

Abstract
The majority of advanced prostate cancer therapies aim to inhibit androgen receptor (AR) signaling. However, AR reactivation inevitably drives disease progression to castration-resistant prostate cancer (CRPC). Here we demonstrate that protein arginine methyltransferase 5 (PRMT5) functions as an epigenetic activator of AR transcription in CRPC, requiring cooperation with a methylosome subunit pICln. In vitro and in xenograft tumors in mice, targeting PRMT5 or pICln suppressed growth of CRPC cells. Full-length AR and AR-V7 transcription activation required both PRMT5 and pICln but not MEP50. This activation of transcription was accompanied by PRMT5-mediated symmetric dimethylation of H4R3 at the proximal AR promoter. Further, knockdown of PRMT5 abolished the binding of pICln (but not vice versa) to the AR proximal promoter region, suggesting that PRMT5 recruits pICln to the AR promoter to activate AR transcription. Differential gene expression analysis in 22Rv1 cells confirmed that PRMT5 and pICln both regulate the androgen signaling pathway. Additionally, PRMT5 and pICln protein expression positively correlated with AR and AR-V7 protein expression in CRPC tissues and their expression was highly correlated at the mRNA level across multiple publicly available CRPC datasets. Our results suggest that targeting PRMT5 or pICln may be explored as a novel therapy for CRPC treatment by suppressing expression of AR and AR splice variants to circumvent AR reactivation.
Keywords: Protein arginine methyltransferase 5 (PRMT5), pICln, androgen receptor (AR), castration-resistant prostate cancer (CRPC), epigenetics
Introduction
Prostate cancer (PC) remains the second leading cause of cancer death in American men (1). The primary cause of PC mortality is the development of metastasis (2). Currently, androgen deprivation therapy (ADT), in combination with either docetaxel or abiraterone acetate, is the first-line treatment for metastatic PC (3). Because the growth of PC cells is dependent on androgen receptor (AR) signaling, suppressing AR signaling via ADT inhibits tumor growth. Despite initial positive response in the majority of patients, ADT eventually fails, leading to the development of castration-resistant PC (CRPC) (4).
AR reactivation drives CRPC progression and occurs via multiple mechanisms (AR gene amplification, expression of ligand-independent splice variants, or mutations of AR, and others) (4). For example, AR splice variant 7 (AR-V7) presents in 18-28% of CRPC tissues (5). AR-V7 expression correlates with poor patients’ prognosis (5). Because AR-V7 lacks the ligand-binding domain, it is constitutively active and can regulate transcription of AR target genes despite castrate levels of androgens (6). Inhibitors that target AR signaling, such as enzalutamide, demonstrate poor outcome towards CRPC that express AR-V7. Moreover, targeting full-length AR (AR-FL) can increase AR-V7 expression, exacerbating the condition (6). Thus, there is an urgent need to develop therapeutic approaches to overcoming AR reactivation. Because we have recently shown that protein arginine methyltransferase 5 (PRMT5) activates AR transcription in hormone-naïve PC (HNPC) (7), we investigated whether PRMT5 also regulates the transcription of AR and AR variants in CRPC.
PRMT5 is a methyltransferase that symmetrically dimethylates arginine residues in histones (H4R3, H3R8, H3R2, and H2AR3) to regulate transcription of target genes (8–11). While PRMT5 is generally considered an epigenetic repressor (8–11), PRMT5 also functions as an epigenetic activator (7,12,13). Although in vitro studies suggest that PRMT5 interacting proteins methylosome protein 50 (MEP50) and methylosome subunit pICln enhance PRMT5 enzymatic activity (14,15), how these proteins cooperate with PRMT5 to regulate gene transcription in vivo remains unknown. MEP50 functions as a critical PRMT5 cofactor facilitating substrate recognition and positioning via interaction with the N-terminal region of PRMT5 to form a heterooctameric complex (14,16). Since both pICln and MEP50 can enhance PRMT5 activity towards SmD3 protein in vitro (17,18), pICln may interact with PRMT5 similarly as MEP50 does to activate PRMT5 methyltransferase activity or alter substrate specificity. Indeed, we recently demonstrated that pICln, but not MEP50, cooperates with PRMT5 to activate transcription of DNA damage response genes (12). Here we provide evidence that PRMT5 promotes the growth of CRPC cells via epigenetic activation of transcription of both AR-FL and AR-V7 in a pICln-dependent, but MEP50-independent, manner. Results from our in vitro and in vivo studies suggest that targeting PRMT5 may present a promising approach for CRPC treatment.
Materials and Methods
Cell lines and reagents
LNCaP, 22Rv1, VCaP, COS-1, and 293T cells were purchased from ATCC (Manassas, VA, USA). LN95 cells were a kind gift from Dr. Jun Luo of Johns Hopkins University. Frozen cultures were recovered and expanded in complete media: LNCaP and 22Rv1 in RPMI1640 (Corning, NY, USA), LN95 was cultured in RPMI1640 without phenol red (Corning, NY, USA), 293T, COS-1, and VCaP in DMEM (Corning, NY, USA) supplemented with 10% FBS (Atlanta Biologicals, Lawrenceville, GA, USA) or for LN95 charcoal-stripped FBS (Corning, NY, USA), 2 mM L-glutamine (Corning, NY, USA), and 100 units/mL penicillin and 100 μg/mL streptomycin (Gibco, Gaithersburg, MD, USA)). Cells were not passaged more than 30 times. Long-term storage, cell authentication and mycoplasma contamination check were described previously (12). Methocel A4M was purchased from Sigma (St. Louis, MO, USA), abiraterone acetate and enzalutamide were purchased from MedChemExpress (Monmouth Junction, NJ, USA).
Bimolecular fluorescence complementation (BiFC)
BiFC plasmids (250 ng each) encoding a protein of interest fused to the N- or C-terminal fragment of the Venus fluorescent protein (VN155 or VC155) and 100 ng of the plasmid encoding the Cerulean fluorescent protein (CFP, as a positive control for transfection) were co-transfected into COS-1 cells and BiFC efficiency (YFP/CFP) was analyzed essentially as described previously (19). For BiFC competition assay, 500 ng of the plasmid encoding a PRMT5 interacting protein (MEP50 or pICln) or empty vector control were co-transfected to analyze the inhibition of PRMT5:MEP50 interaction. Results are presented as median ± SD from three independent biological replicates.
Xenograft tumor growth
Animal experiments were performed in the Biological Evaluation Facility of the Purdue University Center for Cancer Research approved by the Purdue University Animal Care and Use Committee. Six to eight weeks old male non-obese diabetic-Rag1(null)-γ chain(null) (NRG) mice were castrated, and 14 days later 2 x 105 cells of 22Rv1-shPRMT5, or -shpICln, or -shSC in 100 μl of RPMI1640 media were mixed with 100 μl of Matrigel (200 μl total) and injected subcutaneously into the right lower flank (10 mice/group). After tumor volumes reached ~100 mm3, mice were treated with Dox (1 mg/mL in drinking water) to induce the expression of shRNA or treated 5 days/week with ASI in 0.5% Methocel orally (abiraterone acetate 200 mg/kg/day, enzalutamide 25 mg/kg/day), vehicle, or in combination. Tumor growth was measured every 2-3 days, and tumor volume was calculated using ½ × L × W × H without blinding method. When control tumors reached ~2000 mm3, tumors were resected for immunohistochemistry (IHC) analysis.
Clinical data analysis
Gene expression profiles of 34 PC data sets were obtained from Gene Expression Omnibus (GEO) (20), cBioportal (21,22), and Oncomine (23) (Supplementary Table S1) with total of 4624 samples. Gene expression levels were log2 transformed and median centered. Gene expression profiles from cBioportal were downloaded with annotation of “mRNA_median_Zscores”. If one gene had multiple gene expression files in the same dataset, the sum of all corresponding mRNA levels was used. Spearman’s rank correlation coefficients were calculated to evaluate the correlations of specific gene pairs. Wilcoxon Rank Sum test was used to compare the differences between groups for all 34 datasets.
The clinical information and gene expression data for PC (24) were downloaded from cBioPortal (21,22) for the survival analysis. Patients were divided into two groups based on the top and bottom 50% quantile of expression levels for selected genes. Survival probability was computed in R using the survfit function in the R package survival. Kaplan-Meier plots were generated using the ggsurvplot function of package survminer.
Construction of CRPC tissue microarray (TMA) containing samples from 20 patients and HNPC TMA containing samples from 72 patients (32 with BPH, 20 with PC Gleason score 6 and 20 with PC Gleason score⩾7) was described previously (7,25).
Additional Methods
Stable cell line generation, cell proliferation assay, cell cycle analysis, ChIP-qPCR, reverse transcription and qPCR, IHC staining and scoring, RNA-seq analysis (deposited in GEO, accession number GSE154951), western blot, and statistical analysis were performed as described previously (7,12). Detailed procedures for these methods are provided in Supplementary Methods.
Results
PRMT5 promotes growth of CRPC cells via epigenetic activation of AR expression
To determine the role of PRMT5 in CRPC, we analyzed effect of PRMT5 inhibition on the growth of CRPC cell line 22Rv1, which expresses both full-length AR (AR-FL) and AR-V7. Treatment of 22Rv1 cells with our PRMT5 inhibitor BLL3.3, also called CMP5 (7,26), reduced cell proliferation compared to DMSO control (Fig. 1A) and downregulated AR-FL and AR-V7 at protein and mRNA levels (Fig. 1B–D). Another PRMT5 inhibitor JNJ-64619178 (JNJ), which is currently in Phase I clinical trial for Non-Hodgkin lymphoma and solid tumors (27), similarly reduced cell growth and downregulated AR-FL and AR-V7 expression (Fig. 1E–H). To corroborate these findings, we used lentivirus-based shRNA constructs (two separate shRNA constructs per gene) to establish doxycycline (Dox)-inducible PRMT5 knockdown cell lines in 22Rv1 (22Rv1-shPRMT5 and 22Rv1-shPRMT5#2). PRMT5 knockdown inhibited cell growth (Fig. 1I, Supplementary Fig. S1A) and decreased expression of both AR-FL and AR-V7 (Fig. 1J–L, Supplementary Fig. S1B–D) while expression of scramble control (22Rv1-shSC) did not affect cell growth or protein expression (Fig. 1M–P). Moreover, expression of several AR target genes (28) was suppressed upon PRMT5 knockdown (Supplementary Fig. S1E), consistent with the decreased AR expression. These results suggest that PRMT5 regulates cell growth, AR expression, and AR signaling in 22Rv1.
