

Abstract
The aggressive primary brain tumor glioblastoma (GBM) is characterized by aberrant metabolism that fuels its malignant phenotype. Diverse genetic subtypes of malignant glioma are sensitive to selective inhibition of the NAD+ salvage pathway enzyme nicotinamide phosphoribosyltransferase (NAMPT). However, the potential impact of NAD+ depletion on the brain tumor microenvironment has not been elaborated. In addition, systemic toxicity of NAMPT inhibition remains a significant concern. Here we show that microparticle-mediated intratumoral delivery of NAMPT inhibitor GMX1778 induces specific immunological changes in the tumor microenvironment of murine GBM, characterized by upregulation of immune checkpoint PD-L1, recruitment of CD3+, CD4+, and CD8+ T cells, and reduction of M2-polarized immunosuppressive macrophages. NAD+ depletion and autophagy induced by NAMPT inhibitors mediated the upregulation of PD-L1 transcripts and cell surface protein levels in GBM cells. NAMPT inhibitor modulation of the tumor immune microenvironment was therefore combined with PD-1 checkpoint blockade in vivo, significantly increasing the survival of GBM bearing animals. Thus, the therapeutic impacts of NAMPT inhibition extended beyond neoplastic cells, shaping surrounding immune effectors. Microparticle delivery and release of NAMPT inhibitor at the tumor site offers a safe and robust means to alter an immune tumor microenvironment that could potentiate checkpoint immunotherapy for glioblastoma.
Introduction
Dysregulation of metabolism is a hallmark of cancer and is critical for driving the pathogenesis of malignant neoplasia (1, 2). Nicotinamide adenine dinucleotide (NAD+) is an essential co-factor involved in cellular redox reactions (e.g., GAPDH converting NAD+ to NADH), as well as a substrate of NAD+-dependent enzymes such as poly(ADP-ribose)polymerases (PARP) and sirtuins (3). Unlike normal cells, neoplastic cells are dependent on specific NAD+ biosynthesis pathways, and determining their NAD+ metabolic dependencies offers opportunities for cancer therapy (4, 5). We and others have shown that primary brain gliomas from diverse genetic sub-types, genetically characterized by IDH1 R132H or PPM1D mutations and Myc family gene amplification, selectively rely on the salvage NAD+ synthetic pathway, due to epigenetic silencing of NAPRT and glycolytic dependency, respectively (6–8). Accordingly, these gliomas are vulnerable to NAD+ depletion caused by pharmacological inhibition of nicotinamide phosphoribosyltransferase (NAMPT) that metabolizes nicotinamide to nicotinamide mononucleotide (NMN), serving as the rate-limiting enzyme of the NAD+ salvage pathway (6, 7). High NAMPT expression is associated with poorer prognosis in patients with glioblastoma (GBM), the most malignant form of gliomas, and a NAMPT-dependent cellular NAD+ pool is required for an E2F2-driven transcriptional program that maintains the phenotypes of GBM cancer stem cells (9). These findings support the clinical development of NAMPT inhibitors as NAD+-directed treatments for malignant gliomas.
Notably, however, the potential impact of NAMPT inhibition is not limited to neoplastic cells, but also encompasses the surrounding normal cells within a tumor microenvironment. Mounting evidence in the literature indicates a role of NAMPT in modulation of the immune system. Physiologically, increased levels of NAMPT have been well documented in a variety of inflammatory disorders (10), and immune cells have been shown to upregulate NAMPT in response to inflammatory stimuli such as lipopolysaccharide (LPS) (11). A recent study reported that LPS-triggered mitochondrial ROS production in macrophages induced a DNA damage response and NAD+ consumption by PARP, leading to the activation of the NAD+ salvage pathway and inflammatory responses (12). Despite increasing knowledge of NAD+ metabolism in immune cells and inflammation, it remains poorly understood how NAMPT inhibition may alter immunological aspects of the tumor microenvironment. This topic could have substantial relevance for GBM, because while it is heavily infiltrated with immune cells that can represent up to 50% of the tumor cellular volume (13), those cells typically suppress anti-tumor immunity (14). In GBM, tumor-associated macrophages (TAM) with an anti-inflammatory M2 phenotype derived from myeloid-derived suppressor cells (MDSCs) dominate tumor infiltrating immune cells, while T cells with effector activity are sparse. Perhaps reflecting this challenging situation, a randomized clinical trial investigating the immune checkpoint inhibitor anti-PD-1 antibody nivolumab in recurrent GBM did not yield improvements in progression-free or overall survival in patients (15). A better understanding of the immunological consequences of NAMPT inhibition in GBM could identify approaches for therapeutic application of these agents.
Importantly, the clinical development of NAMPT inhibitors for cancer therapy has been hampered by significant adverse events following systemic administration that occur beyond the immediate tumor microenvironment, from normal cell toxicities in organs such as retina, bone marrow, heart and liver (16). Both clinically and preclinically, this has resulted in adverse events that manifest as thrombocytopenia, cardiotoxicity, retinal damage, and cholangitis (16, 17). To overcome this problem, we have previously shown the utility of polymer microparticles (MP) loaded with NAMPT inhibitors that are directly applied to the tumor site in the brain, allowing for local, controlled, and sustained release of the compound in situ (17). The primary goal of the current report is to determine the impact of MP-delivered NAMPT inhibitor on the surrounding tumor microenvironment in a murine model of intracerebral GBM. We discovered distinct alterations in immunological states that are induced by local treatment with NAMPT inhibitor, and exploit this knowledge to provision a new combination immunotherapy strategy for the treatment of GBM.
Materials and Methods
Cells
Human sphere-forming glioma stem-like cells (MGG4, MGG18, MGG23, MGG119, MGG152) that were isolated from patient specimens (6, 7, 18–20) were authenticated by confirming patient-specific genetic alterations by focused sequencing. They were cultured in EF20 medium composed of Neurobasal medium (Thermo Fisher Gibco) supplemented with 3 mM L-Glutamine (Corning Mediatech), 1×B27 supplement (Thermo Fisher Gibco), 0.5×N2 supplement (Thermo Fisher Gibco), 2 μg/ml heparin (Sigma, St Louis, MO), 20 ng/ml recombinant human epidermal growth factor (R&D Systems, Minneapolis, MN), 20 ng/ml recombinant human fibroblast growth factor-2 (PeproTech, Rocky Hill, NJ), and 0.5×penicillin G/streptomycin sulfate/amphotericin B complex (Corning Mediatech) at 37 °C and 5% CO2. To passage cells, neurospheres were dissociated with the NeuroCult Chemical Dissociation Kit (Stem Cell Technologies). Murine GBM cells GL261, obtained from National Cancer Institute, and CT2A, a gift from Dr. Thomas Seyfried, were grown in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal calf serum (FCS, Hyclone). 005 murine GBM stem cells were provided by Dr. Inder Verma and cultured in EF20 medium (21–23). Normal human astrocytes (NHA) were obtained from Dr. E. Antonio Chiocca and grown in DMEM supplemented with 10% FCS. Cells were confirmed to be mycoplasma free (LookOut Mycoplasma kit, Sigma) and used under passage 15. Establishment of cell lines from patients were approved by the Institutional Review Board at MGH.
Synthesis and characterization of sustained release microparticles loaded with GMX1778
Polymeric MP loaded with GMX1778 were synthesized by a method similar to what was previously described (17). Briefly, 30–35 mg poly(lactide) (viscosity −0.55–0.75 dL/g in hexafluoroisopropanol) (Lactel Absorbable polymers, USA) were dissolved in 0.5 mL of chloroform. Five milligrams of GMX1778 were dissolved in 0.5 mL of a solvent mixture containing chloroform and methanol (volume ratio of chloroform to methanol was 4:1). The drug and polymer solutions were mixed and added to 15 mL 2.5%w/v poly(vinyl alcohol) in water. The mixture was homogenized using a Silverson homogenizer at 1500 RPM for 5 minutes. The resultant emulsion was transferred to a round bottom flask and the organic solvents were evaporated under reduced pressure using a rotary evaporator. Following removal of the organic solvent, the MP suspension was passed through a 70 μm mesh size filter. The MP were washed twice with 10%w/v Tween 20 solution and twice with deionized water using centrifugation (4000 RPM, 10 min, 4˚C). After the final wash, the MP were suspended in ~1 mL of deionized water and frozen to −80˚C, and lyophilized for 48 hours. MP were stored at −20˚C until further use. Control MP containing no drug were prepared using an identical method, but without the addition of drug.
