 in SCLC development. Fgfr1 knockout significantly inhibited tumor growth in various SCLC models, notably in human SCLC lines that have previously shown little sensitivity to existing pharmacological inhibitors (8). While previous studies utilizing FGFR1 inhibition in human cell lines have focused on FGFR1 amplification as a biomarker for drug sensitivity, our findings suggest that FGFR1 expression may confer sensitivity as well, including in subtypes that lack FGFR1 amplification.
Elucidating the mechanisms behind tumor dependency on FGFR1 signaling would contribute to our understanding and treatment of SCLC. However, regulators of FGFR1 expression in cancer, whose activity may be used to identify tumor dependency on FGFR1 signaling, remain poorly defined. Our findings from mouse SCLC cells and GEMM models suggest that RBL2 regulates FGFR1 expression, as we observed FGFR1 induction and sensitivity to FGFR1 inhibition in RBL2-deficient cells. However, unlike mouse SCLC cells, RBL2 expression correlates poorly with FGF1 expression and cellular response to FGFR1 inhibition in human SCLC cells (Supplementary Fig. 1A; Supplementary Data 3), suggesting that the relationship between RBL2 and FGFR1 is more complex in human SCLC than in mice and involves additional molecular factors that may vary across different cell lines. For instance, phosphorylation may render RBL2 inactive so that FGFR1 is expressed even in the presence of the tumor suppressor; indeed, RBL2 is phosphorylated in most FGFR1-positive cells (H209, H69, H2141, and H2171) (Supplementary Fig. 1A). Additionally, status of the E2F family of transcription factors may determine FGFR1 expression, as they are variously expressed in SCLC (11, 39, 40). Expression of E2F1, which activates FGFR1 transcription (41), is readily detected in SCLC lines except for H82 that expresses a lower level of FGFR1 (Supplementary Fig. 1A, B). On the other hand, expression of E2F4, which interacts with RBL2 to repress FGFR1 transcription (31), is widespread but notably higher in H82 (Supplementary Fig. 1A, B). Furthermore, at least in the few SCLC lines examined as well as mouse SCLC, phosphorylation of FGFR1 seems to be a better indicator of FGFR1 activity and dependency than the protein level alone (Supplementary Fig. 1A, B). These findings together suggest previously unappreciated heterogeneity in the FGFR1-positive subset of SCLC.
Our findings indicate that PLCG1 is a critical mediator of FGFR1 signaling in SCLC. PLCG1 is a member of the phospholipase C (PLC) family of enzymes. When activated by growth factor receptors, PLCG1 hydrolyzes phosphatidylinositol 4,5-bisphsphate (PIP2) to generate inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG). IP3 then trigger Ca2+ release and signaling while DAG activates protein kinase C (PKC), giving rise to various intracellular signaling cascades that control proliferation, differentiation, morphology, and migration in multiple cell types (42). The functional impact of the interaction between FGFR1 and PLCG1 has remained poorly understood. Almost two decades ago, the first reports establishing their interaction suggested that it is not critical for mitogenesis (43, 44). More recently, PLCG1 has been shown to mediate FGFR1 signaling in maintaining the differentiation capacity of adult neural stem cells (45). Our data show that PLCG1 activation concurs with transcriptomic change related to neural differentiation during FGFR1-driven tumorigenesis, and thus suggest a role for PLCG1 in regulating neuroendocrine differentiation in SCLC. Our findings help add SCLC to the growing list of malignancies in which the activity of PLCG1 is implicated (46–48), suggesting PLCG1 as a potential therapeutic target in SCLC.
Other known transducers of FGFR1 signaling, including ERK1/2, do not appear to participate in FGFR1 signaling in a subset of human cell lines, which is in line with previous studies showing that ERK1/2 and AKT activation varied among SCLC primary tumors and did not strongly correlate with disease-free or overall survival (49, 50). The role of the ERK pathway in FGFR signaling in SCLC remains unclear. A hyperactive RAF-MEK-ERK pathway has been shown to be tumor-suppressive in SCLC cell lines, in contrast to the oncogenic role of this pathway in non-small cell lung cancer cells (10, 11). Further, in a recent study in Rb1/Trp53-GEMM, constitutive FGFR1 activation suppressed the development of SCLC originating from CGRP-positive neuroendocrine cells while promoting the tumor development from keratin 14 (K14)-positive airway epithelial cells (13). These studies suggest that whether FGFR1 and the ERK pathway are oncogenic in SCLC may depend on cell of origin. Importantly, our findings demonstrate an oncogenic role for FGFR1 in SCLC tumors with neuroendocrine origin. We surmised that differences in FGFR1 dependency may be due to the extent of signaling from constitutively active versus wild-type of FGFR1 and the interactions between FGFR1 and its signal transducers.