Figure 1. PRMT5 promotes growth of CRPC cells via epigenetic activation of AR expression.
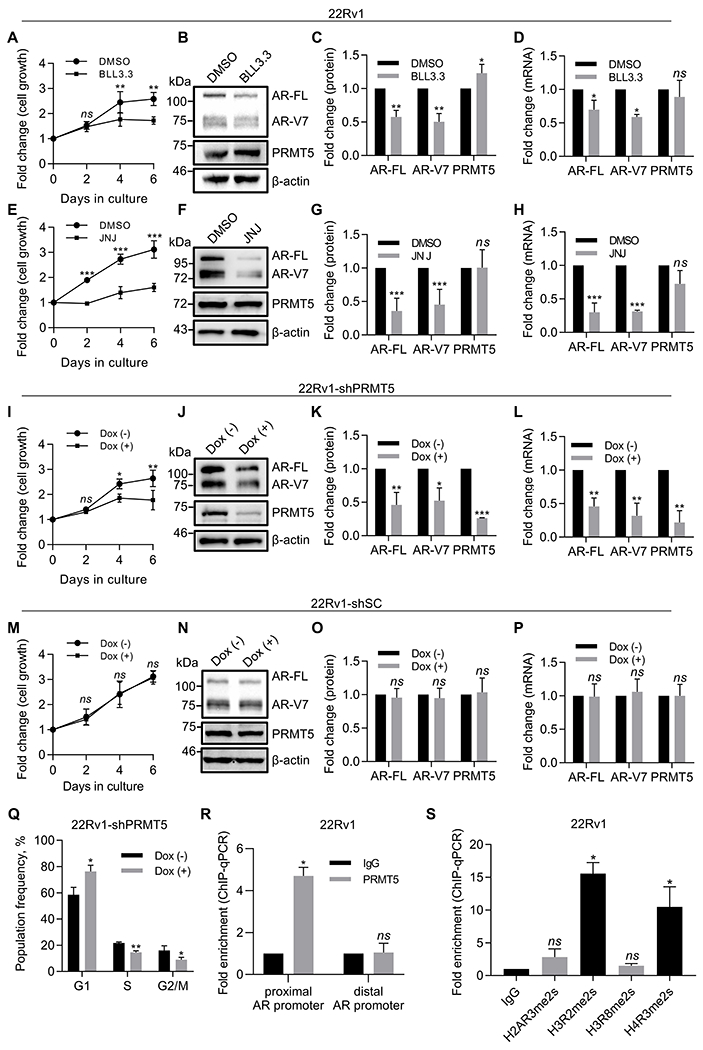
A, Growth curve (MTT assay) of 22Rv1 cells incubated with 10 µM PRMT5 inhibitor (BLL3.3) or equal volume of vehicle (DMSO) for 6 days. B-С, Representative western blot images (B) and quantification (С) of protein expression in cell lysates from Day 6 of A. D, qPCR analysis of gene expression in cells from Day 6 of A. E, Growth curve (MTT assay) of 22Rv1 cells incubated with 10 µM PRMT5 inhibitor (JNJ-64619178, referred to as JNJ) or equal volume of vehicle (DMSO) for 6 days. F-G, Representative western blot images (F) and quantification (G) of protein expression in cell lysates from Day 6 of E. H, qPCR analysis of gene expression in cells from Day 6 of E. I, Growth curve (MTT assay) of 22Rv1 cells with doxycycline-inducible PRMT5 knockdown (22Rv1-shPRMT5) incubated in the presence (Dox (+)) or absence (Dox (–)) of doxycycline for 6 days. J-K, Representative western blot images (J) and quantification (K) of protein expression in cell lysates from Day 6 of I. L, qPCR analysis of gene expression in cells from Day 6 of I. M, Growth curve (MTT assay) of 22Rv1 cells with doxycycline-inducible scramble control expression (22Rv1-shSC) incubated in the presence (Dox (+)) or absence (Dox (–)) of doxycycline for 6 days. N-O, Representative western blot images (N) and quantification (O) of protein expression in cell lysates from Day 6 of M. P, qPCR analysis of gene expression in cells from Day 6 of M. Q, Flow cytometry analysis of cells following PI staining at Day 6 of I (sub-G1 cells were gated out). R, ChIP-qPCR for PRMT5 binding to the proximal or distal AR promoter. S, ChIP-qPCR for the enrichment of the indicated histone methylations on the proximal promote region of AR. For MTT, western blotting, cell cycle, and qPCR analysis, statistical significance of group difference was determined for ‘DMSO vs BLL3.3’, ‘DMSO vs JNJ’, or ‘Dox (–) vs Dox (+)’. For ChIP-qPCR, values were normalized to the corresponding IgG control, and indicated statistical significance of group difference was determined for ‘specific IP vs IgG IP’. For all experiments, results are mean ± SD from 3 independent experiments. For western blotting of AR, the AR N-20 antibody (sc-816, Santa Cruz) was used. Student’s t-test with Welch’s correction was performed to determine statistical significance of group difference. ns P > 0.05, * P < 0.05, ** P < 0.01, *** P < 0.001.
Because AR reactivation occurs via several mechanisms, we next determined whether PRMT5 regulates AR expression in other CRPC models. We reported previously that PRMT5 targeting downregulates AR expression and inhibits growth in C4-2 cells, which model CRPC via AR overexpression (7). We further evaluated the effect of PRMT5 inhibition in VCaP cells which bear AR gene amplification (29) and LN95 cells which express AR splice variants (30) and observed that both PRMT5 inhibitors BLL3.3 and JNJ suppressed cell growth and downregulated AR expression (Supplementary Fig. S1F–M). Cell cycle analysis confirmed that PRMT5 knockdown caused G1 arrest in 22Rv1 (Fig. 1Q), consistent with previous observations (31,32). No significant induction of cell death was observed (Supplementary Fig. S1N). Collectively, our results suggest that PRMT5 promotes cell growth via activation of AR transcription in CRPC cells with AR overexpression, AR gene amplification, or expression of AR splice variants.
To investigate whether PRMT5 regulates AR expression via histone methylation at the AR promoter, we performed chromatin immunoprecipitation (ChIP)-assays in 22Rv1 using PRMT5-specific antibody. PRMT5 bound to the proximal region of the promoter (−493 to −226 bp from transcription start site (TSS)) but not to the distal region (-4481 to -4308 bp from TSS) (Fig. 1R). ChIP-qPCR analysis revealed that H4R3me2s and H3R2me2s were highly enriched at the AR proximal promoter (Fig. 1S). Further, PRMT5 knockdown decreased these enrichments and PRMT5 binding, confirming that PRMT5 methylates H4R3 and H3R2 at the AR promoter (Supplementary Fig. S1O). As observed in LNCaP cells (7), Brg1 and Sp1 also bound to the same region (Supplementary Fig. S1P). Taken together, our findings demonstrate that PRMT5 activates AR transcription in CRPC cells by binding to the proximal region of the AR promoter to methylate histones H4 and H3 in a similar manner as in HNPC cells (7).
MEP50 is not required for PRMT5-mediated activation of AR transcription in CRPC cells
MEP50 is considered a canonical cofactor of PRMT5 (8–11). We next established Dox-inducible MEP50 knockdown stable cell lines in 22Rv1 (22Rv1-shMEP50 and 22Rv1-shMEP50#2) to examine the effect of MEP50 knockdown on AR expression. Unexpectedly, MEP50 knockdown affected neither AR-FL/AR-V7 expression (Fig. 2A–C, Supplementary Fig. S2A–C) nor the expression of AR target genes (Supplementary Fig. S2D). However, MEP50 knockdown de-repressed expression of involucrin (IVL) mRNA (Fig. 2C), confirming that PRMT5/MEP50 represses IVL transcription (33). We also confirmed that MEP50 knockdown had no effect on AR expression in LNCaP cells (Fig. 2D–F). ChIP-qPCR analysis revealed that MEP50 did not bind to the AR proximal promoter in these cell lines (Fig. 2G–H), though MEP50 antibody efficiently immunoprecipitated MEP50 (Supplementary Fig. S2E) and MEP50 bound to the IVL promoter (Fig. 2G–H). Additionally, MEP50 knockdown in 22Rv1 did not significantly change H4R3me2s and H3R2me2s levels at AR proximal promoter (Supplementary Fig. S2F). Notably, MEP50 knockdown decreased the total cellular level of H3R2me2s but did not significantly affect the total level of H4R3me2s (Supplementary Fig. S2G). Contrary to the lack of cell death following PRMT5 knockdown (Supplementary Fig. S1I), MEP50 knockdown induced both cell death (Fig. 2I) and G1-cell cycle arrest (Fig. 2J) in 22Rv1 cells, indicating that PRMT5 and MEP50 might have distinct roles in cell proliferation. Taken together, MEP50 does not appear to participate in the regulation of AR transcription by PRMT5.
Figure 2. MEP50 is not required for PRMT5-mediated activation of AR transcription in CRPC cells.