To determine the loading of GMX1778 in the MP, MP were weighed and drug was extracted overnight with methanol. On the next day, the MP were separated by centrifugation (4000 RPM, 10 minutes), and the supernatant was collected. The supernatant was analyzed using high performance liquid chromatography-UV (HPLC-UV) analysis as described (17). For studies reported in this paper, we prepared two batches of MP, and the drug loading was observed to be 4.1 ± 0.2 %w/w (n = 6) and 5.4 ± 0.6 %w/w (n = 3), which are consistent with our previous work.
For some experiments, MP were co-loaded with GMX1778 and a hydrophobic fluorescent dye, coumarin6. Fluorescent MP were prepared as described above, except that coumarin6 (dissolved in chloroform at a concentration of 10 mg/mL, 0.5 mg) was added to the polymer solution prior to emulsification. Drug loading in fluorescent MP was found to be 3.5 ± 0.6 %w/w (n = 4). To measure the release of GMX1778 in media, MP were dispersed in DMEM media supplemented with 10% fetal bovine serum and 1% penicillin streptomycin (3 mg MPs/ml) and placed in an incubator shaker at 100 RPM and 37C. After 1 day, the dispersions were centrifuged to pellet the MP (4000 RPM, 10 min), and the supernatant was collected. The pellet was dispersed in 1 ml fresh media and the release was continued. On day 4, the dispersions were centrifuged and the supernatant was collected as described before. Equal volumes of supernatant from days 1 and 4 were mixed. One ml of the mixture was lyophilized, while the rest of the mixture was saved for a bioassay. GMX1778 was extracted from the lyophilized mixture with 1 ml methanol. Following extraction, the samples were centrifuged (15000 RPM, 10 min) and the supernatant was analyzed using HPLC. Concentrations obtained from HPLC analysis were used to determine dilutions for bioassays.
Flow cytometry analysis
Cells were collected, washed with PBS, concentration adjusted to 1×106 cells/mL in 100 μl PBS. DAPI was added (100 ng/ml) and Incubated for 5 min at room temperature to stain dead cells. Cells were washed with FACs buffer 3 times and blocked with FcR blocking for 20min at 4C. Antibodies were added at appropriate (1:100) dilution for 60 min at 4C, washed 3 times, and fixed with 4% PFA for 15 min and analyzed with BD LSRII flow cytometry and FlowJo software (BD).
Cell viability assay
Dissociated cells were plated in 96-well plates and treated the following day. Cell viability was measured after treatment by Cell titer-Glo or MTS assay (Promega), according to the manufacturer’s instructions. Results were analyzed in the Prism GraphPad software and IC50 was calculated from the dose–response curve (nonlinear curve fit).
Western blotting
Protein (10–15 μg/lane) was separated by 4–15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (BioRad) and transferred to polyvinylidene difluoride membranes (Bio-Rad) by electro blotting. Membranes were blocked with 5% nonfat skim milk for 1 h at room temperature and then incubated with primary antibodies at 4 °C overnight. Membranes were then washed in TBST (20 mM Tris pH7.5, 150 mM NaCl, 0.1% Tween20) and incubated with appropriate peroxidase-conjugated secondary antibodies (Promega and Cell Signaling Technology) for 1 h at room temperature. Signals were visualized with an ECL Kit (Amersham Bioscience or BioRad).
NAMPT inhibitor treatment of freshly isolated cells harvested from mouse glioblastoma
GL261 GBM tissue was dissected from the brain and cut into 1mm fragments. These fragments in DMEM were transferred to a conical tube, spun at 1100 rpm, and resuspended in Accutase and DNase I (10 U/ml; Promega). After incubation at 37°C for 10 minutes, tissue was triturated, passed through a 40 μm cell strainer to yield a single cell suspension. These cells were spun at 1500 rpm, and the supernatant aspirated. GL261 tumor-derived cells (1× 105) were plated on round cover-glasses placed in 24-well plates and treated with FK866 (100 nM) and GMX1778 (1000 nM) for 24, 48 or 72 h in triplicate. Cells were wash with PBS and fixed in 4% paraformaldehyde. After blocking in 2% BSA in PBS, cells were incubated with primary antibody (Nestin, Arg1, and CD11b) overnight at 4°C in a humidified chamber. After wash with PBS, cells were incubated with secondary antibodies Alexa-488 anti-mouse (1:250; Jackson ImmunoResearch) and Cy3 anti-rabbit (1:250; Jackson ImmunoResearch) for 1 h at room temperature, and mounted on slides with VectaShield (DAPI included, Vector Laboratories). Staining was imaged with a Nikon 90i microscope and quantified.
Immunofluorescence and immunohistochemistry
Paraffin-embedded tissue sections were de-paraffinized in xylene twice 10 min each, followed by gradual rehydration using 100%, 90% and 70% ethanol treatment (5 min each). The samples were heated by microwave in 10mM Na Citrate buffer for 15 min for antigen retrieval, incubated with blocking buffer for 1 hour, and incubated with diluted primary antibody overnight at 4C. After wash with PBS for 5 minutes three times, secondary antibody solution was added at room temperature for 1 hour, followed by wash with PBS three times. Anti-fade DAPI solution (10 ul per well) was applied and cover-glass mounted. For immunofluorescence of nestin or PD-L1 along with visualization of coumarin6 labeled MP, mice were perfused with 4% paraformaldehyde and frozen sections were prepared. Staining was done as described above from the blocking step. For immunohistochemistry, following antigen retrieval, endogenous peroxidase activity was quenched with 3% H2O2 incubation for 5 min. After blocking, slides were incubated with diluted primary antibody overnight at 4C. After PBS washes, ImmPRESS HRP anti-rabbit IgG polymers (Vector) was applied, followed by PBS washes. Signals were generated with DAB (Dako). Nuclei were counterstained with hematoxylin. Staining was imaged with a Nikon 90 microscope and quantified.
NAD+ quantitation
To evaluate qualitative values of NAD+, we used NADH, NAD/NADH-Glo Assay (Promega), according to the manufacturer’s recommendations. Briefly, 1×105 cells were lysed with 400 μl of PBS and then 400 μl of 0.2N NaOH containing 1% Dodecyltrimethylammonium bromide (DTAB) (Sigma-Aldrich) was added. To measure NAD+, 100 μl of 0.4N HCl were added to the lysed cell samples (200 μl) and then heated at 60C for 15 min. After 10 min incubation at room temperature, 0.5M Trizma base buffer (100 μl, Sigma-Aldrich) was added. To measure NADH, lysed cell samples (200 μl) were heated at 60C for 15 min, and then incubated at room temperature for 10 min, followed by adding equal volume of HCl/Trizma solution. Finally, samples were seeded into 96 well plates and incubated with NAD/NADH Glo detection reagent for 60 min. Data were compared to DMSO treated cells and expressed as % control.
Animal Study
Mice anesthetized with pentobarbital were fixed in the stereotactic head frame. A burr hole was drilled on the coronal suture at 2.3 mm lateral (right) from the Bregma. GL261 cells (100,000 cells in 3 μl PBS) were slowly injected into the brain at the depth of 2.5 mm using a Hamilton syringe. The bone wax was used to close the hole and the wound sutured. At day16, blank, GMX1778, coumarin6 or coumarin6 GMX1778 MP were freshly diluted in PBS to adjust the dose to 39 ng GMX1778 (for GMX1778 MP, and equivalent weight of blank MP) or 74 ng GMX1778 (for coumarin6-GMX1778 MP, and equivalent weight of coumarin6 MP, adjusted based on the rate of drug release) in 3 μl, vortexed and injected using a Hamilton syringe into the same location as the tumor cells were implanted. For survival studies, GL261 mice were treated with blank MP or GMX1778 MP (39 ng GMX1778 / 3 μl) at day 6 and anti-PD-1 antibody (10 mg/kg, rat IgG, Bio X Cell, BE0146) or isotype control IgG (10 mg/kg rat IgG) injected i.p. on days 8, 10 and 12. The number of mice was N=6 for the group receiving blank particle and control IgG, and N=7 for the other 3 groups. Mice were followed for neurological symptoms. For immune analysis, GL261 tumor-bearing mice were treated with blank particle or GMX1778 MP (39 ng / 3 μl) at day 14 and anti-PD-1 antibody (10 mg/kg) or isotype control IgG (10 mg/kg rat IgG) injected i.p. on days 16 and 18. Mice were killed on day 20 for preparing paraffin embedded block of the brains. All in vivo procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at MGH.
