

Abstract
Background & Aims:
Primary sclerosing cholangitis (PSC) is a chronic liver disease characterized by peribiliary inflammation and fibrosis. Cholangiocyte senescence is a prominent feature of PSC. Here we hypothesize that extracellular vesicles (EVs) from senescent cholangiocytes influence the phenotype of target cells.
Methods:
EVs were isolated from normal human cholangiocytes (NHCs), cholangiocytes from PSC patients, and NHCs experimentally induced to senescence. NHCs, malignant human cholangiocytes (MHCs), and monocytes were exposed to 108 EVs from each donor cell population and assessed for proliferation, MAPK activation, and migration. Additionally, we isolated EVs from plasma of wild-type and Mdr2−/− mice (a murine model of PSC), and assessed mouse monocyte activation.
Results:
EVs exhibited the size and protein markers of exosomes. The number of EVs released from senescent human cholangiocytes was increased; similarly, the EVs in plasma from Mdr2−/− mice were increased. Additionally, EVs from senescent cholangiocytes were enriched in multiple growth factors, including EGF. NHCs exposed to EVs from senescent cholangiocytes showed increased NRAS and ERK1/2 activation. Moreover, EVs from senescent cholangiocytes promoted proliferation of NHCs and MHCs, findings that were blocked by erlotinib, an EGF receptor inhibitor. Furthermore, EVs from senescent cholangiocytes induced EGF-dependent Interleukin 1-beta and Tumor necrosis factor expression and migration of human monocytes; similarly, Mdr2−/− mouse plasma EVs induced activation of mouse monocytes.
Conclusions:
The data continue to support the importance of cholangiocyte senescence in PSC pathogenesis, directly implicate EVs in cholangiocyte proliferation, malignant progression, and immune cell activation and migration, and identify novel therapeutic approaches for PSC.
Keywords: biliary epithelial cell, cellular senescence, extracellular vesicles, primary sclerosing cholangitis, senescence-associated secretory phenotype
Lay Summary:
Primary sclerosing cholangitis (PSC) is a progressive, incurable biliary disease. We previously reported that biliary epithelial cell senescence (cell cycle arrest and hypersecretion of bioactive molecules) is an important phenotype in PSC. Here we demonstrate that senescent cholangiocytes secrete increased numbers of extracellular vesicles, membrane enclosed secreted structures important for intercellular communication that modify the phenotype of target cells.
Introduction
Primary sclerosing cholangitis (PSC) is a cholestatic liver disease of unknown etiology characterized by a progressive peribiliary fibroinflammatory response that leads to destruction of intrahepatic and/or extrahepatic bile ducts, which may ultimately result in cirrhosis and liver failure. PSC patients have an increased risk of developing cholangiocarcinoma (CCA) underscoring the aggressiveness of this disease [1]. However, no effective therapies exist for the treatment of PSC making it one of few major liver diseases without approved pharmacotherapy [2]. Thus, liver transplant is the only life-saving option [1, 3].
The cholangiocyte (biliary epithelia) response to injurious stimuli plays a critical role in initiating, modulating, and exacerbating PSC disease progression [4, 5]. We’ve previously shown that senescent cholangiocytes are abundant in livers of patients with PSC [6, 7]. Cellular senescence, a state of permanent replicative arrest and apoptosis resistance, can be triggered by a number of stimuli, including reactive oxygen species, strong mitogenic or oncogenic signals, telomere shortening, and/or non-telomeric DNA damage from exogenous or endogenous stressors [8]. Cellular senescence has been implicated in the pathogenesis of multiple chronic and age-related diseases including atherosclerosis, osteoarthritis, and chronic obstructive pulmonary disease [8–10]. Importantly, senescent cells may exhibit detrimental effects through the senescence-associated secretory phenotype (SASP), a cellular feature characterized by the robust secretion of a variety of bioactive molecules including, but not limited to chemokines, cytokines, and growth factors [11]. Through the SASP, senescent cells alter their microenvironment via intercellular communication and initiate injurious cellular responses [8].
Extracellular vesicles (EVs) are membrane-enclosed structures secreted by all cell types in health and disease, and increasingly recognized as a mode of intercellular communication which can affect the local microenvironment (autocrine and paracrine signaling) or act at a distance (endocrine signaling) [17,18]. EVs range in size from 30 nm to 200 μm and are generally classified into three groups, apoptotic bodies, microvesicles, and exosomes, based on their origin, size, and molecular content [12]. Apoptotic bodies are large vesicles, usually greater than 500 nm, released from cells undergoing apoptosis. Microvesicles are formed from protrusions of the plasma membrane ranging in size between 100 nm and 1 μm. Exosomes are the smallest of the three types, ranging between 40 and 150 nm that originate from multivesicular bodies in the endosomal compartment as intraluminal vesicles, and are released via multivesicular body fusion with the plasma membrane. EVs are now recognized as a cargo delivery system that contains cytokines, receptors, miRNAs, and a wide range of other molecules [13]. EVs have been implicated in a myriad of human diseases including cancer, metabolic, and chronic diseases [14], including liver disease, the effects of which are dependent on the molecular contents of the EV cargo [15–18].
EVs are increasingly recognized as important components of SASP [19]. These senescence-associated EVs have been implicated in a variety of pathogenic outcomes including fibrosis, inflammation, tumor promotion, and paracrine senescence [19]. However, the contribution of EVs to cholangiocyte SASP in PSC is unknown; moreover, whether senescent cholangiocyte-derived EVs exhibit distinct features compared to EVs derived from nonsenescent cholangiocytes and how senescent EVs affect communication between cholangiocytes and target cells has not been fully examined. Therefore, we explored the role of senescent cholangiocyte-derived EVs in mediating intercellular signaling. We isolated and characterized EVs from normal human cholangiocytes (NHC), NHC experimentally induced to senescence, and cholangiocytes derived from PSC patient livers. We then examined whether these EVs differentially affected target cell proliferation, activation, and migration. Our cumulative data support that senescent cholangiocytes release increased numbers of EVs with a distinct molecular profile that promotes phenotypic changes in target cells likely via epidermal growth factor (EGF) associated neuroblastoma-ras (NRAS)/mitogen-activated protein kinase 1 and 3 (MAPK3/3, [ERK1/2]) activation.
Methods
Cell Lines and Cell Culture
This study was approved by the Mayo Clinic Institutional Review Board (IRB # 10–005896) following the rules of the 1975 Declaration of Helsinki. Adult participants were prospectively enrolled from the Mayo Clinic Liver Transplant inpatient and outpatient services. The well-characterized NHC cell line derived from normal liver was provided by Dr. Juan Medina (University of Navarra, Pamplona, Spain) [20]. NHCs were cultured in Dulbecco’s modified Eagle’s medium/F12 (Gibco, 11330–032), supplemented with fetal bovine serum (FBS), penicillin/streptomycin (Gibco, 15140–122), adenine (Sigma, A8626), insulin (Sigma, I-6634), epinephrine (Sigma, E-1635), transferrin (Sigma, T-1147), triiodo-L-thyronine (Sigma, T-6397), and hydrocortisone (Sigma, H-0888)]. Exosome-depleted FBS (Gibco, A25904/A27208) was used as appropriate. NHCs were induced to senescence using lipopolysaccharide (LPS, 200 ng/ml), and H2O2 (50nM), as previously described [6]. Cholangiocytes were treated with ionizing radiation, 10-Gy using the RS 2000 Biological Research Irradiator (Rad Source Technologies) and allowed to senesce for 7–10 days prior to EVs isolation. Cholangiocytes were isolated from three PSC patients and cultured as previously described [21]. These patient-derived cholangiocytes were isolated from explant liver tissue from two male (46 and 57 years of age) and one 58-year-old female patient. All had a previous diagnosis of inflammatory bowel disease, and were confirmed negative for CCA. The human monocytic cell line THP-1 (ATCC TIB-202) was cultured in RPMI-1640 plus L-glutamine with 10% FBS. The stellate cell line, LX2, was cultured in RPMI-1640 with 10% FBS. All cell lines were confirmed negative for mycoplasma contamination using MycoAlert mycoplasma detection kit (Lonza, Basel, Switzerland) and authenticated by Short Tandem Repeatanalysis.
EV Isolation and Quantification
NHCs were plated on collagen-coated 7.5 cm polycarbonate membrane plates and cultured for 3 to 4 days. Cells were washed with Dulbecco’s phosphate-buffered saline (PBS) and incubated for 48 hours in media containing exosome-depleted FBS. EVs were isolated from the conditioned media (10 mL) by differential ultracentrifugation [22]. We employed a series of clearing spins (10 min, 300 g and 30 min, 3,000 g), filtered the supernatants through 0.2 μm filters, and centrifuged twice in SW28 or SW41 rotors (90 min, 120,000 g). The EV pellet was resuspended in sterile PBS and either stored at −80°C or used directly for downstream applications.
Plasma Collection from Murine Blood
Blood was collected from wild-type C57BL/6J mice and the ATP-binding cassette, sub-family B member 4 knockout (Abcb4−/− [Mdr2−/−]) mice via cardiac puncture. Samples were spun twice at 3,000 g for 15 minutes to collect plasma. Sterile PBS was then added to each sample (1:10 dilution) before EV isolation using differential ultracentrifugation.
Nanoparticle Tracking Analysis
Nanoparticle tracking analysis (NTA) was performed by diluting EVs in PBS, and analyzing using NanoSight NS300 with NTA 3.0 software (Malvern Instruments) with camera capture setting 12, and process threshold 3 or 4, consistent within each experiment. Samples were run continuously through a flow-cell top-plate using a syringe pump at a rate or 25 μL/min. At least three 60 second videos were recorded to document Brownian motion of particles, and a minimum of 1,000 valid tracks were analyzed.
Western Blot Analysis
Equal number of EVs (measured by NTA) were suspended in 30 μl PBS and 10μl 4x Laemmli lysis buffer (BIO-RAD, #161–0747) and boiled for 10 min. Primary antibodies (1:1000 dilution) used for Western blotting were: rabbit anti-alix (E6P9B; cell signaling); rabbit anti-TSG101 (Ab125011; abcam); rabbit anti-cd9 (D8O1A; cell signaling); rabbit anti-cd81 (D5O2Q; cell signaling). Bands were detected with either SuperSignal West Femto Substrate (ThermoFisher) or Enhanced Chemiluminescent Plus detection system (ECL Plus, Amersham).
Immunoelectron Microscopy
Immunoelectron microscopy was performed by the Mayo Clinic Microscopy and Cell Analysis Core. EV pellets were fixed with 4% paraformaldehyde in PBS, and deposited on formvar/carbon-coated 200 mesh nickle grids. Samples were probed with CD63 primary antibodies (Santa Cruz Biotechnology) and or 10 nm gold-conjugated protein A. Samples were then treated with 1% glutaraldehyde in PBS, and stained in 4% uranyl acetate and 2% methylcellulose. Images were collected using an electron microscope (model 1400, JEOL).
Growth Factor and Cytokine Antibody Array
Antibody arrays (ab134002 [abcam], ARY005B [R&D Systems]) were performed according to manufacturer’s instruction. Briefly, membranes were blocked and incubated overnight at 4°C with equal volumes containing equal number of EVs. Following biotin-conjugated antibody detection of growth factors or cytokines, the membranes were washed, incubated with HRP-streptavidin and processed for chemiluminescent signal detection.