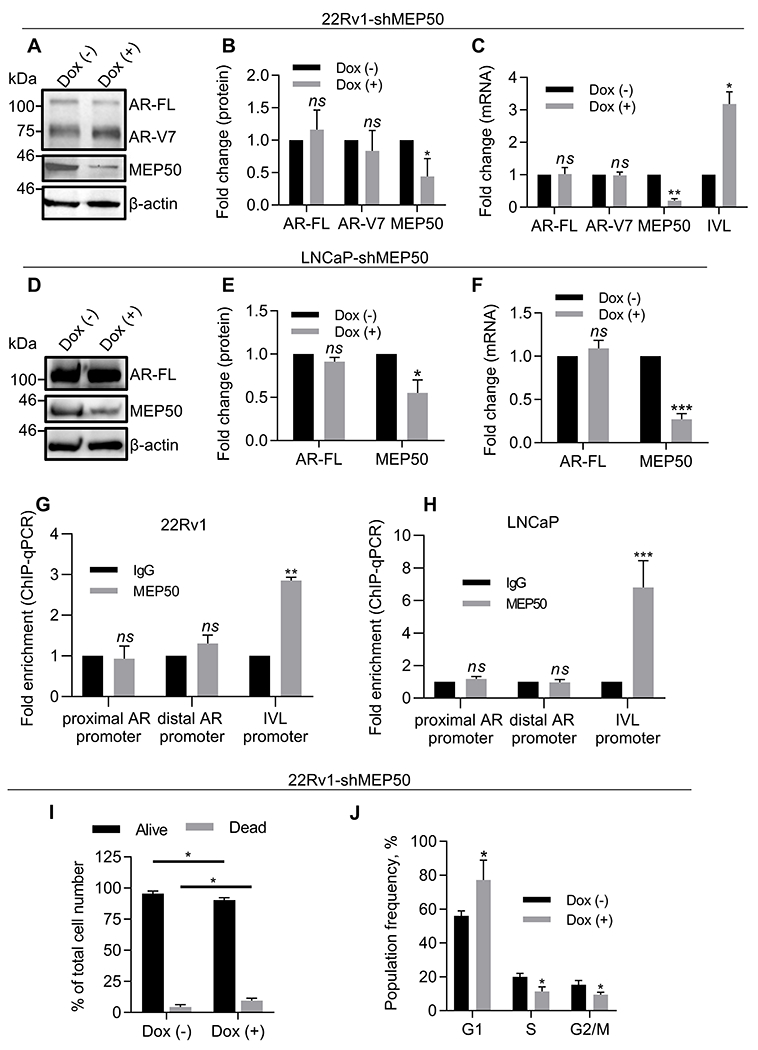
A-B, Representative western blot images (A) and quantification (B) of protein expression in cell lysates of 22Rv1 cells with doxycycline-inducible MEP50 knockdown (22Rv1-shMEP50) incubated in the presence (Dox (+)) or absence (Dox (–)) of doxycycline for 6 days. C, qPCR analysis of gene expression in cells from A. D-E, Representative western blot images (D) and quantification (E) of protein expression in cell lysates of LNCaP cells with doxycycline-inducible MEP50 knockdown (LNCaP-shMEP50) incubated in the presence (Dox (+)) or absence (Dox (–)) of doxycycline for 6 days. F, qPCR analysis of gene expression in cells from D. G-H, ChIP-qPCR assay of MEP50 binding to the proximal AR promoter or control gene IVL promoter was performed with non-specific IgG binding as a control in 22Rv1 (G) and LNCaP (H) cells. I, Trypan blue cell viability analysis in 22Rv1-shMEP50 cells after 6 days of MEP50 knockdown. J, Flow cytometry analysis of cells following PI staining at Day 6 of MEP50 knockdown in 22Rv1-shMEP50 (sub-G1 cells were gated out). For western blotting, cell cycle, cell viability, and qPCR analysis, statistical significance of group difference was determined for ‘Dox (–) vs Dox (+)’. For ChIP-qPCR, values were normalized to the corresponding IgG control, and indicated statistical significance of group difference was determined for ‘specific IP vs IgG IP’. Results are mean ± SD from 3 independent experiments. For western blotting of AR, the AR N-20 antibody (sc-816, Santa Cruz) was used. Student’s t-test with Welch’s correction was performed to determine statistical significance. ns P > 0.05, * P < 0.05, ** P < 0.01, *** P < 0.001.
pICln participates in epigenetic activation of AR transcription
The surprising finding that MEP50 was not involved in AR transcription regulation in HNPC and CRPC cells prompted us to search for PRMT5-interacting proteins that might cooperate with PRMT5 to regulate AR transcription (17,34,35). First, we found that only pICln, but not RIOK1 and COPR5, bound to the same AR proximal promoter region as PRMT5 did (Fig. 3A). Next, we established Dox-inducible pICln knockdown cell lines in 22Rv1 and LNCaP cells (22Rv1-shpICln and LNCaP-shpICln) to further interrogate a role of pICln in AR regulation. Indeed, pICln knockdown significantly suppressed cell proliferation (Fig. 3B and C), inhibited AR expression at both protein and mRNA levels in 22Rv1 (Fig. 3D–F) and LNCaP (Fig. 3G–I), and decreased its binding and the H4R3me2s level at the AR promoter (Fig. 3J). However, pICln knockdown did not affect PRMT5 binding nor the H3R2me2s level (Fig. 3J). Consistently, at the total cellular level, pICln knockdown decreased H4R3me2s but did not affect H3R2me2s (Supplementary Fig. S3A). In contrast, PRMT5 knockdown decreased pICln binding to the AR promoter (Fig. 3K). These results suggest that PRMT5 recruits pICln to the AR promoter, and PRMT5/pICln interaction is required for H4R3 methylation at the AR promoter. Contrary to PRMT5 knockdown, pICln knockdown induced cell death (Fig. 3L) and G2 cell cycle arrest in 22Rv1 cells (Fig. 3M). Thus, pICln has additional roles in cell proliferation and survival independently of PRMT5. Nonetheless, our results demonstrate that pICln is required for PRMT5-mediated H4R3 methylation to activate AR transcription.
Figure 3. pICln participates in epigenetic activation of AR transcription.
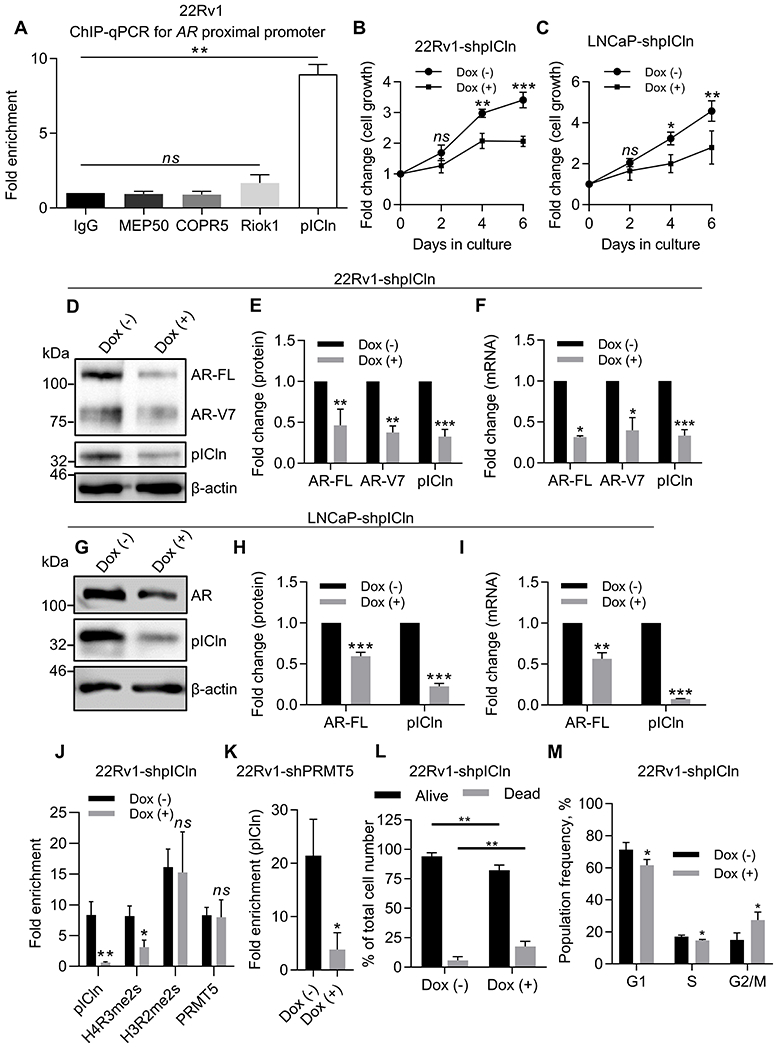
A, ChIP-qPCR assay for binding of PRMT5-interacting proteins to the proximal AR promoter in 22Rv1 cells. Values were normalized to IgG control. B-C, Growth curve (MTT assay) of 22Rv1 (B) or LNCaP (C) cells with Dox-inducible pICln knockdown (shpICln). D-E, Representative western blot images (D) and quantification (E) of protein expression in 22Rv1 after 6 days of pICln knockdown. F, qPCR analysis of gene expression in 22Rv1 after 6 days of pICln knockdown. G-H, Representative western blot images (G) and quantification (H) of protein expression in LNCaP after 5 days of pICln knockdown. I, qPCR analysis of gene expression in LNCaP after 5 days of pICln knockdown. J, ChIP-qPCR assay for pICln and H4R3me2s presence at the proximal AR promoter in 22Rv1 upon pICln knockdown. K, ChIP-qPCR assay for pICln presence at the proximal AR promoter upon PRMT5 knockdown. L, Trypan blue cell viability analysis in 22Rv1-shpICln cells after 6 days of pICln knockdown. M, Flow cytometry analysis of fixed and stained with propidium iodide 22Rv1-shpICln cells after 6 days of pICln knockdown. For MTT, western blotting, cell cycle, and qPCR analysis statistical significance of group difference was determined for ‘Dox (–) vs Dox (+)’. For ChIP-qPCR, values were normalized to the corresponding IgG control, and indicated statistical significance of group difference was determined for ‘specific IP vs IgG IP’ (A) or ‘Dox (–) vs Dox (+)’ (J, K). For all experiments, results are mean ± SD from 3 independent experiments. For western blotting of AR, the AR N-20 antibody (sc-816, Santa Cruz) was used. Student’s t-test with Welch’s correction was performed to determine statistical significance. ns P > 0.05, * P < 0.05, ** P < 0.01, *** P < 0.001.