Quantitative RT-PCR.
GL261 cells were treated with FK866, GMX1778, 3-MA, Everolimus, combination treatment or control cells harvested for RNA extraction with Trizol (Invitrogen) according to the manufacturer’s protocol. cDNA was synthesized using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Quantitative PCR was performed with SYBR green PCR master mix (Applied Biosystems) in a real-time PCR machine (Step One Plus Real-Time PCR System, Applied Biosystems). Beta-actin (Actb) was used as a house-keeping gene control.
Statistical analysis
Experimental results in 2 groups were analyzed and compared using unpaired two-sided Student’s t test. In vivo survival was analyzed by Kaplan–Meier plot and Wilcoxon test (Prism; GraphPad). p< 0.05 was considered statistically significant.
Antibodies used in this study are listed in Supplemental Table 1.
Results
NAMPT inhibitor-induced NAD+ depletion upregulates PD-L1 on mouse and human glioblastoma cells.
We first confirmed that glioma cells genetically defined by mutant IDH1 and Myc family gene amplification are sensitive to pharmacological inhibition of NAMPT, as previously shown (6, 7)(Supplementary Fig. S1A, B). In more extended panel testing, we observed that murine GBM cell lines (IDH1 wild-type) were not very sensitive to NAMPT inhibitors, FK-866 and GMX1778, but responded, with FK-866 being more potent (Supplementary Fig. S1C, D).
We next tested in vitro whether NAMPT inhibitor-induced decrease of NAD+ caused changes in PD-L1 levels in GBM cells. Treatment of GL261 cells in vitro with NAMPT inhibitor FK-866 or GMX1778 decreased intracellular NAD+ levels in a dose dependent manner (Fig. 1A). This reduction of NAD+ was associated with a concomitant increase in cell surface PD-L1 levels (Fig. 1A). We confirmed that NAMPT inhibitor induced upregulation of cell surface PD-L1 on cells that were alive, while the anti-PD-L1 antibody non-specifically bound dead cells (Supplementary Fig. S2A–C). To determine whether NAD+ depletion caused the observed upregulation of PD-L1, we used NMN, the direct catalytic product of NAMPT, to bypass NAMPT inhibition. Supplementation of cultures with NMN rescued NAD+ depletion and reversed the effect of NAMPT inhibitors on PD-L1 (Fig. 1B), suggesting a causal relationship between NAD+ depletion and PD-L1 upregulation. We next tested whether NAMPT inhibitor-induced upregulation of PD-L1 occurred on transcript levels. RT-PCR analysis showed that FK-866 and GMX1778 increased PD-L1 (Cd274) mRNA levels in a time dependent manner in GL261 (Fig. 1C), which was accompanied by increases in the cell surface PD-L1 levels (Fig. 1C). We next confirmed that NAMPT inhibitors upregulated PD-L1 on human glioma cells. IDH1 wild-type (MGG23) and IDH1 R132H (MGG119) patient-derived GBM sphere cells displayed dose-dependent increases in the cell surface PD-L1 levels in response to treatment with FK-866 or GMX1778 (Fig. 1D, E). Both NAMPT inhibitors increased median fluorescence intensity in mouse and human GBM cells, suggesting an increase of cell surface PD-L1 levels per cell (Supplementary Fig. S3A–C). Together, these data show that NAMPT inhibitor-mediated depletion of intracellular NAD+ increases PD-L1 at transcript and protein levels in GBM.
Figure 1. NAMPT inhibitor-induced NAD+ depletion upregulates PD-L1 on mouse and human glioblastoma cells.
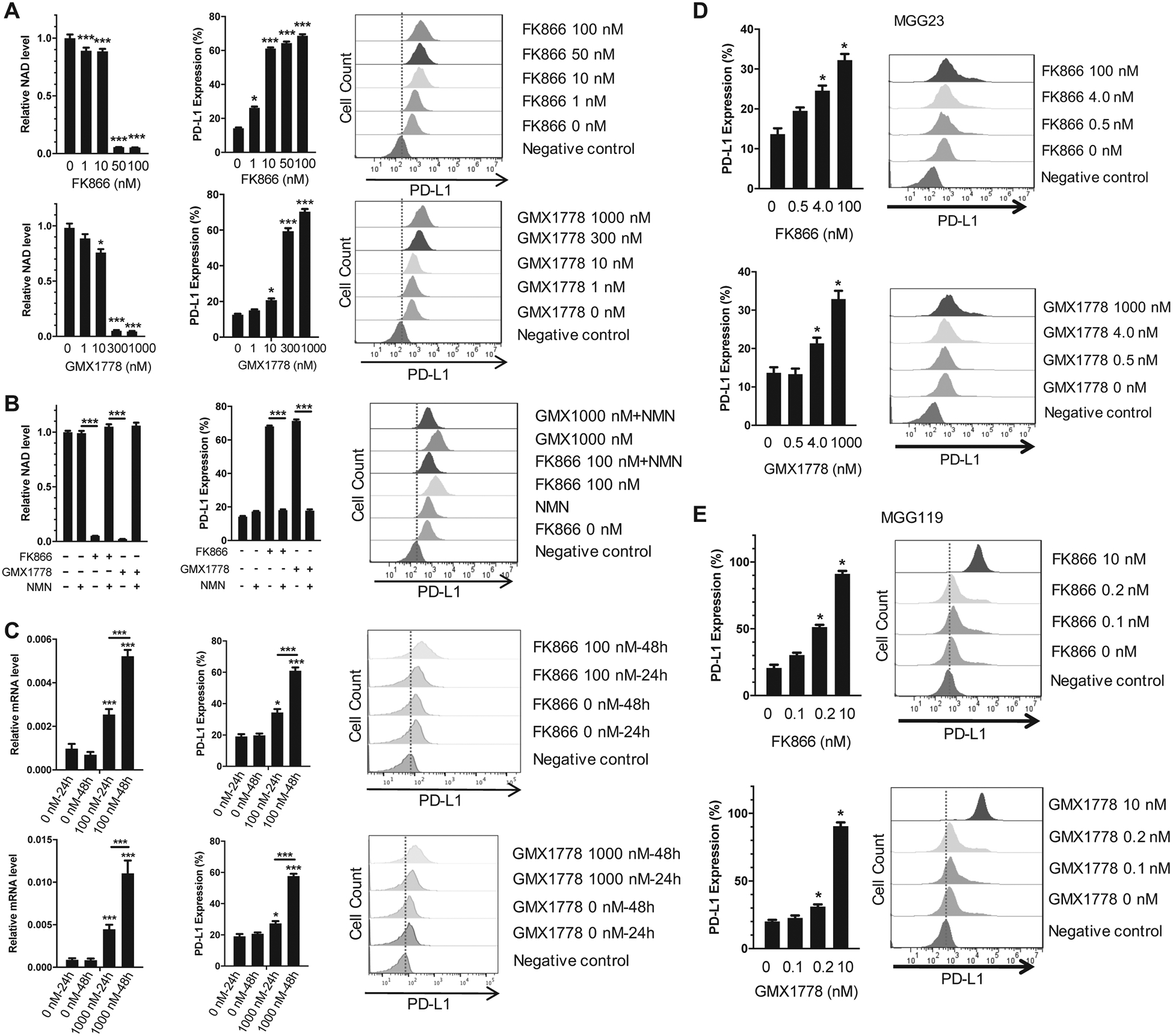
A, Left, NAD+ assay showing treatment with FK866 and GMX1778 for 72 h induced a decrease in NAD+ levels in GL261 cells. Middle and right, Flow cytometry analysis showing dose dependent upregulation of PD-L1 levels in NAMPT inhibitor-treated GL261 cells after 72 h. *p<0.05, ***p<0.01 for the difference between drug-treated and untreated cells. B, Nicotinamide mononucleotide (NMN, 1 mM) reverses NAD+ levels (Left) and PD-L1 levels (Middle and right) in GL261 cells treated with NAMPT inhibitors for 72 h. FK-866, 100 nM; GMX1778, 1000 nM. ***p<0.01 for the difference between indicated two groups. C, Left, quantitative RT-PCR showing upregulation of PD-L1 (Cd274) mRNA by FK866 (upper) and GMX1778 (lower) in a time dependent manner. Middle and right, Flow cytometry confirms corresponding PD-L1 upregulation. D and E, Dose-dependent upregulation of cell surface PD-L1 after 72-h exposure to NAMPT inhibitors FK866 (upper panels) and GMX1778 (lower panels) in MGG23 GBM (IDH wild-type) cells (D) and MGG119 GBM (IDH1-mutant) cells (E). *p<0.05, ***p<0.01 for the difference between drug-treated and untreated cells, and between two groups indicated. Error bars, SEM. Drug doses include IC10 (10% inhibitory concentration), IC50 and IC80 or IC90.