Cellular Proliferation Assay
NHCs and malignant human cholangiocytes (MHCs) were seeded in 96 well plates. Cells were incubated with EVs (1e8/mL) isolated from NHCs, LPS- induced senescent cholangiocytes, or PSC patient-derived cholangiocytes for 24 hours. CellTiter AqueousOne Solution Reagent was added to culture wells and incubated for 1 hour. The formazan product was measured by recording absorbance at 490 nm. In experiments using pharmacologic inhibitors, cells were pre-incubated with the inhibitors for 1 hour.
NRAS Activation Assay
NRAS activation was determined as reported previously [23]. Lysates were diluted to equal protein concentration as determined by Bradford Analysis. Raf-RBD beads (Cytoskeleton) were added and samples were incubated at 4°C, centrifuged, washed, and resuspended in Laemmli Sample buffer (Bio-Rad). The samples were run on a Tris-HCl gel, transferred to a nitrocellulose membrane, and immunoblotted with a primary antibody to NRAS (Santa Cruz) and appropriate secondary antibody (LiCor). The membrane was visualized using LiCor’s Odyssey infrared imaging system. Ponceau Red staining for total GST-RBD was used as loading control.
Cellular Migration Assay
MHCs and THP-1 were seeded in the top wells of Cell Biolabs CytoSelect 96-well Cell Migration Assay Kit equipped with a polycarbonate membrane (5 μm pore size; Cell Biolabs Inc). Equal numbers of EVs were diluted into an equal volume of EV-depleted media and were added to the lower portion of each well. The cells were incubated at 37°C for 24 hours. Migratory cells were washed, detached, and detected by fluorometry (480/525nm) using CyQuant GR Dye. In experiments where pharmacologic inhibitors were used, cells were pre-incubated with inhibitors for 1 hour.
Macrophage Activation Assay
THP-1 cells were plated at equal numbers in 6-well plates (~5 × 105 cells/well). After 2 hours, media control (THP-1 media depleted of EVs) or EVs (1e8/mL) were added and cells were cultured for an additional 24 hours and total RNA was collected.
Reverse Transcription-Quantitative Polymerase Chain Reaction
Total RNA extraction was performed using TRIzol Reagent (Thermo Fisher). Reverse transcription was performed starting from 500 ng of total RNA. Reverse transcription-quantitative polymerase chain reaction (RT-qPCR) was performed in a volume of 10 μl containing 10 ng cDNA/reaction, at the following cycling conditions: 95°C for 5 min (activation); 95°C for 10 seconds, 60°C for 10 seconds, 72°C for 30 seconds (40 cycles, amplification); 95°C for 5 seconds, 65℃ for 1 min, followed by dissociation curve analysis. Target gene expression was calculated using the ΔΔCt method and expression was normalized to 18s expression levels.
Animal Studies
This study was approved by the Mayo Clinic Institutional Review Board and the Institutional Animal Care and Use Committee. C57BL/6J background Mdr2−/− mice were obtained as a gift from Dr. Oude Elferink (Tytgat Institute, Amsterdam, Netherlands). C57BL/6J background (wild-type) mice were obtained from Jackson Labs. Mice were housed at the Mayo Clinic animal care facility with a standard 12:12 hour light/dark cycle and ad libitum access to water and standard rodent diet. Mice were sacrificed under general anesthesia at 60 days of age. Serum plasma was collected and processed for EV isolation as described above.
In-vivo EV Uptake Assays
DiO dye (Thermo Fisher) was added to the pellet of EVs isolated from plasma. The EVs were resuspended in PBS and ultracentrifuged twice for 90 minutes at 100,000 g at 4°C. The labeled EVs were resuspended in sterile PBS solution and injected into the portal vein of C57BL/6J mice. Livers were harvested and incubated in modified H69 media at 37°C for 1 hour. The livers were then processed for cryosectioning and immunofluorescence.
Statistical Analysis
All data are reported as the mean (or fold change in mean) ± standard error of the mean from a minimum of three independent experiments. Statistical analyses were performed with Student t test or analysis of variance when appropriate. P<0.05 was considered statistically significant.
Results
Cultured Senescent Cholangiocytes Secrete Increased Numbers of EVs with Features Consistent with Exosomes
We performed next generation RNA sequencing on NHCs and cholangiocytes derived from three patients with PSC. We performed a functional enrichment analysis (FunRich Version 3.0 [24]) of this data set using genes expressed at a level of at least 5 reads per kilobase of transcript, per million mapped reads in NHC, and upregulated more than 2-fold in PSC cholangiocytes compared to NHC. Based on these criteria, the cholangiocytes isolated from the 3 PSC patients (PSC-1, PSC-2, and PSC-3) exhibited increased expression of 866, 1,206, and 877 genes compared to NHC, respectively. Analysis of these upregulated genes identified significant enrichment for the “cellular components” categories of exosome, extracellular, and plasma membrane (Figure 1A). These data suggest that PSC-derived cholangiocytes exhibit upregulated expression of genes associated with cellular components suited for intercellular communication. We next looked specifically for known exosomal markers in the genes upregulated in PSC patient-derived cholangiocytes by comparing these genes to a data set, collated by Vesiclepedia (microvesicles.org) [25], of the top 100 proteins identified in EVs. The cholangiocytes isolated from the 3 PSC patients exhibited increased expression of 10, 14, and 15 EV-associated genes found in the top 100 EV protein list, including CD63 and CD81 (Supplementary Table 1). We next verified, by RT-qPCR, the upregulated expression of both CD63 and CD81 in the PSC patient-derived cholangiocytes. We found increased messenger RNA (mRNA) of both of these exosomal markers compared to NHC (Figure 1B). These results demonstrate that PSC-derived cholangiocytes not only exhibit increased expression of genes associated with exosome biogenesis and release, but upregulated genes that are known components of these EVs. Extending these RNA sequencing (RNA-seq) data, we isolated and characterized EVs from NHCs, PSC patient-derived cholangiocytes, and NHC induced to senescence with LPS [6]. LPS-induced and PSC patient-derived cholangiocytes have been previously characterized for cellular senescence [21]. Here, we further demonstrate that both LPS-induced and PSC patient-derived cholangiocytes used in this study exhibit increased levels of p16, p21, and SA-β-gal enzymatic activity (Supplementary Figure1). Immunoelectron microscopy analysis revealed that EVs isolated from NHCs, LPS-induced and PSC patient-derived senescent cholangiocytes exhibited the typical size (~100 nm) and morphology of exosomes (eg, deflated football) and were positive for the exosomal marker CD63 (Figure 2A). Immunoblot analysis confirmed that EVs isolated from the three cellular populations were positive for the exosomal markers ALIX, TSG101, and CD9 (Figure 2B). NTA revealed that the size distribution of EVs isolated from the three cellular populations was similar and had a mode size of 120 nm, consistent with exosomes (Figure 2C and D). The number of released EVs was increased in LPS-induced senescent cholangiocytes (~7-fold) and PSC-derived EVs (~6-fold) compared to the number of EVs released from NHCs (Figure 2C and E). Gamma irradiation (IR) and chronic exposure to hydrogen peroxide (H2O2) are additional, well-studied inducers of cellular senescence [8]. We therefore determined whether EV secretion is induced in H2O2- or IR-induced senescent cholangiocytes. While size did not differ, we observed an increased number of small EVs (~ 5-fold) in H2O2- and IR-induced senescent cholangiocytes (Figure 2D and E). These data demonstrate that senescent cholangiocytes secrete increased numbers of EVs compared to NHCs, and support that EVs isolated from both experimentally-induced senescent human cholangiocytes and PSC patient-derived cholangiocytes, are enriched in exosomes.
Figure 1. Primary sclerosing cholangitis (PSC) patient-derived cholangiocytes are enriched in exosomes.
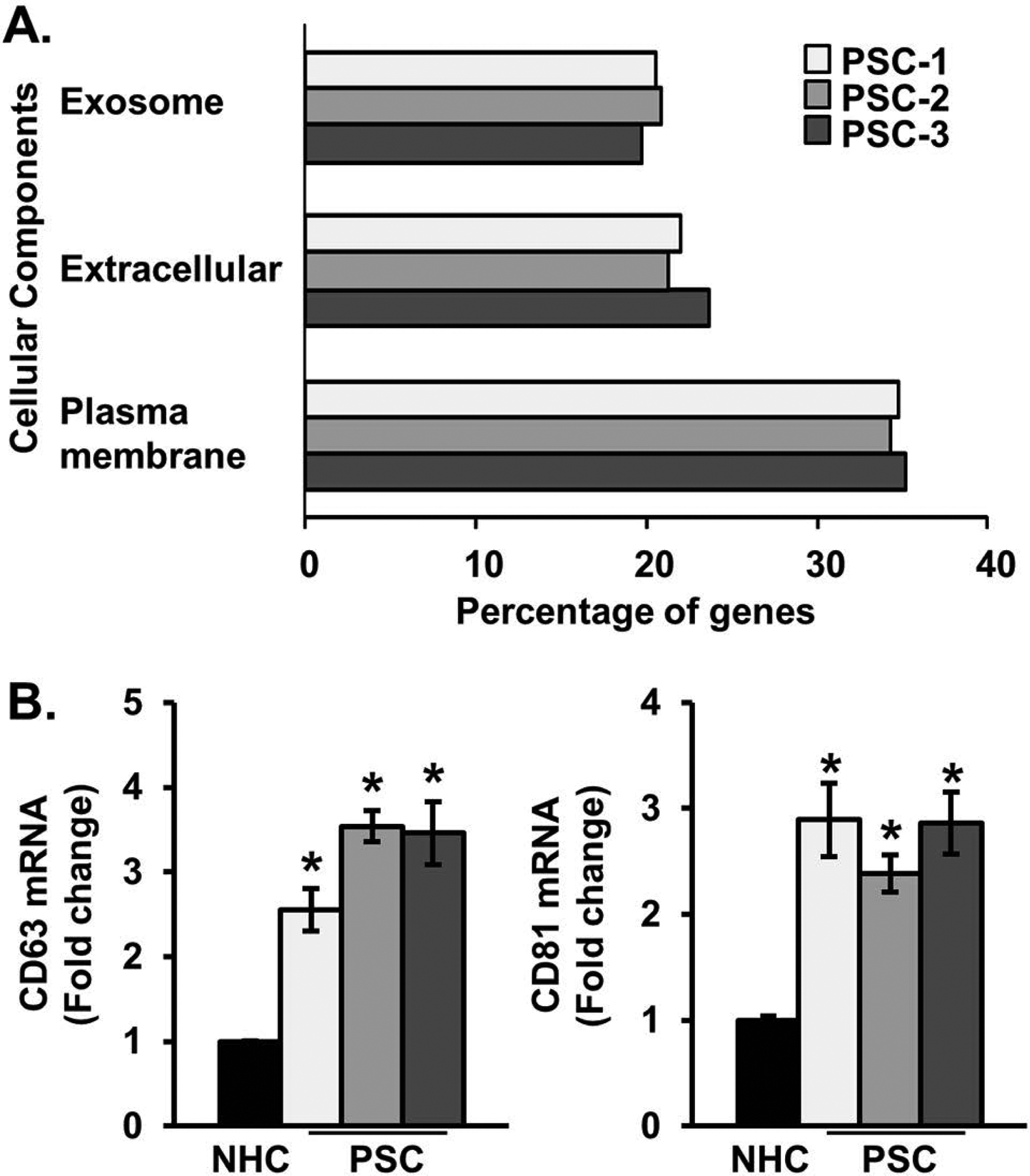
A, RNA sequencing was performed on one normal human cholangiocyte (NHC) and three PSC patient-derived short-term cholangiocyte cultures. Genes expressed at a level of at least 5 copies per million in NHC, and upregulated more than 2-fold in PSC cholangiocytes were used for functional enrichment analysis. Based on these criteria, the cholangiocytes isolated from the three PSC patients (PSC-1, PSC-2, and PSC-3) exhibited increased expression of 866, 1,026, and 877 genes compared to NHC, respectively. We identified consistent enrichment in the cellular components categories of exosome, extracellular, and plasma membrane. Bars show percentage, ie, number of upregulated genes in each category/total number of upregulated genes. The hypergeometric P value test was performed for each PSC patient-derived sample in each category and ranged from 9.5E-19 - 2.0E-05. B, Messenger RNA (mRNA) expression of exosomal markers (CD63 and CD81) are elevated in PSC patient-derived cholangiocytes. Bars represent mean fold-change of per million mapped reads (RPKM) vs NHC (±) standard error of the mean (SEM); n=3. *P<0.05 vs NHC.