To investigate whether pICln binds to the N-terminal region of PRMT5 as MEP50 does (14,16), we utilized the bimolecular fluorescence complementation (BiFC) assay (19). Co-expression of PRMT5(NT292)-VN155 and VC155-pICln in COS-1 cells resulted in YFP fluorescence, indicating that pICln interacted with N-terminal fragment of PRMT5 (Supplementary Fig. S3B). To determine whether pICln might bind to a similar site in PRMT5 as MEP50 does, we performed the BiFC competition assay, in which VN155-PRMT5 and VC155-MEP50 were co-expressed with MEP50 or pICln. Indeed, overexpression of MEP50 or pICln similarly decreased BiFC efficiency of the PRMT5-MEP50 BiFC interaction (Supplementary Fig. S3C–E), suggesting that pICln may indeed function as a cofactor by binding to the N-terminus of PRMT5 like MEP50 (16).
We next aimed to check whether the anti-proliferative effect of PRMT5 or pICln knockdown is mediated through the regulation of AR expression. For this purpose, we performed AR rescue assays. We transfected 22Rv1-shPRMT5, 22Rv1-shpICln, and 22Rv1-shSC cells with the plasmid encoding FLAG-AR expression or the empty vector control. Remarkably, exogenously expressed AR completely restored cell proliferation in 22Rv1-shpICln and 22Rv1-shPRMT5 cells but did not affect cell proliferation in 22Rv1-shSC (Supplementary Fig. S4A–D) as observed previously in LNCaP-shPRMT5 cells (7). This observation suggests that the inhibition of 22Rv1 cell proliferation upon PRMT5 or pICln knockdown is primarily mediated through downregulation of AR expression.
PRMT5 and pICln regulate the AR signaling independently of MEP50
The above results suggest distinct regulatory roles of PRMT5, MEP50, and pICln in cell proliferation, cell cycle progression, and cell death. To further understand their roles in genome-wide gene regulation, we performed RNA-seq of 22Rv1 cells with and without knockdown of PRMT5, MEP50, or pICln. We identified 6,730 out of 23,334 genes which had at least one differentially expressed transcript (DET) upon PRMT5 knockdown, including 3,426 genes with upregulated transcripts and 3,304 genes with downregulated ones (Fig. 4A). Following MEP50 knockdown, 447 upregulated and 626 downregulated differentially expressed genes (DEGs) overlapped with the PRMT5-knockdown DEGs (Fig. 4A). Notably, pICln knockdown led to more overlapped genes with the PRMT5-knockdown DEGs, including 1,033 upregulated and 1,361 downregulated genes (Fig. 4A). To confirm the regulation of the AR signaling by PRMT5 and pICln, we analyzed the enrichment of different sets of DEGs involved in AR signaling pathway, Gene Ontology GO:0030521. Consistently, genes of this pathway were significantly over-represented among PRMT5- and pICln-knockdown DEGs but not among MEP50-knockdown DEGs (Fig. 4B). Compared to fold changes (in log scale with base 2) of selected AR signaling pathway DEGs identified by mRNA-seq (left panel in Fig. 4C), qPCR analysis confirmed that PRMT5 and pICln, but not MEP50, similarly regulate the expression of these genes (right panel in Fig. 4C). These results suggest that PRMT5 and pICln co-regulate AR signaling in a MEP50-independent way.
Figure 4. PRMT5 and pICln regulate the AR signaling independently of MEP50.
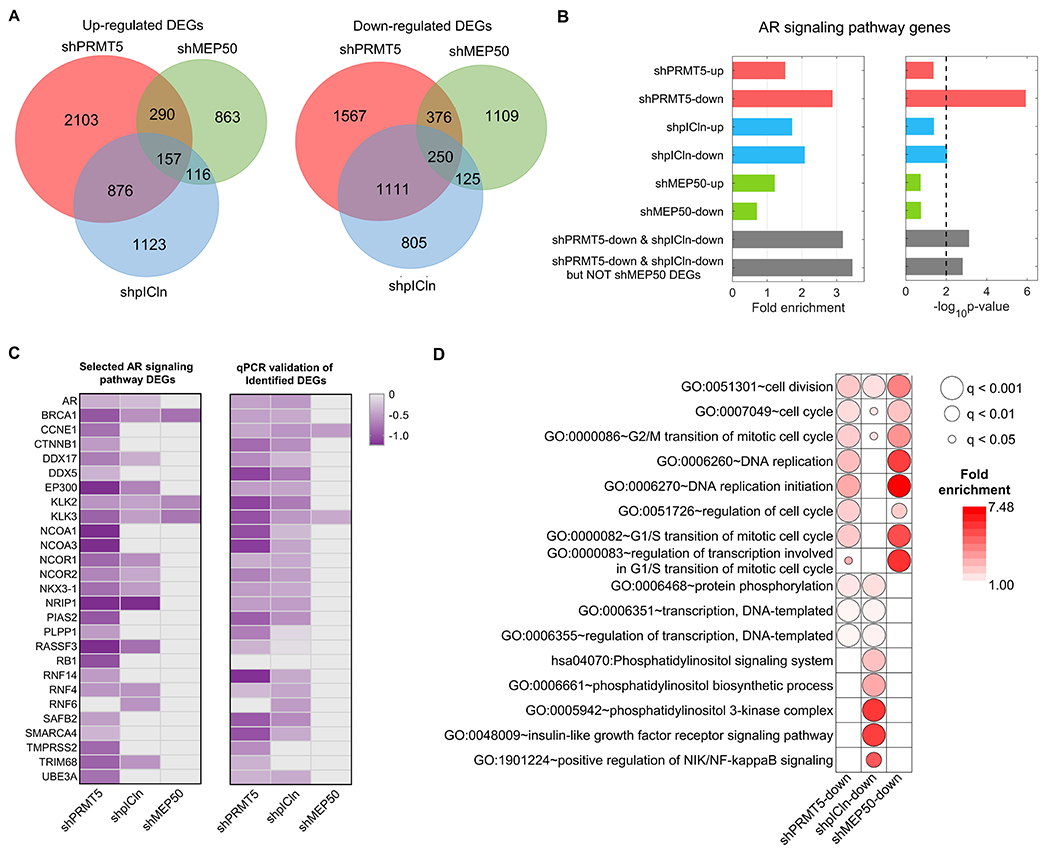
A, RNA-seq analysis of 22Rv1 cells upon 6 days of shRNA-mediated knockdown of PRMT5 (shPRMT5), MEP50 (shMEP50), or pICln (shpICln). Venn diagrams indicate overlap of upregulated and downregulated DEGs among three experiments. B, Presence of AR signaling pathway (GO:0030521) among identified up- and downregulated DEGs. C, Heatmap indicating expression fold change (FC, log2) of individual AR signaling pathway genes down-regulated upon knockdown of PRMT5 (shPRMT5), MEP50 (shMEP50), and pICln (shpICln) from RNA-seq analysis (left panel) and qPCR validations (right panel). D, Gene ontology (GO) analysis of DEGs that were downregulated upon knockdown of PRMT5 (shPRMT5-down), pICln (shpICln-down), and MEP50 (shMEP50-down). Presented are selected GO terms significantly enriched in the DEG sets related to cell-cycle regulation, DNA replication, transcription, and phosphorylation. The color of each dot indicates the fold enrichment for the GO term, whereas the size of the dot indicates q-value (FDR-corrected p-value) of statistical significance of the enrichment.
The GO enrichment analysis explored many GO terms and KEGG pathways significantly enriched in DEGs downregulated after the knockdown of PRMT5, pICln or MEP50 (Fig. 4D). For example, GO:0051301 “cell division”, GO:0007049 “cell cycle”, and GO:0000086 “G2/M transition of mitotic cell cycle” were shared by repressed genes after PRMT5/pICln/MEP50 knockdown. GO functions associated with G1/S phase regulation were notably over-represented in DEGs downregulated by PRMT5- or MEP50-knockdown, but not in the pICln-knockdown group, for instance, GO:0000082 “G1/S transition of mitotic cell cycle”, GO:0000083 “regulation of transcription involved in G1/S transition of mitotic cell cycle”, and GO:0006270 “DNA replication” among others. This was consistent with our cell cycle analysis results (Fig. 1Q, Fig. 2J, Fig. 3M). However, we noticed that PRMT5- or MEP50-knockdown DEGs can be different, even though they were associated with the same GOs or KEGG pathways. But PRMT5 and pICln tended to mediate the same DEGs involved in some GOs and KEGG pathways, e.g. GO:0006351 “transcription, DNA-templated” and GO:0006355 “regulation of transcription, DNA-templated” (Fig. 4D). Interestingly, pICln appears to have additional roles in regulating phosphatidylinositol signaling and NF-κB signaling in PC independently of PRMT5 (Fig. 4D). Taken together, our genome-wide gene expression analysis confirms the role of PRMT5/pICln in AR signaling in PC and reveals distinct regulatory roles of PRMT5, MEP50, and pICln in various cellular processes such as cell cycle progression.