Local treatment with GMX1778 microparticles upregulates PD-L1 in glioblastoma in vivo.
Next, to characterize the immunological effect of NAMPT inhibitor treatment in intracerebral GBM in vivo, we tested MP delivery of the NAMPT inhibitor GMX1778 directly into the tumor, an approach that can circumvent adverse events caused by systemic treatment with NAMPT inhibitors (16, 17). Intratumoral injection of poly(lactide) MP co-loaded with the NAMPT inhibitor and green-fluorescent dye coumarin6 (coumarin6-GMX1778 MP) (Fig. 2A upper left) into established orthotopic GL261 GBM tumors resulted in wide-spread distribution (approximately 1.3mm in width) of the MP within the tumor, marked by nestin, in vivo (Fig. 2A). We next examined the immunological consequences in the tumor microenvironment following local treatment with GMX1778-containing MP in this model. Initially, we focused on potential changes in PD-L1 as one of the most clinically relevant immune checkpoints. Four days after intratumoral injection of GMX1778 MP, there was a significant increase in the number of PD-L1-positive cells within GL261 tumors as compared to two controls, PBS and blank MP not containing drug (Fig. 2B, C). Co-immunofluorescence of PD-L1 and glioma marker nestin or macrophage marker Arg1 showed that upregulation of PD-L1 occurred in both GBM cells and glioma-infiltrating macrophages (Fig. 2D, E). Thus, local MP delivery of NAMPT inhibitor induces upregulation of immune checkpoint PD-L1 in murine GBM. We next tested coumarin6-GMX1778 MP to examine if distribution of NAMPT inhibitor MP and that of PD-L1+ cells correlate in the tumor. In vitro, coumarin6-GMX1778 MPs did release GMX-1778, but to a lower degree compared with GMX-1778 MP (Supplementary Fig. S4A). GMX1778 released from coumarin6-GMX1778 MP and GMX1778 MP was comparably active in the cytotoxicity to GL261 cells (Supplementary Fig. S4B). In vivo, scattered fluorescent signals of coumarin6 were noted within GL261 GBM 4 days after intratumoral injections of coumarin6-GMX1778 MP or coumarin6 MP (Supplementary Fig. S4C). Coumarin6-GMX1778 MPs significantly induced tumor PD-L1 expression as compared with coumarin6 MP (Supplementary Fig. S4C, D), recapitulating the result with GMX1778 MP. There was no statistically significant difference in PD-L1 immuno-positivity between tumor areas with and without coumarin6 fluorescence signals (Supplementary Fig. S4C, D).
Figure 2. Local treatment with GMX1778 microparticles upregulates PD-L1 in glioblastoma in vivo.
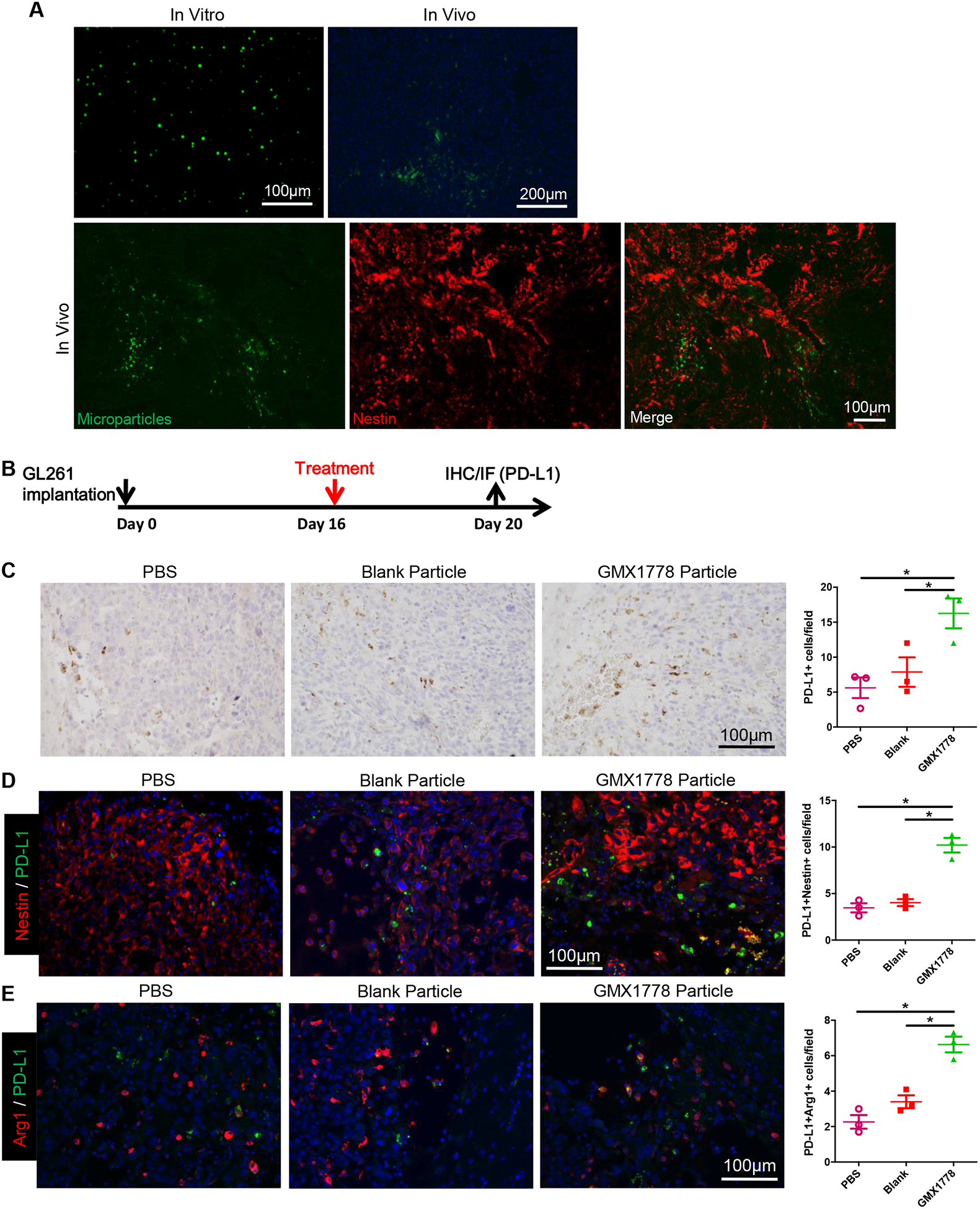
A, Three ul microliter of coumarin-6/GMX1778 loaded microparticles (MP) (Upper left, In vitro) were injected into intracranial GL261 tumors, and distribution of MP was observed under fluorescence microscopy (Upper right, In vivo). Lower panels, immunofluorescence for glioblastoma (GBM) marker nestin (red) revealed MP spread within the GL261 brain tumor. B, Mice bearing GL261 GBM were treated with intratumoral injections of PBS, blank MP or GMX1778 MP on day 16, and the brains were collected on day 20 (N=3/group). C, Representative microscopic pictures of immunohistochemistry for PD-L1, and quantification on the right. D and E, double immunofluorescence for PD-L1 (green) and Nestin (red) (D) and PD-L1 (green) and Arg1 (red) (E). Representative picture from each treatment group is presented. Number of double positive cells for each treatment group is plotted on the right (D, E). *, P<0.05.
NAMPT inhibitor-induced autophagy underlies an increase in PD-L1 mRNA and protein levels.