Figure 2. Cultured senescent cholangiocytes secrete an increased number of extracellular vesicles (EVs).

A, EVs isolated from normal and senescent cholangiocytes (lipopolysaccharide- [LPS-] induced, and primary sclerosing cholangitis [PSC]-patient derived) were examined by transmission electron microscopy (EM) and immunogold electron microscopy (IEM). The purified vesicles exhibited the typical “deflated football” exosome morphology (inset) and were positive for the exosomal marker, CD63. B, Immunoblotting on whole cell (WCL) and isolated EV lysates. Analysis indicated that EV samples are positive for exosomal markers, Alix, TSG101, and CD9, and are negative for the cytoskeletal protein, actin. C, Nanoparticle tracking analysis (NTA) of NHC (dotted line), LPS-induced (dashed line) and PSC patient-derived senescent cholangiocytes (solid line) D, and E, Quantitation of NTA showed similar EV size between NHCs and LPS-, H202-, irradiation (IR)-induced or PSC patient-derived senescent cholangiocytes. However, the number of EVs, normalized to total cell count, was increased in the senescent cholangiocytes. Bars represent mean (±) standard error of the mean (SEM); n=7–10. *P<0.001.
EVs from Senescent Cholangiocytes are Enriched in Growth Factors
Next, we asked whether senescent cholangiocyte-derived EVs exhibit a distinct protein profile. The SASP includes, but is not limited to, cytokines, chemokines, and growth factors. While a cytokine/chemokine array analysis of EVs showed little positivity for any cytokine/chemokine, and did not show differences in expression between senescent cholangiocytes (LPS- or IR-induced) and NHCs (data not shown), we found, using a growth factor antibody array that EVs from PSC patient-derived and LPS-induced senescent cholangiocytes were enriched in multiple growth factors (~2-fold) compared to NHC-derived EVs including: amphiregulin, EGF, and fibroblast growth factor 7 (Figure 3A). These findings, coupled with the observation that increased EVs are released from senescent cells, are consistent with EVs functioning as a component of the cholangiocyte SASP and intercellular communication.
Figure 3. Senescent cholangiocyte-derived extracellular vesicles (EVs) are enriched in growth factors.
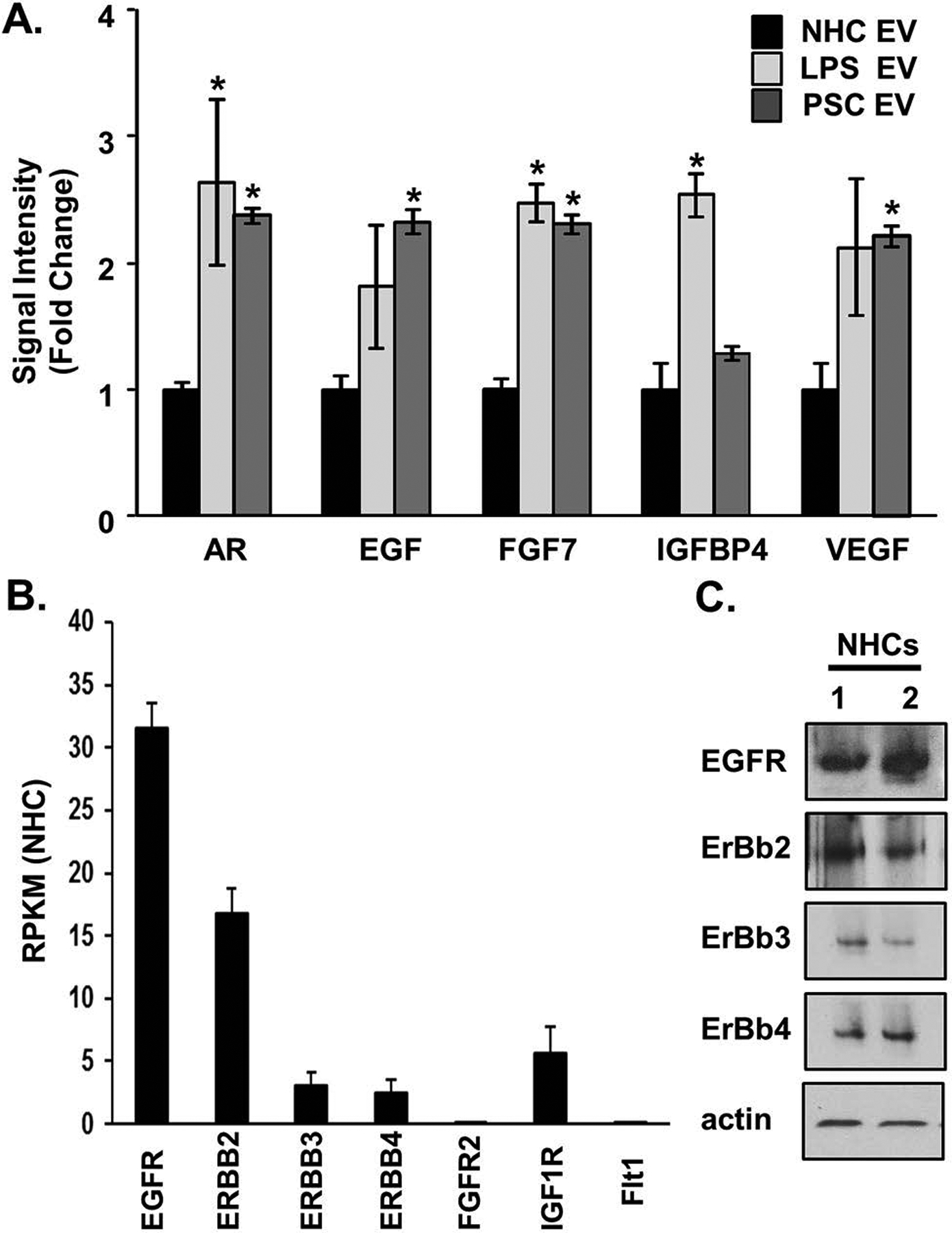
A, Equal numbers of normal and senescent cholangiocyte-derived EVs were harvested for protein analysis using a growth factor array (Abcam). Bars represent mean (±) standard error of the mean (SEM); n=3.The primary sclerosing cholangitis (PSC) values represent the mean of 3 PSC patient samples. Values represent a fold change versus EVs derived from normal human cholangiocytes (NHCs). *P<0.05. B, RNA-sequencing data on NHCs, shown as reads per Kilobase of transcript, per million mapped reads (RPKM) from 3 biological replicates revealed increased expression of the epidermal growth factor receptors (EGFRs), EGFR and Erb-B2 receptor tyrosine kinase 2 (ERBB2), compared to the fibroblast growth factor receptor (FGFR2), the insulin-like growth factor 1 receptor (IGF1R), and FMS-related tyrosine kinase 1 (FLT1, ie, VEGFR-1). C, Immunoblotting analysis on NHCs (2 biological replicates) showed positive protein expression of the EGF receptors.
Senescent Cholangiocyte EVs Induce EGF-Dependent Proliferation of Bystander NHCs
We next utilized our RNA-seq data set to determine whether NHCs express receptors for the EV-enriched growth factors. The RNA-seq data confirmed that two members of EGF receptor (EGFR) family, EGFR and ERBB2 receptor tyrosine kinase 2, are highly expressed in NHC, while the receptors for the other upregulated exosome-associated growth factors are minimally expressed in NHC (Figure 3B). Additionally, using immunoblotting, we confirmed EGFR family protein expression in cultured NHCs (Figure 3C). We previously demonstrated that cholangiocyte EGFR activation promotes cholangiocyte proliferation and the activation of the small GTP-binding protein, NRAS [26]. We therefore assessed whether senescence-associated EVs promoted bystander NHC proliferation via activation of EGFR family members. NHCs were treated with EVs (1e8/mL) isolated from NHC, PSC, and LPS-induced senescent cholangiocytes in the presence or absence of the pharmacologic EGFR inhibitor, erlotinib (2 nM). At the 24-hour time point, NHCs treated with LPS-induced and PSC patient-derived senescent cell EVs, but not NHC-derived EVs, exhibited increased proliferation, as assessed by MTS assay a process blocked by the presence of erlotinib (Figure 4A). This experiment was repeated and the results confirmed using the Celigo Imaging Cytometer and automated calculation of monolayer confluency (Supplementary Figure 2A). Moreover, we observed that both H2O2-induced and IR-induced senescent cholangiocyte EVs induced a similar, EGF-dependent, increase in proliferation of NHC, as assessed by MTS assay (Supplementary Figure 2B). These data support that growth factor enriched EVs from senescent cholangiocytes promote bystander NHC proliferation via activation of EGFR family members.
Figure 4. Senescent cholangiocyte-derived extracellular vesicles (EVs) induce proliferation of bystander cholangiocytes.