Taken together, our identification of two novel players in in FGFR1 signaling, PLCG1 and RBL2, shed light on the mechanism of FGFR1-driven SCLC development and homeostasis (Fig. 7C), and may facilitate the development of biomarkers for SCLC subtypes appropriate for FGFR1-targeted therapies, as well as targeted therapeutic strategies for SCLC.
Supplementary Material
Statement of Significance:
This study identifies RBL2 and PLCG1 as critical components of amplified FGFR1 signaling in SCLC, thus representing potential targets for biomarker analysis and therapeutic development in this disease.
Acknowledgements
We thank Drs. Anton Berns, Tyler Jacks, Julien Sage, Juha Partanen, and David Ornitz for sharing Trp53 lox, Rb1lox, Rbl2lox, Fgfr1lox and Fgfr2lox mice, respectively. We thank Jenny Hsu and Julien Sage for comments on the manuscript. This work was supported by NIH/NCI R01CA194461; U01CA224293 and American Cancer Society RSG-15-066-01-TBG (K.-S. Park) and Brain Pool Program through the National Research Foundation funded by Korea Ministry of Science NRF-2019H1D3A2A01060387 (D.-W. Kim). We also thank the Research Histology Core and the Genome Analysis and Technology Core at the UVA Cancer Center and Biostatistics and Bioinformatics Shared Resources at the H. Lee Moffitt Cancer Center & Research Institute, which were supported by NCI P30CA044579 and P30CA076292.
Footnotes
Conflict of Interest: The authors declared no potential financial conflicts of interest to disclose.
References
- 1.Byers LA, Rudin CM. Small cell lung cancer: where do we go from here? Cancer 2015;121:664–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peifer M, Fernandez-Cuesta L, Sos ML, George J, Seidel D, Kasper LH, et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet 2012;44:1104–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.George J, Lim JS, Jang SJ, Cun Y, Ozretic L, Kong G, et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015;524:47–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Voortman J, Lee JH, Killian JK, Suuriniemi M, Wang Y, Lucchi M, et al. Array comparative genomic hybridization-based characterization of genetic alterations in pulmonary neuroendocrine tumors. Proc Natl Acad Sci U S A 2010;107:13040–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang L, Yu H, Badzio A, Boyle TA, Schildhaus HU, Lu X, et al. Fibroblast Growth Factor Receptor 1 and Related Ligands in Small-Cell Lung Cancer. J Thorac Oncol 2015; 10:1083–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Desai A, Adjei AA. FGFR Signaling as a Target for Lung Cancer Therapy. J Thorac Oncol 2016;11:9–20 [DOI] [PubMed] [Google Scholar]
- 7.Pardo OE, Latigo J, Jeffery RE, Nye E, Poulsom R, Spencer-Dene B, et al. The fibroblast growth factor receptor inhibitor PD173074 blocks small cell lung cancer growth in vitro and in vivo. Cancer Res 2009;69:8645–51 [DOI] [PubMed] [Google Scholar]
- 8.Sos ML, Dietlein F, Peifer M, Schottle J, Balke-Want H, Muller C, et al. A framework for identification of actionable cancer genome dependencies in small cell lung cancer. Proc Natl Acad Sci U S A 2012;109:17034–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thomas A, Lee JH, Abdullaev Z, Park KS, Pineda M, Saidkhodjaeva L, et al. Characterization of fibroblast growth factor receptor 1 in small-cell lung cancer. J Thorac Oncol 2014;9:567–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ravi RK, Weber E, McMahon M, Williams JR, Baylin S, Mal A, et al. Activated Raf-1 causes growth arrest in human small cell lung cancer cells. J Clin Invest 1998;101:153–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ravi RK, Thiagalingam A, Weber E, McMahon M, Nelkin BD, Mabry M. Raf-1 causes growth suppression and alteration of neuroendocrine markers in DMS53 human small-cell lung cancer cells. Am J Respir Cell Mol Biol 1999;20:543–9 [DOI] [PubMed] [Google Scholar]
