

Abstract
Rapidly developing single-cell sequencing analyses produce more comprehensive profiles of genomic, transcriptomic, and epigenomic heterogeneity present in tumor subpopulations than traditional bulk sequencing analyses. Moreover, single-cell techniques allow a tumor’s response to drug exposure to be more thoroughly investigated. Deep learning models have successfully extracted features from complex bulk sequence data to predict drug responses. Here, we review recent innovations in single-cell technologies and deep learning-based approaches related to drug sensitivity predictions. We believe that using insights from bulk sequence data, deep transfer learning would facilitate the application of single-cell data to train superior deep learning-based drug prediction models.
Keywords: drug response, deep learning models, single-cell technologies, deep transfer learning framework
Overview of Current Research Methodologies for the Investigation of Drug Resistance
Treatments for cancers have undergone major advancements with the development of molecularly targeted therapy, immunotherapy, chemotherapy, and radiotherapy [1]. Targeted drugs—monoclonal antibodies and small molecules—used by molecularly targeted therapies achieve the highest level of cytotoxicity in tumors as they are able to precisely target cancer cells [2]. The availability of these treatments has dramatically improved patient prognoses. Moreover, if the feature targeted by drug treatment is shared by all the tumor’s subpopulations, the targeted drugs can yield complete remission of the disease. However, cancerous tumors are rarely homogenous. They are comprised of diverse cell subpopulations demarcated by distinct genomes and transcriptomes, each of which can yield a unique response and sensitivity to a given drug [3]. As a result, heterogeneous cancer subpopulations with extraordinarily dynamic characteristics often exhibit resistance to single-drug treatments, preventing the complete eradication of the disease [4]. After the vast majority of the tumor has been wiped out, a small number of remaining cancerous cells, called the minimal residual disease (MRD, see Glossary), survive and continue proliferating [4]. The inevitable relapse features a disease now largely resistant to the initial treatment. In addition to some tumor subpopulations possessing an inherent resistance to select treatments, cancer cells can also acquire resistance via multiple mechanisms such as drug inactivation, target alternation, and drug efflux [1, 5, 6]. Insensitivity to treatment is now responsible for up to 90% of cancer-related deaths [7]. Thus, it is imperative to increase our understanding of the mechanisms by which resistance is propagated and accurately predict which drug combination will be the most effective against specific cancers.
High-throughput sequencing techniques like DNA-sequencing, RNA-sequencing, assay for transposase-accessible chromatin with high-throughput sequencing (ATAC-seq), and Chromatin immunoprecipitation with high-throughput sequencing (ChIP-seq) can characterize the genomic, transcriptomic, and epigenetic landscapes of tumors. These profiles are indispensable to gleaning insight into resistant tumors. For example, the whole-genome characterization of resistant ovarian cancer by DNA-seq directly led to the identification of tumor repressors that, when inactivated, resulting in drug resistance [8]. The advancement of single-cell techniques, including single-cell DNA-sequencing (scDNA-seq) and single-cell RNA-sequencing (scRNA-seq), has enabled scientists to analyze the genomic and transcriptomic profiles of individual cells. Such innovation allows researchers to better investigate cancer heterogeneity and deduce the culprits of drug resistance. Moreover, the findings produced by studying these concepts facilitate drug sensitivity predictions for independent cancer subpopulations. For example, the key regulators (KDM5A/B) of therapeutic resistance in breast cancers subsets have been identified via scRNA-seq and bulk ChIP-seq [9].
Deep learning (DL) models have also successfully predicted drug responses (Figure 1, Key Figure). However, vast amounts of genomic and transcriptomic data are required to produce meaningful and generalizable DL prediction tools. Many DL models have benefited from the vast libraries of drug-, protein- and gene-related data from many disease (sub)types that are available in the public domain. Specifically, databases such as The Cancer Genome Atlas, Genomics of Drug Sensitivity in Cancer [10], The Cancer Cell Line Encyclopedia [11], Cancer Target Discovery and Development [12], and the University of California, Santa Cruz TumorMap [13] are all commonly utilized to train DL models.
Figure 1. Key Figure. Combination of Single-cell and DL models in drug sensitivity prediction.
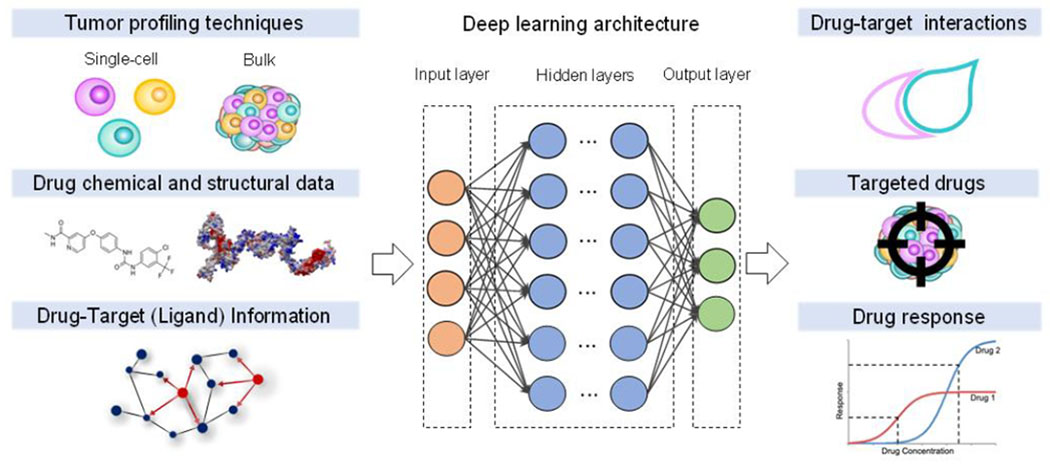
DL models are typically trained on tumor profiles, chemical and structural information of drugs and drug-target data. Extracting high-dimensional features through multi-layer perceptrons, DL models can infer drug-target interactions, purpose new drugs, and predict drug resistance.
Even though single-cell sequencing can enhance the resolution by which heterogeneity is studied, the data generated by these techniques are of large volume and high complexity. The copious number of cells increases the dimensionality and scale of data. Both of which make it challenging to use the single-cell sequence data to connect the distinct modalities they encode and construct heterogeneous biological networks. However, recent work in classifying cell types has highlighted DL architectures which can process the large-scale single-cell data and effectively extract high dimensional features. Yet, the small amount of data available at the single-cell resolution currently precludes the active development of DL models for drug-related predictions.
DL Models Can Accurately Predict Drug-Target Interactions and Drug Sensitivity
Using publicly available data, DL models have boasted modest success in predicting a novel cell type’s sensitivity to drug treatment options. DL models use artificial networks to emulate biological neural networks and learn patterns from the source data. DL has gained popularity due to its ability to analyze and extract insights from exceedingly large amounts of unstructured or unlabeled data [14]. The insights derived from these networks can be built into models that are used for a variety of tasks, including data denoising, cell clustering, phenotype prediction, and image processing. The unstructured and multi-dimensional datasets produced by high-throughput sequencing has been utilized by DL models to predict drug sensitivities in cancer and other diseases [15]. Numerous DL models have been developed to make these predictions. The most common DL classes of models tasked with evaluating anticancer treatment options have been Deep Neural Networks (DNN), Convolutional Neural Network (CNN), Recurrent Neural Networks (RNN), and Graph Convolutional Networks (GCN) (Box 1, Figure 1A). Specifically, Table 1 presents a non-exhaustive overview of DL approaches that have been utilized to address current limitations in predicting drug efficacy.
TEXT BOX.
Box 1. Deep learning models and frameworks.
Four deep learning models that have been applied in drug prediction are DNN, CNN, RNN, and GCN (Box 1, Figure IA). These are discussed below:
DNN is a feed-forward neural network consisting of input, hidden, and output layers that are densely connected. For drug sensitivity prediction, we would like to construct a function from tumor drug profiles to sensitivity. During the model training, those hidden layers are able to perform data abstractions and transformations, which turn out to be parameters in the function.
CNN operates in a manner inspired by the human visual cortex and consists of convolutional (feature extraction) and pooling (dimension reduction) layers. The convolution and pooling layers help to extract all information in datasets without consuming too much time and computational sources.
RNN is a type of artificial neural network where connections between nodes form a directed graph along a temporal sequence.
GCN is a type of neural network architectures that can leverage the graph structure and aggregate node information from neighbors in a convolutional fashion. It has natural advantages when dealing with graph-based data.
Further, two frameworks, DTL and VAE, can be combined with DL models (Box 1, Figure IB). DTL transfers store knowledge gained while solving one problem and applying it to a different but related problem. VAE consists of an encoder, a decoder, and a loss function. The encoding distribution is regularized by a standard deviation vector and a mean vector during the training in order to ensure that its latent space has good properties to generate new data.