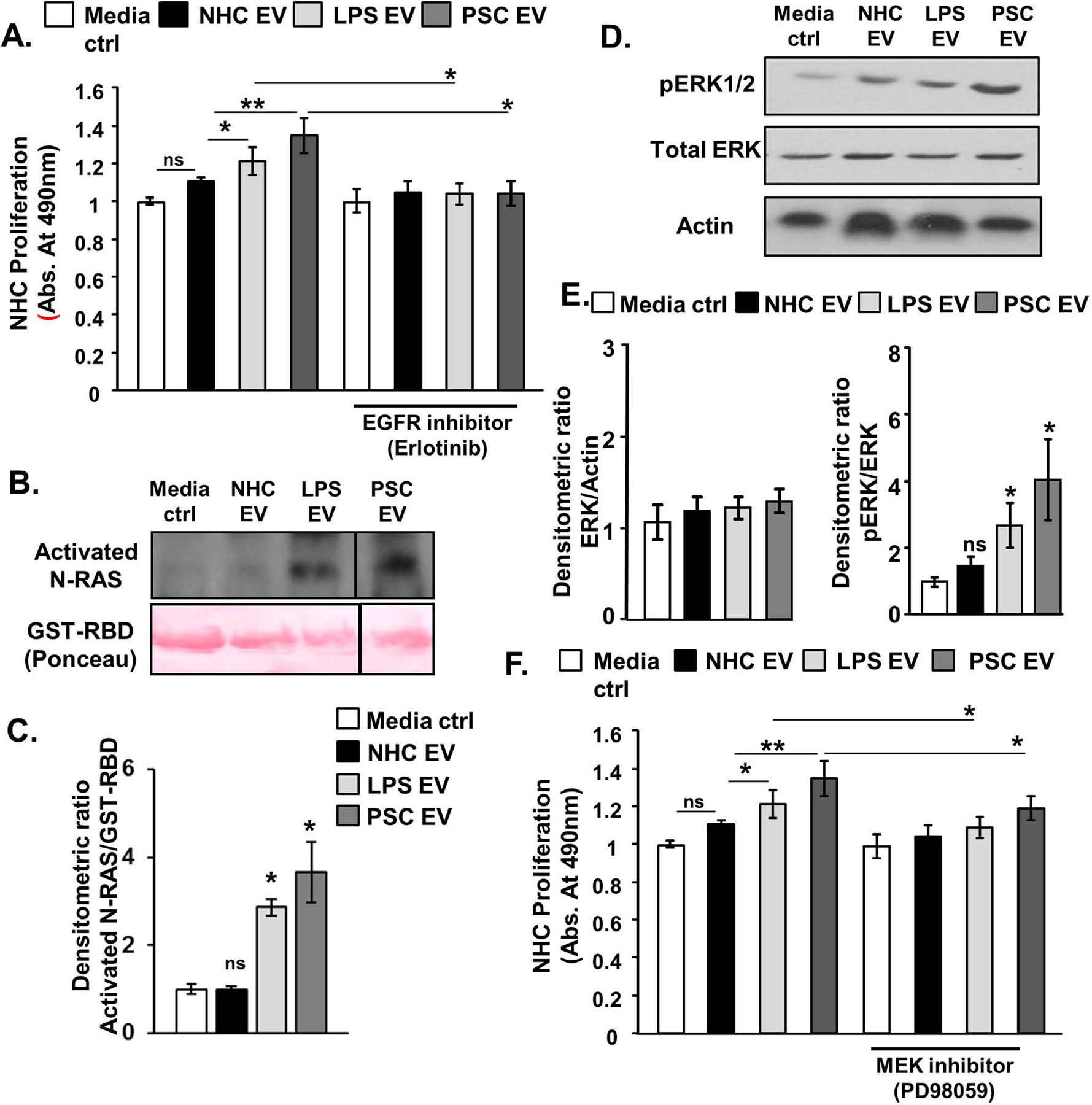
A, MTS proliferation assay; equal number of normal and senescent cholangiocytes derived EVs were applied to normal human cholangiocytes (NHCs) in the presence of absence of the epidermal growth factor receptor inhibitor, erlotinib. Cellular proliferation was measured after 24 hours incubation. The observed increase in proliferation in NHCs treated with senescent derived EVs is abrogated in the presence of erlotinib. B and C, NRAS activation assay and semi-quantitative densitometric analysis demonstrated that EVs isolated from senescent cholangiocytes (1e8/ml) promoted NRAS activation. D and E, Immunoblotting analysis and semi-quantitative densitometric analysis indicated that senescent cholangiocytes derived EVs (1e8/ml) induced increased ERK1/2 phosphorylation. F, MTS proliferation assay; equal number of normal and senescent cholangiocytes derived EVs were applied to NHCs in the presence of absence of the (MEK inhibitor, PD98059. Cellular proliferation was measured after 24 hours incubation. The observed increase in proliferation in NHCs treated with senescent derived EVs is abrogated in the presence of PD98059. For all panels, bars represent mean (±) standard error of the mean (SEM); n=3–5 biological replicates, *P<0.05, **P<0.01.
To determine whether senescent cholangiocyte-associated EVs promote NRAS activation in bystander nonsenescent NHC, we treated NHC with EVs (1e8/mL) isolated from NHC, PSC, and LPS-induced senescent cholangiocytes. Following a 24-hour incubation, NHCs treated with EVs isolated from LPS-induced and PSC patient-derived senescent cholangiocytes showed robust NRAS activation (ie, GTP-bound NRAS) compared to NHCs treated with NHC-derived EVs (Figure 4B, C). In addition, we assessed phosphorylation of ERK1/2, a key downstream mediator of NRAS signaling. Again, NHCs treated with LPS-induced and PSC patient-derived senescent cell EVs, but not NHC-derived EVs, exhibited increased ERK1/2 phosphorylation (Figure 4D, E). Having demonstrated that senescent cholangiocyte-derived EVs promote bystander NHC proliferation in an EGFR-dependent manner, and activate the NRAS/mitogen-activated protein kinase (MAPK) pathway, we assessed whether inhibition of the MAPK kinases, MAP2K1 (MEK1) and MAP2K2 (MEK2) prevented bystander NHC proliferation. We incubated bystander NHCs with a pharmacologic inhibitor of MEK1 and MEK2 (PD98059). These cells were then treated with normal and senescent EVs for 24 hours. PD98059 reduced the proliferative effect of senescent EVs on bystander NHCs (Figure 4F). These data support that EVs derived from senescent cholangiocytes promote proliferation in target cholangiocytes via EGFR activation and signaling through the NRAS/MAPK pathway.
Prolonged Incubation with Senescent Cholangiocyte EVs Induces Cellular Senescence of Bystander NHC
In previous work, we demonstrated that short-term treatment of NHC with an injurious stimulus (eg, LPS) promotes a proliferative phenotype, while persistent insult (eg, 10-day LPS treatment) induces NHC senescence in an NRAS-dependent manner [6]. Hence, having demonstrated that short-term incubation of bystander NHC with EVs derived from LPS-induced and PSC-patient-derived senescent cholangiocytes induced proliferation and activation of NRAS, we assessed whether long-term incubation with senescence-associated EVs promoted bystander NHC cellular senescence. NHCs were treated every other day for 10 days with EVs (1e8/mL) isolated from NHC, PSC-derived, and LPS-induced senescent cholangiocytes. Treatment of bystander NHC with both LPS-induced and PSC-derived EVs resulted in an increase in senescence-associated β-gal (SA-β-gal)-positive NHCs, a marker of cellular senescence, compared to NHCs treated with EVs derived from NHCs (Figure 5A and 5B). Moreover, NHCs treated with EVs isolated from LPS-induced and PSC patient-derived senescent cholangiocytes exhibited increased mRNA expression of senescence-associated cell cycle regulators, p16 and p21, as assessed by real-time PCR (Figure 5C). Furthermore, NHCs treated with EVs isolated from LPS-induced and PSC patient-derived senescent cholangiocytes, but not NHC-derived EVs, induced markers of SASP (IL6, CXCL8 [IL8], IL1B, and MMP2), as assessed by real-time PCR (Figure 5C). These data support that prolonged exposure to senescent cholangiocyte-derived EVs promotes paracrine senescence in target cholangiocytes.
Figure 5. Senescent cholangiocyte-derived extracellular vesicles (EVs) induce cellular senescence of bystander cholangiocytes.
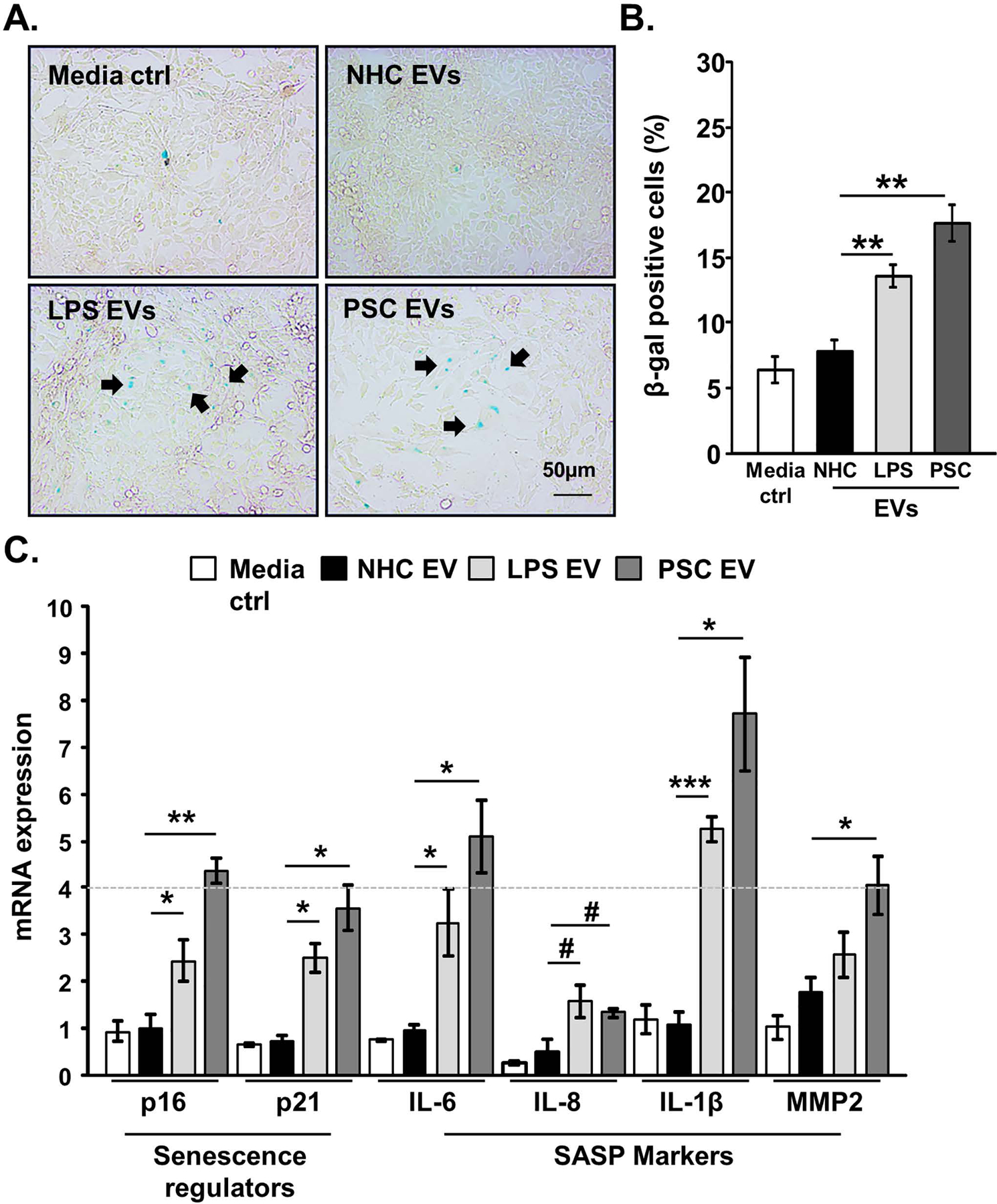
A, Senescence-associated β-galactosidase activity (SA-β-gal) assay; bystander normal human cholangiocytes (NHCs) were incubated with normal and senescent (lipopolysaccharide [LPS]-induced, and primary sclerosing cholangitis [PSC]-derived) cholangiocyte-derived EVs (1e8/ml) every other day for 10 days. SA-β-gal activity was assessed using β-galactosidase assay. Arrows indicate representative SA-β-gal positive NHCs. B, Quantification of the SA-β-gal assay; data presented as percentage of SA-β-gal positive cells per total cell count. The PSC patient-derived data represent the average of 3 replicates of 3–5 separate PSC patient samples. C, Induction of senescence was assessed by measuring messenger RNA (mRNA) expression (RT-qPCR) of various cellular senescence markers. Bars represent mean (±) standard error of the mean (SEM); n=3–5. *P<0.05, **P<0.01, #P=0.07.