PRMT5 and pICln expression positively correlates with AR in CPRC patients
To investigate the clinical relevance of our findings, we examined the expression of AR, PRMT5, pICln, and MEP50 in HNPC and CRPC tissues. Nuclear PRMT5 and pICln expression was the highest in CRPC tissues with elevated AR expression (Fig. 5A), and nuclear PRMT5, pICln, and MEP50 expression correlates positively with AR expression (Fig. 5B). In general, correlation with AR expression was higher for PRMT5 and pICln compared to MEP50 (Fig. 5B, Supplementary Fig. S5A–D). Notably, when samples were stratified by top and bottom 50% of AR staining (ARhigh and ARlow), the nuclear PRMT5/pICln expression was lower in ARlow tissues compared to ARhigh (Supplementary Fig. S5E and F). However, PRMT5/MEP50 correlation was similar between ARlow and ARhigh groups (Supplementary Fig. S5G–I). Consistent with the nuclear PRMT5/AR expression correlation in HNPC tissues (7), nuclear pICln expression also positively correlated with both nuclear PRMT5 and AR expression in these tissues (Supplementary Fig. S5J and K). These results further suggest that PRMT5 and pICln are strongly associated with higher AR expression.
Figure 5. PRMT5 and pICln expression positively correlates with AR in CPRC patients.
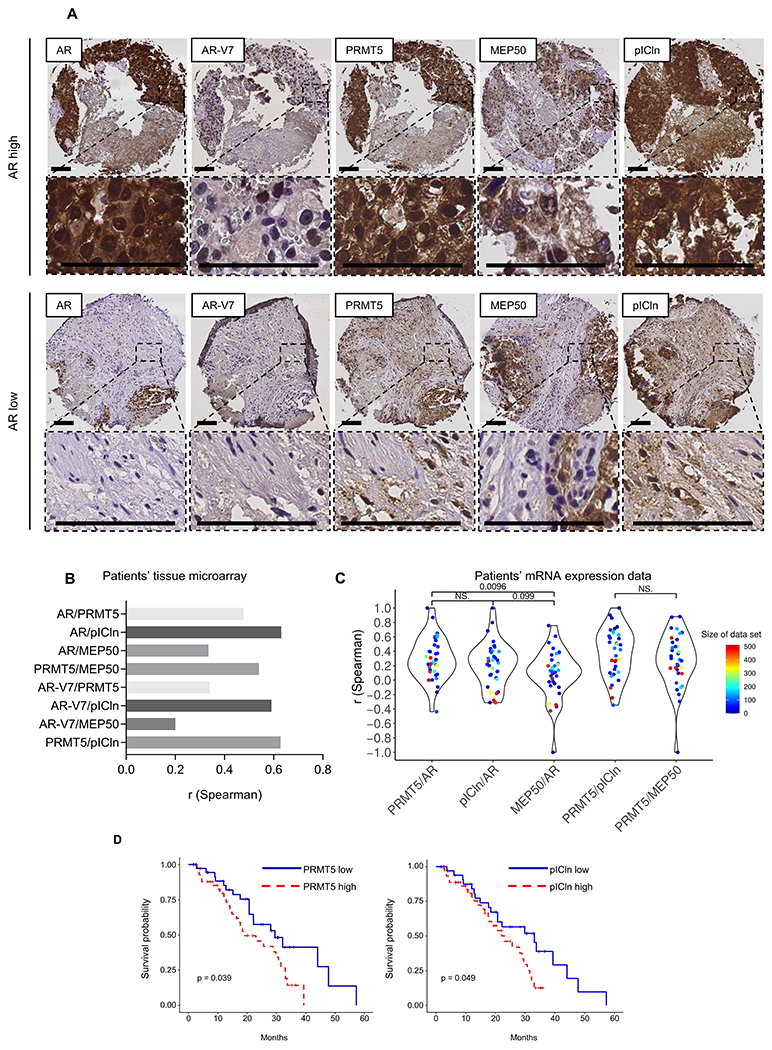
A-B, AR, AR-V7, PRMT5, pICln, and MEP50 protein expressions were analyzed by IHC in metastatic CRPC samples. A, Representative IHC images of AR, AR-V7, PRMT5, pICln, and MEP50 expression. Scale bar indicates 100 μm. B, Spearman correlations of protein-level expression of AR, AR-V7, PRMT5, pICln, and MEP50 in CRPC tissues. C, Spearman correlations of mRNA expression levels between AR, PRMT5, pICln, and MEP50. The mRNA expression data for 4624 patient samples were obtained from 34 published datasets. Each dot denotes one dataset, representing the gene expression correlation between the pair of selected mRNAs. The dot color indicates the sample size of corresponding dataset. D, Kaplan-Meier curves comparing influences of mRNA expression levels of PRMT5 and pICln, respectively, on patients’ survival. Red curves represent patients with high (top 50%) expression of PRMT5 and pICln, whereas blue ones are groups with low (bottom 50%) expression. The mRNA expression and patient survival data were downloaded from the cBioportal SU2C-PCF dataset.
Next, we retrieved 34 datasets from GEO and cBioportal including a total of 3425 HNPC and 1199 CRPC cases with mRNA expression profiles. PRMT5/AR and pICln/AR correlations were significantly higher than MEP50/AR correlation, confirming the role of PRMT5/pICln in AR signaling (Fig. 5C). Interestingly, comparable PRMT5/pICln and PRMT5/MEP50 correlations were observed, consistent with their distinct cellular roles. These results further support our finding that PRMT5 cooperates with pICln to activate transcription of AR in HNPC and CRPC tissues.
We further investigated the relationship of PRMT5 and pICln mRNA expression with patients’ survival in CRPC (24). Notably, patients with high expression of either PRMT5 or pICln had lower survival (Fig. 5D). These findings support that expression levels of PRMT5 and pICln may affect patient outcomes or potential responses to therapy, indicating their role in cancer progression.
Next, we examined whether nuclear-localized PRMT5 promotes cell proliferation. Antonysamy et al. reported that PRMT5 protein tended to aggregate in the absence of its cofactors (16). Thus, we reasoned that overexpression of PRMT5 alone may promote aggregate formation and decrease the cellular amount of active PRMT5 leading to reduced cell proliferation which was previously observed in LNCaP cells by Gu et al. (36). In line with this, we performed overexpression of mutant shRNA-resistant PRMT5 fused with nuclear localization signal (NLS) or nuclear export signal (NES) or without localization signal in LNCaP or 22Rv1 cells on the background of PRMT5 knockdown. Consistent with a previous report by Gu et al., NLS-PRMT5 decreased while NES-PRMT5 promoted cell proliferation in WT cells (Supplementary Fig. S6A, D). Conversely, NLS-PRMT5 promoted while NES-PRMT5 decreased cell proliferation in LNCaP-shPRMT5 or 22Rv1-shPRMT5 (Supplementary Fig. S6A, D). In cells with PRMT5 knockdown, NLS-PRMT5 promoted AR expression at both protein (Supplementary Fig. S6B, E) and mRNA level (Supplementary Fig. S6C, F). These observations further confirm that nuclear-localized PRMT5 promotes cell proliferation and AR expression in PC cells.
Knockdown of PRMT5 or pICln suppresses CRPC tumor growth in mice
To determine whether targeting PRMT5 or its cofactor pICln can suppress the growth of CRPC tumors in vivo, we implanted 22Rv1-shPRMT5, 22Rv1-shpICln, and 22Rv1-shSC cells subcutaneously into male, pre-castrated NRG mice. When the average tumor volumes reached 100 mm3, shRNA expression was induced by Dox treatment, and tumor growth was monitored. PRMT5 or pICln knockdown significantly suppressed tumor growth (Fig. 6A and B), consistent with the suppression of AR expression in xenograft tumors (Fig. 6C). Analysis of cleaved caspase-3 staining suggested slight induction of apoptosis by pICln knockdown but not by PRMT5 knockdown (Fig. 6C–E), confirming our in vitro findings. Ki-67 analysis showed that tumors with either PRMT5 or pICln knockdown had significantly lower proliferative index compared to scramble control (Fig. 6F and G). Taken together, these results demonstrate that PRMT5 and pICln also regulate AR expression and the growth of CRPC tumors in vivo.
Figure 6. Knockdown of PRMT5 or pICln suppresses CRPC tumor growth in mice.
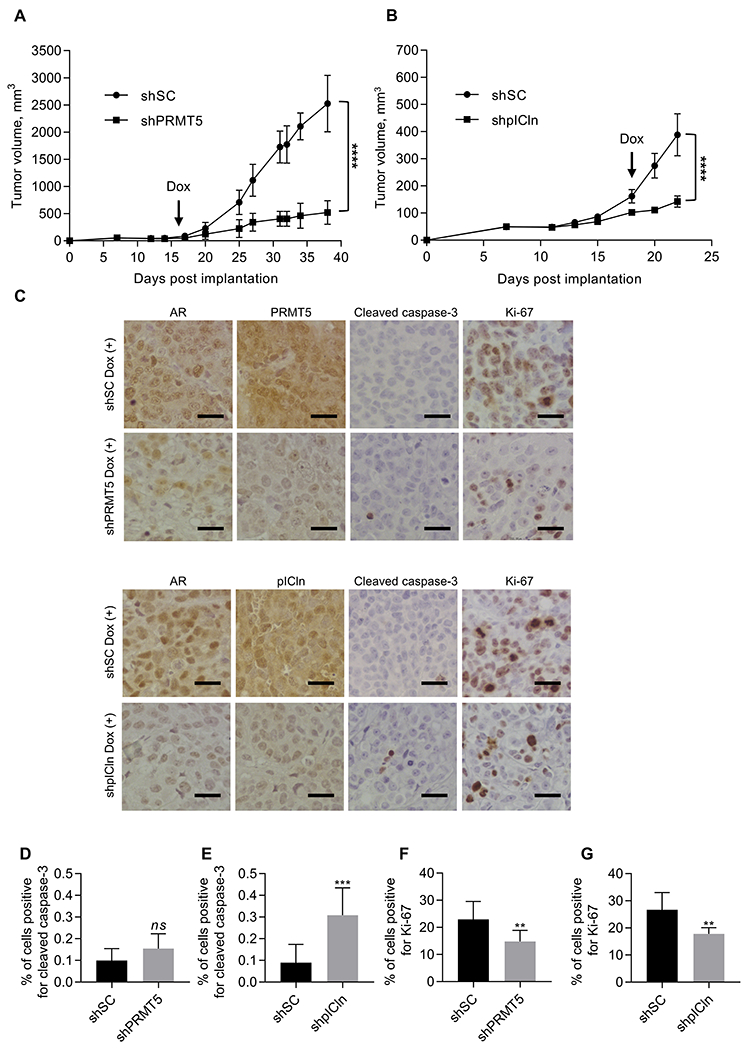
22Rv1 cells with Dox-inducible knockdown of PRMT5 (shPRMT5), pICln (shpICln) or scramble control (shSC) were injected subcutaneously into right flanks of surgically castrated male NRG mice. Tumor-bearing mice were treated with doxycycline in drinking water once tumors reached ~100 mm3. A-B, Tumor growth curves were determined and compared between treatment groups (ANOVA; ****, P < 0.0001). C-G, At the end of treatment, tumors were resected and probed for cleaved caspase-3 and Ki-67 using IHC. Presented are representative images (C) and the quantification of the percentage of positively stained cells out of the total number of cells (D-G). Scale bar indicates 40 μm. Results are mean ± SD (n = 10 per group). D-G, Student’s t-test was performed to determine statistical significance. ns P > 0.05, ** P < 0.01, *** P < 0.001.