- 12.Ornitz DM, Itoh N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip Rev Dev Biol 2015;4:215–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferone G, Song JY, Krijgsman O, van der Vliet J, Cozijnsen M, Semenova EA, et al. FGFR1 Oncogenic Activation Reveals an Alternative Cell of Origin of SCLC in Rb1/p53 Mice. Cell Rep 2020;30:3837–50 e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meuwissen R, Linn SC, Linnoila RI, Zevenhoven J, Mooi WJ, Berns A. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell 2003;4:181–9 [DOI] [PubMed] [Google Scholar]
- 15.Gazdar AF, Savage TK, Johnson JE, Berns A, Sage J, Linnila RI, et al. The comparative pathology of genetically engineed mouse models for neuroendocrine carcinomas of the lung. J Thorac Oncol 2015;10:553–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim DW, Wu N, Kim YC, Cheng PF, Basom R, Kim D, et al. Genetic requirement for Mycl and efficacy of RNA Pol I inhibition in mouse models of small cell lung cancer. Genes Dev 2016;30:1289–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trokovic R, Trokovic N, Hernesniemi S, Pirvola U, Vogt Weisenhorn DM, Rossant J, et al. FGFR1 is independently required in both developing mid- and hindbrain for sustained response to isthmic signals. EMBO J 2003;22:1811–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu K, Xu J, Liu Z, Sosic D, Shao J, Olson EN, et al. Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development 2003;130:3063–74 [DOI] [PubMed] [Google Scholar]
- 19.DuPage M, Dooley AL, Jacks T. Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat Protoc 2009;4:1064–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 2009;25:1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 2015;31:166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McFadden DG, Papagiannakopoulos T, Taylor-Weiner A, Stewart C, Carter SL, Cibulskis K, et al. Genetic and clonal dissection of murine small cell lung carcinoma progression by genome sequencing. Cell 2014;156:1298–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim YH, Girard L, Giacomini P, Wang P, Hernandez-Boussard T, et al. Combined microarray analysis of small cell lung cancer reveals altered apoptotic balance and distinct expression signatures of MYC family gene amplification. Oncogene 2006;25:130–8 [DOI] [PubMed] [Google Scholar]
- 25.Schaffer BE, Park KS, Yiu G, Conklin JF, Lin C, Burkhart DL, et al. Loss of p130 accelerates tumor development in a mouse model for human small-cell lung carcinoma. Cancer Res 2010;70:3877–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park KS, Martelotto LG, Peifer M, Sos ML, Karnezis AN, Mahjoub MR, et al. A crucial requirement for Hedgehog signaling in small cell lung cancer. Nat Med 2011;17:1504–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sen T, Tong P, Stewart CA, Cristea S, Valliani A, Shames DS, et al. CHK1 Inhibition in Small-Cell Lung Cancer Produces Single-Agent Activity in Biomarker-Defined Disease Subsets and Combination Activity with Cisplatin or Olaparib. Cancer Res 2017;77:3870–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arman E, Haffner-Krausz R, Gorivodsky M, Lonai P. Fgfr2 is required for limb outgrowth and lung-branching morphogenesis. Proc Natl Acad Sci U S A 1999;96:11895–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yuan T, Klinkhammer K, Liu H, Gao S, Yuan J, Hopkins S, et al. Temporale Expression of Fgfr1 and 2 During Lung Development, Homeostasis, and Regeneration. Front Pharmacol 2020;11:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pearson A, Smyth E, Babina IS, Herrera-Abreu MT, Tarazona N, Peckitt C, et al. High-Level Clonal FGFR Amplification and Response to FGFR Inhibition in a Translational Clinical Trial. Cancer Discov 2016;6:838–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parakati R, DiMario JX. Dynamic transcriptional regulatory complexes, including E2F4, p107, p130, and Sp1, control fibroblast growth factor receptor 1 gene expression during myogenesis. J Biol Chem 2005;280:21284–94 [DOI] [PubMed] [Google Scholar]