We previously reported that NAMPT inhibitor induced autophagy in IDH1 mutant human glioma cells (7). We therefore explored the potential mechanistic role of autophagy in NAMPT inhibitor-induced PD-L1 upregulation in the IDH1 wild-type GL261 cells. Western blot for the autophagy marker LC3 showed induction of autophagy in GL261 cells at 48 hours following exposure to FK866 and GMX1778 (Supplementary Fig. S5A). Co-treatment of GL261 cells with autophagy inhibitor, 3-methyladenine (3-MA), and FK-866 or GMX1778 reduced LC3-II generation and significantly abrogated the PD-L1 upregulation caused by NAMPT inhibition at both mRNA and protein levels (Supplementary Fig. S5A, B), suggesting a potential causal contribution of autophagy to this upregulation.
Local treatment with GMX1778 microparticles activates immune microenvironment in glioblastoma.
The increase of PD-L1-positive TAMs after treatment with GMX1778 MP (Fig. 2E) revealed the impact of NAD+ depletion on the tumor microenvironment. We further investigated immunological changes in the tumor microenvironment following intratumoral injection of GMX1778 MP into orthotopic GL261 GBM. We found that infiltration of CD3+, CD4+, and CD8+ T cells within the tumor was increased 4 days after injection of GMX1778 MP, compared to blank MP and PBS; however, GMX1778 MP lowered the number of cells labeled with the regulatory T cell (Treg) marker Foxp3 (Fig. 3A–D). CD68+ macrophages/microglia and iNOS-positivity that marks macrophages with an M1 (inflammatory and anti-tumoral) phenotype were increased by the treatment with NAMPT inhibitor-releasing MP (Fig. 3E, F). In contrast, Arg1-positive cells, representing macrophages of an M2 anti-inflammatory and pro-tumoral phenotype, were significantly decreased following GMX1778 MP injections into the tumor (Fig. 3G). Double staining of Arg1 and CD68 revealed that GMX1778 MP-treated tumors had an increase in Arg1−/CD68+ cells and a tendency of decreased Arg1+/CD68− cells (Supplementary Fig. S6).
Figure 3. Local treatment with GMX1778 microparticles activates immune microenvironment in glioblastoma.
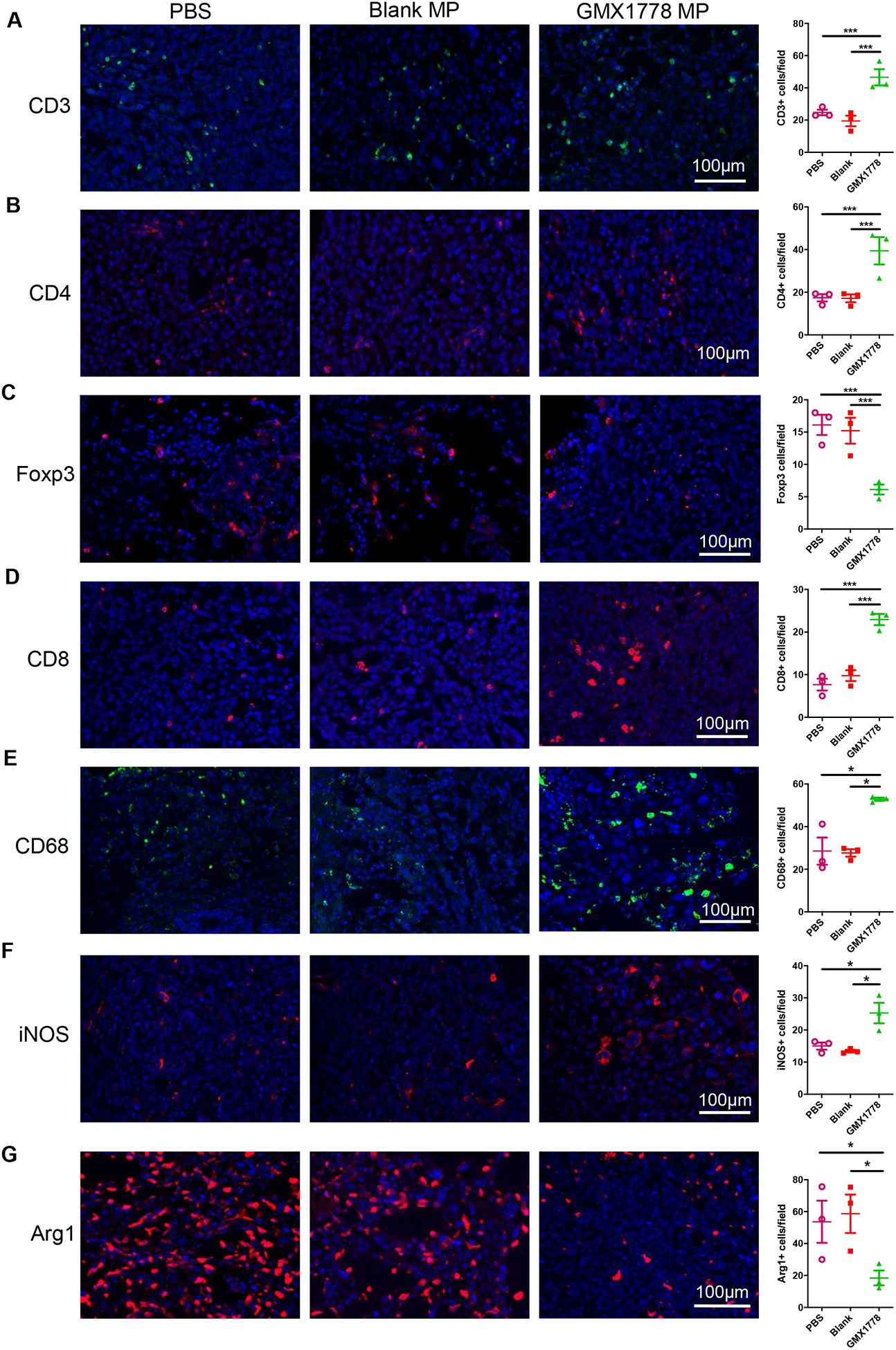
Immunofluorescence for CD3 (A), CD4 (B), Foxp3 (C), CD8 (D), CD68 (E), iNOS (F), and Arg1 (G) in orthotopic GL261 glioblastoma treated with PBS, blank microparticles (MP) and GMX1778 MP. Brains were removed 4 days after treatment of 16-day-old tumors. *, P<0.05; ***, P<0.01.
NAMPT inhibitor decreased glioblastoma-associated M2 macrophages in vitro.
We next examined the direct effect of NAMPT inhibitors on GBM-associated, M2-polarized macrophages. Acutely dissociated cells derived from freshly excised GL261 GBM were cultured and treated with NAMPT inhibitors. We used immunofluorescence to analyze changes in GBM cell marker nestin, macrophage marker CD11b, and M2 marker Arg1 over 3 days of culture. As expected, NAMPT inhibitors potently inhibited the growth of nestin-positive tumor cells (Fig. 4A, Supplementary Fig. S7A). Additionally, NAMPT inhibition resulted in a significant decrease in cells positive for Arg1 and CD11b (Fig. 4B, C, Supplementary Fig. S7B, C), which was accompanied by depletion of total cells stained with DAPI (Supplementary Fig. S7D). Thus, NAMPT inhibitor depleted GBM-associated, M2-polarized macrophages, in line with what was observed in vivo (Fig. 3G). Thus, notwithstanding its cell-autonomous effect on PD-L1 on tumor cells, local treatment with GMX1778 MP additionally had distinct influences on multiple components in the immune tumor microenvironment, overall modifying towards reversal of the immunosuppressive state in murine GBM.
Figure 4. NAMPT inhibitor is cytotoxic to glioblastoma-associated macrophages.