Box 1, Figure I. The scheme of deep learning models and frameworks.
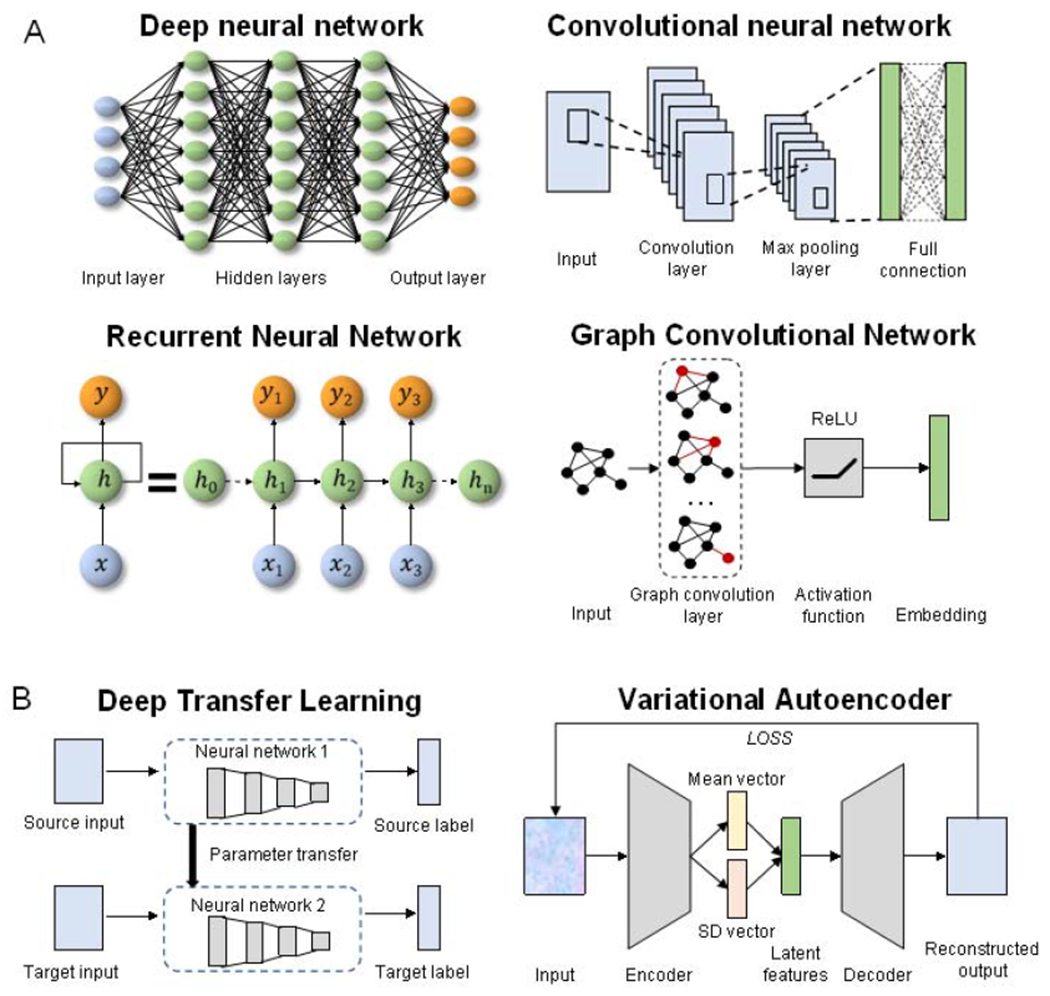
(A) Four deep learning models that have been applied in drug prediction, including DNN, CNN, RNN, and GCN. (B) Two frameworks that can be combined with DL models.
Table 1.
Deep learning approaches and examples in predicting drug treatment efficacy.
| Model | Tools* | Input Data | Purpose | Ref |
|---|---|---|---|---|
| Drug-Target Interactions | ||||
| DBN | DeepDTIsi | Drug-target pairs information, drug structure, and protein sequence | The probability of interaction for any provided drug-target pair was inferred by DeepDTI based on external, experimental drug-target pairs. Among the top ten predicted drug-target interactions, four had been previously reported, and one was found to have a low binding affinity to the glucocorticoid receptor. | [17] |
| DNN | DeepCPIii | Drug structure and protein sequence for drug-target pairs | Drug-target interactions were predicted by DeepCPI. The inferred interactions between small molecules and glucagon-like peptide-1 receptor, glucagon receptor, and vasoactive intestinal peptide receptor were experimentally validated. | [18] |
| DNN | deepDTnetiii | Drug-target pairs information, drug similarity, and target similarity | deepDTnet can identify targets of known drugs using a heterogeneous drug–gene–disease network embedding 15 types of chemical, genomic, phenotypic, and cellular network profiles. A new, direct inhibitor of human retinoic-acid-receptor-related orphan receptor-gamma t, topotecan is predicted by deepDTnet, and then experimentally validated by authors. | [26] |
| CNN | DeepConv-DTIiv | Raw protein sequences and drug-target pair data | Local residue patterns from proteins in drug-target interactions were captured by DeepConv-DTI via the convolution on various lengths of amino acid subsequences. This model achieved higher accuracy than DBN-based DeepDTI and CNN-based DeepDTA. | [20] |
| CNN | DEEPScreenv | 2-D structure of compounds and protein structure | Drug-target interactions were predicted based on 704 target proteins and the 2-D structure of compounds. JAK proteins were predicted by the model as new targets of drug cladribine and experimentally validated in vitro. | [84] |
| CNN | AtomNet | 3-D structure of target proteins and small molecules | Applying local convolutional filters to extract the target’s structural information, AtomNet successfully predicted new active molecules for targets like wee1 and 1qzy which previously had no known modulators. | [21] |
| GCN | DeepChemvi | Compound structure | GCN and long short-term memory were employed to optimize small molecule-based drug discovery by predicting the toxicity and bioactivity of candidate drugs using their structural data. | [23] |
| RNN CNN GCN DTL | DeepAffinityvii | Raw protein sequences and compound sequences (2-D information) | Bidirectional RNN was utilized to capture nonlinear joint dependencies among either protein residues or compound atoms that are sequentially distant. DeepAffinity unified RNN-CNN/RNN-GCN to predict drug-target interactions. The model outperformed conventional models in achieving relative error in the half-maximal inhibitory concentration within five-fold for test cases and 20-fold for protein classes not included in training. | [19] |
| DNN | Deep-AmPEP30viii | Genomic sequence data and known AMP sequences | Antimicrobial peptides to treat a variety of diseases such as cancer and infections were identified based on known AMP sequences. One peptide (FWELWKFLKSLWSIFPRRRP) the model produced proved to have the same anti-bacteria efficacy as ampicillin. | [24] |
| Drug Repurposing | ||||
| DNN | - | Transcriptional response to drug exposure | The therapeutic categories of drugs were exclusively identified from transcriptional profiles. 26,420 drug perturbation samples were analyzed for three cell lines and then assigned to one of twelve therapeutic categories. | [29] |
| VAE | deepDRix | Drug-disease pairs and drug’s chemical information | Fourteen candidates of the top 20 drug candidates to treat Parkinson’s disease predicted by deepDR were validated by previous studies. | [28] |
| Drug Response | ||||
| DNN | RefDNNx | Drug’s structure and gene expression data prior to drug exposure | RefDNN learned representations for a high-dimensional gene expression vectors and a molecular structure vectors of drugs, to predict drug response, then labeled and identified biomarkers contributing to drug resistance. Among the top ten genes identified by RefDNN, six (high expression patterns of MYOF, UBC, NQO1, and LGALS3 and low expression patterns of RACK1 and RPS23) were experimentally proven to be associated with nilotinib resistance. | [30] |
| DNN VAE | DeepDR | Genomic and transcriptomic profiles before and after drug treatments | The drug response of tumors was predicted from integrated genomic profiles. Specifically, DeepDR improves the prediction of drug response and identification of novel therapeutic options. The model was applied to predict drug response in 9059 tumors from 33 cancer types. The resulting predictions include known therapies, such as EGFR inhibitors in non-small cell lung cancer and tamoxifen in breast cancer, as well as novel drug targets, such as vinorelbine for TTN-mutated tumors. | [33] |
| MLP RNN CNN | - | HIV genome sequence and drug sensitivity data | HIV-related drug resistance was predicted by three deep learning models. Of the 20 most important features predicted by the models, 18, 9, and 16 known drug resistance mutations positions were identified by using CNN, MLP, and RNN models, respectively. | [32] |
| VNN | Dcellxi | Genotype data | The phenotypic resulting from individual gene perturbations in eukaryotic cells was transparently simulated. During the simulation, 80% of the importance of growth prediction is captured by 484 subsystems. | [31] |
| Drug-Drug Interactions | ||||
| DNN | DeepSynergyxii | Drug chemical data and transcriptional data | DeepSynergy distinguished different cancer cell lines and found specific drug combinations to maximal efficacy on a given cancer cell line through the incorporation of genomic information with compound information. | [34] |
| GCN | Decagonxiii | Drug-drug interaction, protein-drug interaction, protein-protein interaction, and side effects | Decagon constructed a large two-layer multimodal graph of protein-protein interactions, drug-target interactions, and drug-drug interactions to predict the potential side effects of drug pairs. Decagon accurately predicted polypharmacy side effects, outperforming baselines by up to 69%. It had the best performance in modeling side effects with strong apparent molecular underpinnings; for example, in Mumps, Carbuncle. | [85] |
| Other | ||||
| CNN | DeepMACTxiv | 3D image data | DeepMACT performed image recognition to track the biodistribution of antibody-based agents. Trained on an MDA-MB-231 cancer cell-based tumor model, DeepMACT has 80% accuracy to detect metastasis. | [86] |
The superscript numbers refer to the websites in the Resource section.