Senescent Cholangiocyte EVs Induce Proliferation and Migration of MHCs (HuCCT1) via EGFR
The risk of developing CCA is 400 times higher in PSC patients than in the general population [27]. Senescent cells promote tumorigenesis through secretion of cytokines, chemokines, and various SASP molecules [11]. Here, we assessed whether senescent EVs (PSC-derived, and LPS-induced) promoted MHC proliferation. We treated MHCs with EVs (1e8/mL) derived from NHC, LPS-induced, and PSC-derived senescent cholangiocytes. At the 24-hour time point MHCs treated with LPS-induced and PSC patient-derived senescent cell EVs exhibited increased proliferation as assessed by MTS proliferation assay; however, treatment of MHC with erlotinib (2 nM) blocked EV-induced proliferation (Figure 6A). Moreover, we observed that both H2O2-induced and IR-induced senescent cholangiocyte EVs induced a similar, EGF-dependent, increase in proliferation of MHC, as assessed by MTS assay (Supplementary Figure 2C). Next, we assessed whether EVs induced migration of MHCs using a coculture transwell system. We found that EVs (1e8/mL) from LPS-induced and PSC patient-derived senescent cholangiocytes promoted increased migration (1.6, and ~2-fold, respectively); yet, pretreatment of MHC with erlotinib blocked EV-induced migration (Figure 6B). We confirmed this data using a wound healing assay [28]. We found that the migration rate of MHC was increased (~2-fold) upon treatment with LPS-induced and PSC patient-derived senescent cell EVs, but not NHC-derived EVs. Migration was blocked when MHC were pretreated with erlotinib (Supplementary Figure 3A and 3B). EGFR expression was confirmed in MHC (Hucct1), and additional target cells used in this study [hepatic stellate cells (LX2), and monocytes (THP-1)] by immunoblot (Supplementary Figure 3C). These results support that EVs derived from PSC and LPS-induced senescent cholangiocytes promote MHC proliferation and metastatic potential.
Figure 6. Senescent cholangiocyte-derived extracellular vesicles (EVs) promote phenotypic changes in target cells.
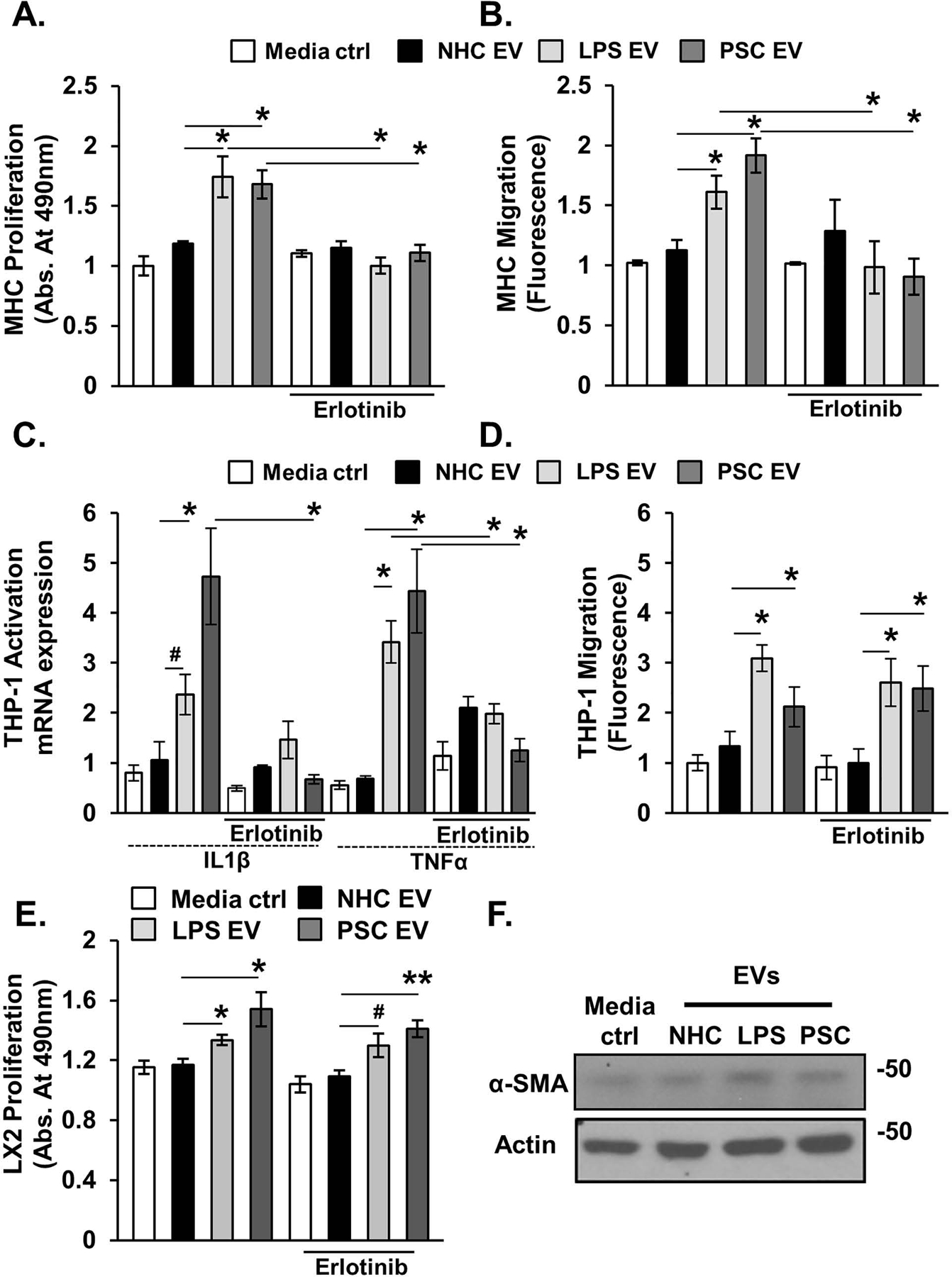
Equal number of normal and senescent cholangiocyte-derived EVs were applied to MHCs (A&B, Hucct1), human monocytes (C&D, THP-1), and hepatic stellate cells (E&F, LX2) for 24 hours incubation period (+/− erlotinib). A, MTS proliferation assay. EVs isolated from senescent cholangiocytes induced an increase in the target MHCs proliferation. Incubation with erlotinib prevented senescent EV-induced increased proliferation. B, Migration was measured using a colorimetric assay (Cell Biolabs). Senescent-derived EVs promoted MHC migration, while erlotinib inhibited this effect. C, Activation of THP-1 human monocytes was assessed by measuring monocytes messenger RNA (mRNA) expression levels of IL1B and TNF. Senescent-derived EVs promoted expression of these proinflammatory genes, while erlotinib treatment blunted this response. D, Migration of THP-1. Senescent-derived EVs promoted THP-1 migration and erlotinib had no effect on senescent EV-induced THP-1 migration. E, MTS proliferation assay. Senescent cholangiocyte-derived EVs induced proliferation of LX2. F, Immunoblotting analysis of α-SMA (stellate cells activation marker) showed no difference in expression upon incubation with senescent-derived EVs. Bars represent mean (±) standard error of the mean (SEM); n=3. *P<0.05, **P<0.01, #p=0.07.
Senescent Cholangiocyte EVs Induce EGF-Dependent Activation and EGF-Independent Migration of Human Monocytes
Macrophages accumulate around bile ducts in PSC-patients and the Mdr2−/− murine model of PSC [29, 30], and human monocytes are activated by conditioned media from senescent cholangiocytes. We therefore assessed, by RT-qPCR, expression levels of proinflammatory cytokines IL1B and TNF in human monocytes (THP-1) incubated with EVs from NHC, LPS-induced and PSC-patient derived senescent cholangiocytes in the presence or absence of erlotinib. LPS-induced and PSC patient-derived EVs induced IL1B and TNF expression, yet, pretreatment with erlotinib diminished this response (Figure 6C). We next assessed whether EVs derived from LPS-induced or PSC patient-derived senescent cholangiocytes promoted THP-1 migration using a two chamber co-culture system. EVs from senescent cholangiocytes increased the migration of THP-1 (~2 – 4-fold) compared to EVs derived from NHCs; pretreatment with erlotinib did not alter migration of THP-1 cells in response to senescent cell derived EVs (Figure 6D). We also assessed whether senescent EVs (PSC-derived, and LPS-induced) promoted proliferation of the hepatic stellate cell line, LX2. Hepatic stellate cells are the main source of extracellular matrix production in liver injury [31]. We treated LX2 with EVs (1e8/mL) derived from NHC, LPS-induced, and PSC-derived senescent cholangiocytes. LX2 treated with LPS-induced and PSC patient-derived senescent cell EVs, but not NHC-derived EVs, exhibited increased proliferation as assessed by MTS proliferation assay (Figure 6E). Treatment of LX2 with erlotinib (2 nM) did not block EV-induced proliferation (Figure 6E). Moreover, LPS-induced and PSC patient-derived senescent cell EVs did not induce activation of LX2 as assessed by measuring protein expression of α-SMA (Figure 6F). Together, these data support that EVs derived from LPS-induced and PSC patient-derived senescent cholangiocytes induce THP-1 proinflammatory gene expression, and that this response is blunted in the presence of the EGFR inhibitor, erlotinib. Moreover, the data support that senescent cell-derived EVs promote monocyte/macrophage migration and hepatic stellate cell proliferation independent of the EGFR.
EVs are Increased in Serum From a Murine Model of PSC, and Induce Activation of Mouse Monocytes
We next explored the role of extracellular vesicles in the Mdr2−/− murine model of PSC. This model, exhibits peribiliary inflammation and fibrosis, and an increased number of senescent cholangiocytes [32]. Circulating EVs were isolated from four month-old male wild-type and Mdr2−/− mouse plasma, and quantified by NTA. Mdr2−/− mice had an approximate 2-fold increase in small plasma EVs compared to wild-type mice (Figure 7A and 7B). Immunoblot analysis indicated that circulating EVs from wild type and Mdr2−/− mice exhibited the exosomal markers Alix, TSG101, and CD81 (Figure 7C). Next, we assessed whether circulating EVs induced macrophage activation. Mouse monocytes (RAW cells) were incubated with wild-type and Mdr2−/− -derived EVs for 24 hours, and activation was assessed by RT-PCR of proinflammatory cytokines Il1b and Tnf. Circulating EVs from Mdr2−/− mice significantly increased Tnf and Il1b mRNA expression compared to wild-type EVs (Figure 7D). Furthermore, to demonstrate EV uptake by monocytes in vivo, we fluorescently labeled wild-type and Mdr2−/− plasma-derived EVs, injected them into the portal vein of wild-type mice, and incubated the livers in culture media at 37℃ for 1 hour. We performed immunofluorescence on frozen sections and found that labeled EVs colocalize with the monocyte marker CD68 (Figure 7E). These results are consistent with EVs playing a central role in cell to cell communication in PSC including macrophage activation.