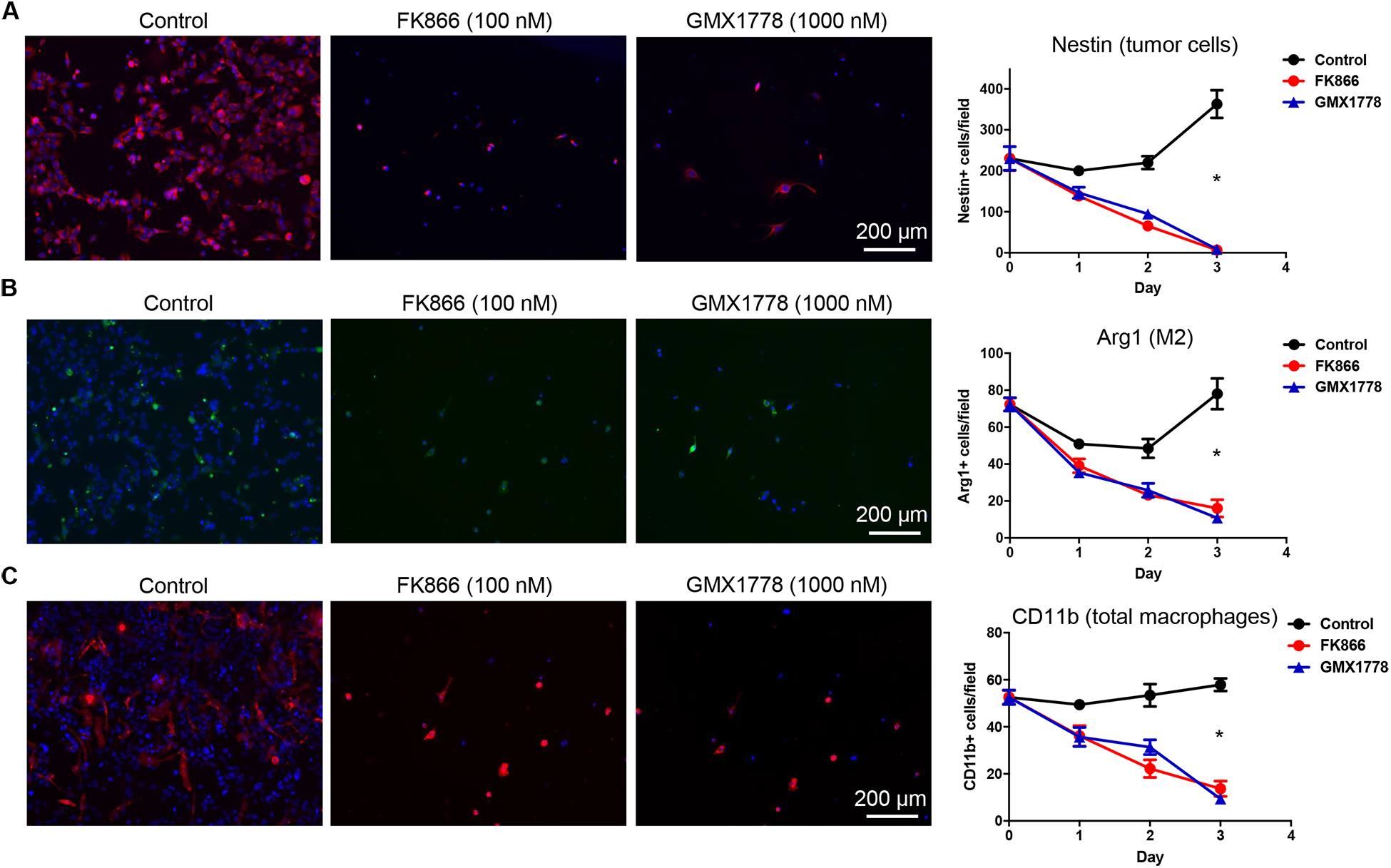
Freshly isolated cells from GL261 glioblastoma tissue were subjected to culture and untreated or treated with NAMPT inhibitors in vitro (FK866, 100 nM; GMX1778, 1000 nM). Cells were stained for nestin (A), Arg1 (B) and CD11b (C) at 24, 48 and 72 hours after treatment. Positive cells were quantified. Error bars, SEM. *, P<0.05. Here representative microscopic pictures on Day 3 (72 h) are shown. See Supplementary Fig. S7 for staining on Day 0, 1 and 2. *, P<0.005 (Control versus FK866 and Control versus GMX1778).
Combination therapy of GMX1778 microparticles and systemic anti-PD-1 enhances therapeutic efficacy.
Our observation of NAMPT inhibitor-mediated upregulation of PD-L1 in GBM offered a rationale for a therapeutic combination of local NAMPT inhibition and blockade of PD-L1/PD-1 axis. We tested a sequential treatment of local delivery of GMX1778 MP followed by systemic administration of anti-PD-1 (3 doses in 5 days), and compared this to each mono-therapies with GMX1778 MP or anti-PD-1 as well as to control (Fig. 5A). Kaplan-Meier analysis of animal survival showed that combination treatment of GMX1778 MP and anti-PD-1 significantly extended survival as compared to either mono-therapy or the control (Fig. 5B). One animal in the combination therapy survived 100 days and was confirmed tumor-free. Treatments were well-tolerated as there was no significant loss of body weight (Fig. 5C). Interestingly, upregulation of PD-L1 in the tumor induced by GMX1778 MP was no longer observed when GMX1778 MP was followed by anti-PD-1 immunotherapy (Fig. 5D, E, Supplementary Fig. S8). Immunofluorescence analysis revealed that combination therapy significantly increased tumor-infiltrating CD8+ T cells, not total CD3+ and CD4+ T cells, over GMX1778 MP monotherapy (Fig. 5F–H, Supplementary Fig. S9). There was no significant difference in the number of GranzymeB+ cells between the treatment groups (Supplementary Fig. S10). Combination therapy did not significantly alter the levels of CD68+, iNOS+, and Arg1+ TAM fractions that were altered by GMX1778 (Fig. 5I–K, Supplementary Fig. S9). Thus, combination therapy of GMX1778 MP and systemic anti-PD-1 enhances CD8+ T cell recruitment and therapeutic efficacy.
Figure 5. Combination therapy of GMX1778 microparticles and systemic anti-PD-1 enhances efficacy.
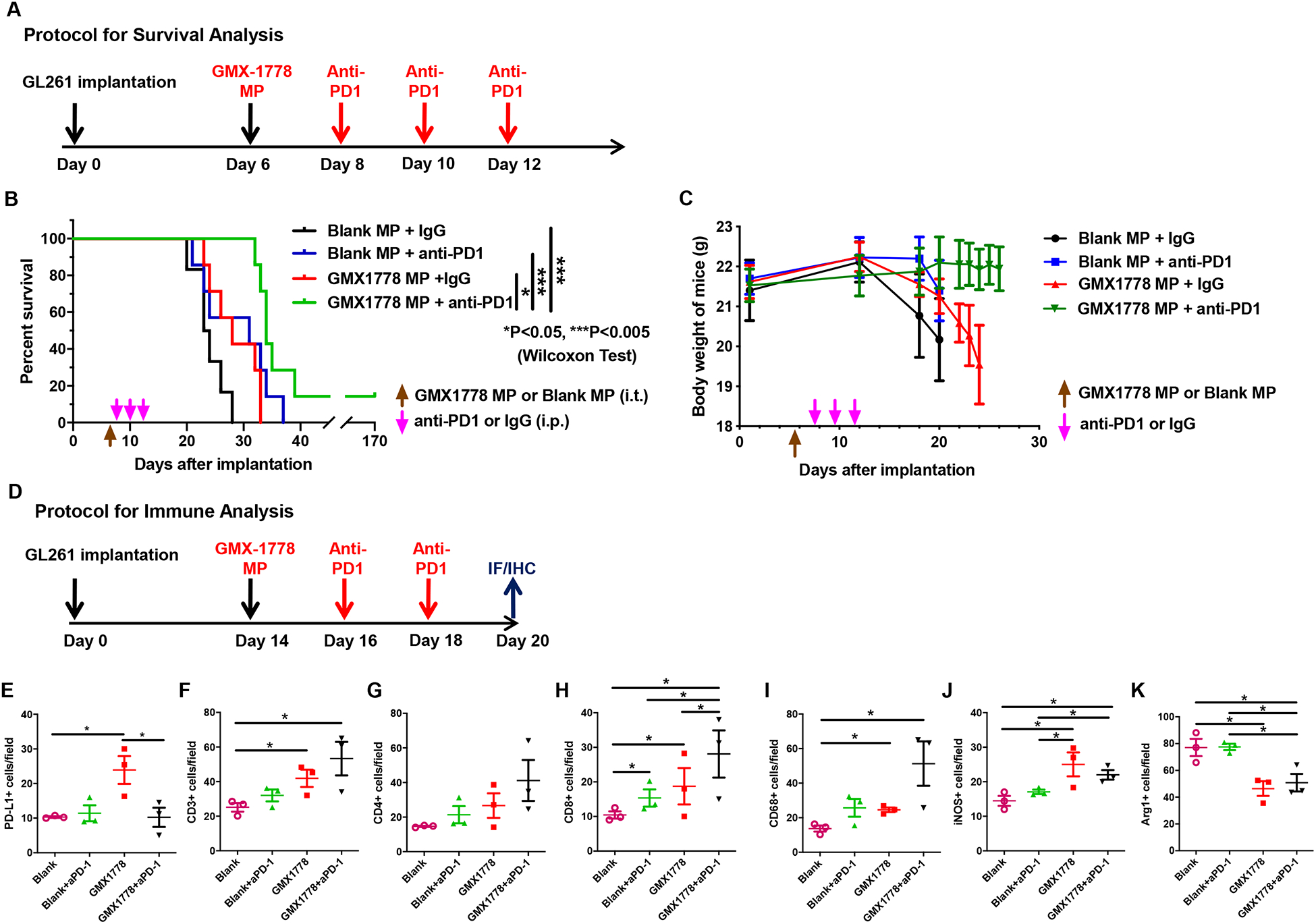
A, Experimental protocol for survival analysis. Microparticles, MP; anti-PD-1, aPD1. B, Kaplan-Meier survival plots of mice bearing orthotopic GL261 tumors treated with or without GMX1778 microparticles (intratumoral) and anti-PD-1 antibody (systemic). C, Changes in body weight of animals in the survival study. D, Experimental protocol for immune profiling analysis. E-K, Immunohistochemical (for PD-L1) or immunofluorescence (all others) analysis of a panel of immune markers in GL261 glioblastoma treated with or without GMX1778 microparticles (intratumoral) and anti-PD-1 antibody (systemic). E, PD-L1. F, CD3. G, CD4. H, CD8. I, CD68. J, iNOS. K, Arg1. Blank, Blank microparticles; GMX1778, GMX1778 microparticles; aPD-1, anti-PD-1. *, P<0.05. See data of PD-L1 immunohistochemistry in Supplemental Fig. S8 and immunofluorescence in Supplemental Fig. S9.