As the table highlights, DNN is one of the most popular DL architectures used for predicting drug-target interactions (DTIs) using both existing and novel molecules. For example, DeepDTIs employed a Deep Belief Network (DBN) model to extract features from drug-target data in the DrugBank database [16] and predicted the interaction likelihood of drug-target pairs [17]. However, DeepDTIs creates inferences that introduce unwanted noise into the model, reducing its performance. Three additional DL models have since been built, claiming to outperform DeepDTIs: (i) DeepCPI, another DNN-based model, showed superior predictive performance and scalability for large-scale compound affinity data [18]; (ii) DeepAffinity predicts compound-protein interaction but utilizes RNN and GCN model to capture long-term, nonlinear dependencies among residues/atoms in proteins/compounds [19]. DeepAffinity also employs a deep transfer learning (DTL) framework to facilitate the enhancement in predicting DTIs from limited labeled protein-compound interaction data compared to DeepDTIs; and DeepConv-DTI captures local residue patterns of proteins participating in DTIs and detect the binding sites of DTIs [20]. DeepConv-DTI uses a CNN model which is also quite successful at extracting meaningful features from sparse interactions, making them an ideal model for predicting DTIs. One reason for DeepConv-DTIs enhanced performance over DeepDTIs is that DeepConv-DTIs CNN architecture does not require entire structure or sequence data to learn DTIs [17, 20]. In other words, DeepDTIs model learns from sequences not involved in DTIs; these irrelevant features detract from its prediction accuracy. AtomNet [21] also implements a CNN model to analyze feature locality and hierarchical composition to predict the bioactivity of small molecule DTIs. DEEPScreen takes advantage of CNN’s excellence in image analysis to predict DTI’s from simple 2-D images of compounds [22]. DeepChem does this also, but is built on a GCN model instead [23]. DeepAMPEP30 is another published CNN-based model. Rather than identifying DTIs from known drugs and proteins, this model suggests novel drugs with the potential to target affected cells [24]. Specifically, DeepAMPEP30 proposes short peptide sequences that exhibit optimized antimicrobial properties and can be used as a targeted treatment for a wide breadth of diseases, including bacterial infections and cancers. In a proof-of-concept experiment, some of the proposed peptides were as effective as ampicillin at treating multiple bacterial infections [24].
In addition to predicting DTIs, the aforementioned models demonstrate a capacity for inferring novel drug indications, or novel uses for existing drugs. This practice is referred to as drug repurposing. DeepCPI, for example, highlighted several drugs as candidates to be repurposed for neural pharmacology [18]. One such drug, oxazepam is traditionally used to treat alcohol withdrawal. DeepCPI found that it might also impact intramitochondrial cholesterol transfer, implicating its potential application for the treatment of Alzheimer’s disease [18, 25]. In another attempt to enhance drug repurposing efforts by predicting DTI’s, Zeng et al. proposed deepDTnet: a DNN for graphical representation [26]. The model created a drug-gene-disease network that successfully predicted topotecan—traditionally used to treat ovarian and lung cancers—as a drug repurposing candidate to treat multiple sclerosis. Specifically, deepDTnet found human retinoic-acid-receptor orphan receptor-gamma t, whose overexpression can lead to the development of multiple sclerosis, able to be inhibited by topotecan [26]. Similar to deepDTnet, arbitrary-order proximity embedded deep forest (AOPEDF) is also proposed for drug-gene-disease network prediction using a deep forest model [27]. While this model is not expressly a DL model, we have included it in our discussion as it offered better performance than deepDTnet. Additionally, AOPEDF requires fewer manual adjustments of the parameters controlling the model’s learning process than DNNs, such as deepDTnet, to achieve high performance[27].
While DTI models can be applied to drug repurposing research, other approaches have been devised with this exclusive purpose. For example, a DNN-based model, deepDR [28], employs a Variational Autoencoder (VAE) framework to extract high-dimensional features from low-dimensional representations of drugs and reported drug-disease pairs and infer candidates for repurposing (Box 1 and Figure 1B). Aliper et al. also used a DNN model to classify known drugs into therapeutic categories based on the effect that drug exposure has on gene expression [29]. The team found that ‘misclassified’ drugs might not be misclassified, but rather represent novel drug indications. Two such ‘misclassified’ drugs, otenzepad and pinacidil, support their hypothesis [29]. These drugs were classified as central nervous system drugs; however, to date, both exclusively treat cardiovascular conditions. That said, previous studies provide evidence that confirms both drugs might also impact brain function. For instance, while pinacidil targets K-channels in the treatment of cardiovascular diseases, K-channels are also extremely prevalent in the brain and required for signal transduction [29]. The prevalence of K-channels in the brain would make pinacidil an excellent candidate to treat certain neural conduction disorders.
Once novel drug treatment options have been identified, it becomes necessary to evaluate and predict a disease’s response and sensitivity to each treatment. Multiple DL models, including RefDNN, have been produced to make these predictions. RefDNN identified drugs capable of improving hepatocellular carcinoma’s (HCC) sensitivity to sorafenib, HCC’s only approved treatment [30]. Sorafenib typically elicits only a modest response in HCCs; however, the proposed drugs were experimentally validated to either synergize with sorafenib or target HCC’s other key regulatory pathways. Dcell, another DL model, predicts drug responses via a Visible Neural Network (VNN). While it is a unique approach, the VNN provided Ma et al. with an opportunity to glean novel biological perspectives [31]. Dcell predicts the impact modulating gene function has on cell phenotypes and depicts the underlying mechanisms and pathways, through an easily interpreted hierarchal visualization [31]. Dcell is a notable improvement in terms of the transparency and interpretability of DL models.
While more traditional DL models have enabled researches to glean biological insights and predict drug responses, these models are not transparent. The underlying mechanisms producing these insights often remain obscured by the black boxes through which DL models make their predictions until they are ascertained by either in vitro or in vivo experimentation. DL models have also been limited by the suboptimal accuracy of drug sensitivity predictions in diseases with high mutation rates, including HIV and cancers [32]. This is owing, in part, to a lack of relevant data needed for DL models to create generalizable inferences [30, 32, 33]. For these hypermutable disorders, heterogeneous disease states can also influence the accuracy of DL models. Currently, many DL models are trained using bulk sequence data, which has an insufficient resolution of cells to effectively analyze complex heterogeneity. Models, such as DeepSynergy, which attempt to identify combinations of drugs that maximize treatment sensitivity in all tumor subpopulations while minimizing systemic side effects, could indubitably benefit from the enhanced resolution [34].
Single-cell Technologies Discern Heterogeneity’s Effect on Drug Resistance
Cellular heterogeneity inferred from single-cell data can greatly facilitate the accurate prediction of drug sensitivity and help the design of combinational drug. While still a relatively new technology, researchers are using single-cell technologies with an increasing frequency to study heterogeneous disorders. This increased popularity is owing to single-cell sequencing’s capacity to capture subtle differences in genomic and transcriptional states of heterogeneous subpopulations; whereas, bulk sequencing merely produces an aggregate estimate of gross cellular features [35]. Moreover, the high-resolution data provided by single-cell technologies allow researchers to harness single-cell sequencing [36] to individually profile the genomic and transcriptomic heterogeneity of cells within tumor subpopulations. Gene subnetworks identified from scRNA-seq profiles can be highly correlated with a patient’s survival and drug response to cancer [37]. A non-exhaustive list of the recent applications of available single-cell technologies has been highlighted in Table 2 to provide examples of what methodologies scientists have at their disposal to investigate the intricacies of single-cell environments.