Figure 7. Circulating extracellular vesicles (EVs) are increased in the Mdr2−/− murine model of primary sclerosing cholangitis (PSC).
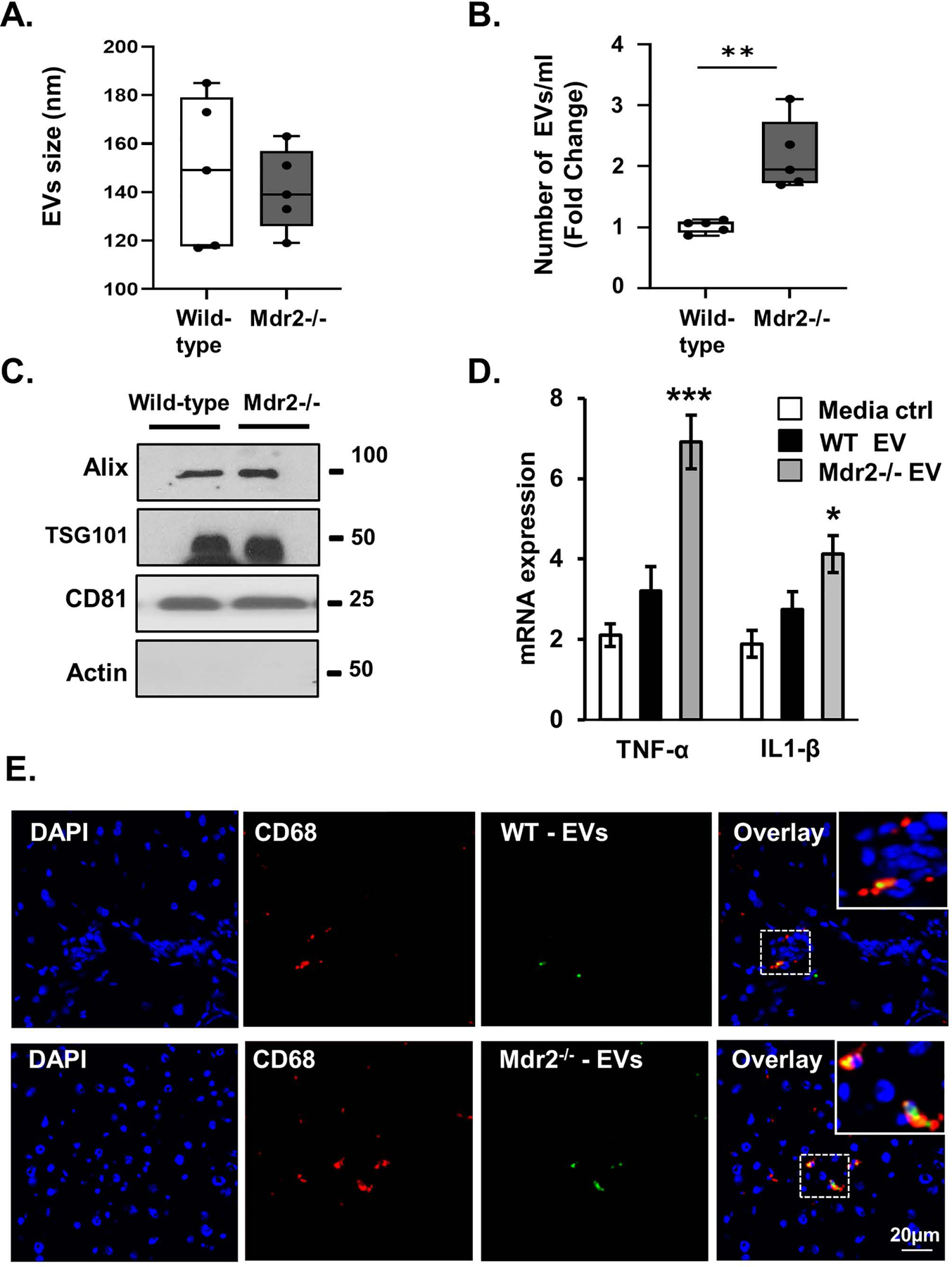
A and B, Characterization of wild-type C57BL/6J and Mdr2−/− mouse plasma-derived EVs, vesicle size, and number using nanoparticle tracking analysis (NTA) S300. The EVs isolated from wild-type and Mdr2−/− were similar size, yet more EVs were detected in the plasma of Mdr2−/− mice compared to wild-type mice. Data represent size and number from 5 mice per group. C, Western blotting analysis indicated that wild-type and Mdr2−/− mouse-derived EVs were positive for the exosome marker Alix, TSG101, and CD81. D, Equal numbers of wild-type and Mdr2−/− mouse-derived EVs (1e8/ml) were applied to a mouse monocyte cell line (RAW). Activation after 24-hours incubation was assessed by measuring monocyte messenger RNA (mRNA) expression levels (RT-qPCR) of tumor necrosis factor (Tnf) and interleukin 1 beta (Il1b). E, In vivo EV uptake assay reveals that Dio dye-labeled plasma-derived EVs colocalize with hepatic monocytes. EVs were collected from wild-type (WT) or Mdr2−/− mouse plasma and labeled with DiO dye. Immunofluorescence reveals colocalization of CD68 (red, monocytes marker) and plasma EVs (green, labeled with DiO dye) in liver tissue of WT (upper panel) and Mdr2−/− (lower panel) mice. Bars represent mean (±) standard error of the mean (SEM); n=3. *P<0.05, **P<0.01.
Discussion
The major findings described here relate to intercellular communication between senescent cholangiocytes and target cells via EVs likely enriched in exosomes and the relevance of this communication to PSC pathogenesis (summarized in Figure 8). More specifically, compared to EVs from nonsenescent cholangiocytes, EVs from patient-derived or experimentally induced senescent cholangiocytes are increased in number, contain different cargo, and have different functional effects on target cells. Our in vitro studies show that EVs from senescent cholangiocytes: i) exhibit increased content of growth factors, including EGFR ligands; ii) promote proliferation of bystander NHCs via EGFR activation and downstream NRAS/MAPK signaling; iii) induce cellular senescence of bystander NHCs; iv) promote migration and proliferation of MHCs via EGFR; and v) promote EGFR-dependent macrophage proinflammatory activation and EGFR-independent macrophage migration. Our in vivo studies reveal that Mdr2−/− mice exhibit increased circulating EVs (enriched in exosomes) compared to control C57BL/6J mice, suggesting that biliary damage associated with the Mdr2−/− phenotype promotes endocrine release of EVs. Moreover, these Mdr2−/− -derived EVs, but not EVs from C57BL/6J mice, promote proinflammatory activation of cultured mouse macrophages. Together these data support that EVs released from senescent cholangiocytes are an important component of the SASP, promote paracrine phenotypic alterations in nonsenescent normal and malignant cholangiocytes and endocrine mediated changes in macrophages via upregulated release of EGFR ligand enriched EVs, and are likely involved in peribiliary inflammation associated with PSC.
Figure 8. Working model of intercellular communication between senescent cholangiocytes and target cells.
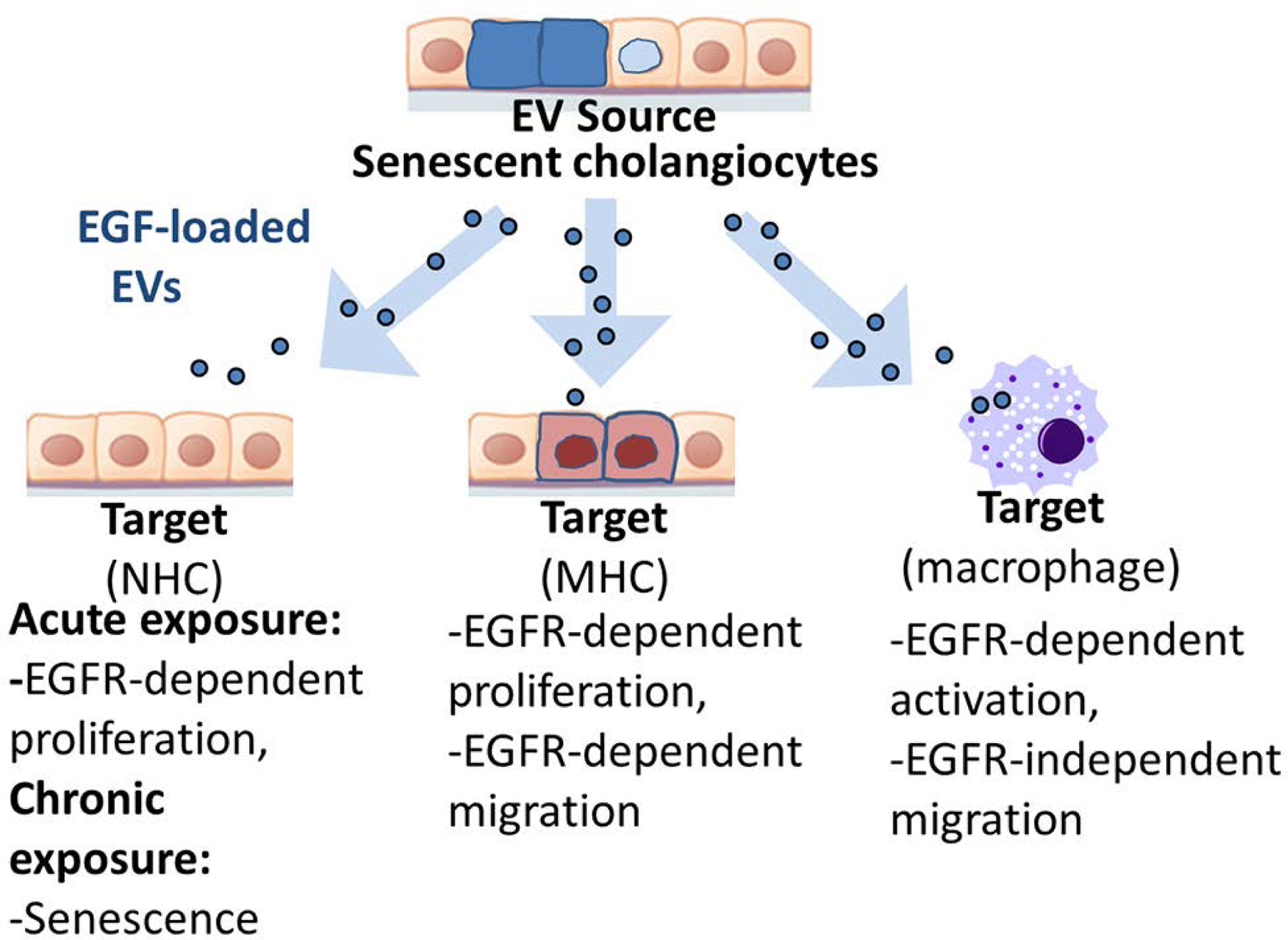
Senescent cholangiocytes release increased numbers of EVs that are likely enriched in exosomes. Compared to EVs from nonsenescent cholangiocytes, EVs from patient-derived or experimentally induced senescent cholangiocytes are increased in number, enriched in growth factors including epidermal growth factor receptor (EGFR) ligands, and have different functional effects on target cells.