Discussion
In this work, we show that local therapy with NAMPT inhibitor MP dramatically alters the tumor immunologic microenvironment in GBM. Specifically, we identified: 1) upregulation of PD-L1 on tumor cells and macrophages; 2) Recruitment of CD3+, CD4+ and CD8+ T cells; and 3) M2 to M1 polarization of TAMs. We further show that these changes enable rational combination therapy of NAMPT inhibitor MPs with the blockade of PD-L1/PD-1 axis, which produced increased efficacy.
The upregulation of PD-L1 levels on the cell surface was cell autonomous and did not require cytokine production from immune cells as we showed a clear dose-dependent increase of PD-L1 levels across a cohort of human and murine GBM cells in vitro. A variety of molecular mechanisms have been described with which exogenous stimuli or oncogenic signaling regulate cellular PD-L1 levels (24–26). Our data demonstrate the importance of NAD+ depletion in the upregulation of PD-L1 in GBM cells, and increases in transcript and protein levels suggested transcriptional regulation. Our data also supports that NAMPT inhibitor-induced autophagy plays a role, but partial reversal with 3-MA suggests the existence of other mechanisms. Previously reported transcriptional factors that regulate the expression of PD-L1 include STATs, NF-kB, AP-1, and HIF1α (25, 26). Molecular mechanisms linking autophagy and PD-L1 need to be studied in future studies.
Intratumoral injections of GMX1778 MP altered the number and phenotype of immune cells that infiltrated the tumor in the brain. Both innate macrophages and adaptive T cells were impacted. Despite the increase in immune checkpoint PD-L1, overall changes in tumor-infiltrating immune cells were in the direction of reversal of immunosuppression as CD8+ T cells and iNOS+ macrophages increased and Arg1+ macrophages decreased. Our observation that NAMPT inhibition in GBM promoted M2 to M1 polarization of TAM is intriguing, as NAMPT is induced in M1 macrophages during a pro-inflammatory state, presumably to support glycolysis (10), and inhibition of salvage synthesis of NAD+ impairs macrophage phagocytosis (27) and TNFα production (28). However, recent work by Travelli and colleagues showed that NAMPT promoted the mobilization of MDSCs via SIRT1-mediated inactivation of HIF1α-CXCR4 axis (29), providing a potential link between NAMPT and suppressive TAM. The role of NAMPT NAD+ salvage in T cell function, on the other hand, is understudied and unclear. Impact of intracellular NAMPT inhibition on anti-tumor T cell immunity is unknown. In murine models of experimental autoimmune encephalomyelitis (EAE), apparently conflicting roles of NAMPT in T cell functions have been reported. FK-866 inhibition of NAMPT induced NAD+ depletion in activated T cells and reduced demyelination and neurological symptoms (30), while treatment with NAD+ protected against EAE by regulating CD4+ T cell differentiation, inducing Th1 CD4+ T cells to secrete immunosuppressive Th2 cytokine IL-10 (31). The beneficial changes in T cell phenotypes induced by GMX1778 MP treatment in our murine GBM could be a consequence of direct NAD+ depletion in T cells or secondary to the change in innate immune cells or PD-L1 levels.
Immunotherapy for GBM has been challenged by its characteristic immuno-suppressive tumor microenvironment. The Phase III part of the CheckMate143 trial evaluating the efficacy and safety of nivolumab versus bevacizumab in patients with recurrent GBM did not reveal survival benefits of nivolumab (15, 32). Recently, neoadjuvant use of anti-PD-1 antibodies for recurrent GBM have been reported to increase overall survival compared with adjuvant use (33, 34), suggesting the potential of boosting the activity of immune checkpoint blockade if an appropriate intervention of the tumor microenvironment, in this case by surgical resection, is accompanied. Here, we show that metabolic perturbation enhances checkpoint immunotherapy in a mouse model of GBM. Local NAMPT inhibition followed by PD-1 inhibitor was efficacious in prolonging survival while both monotherapies had limited activity. Our immune profiling studies showed that NAMPT inhibition-mediated alteration of the tumor microenvironment, i.e., upregulation of PD-L1, recruitment of T cells and M2 to M1 polarization, set a stage that invigorated the immunotherapy targeting the PD-L1/PD-1 axis. Consistent with our findings, combination treatment with an anti-PD-1 antibody and NAMPT inhibitor MV87 displayed a better activity in a murine fibrosarcoma model as compared with the single treatments, via NAMPT inhibitor-mediated suppression of MDSCs (29). Although this combination therapy appeared to be feasible in this work, the concern remains that known adverse effects of systemic administration of NAMPT and anti-PD-1 inhibitors could additively increase the likelihood of organ toxicities. We propose that use of MP delivery of NAMPT inhibitor offers a translational methodology to circumvent such problems by focally constraining the pharmacological activity to the tumor site in the brain (17). While the dispersal of the therapeutic released from MP could extend beyond the area of MP injection, optimizing delivery efficiency, such as the use of convection-enhanced delivery, has the potential to improve therapeutic efficacy in infiltrative gliomas. On the other hand, delivery of MP to all tumor cells may not be critical if NAMPT inhibitor based combination strategies can induce immune mediated effects that are capable of eliminating infiltrating tumor cells.
In conclusion, we report that localized metabolic perturbation with NAMPT inhibitor induces a dramatic alteration in the micro-environmental immune landscape in GBM. Combining MP delivery of NAMPT inhibitor and immune checkpoint blockade offers a mechanistically rational combination approach that exploits the convergence of metabolism and immunity for therapeutic gain. Further exploration of such a strategy to improve immunotherapy response in GBM patients is warranted.
Supplementary Material
Statement of Significance.
Microparticle-mediated local inhibition of NAMPT modulates the tumor immune microenvironment and acts cooperatively with anti-PD-1 checkpoint blockade, offering a combination immunotherapy strategy for the treatment of GBM.
Acknowledgments
Funding support: NIH R01CA227821 (D. Cahill and H. Wakimoto). G. Traverso was supported by R01CA227821 (NIH) and funding from the Department of Mechanical Engineering at MIT. A. Kirtane was supported by the PhRMA foundation postdoctoral fellowship.