Table 2.
Overview of available single-cell technologies used to study drug resistance in heterogeneous disorders.
| Technology | Purpose | Ref |
|---|---|---|
| Targeted drugs | ||
| cDNA-seq | Single-cell DNA sequencing (scDNA-seq) was performed on 510 circulating tumor cells and 189 leukocytes. Microheterogeneity analysis of individual CTCs discerned there existed cells prior to drug exposure that was resistant to ERBB2-targeted therapies. | [40] |
| scRNA-seq bulk ATAC-seq | Single-cell RNA-sequencing (scRNA-seq) of paired drug naïve and resistant acute myeloid leukemia patient samples highlighted regulators of enhancer function as important modulators of the resistant cell state. The inhibition of Lsd1 facilitated the binding of the pioneer factor and cofactor to nucleate new enhancers, overcoming stable epigenetic-derived resistance. | [45] |
| CROP-seq | Pooled CRISPR screening was combined with single-cell RNA sequencing to facilitate high-throughput functional dissection of complex regulatory mechanisms and heterogeneous cell populations. ETS1, RUNX1, and GATA3 were found to be essential for Jurkat T-cell function. | [66] |
| Single-cell FISH | Single-cell FISH visualized transcriptional variability at the single-cell level which was used to predict drug resistance development. It was found that the addition of drugs induces epigenetic reprogramming in certain cells, converting a transient transcriptional state to a stable one. Reprogramming began with a loss of SOX70-mediated differentiation followed by activation of new signaling pathways, partially mediated by Jun-AP-1 and TEAD transcription factors. | [43] |
| sci-Plex scRNA-seq | ‘Nuclear hashing’ was used to quantify global transcriptional responses in thousands of independent perturbations at a single-cell resolution. sci-Plex was employed to screen three cancer cell lines exposed to 188 compounds. Approximately 650,000 single-cell transcriptomes across ~5,000 independent samples were profiled in one experiment. The similarity in single-cell transcriptomes treated with distinct compounds highlighted drugs that target convergent molecular pathways. | [35] |
| scRNA-seq | A candidate tumor cell subgroup associated with anti-cancer drug resistance was identified using scRNA-seq on viable patient-derived xenograft (PDX) cells. 50 tumor-specific single-nucleotide variations were observed to be heterogeneous in individual PDX cells after performing scRNA-seq on 34 PDX tumor cells from a lung adenocarcinoma patient. PDX cells that survived in vitro anti-cancer drug treatment displayed transcriptome signatures consistent with the group characterized by KRAS(G12D). | [39] |
| scRNA-seq | 4645 cells isolated from 19 patients, including malignant, immune, stromal, and endothelial cells, were profiled using scRNA-seq. Malignant cells within the same tumor displayed transcriptional heterogeneity associated with drug-resistance. Analysis of tumor-infiltrating T-cells revealed exhaustion mechanisms were connected to T cell activation and clonal expansion, and their variability across patients. | [41] |
| RT–qPCR | Single-cell RT-qPCR of the luminal-type breast cancer cell line MCF7 and its derivatives, including docetaxel-resistant cells identified that in the drug-resistant cells, epithelial-to-mesenchymal transition and stemness-related genes were upregulated and cell-cycle-related genes were downregulated. Both were primarily regulated by LEF1. | [46] |
| Microfluidic Platform | An integrated microfluidic platform was built to construct single-cell arrays that could analyze drug resistance. A proof-of-concept experiment was implemented by determining the vincristine resistance of single glioblastoma cells with different biomechanical properties. The results indicated that the biomechanics of tumor cells has significant implications for cell drug resistance | [87] |
| MULTI-seq | MULTI-seq: multiplexing using lipid-tagged indices for single-cell and single-nucleus RNA sequencing. MULTI-seq reagents were shown to be able to barcode any cell type or nucleus from any species so long as there was an accessible plasma membrane. | [88] |
| Single-cell barcoding | Transient transfection with short barcode oligonucleotides simultaneously analyzed multiple samples with scRNA-seq. The accuracy of the method was validated and its ability to identify multiplets and negatives was confirmed by analyzing samples from a 48-plex drug treatment experiment. | [74] |
| Immunotherapy | ||
| TCR-seq scRNA-seq | By performing single-cell RNA sequencing on 5,063 single T-cells and coupled TCR-seq, 11 T-cell subsets were distinguished based on their molecular and functional properties which also delineated their developmental trajectory. The gene layilin, which was found to be upregulated on both activated CD8+ T-cells and Tregs, represses the CD8+ T-cell functions in vitro. | [58] |
| LIBRA-seq (scBCR-seq) | The antigen specificity of thousands of B-cells from two HIV-infected subjects was mapped. The predicted antigen specificities were confirmed for a number of HIV- and influenza-specific antibodies. | [60] |
| RAGE-seq (scTCR-seq+scBCR-seq) | 7138 cells sampled from the primary tumor and draining lymph node of breast cancer were used to infer B-cell clonal evolution and identify alternatively spliced BCR transcripts. | [61] |
| Perturb-seq | Perturb-seq was performed on 200,000 immune cells, identifying transcription factors that regulate the response of dendritic cells to lipopolysaccharide. Perturb-seq was shown to accurately identify individual gene targets, gene signatures, and cell states affected by both individual perturbations and their genetic interactions. | [67] |
| scRNA-seq | By combing single-cell RNA-seq and T cell receptor (TCR) analysis, TCR signal intensity was found to not affect resting/activated Treg proportions but activated Treg programs | [57] |
Traditionally, single-cell technologies have primarily been utilized to characterize distinct cell types [9, 38]. scRNA-seq has been the obvious candidate for such tasks as distinct cell types possess unique transcriptional profiles [38]. Both scRNA-seq and scDNA-seq have been employed to characterize the distinct cell subpopulations within tumors [38–42]. ScDNA-seq is beneficial when attempting to discern genomic heterogeneity amongst cancers. For example, Yang and colleagues used scDNA-seq analysis to highlight variants in genes, including MLL2, as key drivers in the growth and survival of bladder cancer stem cells [42]. Likewise, the transcriptional variation identified through scRNA-seq is vital to understanding the mechanism underlying drug resistance. Kim et al. found scRNA-seq could characterize and predict the most aggressive tumor subpopulations in lung adenocarcinomas [39]. Of note, when they compared the results of their single-cell approach to conventional bulk tumor analysis, they discovered that the transcriptional profiles of the resistant subpopulations are often masked by more prominent subpopulations, obscuring meaningful insights. This highlights the necessity to transition from bulk methods to single-cell analyses when investigating heterogeneous cells. A consideration of circulating tumor cells (CTCs) also strengthens this notion. It is suggested that clinicians may benefit from using scDNA-seq to detect and profile CTCs in peripheral blood to stage cancers and monitor their progression [40, 43]. Even in patients with advanced metastatic cancers, CTCs are only present at a magnitude of fewer than 100 cells per the 5 million total cells in 1mL of whole blood [44]. Single-cell technologies now have the sensitivity to accurately detect CTCs at this extremely low concentration, which is something that bulk analyses will never be able to replicate.
Being able to successfully distinguish unique cells, the focus of single-cell studies has shifted from merely characterizing cell types (Figure 2A) to elucidating the biological mechanisms responsible for the development of drug resistance in previously characterized, resistant subpopulations. Drug-resistance can be caused by genetic and non-genetic factors. Genetic resistance originates when a heritable mutation is introduced to a cell’s DNA, granting it and its progeny the resistance to current drug treatment [40, 42]. Using scDNA-seq, genetic alterations can be quickly identified, and the treatment can be adjusted to target the novel subpopulation [40, 42]. In drug-resistant cell lineages without a novel mutation, discerning how the resistance developed is a more challenging endeavor.
Figure 2. Prediction of drug sensitivity at the single-cell level.