Targeting PRMT5 overcomes resistance to ASI treatment in CRPC cells and tumors
As resistance to androgen signaling inhibitor (ASI) treatment remains a clinical challenge for CRPC, we examined whether targeting PRMT5 can overcome the resistance to ASI. Since intracellular androgen synthesis by PC cells is one of the AR reactivation mechanisms in CRPC, and 22Rv1 produces CYP17A1 (37), we also treated 22Rv1 cells with the CYP17A1 inhibitor abiraterone. First, we performed MTT assay using either PRMT5 enzymatic inhibitors (BLL3.3 or JNJ-64619178) or ASI (abiraterone or enzalutamide) alone, in combination, or vehicle (DMSO). Notably, the combinational treatment decreased cell growth more effectively than either of drugs alone (Fig. 7A). However, using the Chou-Talalay method and software CompuSyn (http://www.combosyn.com/) to analyze the drug interaction, the combinational indexes for BLL3.3/abiraterone and BLL3.3/enzalutamide pair were 0.91 and 0.92, and for JNJ-64619178/abiraterone and JNJ-64619178/enzalutamide were 0.94 and 0.91 (Supplementary Fig. S7), respectively, indicating that PRMT5 inhibition in combination with ASI can achieve additive effect.
Figure 7. Targeting PRMT5 overcomes resistance to ASI treatment in CRPC cells and tumors.
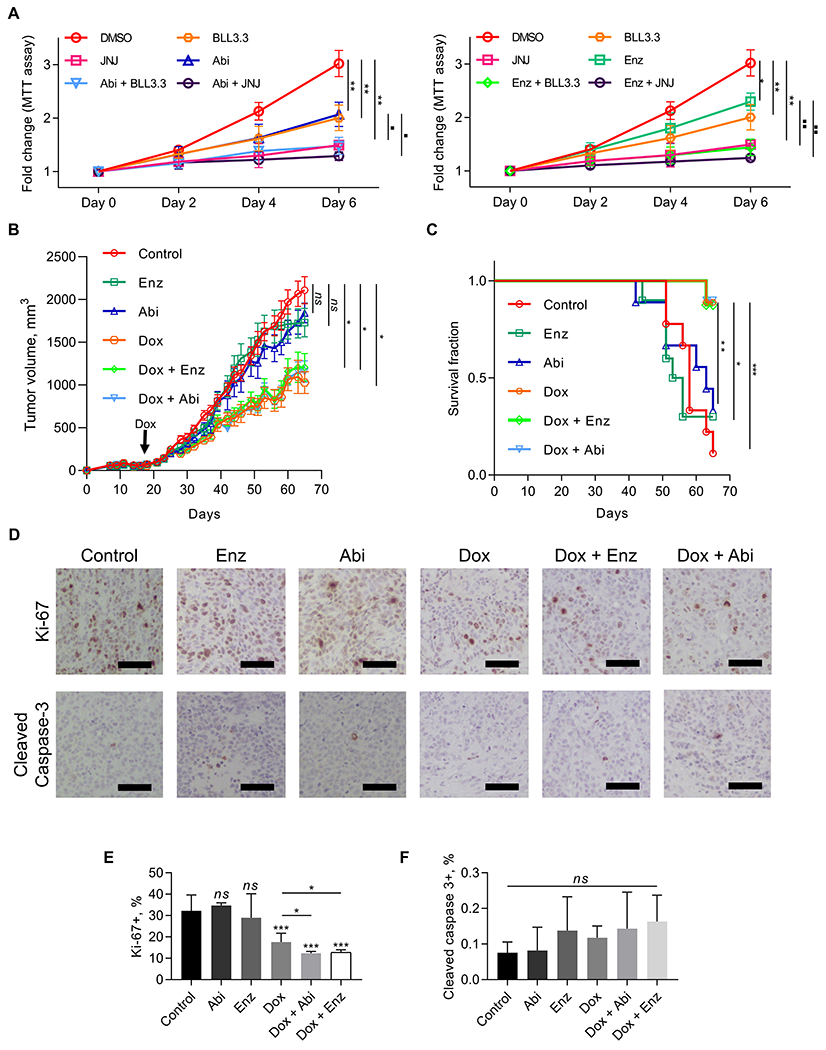
A, Growth curve (MTT assay) of 22Rv1 cells incubated with 10 µM PRMT5 inhibitor (BLL3.3 or JNJ-64619178, referred to as JNJ) or 10 µM of either abiraterone acetate (Abi) or enzalutamide (Enz), or equal volume of vehicle (DMSO) for 6 days. Cell proliferation assays were performed at the indicated time points, and OD550 values were normalized to values from Day 0 for each cell line. ANOVA test with Welch’s correction was performed to determine statistical significance. Stars represent significant difference with DMSO group, squares represent significant difference of “Abi” vs “Abi + BLL3.3”, “Abi vs Abi + JNJ”, “Enz” vs “Enz + BLL3.3”, or “Enz vs Enz + JNJ” groups. Results are mean ± SD from 3 independent experiments. B, 22Rv1 cells with Dox-inducible knockdown of PRMT5 were injected subcutaneously into right flanks of surgically castrated male NRG mice. Once tumors reached ~100 mm3, tumor-bearing mice were treated with doxycycline in drinking water, or abiraterone acetate per oral 200 mg/kg/day, or enzalutamide 25 mg/kg/day, or combination. Tumor growth curves were determined and compared between groups (ANOVA; *, P < 0.05). C, Survival of tumor–bearing mice is represented as Kaplan–Meier plot. D-F, At the end of treatment, tumors were resected and probed for cleaved caspase-3 and Ki-67 using IHC. Shown are representative images of IHC staining (D) and the quantified percentage of positively stained cells out of the total number of cells counted (E, F). Results are mean ± SD (n = 10 per group). Student’s t-test was performed to determine statistical significance of difference vs “Control” group. ns P > 0.05, *** P < 0.001, **** P < 0.0001.
Next, we implanted 22Rv1-shPRMT5 cells in the castrated male mouse to evaluate the observed in vitro effect. After average tumor volumes reached 100 mm3, PRMT5 knockdown was initiated (Dox), or treatment with ASI (abiraterone acetate or enzalutamide) started. Consistent with previous findings that 22Rv1 xenografts tumors in mice are resistant to ASI (38,39), treatment of mice with either drug alone did not affect tumor growth (Fig. 7B and C). However, PRMT5 knockdown significantly suppressed tumor growth and showed better survival. Although combinational treatment was not more effective than PRMT5 knockdown alone in terms of tumor growth suppression (Fig. 7B), Ki-67 analysis suggested that combination of PRMT5 knockdown with ASI showed a better inhibition of tumor cell proliferation (Fig. 7D, E). Thus, PRMT5 targeting alone is effective to overcome the resistance of CRPC tumors to ASI in mice. The lack of additive effect of PRMT5 knockdown and ASI on tumor growth in the xenograft model is likely due to the fact that PRMT5 knockdown and ASI both act on the same AR signaling pathway. Alternatively, incomplete knockdown of PRMT5 in xenograft tumors may be an attributing factor (Supplementary Fig. S8). Analysis of cleaved caspase-3 staining suggested no significant induction of apoptosis by ASI treatment or PRMT5 knockdown (Fig. 7F), confirming our in vitro findings. Taken together, these results suggest that PRMT5 targeting is an effective treatment approach for ASI-resistant CRPC.
Discussion
PRMT5 regulates AR signaling through multiple mechanisms
PRMT5 has emerged as a putative oncogene in multiple human cancers (8). Although earlier studies suggested that PRMT5 promotes proliferation of cancer cells via epigenetic repression of tumor suppressors (8–11), we recently reported that PRMT5 epigenetically activates AR transcription in HNPC (7). Because AR drives PC development and progression, we investigated whether PRMT5 also regulates AR expression in CRPC. We present evidence demonstrating that PRMT5 activates transcription of AR and AR-V7 in multiple CRPC cell lines. First, knockdown or pharmacological inhibition of PRMT5 in several CRPC cell lines (22Rv1, VCaP, C4-2 and LN95) reduced the expression of AR and AR-V7 at both mRNA and protein levels. Second, PRMT5 bound to the proximal promoter of AR and methylated H4R3 and H3R2. Third, transcriptomic analysis confirmed that PRMT5 regulated AR signaling in CRPC cells. Finally, PRMT5 expression positively correlates with the expression of AR and AR-V7 in CRPC tissues. Collectively, PRMT5 is overexpressed in PC tissues, and PRMT5-driven regulation of AR transcription is conserved in HNPC and CRPC cells.