Footnotes
Conflict of interest statement
D.P.C. has received travel and speaking fees from Merck, and consulting fees from Lilly. A.R.K., G.T., D.P.C. are co-inventors on a provisional patent application describing formulations of NAMPTi delivered through microparticle formulations. Complete details of all relationships for profit and not for profit for G.T. can be found at https://www.dropbox.com/sh/szi7vnr4a2ajb56/AABs5N5i0q9AfT1IqIJAE-T5a?dl=0.
References
- 1.Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016;23:27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nat Rev Drug Discov. 2011;10:671–84. [DOI] [PubMed] [Google Scholar]
- 3.Garten A, Schuster S, Penke M, Gorski T, de Giorgis T, Kiess W. Physiological and pathophysiological roles of NAMPT and NAD metabolism. Nat Rev Endocrinol. 2015;11:535–46. [DOI] [PubMed] [Google Scholar]
- 4.Chowdhry S, Zanca C, Rajkumar U, Koga T, Diao Y, Raviram R, et al. NAD metabolic dependency in cancer is shaped by gene amplification and enhancer remodelling. Nature. 2019;569:570–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kennedy BE, Sharif T, Martell E, Dai C, Kim Y, Lee PW, et al. NAD(+) salvage pathway in cancer metabolism and therapy. Pharmacol Res. 2016;114:274–83. [DOI] [PubMed] [Google Scholar]
- 6.Tateishi K, Iafrate AJ, Ho Q, Curry WT, Batchelor TT, Flaherty KT, et al. Myc-Driven Glycolysis Is a Therapeutic Target in Glioblastoma. Clin Cancer Res. 2016;22:4452–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tateishi K, Wakimoto H, Iafrate AJ, Tanaka S, Loebel F, Lelic N, et al. Extreme Vulnerability of IDH1 Mutant Cancers to NAD+ Depletion. Cancer Cell. 2015;28:773–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fons NR, Sundaram RK, Breuer GA, Peng S, McLean RL, Kalathil AN, et al. PPM1D mutations silence NAPRT gene expression and confer NAMPT inhibitor sensitivity in glioma. Nat Commun. 2019;10:3790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gujar AD, Le S, Mao DD, Dadey DY, Turski A, Sasaki Y, et al. An NAD+-dependent transcriptional program governs self-renewal and radiation resistance in glioblastoma. Proc Natl Acad Sci U S A. 2016;113:E8247–E56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Travelli C, Colombo G, Mola S, Genazzani AA, Porta C. NAMPT: A pleiotropic modulator of monocytes and macrophages. Pharmacol Res. 2018;135:25–36. [DOI] [PubMed] [Google Scholar]
- 11.Grahnert A, Grahnert A, Klein C, Schilling E, Wehrhahn J, Hauschildt S. Review: NAD +: a modulator of immune functions. Innate Immun. 2011;17:212–33. [DOI] [PubMed] [Google Scholar]
- 12.Cameron AM, Castoldi A, Sanin DE, Flachsmann LJ, Field CS, Puleston DJ, et al. Inflammatory macrophage dependence on NAD(+) salvage is a consequence of reactive oxygen species-mediated DNA damage. Nat Immunol. 2019;20:420–32. [DOI] [PubMed] [Google Scholar]
- 13.Charles NA, Holland EC, Gilbertson R, Glass R, Kettenmann H. The brain tumor microenvironment. Glia. 2012;60:502–14. [DOI] [PubMed] [Google Scholar]
- 14.Nduom EK, Weller M, Heimberger AB. Immunosuppressive mechanisms in glioblastoma. Neuro Oncol. 2015;17 Suppl 7:vii9–vii14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reardon DAea. RANDOMIZED PHASE 3 STUDY EVALUATING THE EFFICACY AND SAFETY OF NIVOLUMAB VS BEVACIZUMAB IN PATIENTS WITH RECURRENT GLIOBLASTOMA: CHECKMATE 143. Neuro Oncol. 2017;19:ⅲ21. [Google Scholar]
- 16.Sampath D, Zabka TS, Misner DL, O’Brien T, Dragovich PS. Inhibition of nicotinamide phosphoribosyltransferase (NAMPT) as a therapeutic strategy in cancer. Pharmacol Ther. 2015;151:16–31. [DOI] [PubMed] [Google Scholar]
- 17.Shankar GM, Kirtane AR, Miller JJ, Mazdiyasni H, Rogner J, Tai T, et al. Genotype-targeted local therapy of glioma. Proc Natl Acad Sci U S A. 2018;115:E8388–E94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wakimoto H, Kesari S, Farrell CJ, Curry WT Jr., Zaupa C, Aghi M, et al. Human glioblastoma-derived cancer stem cells: establishment of invasive glioma models and treatment with oncolytic herpes simplex virus vectors. Cancer Res. 2009;69:3472–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wakimoto H, Mohapatra G, Kanai R, Curry WT Jr., Yip S, Nitta M, et al. Maintenance of primary tumor phenotype and genotype in glioblastoma stem cells. Neuro Oncol. 2012;14:132–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wakimoto H, Tanaka S, Curry WT, Loebel F, Zhao D, Tateishi K, et al. Targetable signaling pathway mutations are associated with malignant phenotype in IDH-mutant gliomas. Clin Cancer Res. 2014;20:2898–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheema TA, Wakimoto H, Fecci PE, Ning J, Kuroda T, Jeyaretna DS, et al. Multifaceted oncolytic virus therapy for glioblastoma in an immunocompetent cancer stem cell model. Proc Natl Acad Sci U S A. 2013;110:12006–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marumoto T, Tashiro A, Friedmann-Morvinski D, Scadeng M, Soda Y, Gage FH, et al. Development of a novel mouse glioma model using lentiviral vectors. Nat Med. 2009;15:110–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saha D, Martuza RL, Rabkin SD. Macrophage Polarization Contributes to Glioblastoma Eradication by Combination Immunovirotherapy and Immune Checkpoint Blockade. Cancer Cell. 2017;32:253–67 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen X, Zhang L, Li J, Li Y, Wang Y, Xu ZX. Recent Findings in the Regulation of Programmed Death Ligand 1 Expression. Front Immunol. 2019;10:1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi Y. Regulatory mechanisms of PD-L1 expression in cancer cells. Cancer Immunol Immunother. 2018;67:1481–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Y, Wang H, Yao H, Li C, Fang JY, Xu J. Regulation of PD-L1: Emerging Routes for Targeting Tumor Immune Evasion. Front Pharmacol. 2018;9:536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Venter G, Oerlemans FT, Willemse M, Wijers M, Fransen JA, Wieringa B. NAMPT-mediated salvage synthesis of NAD+ controls morphofunctional changes of macrophages. PLoS One. 2014;9:e97378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Al-Shabany AJ, Moody AJ, Foey AD, Billington RA. Intracellular NAD+ levels are associated with LPS-induced TNF-alpha release in pro-inflammatory macrophages. Biosci Rep. 2016;36:e00301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Travelli C, Consonni FM, Sangaletti S, Storto M, Morlacchi S, Grolla AA, et al. Nicotinamide Phosphoribosyltransferase Acts as a Metabolic Gate for Mobilization of Myeloid-Derived Suppressor Cells. Cancer Res. 2019;79:1938–51. [DOI] [PubMed] [Google Scholar]
- 30.Bruzzone S, Fruscione F, Morando S, Ferrando T, Poggi A, Garuti A, et al. Catastrophic NAD+ depletion in activated T lymphocytes through Nampt inhibition reduces demyelination and disability in EAE. PLoS One. 2009;4:e7897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tullius SG, Biefer HR, Li S, Trachtenberg AJ, Edtinger K, Quante M, et al. NAD+ protects against EAE by regulating CD4+ T-cell differentiation. Nat Commun. 2014;5:5101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Desai K, Hubben A, Ahluwalia M. The Role of Checkpoint Inhibitors in Glioblastoma. Target Oncol. 2019;14:375–94. [DOI] [PubMed] [Google Scholar]
- 33.Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med. 2019;25:477–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schalper KA, Rodriguez-Ruiz ME, Diez-Valle R, Lopez-Janeiro A, Porciuncula A, Idoate MA, et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat Med. 2019;25:470–6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.