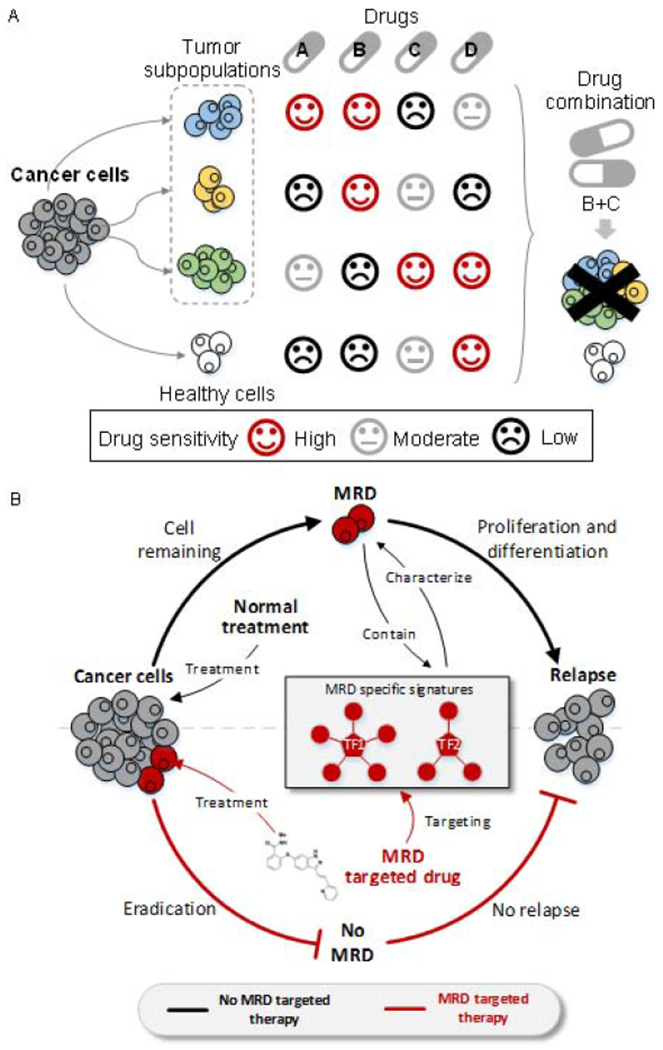
(A) Tumor subpopulations maintain diverse sensitivity to different drugs. Single usage of a drug may obtain less treatment efficiency. Knowing drug sensitivity at the single-cell level can guide the development of combination treatment that maximizes the efficiency of killing tumor cells while minimizes damage to healthy cells. (B) The MRD will proliferate and differentiate into a new tumor population which induces cancer relapse. The understanding of specific signatures characterized in MRD cells can help to discover novel drugs that specifically target MRD. MRD-targeted drug(s) administered in combination with conventional treatments can cure cancer and prevent relapse.
Previous research using single-cell sequencing identifies two general mechanisms by which non-genetic resistance may develop. The first mechanism involves the development of a transient resistance originating from cells within a tumor in a ‘persister’ state [39]. Simply put, persister cells exhibit atypical growth and metabolism enabling a higher tolerance against drug treatment. Interestingly, following drug withdrawal, persister cells lose their resistance and can be eradicated by the original drug treatment [39]. The other mechanism results in cells entering a transcriptionally stable end state. Such cells exhibit sustained resistance in lieu of spontaneous re-sensitization following drug withdrawal [39, 45].
Unfortunately, the mechanism driving this sustained resistance has remained far less elucidated, precluding informed drug selection. However, two hypotheses have been posited to explain the development of sustained resistance, respectively. The first hypothesis involves a rare subpopulation of resistant cells existing in the drug-naive tumor [46]. Administration of a drug treatment selects for these resistant cells while eradicating sensitive cell populations [43]. The continued proliferation of the MRD posttreatment leads to an inevitable relapse (Figure 2B). Indeed, single-cell RT-qPCR has confirmed that, in some breast cancers, drug-resistant subpopulations existed prior to drug exposure [46]. Finding the biomarkers of these refractory subpopulations during initial treatment improves outcomes by informing initial drug selections to target all subpopulations [40]. The other hypothesis involving acquired resistance is seemingly the more common phenomenon. Tumor subpopulations that acquire sustained resistance display transcriptional plasticity during drug exposure [43, 45]. Mathematical modeling, using single-cell data, has suggested that the existence of spatio-temporal heterogeneity might drive transcriptional plasticity towards the sustained resistance state during drug treatment [47]. In turn, changes in gene expression, identified by scRNA-seq, scATAC-seq, and fluorescence in situ hybridization (FISH), modulates pathways controlling functions such as epigenetic remodeling and immune response to initiate and propagate resistance [35, 43, 45, 48–51]. ScRNA-seq, combined with scATAC-seq, implicated chromatin modulation in the continuation of sustained resistance [45]. Chromatin enables proliferation through its retention of acetate, a significant source of nutrition for tumor cells [35, 52]. In selected acute myeloid lymphomas, resistance is initiated through the recruitment of novel enhancers to enable continued expression of genes vital to the disease’s survival [45]. When cancer cells circumvent gene inhibition through the use of alternative enhancers, different transcriptional factors are then also required for continued expression of these genes. This suggests that cancer cells can switch pathways by which key regulators of tumor survival are expressed. Single-cell sequencing also provides an assessment of the immunological contribution to drug-resistance in tumors. CD8+ T-cells are recruited to the tumor by macrophages [50]. Single-cell T-cell Receptor sequencing (scTCR-seq) assays found the expression of genes such as PD1 by tumor cells results in the exhaustion of tumor-infiltrating T-cells [48, 50]. This hampers many of the typical responses from effector T-cells, including cytotoxicity [41,49].
Novel insights obtained from mechanistic analyses have informed current research trends and led to the development of additional technologies to enhance the evaluation of drug response at the single-cell level. Regarding the immune system’s contribution, a novel therapeutic approach called immune checkpoint blockade (ICB) has been employed [48, 53–55]. ICB drugs both stimulate T-cell infiltration and block the molecules inducing T-cell exhaustion [41]. ICB therapies have demonstrated surprising efficacy in re-sensitizing exhausted T-cell tumor infiltrate and, in doing so, represent a remarkable step for immuno-oncology [56–58]. Similar pursuits, including the utilization of single-cell B-cell receptor sequencing (scBCR-seq) to study antigen specificity relevant to antibody therapies, have proven that immuno-oncology is a viable treatment option deserving extensive study [59–65]. Drug discovery methods have also been adapted to utilize single-cell technologies. For example, the single-cell barcoding technique together with CRISPR-Cas9 is employed to introduce gene perturbations into individual cells and evaluate the transcriptional implications of these perturbations in a high-throughput manner [45, 66–68]. As a result, it is possible to associate phenotypes with genetic and transcriptional perturbations at the single-cell level. Such insights will be harnessed in the future to make predictions regarding drug responses.
Challenges and Future Perspectives in Single-Cell Based Drug Prediction
Given that single-cell sequencing remains a relatively nascent field, there is still immense potential for growth and discovery. For scRNA-seq, signal drop-outs remain a major issue, where a gene is observed at a low or moderate expression level in one cell but detected as zero [69]. Imputation and normalization have been utilized to correct this issue as well as batch effects with modest success [70]. Batch effects are differences in measurements that arise from variations in non-biological factors, such as laboratory conditions, reagents, or instruments. Both the dropouts and batch effects introduce noise to the real values, leading to distorted drug response readouts or misclassified cell types. DL models have been proven to be an outstanding way to extract accurate high-dimensional features from sequencing data and infer the intrinsic gene relations, especially in large datasets at the single-cell level [71–73]. Recovering true values and removing batch effects enables more accurate cell type annotations and more efficient integration of large datasets from different samples and sequencing runs which improves the drug response predictions.
On the other hand, barcoding methodologies have also been developed to further overcome the challenges introduced by the technical zero and batch effect [35, 69, 74]. The use of sample-specific barcodes in scRNA-seq enables multiplexing that reduces batch effects. One such barcoding technique, MULTI-seq, claims to even address dropout issues by distinguishing between low-RNA expression and low-quality cells [69]. Another limitation of scRNA-seq is that the assumption mRNA levels are correlated with translations is not always true. FISH and Western blot assays have confirmed multiple instances in which overexpression of genes was accompanied by little to no subsequent translation [45, 46]. Unfortunately, while fields such as proteomics and metabolomics have begun to implement single-cell technologies, they have not progressed rapidly enough [75]. As such, they are not currently scalable, and it would not be feasible to employ them in conjunction with high-throughput techniques such as scRNA-seq to explore this phenomenon.
Facilitating Single-cell Level Drug Predictions Using Deep Transfer Learning
DL models have previously been utilized in single-cell clustering analysis, batch effect correction, and denoising [76–79], yet, has not been used for DTIs prediction or drug responses. As single-cell sequencing is still in its infancy relative to bulk sequencing, there does not yet exist vast public repositories for drug-related single-cell data, limiting the training power of DL models. To overcome such limitation, we could borrow the information contained in bulk sequencing data and transfer to the single-cell level.