- 32.Jia D, Augert A, Kim DW, Eastwood E, Wu N, Ibrahim AH, et al. Crebbp Loss Drives Small Cell Lung Cancer and Increases Sensitivity to HDAC Inhibition. Cancer Discovery 2018;8:1422–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pardo OE, Acaro A, Salerno G, Tetley TD, Valovka T, Gout I, et al. Novel cross talk between MEK and S6K2 in FGF-2 induced proliferation of SCLC cells. Oncogene 2001; 20:7658–67 [DOI] [PubMed] [Google Scholar]
- 34.Wang K, Ji W, Yu Y, Li Z, Niu X, Xia W, et al. FGFR1-ERK1/2-SOX2 axis promotes cell proliferation, epithelial-mesenchymal transition, and metastasis in FGFR1-amplified lung cancer. Oncogene 2018;37:5340–54 [DOI] [PubMed] [Google Scholar]
- 35.Bleasdale JE, Bundy GL, Bunting S, Fitzpatrick FA, Huff RM, Sun FF, et al. Inhibition of phospholipase C dependent processes by U-73122. Adv Prostaglandin Thromboxane Leukot Res 1989;19:590–3 [PubMed] [Google Scholar]
- 36.Ohkubo Y, Uchida AO, Shin D, Partanen J, Vaccarino FM. Fibroblast growth factor receptor 1 is required for the proliferation of hippocampal progenitor cells and for hippocampal growth in mouse. J Neurosci 2004;24:6057–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Denny SK, Yang D, Chuang CH, Brady JJ, Lim JS, Gruner BM et al. Nfib Promotes Metastasis through a Widespread Increase in Chromatin Accessibility. Cell 2016;66:328–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang D, Qu F, Cai H, Chuang CH, Lim JS, Jahchan N et al. Axon-like protrusions promote small cell lung cancer migration and metastasis. Elife 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brown KC, Witte TR, Hardman WE, Luo H, Chen YC, Carpenter AB, et al. Capsaicin displays anti-proliferative activity against human small cell lung cancer in cell culture and nude mice models via E2F pathway. PLoS One 2010;5:e10243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Coe BP, Thu KL, Aviel-Ronen S, et al. Genomic deregulation of the E2F/Rb pathway leads to activation of the oncogene EZH2 in small cell lung cancer. PLoS One. 2013;8(8):e71670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kanai M, Tashiro E, Maruki H, Minato Y, Imoto M. Transcriptional regulation of human fibroblast growth factor receptor 1 by E2F-1. Gene. 2009;438:49–56. [DOI] [PubMed] [Google Scholar]
- 42.Koss H, Bunney TD, Behjati S, Katan M. Dysfunction of phospholipase Cg in immune disorders and cancer. Trends Biochem Sci 2014;39:603–11. [DOI] [PubMed] [Google Scholar]
- 43.Peters KG, Marie J, Wilson E, Ives HE, Esobedo J, Del Rosario M, et al. Point mutation of an FGF recentor abolishes phosphatidylinositol turnover and Ca2+ flux but nor mitogenesis. Nature 1992;358:678–681 [DOI] [PubMed] [Google Scholar]
- 44.Mohammadi M, Dionne CA, Li W, Li N, Spival T, Honegger M, et al. Point mutation in FGF receptor eliminates phosphatidylinositol hydrolysis without affecting mitogenesis. Nature 1992;358:681–684. [DOI] [PubMed] [Google Scholar]
- 45.Vaque JP, Gomex-Lopez G, Monsalvez V, Valera I, Martinez N, Perez C, et al. PLCG1 mutations in cutaneous T-cell lymphomas. Blood 2014;123:2034–43. [DOI] [PubMed] [Google Scholar]
- 46.Kunze K, Spieker T, Gamerdinger U, Nau K, Berger J, Dreyer T, et al. Arecurrent activating PLCG1 mutation in cardiac angiosarcomas increases apoptosis resistance and invasiveness of endothelial cells. Cancer Res 2014;74:6173–83. [DOI] [PubMed] [Google Scholar]
- 47.Gouaze-Andersson V, Delmas C, Taurance M, Martinex-Gala J, Evrard S, Mazoyer S, et al. FGFR1 Induces Glioblastoma Resistance through the PLCγ1/Hif1α Pathway. Cancer Res 2016;76:3036–44. [DOI] [PubMed] [Google Scholar]
- 48.Tang W, Zhou Y, Sun D, Dong L, Xia J, Yang B. Oncogenic role of phospholipase C-γ1 in progression of hepatocellular carcinoma. Hepatol Res 2019;49:559–569. [DOI] [PubMed] [Google Scholar]
- 49.Blackhall FH, Pintillie M, MIchael M, Leighl N, Feld R, Tsao MS, et al. Expression and prognostic significance of kit, protein kinase B, and mitogen-activated protein kinase in patients with small cell lung cancer. Clin Cancer Res 2003;9:2241–7. [PubMed] [Google Scholar]
- 50.Schmid K, Bago-Horvath Z, Berger W, Haitel A, Cejka D, Werzowa J, et al. Dual inhibition of EGFR and mTOR pathways in small cell lung cancer. Br J Cancer 2010;103:622–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.