PRMT5 may also regulate AR signaling through a non-epigenetic mechanism (40,41). TMPRSS2-ERG fusion is present in ~50% of PC cases, and AR-driven expression of this fusion promotes PC growth (42). In TMPRSS2-ERG-positive VCaP cells, mass spectrometry identified PRMT5 as an interacting protein of ERG (41). Mechanistically, ERG mediated both the methylation of arginine 761 on AR by PRMT5 and the recruitment of PRMT5 to the AR target gene promoters. PRMT5-catalyzed methylation of AR attenuated AR binding to a subset of AR target genes, resulting in transcriptional repression of genes associated with prostatic epithelium differentiation. Thus, PRMT5 promoted cell proliferation in TMPRSS2-ERG-positive cells. However, PRMT5 knockdown did not inhibit growth of TMPRSS2-ERG-negative 22Rv1 cells. This contrasts with our observations that both pharmacological inhibition and knockdown of PRMT5 significantly decreased proliferation of several CRPC cell lines, including TMPRSS2-ERG-negative 22Rv1 and C4–2. This discrepancy could be due to the use of heterogeneous pool of shRNA-expressing cells in their study whereas we used single-cell-derived stable clones that express shRNAs targeting different regions of PRMT5. In another study, ectopic overexpression of either PRMT5 or catalytically inactive PRMT5(R368A) mutant in TMPRSS2-ERG-negative PC3 cells enhanced luciferase activity of an androgen-responsive element-containing luciferase reporter (40), suggesting that PRMT5 might also function as a co-activator of AR independent of its methyltransferase activity. As PRMT5 may regulate PC cell growth via direct regulation of AR signaling and indirect modulation of other AR regulators, future investigation of these additional mechanisms will provide a full picture of PRMT5-driven regulation of PC cell growth. Further, genetic analysis for the role of PRMT5 in PC development and progression in mouse models will further validate PRMT5 as a therapeutic target for CRPC.
PRMT5 interacts with pICln to epigenetically activate AR transcription independently of MEP50
The finding that MEP50, a canonical cofactor of PRMT5 (8), did not participate in PRMT5 regulation of AR transcription in LNCaP and 22Rv1 cells was surprising. This led to the discovery of pICln as a potential cofactor of PRMT5 to activate AR transcription. Transcriptomic analysis of PRMT5, pICln and MEP50 target genes further confirmed that pICln, but not MEP50, cooperates with PRMT5 to regulate AR signaling in CRPC tissues (Fig. 4). Interestingly, pICln, but not MEP50, also cooperates with PRMT5 to activate transcription of multiple DNA damage response genes upon ionizing radiation (IR) (12). Thus, pICln rather than MEP50 might be required for the activation of PRMT5 target genes. In contrast, MEP50 might form a complex with PRMT5 and pICln to repress gene transcription. For example, IVL promoter was co-occupied by PRMT5, MEP50, and pICln, and knockdown of either MEP50 or pICln increased IVL expression (Fig. 2C) (12). Future studies may examine whether the co-occupancy of target gene promoters by PRMT5 and PRMT5-interacting proteins, e.g. MEP50, pICln, RIOK1, and COPR5, determines the transcriptional activation versus repression.
Several studies also demonstrated that PRMT5 may activate transcription of individual genes in a variety of tissues and conditions (12,39–47). Consistent with recent transcriptomic analysis in LNCaP cells showing that majority of identified DEGs (1136 out of 2035) was downregulated upon PRMT5 knockdown (12), similar number of upregulated and downregulated DEGs was identified in this study upon PRMT5 knockdown in 22Rv1 cells. Thus, PRMT5 likely functions as an epigenetic activator or repressor for different target genes. This notion is also supported by two recent transcriptomic studies in lung cancer cells A549 (48) and leukemia cells MOLM-13 (13). Because PRMT5 interacts with many chromatin remodelers (9), future studies focusing on the interplay between PRMT5 and PRMT5-interacting proteins including cofactors and other chromatin remodelers will likely shed new light on the epigenetic mechanism of PRMT5-mediated transcriptional regulation of target gene expression.
Targeting PRMT5 as a novel approach for PC treatment
Although targeting AR signaling remains a mainstay of CRPC treatment (4), the inevitable emergence of resistance via AR reactivation limits the therapeutic efficacy of ASI (5,6). Targeting AR protein expression instead may provide an alternative approach for the CRPC treatment and potentially overcome multiple AR reactivation mechanisms. In fact, targeting AR expression by promoting AR degradation effectively suppressed PC cell growth in several pre-clinical studies (49,50). One of the AR degraders utilizing PROTAC technology is in a Phase I clinical trial (51).
Given that epigenetic landscapes of CRPC and HNPC are largely distinct (52), the conserved role of PRMT5 in epigenetic activation of AR transcription in HNPC and CRPC is interesting and significant (7). As the vast majority of HNPC and CRPC demonstrate dependency on the AR signaling, targeting PRMT5 may offer an alternative or even more effective treatment for both HNPC and CRPC. In fact, PRMT5 targeting alone effectively suppressed CRPC growth (Fig. 7). Additionally, since AR reactivation promotes resistance to the next generation ASI, we explored whether targeting PRMT5 can overcome this resistance. Indeed, PRMT5 inhibition in combination with ASI showed additive suppression of CRPC cell growth in vitro. Interestingly, PRMT5 knockdown alone showed suppression of 22Rv1 xenograft tumor growth in mice as well as combination treatments (Fig. 7B, C), indicating that targeting PRMT5 might be an effective approach to overcoming resistance to ASI.
Three PRMT5 inhibitors are currently in clinical trials for leukemia and solid tumors (clinicaltrials.gov). As PRMT5 is an essential gene in normal organism processes, such as hematopoiesis and keratinocyte differentiation (53,54), targeting PRMT5 may cause adverse effects. If so, targeted prostate-specific membrane antigen-based delivery of PRMT5 inhibitors will likely provide an alternative to suppress AR expression in PC specifically (55). Alternatively, targeting PRMT5/pICln interaction may provide another promising approach for both HNPC and CRPC by suppressing or even eliminating AR expression.
Supplementary Material
Significance.
This study provides evidence that targeting PRMT5 can eliminate expression of AR and can be explored as a novel therapeutic approach to treat metastatic hormone-naïve and castration-resistant prostate cancer.
ACKNOWLEDGEMENTS
This work was supported by the following grants: U.S. Army Medical Research Acquisition Activity (W81XWH-16–10394), NCI RO1CA212403, Purdue University Center for Cancer Research Small Grants, Purdue Research Foundation Research Grant, Indiana University Simon Comprehensive Cancer Center (P30CA082709), Purdue University Center for Cancer Research (P30CA023168), Walther Cancer Foundation, and Indiana University Precision Health Initiative (PHI). J. L. Owens was supported by the Indiana Clinical and Translational Sciences Institute (CTSI) Pre-Doctoral Fellowship, which was made possible with partial support from Grant Numbers TL1 TR001107, TL1 TR002531, UL1 TR001108, and UL1 TR002529 (A. Shekhar, PI) from the NIH, National Center for Advancing Translational Sciences, Clinical and Translational Sciences Award. A. M. Asberry was supported by NIH T32 Grant NIH T32GM125620. We also thank Sandra Torregrosa-Allen and Melanie P. Currie for their technical support of animal studies.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest are disclosed.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30. [DOI] [PubMed] [Google Scholar]
- 2.Loh KP, Mohile SG, Kessler E, Fung C. Treatment of Metastatic Prostate Cancer in Older Adults. Curr Oncol Rep. 2016;18:63. [DOI] [PubMed] [Google Scholar]
- 3.Morris MJ, Bryan Rumble R, Basch E, Hotte SJ, Loblaw A, Rathkopf D, et al. Optimizing anticancer therapy in metastatic non-castrate prostate cancer: American society of clinical oncology clinical practice guideline. J Clin Oncol. 2018;36:1521–39. [DOI] [PubMed] [Google Scholar]
- 4.Chandrasekar T, Yang JC, Gao AC, Evans CP. Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl Androl Urol. 2015;4:365–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sciarra A, Gentilucci A, Silvestri I, Salciccia S, Cattarino S, Scarpa S, et al. Androgen receptor variant 7 (AR-V7) in sequencing therapeutic agents for castratrion resistant prostate cancer: A critical review. Medicine (Baltimore). 2019;98:e15608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu C, Brown LC, Antonarakis ES, Armstrong AJ, Luo J. Androgen receptor variant-driven prostate cancer II: advances in laboratory investigations. Prostate Cancer Prostatic Dis. 2020;3:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deng X, Shao G, Zhang H- T, Li C, Zhang D, Cheng L, et al. Protein arginine methyltransferase 5 functions as an epigenetic activator of the androgen receptor to promote prostate cancer cell growth. Oncogene. 2016;36:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stopa N, Krebs JE, Shechter D. The PRMT5 arginine methyltransferase: many roles in development, cancer and beyond. Cell Mol Life Sci. 2015;72:2041–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karkhanis V, Hu YJ, Baiocchi RA, Imbalzano AN, Sif S. Versatility of PRMT5-induced methylation in growth control and development. Trends Biochem Sci. 2011;36:633–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shailesh H, Zakaria ZZ, Baiocchi R, Sif S. Protein arginine methyltransferase 5 (PRMT5) dysregulation in cancer. Oncotarget. 2018;9:36705–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang Y, Bedford MT. Protein arginine methyltransferases and cancer. Nat Rev Cancer. 2013;13:37–50. [DOI] [PubMed] [Google Scholar]