DTL preserves previously learned features and trained parameters to apply to the testing dataset with a similar problem. This has been shown to improve the prediction performance, especially when there is limited data available for the new task and the model was trained originally on large amounts of data. We could first train out similar features from both bulk and single-cell data by passing a discriminator covered in a generative adversarial network. A DTL framework can then be applied to transfer known drug-feature relationship at the bulk level to individual cells, resulting in predicted drug sensitivities for each cell type (Figure 3A). Such pursuits promise to yield significant advancements in the treatment of heterogeneous disorders and in turn will improve the prognosis of patients afflicted by these disorders. In a more advanced way, bulk data can be used to regularize inferences transferred between two single-cell data to improve the accuracy of drug sensitivity prediction (Figure 3B).
Figure 3. Potential applications of DTL framework on single-cell data for drug sensitivity prediction.
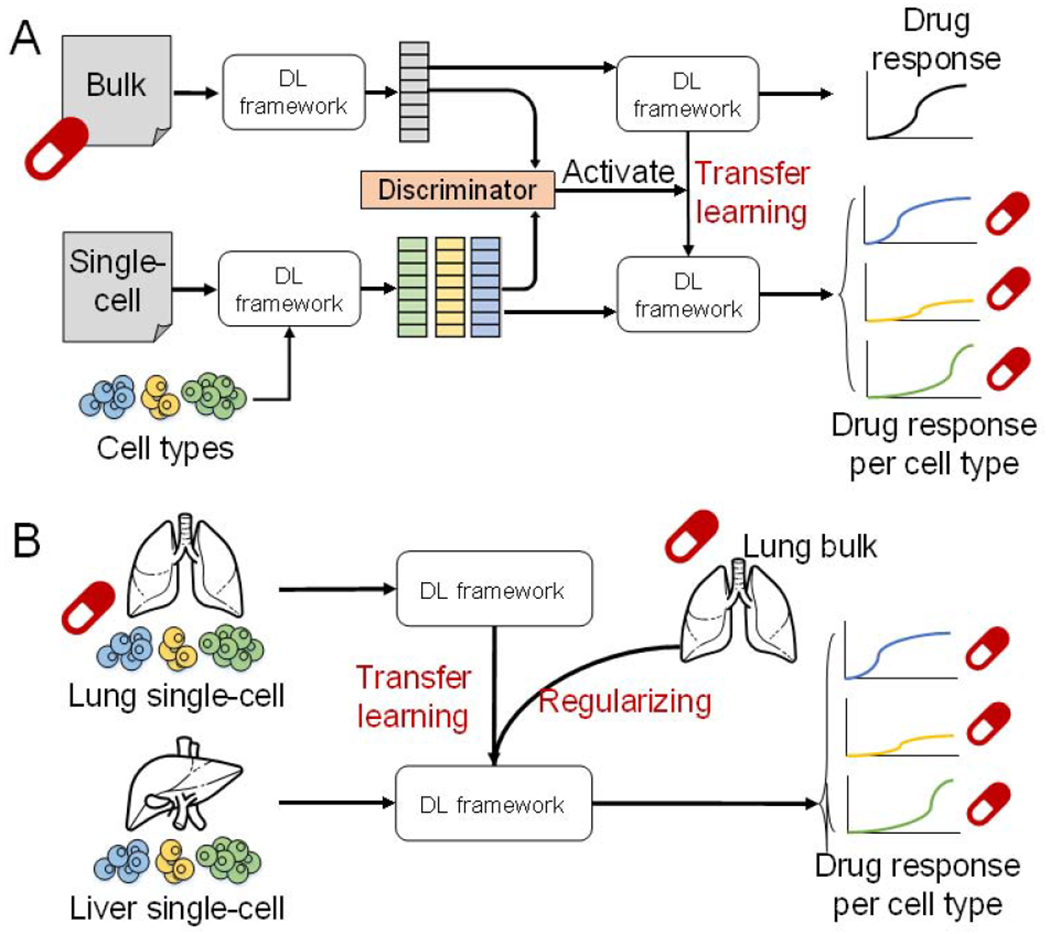
(A) the combination usage of generative adversarial network and DTL framework transfers the drug sensitivity known at the bulk level to the single-cell level. (B) A more advanced application of DTL would transfer drug sensitivity between two single-cell data and use bulk level information as a regularizer to constrain the DL parameters.
Integration of Single-cell Multi-omics Data for Drug prediction
Single-cell multi-omics (scMulti-omics) technologies simultaneously measure multiple modalities within an individual cell, including features from genomics, epigenomics, transcriptomes, and proteomics. Such approaches profile cell behavior and identity more comprehensively than previous individual methodologies. When multiple omics profiles are used in conjunction, one profile can recover the cellular characteristics— such as DNA methylation, gene expression, chromatin accessibility, or protein abundance—that might be lost by another sequencing technique. Furthermore, scMulti-omics can validate conclusions drawn from other omics profiles as well. The DL based integrative analysis could accurately answer biological questions, including tumor type classification and prognostication prediction [80, 81]. Thus, a unified multi-modal learning framework can be expected to incorporate the integrative analysis of scMulti-omics data, protein structure, drug structure, and side effect information. Such a framework can build intrinsic links between genomic variations and phenotypic phenomena induced by drugs and thus enhance the accuracy and efficiency of drug sensitivity prediction. However, there is an immense computational burden that must be addressed and overcome. While there are several methods available to help integrate and analyze scMulti-omics data, including factorization, Bayesian modeling, and network-based modeling, integrating the data from two or more technologies only exacerbates computational issues encountered during analysis [82, 83]. Another challenge is related to the analytical capabilities of integrative tools. Existing computational methods cannot simultaneously perform functions such as identifying cis-regulatory motifs, finding cell-type-specific regulons, and inferring gene regulatory networks. A robust benchmarking pipeline of integrative scMulti-omics analytic methods is needed to rectify this. In addition, there is also room for improvement in DL models. The attention mechanisms can be embedded into DL models to make more accurate inferences.
Concluding Remarks
The variability in the genomic and transcriptomic profiles of heterogenous tumor subpopulations prevents the development of an effective drug regimen for cancer patients. Most targeted cancer therapies and drugs exhibit diverse responses in patients, leading to low cure rates and high relapse rates. It is impossible to experimentally test and validate drug responses in vivo. As such, determining how to accurately and effectively assess drug responses in silico is critical for continued drug development. By training and applying advanced DL models, scientists can rapidly predict potential drug targets for novel treatment, simulate drug response under millions of conditions, and discover new purposes for the existing drugs. We reviewed six DL model types and summarized the application of DL-based tools for drug discovery and in predicting drug responses. Using these DL models, researchers have successfully improved how the accuracy of the predictions they make.
However, accuracy is inherently limited by the resolution of data generated from conventional bulk sequencing methods. One trend in alleviating this limitation is the transition to using advanced single-cell techniques for drug response predictions. Single-cell technologies comprehensively profile the heterogeneity of cancer cells to identify targeted treatment options and assess the risk of developing drug resistance.
Due to the high dimensionality and large sample sizes of single-cell data, DL models are naturally well-suited for single-cell analyses. However, currently, the quantity of available benchmarked, drug-related, single-cell data limits the application power of DL models. To fully address the limitations of both single-cell sequencing and DL models and to maximize their functionality as it pertains to predicting drug responses, several key points must be considered (See Outstanding Questions). We further propose that the substantial application of DL, especially the DTL framework, presents an immediate solution to enable single-cell informed drug response predictions by first learning drug-target information from bulk data. We believe that, while admittedly there is a long way to go, eventually, the combination of DL and single-cell technologies will reshape how drug development and target therapies are conducted.
Outstanding Questions.
How to choose an appropriate deep learning model when dealing with several data categories?
How to incorporate the analysis of drug-target interaction, structural information, and sequencing data using the DL transfer learning model?
What algorithm would help to solve the “black box” problem in artificial intelligence and DL?
Other than drug sensitivity prediction and drug-target interaction, what field will benefit from the DL transfer learning?
How will single-cell multi-omics provide insights into heterogeneous responses across the tumor subpopulations?
What methods we could use to minimize the loss during transferring bulk data knowledge to single-cell analysis?
Highlights.
A comprehensive understanding of heterogeneous tumor subpopulations will benefit the drug sensitivity prediction and combination drug treatment design.
Deep learning models are powerful and extensively used in drug sensitivity prediction and drug-target interaction inference.
Single-cell sequencing techniques offer precise and accurate profiling of tumor subpopulations and reveal the subtle difference in their response to drug treatments.
Applying deep transfer learning to predict drug sensitivity allows us to not only take advantage of prior knowledge obtained from massive bulk sequencing data but also utilize the heterogeneous landscapes generated by single-cell sequencing techniques.
The integration of single-cell multi-omics data for drug sensitivity prediction using transfer learning methods poses a special challenge.