EVs contain a variety of molecular components (noncoding RNA, protein, lipids), and can mediate physiologic and pathophysiologic processes in target cells both through molecules present on their surface and internal cargo delivered into the cytoplasm upon fusion with or endocytosis by target cells [33]. The molecular composition of EVs can modulate signaling pathways of recipient cells resulting in proinflammatory responses [16], cell proliferation [34], cell migration [35], and cell regeneration and degeneration [36], often in a cell-specific manner. Senescent cells secrete robust amounts of intercellular signaling mediators through the SASP [37], and our data support the concept that EVs released from senescent cholangiocytes are an important component of the cholangiocyte SASP.
The SASP is a feature of senescent cells and has been shown to cause detrimental consequences in multiple chronic diseases by communication with cells locally (ie, autocrine and paracrine) and remotely (ie, endocrine) (12). We have shown that cellular senescence and SASP are prominent features of cholangiocytes from patients with PSC, and SASP is increasingly thought to play a pathophysiologic role in biliary disease [6, 38]. In work described here, we observed that senescent cholangiocytes, either derived from PSC patient livers or experimentally-induced to senescence by LPS and other cellular stressors (eg, H2O2 and irradiation), secrete increased numbers of EVs compared to normal, nonsenescent human cholangiocytes. In addition, these EVs were enriched in EVs with the size and markers consistent with exosomes, suggesting that exosomes are a prominent component of the EVs released from senescent cholangiocytes. These data are consistent with reports using other cell types that under acute or chronic stress [15, 16, 39], or those induced to senescence [40, 41], cells release increased quantities of EVs. Furthermore, we examined the composition of senescent cholangiocyte EVs, and found that they were enriched in growth factors including amphiregulin, EGF, and IGFBP4. Our findings are in line with published proteomics analyses of the SASP from senescent cells, showing enrichment in growth factors (ie, PDGF, IGFBP4) [42, 43]. Importantly, our data extend these studies by focusing on the functional consequences of the interaction of senescent cell derived EVs on potential target cells.
EVs from senescent cholangiocytes induced proliferation of both bystander normal and MHCs through EV-mediated EGFR/MAPK signaling. The proliferative influence on nonmalignant cholangiocytes may have implications in proliferative aspects of ductular reactive cholangiocytes and wound healing. Moreover, senescent cholangiocyte derived EVs promoted migration of malignant cholangiocytes, suggesting that EVs may influence the metastatic potential of these cells. In addition to the proliferative and migratory effects of senescent EVs on cholangiocytes, we previously demonstrated the importance of NRAS and ERK activation on persistent LPS-induced cultured NHC senescence [6]. Here we demonstrate that persistent treatment of cultured NHCs with EVs derived from senescent cholangiocytes promotes senescence in NHC, implicating a role of senescent cell derived EVs in the paracrine induction of senescence. Hence, acute exposure of nonsenescent cholangiocytes to senescent cholangiocyte-derived EVs promotes a proliferative response (ie, cholangiocyte reactivity), while chronic, long-term exposure induces a senescent cholangiocyte phenotype. These results are similar to what we have observed in our in vitro model of LPS-induced NHC senescence [6].
In a previous study, we demonstrated that macrophages accumulate around bile ducts in PSC-patients and the Mdr2−/− murine model of PSC [30]. Hence, we asked whether senescent cholangiocyte-derived EVs influenced macrophage activation or migration. We first observed that EVs derived from senescent cholangiocytes induced both activation and migration of human monocytes. We observed that activation was mediated by EGFR activation, which is consistent with the described role of EGFR signaling pathway in macrophage activation [44]. In contrast, EVs from each of the senescent cholangiocyte populations, but not those derived from nonsenescent NHCs, promoted THP-1 migration in an EGFR-independent manner, a process that may involve other components of the senescent cholangiocyte SASP present in these EVs. We extended these findings to explore the role of EVs in the Mdr2−/− murine model of PSC, which exhibits an increased number of senescent cholangiocytes [32]. We detected increased EVs in Mdr2−/− plasma, and these EVs induced activation of mouse monocytes in our in vitro system. It should be noted, however, that this method does not identify the source of the upregulated EVs. We propose that this increase is at least in part due to the increased secretion of EVs from senescent cholangiocytes associated with the Mdr2−/− model. Recent reports also described both detrimental and beneficial effects of exosomes in the Mdr2−/− murine model. Indeed, it was demonstrated that cholangiocyte-derived exosomes mediate transfer of a long noncoding RNA, H19, into hepatocytes and promote cholestatic injury [45], while stem cell derived-EVs reduced ductular reaction and fibrosis [46]. These data are consistent with EVs playing a central role in intercellular signaling and highlight that the source of the EVs influences outcomes in target cells.
Together, our data support that EVs are an essential component of the cholangiocyte SASP. Our findings also suggest that EVs from senescent cholangiocytes may influence both the proliferative cholangiocyte niche (ie, ductular reactive), at least in part through EGFR activation, as well as promote the nonproliferative, senescent niche, both of which are implicated in the pathogenesis of cholestatic disease [5]. We further demonstrate that target malignant cholangiocyte proliferation and migration are enhanced by senescent cholangiocyte EVs, again through an EGFR-dependent manner. These data suggest an integral role of senescent EV-associated EGF ligands in influencing target cholangiocyte phenotypes. Finally, our data support a role for senescent cell-derived EVs in modifying the local peribiliary microenvironment through the recruitment of myeloid lineage cells (eg, macrophages). Hence, our results provide further insight into the pathogenesis of PSC and provide direction for targeted therapeutics aimed at SASP in general and EVs in particular in cholangiocyte and target cell communication. Ongoing studies aim to further clarify senescent cholangiocyte EV composition, and define the mechanisms regulating upregulated EV release.
Supplementary Material
Figure 2S. EVs-derived from senescent cholangiocytes induce EGFR-dependent proliferation of target cells. A, Celigo Imaging Cytometer; equal number of normal and senescent cholangiocytes derived EVs were applied to NHCs (+/− erlotinib). Cellular growth was measured by calculating percent confluency of the cultured cells after 24 hours incubation. The observed increase in proliferation in NHCs treated with senescent derived EVs was blunted by Erlotinib. B, and C, MTS proliferation assay. Equal number of normal and senescent cholangiocyte-derived EVs were applied to NHCs (B) and MHCs (C, Hucct1) (+/− erlotinib). Cellular proliferation was measured after 24 hours incubation. Bars represent mean (±) standard error of the mean (SEM); n=3–5 biological replicates, *p<0.05, **p<0.01.
Figure 3S. EVs-derived from senescent cholangiocytes induced migration of malignant human cholangiocytes. A. Wound healing assay. Equal number of EVs isolated from NHCs, LPS-induced, and PSC-derived were applied to Hucct1 (+/− erlotinib). The rate of migration was measured after 8 hour time point. B. Quantification of wound healing assay. Data presented as a migration rate (μm/8 hours). Bars represent migration rate (±) standard error of the mean (SEM); n=3–5 biological replicates, *p<0.01. C. Immunoblotting analysis shows positive expression of EGFR in Hucct1, LX-2, and THP-1 cell lines.
Figure 1S. LPS-induced and PSC patient-derived cholangiocytes express increased markers of cellular senescence. A. Immunofluorescence of Cytokeratin 19 CK19 (red) and p21 (green) in untreated NHCs, LPS-induced senescent cholangiocytes, and PSC patient-derived cholangiocytes. B. Senescence-associated β-galactosidase activity (SA- β-gal) assay. Increased SA- β-gal activity indicated by blue color. C. Quantification of the SA-β-gal assay. Data are presented as percentage of SA-β-gal positive cells per total cell count. D. Immunoblotting analysis indicate that senescent cholangiocytes (LPS-induced, and PSC-derived) have increased p16 expression. E. Quantification of p16 protein expression. Data are presented as fold change (p16/actin). Bars represent mean (±) standard error of the mean (SEM); n=3. *p<0.05.
Table 1S. Upregulated genes from PSC patient-derived cell lines identified in the top 100 extracellular vesicle markers collated by Vesiclepedia (microvesicles.org). Each column shows genes that are upregulated in each of the PSC patient-derived cholangiocyte cell isolates (PSC-1, PSC-2, and PSC-3) that were also in the top 100 proteins found in EVs.
Acknowledgments:
We thank Dr. Brian A. Davies for his insights and input. We also acknowledge Debbie Hintz for assistance in preparation of the manuscript.
Financial Support: This work was supported by National Institutes of Health Grant DK57993 (to N.F.L), the Mayo Foundation, PSC Partners Seeking a Cure, the Clinical and Optical Microscopy Cores of the Mayo Clinic Center for Cell Signaling in Gastroenterology (P30DK084567) and the Chris M. Carlos and Catharine Nicole Jockisch Carlos Endowment Fund in Primary Sclerosing Cholangitis (PSC).
Abbreviations
- CCA
cholangiocarcinoma
- DMEM
Dulbecco’s modified Eagle’s medium
- EGFR
epidermal growth factor receptor
- ERK
extracellular signal-regulated kinase
- EVs
extracellular vesicles
- FBS
fetal bovine serum
- H2O2
hydrogen peroxide
- IACUC
Institutional Animal Care and Use Committee
- IF
immunofluorescence
- IL
interleukin
- IR
irradiation
- LPS
lipopolysaccharide
- LT
liver transplantation
- MAPK
mitogen-activated protein kinase
- MHC
malignant human cholangiocytes
- mRNA
messenger RNA
- MVB
multivesicular body
- NHC
normal human cholangiocyte
- NTA
nanoparticle tracking analysis
- PBC
primary biliary cirrhosis
- PBS
phosphate buffered saline
- PCR
polymerase chain reaction
- PSC
primary sclerosing cholangitis
- RNA-seq
RNA sequencing
- SASP
senescence-associated secretory phenotype
- SA-β-gal
senescence-associated β-gal
- TLR
toll-like receptor
- THP-1
Tohoku Hospital Pediatrics-1
- TNF
tumor necrosis factor
- TSG101
tumor susceptibility gene 101
Footnotes
Conflict of interest: The authors have no conflict of interest related to the manuscript.