- 12.Owens JL, Beketova E, Liu S, Tinsley SL, Asberry AM, Deng X, et al. PRMT5 Cooperates with pICln to Function as a Master Epigenetic Activator of DNA Double-Strand Break Repair Genes. iScience. 2020;23:100750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu F, Xu Y, Lu X, Hamard P- J, Karl DL, Man N, et al. PRMT5-mediated histone arginine methylation antagonizes transcriptional repression by polycomb complex PRC2. Nucleic Acids Res. 2020;48:2956–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ho MC, Wilczek C, Bonanno JB, Xing L, Seznec J, Matsui T, et al. Structure of the Arginine Methyltransferase PRMT5-MEP50 Reveals a Mechanism for Substrate Specificity. PLoS One. 2013;8:p.e57008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meister G, Eggert C, Bühler D, Brahms H, Kambach C, Fischer U. Methylation of Sm proteins by a complex containing PRMT5 and the putative U snRNP assembly factor pICln. Curr Biol. 2001;11:1990–4. [DOI] [PubMed] [Google Scholar]
- 16.Antonysamy S, Bonday Z, Campbell RM, Doyle B, Druzina Z, Gheyi T, et al. Crystal structure of the human PRMT5:MEP50 complex. Proc Natl Acad Sci U S A. 2012;109:17960–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pesiridis GS, Diamond E, Van Duyne GD. Role of pICLn in methylation of Sm proteins by PRMT5. J Biol Chem. 2009;284:21347–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Friesen WJ, Wyce A, Paushkin S, Abel L, Rappsilber J, Mann M, et al. A novel WD repeat protein component of the methylosome binds Sm proteins. J Biol Chem. 2002;277:8243–7. [DOI] [PubMed] [Google Scholar]
- 19.Kodama Y, Hu CD. Bimolecular Fluorescence Complementation (BiFC) Analysis of Protein-Protein Interaction. How to Calculate Signal-to-Noise Ratio. Methods Cell Biol. 2013;113:107–21. [DOI] [PubMed] [Google Scholar]
- 20.Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, et al. NCBI GEO: Archive for functional genomics data sets - Update. Nucleic Acids Res. 2013;41:D991–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio Cancer Genomics Portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:PL1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, et al. ONCOMINE: A Cancer Microarray Database and Integrated Data-Mining Platform. Neoplasia. 2004;6:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abida W, Cyrta J, Heller G, Prandi D, Armenia J, Coleman I, et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc Natl Acad Sci U S A. 2019;166:11428–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu Y, Song Q, Pascal LE, Zhong M, Zhou Y, Zhou J, et al. DHX15 is up-regulated in castration-resistant prostate cancer and required for androgen receptor sensitivity to low DHT concentrations. Prostate. 2019;79:657–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alinari L, Mahasenan K V., Yan F, Karkhanis V, Chung JH, Smith EM, et al. Selective inhibition of protein arginine methyltransferase 5 blocks initiation and maintenance of B-cell transformation. Blood. 2015;125:2530–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu T, Millar H, Gaffney D, Beke L, Mannens G, Vinken P, et al. Abstract 4859: JNJ-64619178, a selective and pseudo-irreversible PRMT5 inhibitor with potent in vitro and in vivo activity, demonstrated in several lung cancer models. Cancer Res. 2018;78:4859. [Google Scholar]
- 28.Hu R, Lu C, Mostaghel EA, Yegnasubramanian S, Gurel M, Tannahill C, et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012;72:3457–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu W, Xie CC, Zhu Y, Li T, Sun J, Cheng Y, et al. Homozygous deletions and recurrent amplifications implicate new genes involved in prostate cancer. Neoplasia. 2008;10:897–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu LL, Xie N, Sun S, Plymate S, Mostaghel E, Dong X. Mechanisms of the androgen receptor splicing in prostate cancer cells. Oncogene. 2014;33:3140–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pal S, Vishwanath SN, Erdjument-Bromage H, Tempst P, Sif S. Human SWI/SNF-Associated PRMT5 Methylates Histone H3 Arginine 8 and Negatively Regulates Expression of ST7 and NM23 Tumor Suppressor Genes. Mol Cell Biol. 2004;24:9630–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scoumanne A, Zhang J, Chen X. PRMT5 is required for cell-cycle progression and p53 tumor suppressor function. Nucleic Acids Res. 2009;37:4965–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saha K, Adhikary G, Eckert RL. MEP50/PRMT5 Reduces Gene Expression by Histone Arginine Methylation and this Is Reversed by PKCδ/p38δ Signaling. J Invest Dermatol. 2016;136:214–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guderian G, Peter C, Wiesner J, Sickmann A, Schulze-Osthoff K, Fischer U, et al. RioK1, a New Interactor of Protein Arginine Methyltransferase 5 (PRMT5), Competes with pICln for Binding and Modulates PRMT5 Complex Composition and Substrate Specificity. J Biol Chem. 2011;286:1976–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lacroix M, Messaoudi S El, Rodier G, Le Cam A, Sardet C, Fabbrizio E. The histone-binding protein COPR5 is required for nuclear functions of the protein arginine methyltransferase PRMT5. EMBO Rep. 2008;9:452–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gu Z, Li Y, Lee P, Liu T, Wan C, Wang Z. Protein arginine methyltransferase 5 functions in opposite ways in the cytoplasm and nucleus of prostate cancer cells. PLoS One. 2012;7:e44033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Giatromanolaki A, Fasoulaki V, Kalamida D, Mitrakas A, Kakouratos C, Lialiaris T, et al. CYP17A1 and Androgen-Receptor Expression in Prostate Carcinoma Tissues and Cancer Cell Lines. Curr Urol. 2019;13:157–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sekhar KR, Wang J, Freeman ML, Kirschner AN. Radiosensitization by enzalutamide for human prostate cancer is mediated through the DNA damage repair pathway. PLoS One. 2019;14:e0214670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu C, Armstrong C, Zhu Y, Lou W, Gao AC. Niclosamide enhances abiraterone treatment via inhibition of androgen receptor variants in castration resistant prostate cancer. Oncotarget. 2016;7:32210–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hosohata K, Li P, Hosohata Y, Qin J, Roeder RG, Wang Z. Purification and identification of a novel complex which is involved in androgen receptor-dependent transcription. Mol Cell Biol. 2003;23:7019–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mounir Z, Korn JM, Westerling T, Lin F, Kirby CA, Schirle M, et al. ERG signaling in prostate cancer is driven through PRMT5-dependent methylation of the androgen receptor. Elife. 2016;5:e13964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu J, Yu J, Mani RS, Cao Q, Brenner CJ, Cao X, et al. An Integrated Network of Androgen Receptor, Polycomb, and TMPRSS2-ERG Gene Fusions in Prostate Cancer Progression. Cancer Cell. 2010;17:443–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.LeBlanc SE, Konda S, Wu Q, Hu Y- J, Oslowski CM, Sif S, et al. Protein Arginine Methyltransferase 5 (Prmt5) Promotes Gene Expression of Peroxisome Proliferator-Activated Receptor γ2 (PPARγ2) and Its Target Genes during Adipogenesis. Mol Endocrinol. 2012;26:583–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang B, Dong S, Zhu R, Hu C, Hou J, Li Y, et al. Targeting protein arginine methyltransferase 5 inhibits colorectal cancer growth by decreasing arginine methylation of eIF4E and FGFR3. Oncotarget. 2015;6:22799–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fan Z, Kong X, Xia J, Wu X, Li H, Xu H, et al. The arginine methyltransferase PRMT5 regulates CIITA-dependent MHC II transcription. Biochim Biophys Acta - Gene Regul Mech. 2016;1859:687–96. [DOI] [PubMed] [Google Scholar]
- 46.Tarighat SS, Santhanam R, Frankhouser D, Radomska HS, Lai H, Anghelina M, et al. The dual epigenetic role of PRMT5 in acute myeloid leukemia: gene activation and repression via histone arginine methylation. Leukemia. 2015;30:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen H, Lorton B, Gupta V, Shechter D. A TGFβ-PRMT5-MEP50 axis regulates cancer cell invasion through histone H3 and H4 arginine methylation coupled transcriptional activation and repression. Oncogene. 2017;36:373–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Serio J, Ropa J, Chen W, Mysliwski M, Saha N, Chen L, et al. The PAF complex regulation of Prmt5 facilitates the progression and maintenance of MLL fusion leukemia. Oncogene. 2018;37:450–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cheng MA, Chou F- J, Wang K, Yang R, Ding J, Zhang Q, et al. Androgen receptor (AR) degradation enhancer ASC-J9 ® in an FDA-approved formulated solution suppresses castration resistant prostate cancer cell growth. Cancer Lett. 2018;417:182–91. [DOI] [PubMed] [Google Scholar]
- 50.Vanaja DK, Mitchell SH, Toft DO, Young CYF. Effect of geldanamycin on androgen receptor function and stability. Cell Stress Chaperones. 2002;7:55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mullard A First targeted protein degrader hits the clinic. Nat Rev Drug Discov. 2019;18:237–9. [DOI] [PubMed] [Google Scholar]
- 52.Braadland PR, Urbanucci A. Chromatin reprogramming as an adaptation mechanism in advanced prostate cancer. Endocr Relat Cancer. 2019;26:R211–35. [DOI] [PubMed] [Google Scholar]
- 53.Tan DQ, Li Y, Yang C, Li J, Tan SH, Chin DWL, et al. PRMT5 Modulates Splicing for Genome Integrity and Preserves Proteostasis of Hematopoietic Stem Cells. Cell Rep. 2019;26:2316–28. [DOI] [PubMed] [Google Scholar]
- 54.Kanade SR, Eckert RL. Protein arginine methyltransferase 5 (PRMT5) signaling suppresses protein kinase Cδ- and p38δ-dependent signaling and keratinocyte differentiation. J Biol Chem. 2012;287:7313–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wüstemann T, Haberkorn U, Babich J, Mier W. Targeting prostate cancer: Prostate-specific membrane antigen based diagnosis and therapy. Med Res Rev. 2019;39:40–69. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.