Acknowledgment
This work was supported by awards R35-GM126985, R01-GM131399, and R01-DE025447 from the National Institute of General Medical Sciences of the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Science Foundation and the National Institutes of Health.
GLOSSARY
- Assay for transposase-accessible chromatin with high-throughput sequencing (ATAC-seq)
It is used to determine chromatin accessibility across the genome
- Batch effect
External factors associated with experiments will influence the produced data and result in inaccurate conclusions. It represents the systematic technical differences when samples are processed and measured in different batches
- Bulk sequencing
It examines the sequence of information from bulk samples, usually many cells
- Cell-type-specific regulon
The full complement of transcriptional targets that are regulated by a protein. These can include either direct physical targets, transcription factors, and cofactors, or indirect targets for signal transduction
- Chromatin immunoprecipitation with high-throughput sequencing (ChIP-seq)
It is used to identify genome-wide DNA binding sites for transcription factors and other proteins
- Combinational drug treatment
It uses more than one drug to treat diseases, which usually reduces the development of drug resistance
- Deep Belief Network (DBN)
DBN is constructed on several layers of Restricted Boltzmann machine
- Deep learning
An AI function that mimics the workings of the human brain in processing unstructured data through many layers of neural networks
- Drug efflux
Cells express efflux pumps which are able to move drug out of a cell
- Drug inactivation
Cancer cells may express enzymes to digest or modify drugs, which leads to the loss of function
- Drug resistance
The reduction in effectiveness of a medication, such as an antimicrobial or an antineoplastic, in treating disease
- Drug response
The pharmacodynamic (PD) response to a drug; this includes all the effects of the drug on any physiological and/or pathological processes
- Drug sensitivity
The concentration of a drug that inhibits cell growth
- Fluorescent in situ hybridization (FISH)
FISH is a molecular technique that uses fluorescent probes that can specifically bind to DNA/RNA/proteins to visualize the location of those targets
- Immune checkpoint blockade (ICB)
Immune checkpoints are accessory molecules that regulate the activation and silencing of T cells. ICB can release inherent limits on the activation and maintenance of T cell effector function by inhibiting the immune checkpoints
- Minimal residual disease (MRD)
A small number of cancer cells that survive the treatment and usually result in relapse
- Single-cell B/T-cell receptor sequencing (scBCR/scTCR-seq)
scBCR/scTCR-seq is a genomics approach to analyze B/T cell receptors uniquely expressed on B/T cell surface. The diverse range of BCRs/TCRs expressed by the total B/T cell population of an individual is termed as B/T cell repertoire
- Single-cell sequencing
It examines the sequence of information from individual cells with optimized next-generation sequencing technologies, providing a higher resolution of cellular differences
- Target alternation
Cancer cells may downregulate the expression of proteins or modify the proteins which are targeted by drugs
- Visualized neural network (VNN)
It can simulate not only the system function but also the system structure
- Whole-exome sequencing
It will only sequence the coding regions of genes and is only able to identify variants found in these coding regions
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclaimer Statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Resources:
Lab webpage: https://u.osu.edu/bmbl/
References:
- 1.Siegfried Z and Karni R (2018) The role of alternative splicing in cancer drug resistance. Current Opinion in Genetics & Development 48, 16–21. [DOI] [PubMed] [Google Scholar]
- 2.Lee YT et al. (2018) Molecular targeted therapy: Treating cancer with specificity. European Journal of Pharmacology 834, 188–196. [DOI] [PubMed] [Google Scholar]
- 3.Dagogo-Jack I and Shaw AT (2018) Tumour heterogeneity and resistance to cancer therapies. Nature Reviews Clinical Oncology 15 (2), 81–94. [DOI] [PubMed] [Google Scholar]
- 4.Luskin MR et al. (2018) Targeting minimal residual disease: a path to cure? Nature reviews. Cancer 18 (4), 255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Konieczkowski DJ et al. (2018) A Convergence-Based Framework for Cancer Drug Resistance. Cancer cell 33 (5), 801–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Panda M and Biswal BK (2019) Cell signaling and cancer: a mechanistic insight into drug resistance. Mol Biol Rep 46 (5), 5645–5659. [DOI] [PubMed] [Google Scholar]
- 7.Wang X et al. (2019) Drug resistance and combating drug resistance in cancer. Cancer Drug Resistance 2 (2), 141–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Patch A-M et al. (2015) Whole–genome characterization of chemoresistant ovarian cancer. Nature 521 (7553), 489–494. [DOI] [PubMed] [Google Scholar]
- 9.Hinohara K et al. (2018) KDM5 Histone Demethylase Activity Links Cellular Transcriptomic Heterogeneity to Therapeutic Resistance. Cancer cell 34 (6), 939–953.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang W et al. (2012) Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Research 41 (D1), D955–D961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barretina J et al. (2012) The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483 (7391), 603–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aksoy BA et al. (2017) CTD2 Dashboard: a searchable web interface to connect validated results from the Cancer Target Discovery and Development Network. Database (Oxford) 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Newton Y et al. (2017) TumorMap: Exploring the Molecular Similarities of Cancer Samples in an Interactive Portal. Cancer Research 77 (21), e111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.LeCun Y et al. (2015) Deep learning. Nature 521 (7553), 436–44. [DOI] [PubMed] [Google Scholar]
- 15.Eraslan G et al. (2019) Deep learning: new computational modelling techniques for genomics. Nature Reviews Genetics 20 (7), 389–403. [DOI] [PubMed] [Google Scholar]
- 16.Wishart DS et al. (2018) DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res 46 (D1), D1074–d1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wen M et al. (2017) Deep-Learning-Based Drug–Target Interaction Prediction. Journal of Proteome Research 16 (4), 1401–1409. [DOI] [PubMed] [Google Scholar]
- 18.Wan F et al. (2019) DeepCPI: A Deep Learning-based Framework for Large-scale in silico Drug Screening. Genomics Proteomics Bioinformatics 17 (5), 478–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karimi M et al. (2019) DeepAffinity: Interpretable Deep Learning of Compound-Protein Affinity through Unified Recurrent and Convolutional Neural Networks. Bioinformatics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee I et al. (2019) DeepConv-DTI: Prediction of drug-target interactions via deep learning with convolution on protein sequences. PLoS Comput Biol 15 (6), e1007129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wallach I et al. (2015) AtomNet: A Deep Convolutional Neural Network for Bioactivity Prediction in Structure-based Drug Discovery. ArXiv abs/1510.02855. [Google Scholar]
- 22.Rifaioglu AS et al. (2018) DEEPScreen: High Performance Drug-Target Interaction Prediction with Convolutional Neural Networks Using 2-D Structural Compound Representations. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Altae-Tran H et al. (2017) Low Data Drug Discovery with One-Shot Learning. ACS Central Science 3 (4), 283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yan J et al. (2020) Deep-AmPEP30: Improve Short Antimicrobial Peptides Prediction with Deep Learning. Mol Ther Nucleic Acids 20, 882–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Torres S et al. (2019) Mitochondrial Cholesterol in Alzheimer’s Disease and Niemann–Pick Type C Disease. Frontiers in Neurology 10 (1168). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zeng X et al. (2020) Target identification among known drugs by deep learning from heterogeneous networks. Chemical Science 11 (7), 1775–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zeng X et al. (2020) Network-based prediction of drug-target interactions using an arbitrary-order proximity embedded deep forest. Bioinformatics 36 (9), 2805–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zeng X et al. (2019) deepDR: a network-based deep learning approach to in silico drug repositioning. Bioinformatics 35 (24), 5191–5198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aliper A et al. (2016) Deep Learning Applications for Predicting Pharmacological Properties of Drugs and Drug Repurposing Using Transcriptomic Data. Molecular Pharmaceutics 13 (7), 2524–2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Choi J et al. (2020) RefDNN: a reference drug based neural network for more accurate prediction of anticancer drug resistance. Scientific Reports 10 (1), 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma J et al. (2018) Using deep learning to model the hierarchical structure and function of a cell. Nat Methods 15 (4), 290–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Steiner MC et al. (2020) Drug Resistance Prediction Using Deep Learning Techniques on HIV-1 Sequence Data. Viruses 12 (5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chiu Y-C et al. (2019) Predicting drug response of tumors from integrated genomic profiles by deep neural networks. BMC medical genomics 12 (Suppl 1), 18–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Preuer K et al. (2018) DeepSynergy: predicting anti-cancer drug synergy with Deep Learning. Bioinformatics 34 (9), 1538–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Srivatsan SR et al. (2020) Massively multiplex chemical transcriptomics at single-cell resolution. Science (6473), 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hata AN et al. (2016) Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nature medicine 22 (3), 262–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peng H et al. (2019) A component overlapping attribute clustering (COAC) algorithm for single-cell RNA sequencing data analysis and potential pathobiological implications. PLoS Comput Biol 15 (2), e1006772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stuart T et al. (2019) Comprehensive Integration of Single-Cell Data. Cell 177 (7), 1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim KT et al. (2015) Single-cell mRNA sequencing identifies subclonal heterogeneity in anti-cancer drug responses of lung adenocarcinoma cells. Genome Biol 16 (1), 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Polzer B et al. (2014) Molecular profiling of single circulating tumor cells with diagnostic intention. EMBO Molecular Medicine 6 (11), 1371–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tirosh I et al. (2016) Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352 (6282), 189–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang Z et al. (2017) Single-cell Sequencing Reveals Variants in ARID1A, GPRC5A and MLL2 Driving Self-renewal of Human Bladder Cancer Stem Cells. Eur Urol 71 (1), 8–12. [DOI] [PubMed] [Google Scholar]