REFERENCES
- [1].Lazaridis KN, LaRusso NF. Primary Sclerosing Cholangitis. N Engl J Med 2016; 375(12): 1161–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Bashir MR, Muir AJ. Great Expectations - Can Magnetic Resonance Elastography Accelerate Progress in Primary Sclerosing Cholangitis Research? Clin Gastroenterol Hepatol 2019. [DOI] [PubMed] [Google Scholar]
- [3].O’Hara SP, Tabibian JH, Splinter PL, LaRusso NF. The dynamic biliary epithelia: molecules, pathways, and disease. J Hepatol 2013; 58(3): 575–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].O’Hara SP, Karlsen TH, LaRusso NF. Cholangiocytes and the environment in primary sclerosing cholangitis: where is the link? Gut 2017; 66(11): 1873–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Guicciardi ME, Trussoni CE, LaRusso NF, Gores GJ. The Spectrum of Reactive Cholangiocytes in Primary Sclerosing Cholangitis. Hepatology 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Tabibian JH, O’Hara SP, Splinter PL, Trussoni CE, LaRusso NF. Cholangiocyte senescence by way of N-ras activation is a characteristic of primary sclerosing cholangitis. Hepatology 2014; 59(6): 2263–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Moncsek A, Al-Suraih MS, Trussoni CE, O’Hara SP, Splinter PL, Zuber C, et al. Targeting senescent cholangiocytes and activated fibroblasts with B-cell lymphoma-extra large inhibitors ameliorates fibrosis in multidrug resistance 2 gene knockout (Mdr2(−/−)) mice. Hepatology 2018; 67(1): 247–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Campisi J, D’adda Di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 2007; 8(9): 729–40. [DOI] [PubMed] [Google Scholar]
- [9].Childs BG, Durik M, Baker DJ, Van Deursen JM. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med 2015; 21(12): 1424–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Tchkonia T, Morbeck DE, Von Zglinicki T, Van Deursen J, Lustgarten J, Scrable H, et al. Fat tissue, aging, and cellular senescence. Aging Cell 2010; 9(5): 667–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Tchkonia T, Zhu Y, Van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest 2013; 123(3): 966–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. The Journal of cell biology 2013; 200(4): 373–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Chevillet JR, Kang Q, Ruf IK, Briggs HA, Vojtech LN, Hughes SM, et al. Quantitative and stoichiometric analysis of the microRNA content of exosomes. Proceedings of the National Academy of Sciences of the United States of America 2014; 111(41): 14888–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Huang-Doran I, Zhang CY, Vidal-Puig A. Extracellular Vesicles: Novel Mediators of Cell Communication In Metabolic Disease. Trends in endocrinology and metabolism: TEM 2016. [DOI] [PubMed] [Google Scholar]
- [15].Ibrahim SH, Hirsova P, Tomita K, Bronk SF, Werneburg NW, Harrison SA, et al. Mixed lineage kinase 3 mediates release of C-X-C motif ligand 10-bearing chemotactic extracellular vesicles from lipotoxic hepatocytes. Hepatology 2016; 63(3): 731–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hirsova P, Ibrahim SH, Krishnan A, Verma VK, Bronk SF, Werneburg NW, et al. Lipid-Induced Signaling Causes Release of Inflammatory Extracellular Vesicles From Hepatocytes. Gastroenterology 2016; 150(4): 956–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bala S, Petrasek J, Mundkur S, Catalano D, Levin I, Ward J, et al. Circulating microRNAs in exosomes indicate hepatocyte injury and inflammation in alcoholic, drug-induced, and inflammatory liver diseases. Hepatology 2012; 56(5): 1946–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Li L, Masica D, Ishida M, Tomuleasa C, Umegaki S, Kalloo AN, et al. Human bile contains microRNA-laden extracellular vesicles that can be used for cholangiocarcinoma diagnosis. Hepatology 2014; 60(3): 896–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kadota T, Fujita Y, Yoshioka Y, Araya J, Kuwano K, Ochiya T. Emerging role of extracellular vesicles as a senescence-associated secretory phenotype: Insights into the pathophysiology of lung diseases. Mol Aspects Med 2018; 60: 92–103. [DOI] [PubMed] [Google Scholar]
- [20].Joplin R, Strain AJ, Neuberger JM. Immuno-isolation and culture of biliary epithelial cells from normal human liver. In Vitro Cell Dev Biol 1989; 25(12): 1189–92. [DOI] [PubMed] [Google Scholar]
- [21].Tabibian JH, Trussoni CE, O’Hara SP, Splinter PL, Heimbach JK, LaRusso NF. Characterization of cultured cholangiocytes isolated from livers of patients with primary sclerosing cholangitis. Lab Invest 2014; 94(10): 1126–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Thery C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol 2006; Chapter 3: Unit 3 22. [DOI] [PubMed] [Google Scholar]
- [23].O’Hara SP, Splinter PL, Trussoni CE, Gajdos GB, Lineswala PN, LaRusso NF. Cholangiocyte N-Ras protein mediates lipopolysaccharide-induced interleukin 6 secretion and proliferation. J Biol Chem 2011; 286(35): 30352–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Atkin-Smith GK, Tixeira R, Paone S, Mathivanan S, Collins C, Liem M, et al. A novel mechanism of generating extracellular vesicles during apoptosis via a beads-on-a-string membrane structure. Nat Commun 2015; 6: 7439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Pathan M, Fonseka P, Chitti SV, Kang T, Sanwlani R, Van Deun J, et al. Vesiclepedia 2019: a compendium of RNA, proteins, lipids and metabolites in extracellular vesicles. Nucleic Acids Res 2019; 47(D1): D516–D19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Trussoni CE, Tabibian JH, Splinter PL, O’Hara SP. Lipopolysaccharide (LPS)-Induced Biliary Epithelial Cell NRas Activation Requires Epidermal Growth Factor Receptor (EGFR). PLoS One 2015; 10(4): e0125793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Boonstra K, Weersma RK, Van Erpecum KJ, Rauws EA, Spanier BW, Poen AC, et al. Population-based epidemiology, malignancy risk, and outcome of primary sclerosing cholangitis. Hepatology 2013; 58(6): 2045–55. [DOI] [PubMed] [Google Scholar]
- [28].Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc 2007; 2(2): 329–33. [DOI] [PubMed] [Google Scholar]
- [29].Cameron RG, Blendis LM, Neuman MG. Accumulation of macrophages in primary sclerosing cholangitis. Clin Biochem 2001; 34(3): 195–201. [DOI] [PubMed] [Google Scholar]
- [30].Guicciardi ME, Trussoni CE, Krishnan A, Bronk SF, Lorenzo Pisarello MJ, O’Hara SP, et al. Macrophages contribute to the pathogenesis of sclerosing cholangitis in mice. J Hepatol 2018; 69(3): 676–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Mederacke I, Hsu CC, Troeger JS, Huebener P, Mu X, Dapito DH, et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun 2013; 4: 2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Tabibian JH, O’Hara SP, Trussoni CE, Tietz PS, Splinter PL, Mounajjed T, et al. Absence of the intestinal microbiota exacerbates hepatobiliary disease in a murine model of primary sclerosing cholangitis. Hepatology 2016; 63(1): 185–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Gangoda L, Boukouris S, Liem M, Kalra H, Mathivanan S. Extracellular vesicles including exosomes are mediators of signal transduction: are they protective or pathogenic? Proteomics 2015; 15(2–3): 260–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kim HK, Song KS, Chung JH, Lee KR, Lee SN. Platelet microparticles induce angiogenesis in vitro. Br J Haematol 2004; 124(3): 376–84. [DOI] [PubMed] [Google Scholar]
- [35].Shabbir A, Cox A, Rodriguez-Menocal L, Salgado M, Van Badiavas E. Mesenchymal Stem Cell Exosomes Induce Proliferation and Migration of Normal and Chronic Wound Fibroblasts, and Enhance Angiogenesis In Vitro. Stem Cells Dev 2015; 24(14): 1635–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Maumus M, Jorgensen C, Noel D. Mesenchymal stem cells in regenerative medicine applied to rheumatic diseases: role of secretome and exosomes. Biochimie 2013; 95(12): 2229–34. [DOI] [PubMed] [Google Scholar]
- [37].Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 2010; 5: 99–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Zhou T, Wu N, Meng F, Venter J, Giang TK, Francis H, et al. Knockout of secretin receptor reduces biliary damage and liver fibrosis in Mdr2(−/−) mice by diminishing senescence of cholangiocytes. Lab Invest 2018; 98(11): 1449–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Vella LJ, Hill AF, Cheng L. Focus on Extracellular Vesicles: Exosomes and Their Role in Protein Trafficking and Biomarker Potential in Alzheimer’s and Parkinson’s Disease. Int J Mol Sci 2016; 17(2): 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Takasugi M, Okada R, Takahashi A, Virya Chen D, Watanabe S, Hara E. Small extracellular vesicles secreted from senescent cells promote cancer cell proliferation through EphA2. Nat Commun 2017; 8: 15729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lehmann BD, Paine MS, Brooks AM, Mccubrey JA, Renegar RH, Wang R, et al. Senescence-associated exosome release from human prostate cancer cells. Cancer Res 2008; 68(19): 7864–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell 2014; 31(6): 722–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Severino V, Alessio N, Farina A, Sandomenico A, Cipollaro M, Peluso G, et al. Insulin-like growth factor binding proteins 4 and 7 released by senescent cells promote premature senescence in mesenchymal stem cells. Cell Death Dis 2013; 4: e911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hardbower DM, Singh K, Asim M, Verriere TG, Olivares-Villagomez D, Barry DP, et al. EGFR regulates macrophage activation and function in bacterial infection. J Clin Invest 2016; 126(9): 3296–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Li X, Liu R, Huang Z, Gurley EC, Wang X, Wang J, et al. Cholangiocyte-derived exosomal long noncoding RNA H19 promotes cholestatic liver injury in mouse and humans. Hepatology 2018; 68(2): 599–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Mcdaniel K, Wu N, Zhou T, Huang L, Sato K, Venter J, et al. Amelioration of Ductular Reaction by Stem Cell Derived Extracellular Vesicles in MDR2 knockout mice via let-7 microRNA. Hepatology 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure 2S. EVs-derived from senescent cholangiocytes induce EGFR-dependent proliferation of target cells. A, Celigo Imaging Cytometer; equal number of normal and senescent cholangiocytes derived EVs were applied to NHCs (+/− erlotinib). Cellular growth was measured by calculating percent confluency of the cultured cells after 24 hours incubation. The observed increase in proliferation in NHCs treated with senescent derived EVs was blunted by Erlotinib. B, and C, MTS proliferation assay. Equal number of normal and senescent cholangiocyte-derived EVs were applied to NHCs (B) and MHCs (C, Hucct1) (+/− erlotinib). Cellular proliferation was measured after 24 hours incubation. Bars represent mean (±) standard error of the mean (SEM); n=3–5 biological replicates, *p<0.05, **p<0.01.
Figure 3S. EVs-derived from senescent cholangiocytes induced migration of malignant human cholangiocytes. A. Wound healing assay. Equal number of EVs isolated from NHCs, LPS-induced, and PSC-derived were applied to Hucct1 (+/− erlotinib). The rate of migration was measured after 8 hour time point. B. Quantification of wound healing assay. Data presented as a migration rate (μm/8 hours). Bars represent migration rate (±) standard error of the mean (SEM); n=3–5 biological replicates, *p<0.01. C. Immunoblotting analysis shows positive expression of EGFR in Hucct1, LX-2, and THP-1 cell lines.
Figure 1S. LPS-induced and PSC patient-derived cholangiocytes express increased markers of cellular senescence. A. Immunofluorescence of Cytokeratin 19 CK19 (red) and p21 (green) in untreated NHCs, LPS-induced senescent cholangiocytes, and PSC patient-derived cholangiocytes. B. Senescence-associated β-galactosidase activity (SA- β-gal) assay. Increased SA- β-gal activity indicated by blue color. C. Quantification of the SA-β-gal assay. Data are presented as percentage of SA-β-gal positive cells per total cell count. D. Immunoblotting analysis indicate that senescent cholangiocytes (LPS-induced, and PSC-derived) have increased p16 expression. E. Quantification of p16 protein expression. Data are presented as fold change (p16/actin). Bars represent mean (±) standard error of the mean (SEM); n=3. *p<0.05.
Table 1S. Upregulated genes from PSC patient-derived cell lines identified in the top 100 extracellular vesicle markers collated by Vesiclepedia (microvesicles.org). Each column shows genes that are upregulated in each of the PSC patient-derived cholangiocyte cell isolates (PSC-1, PSC-2, and PSC-3) that were also in the top 100 proteins found in EVs.