- 43.Shaffer SM et al. (2017) Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 546 (7658), 431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shen Z et al. (2017) Current detection technologies for circulating tumor cells. Chemical Society reviews 46 (8), 2038–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bell CC et al. (2019) Targeting enhancer switching overcomes non-genetic drug resistance in acute myeloid leukaemia. Nat Commun 10 (1), 2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Prieto-Vila M et al. (2019) Single-Cell Analysis Reveals a Preexisting Drug-Resistant Subpopulation in the Luminal Breast Cancer Subtype. Cancer Res 79 (17), 4412–4425. [DOI] [PubMed] [Google Scholar]
- 47.Pérez-Velázquez J and Rejniak KA (2020) Drug-Induced Resistance in Micrometastases: Analysis of Spatio-Temporal Cell Lineages. Front Physiol 11, 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Balança CC et al. (2020) Dual Relief of T-lymphocyte Proliferation and Effector Function Underlies Response to PD-1 Blockade in Epithelial Malignancies. Cancer Immunol Res. [DOI] [PubMed] [Google Scholar]
- 49.Fairfax BP et al. (2020) Peripheral CD8(+) T cell characteristics associated with durable responses to immune checkpoint blockade in patients with metastatic melanoma. Nat Med 26 (2), 193–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.House IG et al. (2020) Macrophage-Derived CXCL9 and CXCL10 Are Required for Antitumor Immune Responses Following Immune Checkpoint Blockade. Clin Cancer Res 26 (2), 487–504. [DOI] [PubMed] [Google Scholar]
- 51.Wang Q et al. (2019) Single-cell profiling guided combinatorial immunotherapy for fast-evolving CDK4/6 inhibitor-resistant HER2-positive breast cancer. Nature Communications 10 (1), 3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schug ZT et al. (2016) The metabolic fate of acetate in cancer. Nature Reviews Cancer (11), 708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jerby-Arnon L et al. (2018) A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 175 (4), 984–997 e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yao C et al. (2019) Single-cell RNA-seq reveals TOX as a key regulator of CD8(+) T cell persistence in chronic infection. Nat Immunol 20 (7), 890–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kurtulus S et al. (2019) Checkpoint Blockade Immunotherapy Induces Dynamic Changes in PD-1(-)CD8(+) Tumor-Infiltrating T Cells. Immunity 50 (1), 181–194.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McDaniel JR et al. (2016) Ultra-high-throughput sequencing of the immune receptor repertoire from millions of lymphocytes. Nature Protocols 11 (3), 429–442. [DOI] [PubMed] [Google Scholar]
- 57.Zemmour D et al. (2018) Single-cell gene expression reveals a landscape of regulatory T cell phenotypes shaped by the TCR. Nature Immunology 19 (3), 291–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zheng C et al. (2017) Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 169 (7), 1342–1356.e16. [DOI] [PubMed] [Google Scholar]
- 59.Goldstein LD et al. (2019) Massively parallel single-cell B-cell receptor sequencing enables rapid discovery of diverse antigen-reactive antibodies. Communications Biology 2 (1), 304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Setliff I et al. (2019) High-Throughput Mapping of B Cell Receptor Sequences to Antigen Specificity. Cell 179 (7), 1636–1646.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Singh M et al. (2019) High-throughput targeted long-read single cell sequencing reveals the clonal and transcriptional landscape of lymphocytes. Nat Commun 10 (1), 3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rizzetto S et al. (2018) B-cell receptor reconstruction from single-cell RNA-seq with VDJPuzzle. Bioinformatics 34 (16), 2846–2847. [DOI] [PubMed] [Google Scholar]
- 63.Canzar S et al. (2017) BASIC: BCR assembly from single cells. Bioinformatics 33 (3), 425–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Upadhyay AA et al. (2018) BALDR: a computational pipeline for paired heavy and light chain immunoglobulin reconstruction in single-cell RNA-seq data. Genome Med 10 (1), 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Attaf N et al. (2020) FB5P-seq: FACS-Based 5-Prime End Single-Cell RNA-seq for Integrative Analysis of Transcriptome and Antigen Receptor Repertoire in B and T Cells. Front Immunol 11, 216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Datlinger P et al. (2017) Pooled CRISPR screening with single-cell transcriptome readout. Nature methods 14 (3), 297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dixit A et al. (2016) Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell 167 (7), 1853–1866 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Norman TM et al. (2019) Exploring genetic interaction manifolds constructed from rich single-cell phenotypes. Science, eaax4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McGinnis CS et al. (2019) MULTI-seq: sample multiplexing for single-cell RNA sequencing using lipid-tagged indices. Nature Methods 16 (7), 619–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Arisdakessian C et al. (2019) Deeplmpute: an accurate, fast, and scalable deep neural network method to impute single-cell RNA-seq data. Genome Biology 20 (1), 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chi W and Deng M (2020) Sparsity-Penalized Stacked Denoising Autoencoders for Imputing Single-Cell RNA-Seq Data. Genes (Basel) 11 (5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.He Y et al. (2020) DISC: a highly scalable and accurate inference of gene expression and structure for single-cell transcriptomes using semi-supervised deep learning. Genome Biol 21 (1), 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li X et al. (2020) Deep learning enables accurate clustering with batch effect removal in single-cell RNA-seq analysis. Nat Commun 11 (1), 2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shin D et al. (2019) Multiplexed single-cell RNA-seq via transient barcoding for simultaneous expression profiling of various drug perturbations. Science Advances 5 (5), eaav2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Marx V (2019) A dream of single-cell proteomics. Nature Methods 16 (9), 809–812. [DOI] [PubMed] [Google Scholar]
- 76.Wang T et al. (2019) BERMUDA: a novel deep transfer learning method for single-cell RNA sequencing batch correction reveals hidden high-resolution cellular subtypes. Genome Biol 20 (1), 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lieberman Y et al. (2018) CaSTLe - Classification of single cells by transfer learning: Harnessing the power of publicly available single cell RNA sequencing experiments to annotate new experiments. PLoS One 13 (10), e0205499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mieth B et al. (2019) Using transfer learning from prior reference knowledge to improve the clustering of single-cell RNA-Seq data. Sci Rep 9 (1), 20353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang J et al. (2019) Data denoising with transfer learning in single-cell transcriptomics. Nature Methods 16 (9), 875–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Guo LY et al. (2020) Deep learning-based ovarian cancer subtypes identification using multi-omics data. BioData Min 13, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lee TY et al. (2020) Incorporating deep learning and multi-omics autoencoding for analysis of lung adenocarcinoma prognostication. Comput Biol Chem 87, 107277. [DOI] [PubMed] [Google Scholar]
- 82.Huang S et al. (2017) More Is Better: Recent Progress in Multi-Omics Data Integration Methods. Front Genet 8, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ma A et al. (2020) Integrative Methods and Practical Challenges for Single-Cell Multi-omics. Trends in Biotechnology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rifaioglu AS et al. (2020) DEEPScreen: high performance drug–target interaction prediction with convolutional neural networks using 2-D structural compound representations. Chemical Science 11 (9), 2531–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zitnik M et al. (2018) Modeling polypharmacy side effects with graph convolutional networks. Bioinformatics 34 (13), i457–i466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pan C et al. (2019) Deep Learning Reveals Cancer Metastasis and Therapeutic Antibody Targeting in the Entire Body. Cell 179 (7), 1661–1676.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pang L et al. (2016) Construction of single-cell arrays and assay of cell drug resistance in an integrated microfluidic platform. Lab Chip 16 (23), 4612–4620. [DOI] [PubMed] [Google Scholar]
- 88.McGinnis CS et al. (2019) MULTI-seq: sample multiplexing for single-cell RNA sequencing using lipid-tagged indices. Nature Methods 16 (7), 619–626. [DOI] [PMC free article] [PubMed] [Google Scholar]